RETRACTED: Targeting posttranslational modifications of RIOK1 inhibits the progression of colorectal and gastric cancers
- Key Laboratory of Basic Pharmacology, Ministry of Education, Zunyi Medical College, China
- Zhongshan Hospital of Xiamen University, China
- Medical College of Xiamen University, China
- Xiamen Municipal Key Laboratory of Gastrointestinal Oncology, China
- Xiangya School of Medicine, Central South University, China
- Second Affiliated Hospital of Xinjiang Medical University, China
- Rutgers University, United States
- The First Affiliated Hospital of Harbin Medical University, China
- Hongqi Hospital, Mudanjiang Medical University, China
- Zhongshan Hospital of Xiamen University, Fujian, China
- The First Affiliated Hospital of Anhui Medical University, China
- The Third Affiliated Hospital of Harbin Medical University, China
- Rutgers University, The State University of New Jersey, United States
Abstract
RIOK1 has recently been shown to play important roles in cancers, but its posttranslational regulation is largely unknown. Here we report that RIOK1 is methylated at K411 by SETD7 methyltransferase and that lysine-specific demethylase 1 (LSD1) reverses its methylation. The mutated RIOK1 (K411R) that cannot be methylated exhibits a longer half-life than does the methylated RIOK1. FBXO6 specifically interacts with K411-methylated RIOK1 through its FBA domain to induce RIOK1 ubiquitination. Casein kinase 2 (CK2) phosphorylates RIOK1 at T410, which stabilizes RIOK1 by antagonizing K411 methylation and impeding the recruitment of FBXO6 to RIOK1. Functional experiments demonstrate the RIOK1 methylation reduces the tumor growth and metastasis in mice model. Importantly, the protein levels of CK2 and LSD1 show an inverse correlation with FBXO6 and SETD7 expression in human colorectal cancer tissues. Together, this study highlights the importance of a RIOK1 methylation-phosphorylation switch in determining colorectal and gastric cancer development.
https://doi.org/10.7554/eLife.29511.001Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths and one of the most common malignancies in the Western world (Labianca et al., 2013). Over the last decades, treatment strategies have significantly improved. However, CRC remains a high-risk gastrointestinal malignancy. And the occurrence of metastasis after the operation is the leading cause of poor prognosis for CRC patients (Yoshii et al., 2014). Therefore, it is of paramount importance to decipher the potential mechanisms underlying CRC growth and metastasis, which may contribute to the development of effective therapeutics for treating CRC patients.
Rio (right open reading frame) kinase is a relatively conserved family of atypical serine/threonine kinases (LaRonde-LeBlanc and Wlodawer, 2005). RIOK1 and RIOK2 are two members of the Rio family, named for yeast (S. cerevisiae) Rio1p and Rio2p, respectively (LaRonde-LeBlanc and Wlodawer, 2005). A third member named RIOK3 has greater similarly to RIOK1, but is only known to exist within multicellular eukaryotes (Yuan et al., 2014). Although these kinases contain a kinase fold structurally homologous to eukaryotic serine-threonine protein kinase domains, they lack substrate binding loops, an activation loop and their actual in vivo targets are unknown. (Mendes et al., 2015). Recently, the RNA Polymerase subunit Rpa43 in yeast has been identified as the first Rio1 substrate. Furthermore, Rpa43 is phosphorylated on a tyrosine residue, suggesting that RIO1 might be in fact a dual-specific kinase (Iacovella et al., 2015). RIOK1 knockout is lethal and was identified being an essential gene in yeast (Angermayr et al., 2002). Functionally, the yeast RIOK1 orthologue affects cell cycle progression and ribosomal biogenesis (Widmann et al., 2012). Additionally, RIOK1 is essential for the survival and reproduction of C. elegans (Weinberg et al., 2014; Mendes et al., 2015). However, the role of RIOK1 in multicellular organisms remains poorly understood. Recently, several studies have reported that the RIO kinases function in RTK and PI3K signaling pathway (Read et al., 2013), and are required for the survival of Ras-dependent cancer cells (Luo et al., 2009). One new study reported that RIOK1 was overexpressed in colon cancer cells and promoted cell proliferation in vitro in the context of human CRC (Weinberg et al., 2017). However, the exact mechanism remains unknown.
The posttranslational modification (PTM, such as phosphorylation, ubiquitination, and acetylation) of proteins is well-known to dynamically change protein function by fine-tuning protein stability, localization, or interactions (Jensen, 2006). PTMs of proteins rapidly and reversibly regulate cells in response to different stresses. Therefore, once demonstrated, these PTMs could potentially serve as therapeutic targets (Krueger and Srivastava, 2006). Among various posttranslational modifications, lysine methylation acts as a novel regulatory mechanism to control protein functions (Oudhoff et al., 2013). However, most previous studies have predominantly highlighted histone methylation, until recently accumulating evidence indicates the widespread presence of lysine methylation in nonhistone proteins (Patel et al., 2011). Although there are about 50 lysine methyltransferases in mammals, lysine methylation is primarily catalyzed by a family of protein methyltransferases containing a catalytic SET domain (Dillon et al., 2005). Su(var)3–9, enhancer-of-zeste, trithorax (SET) domain-containing protein 7 (SET7) which is also known as SETD7, SETD9, or SETD7/9, and acts on histone H3K4, has been shown to monomethylate various non-histone proteins including Gli3, FOXO3a, p53, HIF-1α, TAF10, and Suv39h1 (Fu et al., 2016; Kim et al., 2016; Couture et al., 2006; Wang et al., 2013). This methylation produces varied results. For example, methylation of Gli3 by SET7 stabilizes Gli3, whereas methylation of HIF-1α stimulates HIF-1α proteasomal degradation (Fu et al., 2016; Kim et al., 2016). Besides, there is increasing evidence that other PTMs may parallel methylation in the regulation of various biological processes, which provide a sophisticated level of crosstalk (Bigeard et al., 2014). Although RIOK1 can be regulated by phosphorylation (Angermayr et al., 2007), the enzymes responsible for other PTMs of RIOK1 remain elusive.
Given that many kinases are largely regulated by PTMs (Oliveira and Sauer, 2012), we speculate that RIOK1 may be regulated by methylation, or phosphorylation, or ubiquitination. The aims of this project were to examine the PTMs and role of RIOK1 in CRC and GC. Our findings definitely will further deepen the understanding of the crosstalk of PTMs signaling, expand the existing knowledge of the RIOK1, and provide a novel mechanism by which RIOK1 regulates tumorigenesis and metastasis.
Results
High-level RIOK1 is closely associated with an aggressive CRC and poor overall survival
To determine the role of RIOK1 in the pathogenesis of CRC, we firstly investigated the levels of RIOK1 in a group of 5 CRC patient tissues and matched normal tissues. As shown in the Figure 1A, compared with the corresponding non-tumor samples, RIOK1 was strongly upregulated in CRC tissues. The scatter diagram demonstrated the increase of RIOK1 mRNA levels in CRC and metastasis lymph node samples versus normal tissues, with an average 4.03-fold and 6.15-fold increase respectively (Figure 1B). To confirm the increased RIOK1 protein expression in a larger sample group, and correlate this to clinical phenotype, we performed immunohistochemical staining (IHC) on the CRC tissue array comprised of 120 patients. IHC demonstrated that CRC tissues showed higher expression of RIOK1 compared to matched normal tissues (Figure 1C1), and that the percentage of cells expressing RIOK1 were 25%, 52.2%, 67.7%, and 87.8% in cancer stage I, II, III, and IV of CRC, respectively (Figure 1C2), revealing that RIOK1 expression correlates with CRC malignancy. Importantly, Kaplan–Meier analysis indicated that high levels of RIOK1 expression are significantly correlated to overall survival (OS; p=0.003) and disease-free survival (DFS; p=0.001) (Figure 1D, Supplementary file 1). Besides, we also observed an increased expression of RIOK1 in gastric cancer (GC) tissues (Figure 1—figure supplement 1). Collectively, our data show that the RIOK1 expression is frequently upregulated in CRC and GC, and correlated with poor prognosis, suggesting that RIOK1 may function as an oncogene in CRC development.
Figure 1 with 1 supplement see all
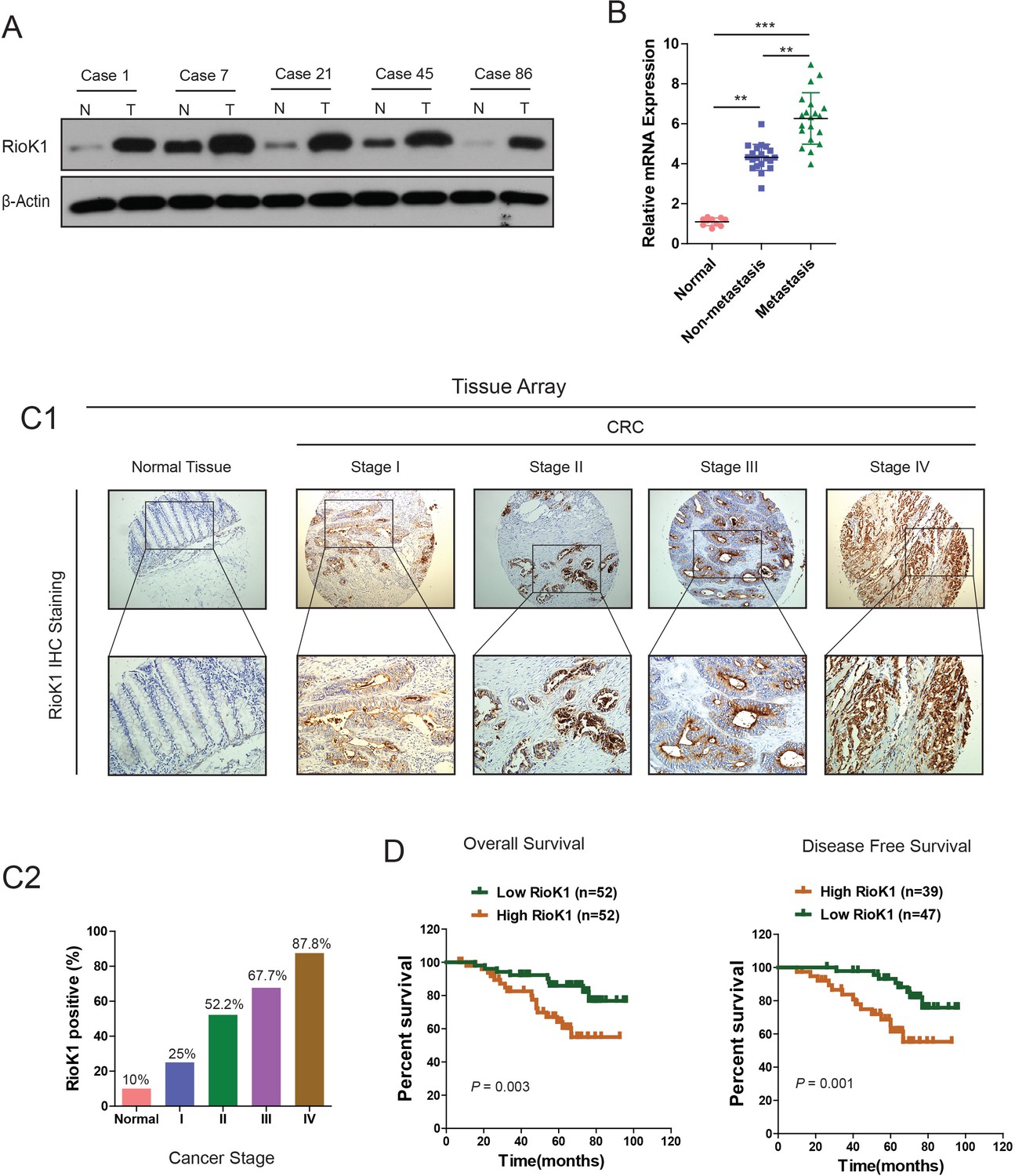
RIOK1 is significantly upregulated in CRC and associated with an aggressive and poor survival.
(A) RIOK1 expression in five paired human CRC biopsies and matched normal mucosa analyzed by Western-blot. (B) Comparison of RIOK1 mRNA expression level in human CRC tissues (with and without metastasis) and matched normal mucosa. RIOK1 mRNA expression was quantified by qPCR and normalized to the matched adjacent normal tissues. (C1) IHC analysis of RIOK1 on a tissue micro array of CRC patients (n = 110) and healthy adjacent tissue (n = 10) using the Allred score. (C2) The IHC signals were scored as 0, 1, 2, and 3; a score ≧1 + indicated positive detection. (D) Kaplan-Meier curves for overall survival and disease free survival of 104 and 86 CRC patients stratified by RIOK1 expression respectively.
RIOK1 promotes the proliferation, invasion, and metastasis of CRC and GC cells in vitro and in vivo
Having observed the association of RIOK1 expression with poor survival in CRC patients, we set out to functionally characterize the effects of RIOK1 on CRC cells. Firstly, we examined the endogenous RIOK1 levels in different CRC cell lines and treated these cell lines respectively using lentivirus-mediated RIOK1-specific short hairpin (sh) RNAs knockdown or by overexpression of a RIOK1 cDNA. It was observed that the levels of endogenous RIOK1 were significantly higher in CRC cell lines than in normal intestinal epithelial cells (IECs) (Figure 2A). Then three RIOK1-specific shRNAs (shRIOK1) were transfected to silence the endogenous RIOK1 expression of CRC cells. According to the western blot analysis, the shRIOK1#1 with the highest inhibition rate was selected for further functional experiments (Figure 2B). Both CCK-8 and colony formation assays revealed that down-regulation of RIOK1 significantly inhibited the proliferation rate of HCT116 cells compared to negative control (Figure 2C and D). As shown in Figure 2G, both migration and invasion were also obviously inhibited in RIOK1 knock-down HCT116 cells. To rule out the possibility of off-target effects, we constructed a shRNA-resistant RIOK1 lentiviral vector, RIOK1Δ, via site-directed mutagenesis. In HCT116-shRIOK1#1 cells, the recovery of RIOK1 expression with this shRNA-resistant vector remarkably restored the high proliferation and metastatic ability of the cells (Figure 2C,D and E). Conversely, overexpression of RIOK1 in RKO had the opposite effects on cell viability (Figure 2F and G), migration and invasion (Figure 2H). Similar data were observed in SW480 cells transfected with shRIOK1#1 and LOVO cells with RIOK1 overexpression respectively (data not shown). In summary, the above results illustrated that RIOK1 might be critical for the proliferation, invasion and metastasis of CRC cells.
Figure 2 with 2 supplements see all
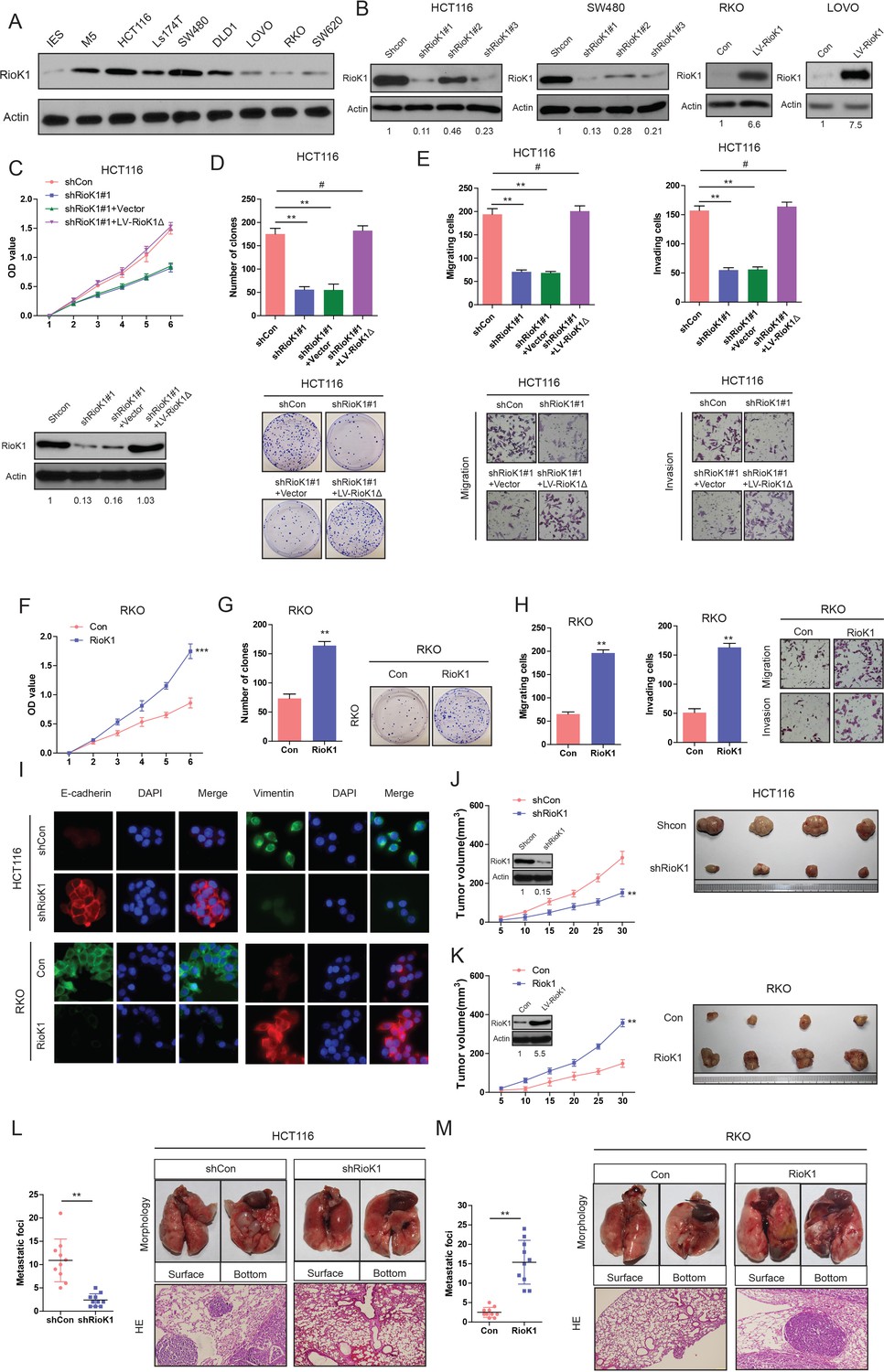
RIOK1 promotes growth and metastasis of CRC cells in vitro and in vivo.
(A) The expression levels of RIOK1 in 7 CRC cell lines and normal intestinal epithelial cells (IECs) were analyzed by Western blot. (B) Western blot for RIOK1 in HCT116 and SW480 cells infected with shRIOK1, or control shRNA lentiviral vector, as well as in RKO and LOVO infected with the RIOK1-expressing or empty control vector (n = 4).And the intensity of the western blot bands was quantified using NIH ImageJ software. (C) Western blot for RIOK1 in the indicated HCT116 cells transfected with shRIOK1#1 or the shRNA-resistant expression construct, RIOK1Δ. RIOK1 expression was recovered in the HCT116-shRIOK1 cells transfected with RIOK1Δ (lower-panel).And the intensity of the western blot bands was quantified using NIH ImageJ software. RIOK1Δ almost restored the proliferative ability of the cells (upper-panel). (D) RIOK1 knockdown inhibited proliferation of HCT116 cells determined by CCK-8 and colony formation assays. (E) RIOK1 knockdown inhibited migration and invasion of HCT116 cells determined by transwell assays. (F) and (G) Ectopic expression of RIOK1 promoted proliferation of RKO cells determined by CCK-8 and colony formation assays. (H) Ectopic expression of RIOK1 promoted migration and invasion of RKO cells determined by transwell assays. (I) Representative immunofluorescence images demonstrated that RIOK1 level has an effect on the expression of EMT proteins in CRC cells. (J) Growth curve of subcutaneous injection of HCT116/Scramble and HCT116/shRIOK1 in NOD/SCID mice (n = 6 per group, left panel). NOD/SCID mice were injected subcutaneously into opposite flanks with 1.5 × 106 control cells and RIOK1-knocked down HCT116 cells. The mice were sacrificed and the tumors were then removed, weighed and compared (right panel). And knockdown of RIOK1 in a xenograft tumor model was analyzed with western blotting, and the intensity of the western blot bands was quantified using NIH ImageJ software. (K) Growth curve of subcutaneous injection of RKO/Vector and RKO/RIOK1 in NOD/SCID mice (n = 6 per group, left panel). The tumors were then removed, weighed and compared (right panel). Over-expression of RIOK1 in a xenograft tumor model was analyzed with western blotting, and the intensity of the western blot bands was quantified using NIH ImageJ software. Effects of RIOK1 Knockdown (L) or overexpression (M) on lung metastasis of indicated cells in NOD/SCID mice (n = 10 per group): the number of metastatic nodules in the lung (left-panel); representative morphological observation of lung metastases (right-upper panel); and histopathological observation of lung sections (right-lower panel). The results are presented as are means ±SD (n = 3 for each panel). Statistical significance was concluded at *p<0.05, **p<0.01, ***p<0.001; # represents no statistical significance.
Interestingly, a notable change was the expression of epithelial-mesenchymal transition (EMT) proteins in RIOK1-overexpressing cells. Cells with relatively high RIOK1 expression displayed a mesenchymal-like phenotype, and the expression of vimentin (a mesenchymal marker) was enhanced, while RIOK1 knockdown strongly upregulated the expression of E-cadherin (an epithelial marker) (Figure 2I).
Importantly, an in vivo tumor formation assay demonstrated that RIOK1 knockdown significantly inhibited the tumorigenesis of CRC cells compared with the control (Figure 2J). Conversely, RIOK1 overexpression enhanced the RKO cell growth rate (Figure 2K). Moreover, down-regulation of RIOK1 significantly decreased CRC metastatic foci in the lung (Figure 2L) whereas overexpression of RIOK1 increased metastatic nodules (Figure 2M). As expected, we observed very similar phenotypes in GC cell lines (Figure 2—figure supplement 1A,B,C,D,E,F and G). Taken together, these results indicate that RIOK1 promotes growth and metastasis of CRC and GC in vitro and in vivo.
RIOK1 promotes CRC and GC cell proliferation and migration through PI3K/AKT pathway
Previous studies in Drosophila have indicated that RIOK1/2 form a complex with mTOR, which promotes glioblastoma initiation via activating Akt signaling pathway (Read et al., 2013). To identify if the functions of elevated RIOK1 expression in CRC and GC cells depend on PI3K/AKT signaling,
CRC and GC cells were incubated with LY294002—an inhibitor of PI3 kinase, or A443654—an inhibitor of Akt kinase. Quantification of the results showed that both proliferation and invasion of RIOK1-transfected cells was significantly reduced in LY294002 or A443654 treated cultures (Figure 2—figure supplement 2A). Meanwhile, treatment of cells with LY294002 or A443654 led to a significant reduction of the levels of Akt phosphorylated at Thr308 and Ser473 (Figure 2—figure supplement 2B). In parallel, E-cadherin and vimentin, which may account for reversal of epithelial-mesenchymal transition (EMT), were down-regulated and up-regulated respectively in inhibitors-treated cells (Figure 2—figure supplement 2B), further demonstrating the involvement of PI3K/AKT cascade in the regulation of CRC and GC cells by RIOK1.
Conversely, overexpression of constitutively active myristylated Akt (myrAkt), abrogated the inhibitory effects of RIOK1 knockdown on proliferation and invasion when compared with of empty vector, or the domain-negative Akt (DN-Akt) transfected cells (Figure 2—figure supplement 2C). And the opposite effects on the levels of phosphorylated AKT and EMT-related protein were observed (Figure 2—figure supplement 2D1 and D2). To confirm the correlation between RIOK1 and PI3K/AKT cascade in CRC and GC, we performed immunohistochemistry (IHC) for RIOK1 and Akt phosphorylated at Serine-473 on a cohort of CRC and GC specimens. It was found that high RIOK1-expressing specimens always showed strong staining for p-Akt-S473 (Figure 2—figure supplement 2E). These findings further confirmed that PI3K/AKT signaling pathway might contribute to the pro-cancer effects of RIOK1 in CRC and GC cells.
RIOK1 interacts with SETD7 in vitro and in vivo
Given that the activity and biologic role of many kinases are largely dependent on its PTMs (Oliveira and Sauer, 2012), and that the previous study implies the involvement of PTMs in regulating the activity and stability of RIOK1 (Angermayr et al., 2002), we sought to decipher whether RIOK1 can be post-translationally modified. To this end, we performed a mass spectrometry analysis of Flag-tagged RIOK1 from the cell lysate of HEK293T. Intriguingly, SETD7 was identified as a RIOK1-interacting protein from LC-MS/MS analysis (Supplementary file 2), suggesting the possible involvement of SETD7 in methylation of RIOK1.
Firstly, we estimated their physical interaction using an immunoprecipitation (IP) assay in cultured cells. The interaction between GFP-tagged-SETD7 and His-tagged-RIOK1 was detected physiologically in RKO cells by using an anti–His antibody followed by Western blot analysis with an anti-GFP antibody (Figure 3—figure supplement 1A). Again, a reciprocal co-IP assay performed by precipitation with an anti-GFP antibody indicated an interaction between SETD7 and RIOK1 (Figure 3—figure supplement 1A). Co-IP assay also confirmed the binding of SETD7 to RIOK1 at endogenous expression levels in RKO cells (Figure 3—figure supplement 1B). The GST pull-down assay was performed to further confirm the SETD7 interacted GST-RIOK1, but not with GST alone (Figure 3—figure supplement 1C).
To further map the domains of SETD7 for RIOK1 binding, we constructed and purified the Full length (FL) and several fragments of GST-SETD7 and then performed a GST pull-down assay. It was observed that both the N-terminal and middle fragments of SETD7 bound to RIOK1 (Figure 3—figure supplement 1D). Next, the regions of RIOK1 for SETD7 binding from the GST pull-down assay were mapped by incubating the FL or fragments of GST-RIOK1 with His-SETD7. His-SETD7 mainly interacted with the FL and the 1–120 aa fragment of GST-RIOK1, but not with the C-terminal fragment of RIOK1 and GST alone (Figure 3—figure supplement 1E). All of the above data indicate that SETD7 directly interacts with RIOK1.
SETD7 monomethylates RIOK1 on lysine 411 in vitro and in vivo
Given that SETD7 has recently been shown to methylate several nonhistone proteins and be preferential for nonhistone proteins, we hypothesized that RIOK1 is methylated by SETD7. To test this idea, we firstly examined whether SETD7 interacts with and methylates RIOK1 in Setd7−/− and Setd7+/+ MEFs. Consistent with findings described above, co-IP of native RIOK1 from Setd7+/+ MEFs cultured at high density confirmed that SETD7 was in a complex with RIOK1 (Figure 3A). When RIOK1 was immunoprecipitated and analyzed by immunoblotting with two distinct anti-methyl lysine antibodies (ab23366, recognizing both mono- and dimethylated lysine, and ab7315, primarily recognizing dimethylated lysine), we found that RIOK1 is monomethylated in Setd7+/+ MEFs but not in Setd7−/− MEFs (Figure 3A). These results suggest that SETD7 may directly monomethylate RIOK1.
Figure 3 with 2 supplements see all

SETD7 monomethylates RIOK1 on lysine 411 in vitro and in vivo.
(A) Anti-RIOK1 immunoprecipitates from high-density Setd7+/+ and Setd7−/− MEFs were immunoblotted for RIOK1, SETD7, methyl-lysine (mono-, dimethylated) (ab23366), and dimethyl-lysine (ab7315) antibodies, respectively. (B) SETD7 methylates RIOK1 in vivo. Stably expressed control, SETD7-WT, or SETD7-H297A HEK293T cells were generated. The cell lysates were then precipitated with an anti–RIOK1 antibody and probed with an anti–pan-me-K antibody to detect methylation of RIOK1 (left panel). Stably expressed shcontrol or sh-SETD7 HEK293T cells were generated. The cell lysates were then precipitated with an anti–RIOK1 antibody and probed with an anti–pan-me-K antibody to detect methylation of RIOK1 (right panel). (C1) An in vitro methylation assay was performed with 3H-SAM, recombinant SETD7, and RIOK1. Autoradiography and Coomassie brilliant blue R-250 staining (CBB) were used to show methylation and protein levels, respectively. (C2) In vitro methylation analysis using indicated proteins. An anti–pan-methyl-lysine antibody (pan-me-K) was used to detect the methylation of RIOK1. CBB was used to show the protein levels. (D) Alignment of the consensus amino acid residues adjacent to lysine targeted by SETD7, and identification of a putative SETD7 methylation site in RIOK1 (upper panel). The consensus SETD7 recognition sequence (middle panel). The lysines targeted for methylation by SET7 in known substrates are shown (lower panel). In each case, the methylated lysine is shown in red. (E) In vitro methylation of various purified RIOK1 mutants by SETD7. (F1) An anti-RIOK1-K411me1-specific antibody is characterized. RIOK1 peptide substrates: 400–414 SKAMEIASQRTKEER(Lane 1), K411me SKAMEIASQRTKmeEER(Lane 2), T410p SKAMEIASQRTpKEER(Lane 3), p: phosphorylation; me: methylation. The above peptides were immunoblotted with indicated antibodies. (F2) GST-RIOK1 (1 μg) was methylated in vitro in the presence or absence of recombinant SETD7 (0.5 μg). The reaction was probed with the anti-RIOK1-K411me1 antibody, in the presence of 10 μg/ml competing wild-type (wt) or mono-methylated K411 peptide (me). (G) The effect of ectopically expressed SETD7 and SETD7-H297A on K411me1 of endogenous RIOK1 in HEK293T cells. (H) Methylation analysis of Flag-RIOK1 and mutants by SETD7 in transfected HEK293T cells. (I) In vitro methylation was performed with GST-RIOK1, S-adenosylmethionine (SAM), recombinant His-SETD7, and His-SET7-H297A. Immunoblotting and CBB were used to show methylation and protein levels, respectively.
To further examine whether the methylation of RIOK1 is mediated by SETD7, HEK293T cell lines were transfected with pcDNA (HEK293T-pcDNA), wild-type SETD7 (HEK293T-SETD7-WT), or methylase-deficient mutant SETD7 (HEK293T-SETD7-H297A), and endogenous RIOK1 from the cell lines was immunoprecipitated and subjected to Western blot analysis with an anti–pan-methyl-lysine antibody. The protein levels of the methylated RIOK1 were significantly increased in the HEK293T-SETD7-WT cell line, but the same phenomenon was not observed in the HEK293T-SETD7-H297A cell line (Figure 3B, left-panel). Conversely, the methylation of RIOK1 was almost abolished in the HEK293T-shSETD7 cells compared with the HEK293T -shcontrol cells (Figure 3B, right-panel). Together, these experiments demonstrate that SETD7 methylates RIOK1 in vivo.
To investigate whether RIOK1 is methylated directly by SETD7, an in vitro methylation assay was performed by incubating SETD7 with 3H-SAM. As shown in Figure 3C1, GST-RIOK1 was methylated by SETD7, but not by a catalytic mutant SETD7-H297A (Figure 3C2), which confirms that SETD7 methylates RIOK1 in vitro.
Next, we sought to identify the SETD7-methylated residue(s) in RIOK1. The amino acid sequence of RIOK1 was compared with the sequences surrounding the SETD7 methylation sites of several known substrates (Figure 3D). It was found that the conserved lysine residue at position 411 and its neighboring residues of RIOK1 strongly conform to the consensus SETD7 recognition sequence designated by K/R-S/T/A-K motif (in which the methylation lysine site is underlined). When the full amino acid sequences of RIOK1 proteins of various species were aligned, it was observed that the identified RIOK1 methylation sequence was evolutionarily conserved (Figure 3D). Moreover, K411R mutation almost completely abolished RIOK1 methylation by SETD7 in vitro, whereas other mutation such as K401R did not affect (Figure 3E). Together, our data suggest that K411 is most likely the major, if not the sole, site within RIOK1 that is methylated by SETD7.
To easily find whether methylation of RIOK1 occurred in cells, we prepared a modification-specific antibody, anti-RIOK1-K411me1, which specifically recognized monomethylated RIOK1-K411 but not the unmethylated peptide (Figure 3F1 and Figure 3—figure supplement 2). This antibody detected methylated GST-RIOK1 after treatment with SETD7, with minimal reactivity against untreated GST-RIOK1 (Figure 3F2). Further, the unmodified RIOK1 peptide poorly prevented the anti-RIOK1-K411me1 antibody binding to methylated GST- RIOK1, whereas the methylated peptide prevented binding of the anti-RIOK1-K411me1 antibody (Figure 3F2).
Using this RIOK1-K411me1-specific antibody, it was observed that ectopic expression of V5- SETD7 in the HEK293T cells led to substantially increased endogenous RIOK1-K411me1, while the enzymatic deficient SETD7-H297A mutant reduced the level of RIOK1-K411me1 (Figure 3G). Furthermore, V5-SETD7 catalyzed K411me1 for Flag-RIOK1-WT and Flag-RIOK1-K401R mutant, but not K411R mutant (Figure 3H). The specificity of anti-RIOK1-K411me1 toward RIOK1 was further confirmed using an in vitro cell-free methylation reaction with purified recombinant GST-RIOK1 and His-SETD7 in the presence of S-adenosylmethionine (Figure 3I). Collectively, these results demonstrate that SETD7 methylates RIOK1 at K411 both in vitro and in vivo.
Methylation of RIOK1 by SETD7 significantly decreases its stability
Lysine methylation is usually linked to the stability of nonhistone, including RelA, DNMT1, Sox2, p53, ERα, RelA, and FoxO3 (Couture et al., 2006; Wang et al., 2013; Estève et al., 2011; Li et al., 2008). Next, we tested whether the methylation of RIOK1 by SETD7 affects its stability in cells. It was found that SETD7 overexpression decreased the expression of RIOK1-WT but not of RIOK1-K411R in HEK293T cells (Figure 4A), and the mRNA levels of RIOK1 were not changed (Figure 4B), suggesting that the methylation of RIOK1 may disrupt its stability. In HEK293T-shcontrol cells, the half-life of endogenous RIOK1 was 4–8 hr, while in HEK293T-sh-SETD7-1 cells it was dramatically prolonged (Figure 4C and D). To evaluate whether this change in the stability of RIOK1 depends on its methylation at K411, the effect of SETD7 on the half-life of RIOK1-WT and –K411R was investigated. Consistently, the half-life of RIOK1-WT was decreased from 4 to 8 hr to <4 hr when RIOK1-WT was coexpressed with SETD7 (Figure 4E and F). Conversely, RIOK1-K411R was more resistant to SETD7-induced degradation, with a much longer half-life (>8 hr) (Figure 4E and F). Collectively, our data demonstrate that the methylation of RIOK1 at K411 by SETD7 reduces its stability.
Figure 4
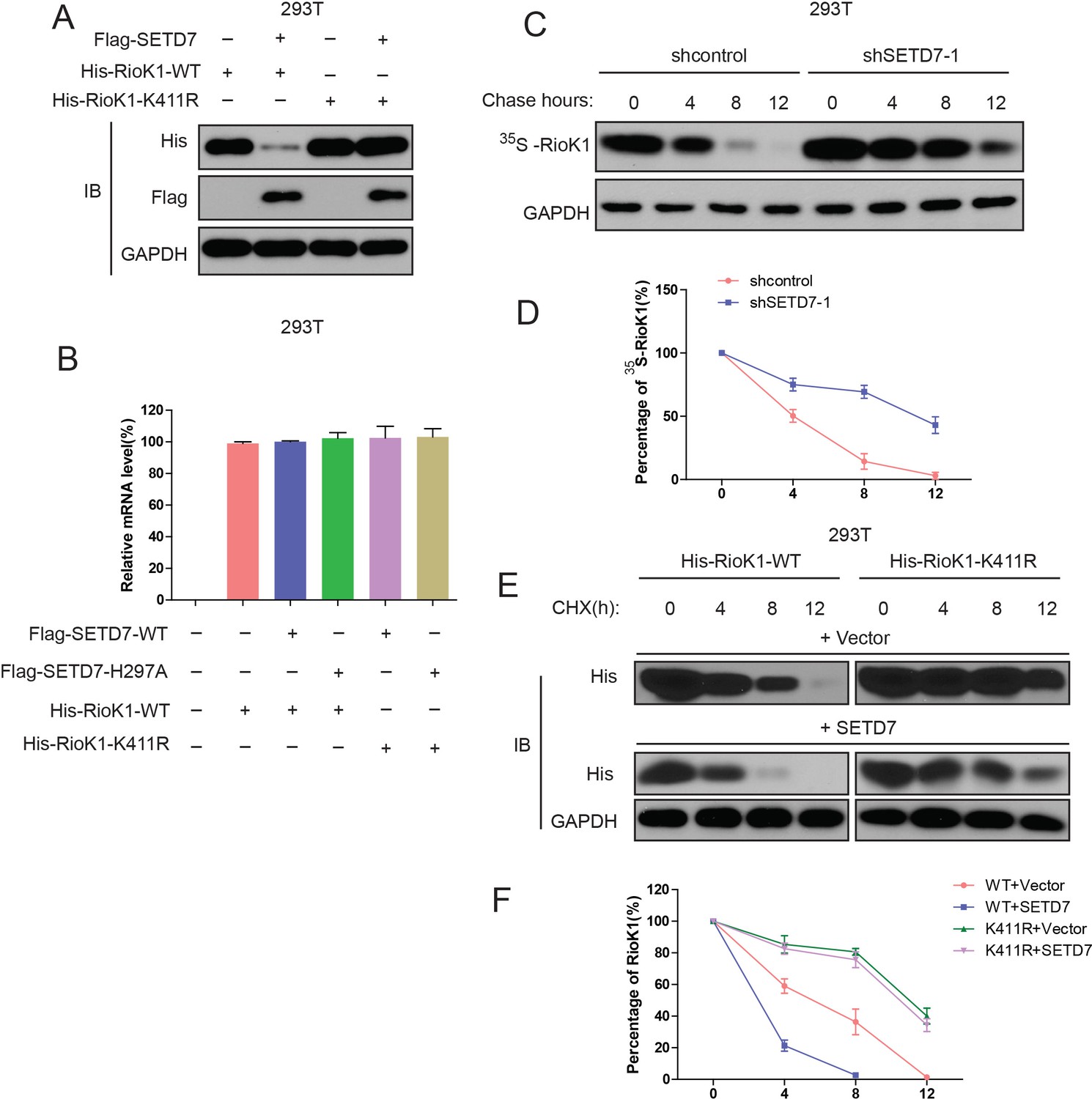
SET7/9 significantly reduces the stability of RIOK1.
(A) His-RIOK1-WT or His-RIOK1-K411R was cotransfected into HEK293T cells for 48 hr with or without Flag-SET7/9. Cell lysates were examined by western blot with indicated antibodies. (B) His-RIOK1-WT was cotransfected with or without Flag-SET7/9 or mutant Flag-SET7/9-H297A into HEK293T cells for 48 hr. Real-time PCR was used to detect the mRNA levels of RIOK1. (C) HEK293T -shcontrol and HEK293T -sh-SET7/9-1 cells were metabolically labeled with [35S]-methionine, as indicated, and followed by chasing with standard medium. Total lysates were examined with anti-RIOK1 antibody. (D) Quantification analysis of endogenous RIOK1 levels in (C). (E) HEK293T cells were transfected with indicated plasmids, and 24 hr later, treated with cycloheximide (CHX), and probed with anti-His antibody. (F) Quantification analysis of the results in (E).
RIOK1 demethylation by LSD1 increases RIOK1 stability
Our mass spectrometric analysis of the RIOK1 immunocomplex from HEK293T cells uncovered that lysine-specific demethylase 1 (LSD1), also known as KDM1A, one component of a BRAF–HDAC complex (BHC) histone deacetylase complex (Iwase et al., 2004), was present in the immunocomplex (Supplementary file 2). Given that LSD1 can demethylate non-histone proteins p53 and Dnmt1 (Jin et al., 2013; Nicholson and Chen, 2009), we hypothesized that LSD1 might reverse SETD7-induced RIOK1 methylation. Indeed, endogenous RIOK1 was observed to be present in LSD1 immunocomplex, and endogenous LSD1 was also present in the RIOK1 immunocomplex in 293T and HCT116 cells (Figure 5A and Figure 5—figure supplement 1). To rule out the possibility of a false-positive interaction resulting from cross-reactivity of antibodies, we demonstrated that Flag-RIOK1 physically associated with HA-LSD1 in 293T cells in the reciprocal co-immunoprecipitation assays (Figure 5B). When GST-RIOK1 and His-LSD1 were mixed, it was observed that GST-RIOK1 especially pulled down His-LSD1 (Figure 5C), indicating that LSD1 directly binds RIOK1.
Figure 5 with 1 supplement see all

SET7/9-mediated methylation of RIOK1 is reversed by LSD1, which increases RIOK1 stability.
(A) Endogenous RIOK1 co-immunoprecipitated reciprocally with endogenous LSD1 from HEK293T cells and subjected to immunoprecipitation followed by immunoblotting with indicated antibodies. (B) HA-RIOK1 co-immunoprecipitated reciprocally with Flag-LSD1. Total cell lysates were extracted from HEK 293T cells transiently cotransfected with expression constructs as indicated and probed with antibodies as indicated. (C) RIOK1 directly interacted with LSD1. Bacterially produced GST-RIOK1 was used to pull down bacterially produced His-LSD1 in vitro. (D) RIOK1 protein levels in Kdm1a+/− and Kdm1a−/− MEFs were compared. (E) Kdm1a−/− MEFs were reconstituted with either WT or a catalytically inactive Mutant (K661A) of LSD1. RIOK1 protein levels were monitored. (F) Kdm1a+/−MEFs were pretreated with pargyline for 12 hr, RIOK1 protein levels were evaluated. (G) Overexpressing LSD1 with MG132 treatment significantly decreases RIOK1 methylation in HEK293T cells. (H) LSD1 reverses SET7/9-dependent RIOK1 methylation. In vitro demethylation assays using purified His-LSD1 were performed and RIOK1 methylation was detected by autoradiography. (I) HEK293T-LV-SETD7 cells transfected with indicated plasmids were treated with CHX (20 μg /ml), collected at the indicated times, and analyzed by western blot. (J) Overexpressing LSD1 with MG132 treatment from HEK293T cells co-transfected with the indicated plasmids. And cell lysates were subjected to pull-down with Ni2+-NTA beads. RIOK1 ubiquitination was assessed by anti-RIOK1 antibody in the presence of MG132.
As expected, compared with the protein levels of RIOK1 in Kdm1a+/− MEFs, those in Kdm1a−/− MEFs were found to be greatly decreased (Figure 5D). Reconstitution experiments with either LSD1 WT or an enzymatically inactive LSD1-K661A mutant in Kdm1a−/− MEFs further indicated that RIOK1 protein levels were restored in only LSD1 WT-reconstituted cells but not in LSD1-K661A mutant-reconstituted cells (Figure 5E). Moreover, treatment of pargyline, a LSD1 inhibitor which blocks LSD1 enzymatic activity, promoted RIOK1 destabilization (Figure 5F).
Then we tested whether LSD1 is responsible for RIOK1 demethylation. Indeed, LSD1 led to RIOK1 demethylation both in vivo and in vitro (Figure 5G and H). Moreover, treatment of cycloheximide (CHX) showed that SETD7 overexpression significantly decreased the half-life of endogenous RIOK1, while ectopic LSD1 expression increased it (Figure 5I). To confirm the involvement of 26S proteasome-dependent degradation pathway in the stability of the RIOK1 protein, we performed RIOK1 ubiquitination assay with SETD7 or LSD1 in the presence of MG132. Unsurprisingly, SETD7 significantly increased RIOK1 ubiquitination whereas the overexpression of LSD1 almost completely abolished the increase in RIOK1 ubiquitination (Figure 5J). Collectively, our results suggest that LSD1-dependent demethylation of RIOK1 significantly stabilizes RIOK1 proteins by reversing SETD7-mediated RIOK1 methylation-dependent degradation by 26S proteasomes.
SETD7 promotes RIOK1 ubiquitination and degradation through the F box protein FBXO6
Next, we set out to identify the E3 ubiquitin ligase that mediates K411me-dependent RIOK1 ubiquitination and degradation. Since both FBXW7 and FBXO6 were present in the RIOK1 immunocomplex (Supplementary file 2), we focused our attention on the Cul1-containing (Skp1-Cul1-F box) SCF E3 ligase complex. We found that knockdown of FBXO6, but not FBXW7, by two different shRNAs increased not only levels of RIOK1 proteins (Figure 6A), but also the stability of the RIOK1 protein in cells treated with CHX (Figure 6B).
Figure 6 with 1 supplement see all
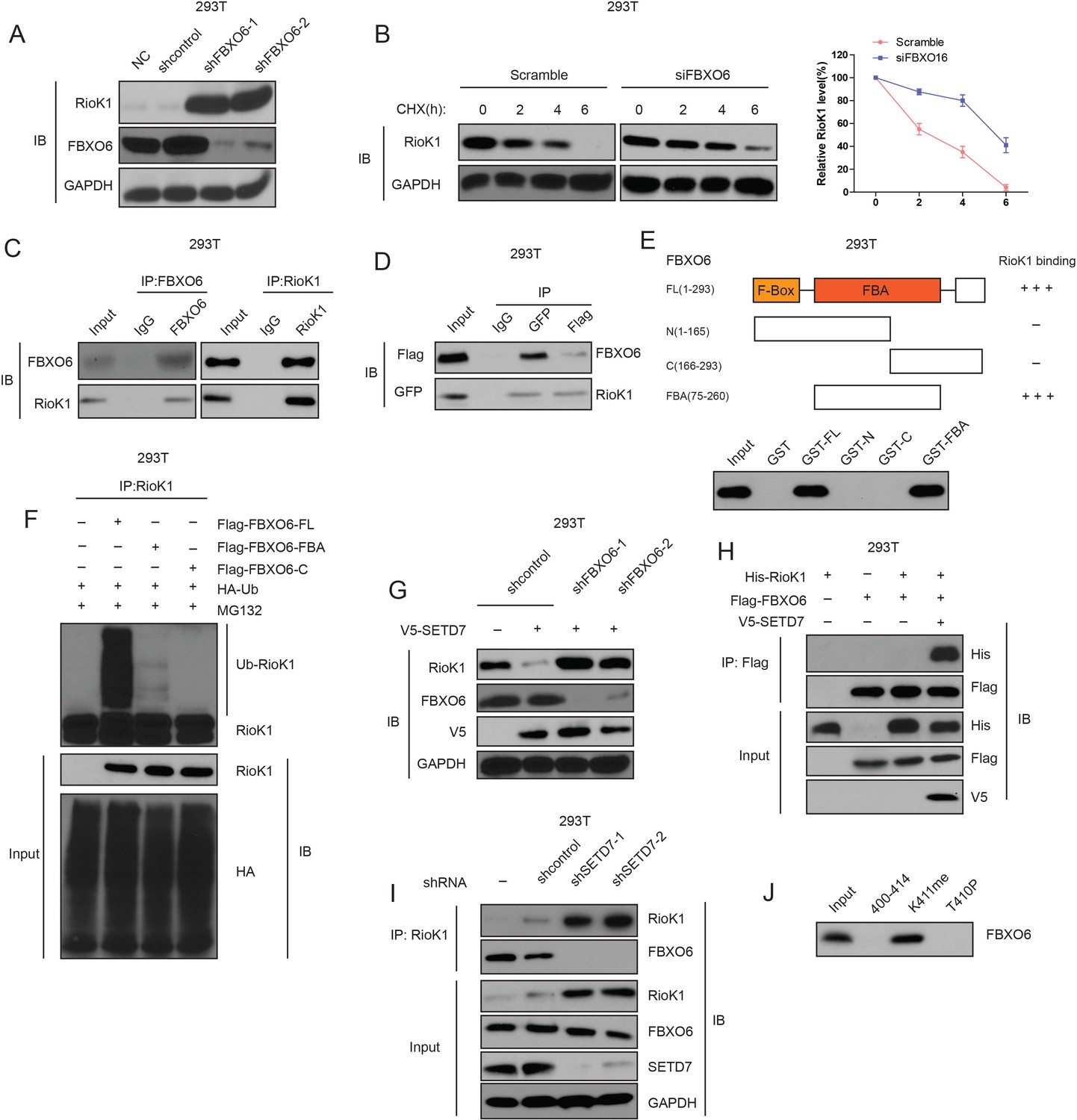
K411 methylation of RIOK1 by SETD7 promotes the interaction between FBXO6 and RIOK1.
(A) The effect of FBXO6 knockdown on the levels of RIOK1 proteins in HEK293T cells. (B) HEK293T cells were transfected with the indicated siRNAs, and after 48 hr, treated with 160 μM CHX, and representative RIOK1 expression result is shown in the left panel. Right panel indicates quantitation of the RIOK1 blots. (C) HEK293T cells were extracted and immunoprecipitated with an anti–FBXO6 (left) or anti-RIOK1 (right) antibody. Western blot analysis was performed with indicated antibodies. (D) Whole-cell lysates of HEK293T cells transfected with GFP-FBXO6 and Flag-RIOK1 were precipitated with an anti-GFP or anti-Flag antibody, and the interactive components were analyzed by Western blot. (E) Binding of several different domains of human FBXO6 to RIOK1. Numbers represent the amino acid residues in human FBXO6. FL, N, C, and FBA represent the full-length, amino terminus, carboxyl terminus, and the FBA domain only of FBXO6, respectively. The extent of the interaction between RIOK1 and FBXO6 domains is indicated by the number of plus signs (+). (F) HEK293T cells were transfected with HA-RIOK1, Myc-tagged FBXO6 FL or mutants with His-ubiquitin for 48 hr. Cells were lysed and blotted with indicated antibodies. (G) Knockdown of FBXO6 blocked V5-SETD7-induced RIOK1 degradation. (H) The interaction between Flag-FBXO6 and HA-RIOK1 was dependent on SETD7. (I) SETD7 knockdown disrupted the interaction between endogenous FBXO6 and RIOK1 in HEK293T cells. (J) The binding of FBXO6 to unmodified, T410p, or K411me RIOK1 aa 400–414 peptides was analyzed using in vitro peptide pull-down assay.
Next, we estimated their physical interaction using an immunoprecipitation assay in cultured cells. Endogenous RIOK1 was immunoprecipitated from 293T cells, and subsequent immunoblotting revealed that FBXO6 was co-immunoprecipitated with RIOK1 (Figure 6C and Figure 6—figure supplement 1). Reverse immunoprecipitation of FBXO6 confirmed the identified interaction with RIOK1 (Figure 6C and Figure 6—figure supplement 1). The interaction between RIOK1 and FBXO6 was also obvious when a co-IP assay was performed in 293T cells with the overexpression of GFP-SETD7 and Flag-RIOK1 (Figure 6D). To further define the RIOK1-FBXO6 interaction, we generated several polypeptides corresponding to different domains of human FBXO6. It was observed that the F box associated (FBA) domain mediates the binding of FBXO6 to RIOK1 (Figure 6E).
To examine the role of FBXO6 in RIOK1 ubiquitination, we transfected 293T cells with HA-RIOK1, His-ubiquitin, and different domains of human FBXO6. Interestingly, poly-ubiquitination of HA-RIOK1 was readily observed in the cells with FBXO6 full length (FL), but was significantly less noticeable in cells cotransfected with the FBXO6-derived FBA (Figure 6F), demonstrating that only the FBXO6 FL protein supported ubiquitination of RIOK1 in vitro. Together, these data suggest that FBXO6 is a ubiquitination E3 ligase of RIOK1.
We next investigated whether FBXO6 mediates SETD7-induced RIOK1 degradation. Western blot analysis confirmed that knockdown of FBXO6 by two different shRNAs blocked RIOK1 degradation induced by SETD7 (Figure 6G), suggest that FBXO6 is required for SETD7-induced RIOK1 degradation.
Next question was how SETD7-induced RIOK1-K411me is functionally linked to FBXO6. When V5-SETD7 was co-expressed, Flag-FBXO6 was found to associate with HA-RIOK1(Figure 6H). Furthermore, Flag-FBXO6 was found to interact with RIOK1 in the HEK293T cellular extracts, however, upon knockdown of SETD7, this interaction was abolished (Figure 6I). Finally, in vitro peptide pull-down assay indicated that the full-length FBXO6 specifically bound to the K411me peptide, not the peptide phosphorylated at T410 (T410p) (Figure 6J). Collectively, our data suggest that SETD7 promotes the interaction between FBXO6 and RIOK1 through its ability to methylate RIOK1 at K411.
Casein kinase 2 (CK2) phosphorylates RIOK1 both in vitro and in vivo
Given that the full-length FBXO6 only bound specifically to the K411me peptide, not the peptide phosphorylated at T410 (T410p), and especially threonine residue at 410 is exactly located within the consensus sequences of SETD7, we reasoned that T410 phosphorylation of RIOK1 might influence the biological functions of SETD7 –mediated K411 methylation of RIOK1. To test this idea, we firstly aimed to identify the kinase responsible for the phosphorylation of threonine residue at 410 (T410) on RIOK1. The residues surrounding T410 of RIOK11 (RTKEER), with acidic residues at +1 and +4, conform to a putative protein kinase CK2 phospho-motif (S/TXXE/D), which makes T410 an optimal site for phosphorylation by CK2 (Figure 7A). Thus, we investigated whether CK2 mediated the phosphorylation of RIOK1 at T410 and what functional relevance this modification had in various cell types (including CRC cells).
Figure 7 with 1 supplement see all
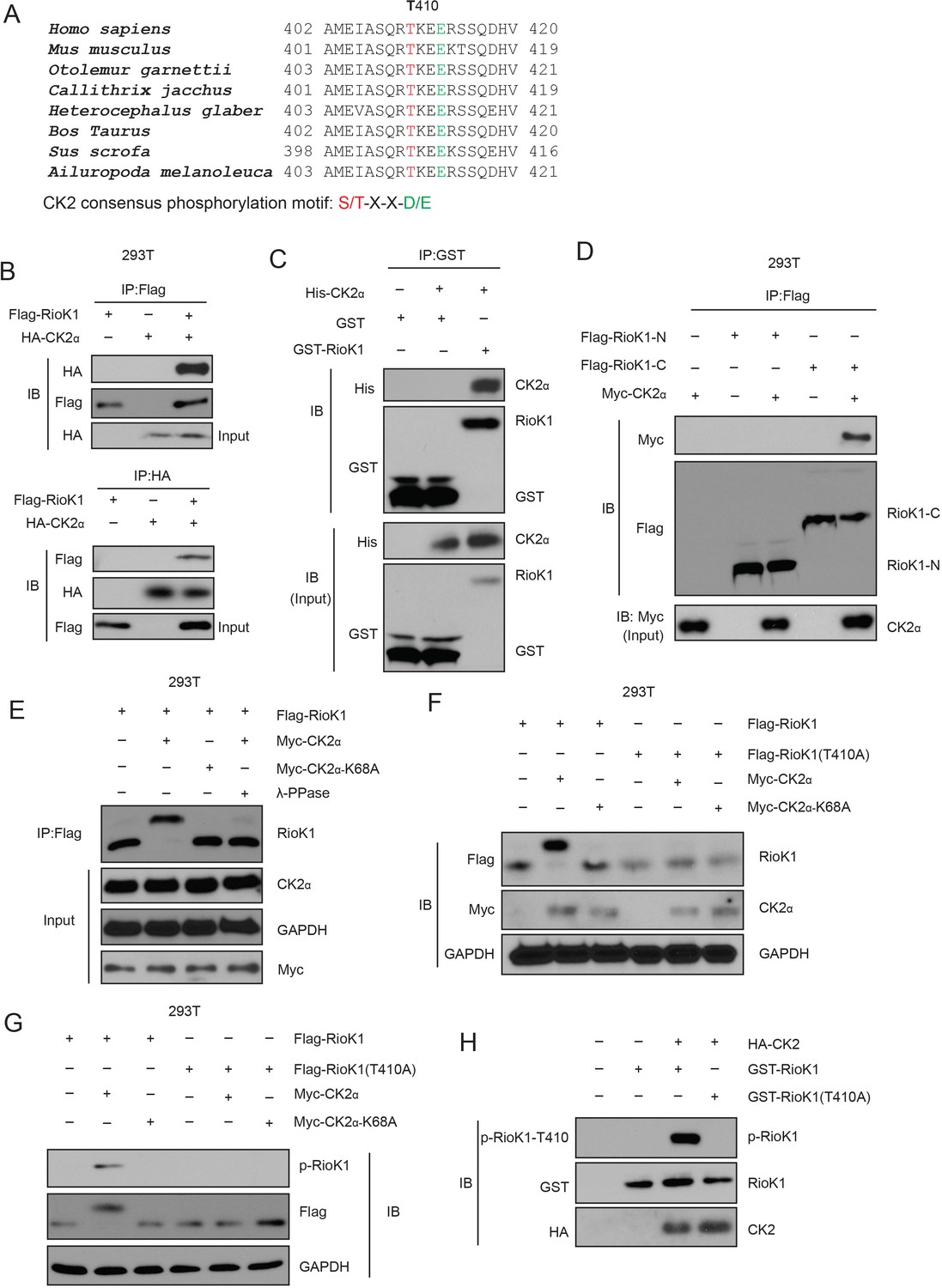
CK2 phosphorylates RIOK1 at T410 both in vitro and in vivo.
(A) RIOK1 consensus phosphorylation sites (shown in red) corresponding to the CK2 consensus motifs S/TXXE/D are presented. These consensus phosphorylation sites are exactly located within the consensus sequences of SETD7. (B) FLAG-RIOK1 was present in the HA-CK2 immunocomplex. Total cell lysates were extracted from 293T cells transiently co-transfected with FLAG-RIOK1 and HA-Vec and HA-CK2α subjected to immunoprecipitation with an anti-HA antibody followed by immunoblotting with indicated antibodies. (C) In vitro glutathione S-transferase (GST)-precipitation assay of no CK2α, or purified His-tagged CK2α combined with GST alone or GST-RIOK1. (D) Immunoprecipitation and immunoblot analysis of lysates of HEK293T cells expressing no plasmid (−) or plasmid encoding the Flag-tagged amino terminus (amino acids 1–242) of RIOK1 (Flag–RIOK1-N) or carboxyl terminus (amino acids 243–568) of RIOK1 (Flag–RIOK1-C), plus Myc-tagged CK2α, probed with anti-Myc and/or anti-Flag. (E) Lysates of HEK293T cells expressing Flag-tagged wild-type RIOK1 and Myc-tagged wild-type CK2α or CK2α-K68A; far right (λ-PPase), was analyzed using Western blot. GAPDH serves as a loading control throughout. (F) Immunoblot analysis of RIOK1 and CK2α in lysates of HEK293T cells expressing Flag-tagged wild-type RIOK1 or RIOK1 (T410A) and Myc-tagged wild-type CK2α or CK2α-K68A. (G) Immunoblot analysis of phosphorylated p-RIOK1-T410 and total RIOK1 and CK2 in lysates of HEK293T cells. (H) In vitro kinase assay of purified recombinant GST-tagged RIOK1 or RIOK1-T410A with HA-tagged CK2.
CK2 is a constitutively active serine/threonine kinase that is ubiquitously expressed (Bollacke et al., 2016). Its tetrameric holoenzyme is composed of two catalytic subunits (CK2α and CK2α’) and two regulatory β subunits (Bollacke et al., 2016). As expected, we confirmed that both HA-tagged CK2α physically associated with FLAG-tagged RIOK1 (Figure 7B). We further found that endogenous RIOK1 in 293T cells co-immunoprecipitated with endogenous CK2α, and with CK2α’ to a lesser extent, and that endogenous CK2α bound to endogenous RIOK1 (Figure 7—figure supplement 1A). Purified GST-CK2α was able to pull down bacterially produced His-RIOK1 (Figure 7C), indicating that the interaction between RIOK1 and CK2 is direct. We further found that the carboxy-terminal domain of RIOK1 is required for its binding to CK2 (Figure 7D).
We next investigated the effect of CK2 on RIOK1. When co-expressed with CK2α, but not CK2α-K68A, a kinase-inactive mutant of CK2α (Lebrin et al., 2001), RIOK1 was found to migrate more slowly only in the presence of CK2α than did RIOK1 on its own by SDS-PAGE (Figure 7E), suggesting that this differential migration was the result of phosphorylation. Threonine 410 residue is conserved in RIOK1 from various species and is homologous to the CK2-phosphorylated substrate motif (Figure 7A). Substitution of the threonine at position 410 with alanine (T410A) abolished the CK2-induced slower migration of RIOK1 by SDS-PAGE (Figure 7F). To further study the phosphorylation of RIOK1 by CK2, we generated a rabbit polyclonal antibody to RIOK1 phosphorylated at T410 (p-T410) by immunizing rabbits with the RIOK1 peptide. We validated the antibody endogenously in two cell lines. Before development with anti-RIOK1 antibodies, these two cell lines were either incubated with or without λ-PPase (Figure 7—figure supplement 1B). Co-expression of wild-type CK2α, not CK2α-K68A, with RIOK1 induced phosphorylation of RIOK1 at T410, and the T410A substitution in RIOK1 eliminated the phosphorylation of RIOK1 by CK2 (Figure 7G). In an in vitro kinase assay, purified RIOK1 was phosphorylated by CK2, but mutant RIOK1 (T410A) did not (Figure 7H). Collectively, our results suggest that CK2 directly phosphorylated RIOK1 at T410.
RIOK1 phosphorylation at T410 by CK2 blocks methylation at K411 by SETD7 and stabilizes RIOK1
Given the close proximity of T410 and K411 residues, we analyzed whether T410p by CK2 could affect K411me by SETD7. Indeed, knockdown of CK2α in HCT116 cells led to a substantial reduction of RIOK1 protein and p-T410, and a significant increase of RIOK1 K411me (Figure 8A). Consistently, in the presence of CK2α-K68A, both RIOK1-p-T410 and total RIOK1 level were less abundant, and RIOK1-K411me1 levels increased (Figure 8B).
Figure 8 with 1 supplement see all
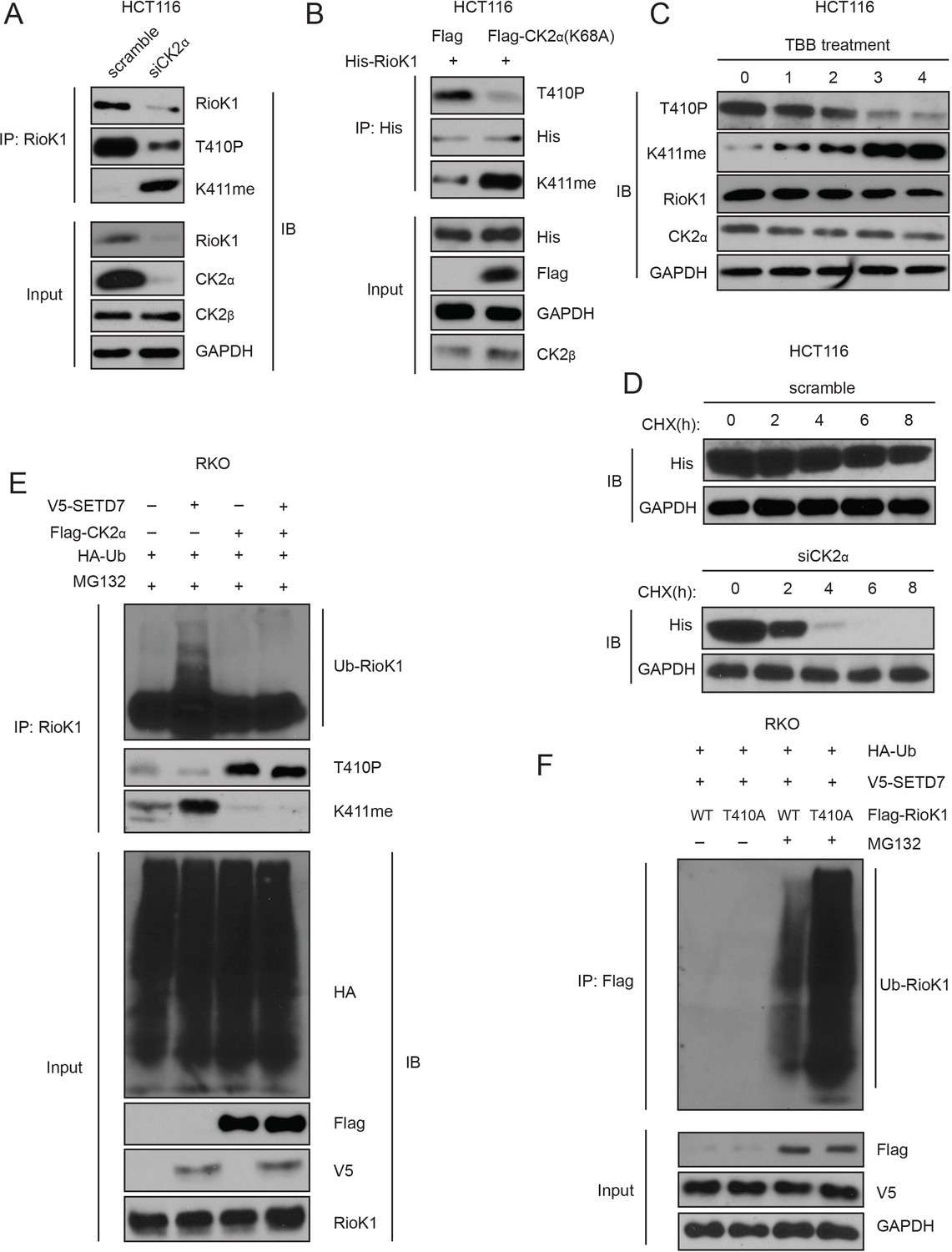
T410 phosphorylation of RIOK1 antagonists SETD7-mediated K411 methylation, which stabilizes RIOK1 in CRC and GC cells.
(A) The effect of knockdown of CK2α in HCT116 cells on the levels of RIOK1 proteins, T410p, and K411me. (B) The effect of the CK2α-K68A mutant on the levels of RIOK1 proteins, T410p, and K411me. (C) The effect of the CK2 kinase inhibitor TBB on the levels of RIOK1 proteins, T410p, and K411me. (D) The effect of CK2 knockdown in HCT116 cells on the RIOK1 protein stability. (E) CK2 inhibits K411me by SETD7 and its consequent RIOK1 ubiquitination in RKO cells. Indicated cells were cotransfected with HA-Ub and treated with MG132 for 12 hr. (F) Wild-type RIOK1 or the RIOK1-K411me1 mutant was overexpressed in cells, along with HA-ubiquitin and V5-SET7, in the presence or absence of the proteasome inhibitor MG132. Cell lysates were analyzed with indicated antibodies.
To confirm that RIOK1 stability is CK2 dependent, HCT116 cells were treated with the CK2 inhibitor TBB (Leung and Shilton, 2015). RIOK1-p-T410 levels substantially decreased within 3 hr of the treatment, while RIOK1-K411me proportionally increased (Figure 8C). Along with the gradual increase of RIOK1-K411me, more degradation of RIOK1 was observed, causing a decrease in total RIOK1 levels. However, the total level of CK2α remained similar (Figure 8C). Additionally, knockdown of CK2α significantly reduced the half-life of RIOK1 (6–8 hr in control versus less than 4 hr) in HCT116 cells (Figure 8D). These data suggest that CK2-induced phosphorylation of RIOK1 at T410 might stabilize RIOK1 by inhibiting K411me by SETD7.
Next, we investigated how CK2 influences RIOK1 ubiquitination. While overexpression of SETD7 in RKO cells led to increased K411me and RIOK1 ubiquitination, overexpression of CK2α significantly increased p-T410 and inhibited RIOK1 ubiquitination (Figure 8E). Importantly, co-expression of CK2α with SETD7 only significantly led to an increase in p-T410, but not K411me, suggesting that p-T410 by CK2 predominates over and inhibits K411me by SETD7 (Figure 8E). Consistent with this idea, CK2α overexpression blocked SETD7-induced RIOK1 ubiquitination (Figure 8E). Therefore, compared to the methylated RIOK1, the phosphorylated species is less likely to be degraded.
Next, we tested whether the T410A mutation promoted ubiquitin-induced degradation of RIOK1. Unsurprisingly, in the presence of MG132, transfection with V5-SETD7 and Flag-RIOK1-WT led to less ubiquitination than transfection with V5-SET7 and Flag-RIOK1-T410A (Figure 8F), suggesting that the T410A mutation promotes RIOK1 methylation which is a substrate for ubiquitinylation.
Collectively, our data demonstrate that p-T410 by CK2 blocks K411me by SETD7, which protects RIOK1 from ubiquitination and degradation. Furthermore, we also proved that CK2α, LSD1, and RIOK1 were in the same immunocomplex (Figure 8—figure supplement 1), suggesting that LSD1-mediated demethylation of RIOK1 affects the stability of RIOK1.
SETD7 –mediated methylation of RIOK1 negatively regulates CRC growth and metastasis, which is reversed by the CK2-induced phosphorylation of RIOK1
To explore the functional significance of both methylation and phosphorylation of RIOK1, we evaluated the impact of an unmethylatable mutation K411R, a non-phosphorylatable mutation T410A, and an unmethylatable plus phosphomimetic mutation (K411R/T410E) of RIOK1 on CRC cell proliferation, migration, invasion, tumor growth and metastasis. MTT assays showed increased growth in cells stably expressing RIOK1-K411R and RIOK1-K411R/T410E, but attenuated growth in cells with RIOK1-T410A compared to cells with RIOK1-WT expression (Figure 9A). The ability to form colonies was dramatically increased in stable transfectants with the expression of RIOK1-K411R and RIOK1-K411R/T410E, compared to RIOK1-WT (Figure 9B and Figure 9—figure supplement 1A). In agreement with these in vitro growth assays, RKO cells with stable expression of RIOK1-K411R and RIOK1-K411R/T410E showed greater tumor growth than cells with RIOK1-WT in a xenograft tumor model, while the stable expression of RIOK1-T410A stable obviously retarded CRC growth (Figure 9C and D).
Figure 9 with 2 supplements see all
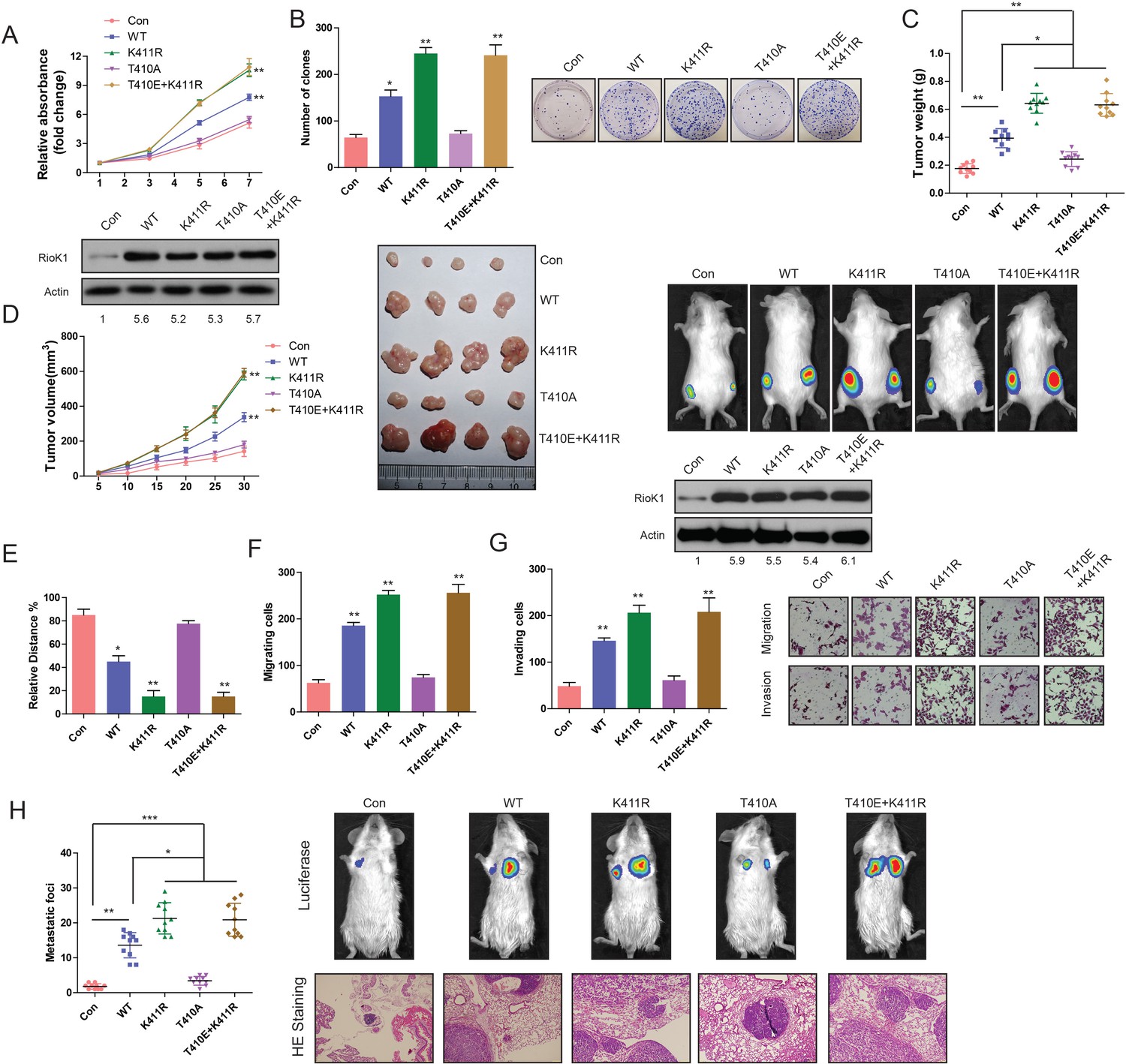
Methylation of RIOK1 at K411 and phosphorylation of RIOK1 at T410 oppositely regulated the CRC growth and metastasis.
(A) MTT assays in RKO cells expressing indicated proteins (upper panel). Western blotting was used to detect expression of RIOK1 in RKO cells using anti-RIOK1 antibody (lower panel), and the intensity of the western blot bands was quantified using NIH ImageJ software. (B) Photography and statistic results of colony formation assays in RKO cells expressing indicated proteins. (C and D) Statistic, tumor growth curves, photography results and representative bioluminescence images of tumor size in RKO cells expressing indicated proteins. Over-expression of RIOK1 in a xenograft tumor model was analyzed with western blotting, and the intensity of the western blot bands was quantified using NIH ImageJ software. (E-G) Migration ability evaluated by wound healing assay (E) and transwell migration assay (F). Invasiveness evaluated by matrigel invasion assay (G) in RKO cells expressing indicated proteins. (H) Statistic results, HE staining and representative bioluminescence images of lung metastasis in RKO cells expressing indicated proteins.
To assess cell migration changes, wound healing and transwell migration assays were performed in CRC cells. As shown in Figure 10E and F, CRC cells with the expression of RIOK1-K411R and RIOK1-K411R/T410E had enhanced migration ability compared to RIOK1-WT. Similarly, the invasion ability of CRC cells stably expressing RIOK1K411R and RIOK1-K411R/T410E was also substantially increased (Figure 9G). In agreement with these in vitro migration assays, stable expression of RIOK1-K411R and RIOK1-K411R/T410E in CRC cells promoted lung metastasis of CRC (Figure 9H). Compared to RIOK1-WT, stable expression of RIOK1-T410A in RKO cells caused a significant reduction of migration, invasion, and metastasis (Figure 9E,F,G and H).
Additionally, all the in vitro and in vivo functional experiments in Figure 9 were repeated in HCT116-shRIOK1 cells stably transfected with RIOK1-T410A, RIOK1-K411R/T410E, RIOK1-T410A, and RIOK1-WT, and showed similar results (Figure 9—figure supplement 1B, C, D, E and F). Together, these data confirm that the methylation and phosphorylation RIOK1 oppositely regulates CRC tumorigenesis and metastasis.
Clinical relevance of RIOK1, SETD7, LSD1, FBXO6 and CK2 Expression in patients with CRC
To determine the clinical relevance of our findings, we firstly checked the expression of phosphorylated-RIOK1 and methylated-RIOK1 in the same tissue in Figure 1A. It was shown that phosphorylated-RIOK1 was positively correlated with RIOK1 expression in CRC cell lines and tissues while methylated-RIOK1 was negatively associated with RIOK1 expression (Figure 9—figure supplement 1G and H). Then, we performed immunohistochemical analysis of 104 primary CRC samples with validated antibodies recognizing RIOK1, SETD7, LSD1, FBXO6, and CK2 proteins. Consistently, we observed an inverse expression pattern between RIOK1 and SETD7 or FBXO6, whereas a positive expression pattern between RIOK1 and LSD1 or CK2 was observed (Figure 9—figure supplement 2A and B). Kaplan–Meier analysis shows that high levels of LSD1 and CK2 in CRC samples correlated with poor overall survival in the patients, whereas low levels of SETD7 and FBXO6 in CRC samples correlated with poor overall survival (Figure 9—figure supplement 2C and D). Therefore, expression levels of these proteins were independent prognostic factors for overall survival and disease-free survival. Together, our data highlight a critical role for RIOK1 phosphorylation and methylation in the clinical aggressiveness of human CRC (Figure 9—figure supplement 2E).
Discussion
In the present study, RIOK1 appears to be important for CRC and GC progression because blockade of RIOK1 decreases tumor growth and metastasis, whereas RIOK1 overexpression causes the opposite effects. Our data are consistent with a recent report that RIOK1 knockdown impaired the growth of the colorectal cancer cell line HCT116, which strongly indicates the oncogenic role of RIOK1 (Weinberg et al., 2017). Furthermore, to our knowledge, we for the first time reveal that RIOK1 is specifically monomethylated at the K411 site by SETD7 in vitro and in vivo. This SETD7-mediated methylation recruits FBXO6 E3 ligase and significantly contributes to RIOK1 instability. However, LSD1-mediated demethylation of RIOK1 at K411 or CK2-mediated phosphorylation of RIOK1 at T410 reverses RIOK1 degradation by FBXO6. Importantly, functional experiments indicate that the RIOK1 demethylation or phosphorylation signals might be a prerequisite for enhanced CRC and GC growth and metastasis. Clinically, our current data indicate that RIOK1 expression is associated with CRC and GC patient prognosis. However, a further test of the PTM antibodies on CRC and GC tissues is needed in the future work. Together, these results provide a novel mechanism through which the crosstalk of PTMs regulates the function of RIOK1, supporting the notion that RIOK1 may serve as a valuable biomarker to monitor human CRC and GC development.
The most striking finding is that a methylation-phosphorylation switch dictates RIOK1 stability and function in CRC and GC. Consistent with our data, a previous study reported a similar methylation-phosphorylation switch in regulating the stability of Sox2. In that paper, AKT1 was found to phosphorylate Sox2 at T118, which blocked K119me by Set7 and stabilized Sox2 (Fang et al., 2014). Independent studies have validated the importance of RIOK1 in cancer cell survival. A cell-based RNAi screen identified RIOK1 required for Ras-mediated cell survival, although this study did not explore the functionality of RIOK1 (Breitkreutz et al., 2010). Additionally, RIOK1 upregulation has been shown to be positively associated with Akt activity in both glioblastoma specimens and cultured cells (Read et al., 2013). Consistent with these notions, in this manuscript, we demonstrate that RIOK1 is upregulated in CRC and GC tumor cells relative to normal control cells, and RIOK1 deficiency decreases tumor cell proliferation, migration, and lung metastasis. Therefore, RIOK1 is a powerful modifier of tumorigenesis. Despite about one decade of study, very little was known about RIOK1 regulation. It has been previously shown that Akt signaling regulates RIO kinase protein stability (Read et al., 2013), but the exact mechanism by which Akt regulates RIOK1 levels remains undetermined. We now provide a novel perspective on the posttranslational regulation of RIOK1 by showing that SETD7-mediated methylation of RIOK1 decreases RIOK1 protein stability, which can be reversed by LSD1. Moreover, we provide ample evidence that CK2-mediated phosphorylation of RIOK1 at K410 reverses RIOK1 degradation by FBXO6 and SETD7. Our data are the first to establish functional connections between the PTMs of RIOK1 in CRC and GC. These results may have broad relevance to other cancers since RIOK1 is strongly expressed in other tumor types (Faraji et al., 2014). Given that RIOK1 creates a feedforward loop that promotes and maintains Akt activity (Read et al., 2013), we hypothesize that there might be a feedback loop between RIOK1 and SETD7 or CK2, and that disruption of these loops is sufficient to trigger apoptosis or chemosensitivity in CRC and GC. This hypothesis awaits substantiation in future studies, which may lead to important new insights into the RIOK1signaling network in both normal and cancer cells.
Compared with the plethora of information about histone methylation, our understanding of non-histone protein methylation is very limited. Although RIOK1 has been shown to directly associates with the protein arginine methyltransferase 5 (PRMT5), RIOK1 was not a substrate of PRMT5 (Guderian et al., 2011). RIOK1 just acts as an adapter protein by recruiting the RNA-binding protein nucleolin to the PRMT5 complex for its symmetrical methylation (Guderian et al., 2011). In the present study, for the first time, we provide evidence that SETD7 and LSD1 methylates or demethylates RIOK1 respectively. One of the main mechanisms by which lysine methylation regulates protein function is effects on protein–protein interaction (Kim et al., 2016; Couture et al., 2006). Indeed, we observed that K411 methylation of RIOK1 promotes the recruitment of FBXO6 to RIOK1 and disrupts the association of CK2 and LSD1 with RIOK1, subsequently leading to RIOK1 degradation. Given that the level of RIOK1 is closely related to diverse cancers (Read et al., 2013; Guderian et al., 2011), our data highlight the significance of understanding this important enzyme in health and disease.
Interplay with other PTMs is another main mechanism by which lysine methylation regulates protein function (Kim et al., 2016; Couture et al., 2006). Therefore, we also examined the phosphorylation and ubiquitination of RIOK1 by CK2 and FBXO6 respectively, which is the second important discovery in this study. Notably, CK2 was previously identified as an interaction partner of the Rio 1 p, a homologs of the human RIOK1 in yeast by MS analysis, and phosphorylation by CK2 leads to moderate activation of Rio1p in vivo and promotes cell proliferation (Angermayr et al., 2007), which is consistent with our data in human CRC and GC. Moreover, we now extend these findings by showing that CK2-mediated phosphorylation of the RIOK1 promoted CRC and GC growth and metastasis by disrupting SETD7-mediated methylation of RIOK1and recruitment of FBXO6 E3 ligase to RIOK1. However, In that study, Six C-terminally located clustered serines were identified as the only CK2 sites present in Rio1p in yeast, and phosphorylation by CK2 renders the Rio 1 p susceptible to proteolytic degradation at the G (1)/S transition, whereas the non-phosphorylated version is resistant (Angermayr et al., 2011), which stands in contrast with our current data. Notably, at first, the C-terminal domain of yeast Rio1p is least conserved in evolution and especially lacks consensus SETD7 motif. Secondly, RIOK1 of higher eukaryotes almost does not contain all six serine residues or CK2 motifs in the corresponding aligned sequences, but has highly conserved threonine residue and positively charged lysine residue next to it. This may provide an explanation for the most puzzling aspect of the effect of CK2 on RIOK1 in yeast and higher eukaryotes.
Recently, it has been demonstrated that LSD1 physically interacts with endogenous CK2 in vivo and is potentially phosphorylated by CK2 on three serine residues (S131, S137, S166) in vitro (Peng et al., 2015). And S137 facilitates RNF168-dependent ubiquitination and recruitment of 53BP1 to the DNA damage sites, which enhances cell survival and proliferation in response to DNA damage (Peng et al., 2015). Thus, it is possible that there exists a feedback loop between LSD1 and CK2 in CRC and GC. Additionally, it needs to be further clarified whether CK2-mediated phosphorylation of the RIOK1 at Thr410 enhances the association of LSD1 with RIOK1, or LSD1-mediated demethylation of RIOK1 increases the binding of CK2 to RIOK1 and phosphorylation.
Previous study suggested that the proteasome has a pivotal role in the regulation of RIOK1 degradation (Guderian et al., 2011). However, no specific ubiquitin complex was previously connected to RIOK1 ubiquitination. Although we can not formally rule out the contribution of other E3 ligase –mediated effects on the functions of RIOK1, we were able to show here that FBXO6 links the activated RIOK1 to the degradation machinery and FBXO6 activity affects the cancer-supportive role of RIOK1 in CRC and GC. Importantly, we show that the ubiquitination of RIOK1 is controlled by molecular pathways (CK2 and LSD1and SETD7) frequently deregulated in CRC and GC (Peng et al., 2015; Lin et al., 2011; Akiyama et al., 2016). In the future, the identification of critical ubiquitination sites will provide new insights into the role and interaction of RIOK1 in cancer.
Clinically, LSD1 and CK2 activation are associated with poor outcome in cancer (Peng et al., 2015; Lin et al., 2011). Interestingly, reduced SETD7 expression or inactivating mutations is significantly correlated with poor patient prognosis in multiple cancers (Akiyama et al., 2016). Recently, FBXO6 has been shown to affect the DNA damage, a process which promotes cell proliferation and survival in response to DNA damage (Zhang et al., 2009). Our data unify these observations and demonstrate that RIOK1 accumulation due to altered SETD7, LSD1, CK2, and FBXO6 expression provides an advantage in CRC and GC cells during disease progression. Indeed, we show a direct correlation between RIOK1 and LSD1, CK2, FBXO6, or SETD7 protein expression to increased metastatic potential and decreased recurrence-free survival of CRC and GC patients. These data suggest that RIOK1 could be a biomarker and a therapeutic target for these diseases. However, it remains to be seen whether the altered function of these enzymes (SETD7, LSD1, CK2, and FBXO6) in different cancers indeed converges on RIOK1 stability.
In summary, we show that RIOK1 is up-regulated in CRC and GC, which results in a significantly reduced survival and a more malignant phenotype by enhancing proliferation and migration of CRC and GC cells. Most importantly, we show for the first time that methylation and phosphorylation control the stability of RIOK1 and its role in CRC and GC progression. Therefore, targeting these PTMs of RIOK1 could be a very promising novel treatment option for CRC and GC.
Materials and methods
Cell culture and transfection
Request a detailed protocolColorectal cancer cell lines LOVO, RKO, M5, LS174T, HCT116, DLD1, SW480, SW620, HEK293T, MKN45 and SGC-7901, MEFs and a normal human intestinal epithelial cells (IECs) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were authenticated by STR profiling according to the cell bank, and have been checked by Universal Mycoplasma Detection Kit (ATCC 30–1012K), and have no mycoplasma contamination. All cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM, Switzerland) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA) and maintained in standard conditions (5% CO2 and 95% atmosphere, 37°C). Using a lentiviral system, stably transfected cells were created. Lentiviral vectors encoding the human RIOK1 gene and its mutants or shRIOK1, or empty vector (LV-control), or Non-specific shRNA were transfected into CRC or GC cells with a multiplicity of infection (MOI) of 40 to 50 in the presence of polybrene (5 μg/ml). At 48 hr after transfection, transfected cells were selected for 2 weeks with 2.5 μg/ml puromycin (Sigma). Pooled populations of knockdown cells and overexpression cells, which were obtained 2 weeks after drug selection without subcloning, were used in both in vitro and in vivo experiments.
Cell proliferation
Request a detailed protocolThe cell proliferation assay was performed with the Cell Counting Kit-8 (CCK-8, Sigma) or MTT assays according to the manufacturer's instructions.
Quantitative PCR
Request a detailed protocolTotal RNA was extracted from tissues or cells using TRIzol reagent (Takara) according to the manufacturer's instructions. RNA reverse transcription to cDNA was performed with a Reverse Transcription Kit (Takara). Quantitative real-time PCR (qRT-PCR) analyses used SYBR Green I (Takara) in triplicate. The results were normalized to the expression of GAPDH. The primer sequences used for qRT-PCR are listed as follows: Human SETD7: 5′-AGTTCTCCAGGGCACGTATG-3′ (forward); 5′-TCTCCAGTCATCTCCCCATC5′−3′ (reversed); Human GAPDH: 5′-ACA GTC AGC CGC ATC TTC TT-3′ (forward) and 5′-GAC AAG CTT CCC GTT CTC AG-3′ (reversed); Human RioK1: 5′-GGCTAAACACAGCAGAGATACC-3′ (forward); 5′-AACTCCCGAGCCTTGGATTCT-3′ (reversed); Lsd1 forward: 5′- CGGCATCTACAAG AGGATAAAACC −3′, Lsd1 reverse: 5′- CGCCAAGATCAGCTACATAGTTTC −3′. Indicated genes’ mRNA expressions were normalized to GAPDH level.
cDNA constructs and RNA interference
Request a detailed protocolThe full-length open reading frames of RioK1 and SETD7 was amplified by PCR using the following primers: RioK1,5′-CGGACGTCGACATATGGACTACCGGCGGCTTCTCATG-3′ and 5′-CTGATGCGGCCGCCTATTTGCCTTTTTTCGTCTTGGC-3′ and confirmed by DNA sequencing. Target DNA fragments were then subcloned into pEGEX6P-1 (GE Healthcare) and pHA vector (, Invitrogen). Different truncated fragments of RioK1 were generated by PCR and subcloned into pEGEX6P-1 and pHA, respectively. The primer for cloning different truncations was as following, (aa 1–120, 5′-GTAGAATTCATGGACTACCGGCG-3′ and 5′-CATCTCGAGTCAATTAATTTTATTCTC-3′; aa 121–242, 5′-GATGCGGCCGCAATTTAGATAAGC-3′ and 5′-CATCTCGAGTCACATTTTCCTAGGGT-3′; and aa 1–242, 5′-CGGACGTCGACATATGGACTACCGGCGGCTTCTCATG-3′ and 5′-CGTGCGGCCGCTCACATTTCCTAGGG-3′), which was described as previously reported (Guderian et al., 2011). Three RioK1 Human shRNA lentiviral constructs (OriGene, TL320739 ) were purchased from OriGene (USA). shRNA-resistant RioK1Δ was made by performing site –directed mutagenesis to introduce five silent mutations (CDS area 551 A > T, 554 T > A, 557 C > T, 566 T > C and 569 T > A) against RioK1 shRNA, primer sequences were listed as follow: Forward sequence:5'-CGG TTG ATA TTG ATT CCG CCA AAA CTC −3'and Reverse sequence: 5'-GAG TTT TTG TGA CGG AAT CAA TAT CAA-3'. The knocking effect of RioK1 was evaluated by western blotting using relevant specific antibodies. The siRNA oligonucleotide duplexes targeting CK2α (sequence: GAUGACUACCAG CUGGUUCdTdT), CK2α’ (sequence: CAGUCUGAGGAGCCGCGAGdTdT), LSD1 3′ UTR (sequence: GCUCUUCUAGCAAUACUAGdTdT). Oligonucleotides the shRNA sequences were as following. For silencing of SETD7: GCAAACTGGCTACCCTTATGT (shSETD7–1); and GGGAGTTTACACTTACGAAGA (shSETD7–2). The shcontrol and sh-LSD1 (shLSD1–1: CACCGCCTGTTTCTGC CATGTAAGGCGAACCTTACATGGCAGAAACAGGC, shLSD1–2: CACCGCCAT GTAAGGAAGGCTCTTCCGAAGAAGAGCCTTCCTTACATGGC). shRioK1#1: GAAGAATGTATCAGGATGCCAGACTTGTC; shRioK1#2: CAGAGCAACTGTAGAACAGGTGTTGGATC; shRioK1#3: AGAACGGTCTAGCCAAGATCATGTGGATG. Specific siRNAs and scrambles were purchased from IBA Nucleic Acids Synthesis, Germany) and transfected with Oligofectamine (Invitrogen) according to the manufacturer’s protocol. The knocking effect of RioK1 was evaluated by western blotting using relevant specific antibodies. SETD7-H297A and RioK1-K411R and K413R mutants were introduced using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. All of the constructs were confirmed by DNA sequencing. The pcDNA-myc/his-SETD7 was created by subcloning SETD7 into pcDNA3.1/myc-His A (Invitrogen). SETD7 siRNA (Ambion, siRNA sequence: 5′‐AGAUAACAUUCGUCAUGGA‐3′) or non‐targeting siRNA (Dharmacon). The plasmid for V5-SETD7 was generated by PCR cloning. Flag-tagged SETD7-WT or -H297A FL and fragment cDNAs were amplified from plasmids by PCR and separately subcloned into pGEX-4T3, EGFP, and pET28 vectors. Human cDNA clones encoding LSD1, CK2α, CK2α’ and CK2β were subcloned into pcDNA3.0 backbone as well as with three copies of HA or FLAG tag at the N-terminus or one copy of MYC tag at the C-terminus. The expression construct of HA-Ub was a gift from Dr.Bing Wang (Rutgers University).
Migration and invasion assays
Request a detailed protocol6.5 mm diameter Boyden chambers with pore size of 8.0 μm (Corning) was used for migration assays. Briefly, the stable cell lines (3.0 × 105 cells per well) were resuspended in the migration medium without FBS, and placed in the upper compartment of transwell chambers. The lower compartment was filled with 500 μl medium containing 10% FBS as a chemoattractant. Cells were fixed in 4% formaldehyde, and stained with 0.1% crystal violet after 24 hr. Ten random fields were counted under a light microscope (Carl Zeiss, Germany). However, cell invasion (3.0 × 105 cells per well) was evaluated in 24-well matrigel-coated invasion chambers after 48 hr.
Purification of RIOK1-binding proteins
Request a detailed protocolRIOK1-binding proteins were purified from extracts of HEK293T cells expressing Flag-tagged RIOK1. Mock purification from HEK293T cells expressing an empty vector was performed as a negative control. The procedure has been described previously (Kim et al., 2016). In brief, The RIOK1-binding proteins were precipitated using Flag M2 agarose beads (Sigma, 100 μl of 50% slurry) from ~100 mg of cell extracts. After overnight incubation at 4°C, the beads were washed three times with a BC150 buffer (1 mM EDTA, 0.05% Nonidet P40, 1 mM dithiothreitol, 0.2 mM PMSF, 20 mM Tris-HCl pH 7.9, 15% glycerol, and 150 mM KCl), and twice with a BC300 buffer, and three times with a TBS buffer. Finally, the bound proteins were eluted, and resolved by SDS–PAGE for LC-MS/MS analysis.
Mass spectrometry
Request a detailed protocolMass spectrometry was performed as described previously (Hong et al., 2014).
In vitro ubiquitination
Request a detailed protocolIn vitro ubiquitination assay was performed according to previous report (Zhang et al., 2009). In Summary, HA-RIOK1 was expressed in 293T cells, pulled down with anti-HA antibodies, and the HA-RIOK1-bound beads were used as the ubiquitination acceptor substrate. Extracts from indicated cells expressing Myc-FBXO6 were immunoprecipitated using protein A-agarose beads coupled with anti-Myc, and treated with hypotonic buffer (2 mM DTT, 0.25 mM EDTA, 20 mM Tris-HCl, pH 7.2, and protease inhibitors), Then we performed in vitro ubiquitination assay in one reaction buffer (5 mM MgCl2, 0.5 mM DTT, 50 mM Tris-HCl, pH 7.5, 2 mM NaF, 10 nM okadaic acid) with 30 μM MG132, 1 μg/ml recombinant Flag-Ub, and 10 μl concentrated indicated cell extracts.
Western blotting and antibodies
Request a detailed protocolCells were harvested and lysed on ice for 30 min in 1 × RIPA as described above. Cell lysates were analyzed by SDS-PAGE and transferred to PVDF memebrane. Membranes were washed and incubated with indicated antibodies at 4°C overnight respectively. Antibody against RIOK1 was raised by injection of recombinant RIOK1 comprising aa 1–242, and affinity-purified on columns with the respective covalently linked antigen. Then membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies (BIO-RAD) according to the manufacturer's instructions. The protein of interest was visualized using ECL Western blotting substrate (Pierce). Or samples were resolved by SDS-PAGE and further analyzed by silver staining and Coomassie staining. Other antibodies are: Anti-HA (F-7, Santa Cruz Biotechnology), anti-FLAG (M2, Sigma-Aldrich), ubiquitin from Santa Cruz, and V5 from Invitrogen. The homemade rabbit anti- SETD7 polyclonal antibody was raised against recombinant GST- SETD7 protein. The mouse anti- SETD7 monoclonal antibody was purchased from Millipore and Cell Signaling. Rabbit polyclonal antibodies used in this study including anti-HA (A190–208A), anti-MYC (A190–205A), anti-LSD1 (A300–215A), (A300–272A), anti-CK2α (A300–198A), anti-CK2α’ (A300–199A), anti-CK2β (A301–984A), anti-GAPDH (A300–643A) were purchased from Bethyl. RIOK1- K119me, p-T410 and 400–414 antibodies were generated by immunizing rabbits with K411-monomethylated, T410-phosphorylated and unmodified 400–414 peptides conjugated with keyhole limpet hemacyanin (KLH), respectively. Briefly, rabbits were immunized by keyhole limpet hemocyanin–conjugated peptides. The sera were tested and collected after four more antigen boosters. To eliminate nonspecific recognition of nonmethylated antigen, the sera were precleaned by incubating with nonmethylated peptide and examining with affinity chromatography. The specific polyclonal antibodies were finally purified by methylated peptide affinity chromatography via CNBr-activated Sepharose 4 Fast Flow (17-0981-01; GE Healthcare). Anti-FBXO6 antibodies were generated against KLH-coupled synthetic peptide corresponding to amino acids 262 to 284 of human FBXO6, which lie very close to the C terminus. Antiserum from peptide-injected rabbits was affinity purified using the AminoLink Plus Immobilization Kit (Pierce).
In vitro protein phosphorylation assays
Request a detailed protocolIn vitro kinase assays were performed as described before (Peng et al., 2015) except that the CK2 kinase buffer contained 1 mM DTT, 1 mM Na3VO4, 10 μM cold ATP, 50 mM HEPES (pH 7.4), 10 mM MgCl2, and 5 μCi [γ-32P]-ATP. The GST pulldown after kinase assay was performed by adding 500 μl of NETN buffer into the reaction after the in vitro kinase assay was completed. In all, 25 μg of RIOK1 peptides were used in similar reactions prior to direct spotting onto nitrocellulose for immunoblot.
Immunofluorescence
Request a detailed protocolFor immunofluorescent staining, indicated cells plated on poly-l-lysine-coated glass coverslips in DMEM and 10% FCS were washed once in PBS, then were fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100 and blocked in 1% BSA in PBS. cells were incubated for 1 hr with indicated primary antibodies at 4°C overnight. Following staining with rhodamine-conjugated secondary anti-rabbit antibodies, cells were analyzed with a 63 × oil immersion lens on a Zeiss Axiovert 200 m microscope. Indicated cells transfected with indicated plasmids were directly analyzed after 48 hr without fixation. The fluorescent staining was recorded using an inverted fluorescence microscope (Leica, Germany).
Patients specimens and tissue microarray
Request a detailed protocolFrom January 2008 to May 2014, a total of 104 pairs of CRC and 20 pairs of GC and non-tumor samples were collected from patients who had CRC or GC resection performed at Third Affiliated Hospital of Harbin Medical University.The samples were used for subsequent RNA extraction or immunohistochemistry (IHC) stained for RIOK1, FBXO6, CK2, LSD1, SETD7. All human materials were obtained with informed consent and approved by the ethics committee of Hospital of Harbin Medical University(Protocol Number: 20150526). The tissue specimens were frozen in liquid nitrogen and stored at −80°C. All tissues were confirmed as adenocarcinoma. CRC tissue microarray (TMA) was purchased form US Biomax (BC051110b) and IHC stained for RIOK1. The slides were scored for the percentage of positive epithelial cells as well as intensity and protein staining was subsequently quantified using the a scoring system based on the Allred score for immunohistochemistry (Allred et al., 1998; Hoekstra et al., 2016). Based on the RIOK1protein expression as defined by Allred score <6 (low) and >6 (high), patients were stratified into two groups: RIOK1 low and high expression groups.
In vivo tumorigenic and metastasis assay
Request a detailed protocolAll animal experiments were performed in accordance with NIH guidelines for the use of experimental animals. Male nonobese/severe combined immunodeficiency (NOD/SCID) mice between 4 and 6 weeks of age, obtained from the Experimental Animal Center of Shanghai Institute for Biological Sciences (SIBS). All animal work was conducted according to Institutional Animal Care Guidelines, and all animal experiments were approved by the ethical committee of the Harbin Medical University (Protocol Number: 20150619). HCT116-pLV-shRIOK1 and HCT116-PLV-shNC and other indicated cells (1 × 107 cells per mouse) were injected subcutaneously into the left or right flank of NOD/SCID mice (n = 10 per group).. After 8 weeks, the remaining mice were sacrificed, and lungs were isolated for examination of the number of metastatic tumors. Tumor volume (V) was estimated from the length, width (w), and height (h) of the tumor. We analyzed primary tumor growth by the formula (length × width2)/2. For the metastasis model, 2 × 106 cells were injected into the tail vein of NOD/SCID mice (n = 10 per group); eight weeks later, the mice were sacrificed, and their lungs were removed and formalin fixed for haematoxylin and eosin (HE) staining.
Clonogenic assay
Request a detailed protocolClonogenic cell survival assay was performed to evaluate CRC and GC cells proliferation potential. In a 10 cm dish, a single cell suspension (250 cells) was added to each dish. Two weeks after plating, single colonies were fixed and stained by haematoxylin. Then colonies were counted and measured using the Nikon Nis Elements for windows software. Each assay was performed three times in triplicate.
Statistical analysis
Request a detailed protocolAll statistical analyses were performed using the SPSS 16.0 statistical software package (Abbott Laboratories, USA). Quantitative values of all experiments are expressed as the mean ±SD. The significance of correlation between the expression of indicated proteins and histopathological factors was determined using Pearson χ2 test. Survival curves were plotted by Kaplan–Meier method and compared by log-rank test. In vitro cell growth assay was tested using factorial design ANOVA. Comparisons between groups were performed with a 2-tailed paired Student's t-test. The correlation between the expression levels of two proteins was determined using Spearman's correlation analysis. In all samples, p<0.05 was considered to be statistically significant. Statistical significance was concluded at *p<0.05, **p<0.01, ***p<0.001; # represents no statistical significance.
Please see the supplementary materials and methods for additional experimental procedures.
References
-
Prognostic and predictive factors in breast cancer by immunohistochemical analysisModern Pathology : An Official Journal of the United States and Canadian Academy of Pathology, Inc 11:155–168.
-
Toward selective CK2alpha and CK2alpha' inhibitors: Development of a novel whole-cell kinase assay by Autodisplay of catalytic CK2alpha'Journal of Pharmaceutical and Biomedical Analysis 121:253–260.https://doi.org/10.1016/j.jpba.2016.01.011
-
Structural basis for the methylation site specificity of SET7/9Nature Structural & Molecular Biology 13:140–146.https://doi.org/10.1038/nsmb1045
-
A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stabilityNature Structural & Molecular Biology 18:42–48.https://doi.org/10.1038/nsmb.1939
-
Characterization of BHC80 in BRAF-HDAC complex, involved in neuron-specific gene repressionBiochemical and Biophysical Research Communications 322:601–608.https://doi.org/10.1016/j.bbrc.2004.07.163
-
Interpreting the protein language using proteomicsNature Reviews Molecular Cell Biology 7:391–403.https://doi.org/10.1038/nrm1939
-
Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeuticsMolecular & Cellular Proteomics : MCP 5:1799–1810.https://doi.org/10.1074/mcp.R600009-MCP200
-
Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-upAnnals of Oncology 24 Suppl 6:vi64–vi72.https://doi.org/10.1093/annonc/mdt354
-
A family portrait of the RIO kinasesJournal of Biological Chemistry 280:37297–37300.https://doi.org/10.1074/jbc.R500013200
-
Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammationThe Journal of Biological Chemistry 283:26771–26781.https://doi.org/10.1074/jbc.M802800200
-
Control of the hippo pathway by Set7-dependent methylation of YapDevelopmental Cell 26:188–194.https://doi.org/10.1016/j.devcel.2013.05.025
-
Expression pattern and first functional characterization of riok-1 in Caenorhabditis elegansGene Expression Patterns 15:124–134.https://doi.org/10.1016/j.gep.2014.05.005
-
The kinase activity of human Rio1 is required for final steps of cytoplasmic maturation of 40S subunitsMolecular Biology of the Cell 23:22–35.https://doi.org/10.1091/mbc.E11-07-0639
-
Factors associated with risk for colorectal cancer recurrence after endoscopic resection of T1 tumorsClinical Gastroenterology and Hepatology 12:292–302.https://doi.org/10.1016/j.cgh.2013.08.008
Article and author information
Author details
Funding
National Natural Science Foundation of China (81602149)
- Xuehui Hong
Natural Science Foundation of Fujian Province (2016J01619)
- Xuehui Hong
Zunyi Medical College Grant
- Zhiyong Zhang
Fujian Provincial Funds for Distinguished Young Scientists (2018D0016)
- Xuehui Hong
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Ethics
Human subjects: All human materials were obtained with informed consent and approved by the ethics committee of Hospital of Harbin Medical University (Protocol Number: 20150526).
Animal experimentation: All animal experiments were performed in accordance with NIH guidelines for the use of experimental animals. Male nonobese/severe combined immunodeficiency (NOD/SCID) mice between 4 and 6 weeks of age, obtained from the Experimental Animal Center of Shanghai Institute for Biological Sciences (SIBS). All animal work was conducted according to Institutional Animal Care Guidelines, and all animal experiments were approved by the ethical committee of the Harbin Medical University (Protocol Number: 20150619).
Copyright
© 2018, Hong et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,613
- views
-
- 446
- downloads
-
- 64
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 64
- citations for umbrella DOI https://doi.org/10.7554/eLife.29511
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
RETRACTED: Targeting posttranslational modifications of RIOK1 inhibits the progression of colorectal and gastric cancers
eLife 7:e29511.
https://doi.org/10.7554/eLife.29511




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}