Insights into the key determinants of membrane protein topology enable the identification of new monotopic folds
- Massachusetts Institute of Technology, United States
- Centro de Investigaciones Biológicas, Spain
Figures
Figure 1 with 1 supplement
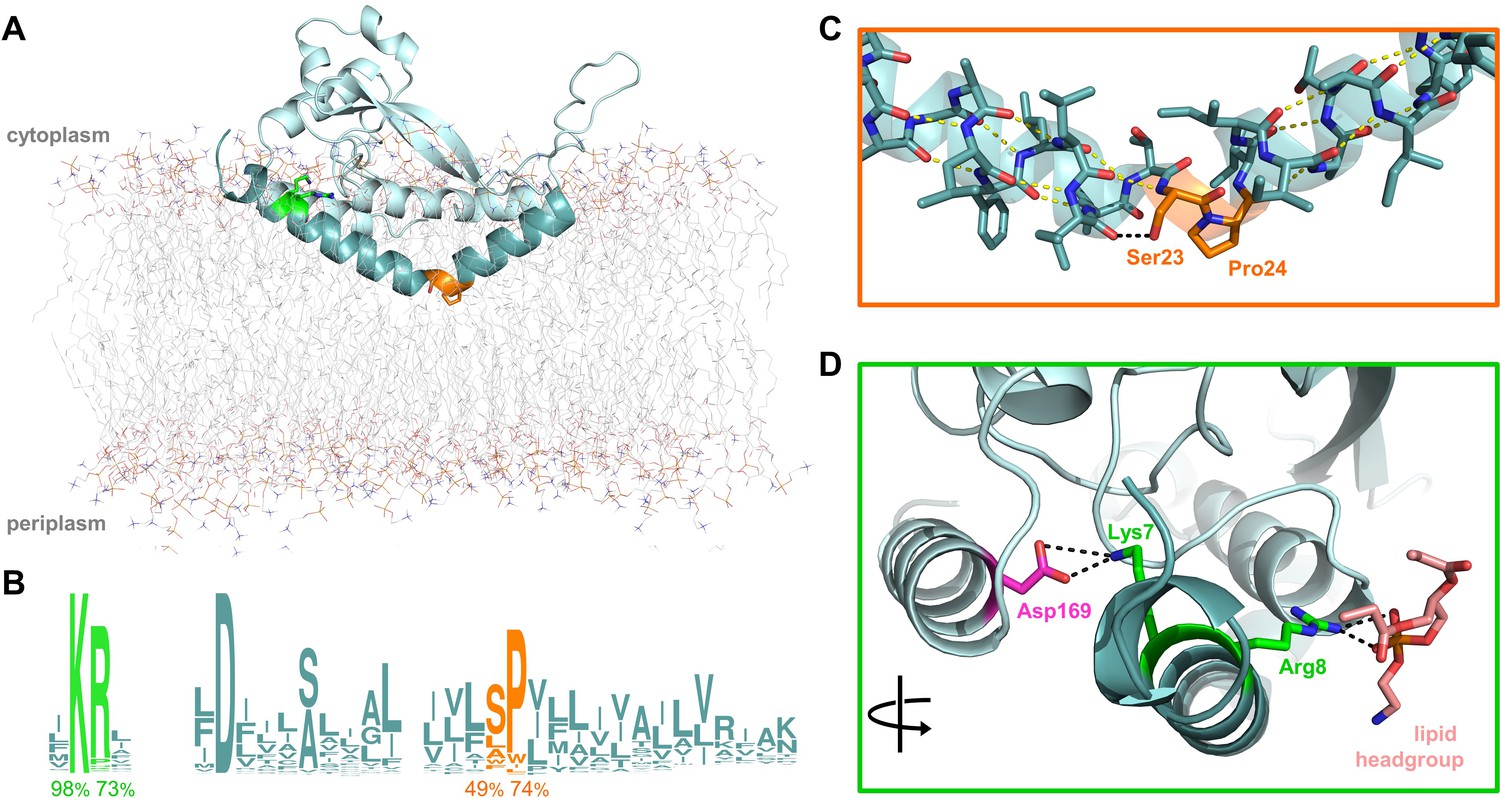
Overview of PglC highlighting the two conserved motifs in the RMH.
(A) Model of PglC from C. concisus in a POPE lipid bilayer. The RMH is shown in teal, Lys7 and Arg8 as green sticks, Ser23 and Pro24 as orange sticks. (B) Sequence logo showing conservation in the RMH domain among PglC homologs. Percent conservation is noted below each residue of interest. Logo generated using weblogo.berkeley.edu. (C) Detailed view of the Ser-Pro motif. Pro24 disrupts the hydrogen-bonding network (yellow dashes) of the RMH backbone. A 2.6 Åhydrogen bond (black dashes) is formed between the hydroxyl side group of Ser23 and the backbone carbonyl of Ile20. (D) Detailed view of the Lys-Arg motif. Lys7 forms a salt bridge with Asp169 (magenta). Arg8 interacts with a the headgroup of a copurified lipid (salmon).
Figure 1—figure supplement 1
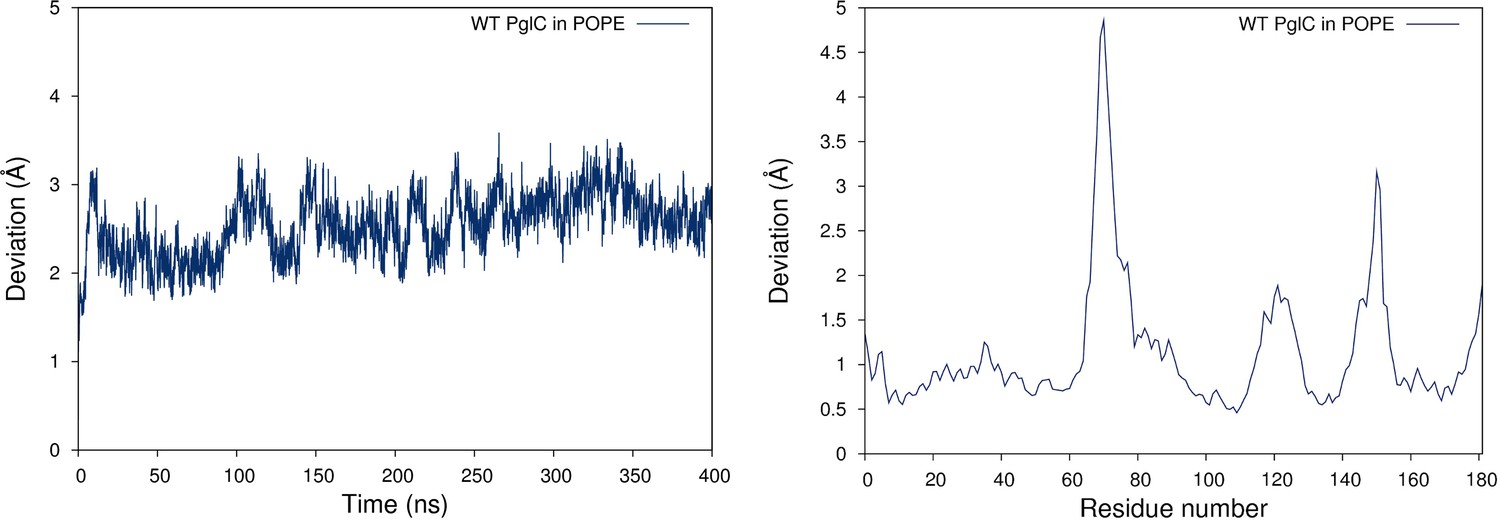
PglC is stable in a model POPE lipid bilayer.
RMSD (left panel) and RMSF (right panel) values measured over 400 ns of MD simulations of PglC in a model POPE membrane.
Figure 2 with 1 supplement
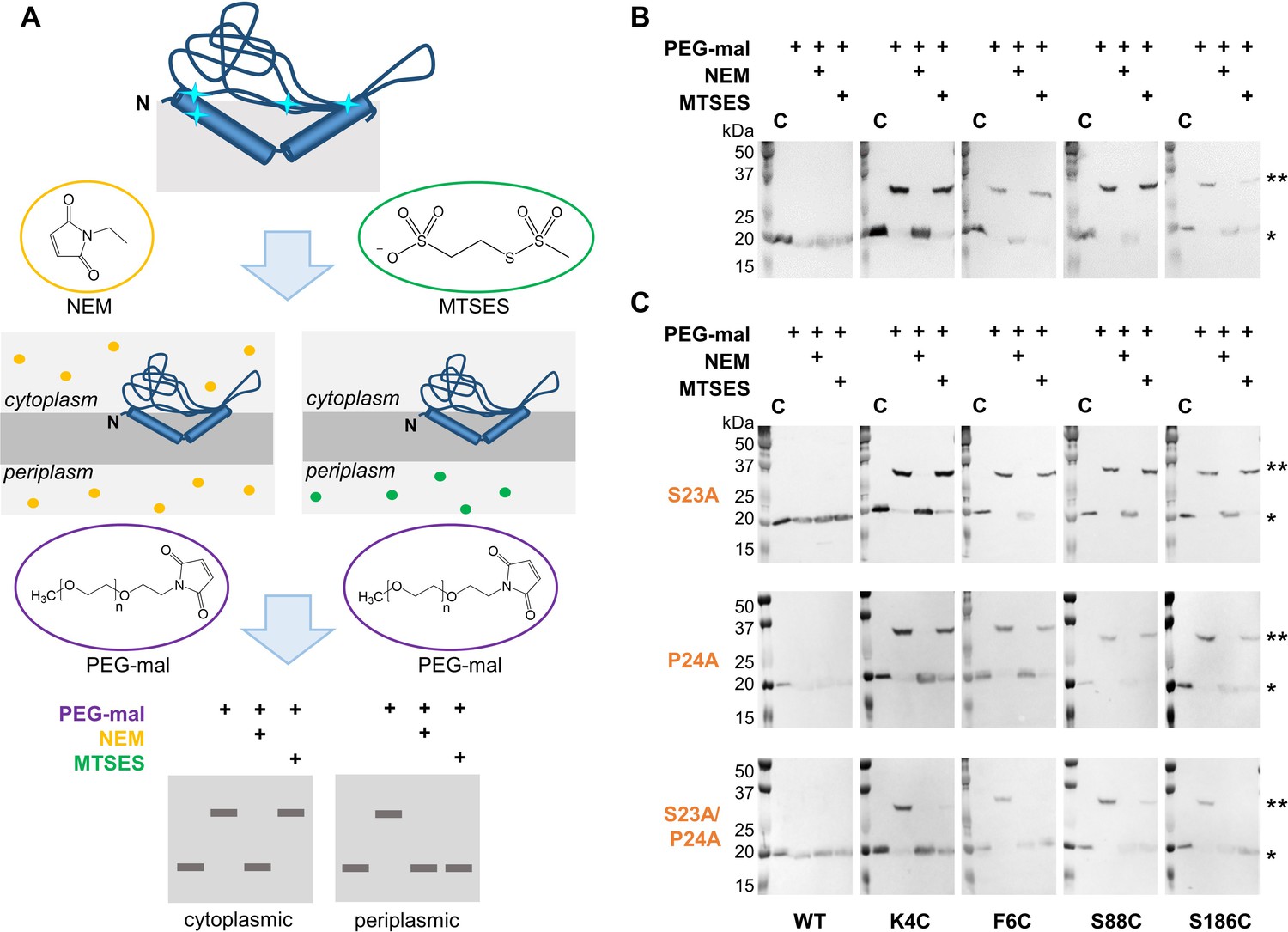
Ser23 and Pro24 act together to enforce the reentrant PglC topology.
(A) Schematic representation of SCAM analysis used to assess the topology of wild-type PglC and variants. Cyan starbursts (top) indicate the location of unique cysteines introduced into PglC. (B) SCAM analysis of wild-type PglC topology (* = native PglC; ** = PglC labeled with PEG-mal; C = control, no PEG-mal labeling). (C) SCAM analysis of S23A, P24A and S23A/P24A PglC variant topologies. All SCAM experiments were performed in duplicate or more. Representative Western blots are shown.
Figure 2—figure supplement 1
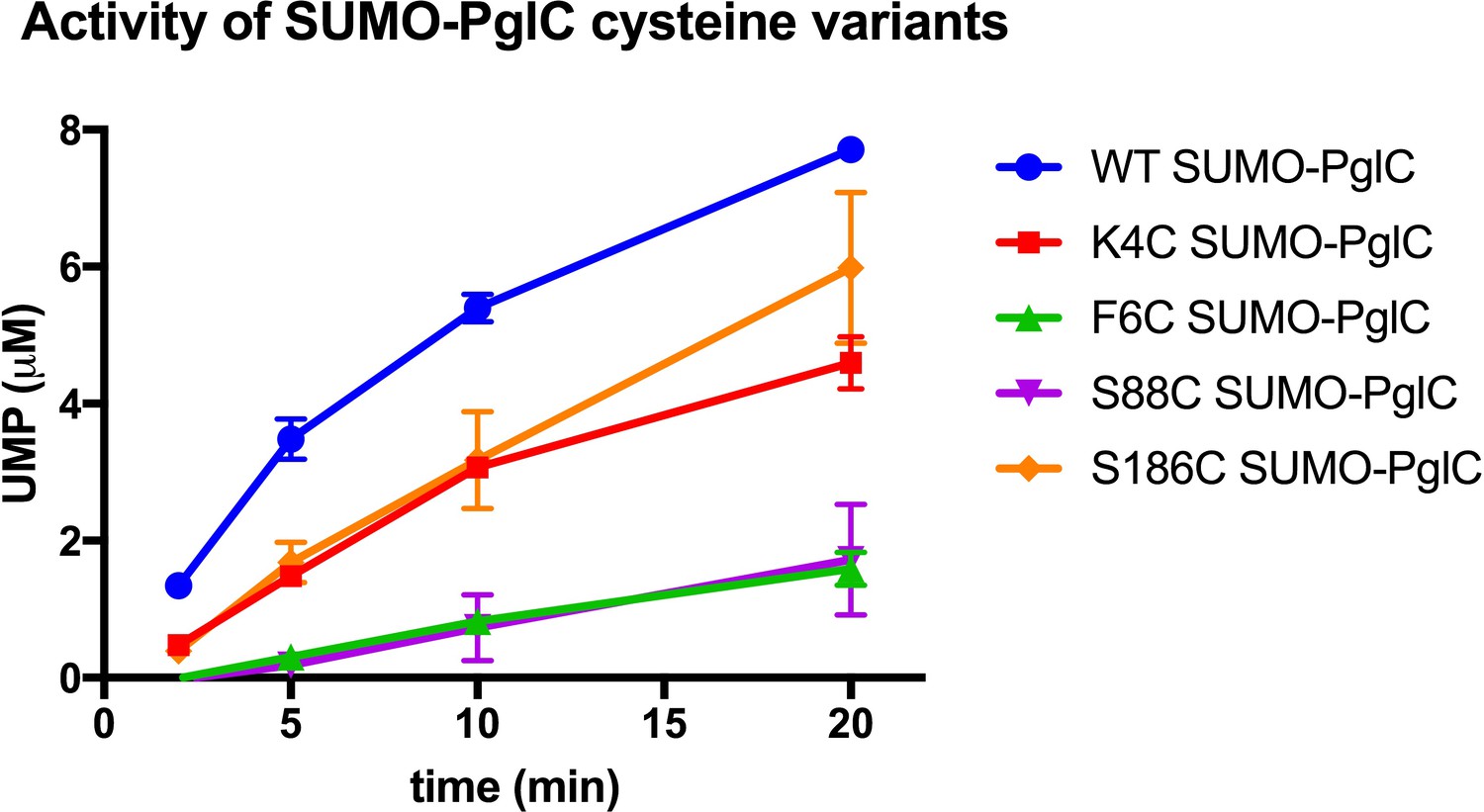
PglC Cys variants used for SCAM analyses.
Activity assays of wild-type SUMO-PglC and K4C, F6C, S88C and S186C SUMO-PglC using UMP-Glo to monitor UMP release. Assays indicate that PglC variants used for SCAM analyses retain 10–50% of native catalytic activity. Error bars are given for mean ± SD, n = 2.
Figure 3 with 1 supplement
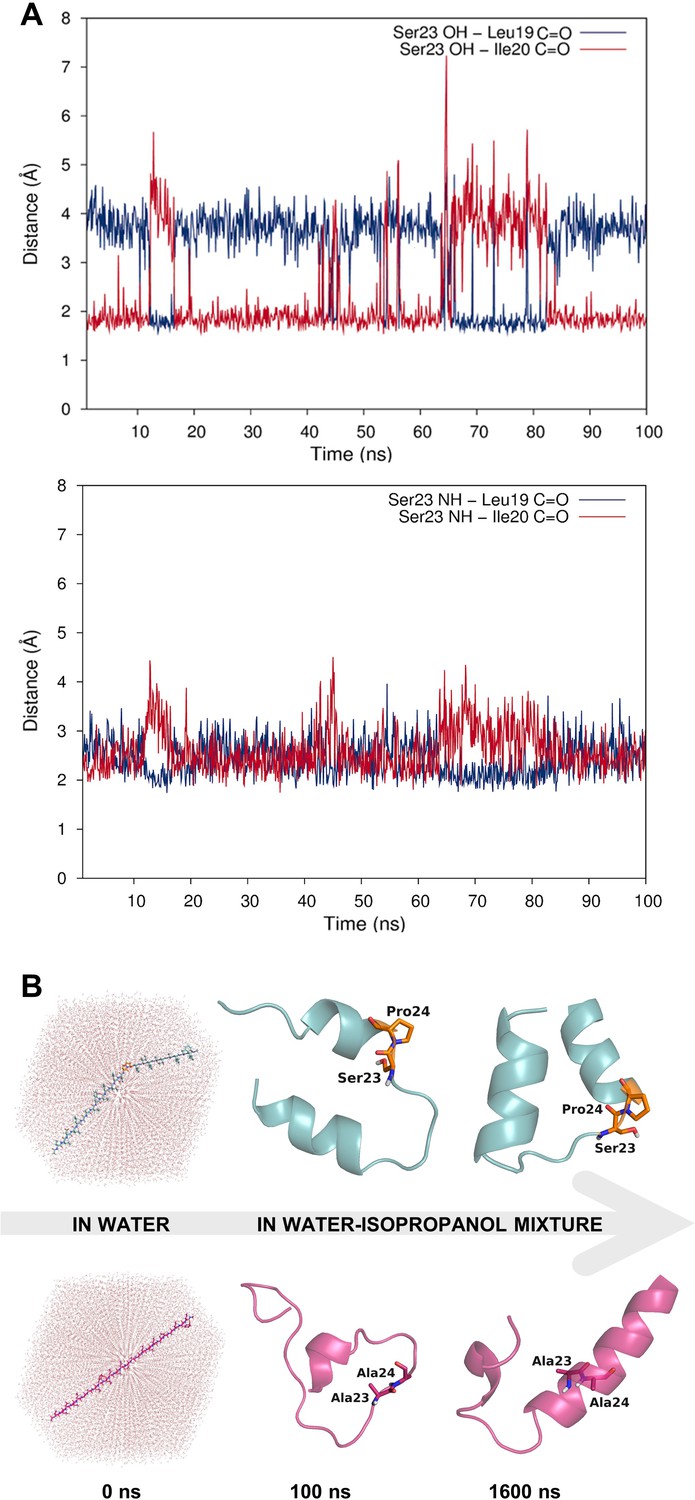
Dynamic hydrogen bonding around the Ser-Pro motif facilitates early folding of the RMH domain.
(A) Dynamic hydrogen bonding between the side chain hydroxyl (top panel) or backbone amide-NH (bottom panel) of Ser23 and the backbone amide-C=O of Leu19 and Ile20 measured during MD simulations of PglC in a POPE lipid bilayer. (B) Peptides corresponding to the RMH domains of wild-type (top) and S23A/P24A PglC (bottom) were folded from an extended conformation (left panel) for 100 ns in water (middle panel), followed by an additional 1500 ns in 20% isopropanol/water (right panel).
Figure 3—figure supplement 1
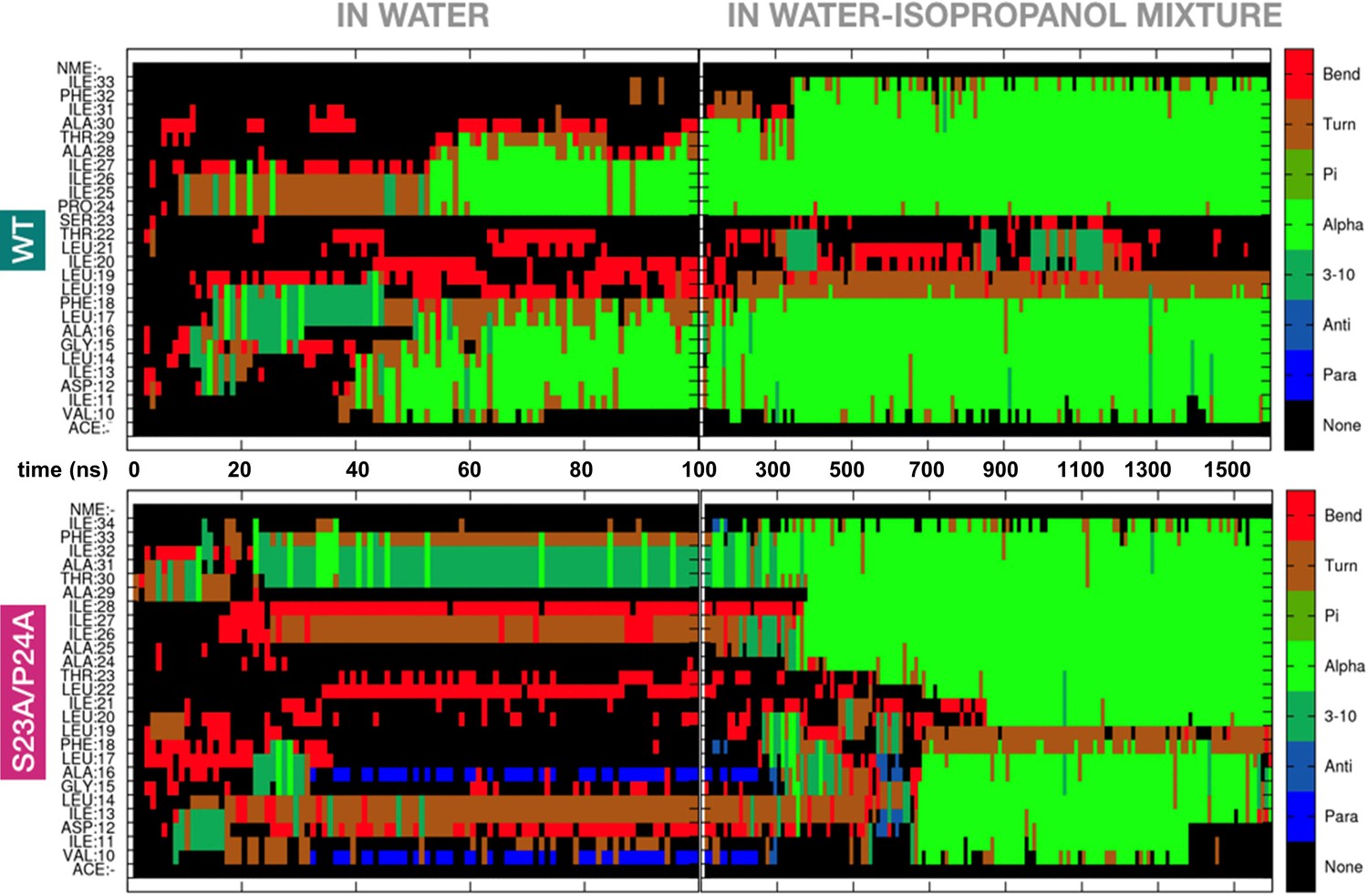
The Ser-Pro motif facilitates formation of the RMH domain.
Appearance of secondary structure in peptides modeling the RMH of wild-type (top panel) and S23A/P24A PglC (bottom panel) during folding simulations in water (for 100 ns) followed by 20% isopropanol/water (for an additional 1500 ns).
Figure 4 with 5 supplements

The Ser-Pro motif contributes to stability of the PglC fold.
(A) Thermal shift analysis of wild-type and S23A/P24A SUMO-PglC. Error bars are given for mean ± SEM, n = 3. (B) Superimposition of frames, taken at 10 ns intervals, along MD simulations of wild-type (top panel) and S23A/P24A (bottom panel) PglC. Colored from blue, t = 0 ns to red, t = 400 ns. PglC is represented as a semi-transparent cartoon and residues Leu21, Leu90 and Val180 as sticks.
Figure 4—figure supplement 1
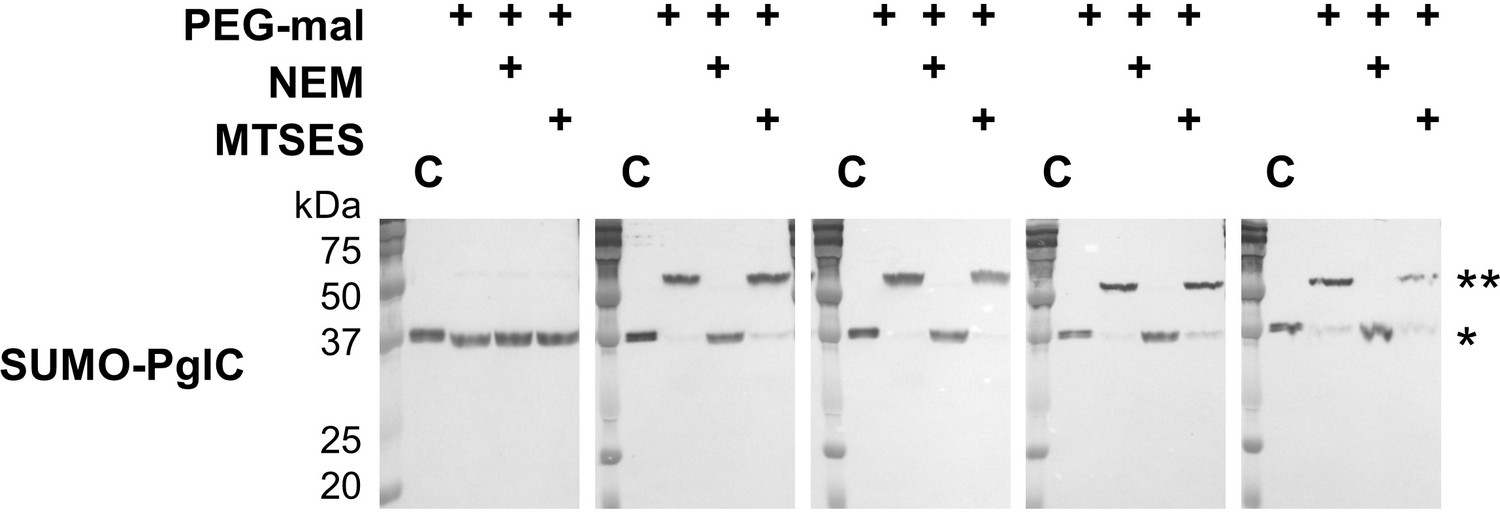
SUMO-PglC shows a reentrant topology similar to native PglC.
SCAM analysis of wild-type SUMO-PglC topology.SCAM experiments were performed in duplicate or more. Representative Western blots are shown.
Figure 4—figure supplement 2
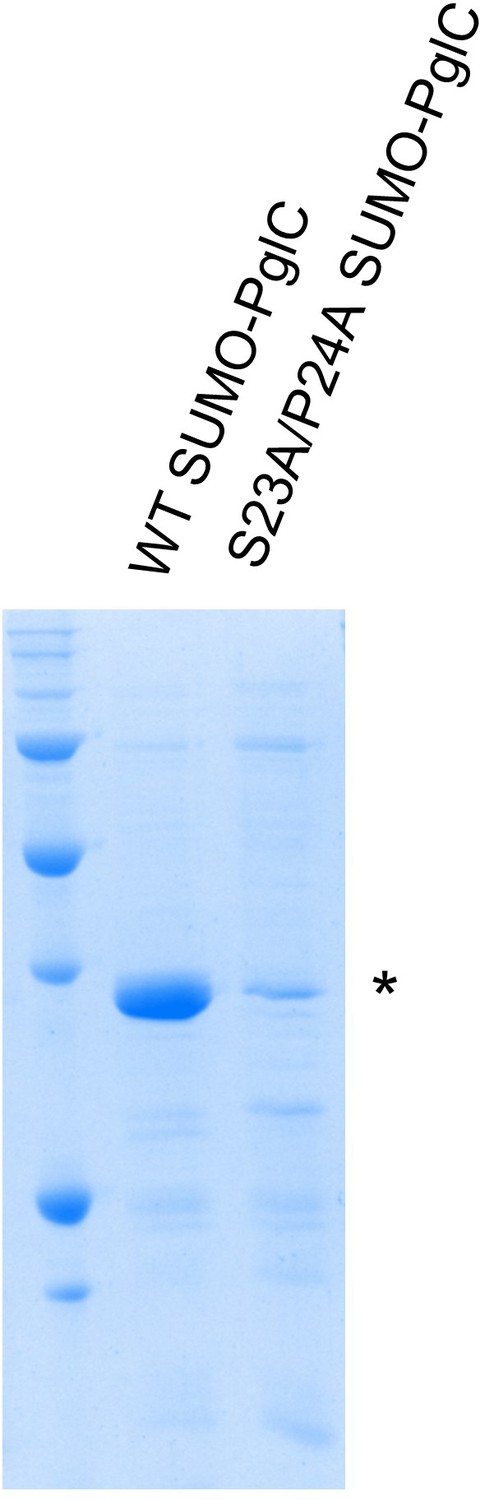
SDS-PAGE analysis of wild-type and S23A/P24A SUMO-PglC.
Comparative SDS-PAGE analysis of wild-type and S23A/P24A SUMO-PglC, Coomassie stain; *=SUMO PglC. Sample loading was normalized by UV absorbance at 280 nm.
Figure 4—figure supplement 3
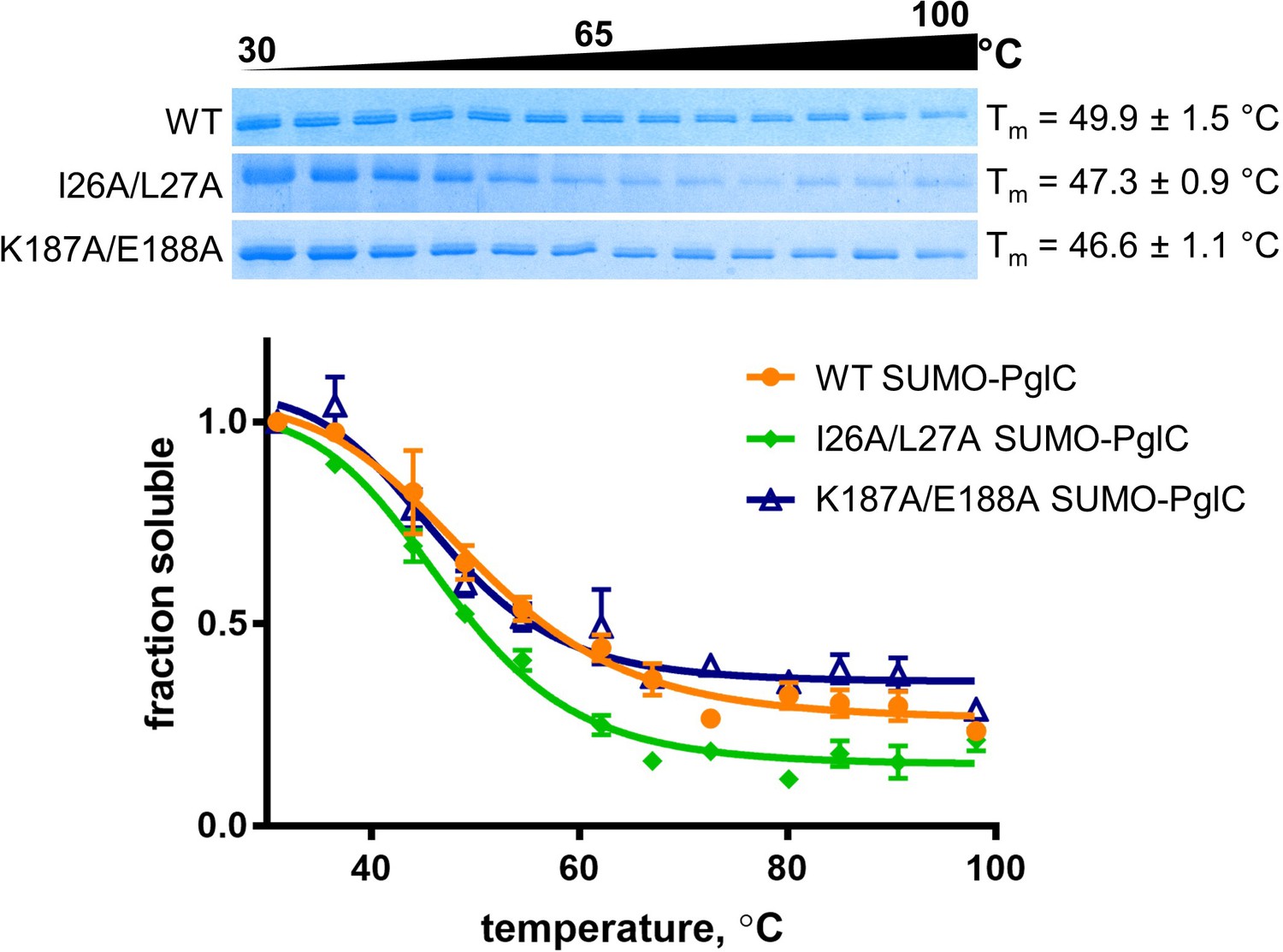
Thermal shift assays of control SUMO-PglC variants.
Thermal shift analysis of wild-type, I26A/L27A and K187A/E188A SUMO-PglC. Error bars are given for mean ± SEM, n = 3.
Figure 4—figure supplement 4
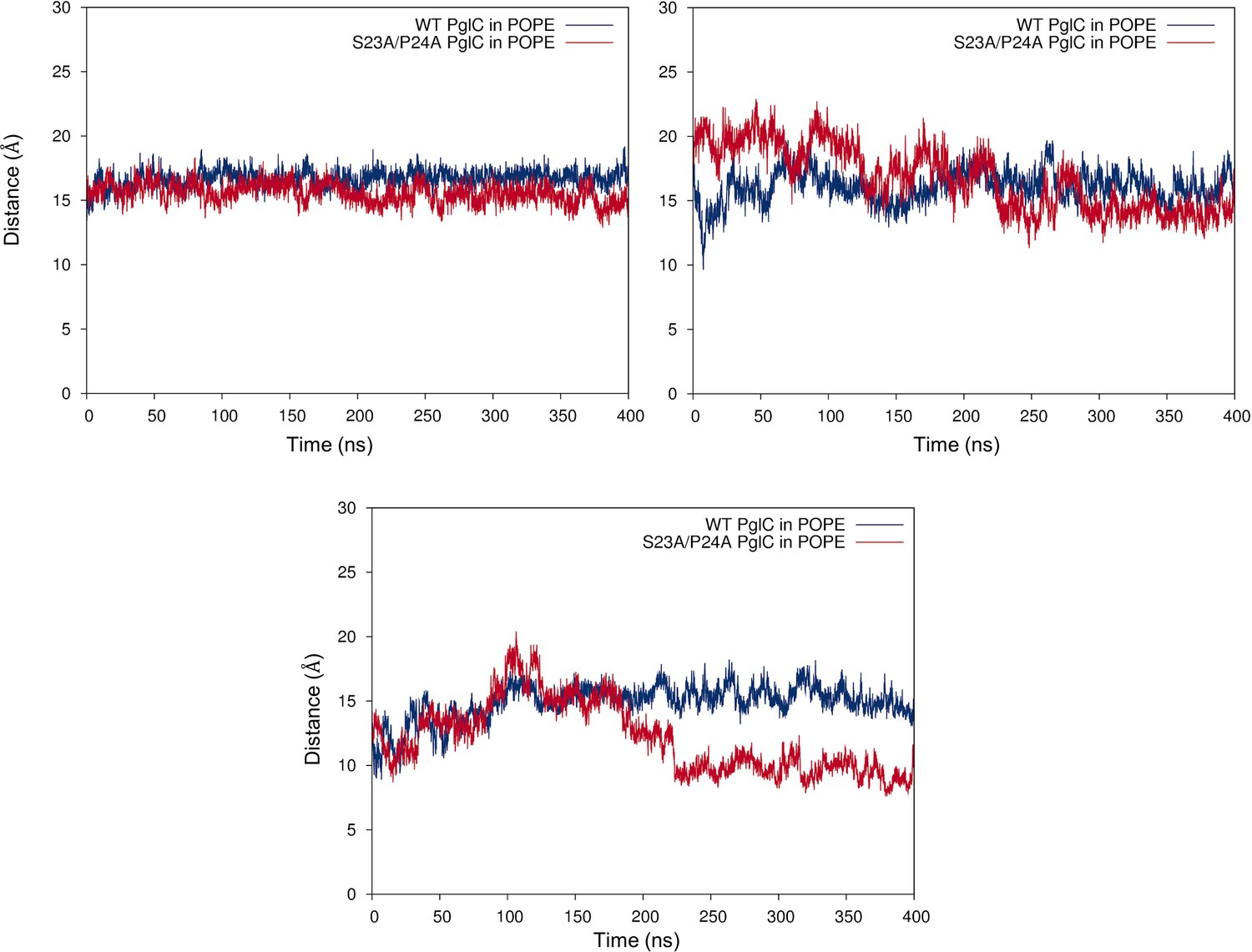
Mutation of the Ser-Pro motif causes a ‘collapse’ of the PglC fold interior.
Cα-Cα distances between Leu21 and Leu90 (top, left panel), Leu21 and Val180 (top, right panel) and Leu90 and Val180 (bottom panel), measured over 400 ns of MD simulations of wild-type and S23A/P24A PglC in a POPE membrane.
Figure 4—figure supplement 5
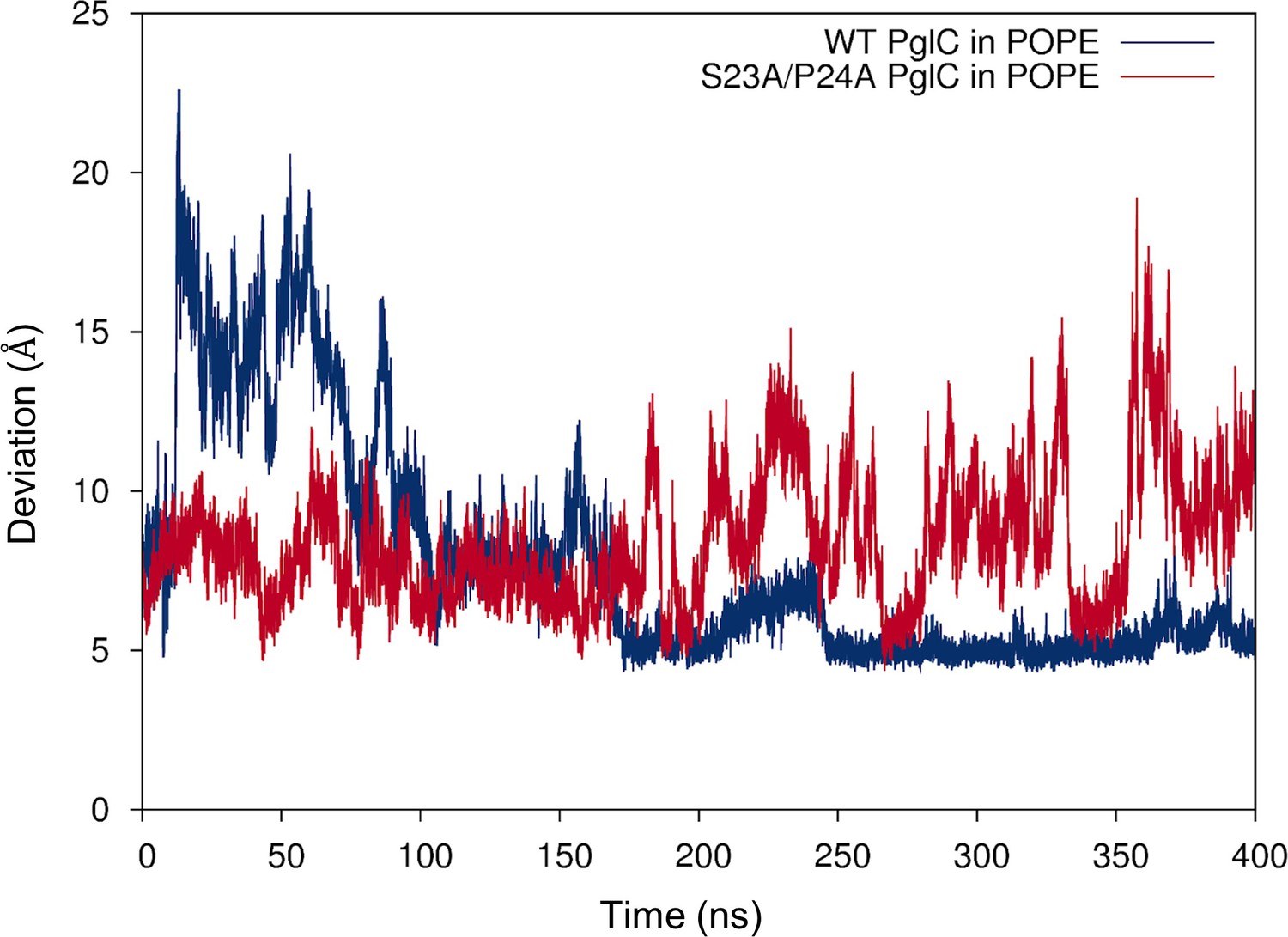
Mutation of the Ser-Pro motif reduces lipid occupancy in the PglC fold interior.
Distance between the geometric centers of the two lipids in the PglC fold interior, measured over 400 ns of MD simulations of wild-type and S23A/P24A PglC in a POPE membrane.
Figure 5

Lys7 and Arg8 additionally contribute to reentrant topology determination.
SCAM analysis of K7A, R8A, K7A/R8A and K7A/R8A/P24A PglC variant topologies (* = native PglC; ** = PglC labeled with PEG-mal; C = control, no PEG-mal labeling). All SCAM experiments were performed in duplicate or more. Representative Western blots are shown.
Figure 6
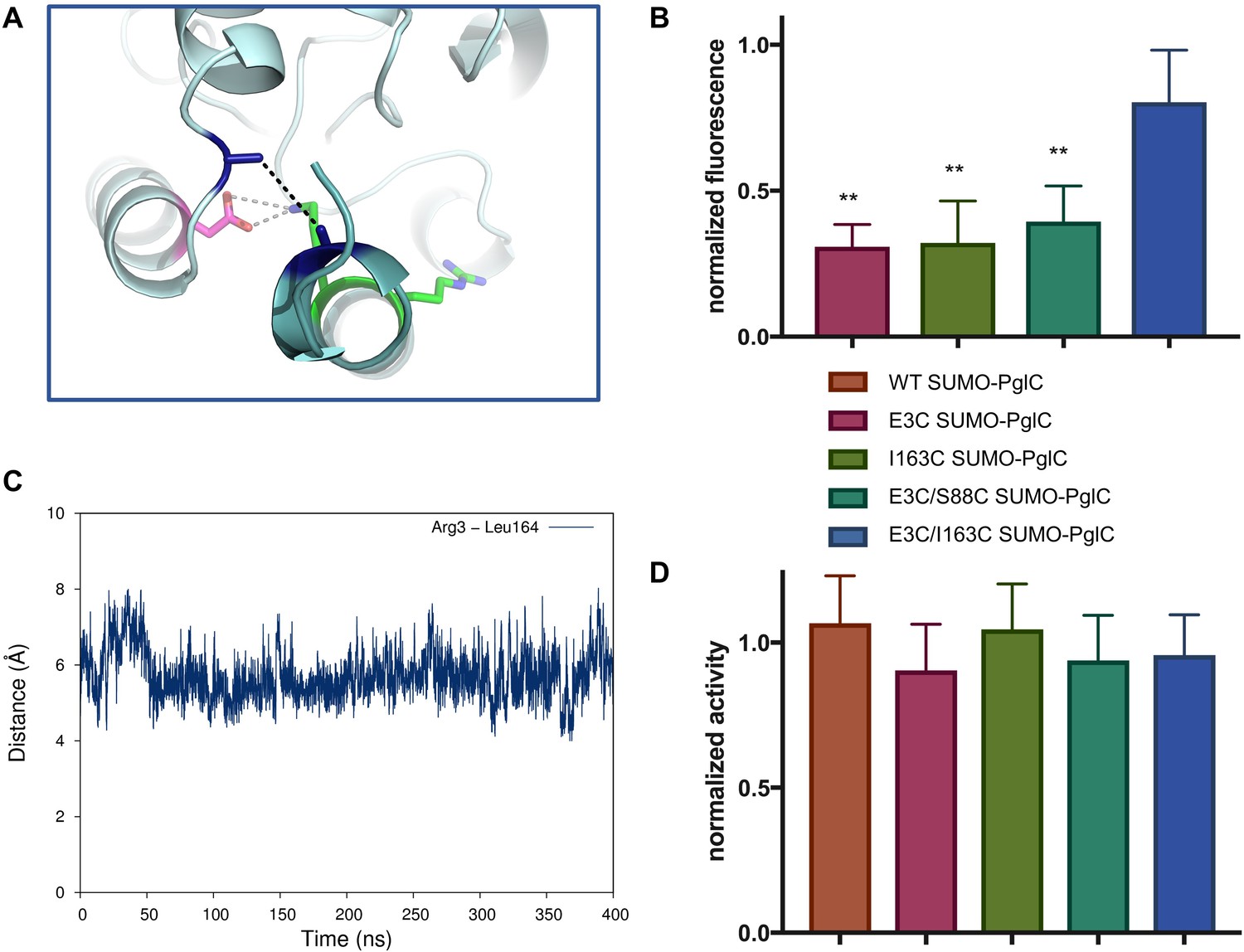
The RMH is held in the observed conformation during catalysis.
(A) Detailed view showing the location of bBBr crosslinking (black dashes). The Cα and Cβ of Arg3 and Leu164 in the structure of PglC from C. concisus are shown as dark blue sticks (the remainder of the side chains is omitted for clarity). The corresponding residues Glu3 and Ile163 in PglC from C. jejuni were substituted with Cys for bBBr crosslinking studies. (B) Fluorescence of SUMO-PglC variants following crosslinking with bBBr, normalized to fluorescence of DTT-quenched samples (quenched samples represent maximum possible fluorescence). Error bars are given for mean ± SD, n = 4 (**p<0.01, Student’s t-test; p-values for each variant are 0.0022 (E3C), 0.0055 (I163C), 0.0091 (E3C/S88C). (C) Distance between Arg3 and Leu164 (measured from the centroid of each residue), over 400 ns of MD simulations of PglC in a POPE membrane. (D) Activity of SUMO-PglC variants following crosslinking with bBBr, normalized to activity following treatment with vehicle. Error bars are given for mean ± SD, n = 3.
Figure 7
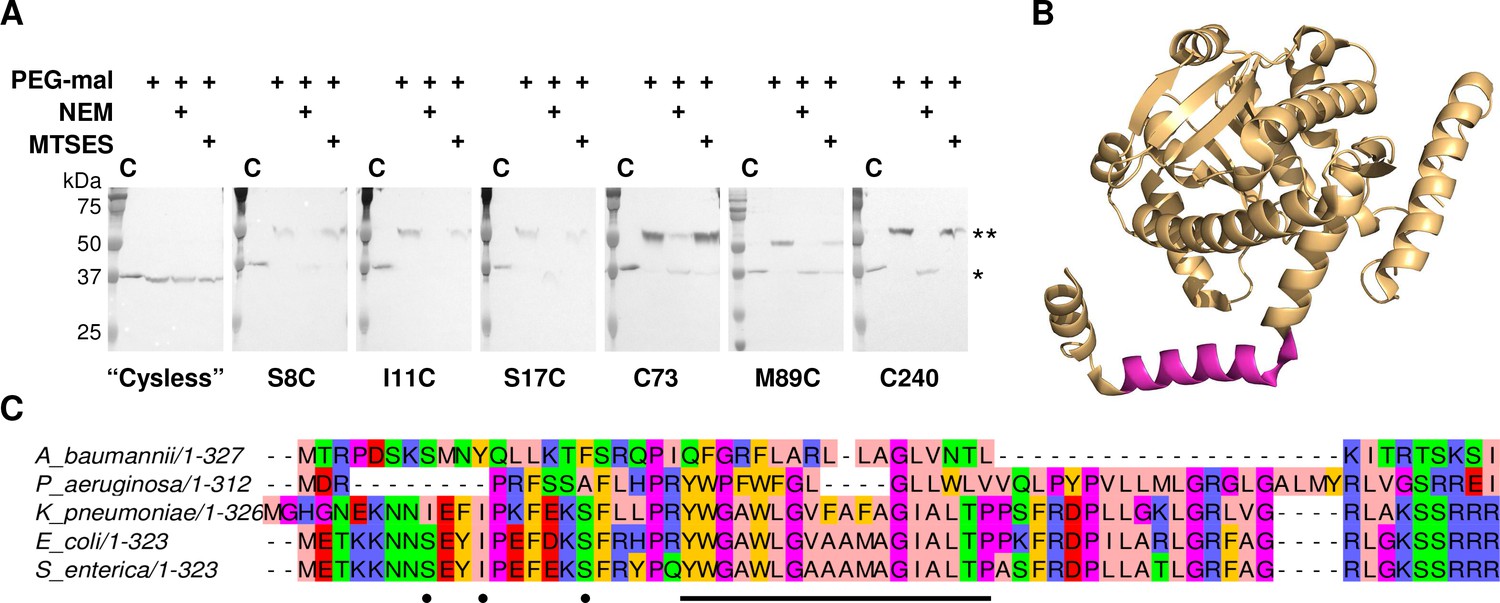
LpxM adopts a reentrant membrane topology.
(A) SCAM analysis on LpxM from E. coli indicates that the fold adopts a reentrant membrane topology rather than the predicted membrane-spanning one (* = native LpxM; ** = LpxM labeled with PEG-mal; C = control, no PEG-mal labeling). All SCAM experiments were performed in duplicate or more. Representative Western blots are shown. (B) The structure of LpxM from A. baumannii (Dovala et al., 2016); PDB 5KN7. The predicted transmembrane domain, as reported for the structure, is shown in pink. (C) Sequence alignment of LpxM from A. baumannii, Pseudomonas aeruginosa, Klebsiella pneumoniae, E. coli and Salmonella enterica. Only the N-terminus is shown. The predicted transmembrane region, corresponding to residues 23–40 of LpxM from E. coli, is underlined with a black bar. Black dots indicate the location of unique cysteines introduced into the N-terminus of LpxM for SCAM analysis.
Figure 8
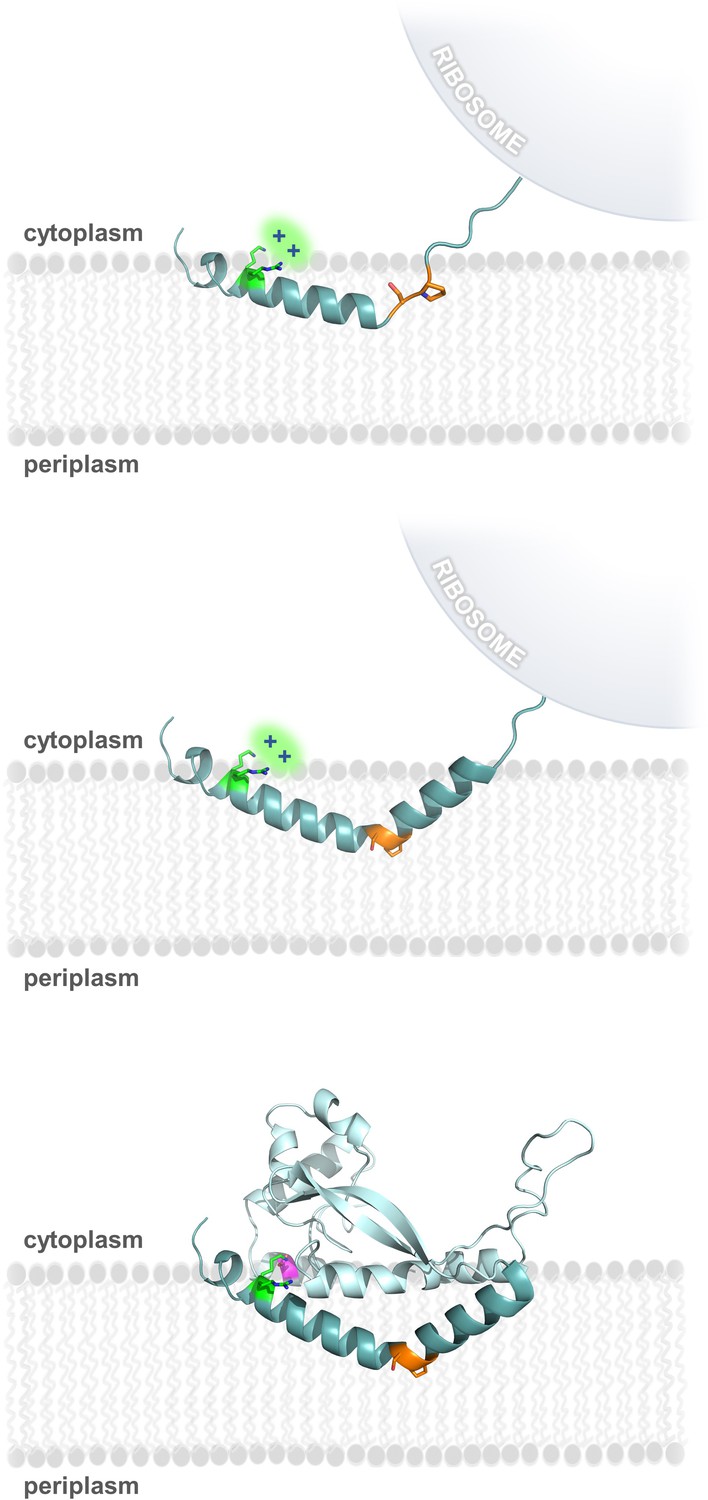
Model of RMH folding and membrane insertion.
The Lys-Arg (green) and the Ser-Pro (orange) motifs facilitate formation of the RMH and insertion into the membrane in an early co-translational event. The positively-charged Lys-Arg motif favors localization of the N-terminus to the cytoplasm (top panel). The Ser-Pro motif creates the characteristic break in the RMH (middle panel), resulting in insertion of the RMH into the membrane in a reentrant topology. Following translation of the globular domain, both motifs further contribute to the overall stability of the PglC fold (bottom panel).
Figure 9
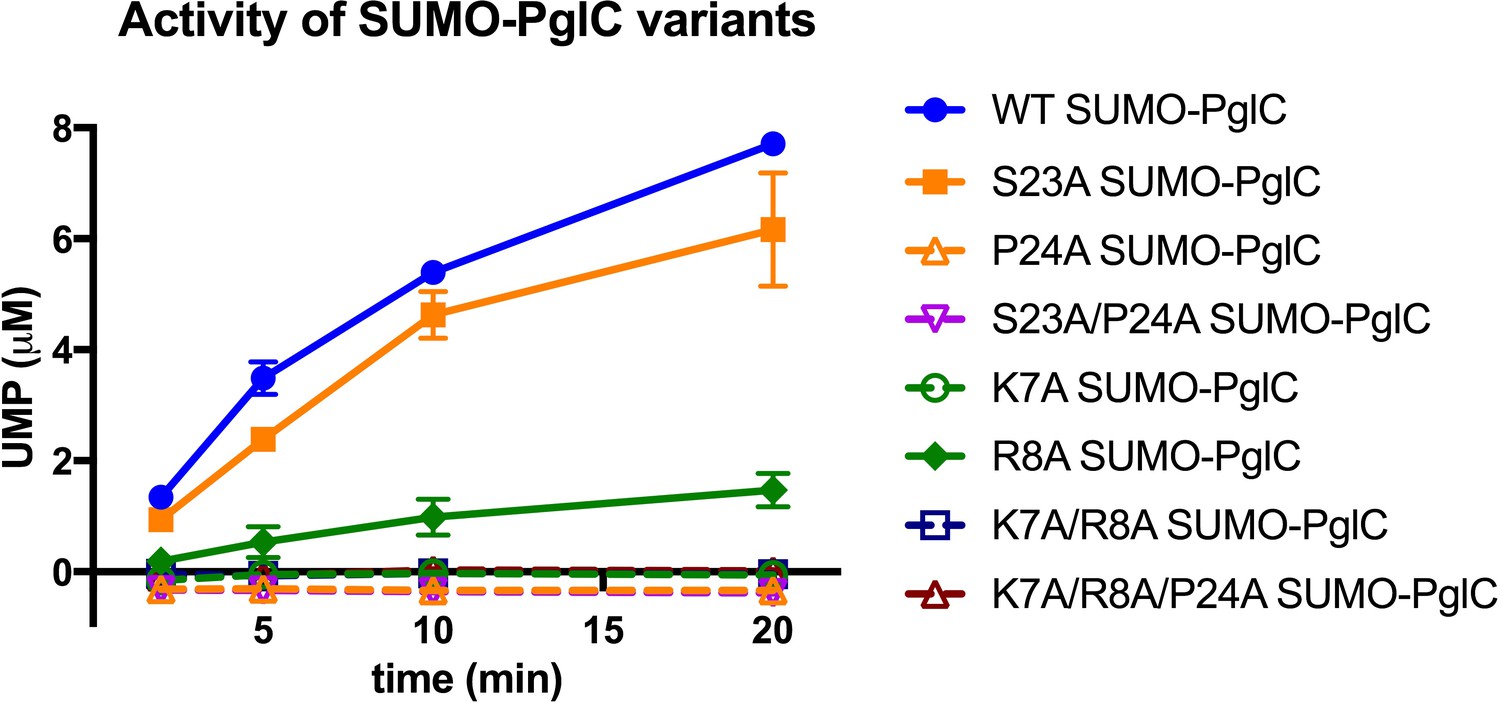
Individual residues differ in their importance for PglC function.
Activity assays of wild-type SUMO-PglC and variants using UMP-Glo to monitor UMP release. Error bars are given for mean ± SD, n = 2.
Tables
Table 1
Candidate reentrant domains identified among predicted bitopic proteins from E. coli.
Sequences were selected from the Membranome database of 196 known and predicted bitopic folds from E. coli, based on the criteria described in the text. Sequences highlighted in red represent the predicted transmembrane domain (TMHMM Server v2.0, http://www.cbs.dtu.dk/services/TMHMM/).
| Name | Function | Sequence of N-terminus (residues 1–50) | |
|---|---|---|---|
| HofN | Putative DNAutilization protein | MNPPINFLPWRQQRRTAFLRRLTGSAEARI | |
| LpxL | Lipid A biosynthesis lauroyltransferase | MTNLPKFSTALLHPRRLGCGLGKLALR | |
| LpxM | Lipid A biosynthesis myristoyltransferase | METKKNNSEYIPEFDKSFRHPRPKFRDPILAR | |
| LpxP | Lipid A biosynthesis palmitoleoyltransferase | MFPQCKFSREFLHPRYWLTRIGAMARP | |
| YihG | Probably acyltransferase | MANLLNKFIMTRILAAITLLKLLLPVP | |
Additional files
-
Supplementary file 1
Corresponding key residues in C. jejuni and C. concisus PglC
- https://doi.org/10.7554/eLife.40889.020
-
Supplementary file 2
Primers used for cloning and mutagenesis of PglC and LpxM variants
- https://doi.org/10.7554/eLife.40889.021
-
Supplementary file 3
Amino acid sequences for peptide folding
- https://doi.org/10.7554/eLife.40889.022
-
Supplementary file 4
Results of covariance analyses performed on sequences in Table 1 using the GREMLIN web-server (http://gremlin.bakerlab.org/).
Interactions between residues of the hydrophobic and globular domains are highlighted in red (>90% probability) and orange (80–90% probability).
- https://doi.org/10.7554/eLife.40889.023
-
Transparent reporting form
- https://doi.org/10.7554/eLife.40889.024
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Insights into the key determinants of membrane protein topology enable the identification of new monotopic folds
eLife 7:e40889.
https://doi.org/10.7554/eLife.40889




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}