Preacinetobactin not acinetobactin is essential for iron uptake by the BauA transporter of the pathogen Acinetobacter baumannii
- Wellcome Trust Centre of Human Genomics, England
- Research Complex at Harwell, Rutherford Laboratory, England
- University of Cagliari, Italy
- The University of St Andrews, United Kingdom
- Jacobs University, Germany
- The Rosalind Franklin Institute, England
Abstract
New strategies are urgently required to develop antibiotics. The siderophore uptake system has attracted considerable attention, but rational design of siderophore antibiotic conjugates requires knowledge of recognition by the cognate outer-membrane transporter. Acinetobacter baumannii is a serious pathogen, which utilizes (pre)acinetobactin to scavenge iron from the host. We report the structure of the (pre)acinetobactin transporter BauA bound to the siderophore, identifying the structural determinants of recognition. Detailed biophysical analysis confirms that BauA recognises preacinetobactin. We show that acinetobactin is not recognised by the protein, thus preacinetobactin is essential for iron uptake. The structure shows and NMR confirms that under physiological conditions, a molecule of acinetobactin will bind to two free coordination sites on the iron preacinetobactin complex. The ability to recognise a heterotrimeric iron-preacinetobactin-acinetobactin complex may rationalize contradictory reports in the literature. These results open new avenues for the design of novel antibiotic conjugates (trojan horse) antibiotics.
https://doi.org/10.7554/eLife.42270.001Introduction
Acinetobacter baumannii is a Gram-negative bacteria that has been identified by the WHO as a critical priority pathogen for new antibiotic development. A. baumannii was highlighted for its increasing role in nosocomial infections and its intrinsic resistance to multiple antibiotics. Several virulence factors have been identified in Acinetobacter species including iron-uptake pathways (Peleg et al., 2012; Harding et al., 2018). Iron is essential for all bacteria, and consequently, human pathogens have to possess an efficient acquisition system to scavenge iron from the host during infection. Gram-negative bacteria secrete small molecules (siderophores), which chelate iron with extremely high affinity. The resulting siderophore-iron complex is recognized by a specific outer-membrane transporter that couples to the TonB system to translocate the complex (and thus iron) into the periplasm where an ABC transporter takes it across the inner membrane (Noinaj et al., 2010).
The conserved and essential nature of the uptake system has drawn interest as a potential drug target. Several siderophore-conjugates have been developed to deliver drugs directly to the periplasm with promising results (Wencewicz and Miller, 2017). The successful compounds in part appear able to overcome the resistance of the bacteria to the antibiotic perhaps by increasing the local concentration. This ‘trojan horse’ strategy is potentially very general and could be used widely to specifically target bacteria and thus avoid the killing of the beneficial microbiome. Three classes of siderophore have been identified as being produced by A. baumannii, acinetobactin (Yamamoto et al., 1994), fimsbactins (Proschak et al., 2013) and baumannoferrins (Penwell et al., 2015) and each would be expected to be transported by distinct TonB-dependent transporters. Analogues of the bis-catechol siderophore fimsbactin A linked to antibiotics such as loracarbef/ciprofloxacin (Wencewicz and Miller, 2013), daptomycin (Ghosh et al., 2018) and cephalosporin (Liu et al., 2018) have proven effective in killing A. baumanni. A. baumanni has been also targeted by directly inhibiting the biosynthesis of acinetobactin (Drake et al., 2010; Neres et al., 2013) and by disrupting the acinetobactin uptake mechanism through the use of an oxidized version of acinetobactin (Bohac et al., 2017).
An important requirement for rational development of a trojan horse strategy is understanding the chemical nature of the siderophore and the structure activity relationship of its translocation, that is what parts are recognised and where cargo can be attached. The acinetobactin system is of particular interest since it has been identified as a virulence factor in mouse infection models (Gaddy et al., 2012). Acinetobactin is synthesized as a precursor, preacinetobactin, which rearranges non-enzymatically and irreversibly at pH >7 to give acinetobactin (Figure 1A) (Sattely and Walsh, 2008; Wuest et al., 2009; Shapiro and Wencewicz, 2016). There have been different reports on whether acinetobactin or preacinetobactin or both are transported into the bacteria (Song et al., 2017; Shapiro and Wencewicz, 2017) complicating rational design. The A. baumannii outer-membrane transporter protein, BauA, that recognises this siderophore has itself been identified as a vaccine candidate (Esmaeilkhani et al., 2016).
Figure 1 with 3 supplements see all
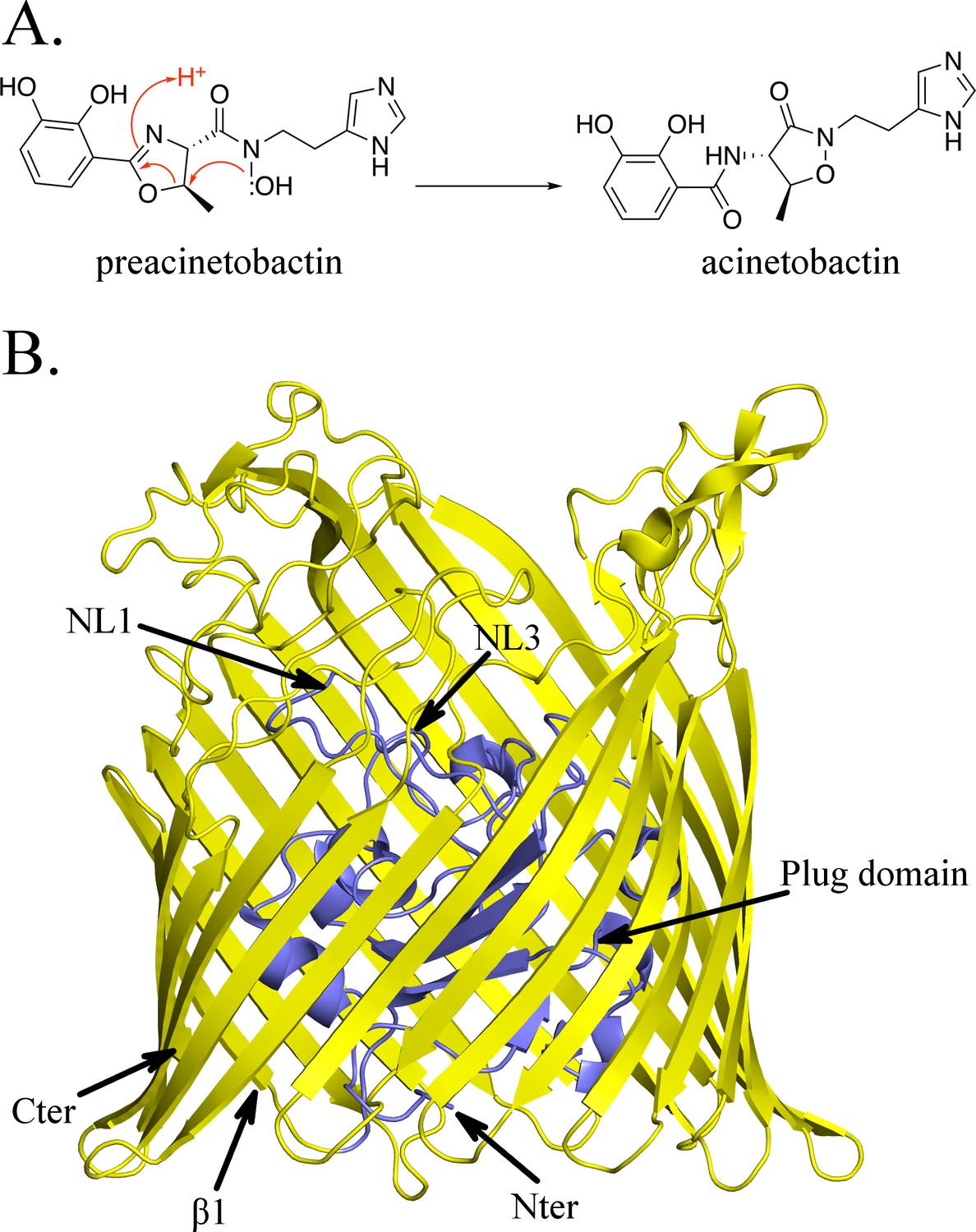
Structure of BauA, the (pre)acinetobactin transporter in A. baumannii.
(A) Schematic representation of the spontaneous isomerization of preacinetobactin into acinetobactin occurring at pH >7. (B) Overall structure of BauA. The N-ter plug domain is colored in blue and the β-barrel in yellow.
We report the structure of the BauA transporter bound to its cognate siderophore. Our structure reveals the molecular basis of recognition and sheds light on the question of which isomer is recognised. We establish that BauA recognises preacinetobactin not acinetobactin. Interestingly, we actually found that a mixed complex of both preacinetobactin and acinetobactin can be bound to the transporter. Under the most likely physiological conditions, low iron and neutral pH, the mixed species would seem the most plausible compound for uptake. Our data provide structural insights that can be used to guide rational design of ‘trojan horse’ antibiotics against A. baumannii.
Results
Structural biology
The apo structure of BauA was solved to a resolution of 1.8 Å by single wavelength anomalous diffraction (SAD) using selenomethionine-labeled proteins. Residues 33 to 703 of the protein are well defined in the electron density map, while the N-terminal region comprising the TonB box is missing, presumably because it is flexible. BauA has the typical outer-membrane TonB-dependent transporter (TBDT) fold with a β-barrel formed by 22 antiparallel strands, several extracellular loops, and a plug domain folded inside of the barrel (Figure 1B). Compared to the other TBDTs, the extracellular loops of the β-barrel form a large open cavity bordered by mainly short and apolar amino acids giving large access to the loops NL1-NL3 of the plug domain (Figure 1—figure supplement 1). Search of the structural database reveals the closest structural homologues are ferrichrome (Locher et al., 1998) (1by5, rmsd of 2.5 Å for 634 residues aligned and 2fcp, 2.4 Å for 631 residues) and enantiopyochelin (Brillet et al., 2011) (3qlb, rmsd of 2.5 Å for 630 residues) transporters. The asymmetric unit contains three monomers arranged around a threefold symmetry axis reminiscent of the porin arrangement (Figure 1—figure supplement 2A) and thus plausibly biologically meaningful. PISA (Krissinel and Henrick, 2007), a computational assessment of oligomeric state in the crystal, suggests a trimer with a high degree of confidence. Biophysical methods (gel filtration and light scattering) do not support a trimeric assembly. Size Exclusion Chromatography coupled to Multi-Angle Light Scattering (SEC-MALS) analysis in C8E4 is consistent with monomer whilst native gel shows the protein runs more closely to a dimer; however, the detergent bound to the protein may increase the apparent weight suggesting a monomer in solution (Figure 1—figure supplement 2B; Figure 1—figure supplement 2C).
We incubated separate native protein crystals with preacinetobactin and acinetobactin compounds (both compounds preloaded with Fe3+); surprisingly in both cases, we obtained the same structure: BauA bound to a heterocomplex Fe3+-preacinetobactin-acinetobactin (1:1:1) (Figure 2A). Given that preacinetobactin is the precursor for acinetobactin in a pH-dependent irreversible rearrangement reaction, it would explain this observation only for the preacinetobactin sample. LC-MS analysis of preacinetobactin in the crystallization conditions shows that in the absence of iron, the majority of preacinetobactin is converted to acinetobactin in 30 min and no preacinetobactin was observed the next day (Figure 3—figure supplement 1C); indicating that the presence of iron stabilizes preacinobactin. On the other hand, we discovered that there was a small amount (~2%) of preacinetobactin in our acinetobactin sample (Figure 3—figure supplement 1A). After full conversion of preacinetobactin into acinetobactin (Figure 3—figure supplement 1B), no crystal structure complex with BauA was obtained with this material.
Figure 2 with 3 supplements see all
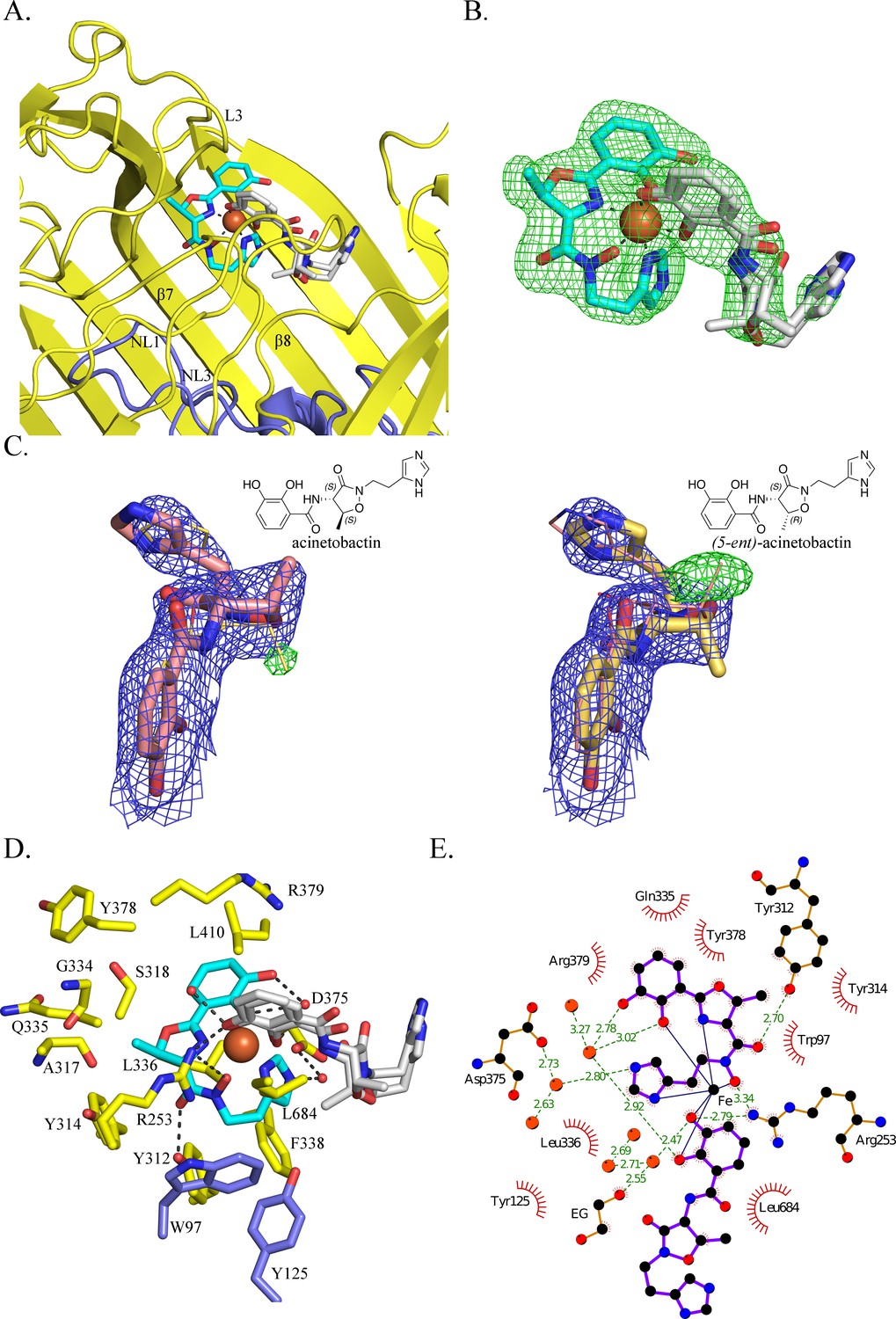
BauA binds a heterocomplex Fe3+, preacinetobactin and acinetobactin.
(A) Overall structure of BauA in complex with Fe3+, preacinetobactin and acinetobactin. The N-ter plug domain is colored in blue and the β-barrel in yellow. Preacinetobactin is shown as sticks with carbon atoms colored cyan and acinetobactin carbon atoms are in white, nitrogen in blue and oxygen in red. The Fe3+ is represented as an orange sphere. Secondary structure elements involved in the binding site have been labeled. (B) FO-FC electron density from the original omit map at 3 σ around Fe3+, preacinetobactin and acinetobactin complex. The color scheme of the Fe3+- siderophores is the same as A. We model the second molecule as a mixture of two diastereomers of acinetobactin. (C) 2FO-FC (blue) and FO-FC (green) electron density maps at 1 and 3 σ, respectively, around acinetobactin. In the left panel, only the acinetobactin (carbon atom represented in pink) has been added in the model before refinement. Position of (5-ent)-acinetobactin is shown in yellow line for information. In the right panel, only (5-ent)-acinetobactin (yellow stick) has been added in the model. Position of acinetobactin is shown in pink line for information. Schematic representation of both compounds has been added in the top corner for each image. (D) Binding site of the siderophores. Residues within 4.0 Å of the siderophores are displayed and hydrogen bonds are shown as black broken lines. Carbon atoms of residues of the β-barrel are in yellow and the ones of the plug domain in blue. (E) Ligplot diagram of Fe3+, preacinetobactin, acinetobactin bound to BauA. Covalent bonds of the siderophore and protein residues are in purple and brown sticks, respectively. Hydrogen bonds are represented by green dashed line and hydrophobic contacts are shown as red semi-circles with radiating spokes. One molecule of ethylene glycol is present in the binding site. Figure prepared with Ligplot (Wallace et al., 1995).
The coordination of the iron is octahedral with four coordination atoms from preacinetobactin (one hydroxy group of the catecholate, the hydroxy of the hydroxamate, the nitrogen of the methyl oxazoline and one nitrogen (N1) of the imidazole group) and two hydroxyls from acinetobactin (the catecholate) (Figure 2B). The Fe3+-preacinetobactin coordination geometry is similar to that observed for one Ga3+ in the gallium complex with anguibactin but the later forms a Ga2L2MeOH2 complex with two methanol molecules bridging the two four coordinate Ga3+ ions (Hossain et al., 1998). The net charge of the Fe3+-preacinetobactin complex assuming three deprotonated catecholates, one deprotonated hydroxamate and Fe3+ would be −1, consistent with stabilization by Arg253. The interaction between the catecholate and the nitrogen of the methyl oxazoline of the acinetobactin and the N1 of the imidazole group and the catecholate of preacinetobactin may play a role in the stabilization of the complex. A water molecule bridges the catecholate oxygen atoms of preacinetobactin.
The protein interacts mainly with the preacinetobactin molecule. The binding site is located inside the β-barrel similar to other siderophore complexes (Locher et al., 1998; Brillet et al., 2011; Chimento et al., 2005; Cobessi et al., 2005a; Cobessi et al., 2005b; Ferguson et al., 1998; Ferguson et al., 2002) (Figure 1—figure supplement 3; Figure 2—figure supplement 1; Figure 2A). The siderophores interact mainly with the residues of the β-barrel and the extracellular loops. BauA recognises the preacinetobactin through two direct hydrogen bonds between the hydroxamate group and Tyr312 of β7 and Arg253 of L3 and one interaction through a bridging water molecule between Asp375 and the nitrogen N2 of the imidazole, respectively (Figure 2D and E). The binding site is predominantly hydrophobic in character with van der Waals interactions involving residues Trp97, Tyr125, Tyr314, Tyr378, Gly334, Gln335, Leu336, Arg379 and Leu684. Although there are no hydrogen bonds between the siderophores and the plug domain, two residues of the plug domain (Trp97 and Tyr125) do make van der Waals interactions with preacinetobactin. The acinetobactin molecule makes only one hydrogen bond (Arg253) and one van der Waals interaction (Leu684). There are no changes in the TonB region as a result of ligand binding. The experimental electron density is most convincingly fitted by a mixture of two diastereomers of acinetobactin (Figure 2C), in an earlier co-crystalliszation experiment the ‘wrong’ diastereomer of acinetobactin was clearer (Figure 2—figure supplement 2F; Figure 2—figure supplement 2G. Consequently, we disfavor explanations for the electron density based on disorder or a mixture of acinetobactin/preacinetobactin (Figure 2—figure supplement 2). However, we find no evidence for the ‘wrong’ diastereomer in any NMR spectra of the starting materials which all match the literature report for the 'correct' diastereomer (Shapiro and Wencewicz, 2016; Takeuchi et al., 2010). Spontaneous chemical interconversion of the diastereomers seems implausible in simple solution. Thus, we are left to suggest the presence of small contaminating amount of the ‘wrong’ diastereomer or chemical conversion under crystallization conditions (protein, buffer, salts, crystal effects etc).
Siderophore binding
Three observations led us to speculate BauA preferentially recognises preacinetobactin; first, the protein ligand contacts are almost all with the preacinetobactin molecule, secondly the reduction of the preacinetobactin impurity to undetectable level gave no co-complex, and finally even a small amount of preacinetobactin as an impurity lead to complex formation. To test our hypothesis, we conducted isothermal titration calorimetry (ITC) experiments with both compounds (Figure 3A and Figure 3—figure supplement 2). These results are unambiguous: the Fe3+-preacinetobactin binds to BauA with nM affinity whilst Fe3+-acinetobactin shows no binding. The titration curves and thus affinity for the Fe3+-preacinetobactin complex and BauA differ depending on the ratio of Fe3+ to siderophore. A stoichiometry of 1:1 Fe3+: preacinetobactin has a Kd of 763 nM, whereas 1:2 Fe3+:preacinetobactin bind with 83 nM affinity. In both cases, the stoichiometry of binding was one Fe3+ to one protein; in neither case did we see any evidence for a biphasic recognition model that has been proposed between enterobactin and FepA (Payne et al., 1997).
Figure 3 with 5 supplements see all
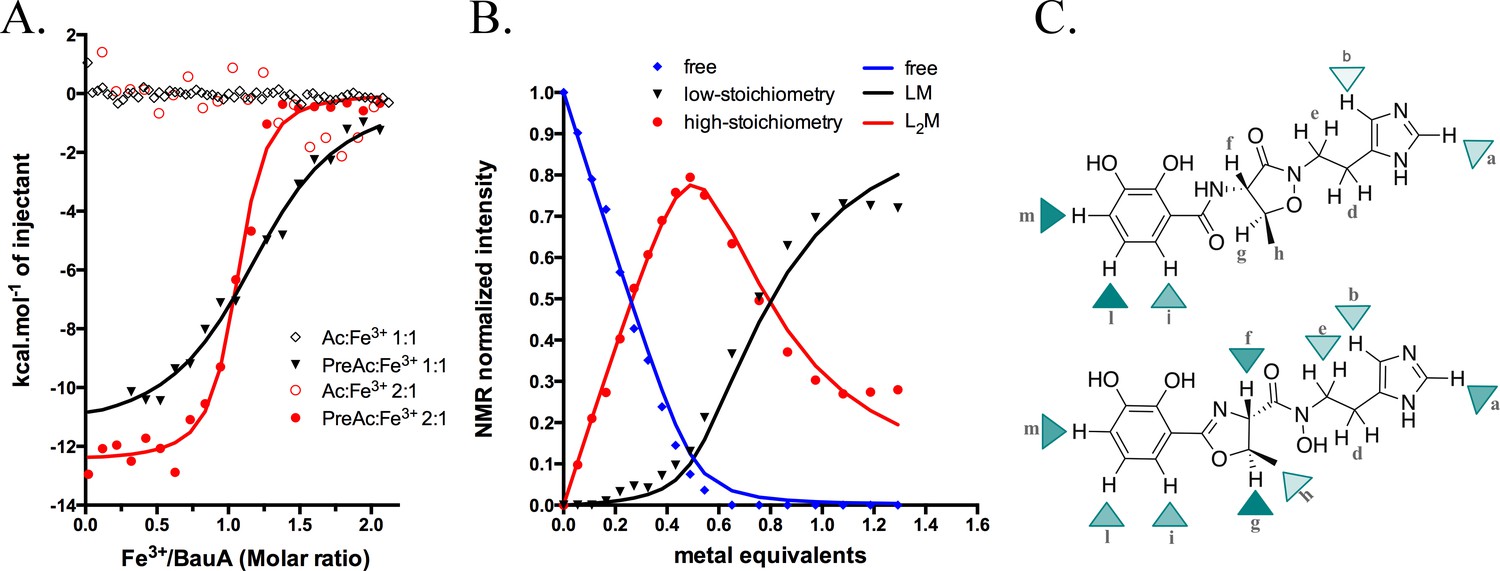
Siderophore iron complex stoichiometry.
(A) Isothermal calorimetry titrations of Fe3+-preacinetobactin (ratio 1:2 in red sphere and 1:1 in black triangle) and Fe3+-acinetobactin (ratio 1:2 in open red circle and 1:1 in open black lozenge). For Fe3+-preacinetobactin, the isotherm fitted with the Origin software gave a Kd of 83 and 763 nM for the ratio 1:2 and 1:1, respectively. With both ratios, no interaction has been detected for Fe3+-acinetobactin. (B) Results of curve fitting for titration of preacinetobactin in acetate buffered saline (ABS) at pH 5.0 (meter reading) with Ga3+. Results are shown for the proton resonance due to the methyl group and the curves correspond to the complexation model with the lowest total square deviation between the calculated (solid lines) and the experimental data (dots). Key: L, ligand; M, metal. (C) Molecular structure of acinetobactin (top) and preacinetobactin (bottom) is shown together with color-coded markers. Cyan triangles were used to represent the chemical shift difference observed in ABS between the resonance of the L2M complex and the corresponding one for the free L in the absence of metal ions (Table 1). Maximum colour intensity was set for the largest shift observed for each case and rescaling the other markers proportionally. Some markers are missing because corresponding resonances were not sufficiently resolved.
Siderophore-metal complex stoichiometry
The different affinities of Fe3+-preacinetobactin observed in ITC prompted us to examine the stoichiometry of the complex. Different molar ratios of metal to preacinetobactin were examined through 1H-NMR by using Ga3+ as a diamagnetic alternative to Fe3+ (Matzanke, 2006) (Figure 3—figure supplement 3). The conversion from preacinetobactin to acinetobactin is much slower at the lower pH used (pH 5) (Shapiro and Wencewicz, 2016), therefore on the timescale of the experiment, we ignored the small amount of acinetobactin. The titration reveals complexation of the free siderophore in the presence of low amounts of metal to give a complex of two siderophores to one metal, with the free siderophore almost disappearing when the metal reaches 0.5 molar equivalent (Figure 3B). Two different new sets of resonances were observed, both with chemical shift distinct from the free siderophore. This magnetic inequivalence clearly indicates two different ion coordination modes for each molecule of preacinetobactin in the ML2 complex. The only exception was the ethyl-imidazole group, which showed one set of resonances the same as free siderophore as well as a new series of different resonances. This is consistent with the imidazole being directly involved in metal coordination in one of the siderophores whilst in the other siderophore the imidazole is uncoordinated. As the amount of metal is further increased, a 1:1 complex of the siderophore with the metal appeared whilst the amount of the 2:1 complex decreased. This indicates there is an equilibrium in which the ML2 complex is favored at low metal concentration but the ML complex is predominant at higher metal concentrations. The formation constants were calculated as Kf(ML)=1.2·105 M−1; Kf(ML2)=7.2·103 M−1. Beyond 1.0 eq. Ga3+, a noticeable deviation between the model and the experimental data can be seen (Figure 3B). This is plausibly due to a fraction of acinetobactin present, which can coordinate some of the ML complexes formed by preacinetobactin giving the mixed complex seen in the structure. Preacinetobactin in these mixed complexes would be magnetically undistinguishable from those in the true ML2 complexes, thus artificially inflating the apparent concentration of this species.
The experiment was repeated with acinetobactin, however, titration was not possible beyond 0.35 molar equivalent of metal due to precipitation. We were able to observe the formation of a complex with concomitant decrease in free siderophore. The decrease in free siderophore versus ion concentration (up to 0.35) is more pronounced for preacinetobactin. NMR shows that metal binding to acinetobactin results in the largest shifts of signals within the catechol group, whereas in preacinetobactin the largest shifts are observed in the central portion of the molecule, where the hydroxamate moiety is located in agreement with the crystal structure (Figure 3C and Table 1).
Table 1
Chemical shift difference (absolute value of ppm in the absence – ppm in the presence of metal ions) is reported for the resonances of both preacinetobactin and acinetobactin during titration.
https://doi.org/10.7554/eLife.42270.016| 1H nuclei† | preacinetobactin‡ | Acinetobactin |
|---|---|---|
| a | 0.34 | 0.07 |
| b | 0.17 | 0.02 |
| d | n.r. | n.r. |
| e | 0.16 | n.r. |
| f | ~0.4§ | n.r. |
| g | 0.56 | n.r. |
| h | 0.12 | n.o. |
| i | 0.27 | 0.15 |
| l | 0.27 | 0.30 |
| m | 0.39 | 0.28 |
-
The titration performed in acetate buffered saline with Ga3+. Reported values correspond to 0.5 and 0.3 eq. Ga3+ in the case of preacinetobactin and acinetobactin, respectively.† Labels refer to Figure 3C.‡ Two resolved resonances were observed for protons g, h, i, l and m, due to two different L2M species (see the main paper). Average values are reported here.§ Complex overlapped with residual water’s resonance. Value estimated by comparison with additional titration performed in DMSO. n.r.: not sufficiently resolved. n.o.: difference not observed.
Discussion
The nature of one of the main siderophore molecules taken up by A. baumannii is important, both for understanding the physiology of this significant pathogen and for the rational design of the so-called ‘trojan horse’ antibiotics. Conflicting reports are extant in the literature as to whether the molecule which is taken up is preacinetobactin or acinetobactin (Song et al., 2017; Shapiro and Wencewicz, 2017). As our own experiments have shown, small amounts of impurities that are hard to eliminate coupled to ready conversion of preacinetobactin to acinetobactin poses significant technical challenges in resolving this problem.
The siderophore is known to be bound from the medium by the integral outer-membrane BauA protein. Structural biology strongly implies that BauA in fact recognizes preacinetobactin and not acinetobactin. In the crystal structure, preacinetobactin fits into a complementary pocket; a simple molecular model shows that the same site would not bind acinetobactin due to extensive van der Waals clashes with Tyr312 (Figure 2—figure supplement 3B). Preacinetobactin wraps around the metal ion by occupying four out of the six possible coordination sites. ITC confirms this hypothesis and demonstrates very tight binding for the Fe3+-preacinetobactin complex but none at all for acinetobactin; thus, for transport, preacinetobactin must be present. We note that our crystallization results show that even small amounts of contaminating preacinetobactin present in our original acinetobactin sample (detected only by careful chemical characterization after the fact), will bind to the protein, a reflection of the large difference in affinity between preacinetobactin and acinetobactin. We observed that in the absence of Fe3+, preacinetobactin rapidly converts to acinetobactin under the crystallization conditions. In our hands, the Fe3+ complex stabilized preacinetobactin by preventing the conversion to acinetobactin. This was also observed by NMR in solution with Ga3+ (Figure 3—figure supplement 4) and it agrees with the literature (Shapiro and Wencewicz, 2016).
We observed a heterotrimeric Fe3+-preacinetobactin-acinetobactin complex in the crystal. The acinetobactin molecule is not itself recognised by the protein, rather it is the favorable coordination of the catechol group to the remaining two sites of Fe3+ that drives complex formation. The lack of recognition would seem supported by our observation that in the crystal structure there is an unnatural diastereomer of acinetobactin playing the role of the second ligand (Figure 2—figure supplement 2; Figure 2C). In contrast, there was no electron density for the other diastereomer of preacinetobactin at the primary site consistent with simple modelling showing it would clash with the protein (Figure 2—figure supplement 3C). The second molecule in the complex, acinetobactin, has arisen from chemical conversion of preacinetobactin, a conversion known to occur rapidly in solution. We have no reason to predict that the conversion is in anyway affected by the crystal environment. The presence of preacinetobactin in the crystal is consistent with its chemical stabilization by Fe3+. Modeling suggests that the catechol of a second molecule of preacinetobactin would be able to bind to the unoccupied iron sites via its catechol without any clash with the protein (Figure 2—figure supplement 3D). We attribute the fact we do not observe it in preacinetobactin-soaked crystals as a consequence of the rapid conversion of catechol only ligated preacinetobactin into acinetobactin.
Based on the 1H-NMR experiment performed with Ga3+, we predict that at siderophore/iron ratio >0.8, the dominant complex is Fe3+-preacinetobactin (Fe3+L) whilst at siderophore/iron ratio <0.6, the Fe3+L2 is dominant. ITC suggests that the Fe3+L2 binds to BauA with higher affinity than the Fe3+L complex (Figure 3—figure supplement 2; Figure 3A).
Combining all our data, we suggest that under physiological conditions such as blood where free iron is very low, it is the heterotrimeric Fe3+-preacinetobactin-acinetobactin complex that is most likely to be bound by BauA and taken up (Figure 4a). Only under acidic conditions (pH <6), where preacinetobactin conversion is slower, is it conceivable that the Fe3+- (preacinetobactin)2 complex could be taken up. This maybe significant as A. baumannii infections do occur in an acidic environment (Peleg et al., 2008; Higgins et al., 2010). Our data cannot rule out the possibility that during uptake, the second ligand, bound only by the catechol group, is removed immediately before translocation. In this scenario only preacinetobactin would be transported but could convert to acinetobactin after transport when iron has been removed. However, in the light of the tendency to form stable ML2 complexes, removal of one L molecule is expected to be energetically unfavorable. The definitive resolution of this question is beyond the scope of this study.
Figure 4
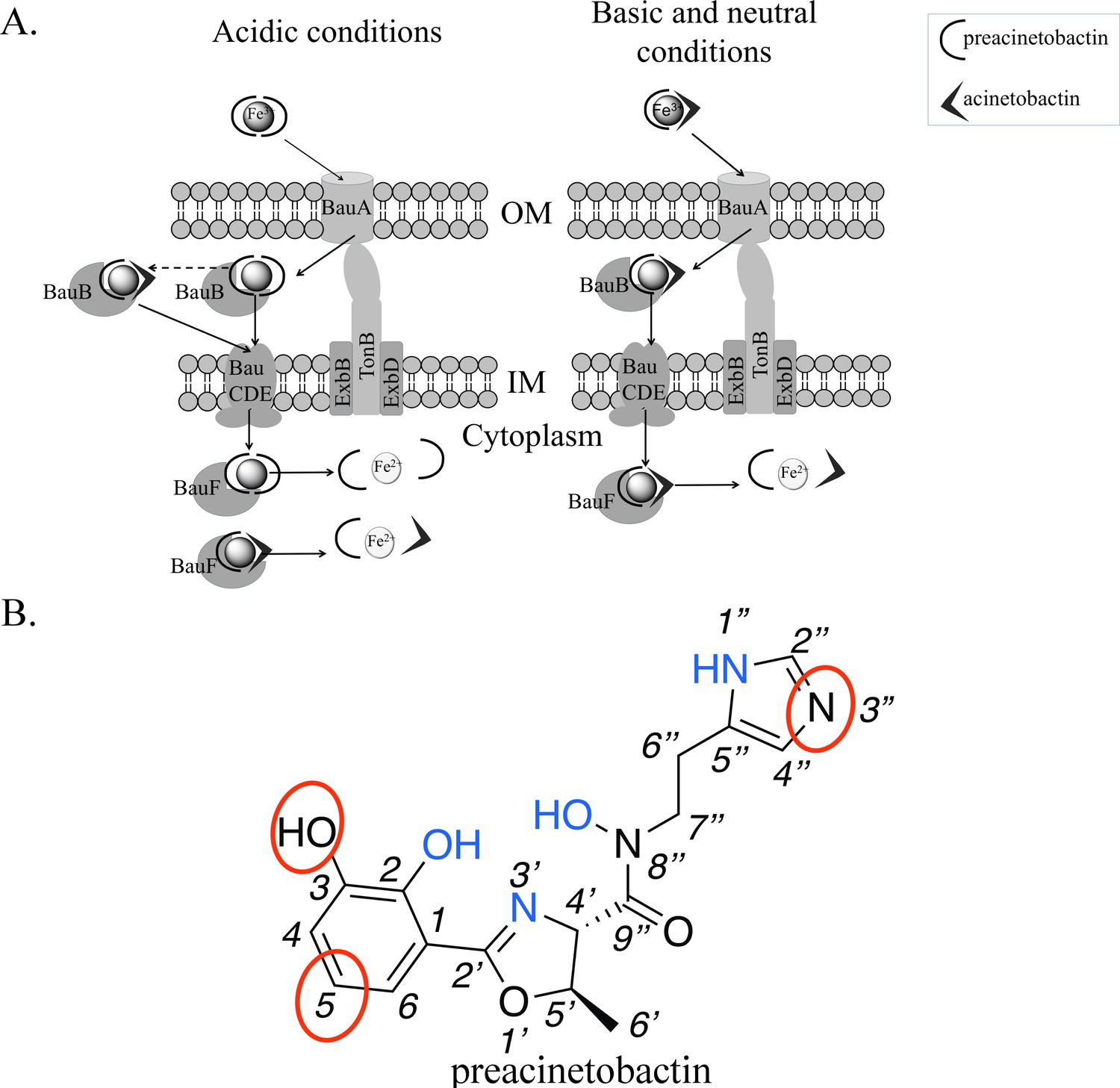
Model of preacinetobactin/acinetobactin uptake and opportunities for design.
(A) In acidic environments, preacinetobactin is stable. Two molecules can chelate Fe3+ and the complex could be stable thus recognised and transported by the TonB dependent transporter, BauA. Once in the periplasm, BauB, a periplasmic binding protein, delivers the Fe3+-siderophore to the inner membrane ABC transport system (BauCDE) to cross the inner membrane. The release of the Fe occurs into the cytoplasm with the help of BauF, a hydrolase. In neutral pH conditions, preacinetobactin isomerizes into acinetobactin. Fe3+ chelation stabilizes one molecule of preacinetobactin but the second molecule isomerizes. The heterotrimeric Fe3+-preacinetobactin-acinetobactin complex is recognised and possibly transported by BauA. (B) Functional groups involved in the Fe3+ chelation are highlighted in blue and the ones which could be modified in the design of siderophore antibiotic conjugates are circled in red.
Our data would appear to resolve the controversy in the literature as to the nature of siderophore taken up by BauA transporter. A complex containing both preacinetobactin and acinetobactin will bind to BauA and may in fact be taken up, but the presence of preacinetobactin is essential. Our work does not eliminate the possibility of another TBDT transporting acinetobactin, but so far none has been identified in A baumannii. It will be interesting to study whether the BauA transporter when it recognises preacinetobactin can ‘pull’ through catechols other than acinetobactin which bind to the two vacant iron coordination sites. The stabilization of preacinetobactin by formation of the tetradentate Fe3+ complex resolves the puzzle as to how a chemically unstable molecule is transported.
The structure outlines two new routes for the rational design of siderophore conjugates that could be taken up via BauA. The first approach is modification of preacinetobactin. The crystal structure suggests the uncoordinated nitrogen atom of the imidazole ring has the potential for modification without affecting recognition. Similarly, one of the oxygens and a carbon of catechol seem plausible candidates for expansion (Figure 4b). Shapiro and Wencewicz (2017) and Song et al. (2017) reported important indications about the role of the different portions of both ligands by comparing the biological activity of multiple derivatives and checking complex formation through UV-vis spectroscopy. These studies confirm the 2-OH of preacinetobactin is essential but changes at the 3 and 5 positions of the catechol are well tolerated (no information on the four position was reported). The second approach would be to design a catechol antibiotic conjugate that forms a high-affinity heterotrimeric complex with iron and preacinetobactin. The structure suggests that there is no protein imposed recognition requirement on the second molecule, this potentially unlocks a diversity of conjugate molecules complementing the fimsbactin A type conjugates (Wencewicz and Miller, 2013; Ghosh et al., 2018; Liu et al., 2018). The third approach would be the design of a molecule that hexacoordinates the iron molecule, whilst preserving the protein preacinetobactin contacts thus binding to BauA. This compound with its much higher affinity would easily outcompete preacinetobactin for iron and could be designed so as to block translocation by BauA.
Materials and methods
Preparation of the substances
Request a detailed protocolAcinetobactin and preacinetobactin were synthesized following the synthetic schemes and the protocols described by Takeuchi et al., 2010. The collected 1H-NMR data were similar to the 1H-NMR described by Takeuchi et al. (2010) and Shapiro et al. (Shapiro and Wencewicz, 2016; Shapiro and Wencewicz, 2017)(Figure 3—figure supplement 3 and 5).
BauA cloning
Request a detailed protocolPosition of the cleavage of the signal peptide of the proteins was predicted with Signal P4.0 (Petersen et al., 2011). The coding sequence of the mature protein BauA (uniprot Q76HJ9) was amplified from the genomic DNA ATCC 19606 using the primers 5'- AGGGGCGCCATGGCTGTTATTGATAATTCAACAAAAACTC and 5'- AATTTGGATCCTTAAAAGTCATATGATACAGATAGCATATACG and Pfu DNA polymerase. PCR product has been digested by NcoI and BamHI restriction enzymes. The gene was cloned with an N-terminal tobacco etch virus (TEV) protease cleavable His7-tag in pTAMAHISTEV vector using NcoI and BamHI restriction sites.
Over-expression in E. coli and purification
Request a detailed protocolThe proteins were over-expressed in E. coli C43 (DE3) cells. Cells were grown at 37°C in LB medium containing 100 μg.ml−1 ampicillin until an OD600 of 0.7 and then induced with 0.4 mM IPTG at 25°C overnight. The proteins were purified following the protocol used for AbPirA as described in Moynié et al. (2017). Isolated outer-membrane pellets were solubilized with 7% octylpolyoxyethylene (octylPOE)) and the proteins were purified with a Ni2+–NTA affinity chromatography followed by the His-tag cleavage by TEV protease and a second IMAC column. Finally, the protein was loaded on a Superdex S200 gel filtration chromatography (10 mM Tris pH 7.5, 150 mM NaCl, 0.45% C8E4) and concentrated to 10 mg.ml−1 before setting up the crystallization plates. Selenomethionine-substituted proteins were produced with the SelenoMethionine Medium Nutrient Mix (Molecular Dimension) in C43(DE3) cells according to the feedback inhibition method (van den Ent et al., 1999). Purification of the protein was the same as the native but with 5 mM β-mercaptoethanol added in all purification buffer.
Structural biology
Request a detailed protocolCrystals of BauA appeared at 20°C after a few days by mixing 1 μl of protein solution (10 mg.ml−1) with 1 μl of reservoir solution containing 10% PEG 8000, 0.1 M Hepes pH 7.5, 0.1M KCl, and 10% ethylene glycol. Crystals were frozen with the same solution containing 26% ethylene glycol. Complex structures were obtained by soaking apo crystals for few hours with mother liquor containing 5 mM Fe3+-acetylacetonate siderophore (preacinetobactin or acinetobactin) in 1:1 ratio (Fe3+: siderophore) before cryoprotection. Subsequent LC-MS analysis showed that the acinetobactin sample had a small amount (~2%) of preacinetobaction present. Preacinetobactin was fully converted overnight into acinetobactin in a 150 mM sodium phosphate pH 8 solution before adding Fe3+ (1:1 ratio) with a ferric ammonium citrate solution (the colour changes from red to blue in 30 min). The ferric complex solution was diluted with the mother liquor to a final concentration of 2 mM before soaking the crystal overnight. No complex has yet been obtained.
Data were collected at the beamlines i02, i03 and i24 at the Diamond light source Oxfordshire. Data were processed with XIA2 (Winter, 2010; Zhang et al., 2006; Sauter et al., 2004; Kabsch, 1993; Evans, 2006). Structure of BauA was solved by SAD with the program AUTO-RICKSHAW (Panjikar et al., 2009). Structures of the complexes have been solved using the apo structure. Models were adjusted with COOT (Emsley and Cowtan, 2004) and refinement was carried out using REFMAC in the CCP4 program suite with TLS parameters (Murshudov et al., 1997). Coordinates and topologies of ligands were generated by PRODRG (Schüttelkopf and van Aalten, 2004). Final refinement statistics are given in Table 2. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (6H7F, 6H7V, 6HCP). The quality of all structures was checked with MOLPROBITY (Chen et al., 2010). Figures were drawn using PYMOL (Schrodinger, 2010).
Table 2
Crystallographic data and refinement statistics
https://doi.org/10.7554/eLife.42270.017| Semet 6H7V | Apo 6HCP | Complex 6H7F | |
|---|---|---|---|
| Data collection | |||
| Space group | C 1 2 1 | C 1 2 1 | C 1 2 1 |
| Cell dimensions | |||
| a, b, c (Å) α, β, γ (°) | 182.3, 220.4, 101.7 90, 98.7, 90 | 180.2, 219.5, 101.4 90, 99.2, 90 | 179.6, 220.7, 101.1 90, 99.2, 90 |
| Resolution (Å) | 139.53–2.54 (2.61–2.54)* | 57.24–1.83 (1.88–1.83)* | 49.93–2.26 (2.30–2.26) |
| Rsym or Rmerge | 0.12 (0.789) | 0.049 (0.665) | 0.108 (0.844) |
| I / σI | 19.3 (3.8) | 17.1 (2.2) | 9.2 (1.6) |
| Completeness (%) | (99.9) 100 | 99.7 (99.6) | 99.8 (99.9) |
| Redundancy | 15.2 (14.1) | 3.8 (3.9) | 3.8 (3.8) |
| CC half | - | - | 0.995 (0.527) |
| Anom completeness | 99.9 (99.9) | - | - |
| Ano multiplicity | 7.5 (6.9) | - | - |
| Refinement | |||
| Resolution (Å) | 139.53–2.54 | 57.24–1.83 | 49.93–2.26 |
| No. of reflections | 123,580 | 322,409 | 171,799 |
| Rwork/Rfree | 0.183/0.213 | 0.156/0.176 | 0.187/0.217 |
| No. of atoms | |||
| Protein | 15,424 | 16,060 | 15,854 |
| Ligand/ion | 201 | 754 | 691 |
| Water | 303 | 1995 | 752 |
| B-factors | |||
| Protein | 44.4 | 33.40 | 39.70 |
| Ligand/ion | 60.4 | 57.70 | 59.22 |
| Water | 36.90 | 45.90 | 36.87 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.010 | 0.008 | 0.008 |
| Bond angles (°) | 1.24 | 1.16 | 0.958 |
-
Each dataset was collected from a single crystal. *Values in parentheses are for highest-resolution shell.
Isothermal microcalorimetry titration
Request a detailed protocolAffinity of BauA for Fe3+-preacinetobactin and Fe3+-acinetobactin were measured by isothermal titration calorimetry using a VP-ITC or a ITC200 instrument (GE Healthcare) at 25 ˚C. Ratio 1:2 and 1:1 of Fe3+: preacinetobactin or acinetobactin have been tested. Solutions of 100 mM of compound and either 100 mM or 50 mM of Fe3+-acetylacetonate in DMSO were diluted with the dialysis buffer (50 mM Sodium phosphate pH 7.5, 50 mM NaCl, 0.8% OctylPOE). Titrations of preacinetobaction were performed using 2 μl injections of 100 μM of Fe3+ and either 100 μM or 200 μM preacinetobactin into 10 μM BauA.
Several conditions have been tested for Fe3+-acinetobactin. Titrations of acinetobaction were performed using 2 μl injections of 100 μM of Fe3+ and 200 μM acinetobactin (1:2) into 10 μM BauA. Titrations of acinetobaction were also performed with higher concentration using 5 μl injections of 370 μM of Fe3+-acinetobactin (1:1) in 20 μM BauA in the same buffer. The heats of dilution were measured by injecting the ligands into the buffer. Titration curves were fitted using Origin software.
LCMS analysis
Request a detailed protocolThe HPLC grade acetonitrile (MeCN) was purchased from Fisher. Aqueous buffers and aqueous mobile-phases for HPLC were prepared using water purified with an Elga Purelab Milli-Q water purification system (purified to 18.2 MΩ.cm) and filtered over 0.45 μm filters.
Analytical RP-HPLC-MS was performed on an Agilent infinity 1260 series equipped with a MWD detector using a Macherey-Nagel Nucleodur C18 column (10 μm x 4.6 × 250 mm) and connected to an Agilent 6130 single quad apparatus equipped with an electrospray ionization source using the following chromatographic system: 1 mL/min flow rate with MeCN and 0.1% TFA in H2O [95% TFA/H2O (5 min), linear gradient from 5% to 25% of MeCN (25 min), 95% MeCN (30 min)] and UV detection at 220 nm.
Nuclear magnetic resonance
Request a detailed protocol1H spectra were acquired at 300K with an Agilent UNITY INOVA spectrometer operating at a Larmor frequency of 500 MHz. Spectra were acquired with a pulse of 6.1 μs (90°), 5 s recycle delay, 48 transients over a spectral window of 6000 Hz. Homonuclear 1H gCOSY was acquired with the same acquisition parameters and sampling each of the 512 increments with 64 transients and 2048 complex points. The same parameters were applied for homonuclear 1H ROESY experiments with both 200 and 300 ms mixing time and applying the MLEV17 scheme for the spin-lock. The 1H chemical shift scale was referenced to the solvent residual resonance.
Titrations were performed in acetate buffered saline (ABS) at pH five prepared in D2O (pD 5.4), at constant ligand concentration in the 2–3 mM range by varying metal concentration (Ga3+), which was added in the form of Ga(ClO4)3. Relative intensities of selected resonances from the same proton group in the different species formed along the titration were measured and normalized for each spectrum. Normalized NMR intensities were analysed by fitting model equations through a Monte-Carlo minimization scheme. The following stepwise equilibria were taken into consideration at constant pH:
(1)
where L is the ligand, M the metal, Kn is the apparent equilibrium constant, n is an integer from 1 to 3, and molar concentrations are indicated with brackets. In addition to these mass-action laws, the following mass-balance relationships must also be applied:
(2)
(3)
where [L0] and [M0] are the total ligand and total metal molar concentration, respectively. Finally, the following quartic equation is obtained for [L], that is the free ligand at equilibrium:
(4)
which was solved numerically with the Newton-Raphson method. A Monte-Carlo scheme was implemented to minimize the sum of square difference between the experimental and calculated data points, by using the Kn as fitting parameters. One parameter was randomly selected at each iteration and randomly varied between −10% and +10%. Only the steps resulting in a decrease of the sum of square difference were accepted, rejecting the last move otherwise. All the datasets pertaining to the different species along the same titration were fitted simultaneously.
Data availability
Diffraction data have been deposited in PDB under the accession codes 6H7F, 6H7V, 6HCP.
-
Protein Data BankID 6H7F. Crystal structure of BauA, the Ferric preacinetobactin receptor from Acinetobacter baumannii in complex with Fe3+-Preacinetobactin-acinetobactin.
-
Protein Data BankID 6H7V. Crystal structure of BauA, the Ferric preacinetobactin receptor from Acinetobacter baumannii.
-
Protein Data BankID 6HCP. Crystal structure of BauA, the Ferric preacinetobactin receptor from Acinetobacter baumannii.
References
-
Rigid oxazole acinetobactin analog blocks siderophore cycling in Acinetobacter BaumanniiACS Infectious Diseases 3:802–806.https://doi.org/10.1021/acsinfecdis.7b00146
-
Pyochelin enantiomers and their outer-membrane siderophore transporters in fluorescent pseudomonads: structural bases for unique enantiospecific recognitionJournal of the American Chemical Society 133:16503–16509.https://doi.org/10.1021/ja205504z
-
Crystal structure of the outer membrane active transporter FepA from Escherichia coliNature structural biology 6:56–63.https://doi.org/10.1038/4931
-
MolProbity: all-atom structure validation for macromolecular crystallographyActa Crystallographica Section D Biological Crystallography 66:12–21.https://doi.org/10.1107/S0907444909042073
-
Comparative structural analysis of TonB-dependent outer membrane transporters: implications for the transport cycleProteins: Structure, Function, and Bioinformatics 59:240–251.https://doi.org/10.1002/prot.20416
-
The Crystal Structure of the Pyoverdine Outer Membrane Receptor FpvA from Pseudomonas aeruginosa at 3.6Å ResolutionJournal of Molecular Biology 347:121–134.https://doi.org/10.1016/j.jmb.2005.01.021
-
Crystal structure at high resolution of ferric-pyochelin and its membrane receptor FptA from Pseudomonas aeruginosaJournal of Molecular Biology 352:893–904.https://doi.org/10.1016/j.jmb.2005.08.004
-
Coot: model-building tools for molecular graphicsActa Crystallographica. Section D, Biological Crystallography 60:2126–2132.https://doi.org/10.1107/S0907444904019158
-
Scaling and assessment of data qualityActa Crystallographica Section D Biological Crystallography 62:72–82.https://doi.org/10.1107/S0907444905036693
-
Uncovering the mechanisms of Acinetobacter Baumannii virulenceNature Reviews Microbiology 16:91–102.https://doi.org/10.1038/nrmicro.2017.148
-
Activity of the investigational fluoroquinolone finafloxacin against ciprofloxacin-sensitive and -resistant Acinetobacter Baumannii isolatesAntimicrobial Agents and Chemotherapy 54:1613–1615.https://doi.org/10.1128/AAC.01637-09
-
Gallium-complex of anguibactin, a siderophore from fish pathogen Vibrio anguillarumJournal of Chemical Crystallography 28:57–60.https://doi.org/10.1023/A:1021782703768
-
Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constantsJournal of Applied Crystallography 26:795–800.https://doi.org/10.1107/S0021889893005588
-
Inference of macromolecular assemblies from crystalline stateJournal of Molecular Biology 372:774–797.https://doi.org/10.1016/j.jmb.2007.05.022
-
A synthetic dual drug sideromycin induces gram-negative bacteria to commit suicide with a gram-positive antibioticJournal of Medicinal Chemistry 61:3845–3854.https://doi.org/10.1021/acs.jmedchem.8b00218
-
Iron Transport: SiderophoresIn Encyclopedia of Inorganic Chemistry. Pp.https://doi.org/10.1002/0470862106.ia117
-
Structure and function of the PiuA and PirA Siderophore-Drug receptors from pseudomonas aeruginosa and Acinetobacter BaumanniiAntimicrobial Agents and Chemotherapy 61:.https://doi.org/10.1128/AAC.02531-16
-
Refinement of macromolecular structures by the maximum-likelihood methodActa Crystallographica Section D Biological Crystallography 53:240–255.https://doi.org/10.1107/S0907444996012255
-
TonB-dependent transporters: regulation, structure, and functionAnnual Review of Microbiology 64:43–60.https://doi.org/10.1146/annurev.micro.112408.134247
-
On the combination of molecular replacement and single-wavelength anomalous diffraction phasing for automated structure determinationActa Crystallographica Section D Biological Crystallography 65:1089–1097.https://doi.org/10.1107/S0907444909029643
-
Biphasic binding kinetics between FepA and its ligandsJournal of Biological Chemistry 272:21950–21955.https://doi.org/10.1074/jbc.272.35.21950
-
PROMALS3D: a tool for multiple protein sequence and structure alignmentsNucleic Acids Research 36:2295–2300.https://doi.org/10.1093/nar/gkn072
-
Acinetobacter Baumannii: emergence of a successful pathogenClinical Microbiology Reviews 21:538–582.https://doi.org/10.1128/CMR.00058-07
-
Structure and biosynthesis of fimsbactins A-F, siderophores from Acinetobacter Baumannii and Acinetobacter baylyiChembiochem : A European Journal of Chemical Biology 14:633–638.https://doi.org/10.1002/cbic.201200764
-
Deciphering key features in protein structures with the new ENDscript serverNucleic Acids Research 42:W320–W324.https://doi.org/10.1093/nar/gku316
-
A latent oxazoline electrophile for N-O-C bond formation in pseudomonine biosynthesisJournal of the American Chemical Society 130:12282–12284.https://doi.org/10.1021/ja804499r
-
Robust indexing for automatic data collectionJournal of Applied Crystallography 37:399–409.https://doi.org/10.1107/S0021889804005874
-
SoftwareThe PyMOL molecular graphics system, version 1.3r1Schrodinger LLC.
-
PRODRG: a tool for high-throughput crystallography of protein-ligand complexesActa Crystallographica Section D Biological Crystallography 60:1355–1363.https://doi.org/10.1107/S0907444904011679
-
Synthesis of acinetobactinChemical &Amp; Pharmaceutical Bulletin 58:1552–1553.https://doi.org/10.1248/cpb.58.1552
-
LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions"Protein Engineering, Design and Selection" 8:127–134.https://doi.org/10.1093/protein/8.2.127
-
Sideromycins as Pathogen-TargetedAntibiotics in AntibacterialsTopics in Medicinal Chemistry, Sideromycins as Pathogen-TargetedAntibiotics in Antibacterials, Springer.
-
xia2 : an expert system for macromolecular crystallography data reductionJournal of Applied Crystallography 43:186–190.https://doi.org/10.1107/S0021889809045701
-
Three siderophores from one bacterial enzymatic assembly lineJournal of the American Chemical Society 131:5056–5057.https://doi.org/10.1021/ja900815w
-
Isolation and structure elucidation of acinetobactin, a novel siderophore from Acinetobacter BaumanniiArchives of Microbiology 162:249–254.https://doi.org/10.1007/s002030050133
-
Automated diffraction image analysis and spot searching for high-throughput crystal screeningJournal of Applied Crystallography 39:112–119.https://doi.org/10.1107/S0021889805040677
Article and author information
Author details
Funding
European Commission (115525)
- Lucile Moynie
- Ilaria Serra
- Mariano A Scorciapino
- Malcolm GP Page
- Matteo Ceccarelli
- James H Naismith
Wellcome Trust (100209/Z/12/Z)
- Lucile Moynie
- James H Naismith
European Commission (115583)
- Lucile Moynié
- James H Naismith
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Basilea Pharmaceutica Ltd as partner of the IMI, Eric Desarbre for the synthesis of acinetobactin and preacinetobactin, Sophie Guillard for the analytical analysis she did to confirm the structures of the substances and A Tortajada for the technical help.
The research leading to these results was conducted as part of the Translocation consortium (www.translocation.eu) and benfitted from support from ND4BB ENABLE Consortium which has received support from the Innovative Medicines Initiatives Joint Undertaking under Grant Agreement Nr 115525 and Nr 115583, resources which are composed of financial contribution from the European Union’s seventh framework programme (FP7/2007–2013) and EFPIA companies in kind contribution. This is work is supported by a Wellcome Trust Investigator (100209/Z/12/Z) award.
Copyright
© 2018, Moynié et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,387
- views
-
- 372
- downloads
-
- 53
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 53
- citations for umbrella DOI https://doi.org/10.7554/eLife.42270
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Preacinetobactin not acinetobactin is essential for iron uptake by the BauA transporter of the pathogen Acinetobacter baumannii
eLife 7:e42270.
https://doi.org/10.7554/eLife.42270




{kind=link}
{kind=link}
{kind=link}
{kind=link}