Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer
- University of California, San Francisco, United States
- Harvard Medical School, United States
Peer review process
This article was accepted for publication as part of eLife's original publishing model.
History
- Version of Record published
- Accepted
- Received
Decision letter
-
Wilbert ZwartReviewing Editor; Netherlands Cancer Institute, Netherlands
-
Jeffrey SettlemanSenior Editor; Calico Life Sciences, United States
-
Wilbert ZwartReviewer; Netherlands Cancer Institute, Netherlands
-
Claus JørgensenReviewer; Cancer Research UK Manchester Institute, United Kingdom
In the interests of transparency, eLife includes the editorial decision letter and accompanying author responses. A lightly edited version of the letter sent to the authors after peer review is shown, indicating the most substantive concerns; minor comments are not usually included.
Thank you for submitting your article "Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer" for consideration by eLife. Your article has been reviewed by two peer reviewers, including Wilbert Zwart as the Reviewing Editor and Reviewer #1, and the evaluation has been overseen by Jeffrey Settleman as the Senior Editor. The following individual involved in review of your submission has also agreed to reveal his identity: Claus Jørgensen (Reviewer #2).
The reviewers have discussed the reviews with one another and the Reviewing Editor has drafted this decision to help you prepare a revised submission.
Summary:
Here, Perera and colleagues describe a novel role of GLI2 in regulating subtype switching, EMT status and RAS dependency in pancreatic cancer.
Pancreatic cancer cells can exist as different subtypes, and this has previously been linked to patient performance and therapeutic sensitivity. However, while there has been a lot of focus on defining subtypes and their potential role in PDA, less is known about the regulators of PDA subtypes. The data presented describing a role for GLI2 in regulating PDA subtype switching are rigorous, experiments are well conducted, key controls are in place and the manuscript is logical and well presented.
Major points:
1) Figure 1D,E: multivariate correction with clinical parameters should be performed; is the difference in survival related to the gene of interest, or to other clinical variables that associate with these factors? Also, do the 2 arms between the different KM curves identify the same patient populations? Are the patients with GLI2 high the same as those with SSH low?
2) Figure 3: the text states that two cell lines were used in these analyses (HPAFII and YAPC), for I can only find the data (especially 3B and D) for the YAPC cell lines. Please confirm findings in several cell lines, and include those data in the paper.
3) Figure 4: same thing here as in Figure 3. In the text, two cell lines are mentioned (KP4 and Panc0327), but the data presented don't show the findings in both cell lines, only one. Please include the data for both cell lines.
4) In Figure 3B, 3D growth analyses (not organoids) is depicted with GLI2 over expression, observing effects on migration. Wouldn't you also expect to see impact on migratory phenotype in Figures 5H and K, when overexpressing GLI2 (H) or the opposite when knocking it down (K)?
https://doi.org/10.7554/eLife.45313.029Author response
Major points:
1) Figure 1D,E: multivariate correction with clinical parameters should be performed; is the difference in survival related to the gene of interest, or to other clinical variables that associate with these factors? Also, do the 2 arms between the different KM curves identify the same patient populations? Are the patients with GLI2 high the same as those with SSH low?
We have performed univariate and multivariate analysis on the data presented in Figure 1D and E as requested. In a univariate analysis, neither GLI2 high versus low expression nor SHH high versus low expression was correlated with clinical variables such as sex, T stage, N stage, or M stage, although, as expected from our prior data, there was a correlation between high SHH and low GLI2 and vice-versa. These data have been included as new panels in Figure 1—figure supplement 1G,H of the revised manuscript. A multivariate analysis of the hazard attributable to SHH or GLI2 status demonstrated that no other variables explained the relationship between SHH or GLI2 and overall or progression free survival. Thus, we conclude that GLI2-high status is an independent prognostic indicator of poor outcomes in PDA. These data have been included as a new supplementary figure and are discussed in the updated text (new Figure 1—figure supplement 2; subsection “Expression of Hh ligands and GLI transcription factors are anti-correlated and predict survival outcomes in PDA”).
2) Figure 3: the text states that two cell lines were used in these analyses (HPAFII and YAPC), for I can only find the data (especially 3B and D) for the YAPC cell lines. Please confirm findings in several cell lines, and include those data in the paper.
We apologize for the oversight. We have now included data demonstrating GLI2-mediated basal-like switching in HPAFII cells (Figure 3—figure supplement 1A, C) and as requested by the reviewer, we provide new data in mouse PDA cells reinforcing these findings (Figure 5—figure supplement 2B). For clarity, we have indicated which cell lines have been used for each experiment within the manuscript text. Consistent with our original data in YAPC cells, ectopic expression of GLI2 in HPAFII cells (HPAFII-GLI2) led to a change in cell morphology involving loss of a compact, cobblestone growth pattern and increased spreading (Figure 3—figure supplement 1C). Additionally, HPAFII-GLI2 cells showed downregulation of epithelial markers, ESRP1, the transcription factor GATA6 – a regulator of the classical subtype of PDA, and SHH and induction of ZEB1 (Figure 3—figure supplement 1A). HPAFII-iGLI2 cells also showed upregulation of the basal-like gene signature and concomitant downregulation of the classical signature (Figure 3E), which are all consistent with data obtained in YAPC cells. Thus, we provide evidence in both YAPC and HPAFII cell lines indicating that ectopic GLI2 expression is sufficient to drive a switch from a classical to a basal-like state.
3) Figure 4: same thing here as in Figure 3. In the text, two cell lines are mentioned (KP4 and Panc0327), but the data presented don't show the findings in both cell lines, only one. Please include the data for both cell lines.
As requested by the Reviewer we provide data in both KP4 and Panc0327 cells to support our finding that GLI2 is required to maintain a basal-like state in PDA. We have also indicated which cell lines have been used for each experiment within the manuscript text. CRISPRCas9 mediated knockout of GLI2 in Panc0327 and KP4 cells caused a switch toward a more epithelial-like morphology in 2D monolayer culture (Figure 4D) and 3D Matrigel growth (KP4; Figure 4—figure supplement 1D). Additionally, GLI2 KO in Panc0327 cells resulted in reduced expression of EMT markers ZEB1, VIM and CDH2 and the stemness factor SOX2 (Figure 4—figure supplement 1C), a decrease in basal-like markers KRT14 and KRT5, and an increase in classical markers ESRP1 and GATA6 (Figure 4—figure supplement 1A) and a coordinated reduction in expression of the basal-like gene signature (Figure 4—figure supplement 1B), consistent with results obtained with KP4 cells (Figure 4A-C). Finally, immunofluorescence staining of Panc0327 and KP4 cells following knockout or knockdown of GLI2 respectively, caused a prominent nuclear re-localization of ESRP1 (Figure 4E and Figure 4—figure supplement 1E). Together these data demonstrate a requirement for GLI2 in maintaining a basal-like state in both Panc0327 and KP4 PDA cell lines.
4) In Figure 3B, 3D growth analyses (not organoids) is depicted with GLI2 over expression, observing effects on migration. Wouldn't you also expect to see impact on migratory phenotype in Figures 5H and K, when overexpressing GLI2 (H) or the opposite when knocking it down (K)?
We thank the reviewers for pointing this out. We have found close concordance between the human and murine cell systems regarding the GLI2-mediated subtype switching, with the sole exception being the lack of changes in cell migration in the mouse cells as noted by the reviewer. For example, consistent with the data in human cells, ectopic GLI2 expression in mouse iKRAS4 cells (iKRASGLI2) induced expression of mesenchymal markers ZEB1 and N-Cadherin, basal markers KRT14 and KRT5, and SOX2 (Figure 5—figure supplement 2B), and upregulated the basallike gene signature (compare Figure 5—figure supplement 1D and Figure 5E). We provide new data showing that expression of GLI2 in YAPC-GLI2 cells is able to rescue growth following suppression of oncogenic KRAS (Figure 5—figure supplement 2D) similar to that observed in iKRASGLI2 mouse PDA cells (Figure 5—figure supplement 2D). As previously observed, YAPC-GLI2 cells additionally show a striking morphological change when grown in 3D culture that is retained following KRAS loss (Author response image 1 bottom panel: images show the effect of stable expression of EV (top) or GLI2 (bottom) on YAPC sphere formation in response to shRNAmediated KRAS knockdown).
Author response image 1
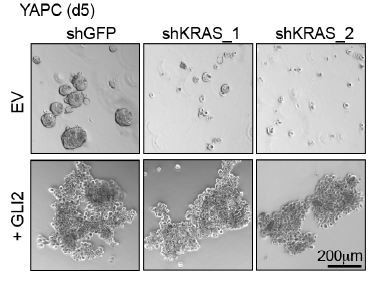
These data support a conserved function for GLI2 in driving a basal-like subtype switch and resistance to mutant KRAS loss in human and mouse PDA cells.
https://doi.org/10.7554/eLife.45313.030Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer
eLife 8:e45313.
https://doi.org/10.7554/eLife.45313




{kind=link}