The orphan receptor GPR88 blunts the signaling of opioid receptors and multiple striatal GPCRs
- Deficits of Reward GPCRs and Sociability, Physiologie de la Reproduction et des Comportements, INRA UMR-0085, CNRS UMR-7247, Université de Tours, Inserm, France
- Biology and Bioinformatics of Signalling Systems, Physiologie de la Reproduction et des Comportements, INRA UMR-0085, CNRS UMR-7247, Université de Tours, France
- Cellular Biology and Molecular Pharmacology of central Receptors, Centre de Psychiatrie et Neurosciences, Inserm UMR_S894 - Université Paris Descartes, Sorbonne Paris Cité, France
- Pompeu Fabra University, Barcelona Biomedical Research Park, Spain
- Douglas Mental Health University Institute, McGill University, Canada
- Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS UMR 7104, Inserm U1258, Université de Strasbourg, 1 rue Laurent Fries, France
Figures
Figure 1 with 2 supplements
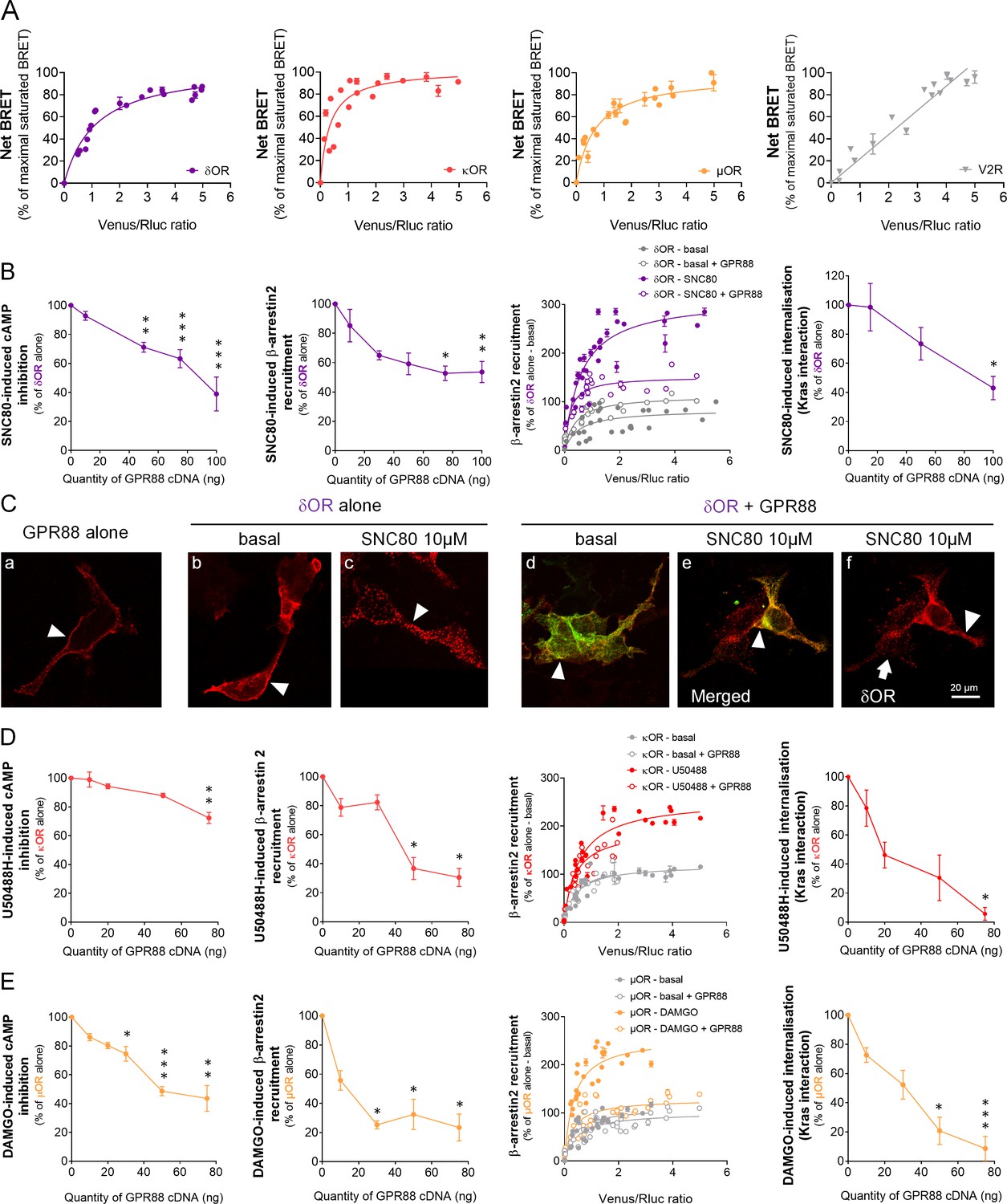
GPR88 comes in close physical proximity to opioid receptors and inhibits their signaling and trafficking in vitro.
(A) BRET1 saturation experiments were performed in transfected HEK293FT cells using constant quantity of GPR88-Rluc8 with increasing amounts of Venus-tagged opioid receptors δOR, κOR and μOR or V2R. Saturated BRET signals indicate close physical proximity (within 10 nm) to the target GPCR, thus possible hetero- or homo-oligomers. Such saturation is not observed with the V2R (right panel), D1 or CXCR4 receptors (see Figure 4), showing that close proximity is not detected for all GPCRs. See reverse constructions for δOR, κOR in Figure 1—figure supplement 1 (B) Co-expressing GPR88 with δOR (wild-type or Rluc8-tagged, 30 ng of cDNA) blunts (left to right panels): SNC80 (δOR agonist, 10 μM)-induced inhibition of cAMP production (cAMP sensor: CAMYEL) (H4,39=28.7, p=0.0000) and Ypet-β-arrestin 2 (β-arr2) recruitment (H5,28=18.1, p=0.0028), Ypet-β-arr2 recruitment at δOR under stimulated but not basal conditions and, finally, SNC80-induced δOR internalization (internalization sensor: Kras-Venus) (H3,12=7,8, p=0.0492) in HEK293FT cells. (C) Confocal microscopy images show (arrow head) that (a) HA-GPR88 (red) is localized at cell surface (non-permeabilized cells), (b) under basal conditions, HA-δOR (red) is localized at cell surface, (c) SNC80-stimulation induces HA-δOR internalization, (d) under basal conditions, HA-δOR (red) and GPR88-Venus (green) co-localize at cell surface, (e) under SNC80 stimulation, GPR88 inhibits δOR internalization, (f) which results in different patterns of HA-δOR (red) distribution in GPR88-Venus (green) expressing versus non-expressing cells (arrow head versus arrow, respectively). (D) Co-expressing GPR88 with κOR (wild-type or Rluc8-tagged, 30 ng of cDNA) decreases (left to right panels): U50488H (κOR agonist, 10 μM)-induced inhibition of cAMP production (CAMYEL) (H4,25=17.9, p=0.0013) and Ypet-β-arr2 recruitment (H4,19=15.9, p=0.0031), Ypet-β-arr2 recruitment at κOR under stimulated but not basal conditions and U50488H-induced κOR internalization (Kras-Venus) (H4,18=13.6, p=0.0087) in HEK293FT cells. (E) Co-expressing GPR88 with µOR (wild-type or Rluc8-tagged, 30 ng of cDNA) inhibits (left to right panels): DAMGO (µOR agonist, 10 μM)-induced inhibition of cAMP production (CAMYEL) (H5,34=27.7, p=0.000) and Ypet-β-arr2 recruitment (H4,23=18.2, p=0.0011), Ypet-β-arr2 recruitment at µOR under stimulated but not basal conditions and DAMGO-induced µOR internalization (Kras-Venus) (H4,23=19.7, p=0.0006) in HEK293FT cells. Inhibition of cAMP production was determined in presence of 250 μM IBMX and 5 μM forskolin. Data are presented as mean ± SEM of n = 3–9 independent experiments (performed in triplicates). BRET1 values are presented as net BRET (normalized as the percentage of maximal BRET values) or induced BRET (normalized as the percentage of maximal BRET values in absence of GPR88) by Venus/Rluc8 BRET ratio. Asterisks: Kruskal-Wallis ANOVA, multiple comparison of mean ranks, *p<0.05, **p<0.01, ***p<0.001. Confocal imaging: representative pictures among n = 10 pictures. Receptor cell surface expression and additional data regarding OR trafficking in presence of GPR88 are displayed in Figure 1—figure supplement 2.
Figure 1—figure supplement 1
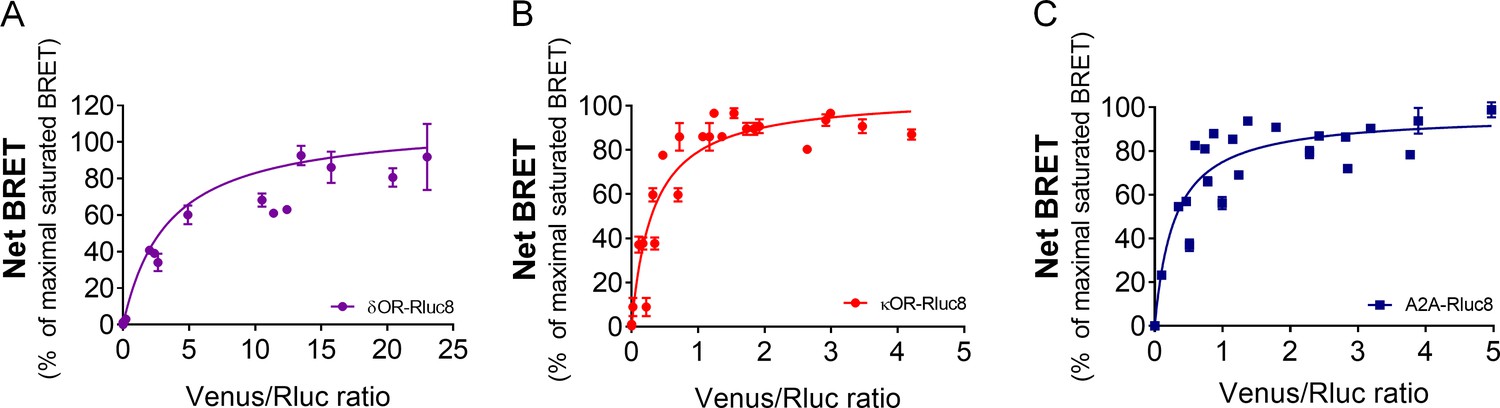
BRET saturation assays using Venus-tagged GPR88 and Rluc8-tagged target receptors.
BRET1 saturation experiments were performed in transfected HEK293FT cells using constant quantity of δOR-Rluc8 (panel A), κOR-Rluc8 (panel B) or A2A-Rluc8 (panel C) with increasing amounts of GPR88 Venus-tagged receptors. Saturated BRET signals indicate close physical proximity (within 10 nm) to the target GPCR, thus possible hetero-oligomers. Data are presented as mean ± SEM of n = 3–4 independent experiments (performed in triplicates). BRET1 values are presented as induced BRET (normalized as the percentage of maximal BRET values in absence of GPR88) by Venus/Rluc8 BRET ratio.
Figure 1—figure supplement 2
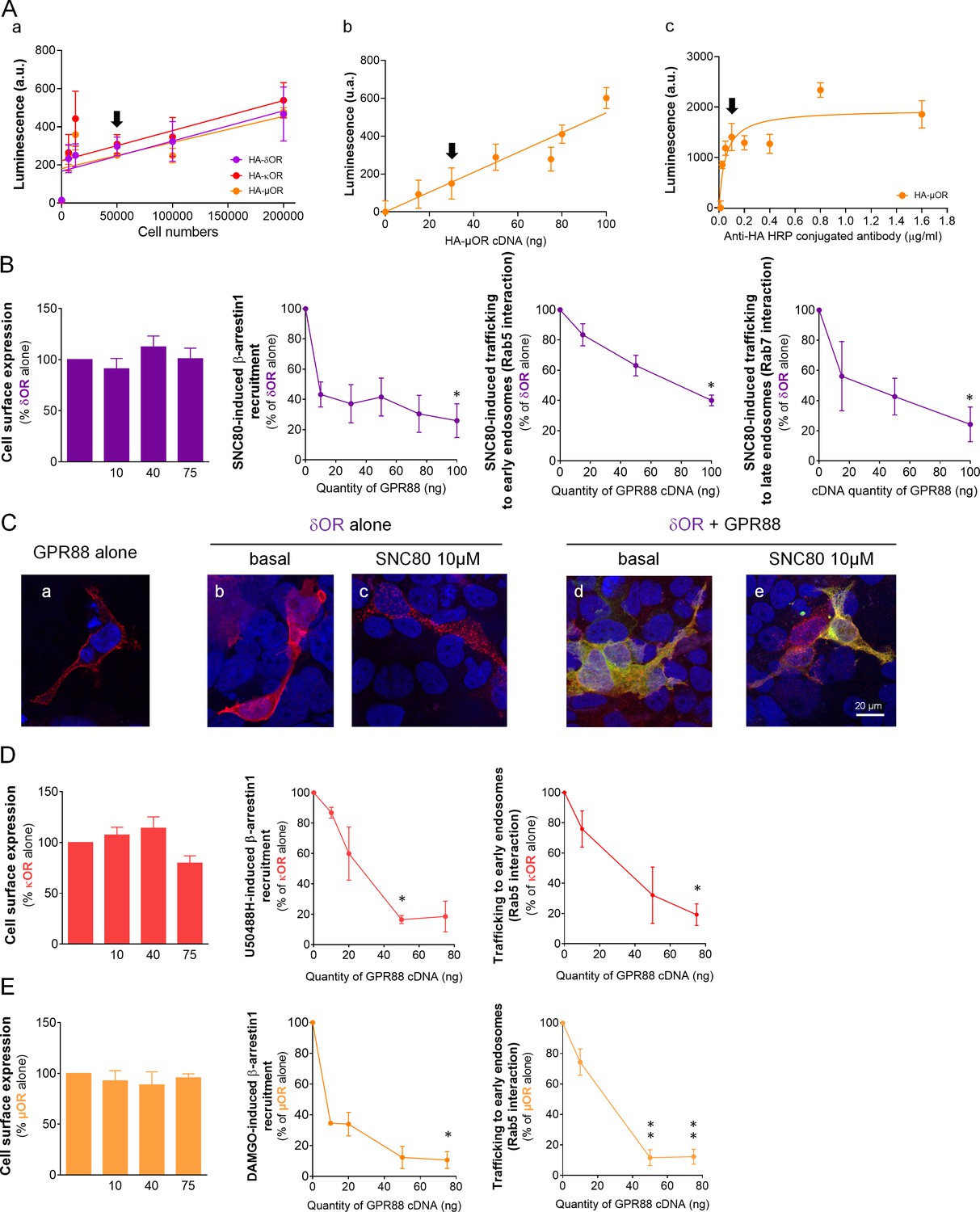
GPR88 inhibits opioid receptor signaling and trafficking in vitro.
(A) We assessed whether the polyclonal anti-HA HRP-conjugated antibody allowed to satisfyingly detect modifications in the cell surface expression of HA-tagged opioid receptors. (a) The antibody displayed a linear response to increasing amounts of HA-tagged GPCR-expressing cells in the 50,000 to 200,000 range (n = 2–4 per receptor, in triplicates). Further tests used 50,000 cells per assay (black arrow). (b) When the cell number was set at 50,000, the anti-HA HRP-conjugated antibody allowed reliable detection of increasing amounts of transfected HA-tagged µOR (n = 4, in triplicates). Further used 30 ng of HA-tagged GPCR cDNA for transfection (black arrow). (c) At the 1:10,000 concentration (black arrow), the antibody allows optimal detection of HA-µOR (n = 1, in triplicates). This concentration was used for further testing. (B) Increasing amounts of GPR88 cDNA did not affect HA-δOR cell surface expression (left panel) but blunted SNC80 (10 µM)-induced Ypet-β-arrestin1 (β-arr1) recruitment and δOR-Rluc8 trafficking (early endosome sensor: Rab5-Venus; late endosome sensor: Rab7-Venus) in HEK293FT cells. (C) Confocal microscopy images show (a) HA-GPR88 expression at cell surface, (b) HA-δOR localized at cell surface, (c) internalized under SNC80-stimulation, (d) HA-δOR (red) and GPR88-Venus (green) co-localized at cell surface, (e) GPR88 inhibition of δOR internalization with cell nuclei stained using DAPI coloration. (D) Co-expressing GPR88 with HA-κOR did not affect κOR cell surface expression but blunted U50488H (10 µM)-induced Ypet-β-arr1 recruitment and κOR trafficking (early endosome sensor: Rab5-Venus) in HEK293FT cells. (E) Co-expressing GPR88 with HA-µOR did not affect µOR cell surface expression but blunted DAMGO (10 µM)-induced Ypet−β-arr1 recruitment and µOR-Rluc8 trafficking (early endosome sensor: Rab5-Venus; late endosome sensor: Rab7-Venus) in HEK293FT cells. Data (B–E) are presented as mean ± SEM of n = 3–5 independent experiments (performed in triplicates). BRET1 values are presented as induced BRET (normalized as the percentage of maximal BRET values in absence of GPR88) by Venus/Rluc8 BRET ratio. Asterisks: Kruskal-Wallis ANOVA, multiple comparison of mean ranks *p<0.05, **p<0.01.
Figure 2 with 2 supplements

GPR88 dampens µOR-mediated signaling in vitro: ERK phosphorylation and effects of µOR expression levels.
(A) Upper panel: in HEK293FT cells, DAMGO stimulated µOR (0.5 µg cDNA) activity, resulting in an increase in the phospho-ERK/ERK total (pERK/tERK) ratio peaking 15 min after DAMGO stimulation; this response was suppressed when GPR88 (1 µg cDNA) was co-expressed with µOR. Lower panel: representative western blotting images. (B) Upper panel: addition of pertussis toxin (PTX, 0.1 ng/µl, overnight) blocked the early phase of DAMGO-induced response, demonstrating its dependence on Gi/o protein recruitment, but failed to inhibit a later component of ERK phosphorylation. GPR88 co-expression completely blocked DAMGO-induced phosphorylation of ERK. Lower panel: representative western blotting images. Levels of phosphorylated-ERK (pERK) and total-ERK (tERK) were normalized to the loading control protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are presented as mean ± SEM of n = 5 independent experiments. Kruskal-Wallis ANOVA, multiple comparison of mean ranks *p<0.05, **p<0.01 (see gels in Figure 2—figure supplement 1 and statistics in Supplementary file 1). (C) Increasing the amount of µOR (wild-type, ng of cDNA) expressed in HEK293FT cells allowed to overcome the inhibitory effects of GPR88 (wild-type) on DAMGO (µOR agonist, 10 μM)-induced inhibition of cAMP production (CAMYEL), and a complete rescue of signaling was observed for the highest doses of µOR cDNA transfected (GPR88 effect: F2,21=86.7, p=0.0018; µOR effect: F7,147=90.9, p=0.0000; GPR88 x µOR interaction: F7,147=3.1, p=0.00030). Although increasing the amount of GPR88 (30 to 50 ng of cDNA) shifted the µOR dose response to the right, it was not able to prevent full restoration of µOR signaling at high doses of µOR. (D) Increasing the amount of µOR (wild-type) expressed in HEK293FT cells allowed only a partial overcoming of the inhibitory effects of GPR88 (wild-type) on DAMGO (µOR agonist, 10 μM)-induced Ypet-β-arr2 recruitment; no further recruitment was detected for doses of µOR cDNA over 50 ng (GPR88 effect: F2,6=86.7, p=0.000037; µOR effect: F6,36=32.3, p=0.0000; GPR88 x µOR interaction: F6,36=15.7, p=0.0000). Increasing the amount of GPR88 (from 30 to 50 ng of cDNA transfected) nearly suppressed β-arr2 recruitment at µOR. Data for 15 ng of µOR cDNA transfected are framed in gray, to allow comparison with Figure 1E. Data are presented as mean ± SEM of n = 3–12 independent experiments (performed in triplicates). BRET1 values are presented as induced BRET (normalized as the percentage of maximal BRET values in absence of GPR88) by Venus/Rluc8 BRET ratio. ANOVA (repeated measure), stars: GPR88 effect, daggers: GPR88xµOR interaction; one symbol: p<0.05, two symbols: p<0.01, three symbols: p<0.001. Effects of GPR88 activation by synthetic agonist Compound 19 on its inhibitory action at µOR signaling in vitro are presented in Figure 2—figure supplement 2.
Figure 2—figure supplement 1

Gels from western blot experiments; ERK phosphorylation assay in HEK293FT cells.
Figure 2—figure supplement 2
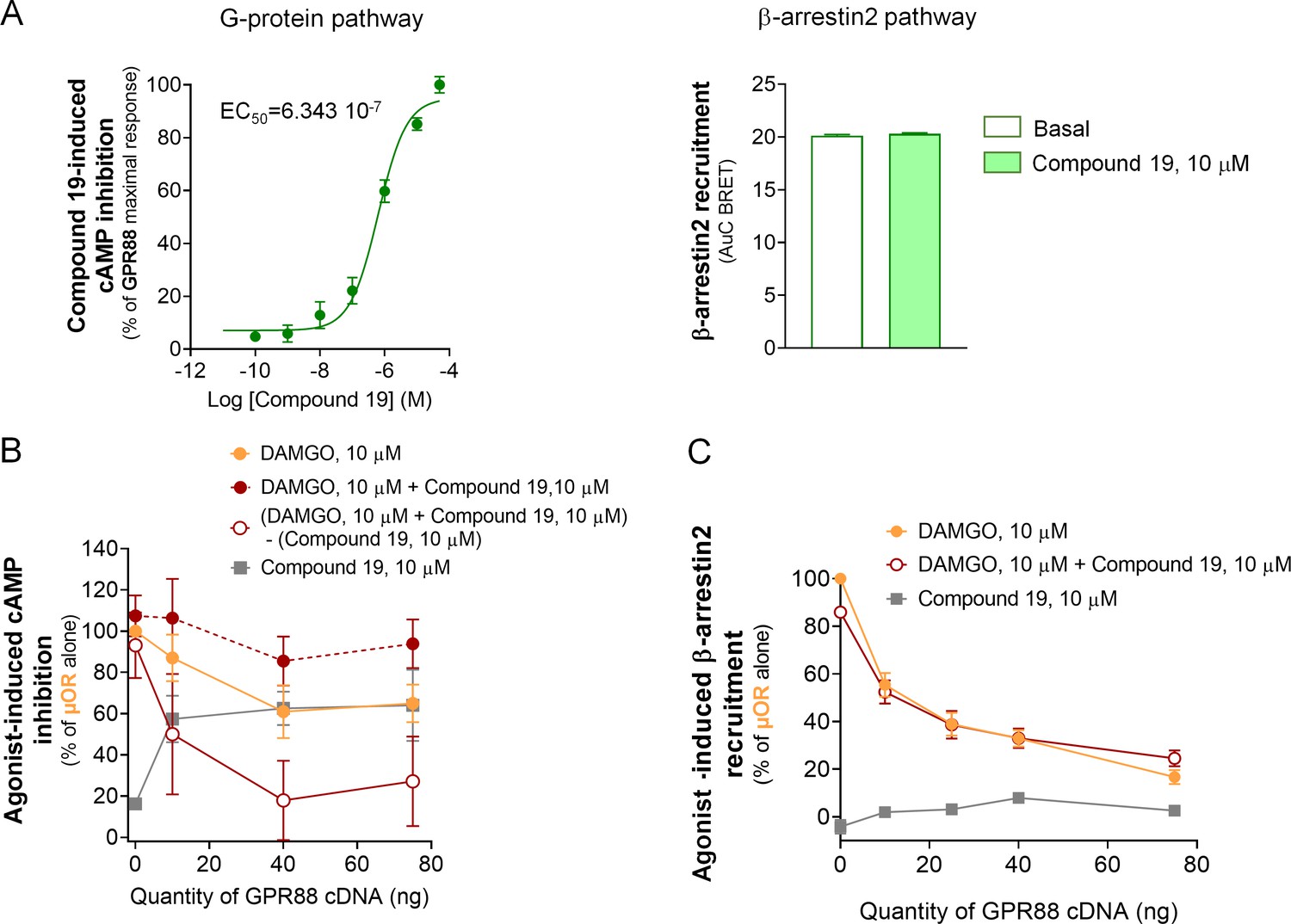
Effects of Compound 19 on GPR88 signaling pathways and GPR88-mediated blunting of G-protein dependent signaling and β-arrestin recruitment by µOR.
(A) Activation of GPR88 by Compound 19 induced a dose-dependent inhibition of cAMP production (cAMP sensor: CAMYEL, Gαi/o GPR88 coupling). Compound 19 failed to induce detectable recruitment of Ypet-β-arr2 by Venus-GPR88 receptor at a high dose of 10 µM. (B) Co-expressing increasing amounts of GPR88 blunted DAMGO (10 µM)-induced inhibition of cAMP production by µOR. In an attempt to visualize the putative effects of GPR88 activation by Compound 19 on this inhibitory action, we subtracted the percentage of cAMP inhibition induced by Compound 19 to the percentage of cAMP inhibition produced by both DAMGO (10 µM) and Compound 19 (10 µM) in presence of µOR and increasing amounts of GPR88. When Compound 19 was added to DAMGO, GPR88 tended to further dampen µOR-induced inhibition of cAMP. (C) Co-expressing increasing amounts of GPR88 with µOR blunted DAMGO (10 µM)-induced Ypet-β-arr2 recruitment at µOR; addition of Compound 19 (10 µM) did not affect this inhibitory effect; Compound 19 had no influence on Ypet-β-arr2 recruitment by µOR in the absence of DAMGO. Data are presented as mean ± SEM of n = 3–4 independent experiments (performed in triplicates). BRET1 values are presented as net BRET (normalized as the percentage of maximal BRET values) or induced BRET (normalized as the percentage of maximal BRET values in absence of GPR88) by Venus/Rluc8 BRET ratio.
Figure 3 with 3 supplements
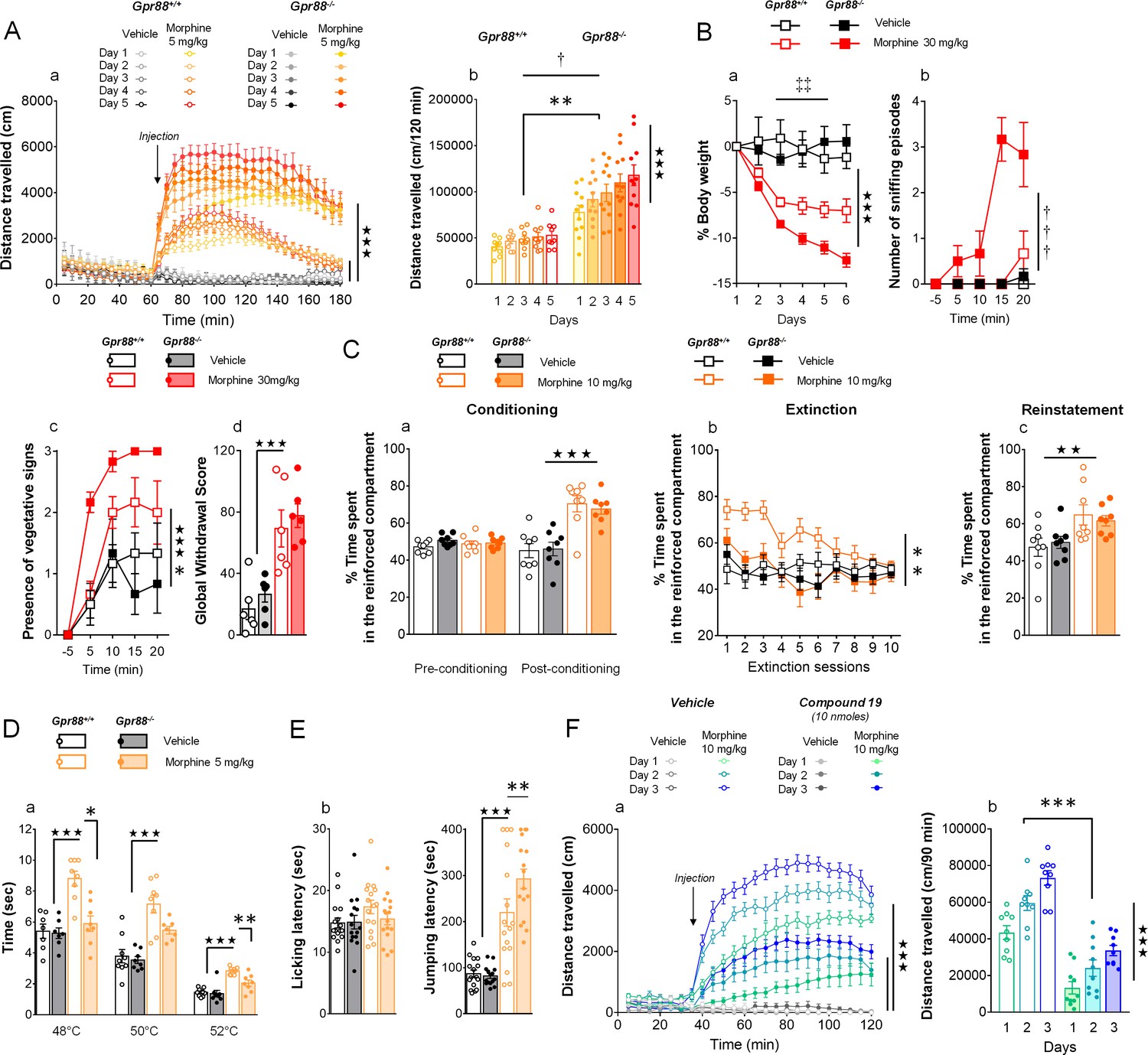
Gpr88 null mice display modified mu-opioid mediated behavioral responses.
(A) In a locomotor sensitization paradigm (n = 5 to 11 mice per treatment and genotype) morphine induced an increase in locomotor activity that sensitized upon repeated administration in Gpr88+/+ and Gpr88-/- mice (panel a); morphine-induced locomotion and sensitization, however, were significantly greater in mutant mice (panel b) (Genotype effect: F1,27=13.5, p=0.0010; Treatment: F1,27=99.9, p=0.0000; Genotype x Treatment interaction: F1,27=14.6, p=0.0007; Day: F4,108=7.9, p=0.0000; Day x Treatment: F4,108=10.2, p=0.0000; Day x Genotype x Treatment = 2.9, p=0.0253); solid stars: treatment effect (two-way ANOVA with one repeated measure – day), asterisks: genotype effect, dagger: Day x Genotype x Treatment interaction. (B) Upon exposure to escalating doses of morphine (n = 6 per treatment and genotype), Gpr88-/- lost more body weight than controls (panel a; Treatment: F1,20=43.7, p=0.0000; Day: F4,80=13.6, p=0.0000; Day x Genotype: F4,80=5.1, p=0.001; Day x Treatment: F4,80=9.9, p=0.0000; body weight was measured daily upon morphine treatment); when withdrawal was triggered by acute naloxone administration (1 mg/kg), mutant animals displayed sniffing episodes (panel b; Genotype/Treatment: F1,20=25.5, p=0.0000; Genotype x Treatment: F1,20=23.1, p=0.0001; Time: F3,60=10.5, p=0.0000; Time x Genotype/Treatment: F3,60=9.3, p=0.0000; Time x Genotype x Treatment: F3,60=8.3, p=0.0001) and vegetative signs of withdrawal (panel c; Genotype: F1,20=5.3, p=0.0324; Treatment: F1,20=5.3, p=0.0324; Time: F3,60=14.4, p=0.0000) quicker than their Gpr88+/+ counterparts, despite similar final withdrawal scores (panel d; Treatment: F1,20=38.9, p=0.0000); solid stars: treatment effect (two-way ANOVA with one repeated measure – day/5 min time bin), double daggers: Time x Genotype interaction, daggers: Time x Genotype x Treatment interaction, asterisk: genotype effect. More withdrawal signs are displayed in Figure 3—figure supplement 1. (C) In a CPP paradigm (n = 8 per treatment and genotype), Gpr88-/-mice acquired preference for a compartment associated to morphine administration (10 mg/Kg) similarly as Gpr88+/+ animals (panel a; Treatment: F1,28=45.8, p=0.0000; Conditioning: F1,28=15.7, p=0.0005; Conditioning x Treatment: F1,28=31.1, p=0.0000); they extinguished this conditioning quicker than wild-type counterparts (panel b; Genotype: F1,28=10.3, p=0034; Treatment: F1,28=6.5, p=0166; Genotype x Treatment: F1,28=5.0, p=0329; Session: F9,252=5.2, p=0.0000; Session x Treatment: F9,252=3.7, p=0.0002) and finally reinstated morphine place preference at comparable levels as the latter (panel c; Treatment: F1,28=9.2, p=0.0052); solid stars: Treatment effect (two-way ANOVA with one repeated measure – day/5 min time bins); asterisks: Genotype effect. (D) In the tail flick test (n = 7–9 per treatment and genotype), Gpr88-/- mice were significantly less sensitive to morphine analgesia at 48°C and 52°C (Genotype: F1,83=22.0, p=0.0000; Treatment: F1,83=84.9, p=0.0000; Temperature: F2,83=162.5, p=0.0000; Genotype x Treatment: F1,83=15.9, p=0.0001; Treatment x Temperature: F2,83=5.4, p=0.0065); (E) in the hot plate test (n = 15–16 per treatment and genotype), morphine-induced analgesia was detected by increased jumping latency in treated animals (right panel); this effect was increased in Gpr88 null mice versus controls (Treatment: F1,59=79.7, p=0.0000; Genotype x Treatment: F1,59=4.0, p=0.0490); solid stars: Treatment effect (two-way ANOVA), asterisks: Genotype x Treatment interaction (Newman Keules post-hoc test). Increased morphine-induced locomotor sensitization in Gpr88 null mice was not associated with modified pERK/tERK ratio in three brain regions (Figure 3—figure supplements 2 and 3). (F) We administered the GPR88 agonist Compound 19 (icv, 10 nmoles) to mice exposed to a morphine-induced locomotor sensitization paradigm (n = 8 to 9 mice per treatment condition). Exposure to morphine (IP, 10 mg/kg) induced an increase in locomotor activity that sensitized upon repeated administration in both vehicle and Compound 19-treated groups (panel a); pharmacological activation of GPR88 drastically reduced morphine-induced locomotor activity but left the amplitude of sensitization unchanged (Morphine effect: F1,30=305.1, p=0.0000; Compound 19: F1,30=55.3, p=0.0000; Day: F2,60=33.1, p=0.0000; Day x Morphine: F2,60=31.6, p=0.0000; Day x Morphine x Compound 19: F2,60=2.1, NS), solid stars: treatment effect (two-way ANOVA with one repeated measure – day), asterisks: genotype effect. Data are presented as mean ± SEM. One symbol: p<0.05, two symbols: p<0.01, three symbols: p<0.001.
Figure 3—figure supplement 1
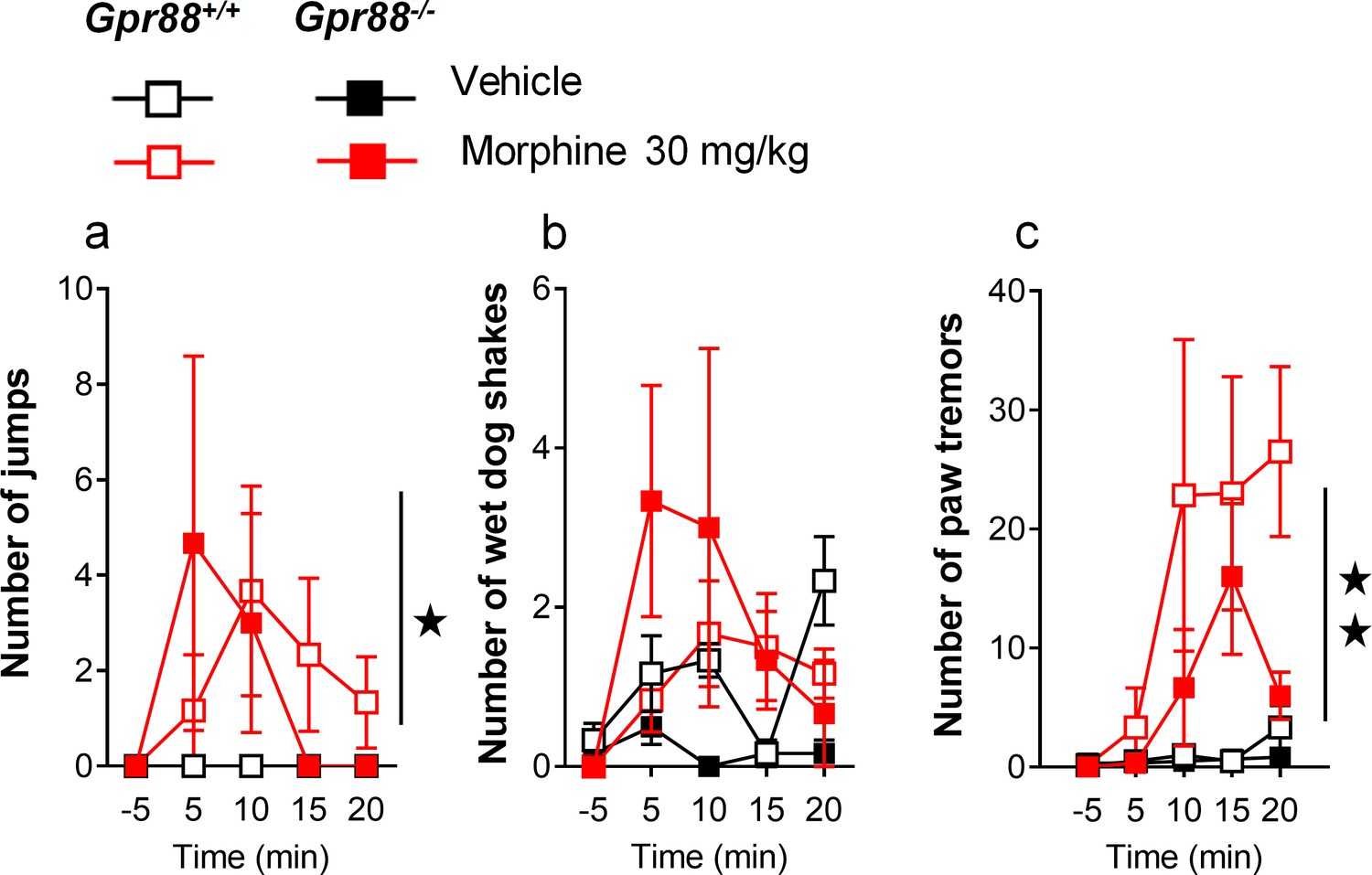
Additional morphine-induced withdrawal signs in Gpr88-/- versus Gpr88+/+ mice.
After an exposure to escalating doses of morphine, Gpr88+/+ and Gpr88-/- mice (n = 6 per treatment and genotype) received an acute injection of naloxone (1 mg/kg) and withdrawal signs were quoted for 20 min. Gpr88 null animals and wild-type controls displayed similar numbers of jumps (panel a; Genotype: F1,20=0.02, p=0.9016; Treatment: F1,20=5.9, p=0.0246), wet dog shakes (panel b; Genotype: F1,20=2.4, p=0.1383; Treatment: F1,20=2.6, p=0.1222) and paw tremors (panel c; Genotype: F1,20=1.8, p=0.1913; Treatment: F1,20=9.0, p=0.0070; Time: F3,60=5.42, p=0.0023; Time x Treatment: F3,60=4.7, p=0054). Data are presented as mean ± SEM. Solid stars: treatment effect (two-way ANOVA with one repeated measure – day/5 min time bin), one symbol: p<0.05, two symbols: p<0.01; three symbols: p<0.001.
Figure 3—figure supplement 2
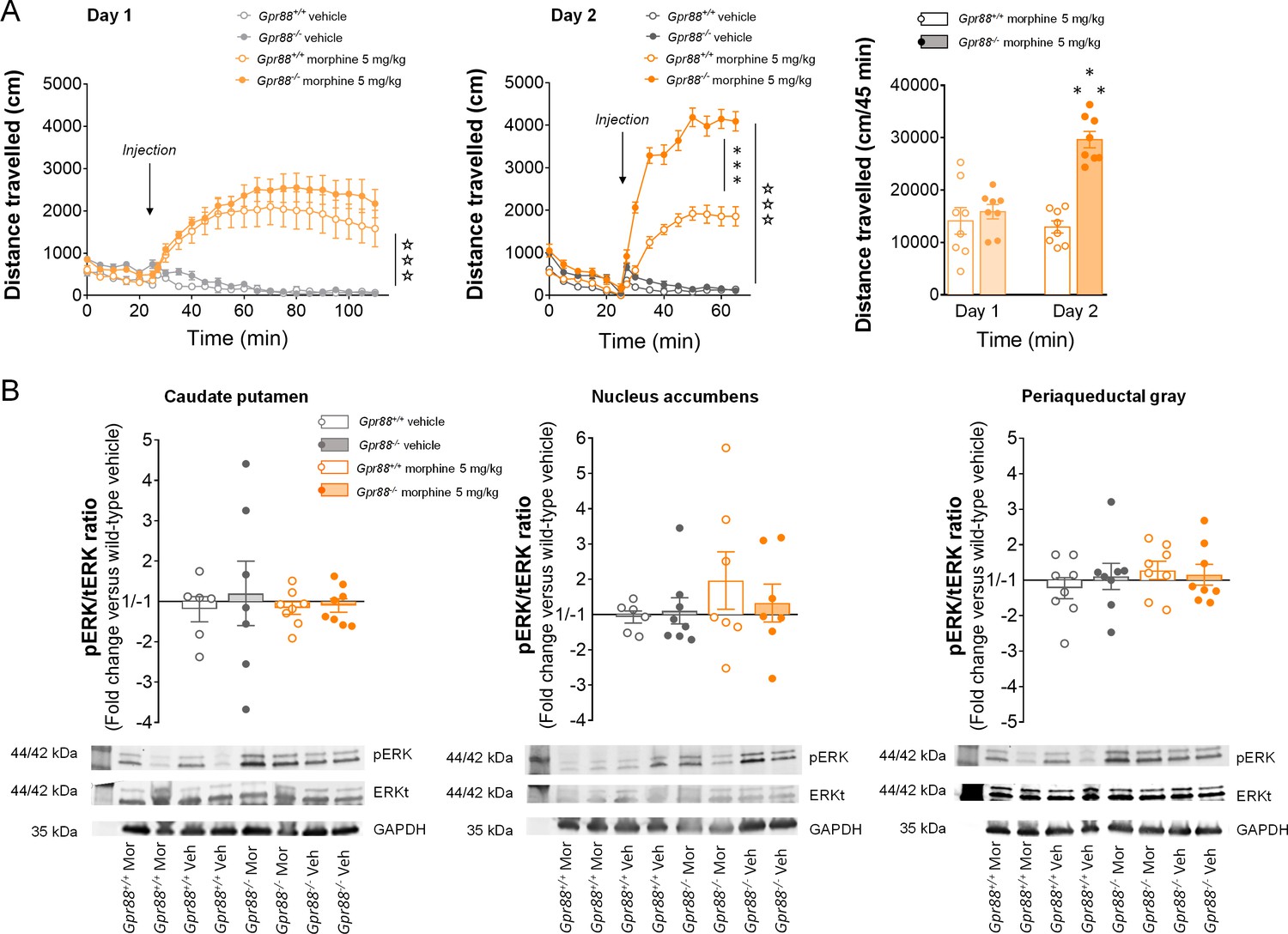
Increased morphine-induced locomotor sensitization in Gpr88 null mice was not associated with modified pERK/tERK ratio in three brain regions.
(A) In a two-day locomotor sensitization paradigm (n = 8 mice per treatment and genotype), morphine (5 mg/kg) induced an increase in locomotor activity (Day 1; Genotype: F1,28=0.83, p=0.3690; Treatment: F1,28=63.4, p=0.0000; Time: F17,176=11.1, p=0.0000) that sensitized after a second administration (Day 2) in Gpr88-/- but not Gpr88+/+ mice (right panel; Genotype: F1,28=78.2, p=0.0000; Treatment: F1,28=365.5, p=0.0000; Genotype x Treatment: F1,28=58.9, p=0.0000; Time: F8,224=76.8, p=0.0000; Time x Genotype: F8,224=7.0, p=0.0000; Time x Treatment: F8,224=139.9, p=0.0000; Time x Genotype x Treatment: F8,224=13.13, p=0.0000). Data are presented as mean ± SEM. Open stars: Time x Treatment interaction, asterisks: Genotype x Treatment (three-way ANOVA with one repeated measure – time bin), three symbols: p<0.001. (B) The mice were sacrificed 60 min after the second morphine challenge for western blot analysis. Upper panels: in contrast with in vitro results, no difference was observed in the pERK/tERK ratio between Gpr88-/- and Gpr88+/+ animals in either striatal regions (caudate putamen: Genotype: F1,25=0.10, p=0.7516; Treatment: F1,25=0.28, p=0.5978; nucleus accumbens: Genotype: F1,24=1.4, p=0.2556; Treatment: F1,24=0.18, p=0.6719), where µOR activation induces morphine sensitization, or in a µOR-expressing region not involved in the sensitization process, the periaqueductal gray (Genotype: F1,28=0.80, p=0.3776; Treatment: F1,28=011, p=0.7378). Lower panels: representative western blotting images. Levels of phosphorylated-ERK (pERK) and total-ERK (tERK) were normalized to the loading control protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are presented as individual values and mean ± SEM of n = 3–4 independent experiments. Two brain punches were lost for striatal regions in the Gpr88+/+ vehicle group.
Figure 3—figure supplement 3
Gels from western blot experiments; ERK phosphorylation assay in brain samples from Gpr88+/+ and Gpr88-/-mice.
Figure 4
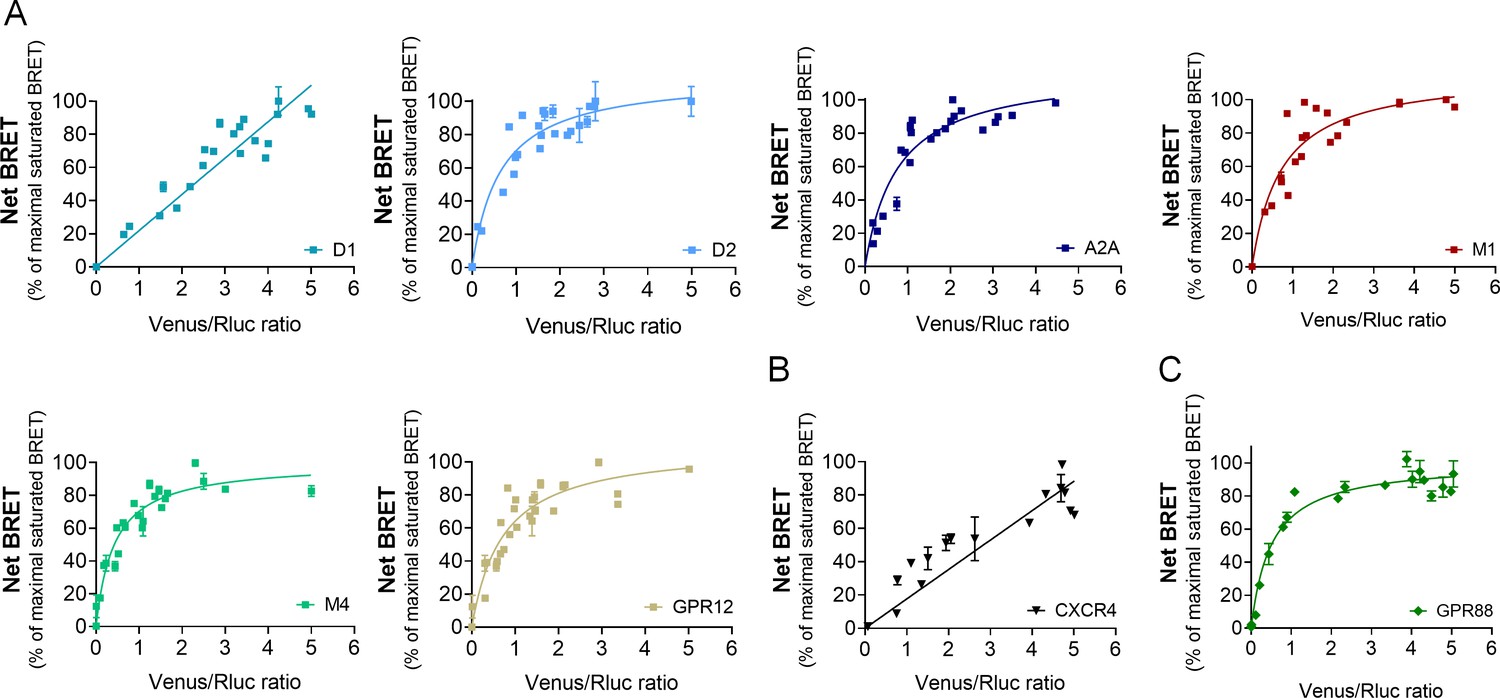
GPR88 comes in close proximity to multiple GPCRs.
BRET1 saturation experiments were performed in transfected HEK293FT cells using constant quantity of GPR88-Rluc8 with increasing amounts of (A) Venus-tagged striatal GPCRs: dopamine D1 and D2, adenosine A2A, muscarinic M1 and M4, and orphan receptor GPR12, (B) Venus-tagged non neuronal GPCRs: chemokine CXCR4 and V2R (see Figure 1). (C) Venus-tagged GPR88. Saturated BRET signals indicate close physical proximity (within 10 nm) to the target GPCR. GPR88 comes in close proximity and possibly forms hetero-oligomers with D2, A2A, M1, M4, GPR12 and itself, but not D1, V2R and CXCR4 receptors. Values (mean ± SEM) from n = 3–4 independent experiments (performed in triplicates) are presented as net BRET (normalized as the percentage of maximal BRET values) by Venus/Rluc8 BRET ratio.
Figure 5
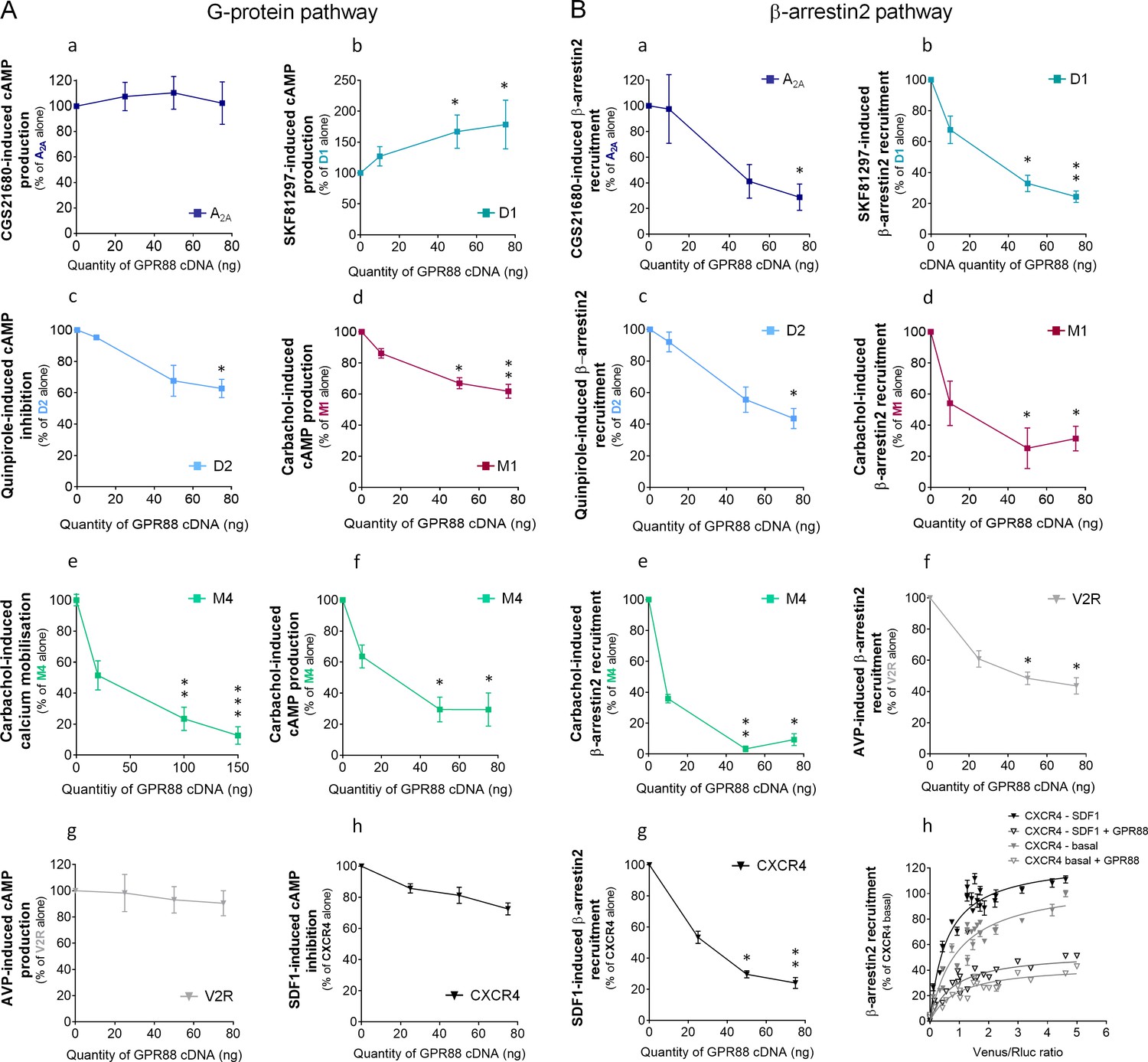
GPR88 biases the signaling of multiple GPCRs.
We evaluated the consequences of GPR88 co-expression on the signaling of adenosine A2A, dopamine D1, dopamine D2, muscarinic M1, muscarinic M4, vasopressin V2R and CXCR4 receptors in HEK293FT cells. BRET1 assay was used to assess the activation of the (A) G protein dependent pathway (cAMP sensor: CAMYEL; calcium sensor: aequorin-GFP) or (B) the recruitment of Ypet β-arrestin 2 (β-arr2) by Venus-tagged receptors in presence of increasing amounts of GPR88 cDNA transfected. (A) GPR88 co-expression blunted the G protein mediated signaling of all GPCRs from which it comes close (panels c-f; dopamine D2: H3,12=9.6, p=0.0223; muscarinic M1: H3,16=13.2, p=0.0041; M4 calcium: H3,35=24.0, p=0.0000; M4 cAMP: H3,12=12.9, p=0.0048), except the adenosine A2A receptor (panel a; H3,12=1.2, p=0.7602). GPR88 had no significant impact on, or even facilitated, G protein dependent signaling of receptors for which we failed to evidence proximity to it, namely D1 (panel b, increased activity in presence of GPR88; H3,20=9.7; p=0.0211), V2R and CXCR4 receptors (panels g,h; V2R: H3,16=1.2, p=0.7602; CXCR4: H3,12=7.6, p=0.0546). (B) In contrast, GPR88 co-expression compromised the ability of all GPCRs tested to recruit Ypet-β-arr2 when activated by their agonist, independently from previously evidenced close proximity to GPR88 (panels a-g; A2A: H3,12=9.7, p=0.0211; D1: H3,12=9.6, p=0.223; D2: H3,16=13.2, p=0.0080; M1: H3,15=10.4, p=0.0151; M4: H3,16=13.2, p=0.0041; V2R: H3,16=10.9, p=0.0123; CXCR4: H3,16=13.2, p=0.0041). Focusing on CXCR4, co-expressing GPR88 diminished the probability of Ypet-β-arr2 recruitment at this receptor under both stimulated and basal conditions (panel h). Specific agonists used to stimulate GPCRs were: CGS21680 (A2A, 10 μM), SKF81297 (D1, 10 µM), quinpirole (D2, 10 µM), carbachol (M1 and M4, 10 µM), AVP (V2R, 10 μM) and SDF1 (CXCR4, 125 µM). Data are presented as mean ± SEM of n = 3–4 independent experiments (performed in triplicates). BRET1 values are presented as induced BRET (normalized as the percentage of maximal BRET values in absence of GPR88) normalized by Venus/Rluc8 BRET ratio. Asterisks: Kruskal-Wallis ANOVA, multiple comparison of mean ranks, *p<0.05, **p<0.01.
Figure 6

GPR88 impedes the recruitment of β-arrestins at other GPCRs independently from physical proximity.
(A) We evaluated the functional consequences of co-expressing CXCR4, κOR and GPR88 with µOR on DAMGO-induced cAMP production and β-arr2 recruitment by µOR. CXCR4 co-expression had no deleterious impact on G-protein-dependent signaling and the recruitment of β-arrestins (cAMP: H3,16=6.6, p=0.0840; β-arr2: H3,16=2.8, p=0.427); in contrast, κOR and GPR88 expression dampened the activation of both, with GPR88 inhibiting cAMP production by µOR more efficiently than κOR (cAMP - κOR: H3,12=10.1, p=0.0176; GPR88: H3,12=10.5, p=0.0145; β-arr2 - κOR: H3,16=13.8, p=0.0031; GPR88: H3,16=13.5, p=0.0036). (B) We then assessed the consequences of co-expressing µOR, κOR and GPR88 with CXCR4 on SDF1-induced cAMP production and β-arr2 recruitment by CXCR4. None of the co-expressed receptors had a significant impact on cAMP production by CXCR4 (μOR: H3,12=7.3, p=0.064; κOR: H3,12=2.6, p=0.4593; GPR88: H3,12=2.9, p=0.4077); as regards β-arr2 recruitment, µOR co-expression had no influence (H3,12=0.75, p=0.8604), whereas both κOR (H3,16=12.7, p=0.0053) and GPR88 (H3,12=9.8, p=0.0203) reduced it, with GPR88 having a more significant influence than κOR at the highest dose transfected. (C) BRET1 saturation experiments evidenced close proximity between µOR and κOR (upper panel), CXCR4 and κOR (middle panel) but not CXCR4 and µOR (lower panel). (D) Ypet-β-arr2 recruitment was higher at CXCR4 and GPR88 than κOR and µOR under basal conditions. (E) Confocal microscopy images show that (a) HA-GPR88 is localized at cell surface and also in the cytosol, following a patchy distribution (permeabilized cells), (b) β-arr2 is expressed in a diffuse pattern near the cell membrane and almost all the cytoplasm and (c) Colocalisation of GPR88 and β-arr2 is clearly seen at the level of cytoplasmic patches, with the intensity of β-arr2 expression appearing lower around the densest GPR88-expressing patches (framed, arrow head). (F) Schematic representation of the putative mechanisms of GPR88 inhibitory action at GPCR signaling. Panel a: GPR88 dampens G-protein-mediated signaling of GPCRs to which it comes close (such as µOR), likely by interfering with G-protein coupling, and inhibits the recruitment of β-arrestins by sequestering them in intracellular compartments. Pharmacological activation of GPR88 may potentiate its inhibitory action on G-protein dependent signaling only. Panel b: when no proximity is detected between GPR88 and the target receptor (such as CXCR4), only the effects on β-arrestin recruitment are observed. Data (A–D) are presented as mean ± SEM of n = 3–4 independent experiments (performed in triplicates). BRET1 values are presented as net BRET (normalized as the percentage of maximal BRET values) or induced BRET (normalized as the percentage of maximal BRET values when the target receptor is expressed alone) by Venus/Rluc8 BRET ratio. Asterisks: Kruskal-Wallis ANOVA, multiple comparison of mean ranks, *p<0.05, **p<0.01. Confocal imaging (E): representative pictures among n = 10 pictures. Legend to Supplementary file 1.
Tables
Key resources table
| Reagent type (species) or resource information | Designation | Source or reference | Identifiers | Additional | |
|---|---|---|---|---|---|
| Chemical compound, drug | Arginine-Vasopressin (AVP) | Tocris Bioscience, Bristol, UK | Cat# 2935/1 | ||
| Chemical compound, drug | Carbamoylcholine chloride | Tocris Bioscience, Bristol, UK | Cat# 2810/100 | ||
| Chemical compound, drug | CGS21680 | Tocris Bioscience, Bristol, UK | Cat# 1063/10 | ||
| Chemical compound, drug | Coelenterazine H substrate | Interchim, Montluçon, France | Cat# R30783 | ||
| Chemical compound, drug | Compound 19 | Kindly synthetized by Domain Therapeutics Dzierba et al., 2015 | |||
| Chemical compound, drug | DAMGO | Tocris Bioscience, Bristol, UK | Cat# 1171/1 | ||
| Chemical compound, drug | Forskolin | Tocris Bioscience, Bristol, UK | Cat# 1099 | ||
| Chemical compound, drug | IBMX | Tocris Bioscience, Bristol, UK | Cat# 2845/50 | ||
| Chemical compound, drug | Ionomycin calcium salt | Tocris Bioscience, Bristol, UK | Cat# 1704/1 | ||
| Chemical compound, drug | Metafectene PRO | Biontex, München, Germany | Cat#T040-5.0 | ||
| Chemical compound, drug | Morphine HCl | Francopia, Antony, France | |||
| Chemical compound, drug | Naloxone | Sigma-Aldrich, Saint-Quentin Fallavier, France | Cat# N7758 | ||
| Chemical compound, drug | Pertussis Toxin | Tocris Bioscience, Bristol, UK | Cat# 3097/50U | ||
| Chemical compound, drug | Quinpirole hydrochloride | Tocris Bioscience, Bristol, UK | Cat# 1061/10 | ||
| Chemical compound, drug | SKF81297 | Tocris Bioscience, Bristol, UK | Cat# 1447/10 | ||
| Chemical compound, drug | SNC80 | Tocris Bioscience, Bristol, UK | Cat# 0764/10 | ||
| Chemical compound, drug | Stromal cell-derived factor 1 (SDF-1) | Tocris Bioscience, Bristol, UK | Cat# 3951 | ||
| Chemical compound, drug | U50488H | Tocris Bioscience, Bristol, UK | Cat# 0495/25 | ||
| Material | Mithras² LB 943 Monochromator Multimode Microplate Reader | Berthold Technologies GmbH and Co. KG, Bad Wildbad, Germany | |||
| Material | MACSQuant10 flow cytometer | Miltenyi, Bergisch Gladbach, Germany | |||
| Material | Trans-Blot Turbo Transfer System | Bio-Rad, Hercules, California, USA. | |||
| Material | Odyssey CLx | LI-COR, Lincoln, Nebraska, USA | |||
| Material | LSM 700 laser scanning confocal microscope | Zeiss, Oberkochen, Germany | |||
| Material | Infrared floor | Videotrack; View Point, Lyon, France | |||
| Material | Computerized CPP boxes | Imetronic, Pessac, France | |||
| Material | Hot plate | Ugo Basile, Gemonio, Italia | |||
| Antibody | GAPDH (14C10) Rabbit mAb | Cell Signaling, Leiden, Netherlands | Cat# 2118 | (1:2000) | |
| Antibody | Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP Rabbit mAb | Cell Signaling, Leiden, Netherlands | Cat# 4370 | (1:2000) | |
| Antibody | p44/42 MAPK (Erk1/2) (3A7) Mouse mAb | Cell Signaling, Leiden, Netherlands | Cat# 9107 | (1:2000) | |
| Antibody | Goat anti-rabbit IRDye800CW | LI-COR, Lincoln, Nebraska, USA | Cat# 926–3221 | (1:15000) | |
| Antibody | Goat Anti-mouse IRDye680CW | LI-COR, Lincoln, Nebraska, USA | Cat# 926–68070 | (1:15000) | |
| Antibody | Goat polyclonal anti-HA HRP-conjugated antibody | Bethyl Laboratories, USA | Cat#A190-138P | (1:10000) | |
| Antibody | Anti-HA tag antibody - ChIP Grade | Abcam, Cambridge, UK | Cat# ab9110 | (1:300) | |
| Antibody | Cy3 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Europe Ltd, Cambridgeshire, Uk | Cat# 711-165-152 | (1:300) | |
| Cell line (Human) | HEK 293FT cell line | ThermoFisher Scientific Inc, Waltham, Massachusetts, USA | Cat# R70007 | ||
| Commercial assay or kit | SuperSignal ELISA Femto Maximum Sensitivity Substrate | ThermoFisher Scientific Inc, Waltham, Massachusetts, USA | (1:10) | ||
| Commercial assay or kit | Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit | Bio-Rad, Hercules, California, USA | Cat# 1704271 | ||
| Commercial assay or kit | DAPI Hardset mounting medium | Vectashield Vector laboratories, Burlingame, California, USA | Cat# H-1500 | ||
| Strain, strain background (Mus musculus) | Gpr88+/+ and Gpr88-/-, hybrid 50% C57BL/6J–50% 129Sv genetic background | Meirsman et al., 2016a | |||
| Strain, strain background (Mus musculus) | C57BL/6JRj | Janvier Labs, Le Genest-Saint-Isle, France |
Additional files
-
Supplementary file 1
Statistical analysis of western blotting data from in vitro experiments (Kruskal-Wallis ANOVA).
- https://cdn.elifesciences.org/articles/50519/elife-50519-supp1-v2.docx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/50519/elife-50519-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The orphan receptor GPR88 blunts the signaling of opioid receptors and multiple striatal GPCRs
eLife 9:e50519.
https://doi.org/10.7554/eLife.50519




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}