Chromatin accessibility dynamics and single cell RNA-Seq reveal new regulators of regeneration in neural progenitors
- Department of Biochemistry, University of Washington, United States
- Oregon Health Sciences Center For Early Detection Advanced Research Center (CEDAR), United States
- Department of Genome Sciences, University of Washington, United States
Figures
Figure 1 with 1 supplement
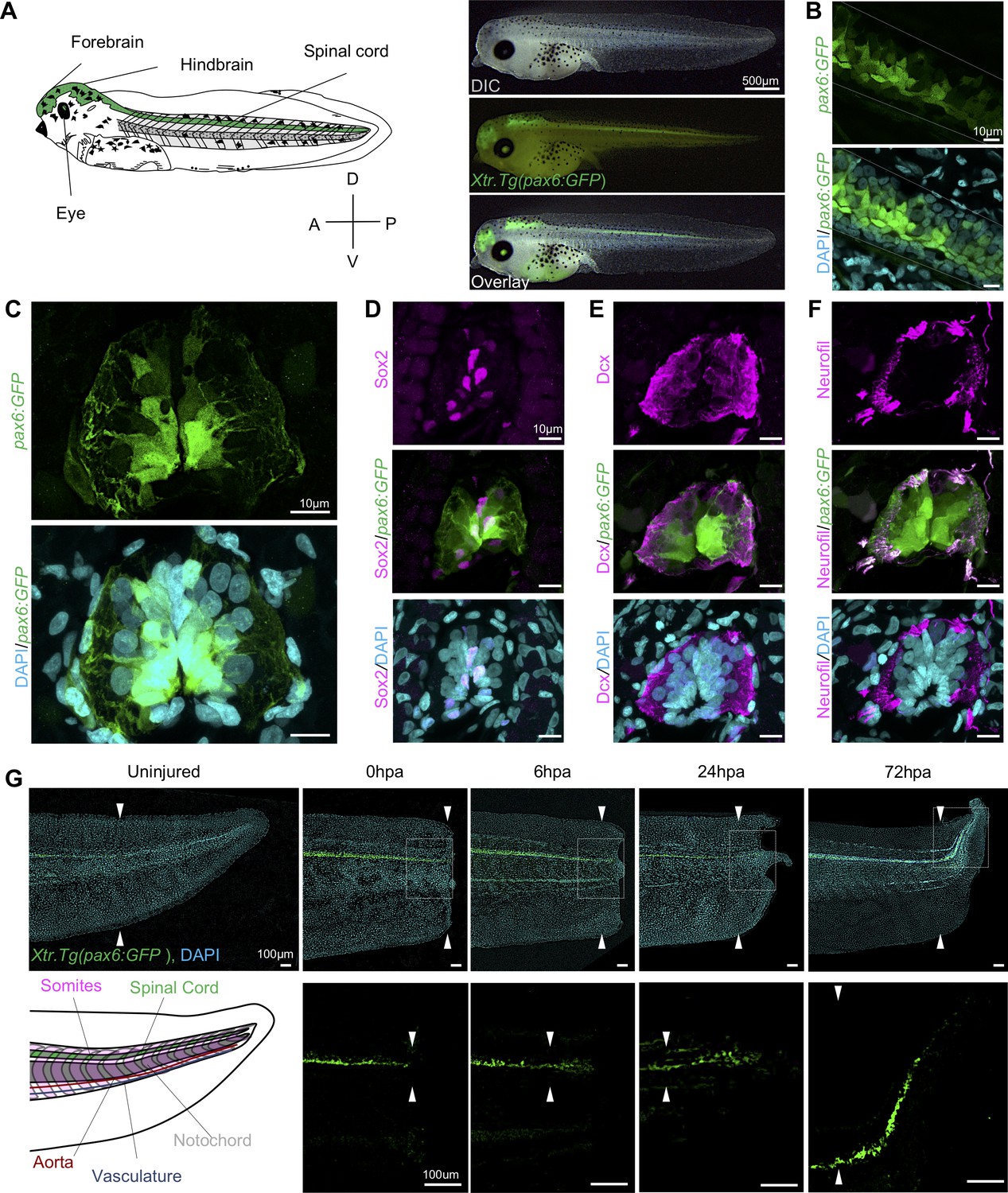
Expression dynamics of pax6:GFP in the regenerating spinal cord.
(A) Left, cartoon depiction of a NF stage 41 tadpoles with known pax6+ domains colored green. Right, Stage 41, transgenic tadpole expressing GFP under the control of pax6 promoter tadpole. For all images in this figure, the green channel represents pax6 reporter GFP fluorescence and the cyan channel represents DAPI staining. (B) Confocal image of a lateral view of a whole-mount transgenic stage 41 tadpole. (C–F) Transverse cryosections through the posterior spinal cord of a transgenic stage 41 tadpole. (D–F) Immunofluorescence images of the spinal cord sections stained with (D) anti-Sox2, (E) anti-Dcx, and (F) anti-neurofilament. (G) Regeneration timecourse of Xtr.Tg(pax6:GFP) tadpole over the first 72 hr following tail amputation. The white box in the top photos correspond with enlarged green channel below. White arrows indicate amputation plane.
Figure 1—figure supplement 1
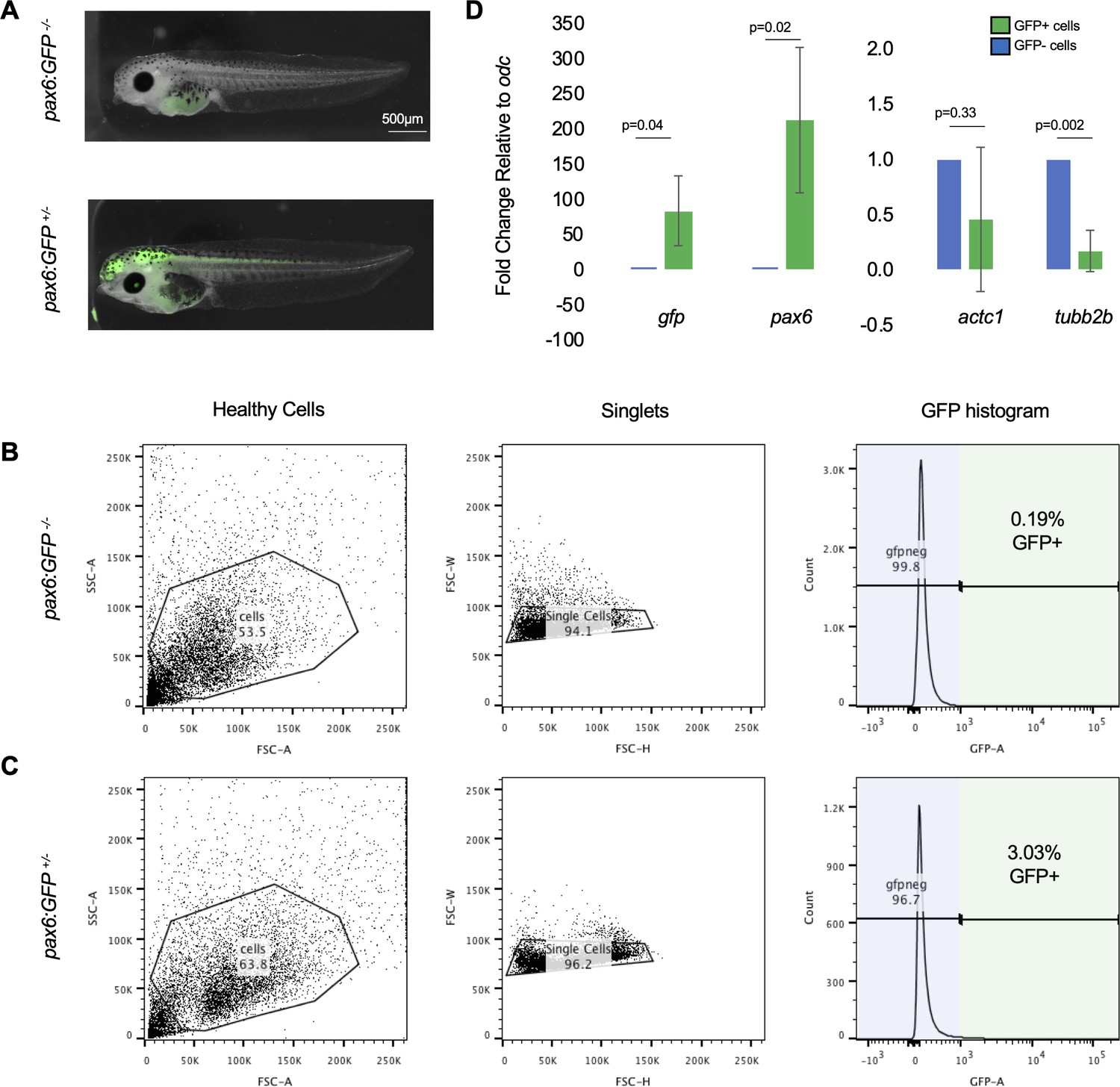
FACS gating was set using wild-type tadpoles to avoid false-positive GFP+ cells.
(A) Representative images of stage 41 (top) wild-type tadpoles and (bottom) Xtr.Tg(pax6:GFP) tadpoles. (B) Representative gating for wild-type tadpoles. Gates were drawn to exclude all cells in the wild-type sample from being part of GFP signal. This gate setting was used to exclude autofluorescence from yolk in tadpole cells. The same gates from (B) were applied in (C) to collect pax6:GFP+ tadpole cells. Cells that fall within the gate represent true GFP+ signal. (D) qRT-PCR on GFP+ and GFP- cells from uninjured tails. GFP was used as a positive control, pax6 was used to assay for NPC marker, actc1 was used as a non-neural marker, and tubb2b was used as a differentiated neural marker.
Figure 2 with 1 supplement
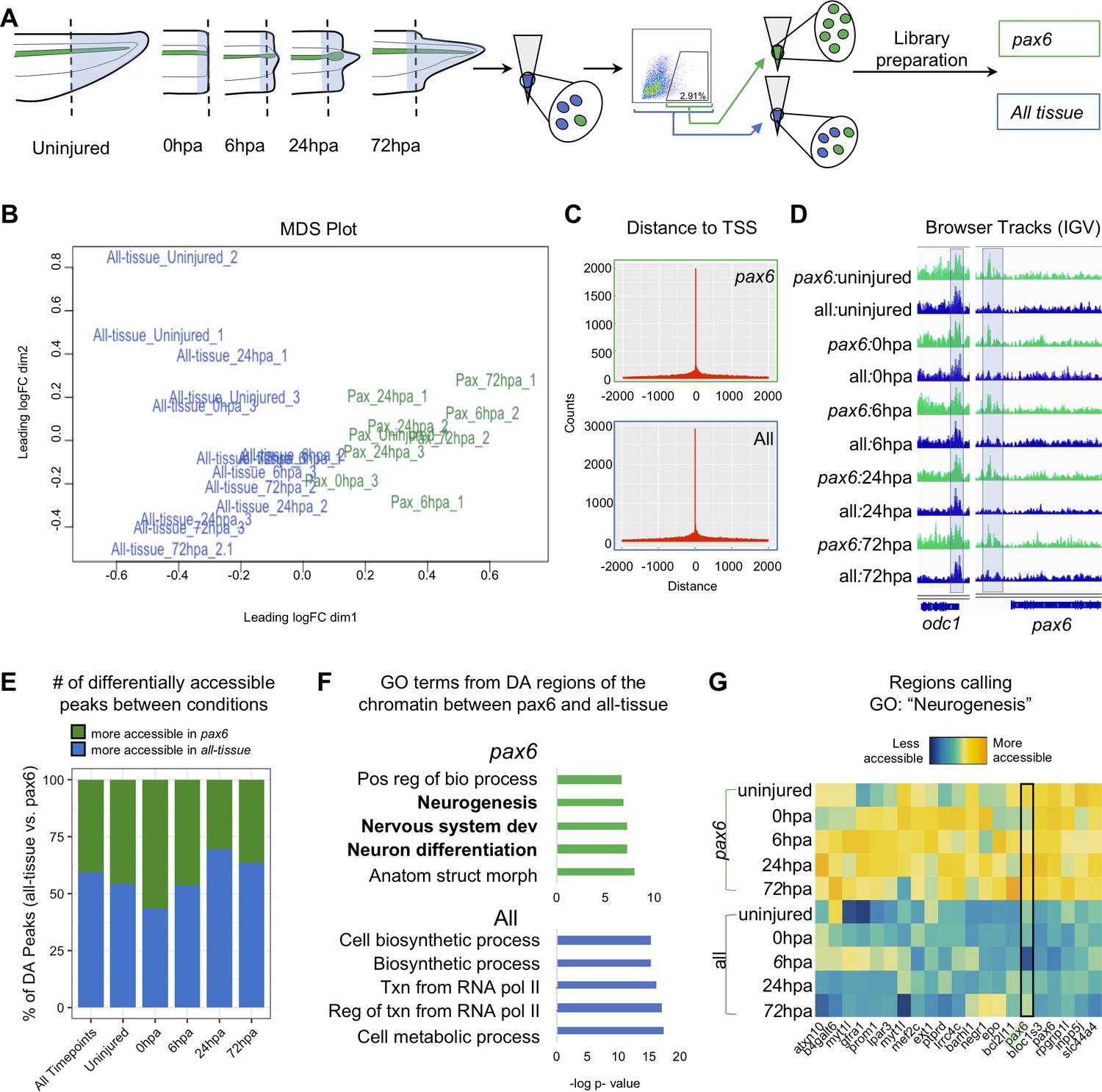
Pax6 ATAC-Seq libraries resolve accessible neural specific regions that were not identified in all-tissue libraries.
(A) Experimental design for FACS isolation of reporter cells for sequencing library preparations. (B) MDS plot of sequenced pax6 and all-tissue ATAC-Seq libraries. (C) Representative histograms showing distance of called peaks to transcription start sites (tss). (D) IGV browser tracks of a house keeping gene, odc and reporter line specific gene, pax6. Shaded bar in odc represents a peak retained in pax6 and all-tissue libraries; shaded bar in pax6 represents a peak called in pax6 libraries, but not in all-tissue. (E) Bar plot showing percent of differentially accessible (DA) peaks that are more accessible in pax6 or all-tissue libraries within an individual timepoint or across all timepoints. (F) Differentially accessible regions were annotated to the nearest TSS and used to call GO:BP terms. Top, GO terms from regions more accessible in pax6 libraries than in all-tissue libraries. Bottom, GO terms from regions more accessible in all-tissue libraries than pax6 libraries. (G) Heat map depicting accessibility of 21/254 randomly sampled peaks that call the GO term: Neurogenesis. pax6 promoter region is boxed.
Figure 2—figure supplement 1
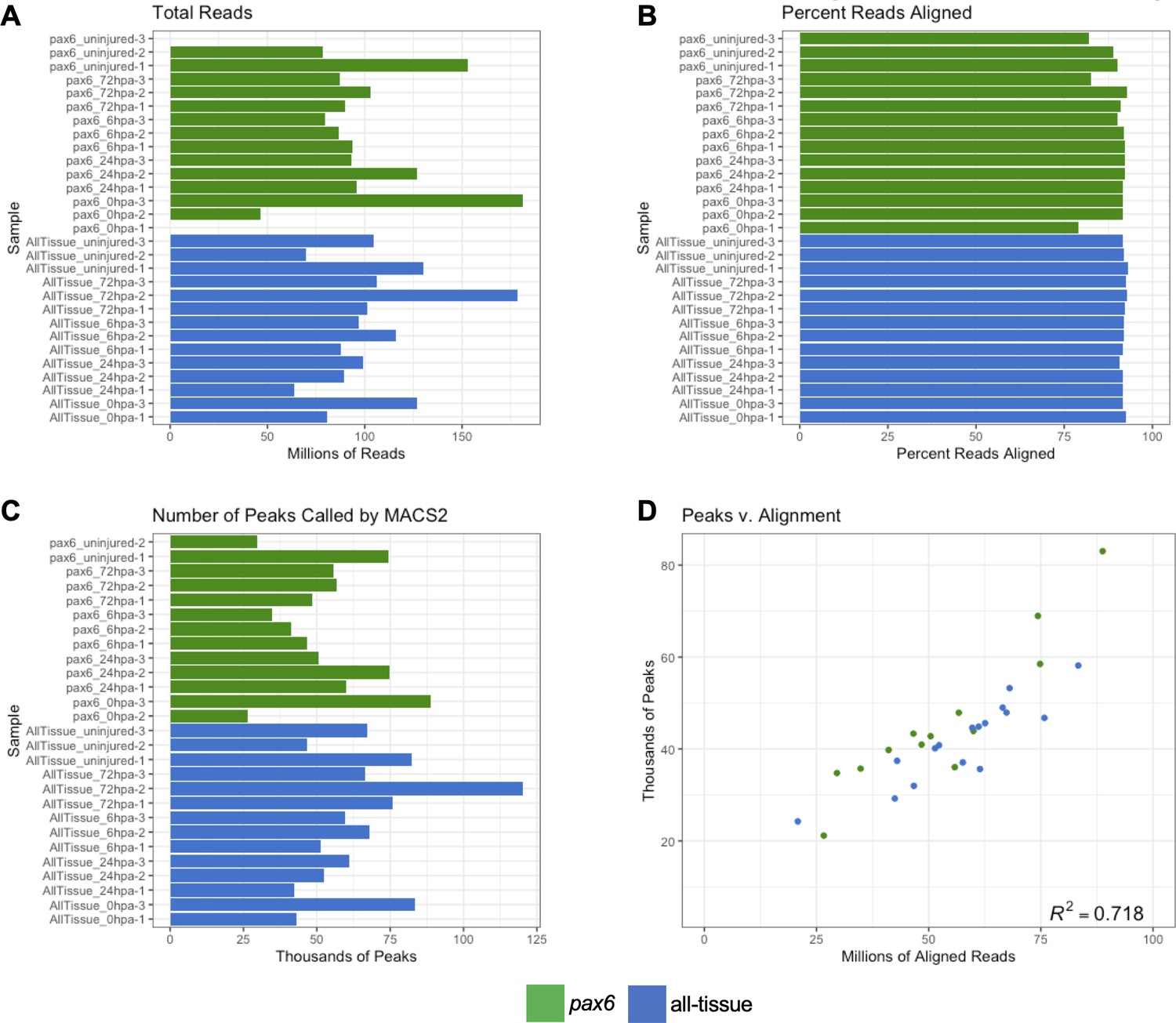
Low input ATAC-Seq libraries meet sufficient quality for downstream analysis.
(a) Barplot of total number of reads sequenced for each library. (b) Barplot of the percent reads aligned to the genome by Bowtie2 (c) Barplot of the number of peaks called from aligned reads by MACS2. (d) Correlation plot of number of aligned reads to number of called peaks. Reads and peaks aligns with correlation coefficient of r2 = 0.785.
Figure 3
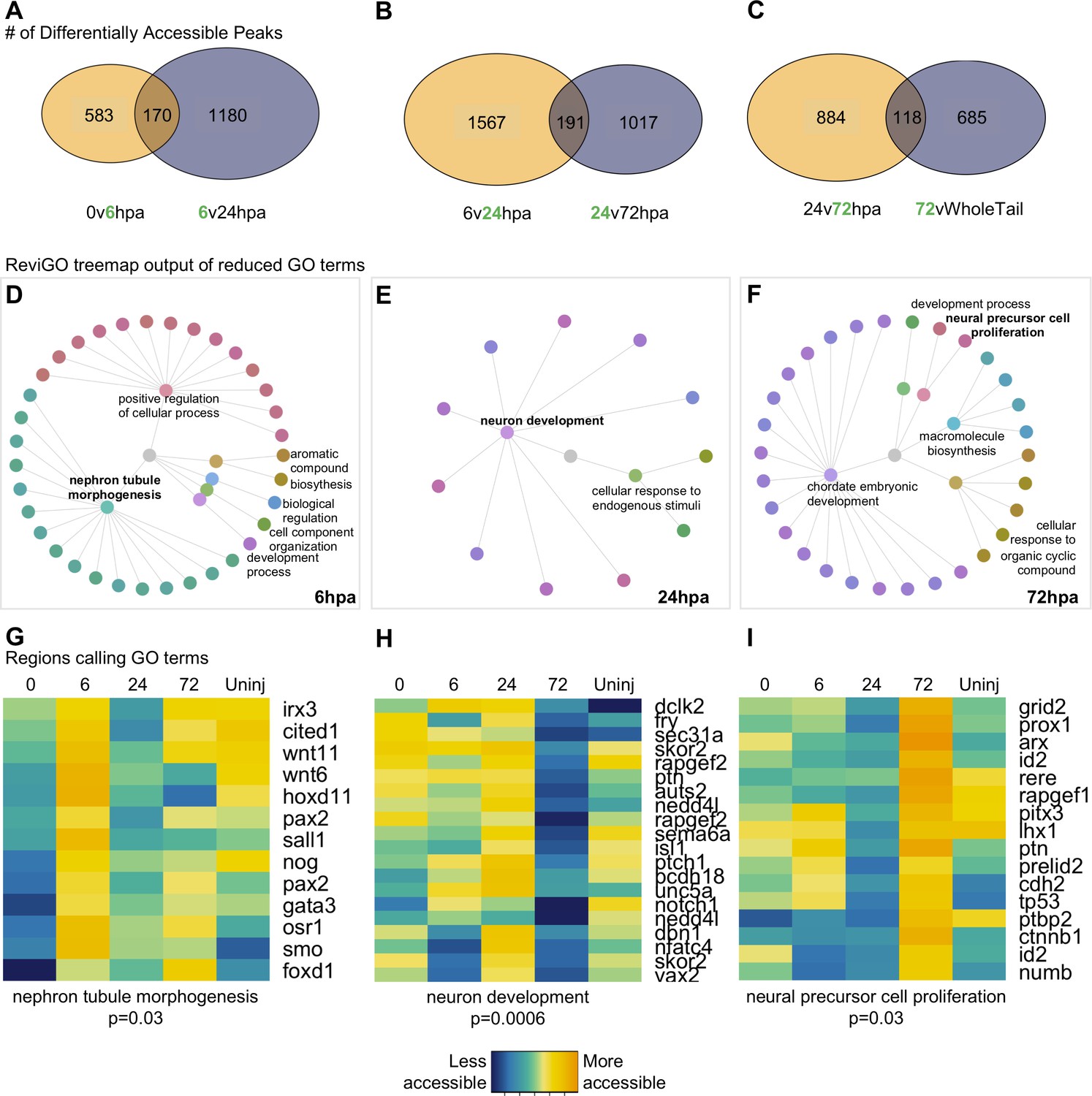
Differential accessibility analysis of pax6 libraries over regenerative time reveals chromatin accessibility prioritizes first tubule morphogenesis, followed by neural differentiation, and later proliferation.
From left to right, data represents the 6hpa timepoint, 24hpa timepoint, and 72hpa timepoint. (A–C) Venn diagrams depicting number of unique and shared peaks called in each of the timepoint contrasts within the pax6 libraries. The intersect represents peaks that were both becoming accessible at a given timepoint, then losing accessibility. The green number in each contrast represents the timepoint that is more accessible. (D–F) All regions represented in (A–C) were used for GO:BP analysis with gProfileR2. ReviGO was then used to reduce redundancy in output GO terms and identify main families of terms. Tree graphs depict the hierarchy of reduced GO list. The central grey circle is the vertex of the graph representing the set of all GO terms included in the data, the second level of circles are the families of GO terms called by ReviGO, and the third level of circles represents GO terms in each family. Terms provided are the family names. (G–I) Heatmap of accessibility of regions that were used to call GO terms: ‘Nephron Tubule Morphogenesis’, ‘Neuron Differentiation’, and ‘Neural Precursor Cell Proliferation’.
Figure 4 with 1 supplement
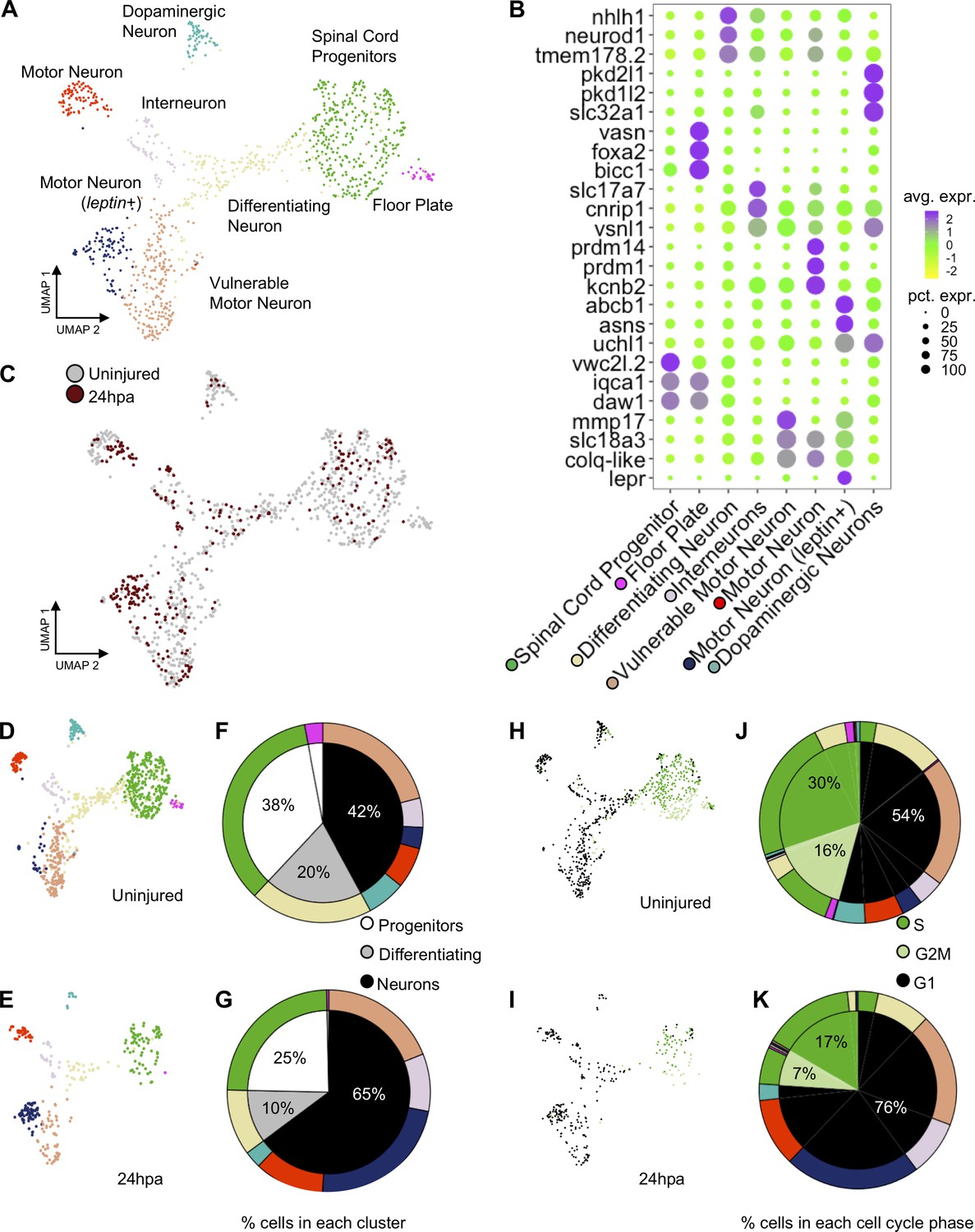
scRNA-Seq of uninjured and 24hpa tails reveals a transcriptional shift to differentiated neuronal types at 24hpa.
(A) UMAP projection of integrated uninjured and 24hpa neural lineage cells. seven distinct clusters were identified. (B) Dot plot of genes identified as differentially expressed between cell clusters. The top three genes exclusively expressed in each of the seven clusters are shown. Color of circle denotes average gene expression across one cluster and size of circle represents percent of cells in each cluster expressing each gene. (C) UMAP projection of neural cells colored by timepoint in the uninjured tails and 24hpa tails. UMAP projections of cell clusters split by timepoint to (D) uninjured and (E) 24hpa. (F–G) Sunburst diagrams showing (inside) the percentage of neural cells that are broadly stem cells, differentiating cells, or differentiated neurons and (outside) percentage of neural cells in each of the seven cell clusters called. Colors on outside of the sunburst chart correlate to cluster colors in (A/B). (H–I) UMAP projections of neural clusters colored by predicted cell cycle phase in the (H) uninjured tail and (I) 24hpa tail. (J–K) Sunburst diagrams showing the percentage of all neural cells predicted to be in each cell cycle phase (inside) and the percentage of each cell cluster predicted to be in each cell cycle phase (outside). (J) represents the distribution in the uninjured tail and (K) represents the distribution in the 24hpa tail. Colors on outside of the sunburst chart correlate to cluster colors in (A/B).
Figure 4—figure supplement 1
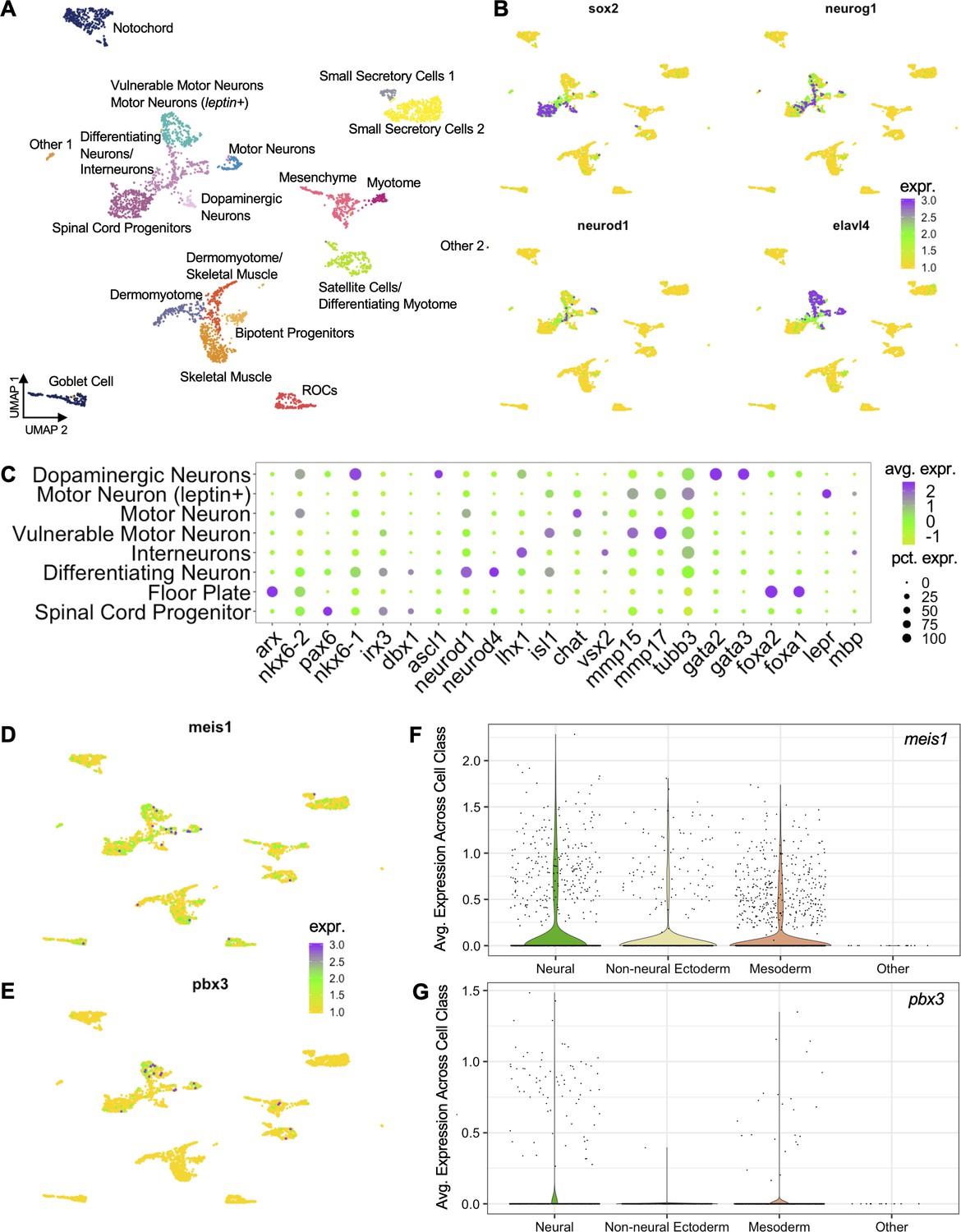
scRNA-Seq analysis of all cells sequenced in uninjured and 24hpa tails.
(A) UMAP dimensionality reduction and cluster assignment of integrated cells from uninjured and 24hpa tails. These cells are a superset of the seven clusters shown in Figure 4. (B) UMAP plots of cell colored by expression of neural lineage markers. Sox2 was used to identify neural stem cells, neurod1 and neurog1 were used to identify differentiating neurons, and elavl4 was used to identify differentiated neurons. 6 cell clusters were identified by these markers as neural lineage clusters. (C) Dotplot showing expression of genes used to determine neural identities as defined by Aztekin et. al. Size of circle represents percentage of cells in cluster expressing gene and color denotes average expression of a gene across a cluster. UMAP plots of cells colored by expression for (D) meis1 and (E) pbx3 across all cells. Violin plots showing distribution of gene expression for (F) meis1 and (G) pbx3. In the violin plots, cell types from (A) were grouped into broader cell lineages.
Figure 5
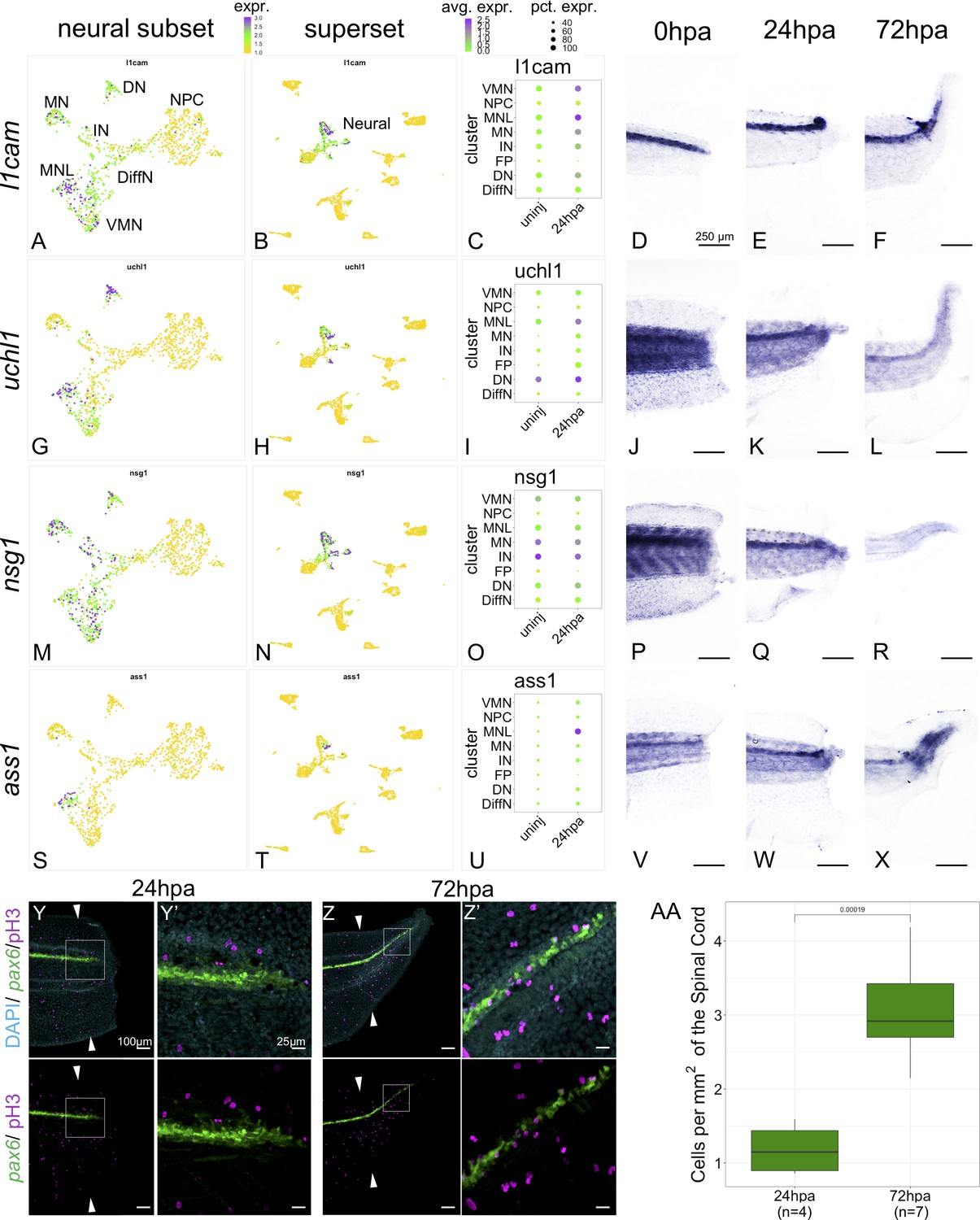
Markers of neuronal differentiation are increased at 24hpa and proliferation increases at 72hpa.
UMAP plots of expression in the neural restricted subset of scRNA-Seq for (A) l1cam, (G) uchl1, (M) nsg1, and (s) ass1. UMAP plots of expression in the superset scRNA-Seq dataset for (B) l1cam, (H) uchl1, (N) nsg1, and (T) ass1. Note that the topology of neural cell clusters is slightly different when subset and reclustered to give high neural cluster resolution (A,G,M,S) than when neural cells are analyzed together with all other cell types (E,K, Q, W), but the same cells and expression data are included in both cases. Dot plots representing average expression of (C) l1cam, (I) uchl1, (O) nsg1, and (U) ass1 per cluster and timepoint. In situ hybridization at 0hpa, 24hpa, and 72hpa for (D–F) l1cam, (J–L) uchl1, (P–R) nsg1, and (V–X) ass1. (Y–Z’) Representative images of 24hpa (Y/Y’) and 72hpa (Z/Z’) regenerated tails from a pax6:GFP transgenic tadpole stained for DAPI (cyan) and mitotic cells with phospho-histone3 (magenta). The white arrows indicate regeneration plane. (Y’) and (Z’) are enlarged images of the boxed areas in (Y) and (Z). (AA) Boxplot representing the number of cells per regenerated spinal cord area (mm2) at 24hpa and 72hpa. Statistics represent t-test performed between the two timepoints. Abbreviations: Neural Progenitor Cell (NPC), Differentiating Neuron (Diff), Interneuron (IN), Vulnerable Motor Neuron (VMN), Dopaminergic Neuron (DN), Motor Neuron Leptin+ (MNL), Motor Neuron (MN)).
-
Figure 5—source data 1
PH3 Cell Count and Regenerated Spinal Cord Area.
- https://cdn.elifesciences.org/articles/52648/elife-52648-fig5-data1-v2.csv
Figure 6
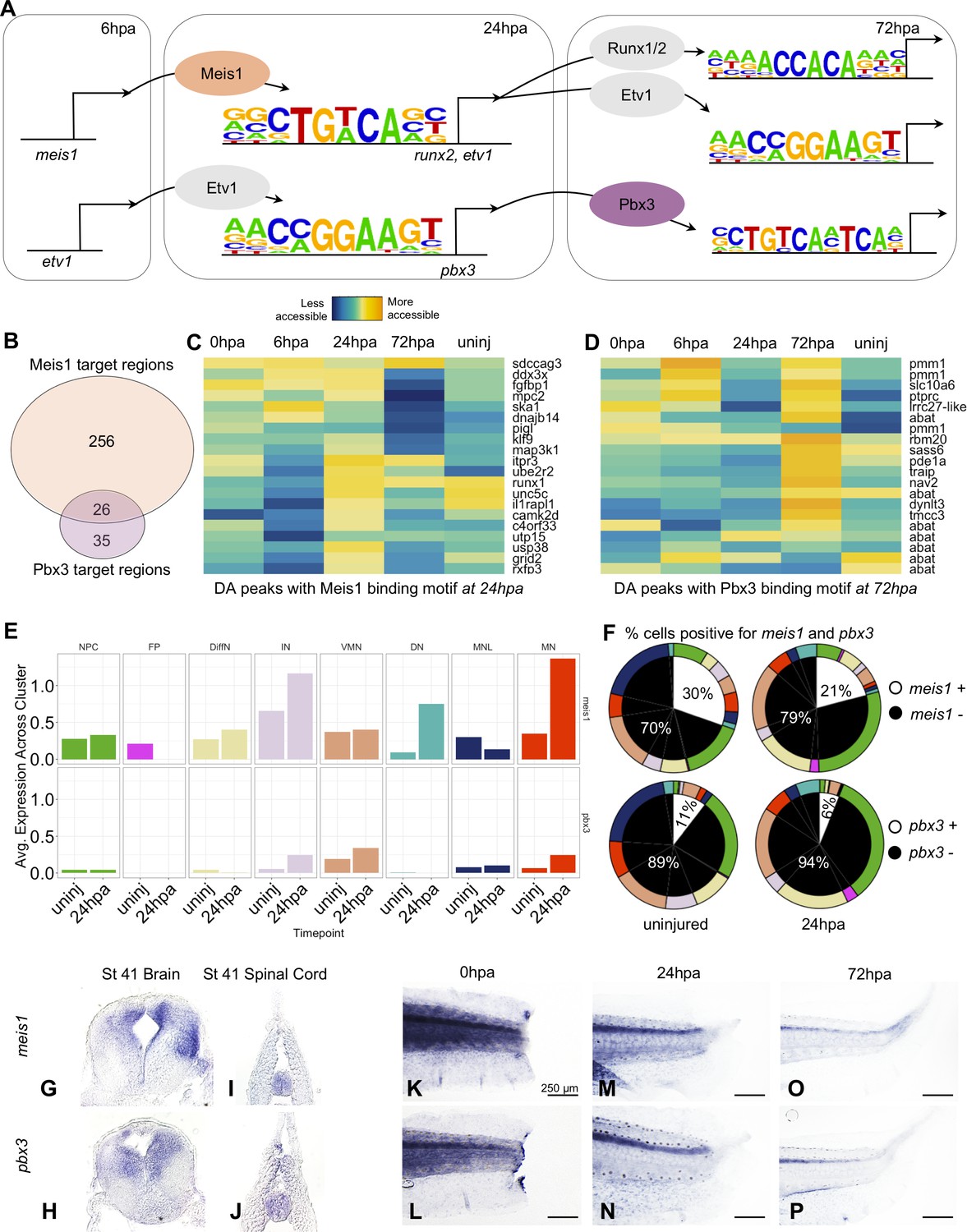
Gene regulatory network prediction identifies Meis1 and Pbx3 as key regulators of neural regeneration.
(A) Predicted gene regulatory networks across regenerative time were derived from integrative analysis between ATAC-Seq and RNA-Seq data (See analysis in Materials and methods). Meis1 and Pbx3 emerged as candidate regulators of regeneration. (B) Venn diagram showing the number of Meis1 and Pbx3 target regions found in differentially accessible regions of the chromatin. The overlap represents regions where both Meis1 and Pbx3 binding motifs were found. (C, D) Heatmaps showing accessibility of 20 differentially accessible regions of the chromatin identified with binding sites for (C) Meis1 or (D) Pbx3. (E) Average gene expression of meis1 and pbx3 in each neural cell cluster in the scRNA-Seq data in the uninjured and 24hpa timepoints. (Abbreviations: Neural Progenitor Cell (NPC), Differentiating Neuron(DiffN), Interneuron (IN), Vulnerable Motor Neuron (VMN), Dopaminergic Neuron (DN), Motor Neuron Leptin+ (MNL), Motor Neuron (MN)). (F) Sunburst diagram showing the percentage of neural cells that are either positive or negative for meis1 or pbx3 gene expression (inside) and the percentage of each neural cluster positive or negative for the genes (outside). (G–P) In situ hybridizations for meis1 and pbx3 in transverse sections of the head (G, H) and tail (I, J) at stage 41. In situ hybridizations for meis1 and pbx3 at (K, L) 0hpa, (M, N) 24hpa, and (O, P) 72hpa.
Figure 7 with 2 supplements
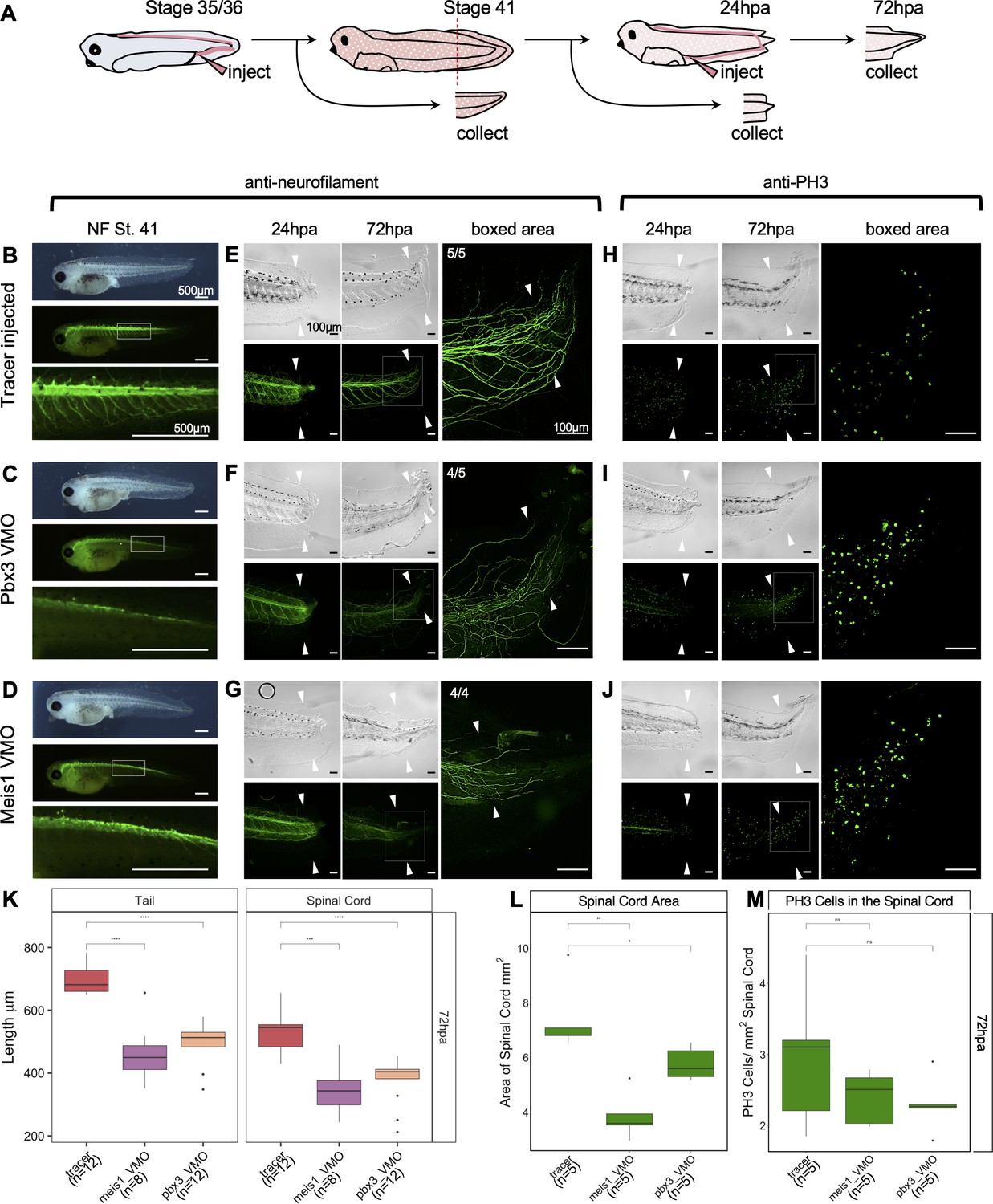
Pbx3 and Meis1 are independently required for successful regeneration of neural tissues and tails in response to injury.
(A) Injection scheme for administering vivo-morpholino. Stage 35 tadpoles were injected with a tracer or vivo-morpholinos (VMO) targeting meis1 or pbx3 and allowed to grow 24 hr to stage 41. Stage 41 tadpoles were amputated and 24hpa regenerates were collected. (B–D) Stage 41, whole-mount tadpoles shown in brightfield and immunostained against neurofilament. The box in the middle image corresponds to the enlarged image below. These images are shown for (B) tracer injected, (C) Meis1 VMO injected, and (D) Pbx3 VMO injected tadpoles. (E–J) 24hpa and 72hpa regenerates were collected and stained for neurofilament (E–G) or PH3 (H–J). Top photos are in DIC and bottom photos are immunostained as indicated. White arrows indicate amputation plane. These images were collected for (E, H) tracer, (F, I) Meis1 VMO, and (G, J) Pbx3 VMO. (K) Regenerated tail and spinal cord lengths were measured and reported in boxplots. (L) Boxplot representing regenerated spinal cord area. (M) Boxplot representing PH3 cells per regenerated spinal cord area. Statistics represent a two-tailed t-test to determine significance between conditions. (ns = not significant, *<0.05, **<0.005, ***<0.0005, ****<0.00005).
-
Figure 7—source data 1
Regenerated Tail Length Data for Vivo-Morphants.
- https://cdn.elifesciences.org/articles/52648/elife-52648-fig7-data1-v2.csv
-
Figure 7—source data 2
PH3 Cell Count and Regnerated Spinal Cord Area for Vivo-Morphants.
- https://cdn.elifesciences.org/articles/52648/elife-52648-fig7-data2-v2.csv
Figure 7—figure supplement 1
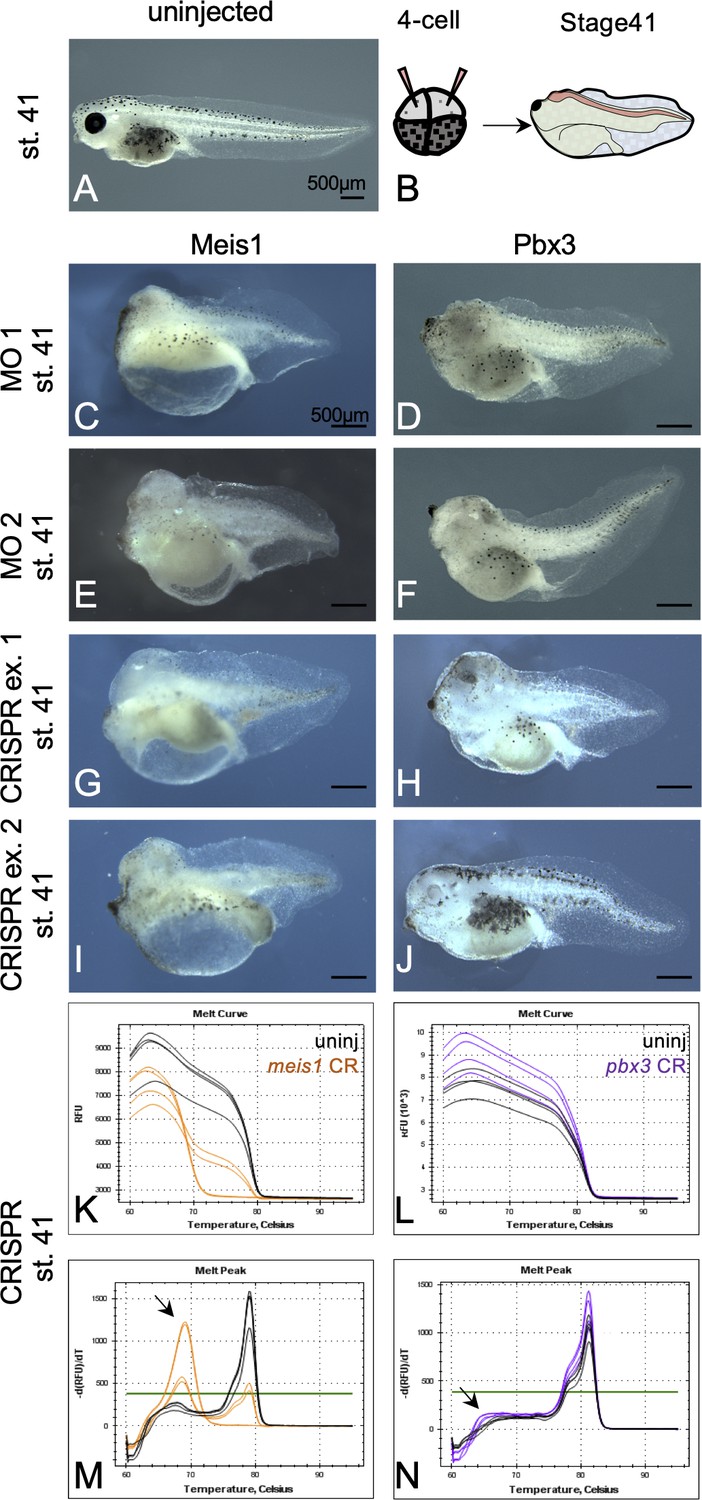
Embryonic Meis1 and Pbx3 morphants are characterized by small heads and missing or small eyes.
(A) Uninjected, stage 41 tadpole. (B) Injection schematic showing injections were made in the two dorsally fated blastomeres at the 4 cell stage. (C–F) Stage 41, morphant tadpoles that were independently injected with two sets of translation blocking morpholinos against (C, E) Meis1 and (D, F) Pbx3. Two different MO sequences were used for each gene. (G–J) Stage 41, mutant tadpoles that were injected with Cas9 and gRNA against (G,I) meis1 and (H,J) pbx3. Two representative animals are shown for each gRNA set; only one gRNA set was used per gene. (K–N) CRISPR mutant tadpoles were analyzed for mutations by high resolution melt (HRM) analysis. Melt curve and peak chart for (K,M) meis1 and (L,N) pbx3 are shown. Black lines depict wild-type siblings from the same clutch whereas orange lines represent meis1 mutants and purple lines depict pbx3 mutants. Black arrowheads indicate the position of curve shift for mutants relative to wild-type siblings.
Figure 7—figure supplement 2
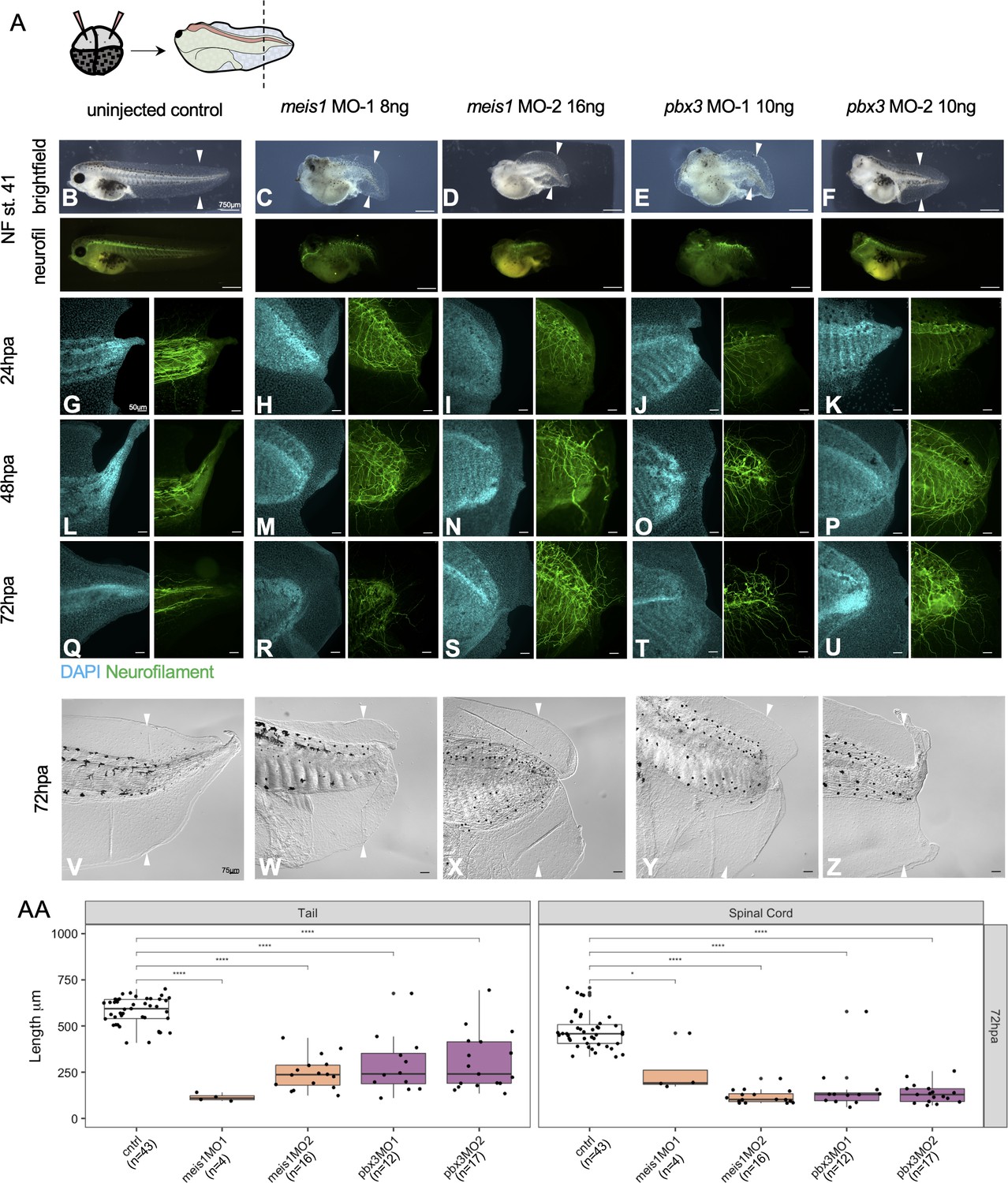
Meis1 and Pbx3 are independently necessary for successful regeneration of the axial tissue and neuronal patterning.
(A) Experimental set up for morpholino injections and amputations. Two sets of morpholinos were injected per gene to dorsally fated blastomeres at the 4 cell stage. Embryos were reared to stage 41 for amputation. Representative images of uninjured stage 41 (B) control, (C, D) Meis1 morphants 1 and 2, and (E, F) Pbx3 morphants 1 and 2. Images are shown in bright field and immunostained for neurofilament. Stage 41 tadpoles were amputated and collected at (G–K) 24hpa, (L–P) 48hpa, and (Q–U) 72hpa. The cyan channel represents tails stained with DAPI and the green channel represents tails immunostained for neurofilament. Scale bar: 75 um. (V–Z) Brightfield images of the 72hpa tails. Scale bar: 50 um. (AA) Boxplots representing length of regenerated tail or spinal cord for each tadpole. Statistics represent a two-tailed t-test to determine significance between conditions. (*<0.05, **<0.005, ***<0.0005, ****<0.00005).
-
Figure 7—figure supplement 2—source data 1
Regenerated Tail Length Data for Embryonic Morphants.
- https://cdn.elifesciences.org/articles/52648/elife-52648-fig7-figsupp2-data1-v2.csv
Figure 8
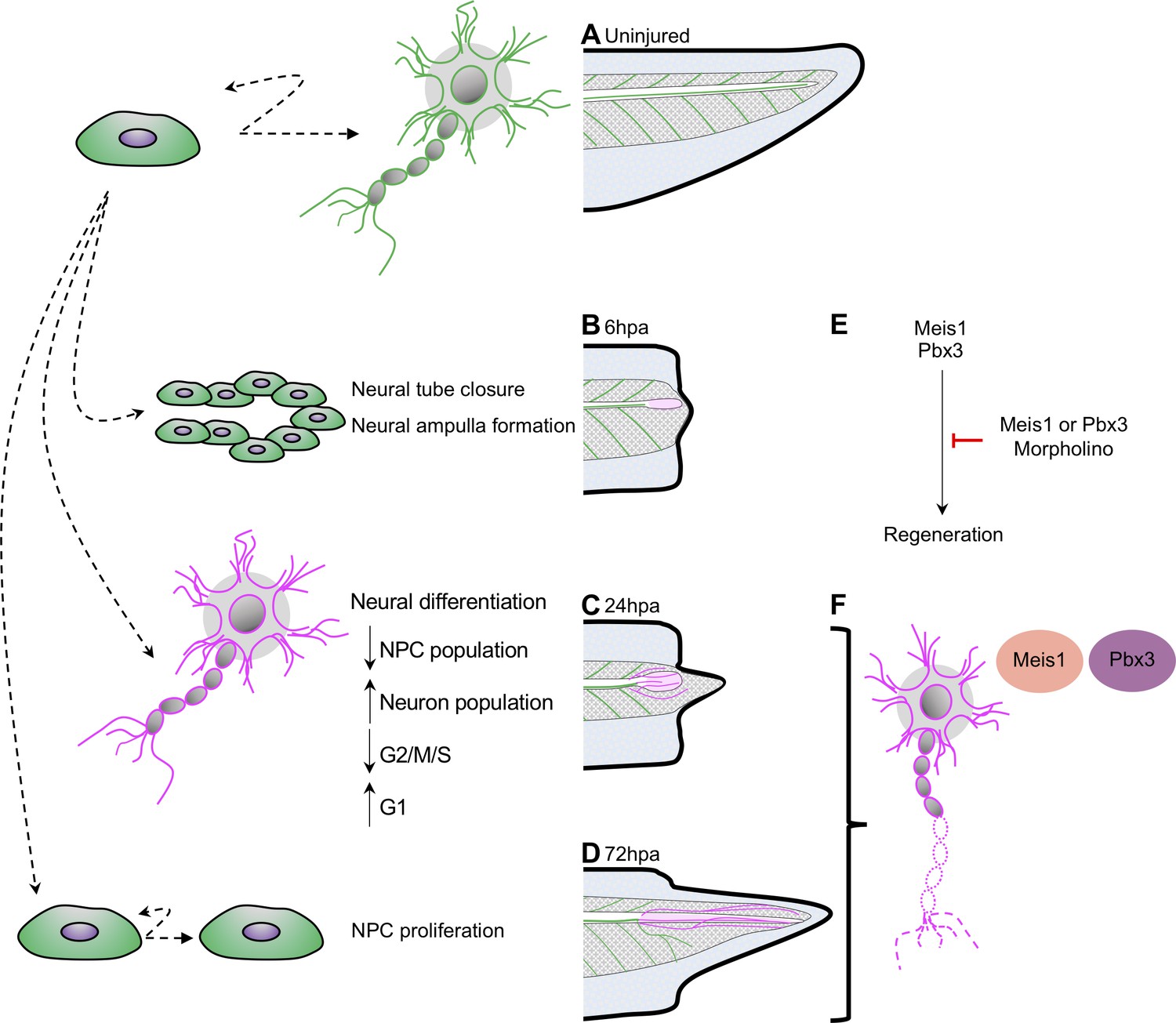
Model for neural progenitor regeneration.
(A) In normal development, neural progenitor cells make cell fate decisions to proliferate or differentiate. From this study we predict from our ATAC-Seq and scRNA-Seq analysis that neural progenitors first prioritize (B) migration and tube morphogenesis at 6hpa, followed by (C) neural differentiation at 24hpa, and then (D) turn on proliferation programs at 72hpa. (E) From integrative analysis of these data sets, we identified meis1 and pbx3 as candidate regulators of NPC regeneration at 24hpa and 72hpa. Loss-of-function experiments suggest Meis1 and Pbx3 are necessary for successful regeneration as well as proper axonal patterning. Magenta shaded spinal cord represents NPCs in regenerated spinal cord. Magenta axons represent axons in the regenerated spinal cord and tail tissue.
Additional files
-
Supplementary file 1
Supplementary output tables.
(a) ATAC-Seq sample preparation details. (b) ATAC-Seq quality control metrics. (c) Pax6 vs. all Tissue gene ontology results (more accessible in pax6 libraries). (d) Pax6 vs. all Tissue gene ontology results (more accessible in all-tissue libraries). (e) 6hpa gene ontology results. (f) 24hpa gene ontology results. (g) 72hpa gene ontology(h) 6hpa ReviGO results. (i) 24hpa ReviGo results. (j) 72hpa ReviGo results. Key Resource Table. Reagents table.
- https://cdn.elifesciences.org/articles/52648/elife-52648-supp1-v2.xlsx
-
Supplementary file 2
Key Resources Table.
- https://cdn.elifesciences.org/articles/52648/elife-52648-supp2-v2.docx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/52648/elife-52648-transrepform-v2.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Chromatin accessibility dynamics and single cell RNA-Seq reveal new regulators of regeneration in neural progenitors
eLife 9:e52648.
https://doi.org/10.7554/eLife.52648




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}