Structural and functional characterization of G protein–coupled receptors with deep mutational scanning
- Department of Chemistry and Biochemistry, UCLA-DOE Institute for Genomics and Proteomics, Molecular Biology Institute, Quantitative and Computational Biology Institute, Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, and Jonsson Comprehensive Cancer Center, UCLA, United States
- MRC Laboratory of Molecular Biology, United Kingdom
- Department of Computer Science, Stanford University, Department of Computer Science, Institute for Computational and Mathematical Engineering, Stanford University, Department of Computer Science, Department of Molecular and Cellular Physiology, Stanford University School of Medicine, Department of Computer Science, Department of Structural Biology, Stanford University School of Medicine, United States
Figures
Figure 1 with 1 supplement
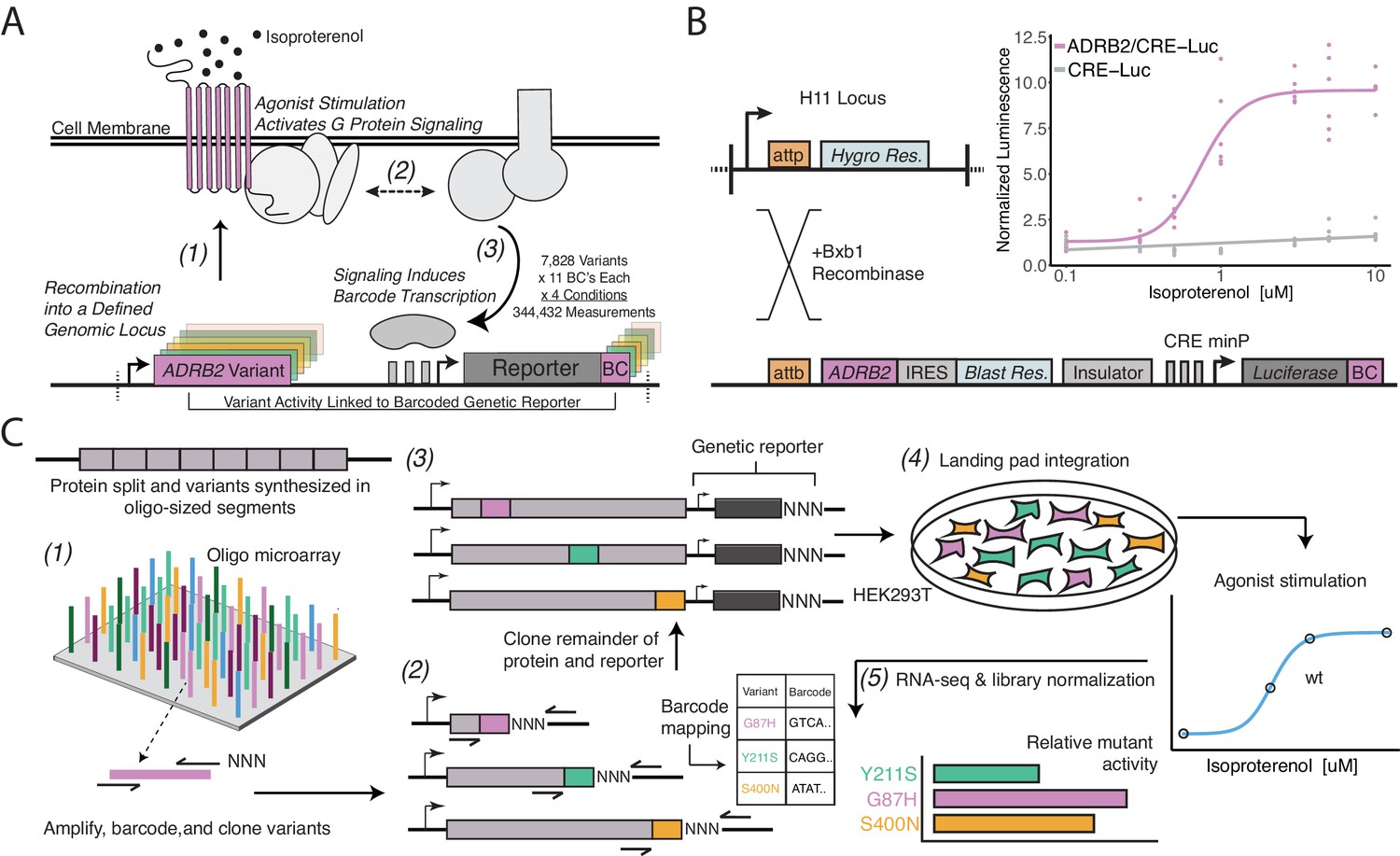
A platform for deep mutational scanning of GPCRs.
(A) Overview of the multiplexed GPCR activity assay. Plasmids encoding ADRB2 variants, a transcriptional CRE reporter of signaling activity, and 15 nucleotide barcode sequences that identify the variant are integrated into a defined genomic locus such that one variant is present per cell. Upon stimulation by isoproterenol, G-protein signaling induces transcription of the CRE genetic reporter and the barcode. Thus, the activity of a given variant is proportional to the amount of barcode mRNA which can be read out in multiplex by RNA-seq. (B) Schematic detailing the recombination of the reporter-receptor expression plasmid into the landing pad locus. Top right: activation of the CRE reporter integrated with (purple) or without (grey) exogenous ADRB2 into the landing pad when stimulated with isoproterenol in ΔADRB2 cells via a luciferase CRE reporter gene assay. (C) Overview of library generation and functional assay. Missense variants are synthesized on an oligonucleotide microarray, the oligos are amplified with random DNA barcode sequences appended, and the variants are cloned into wild-type background vectors. Barcode-variant pairs are mapped with next-generation sequencing and the remaining wild-type receptor and CRE reporter sequences are cloned into the vector. Next, the variant library is integrated en masse into the serine recombinase (Bxb1) landing pad engineered at the H11 locus of ΔADRB2 HEK293T cells. This integration strategy ensures a single pair of receptor variant and barcoded CRE reporter is integrated per cell and avoids crosstalk. After selection, the library is stimulated with various concentrations of the β2AR agonist, isoproterenol. Finally, mutant activity is determined by measuring the relative abundance of each variant’s barcoded reporter transcript with RNA-seq.
Figure 1—figure supplement 1
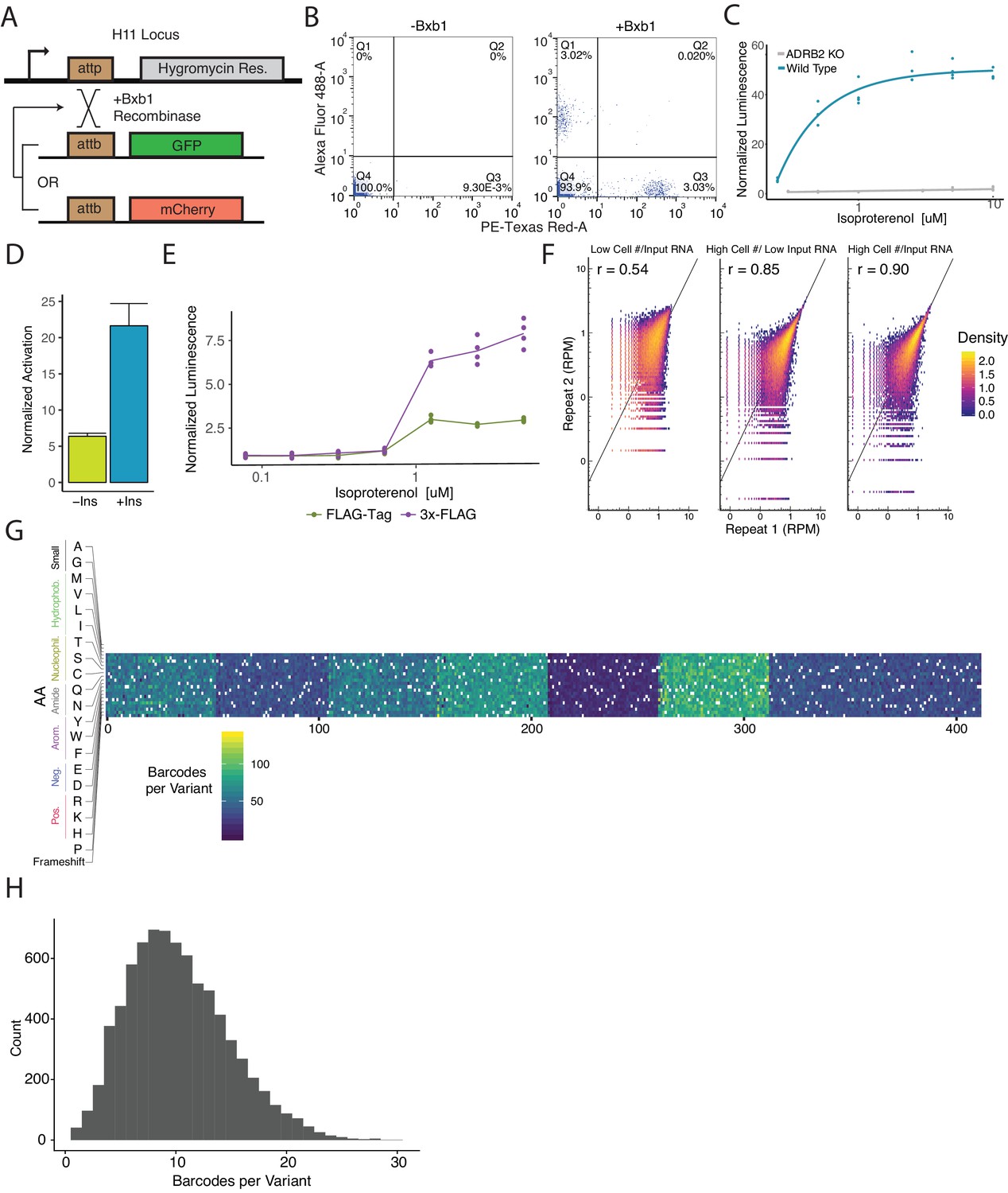
Cellular engineering and reporter optimization for multiplexed assay.
(A) Schematic of experiment to ensure the landing pad is present at single copy in the genome and thus recombine a single donor plasmid per cell. Single copy integration is essential to prevent receptors of variable functionality to activate barcoded reporters mapped to other variants. Upon co-expression of the promoterless GFP and mCherry plasmids with bxb1 recombinase sites, a cell line with a single landing pad will exclusively integrate one cassette. Therefore, cells will be either GFP+ or mCherry+ but never both. (B) Flow cytometry plots detailing the percentage of GFP+ and mCherry+ cells when transfected with an equimolar ratio of promoterless GFP and mCherry expression cassettes with or without Bxb1 recombinase expression. (C) Activation of a cAMP-responsive luciferase reporter gene integrated in the landing pad when stimulated with isoproterenol in a WT or ΔADRB2 background. Activation of the reporter in the WT background emphasizes the importance of generating a ΔADRB2 cell line for the multiplexed assay. (D) Activation of the genetic reporter/ADRB2 expression cassette with or without a CHS4 DNA insulator upstream of the reporter gene integrated in the landing pad when stimulated with isoproterenol. (E) Fold activation of an integrated genetic reporter/ADRB2 expression cassette with a FLAG-tag or 3x-FLAG tag fused to the N-terminus of ADRB2. (F) Alterations to the processes in the multiplexed assay that improve barcode abundance estimate. Initially, we seeded 2,300,000 cells/replicate and processed ~25 µg of total RNA per replicate and observed modest correlation (r = 0.54). We then scaled up to seeding 32,000,000 cells/replicate and correlation improved (r = 0.85). Finally, we began processing ~650 µg of total RNA/replicate and noticed further improvement (r = 0.90). (G) Heatmap displaying the barcodes per variant across the genomically integrated library. (H) Histogram displaying the frequency barcodes per variant across the genomically integrated library.
Figure 2 with 1 supplement
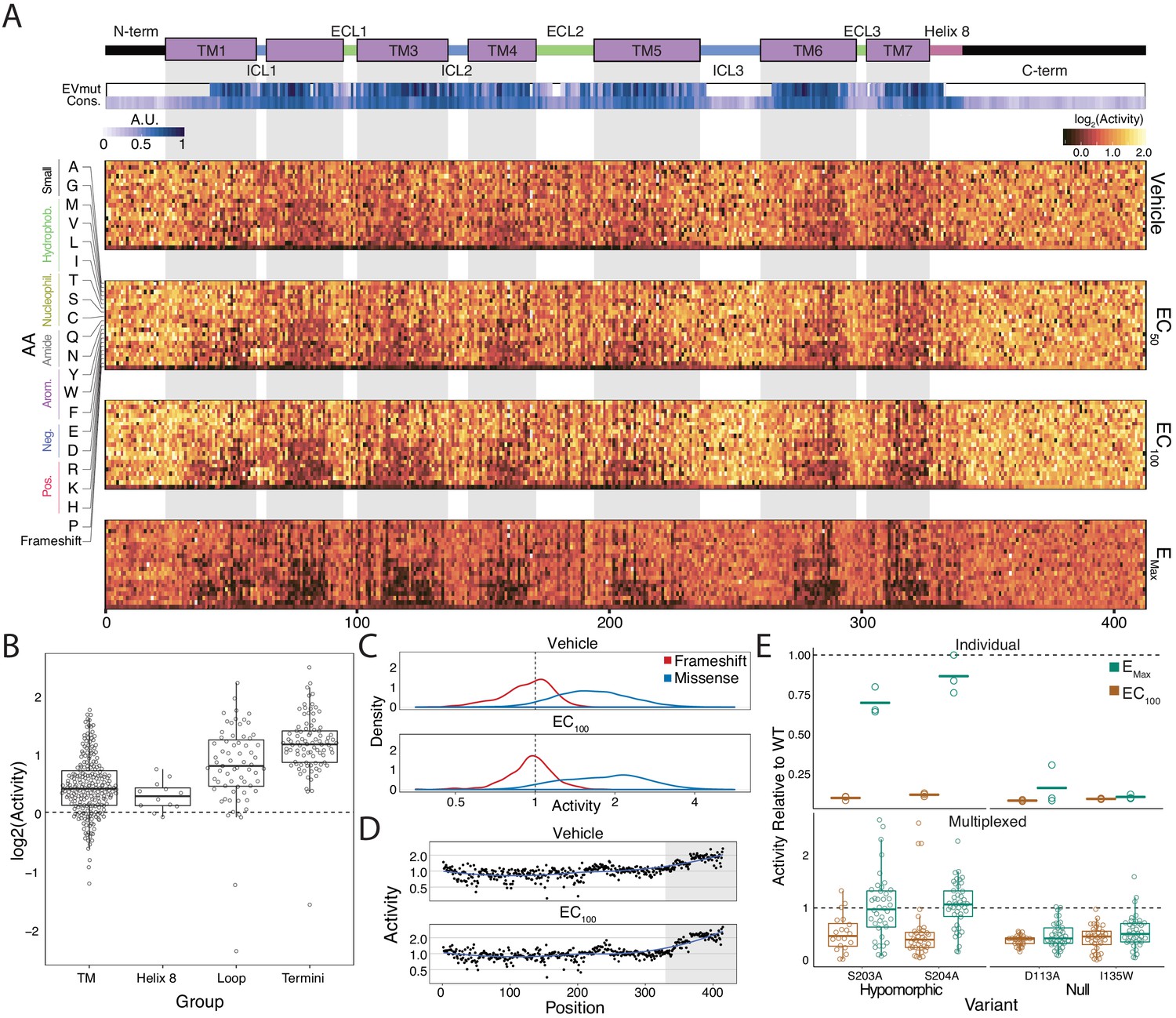
Variant-activity landscape for 7800 missense variants of the β2AR and multiplexed assay validation.
(A) Top: Secondary structure diagram of the β2AR: the N and C termini are black, the transmembrane helices are purple blocks, and the intra- and extracellular domains are colored blue and green, respectively. The EVmutation track (EVmut.) displays the mean effect of mutation at each position as predicted from sequence covariation (Hopf et al., 2017). Conservation track (Cons.) displays the sequence conservation of each residue across 55 β2AR orthologs from the OMA database (Capra and Singh, 2007; Altenhoff et al., 2018). A.U. stands for arbitrary units and the scale for the EVmutation and sequence conservation tracks are individually 0–1 normalized. The shaded guides represent positions in the transmembrane domain. Bottom: The heatmap representation of mutant activity at each agonist condition. Variants are colored by their activity score. relative to the mean frameshift mutation. Activity is the measurement of signaling for each variant relative to the mean frameshift (see methods). (B) The distribution of mutant activity for proline substitutions is significantly different for amino acids that reside in the transmembrane domain/helix eight to those in the flexible loops and termini at EC100 (all p<<0.001 except TM vs Helix 8; Mann-Whitney U). (C) The distribution of frameshift mutant activity (red) is significantly different than the distribution of designed missense mutations (blue) across increasing isoproterenol concentrations (both p<<0.001; Mann-Whitney U). Mean frameshift activity marked with a dashed line. (D) Relative effect of the mean frameshift mutant activity per position is markedly decreased in the unstructured C-terminus of the protein (shaded region) and is consistent across agonist concentration (both p<<0.001; Mann-Whitney U). Blue line represents the LOESS fit. (E) Mutant activity measured individually with a luciferase CRE reporter gene compared to the multiplexed assay at EC100 and EMax isoproterenol induction. Known null mutations (D113A, I135W) have no dose response between EC100 and Emax and are significantly different than synonymous mutants at both concentrations in both systems (all p<<0.001; Wald test). Alternatively, known hypomorphic mutations (S203A, S204A) are significantly different than synonymous mutations at EC100 (all p<<0.001; Wald test), but are not significantly different at Emax (all p>0.01; Wald test). Bars represent mean value in the luciferase data. In the Individual facet, each dot represents a replicate measurement and in the multiplexed facet, each dot represents a different barcode.
Figure 2—figure supplement 1
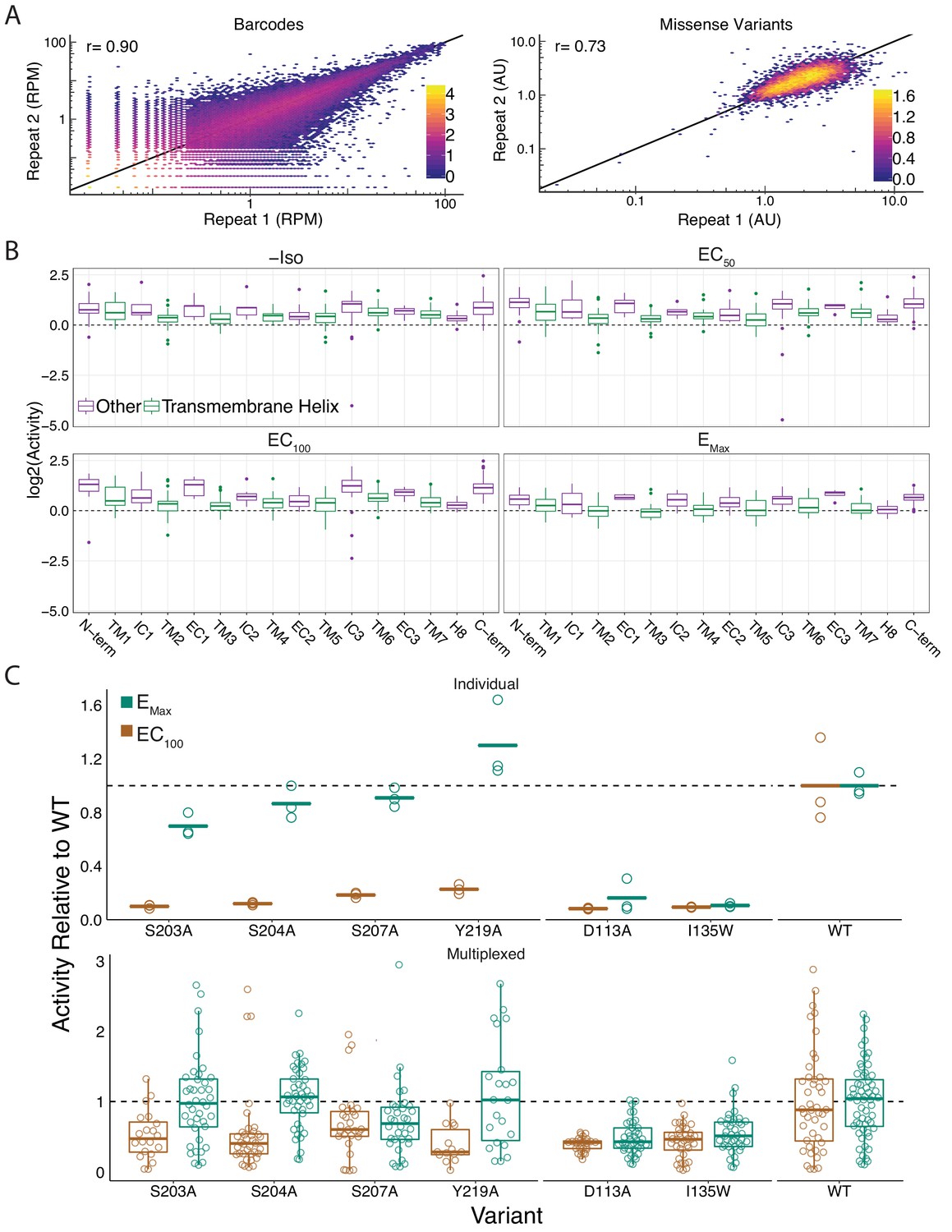
Global metrics of the multiplexed screen.
(A) The measurements between barcodes at the RNA-seq level are correlated (r = 0.89, r = 0.89, r = 0.9, r = 0.87) at all agonist concentrations (0, EC50, EC100, and EMax Iso). Similarly, the mean forskolin-normalized values for each variant are correlated at every concentration as well (r = 0.66, r = 0.69, r = 0.73, r = 0.75). Representative plots from EC100 Iso shown. Bars represent log10 counts per hex-bin. (B) Activity of proline mutations stratified by domain reveals a proline sensitivity in the transmembrane domain across all agonist conditions. (C) Mutant activity measured individually with a luciferase reporter gene compared to the multiplexed assay at EC100 and EMax isoproterenol induction. Known hypomorphic mutations (S203A, S204A, S207A, Y219A) are significantly different than WT at EC100 (all p<<0.001 except S207A in multiplex - p=0.009; Wald test), but recover to near WT activity at Emax (all p>0.01; Wald test) in both systems. Alternatively, known null mutations (D113A, I135W) are significantly different than WT regardless of the drug concentration (all p<<0.001; Wald test). Bars represent mean value in luciferase data.
Figure 3 with 1 supplement
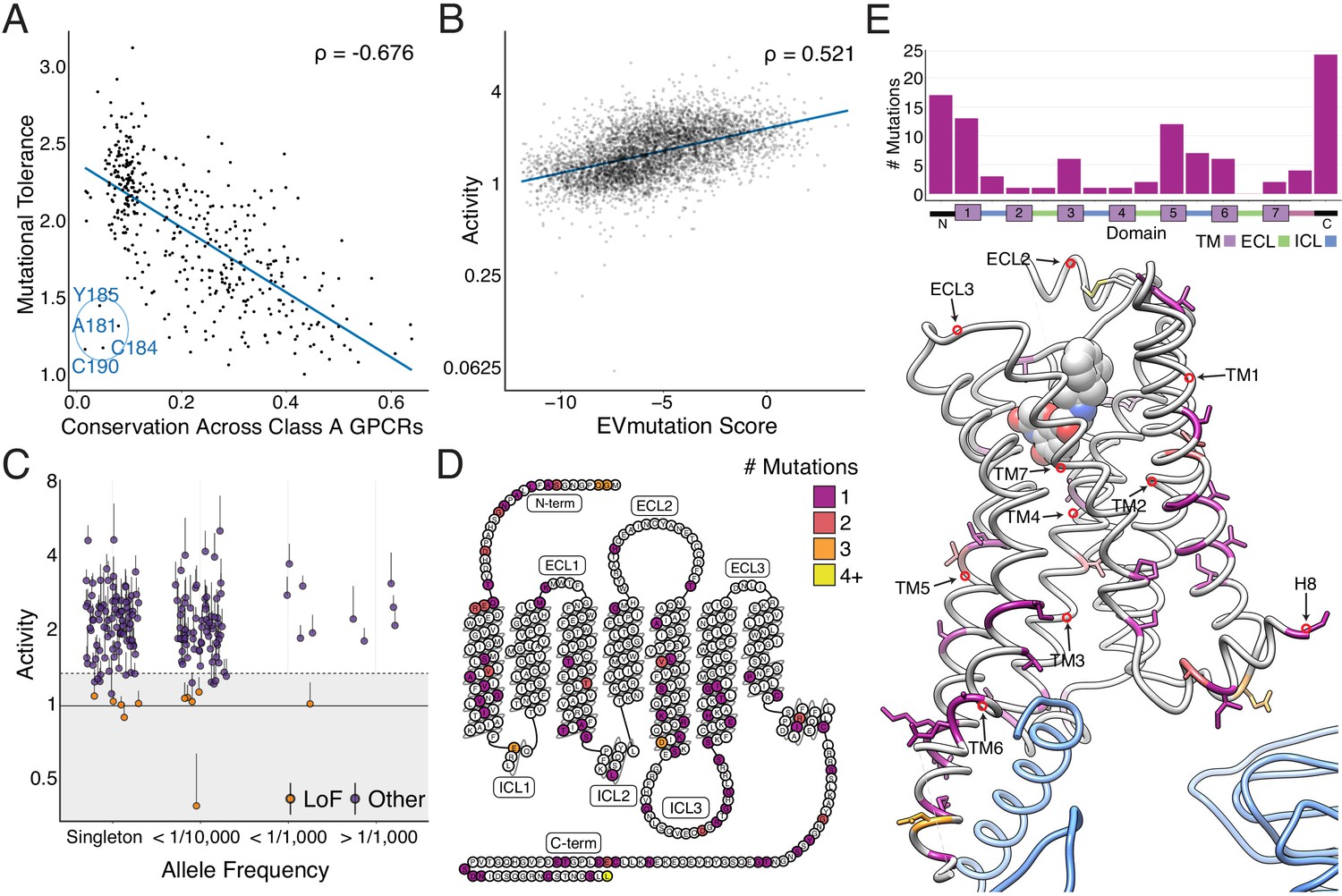
Individual mutations and residues reveal evolutionary and structural insights into β2AR function.
(A) Positional conservation across Class A GPCRs correlates with mutational tolerance (Spearman's ρ = −0.676, Pearson’s r = −0.681), the mean activity of all amino acid substitutions per residue at each agonist concentration, at EC100. However, four of the least conserved positions (C190, C184, A181, Y185) are highly sensitive to mutation and are located in ECL2, suggesting this region is uniquely important to the β2AR. The blue line is a simple linear regression. (B) Individual mutant activity correlates with EVmutation (Spearman's ρ = 0.521, Pearson’s r = 0.480) at EC100. The blue line is a simple linear regression. (C) Activity of individual mutants present in the human population from the gnomAD database stratified by allele frequency. Mutations are classified as potential loss of function (LoF) mutations (orange) are classified as such (shaded region) if the mean activity at EC100 plus the standard error of the mean (upper error bar) is more than two standard deviations below mean frameshift mutant activity (dashed line). (D) The distribution of the 100 most basally activating mutations across the β2AR snake plot reveals a clustering of mutants in the termini, TM1, TM5, and TM6. (E) Top: Distribution of the 100 most basally activating mutations stratified by domain. Bottom: The distribution of the 100 most basally activating mutations across the β2AR 3D structure (PDB: 3SN6). These positions (colored as in D) are concentrated on the surface of the β2AR (Gɑs shown in blue).
Figure 3—figure supplement 1
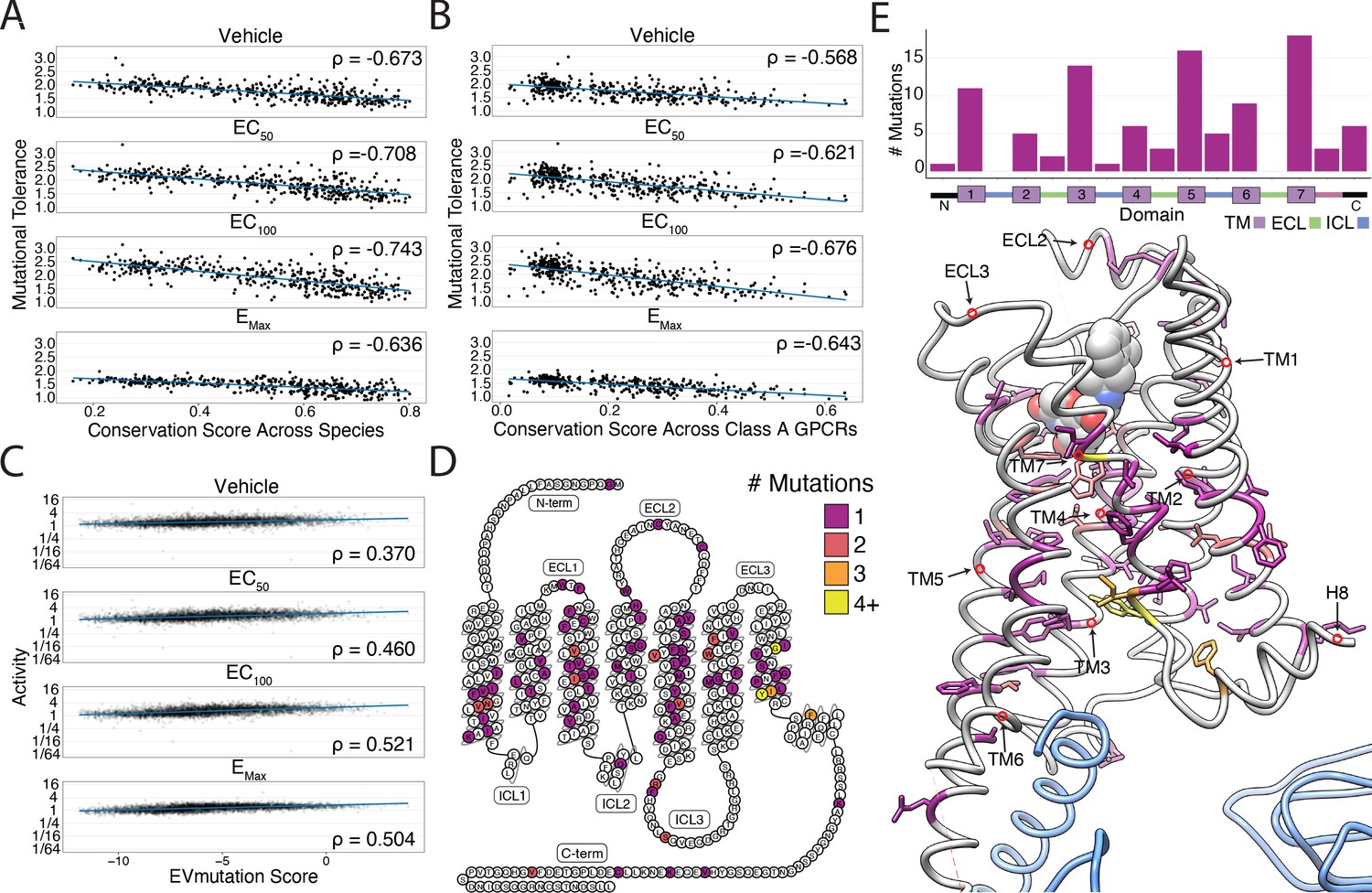
Correlation with sequence conservation and covariation and analysis of individual mutations.
(A) Mutational tolerance is highly correlated with species-level sequence conservation and is maximized at EC100 (Spearman's ρ = −0.673, Pearsons r = −0.65; ρ = −0.71, r = −0.69; ρ = −0.74, r = −0.73; ρ = −0.64, r = −0.62; for -Iso, EC50 Iso, EC100 Iso, and EMax Iso, respectively; all p<<0.0001). Here, we calculated sequence conservation using the Jensen-Shannon divergence from a multiple alignment of 55 ADRB2 orthologs from the OMA database. The blue line is a simple linear regression. (B) Similarly, mutational tolerance of the individual positions is highly correlated with sequence conservation across Class A GPCRs and is also maximized at EC100 (ρ = −0.57, r = −0.57; ρ = −0.62, r = −0.63; ρ = −0.68, r = −0.68; ρ = −0.64, r = −0.65 for -Iso, EC50 Iso, EC100 Iso, and EMax Iso, respectively; all p<<0.0001). The blue line is a simple linear regression. (C) Activity measurements for individual substitutions correlates with predictions from EVMutation, and is maximized at EC100 (ρ = 0.37, r = 0.33; ρ = 0.46, r = 0.41; ρ = 0.52, r = 0.48; ρ = 0.50, r = 0.49; all p<<0.0001). The blue line is a simple linear regression. (D) The location of the 100 most deleterious mutations by activity score at EMax Iso on the β2AR snake plot. Mutations are concentrated in the transmembrane domain. (E) Top: Distribution of the 100 most deleterious mutations by activity score at EMax Iso stratified by domain. Bottom: Location of these mutants on the 3D structure of the β2AR. These positions (colored as in D) tend to face into the core of the β2AR (PDB: 3SN6; Gs in blue).
Figure 4 with 1 supplement
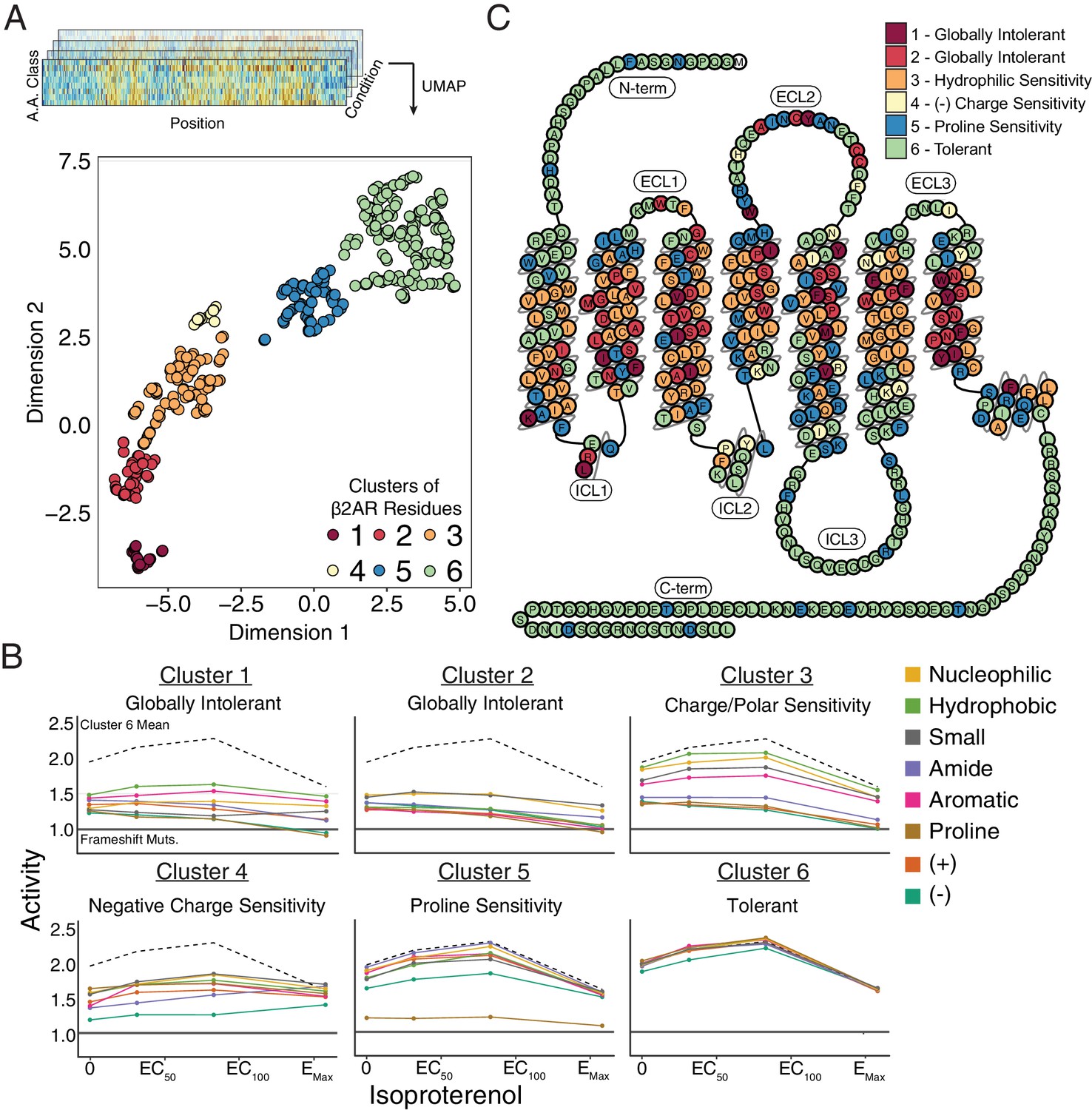
Unsupervised learning segregates residues into clusters with distinct responses to mutation.
(A) Amino acids were segregated into classes based on their physicochemical properties and mean activity scores were reported by class for each residue. With Uniform Manifold Approximation and Projection (UMAP) a 2D representation of every residue’s response to each mutation class across agonist conditions was learned. Each residue is assigned into one of six clusters using HDBSCAN (see Figure 4—figure supplement 1). (B) Class averages for each of these cluster reveal distinct responses to mutation. The upper dashed line represents the mean activity of Cluster 6 and the lower solid line represents the mean activity of frameshift mutations. (C) A 2D snake plot representation of β2AR secondary structure with each residue colored by cluster identity.
Figure 4—figure supplement 1
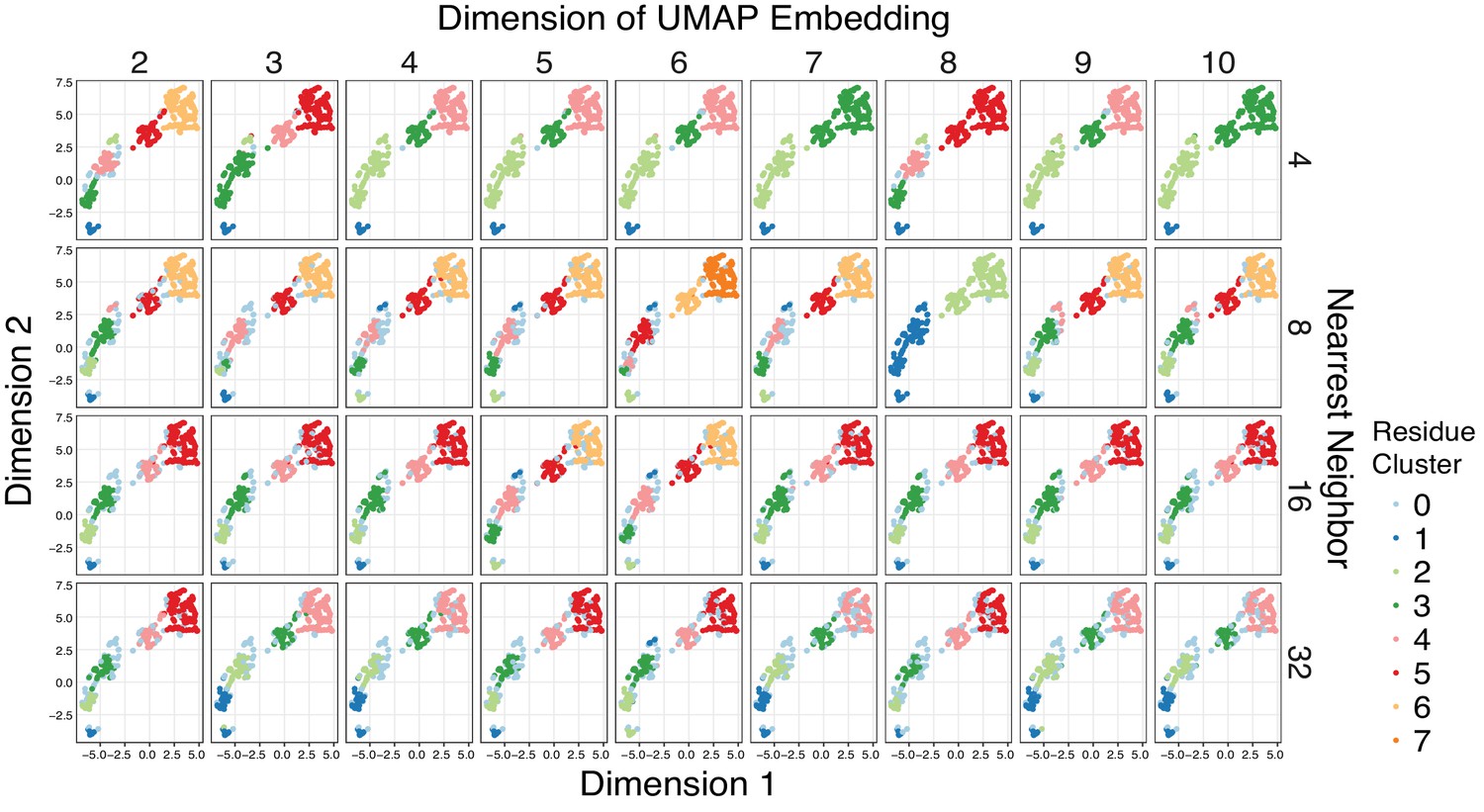
Cluster assignment is robust across different UMAP embeddings.
Given the high dimensionality of the mutational responses, Uniform Manifold Approximation and Projection (UMAP) (McInnes and Healy, 2018) was used to learn lower dimension representations of the all the mutational data across agonist conditions summarized by amino acid class before clustering the output with HDBSCAN (minimum cluster size = 10) (Campello et al., 2013). To ensure that the clustering results are not biased by a particular UMAP embedding, a hyperparameter search was run over the dimension and nearest neighbor parameters of UMAP. The HDBSCAN cluster assignments were plotted on a 2D UMAP embedding to ease visualization. Points that HDBSCAN does not assign to a cluster are colored powder blue. Groups of residues reliably cluster together regardless of the UMAP embedding, and residues were assigned to one of six distinct clusters.
Figure 5 with 2 supplements
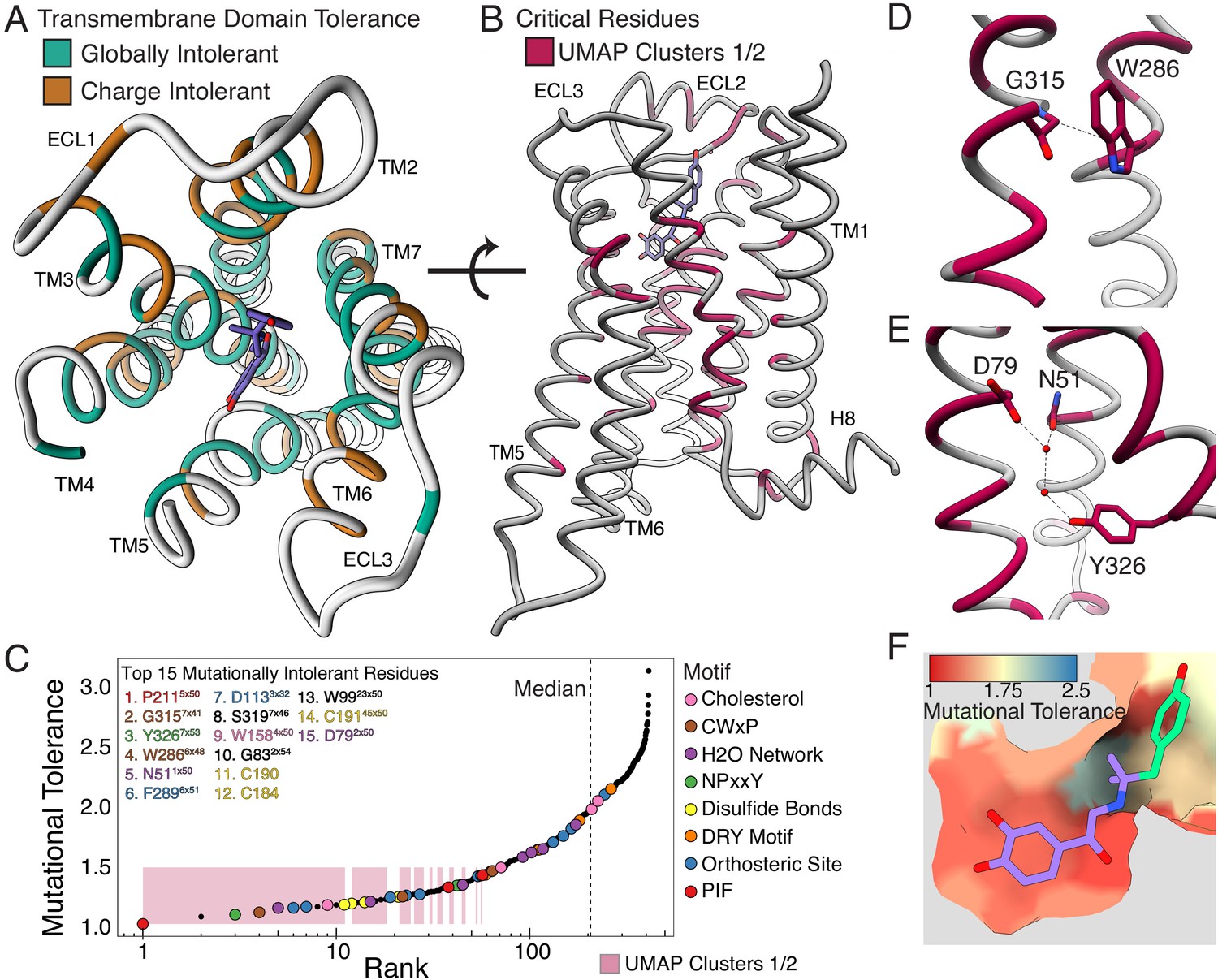
Mutational tolerance elucidates broad structural features and critical residues of the β2AR.
(A) Residues within the transmembrane domain colored by their tolerance to particular classes of amino acid substitution. Teal residues are intolerant to both hydrophobic and charged amino acids (globally intolerant), and brown residues are tolerant to hydrophobic amino acids but intolerant to charged amino acids (charge intolerant). The charge-sensitive positions’ side chains are enriched pointing into the membrane, while the globally intolerant positions’ side chains face into the core of the protein (see Figure 5—figure supplement 1). (B) The crystal structure of the hydroxybenzyl isoproterenol-activated state of the β2AR (PDB: 4LDL) with residues from the mutationally intolerant Clusters 1 and 2 highlighted in maroon. (C) 412 β2AR residues rank ordered by mutational tolerance at the EC100 isoproterenol condition. Residues in known structural motifs (colored points) are significantly more sensitive to mutation than other positions on the protein (p<<0.001). Dashed line demarcates the median of the ranking. The top 15 mutationally intolerant residues are listed and colored by motif association. (D-F) Selected vignettes of residues from the mutationally intolerant UMAP clusters and ranking. (D) W2866x48 of the CWxP motif and the neighboring G3157x41 are positioned in close proximity. Substitutions at G3157x41 are likely to cause a steric clash with W2866x48 (PDB: 4LDL). (E) An inactive-state water-mediated hydrogen bond network (red) associates N511x50 and Y3267x53 (PDB: 2RH1). Disruption of this network may destabilize the receptor. (F) The ligand-bound orthosteric site surface colored by mutational tolerance. Receptor-ligand contacts with the catecholamine head (present in agonist used in assay) are more intolerant to mutation than those in the hydroxybenzyl tail (not present in agonist used in assay) of the isoproterenol analog depicted in this crystal structure (PDB: 4LDL).
Figure 5—figure supplement 1
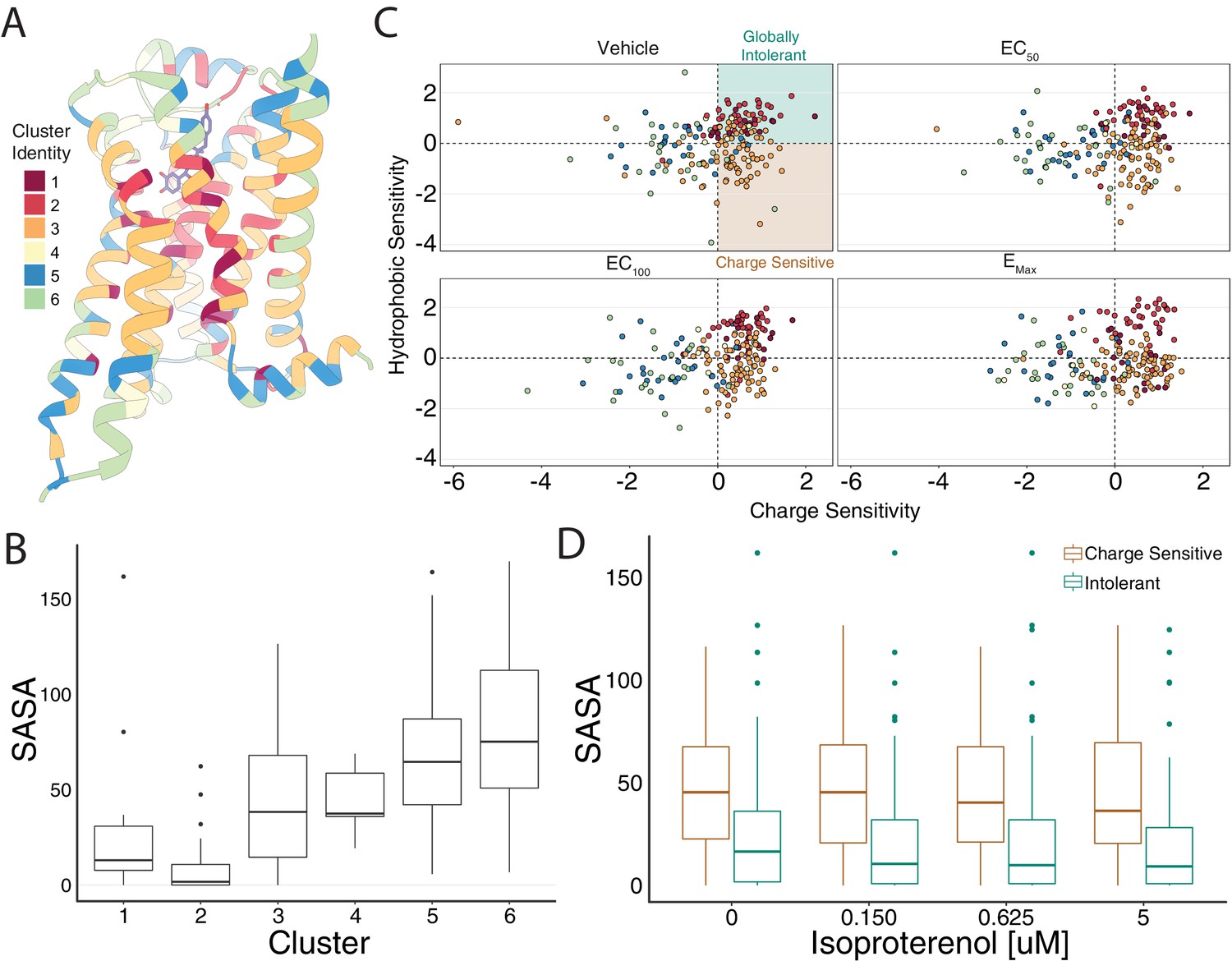
Mutational profile suggests side chain orientation and environment.
(A) The crystal structure of the hydroxybenzyl isoproterenol-activated state of the β2AR (PDB: 4LDL) with residues colored by UMAP cluster identity. (B) Distributions of solvent-accessible surface area (SASA) for each cluster at EC100. (C) Hydrophobic versus charge sensitivity across all drug conditions. Points are colored by cluster identity. Residues are defined as globally intolerant to substitution if their hydrophobic and charge sensitivity is greater than 0. Similarly, residues are defined to be uniquely charge sensitive if their hydrophobic sensitivity is less than one and their charge sensitivity is greater than 1 (see Materials and methods). (D) The median SASA is significantly higher for positions in the charge-sensitive clusters when compared to the ones in the intolerant clusters across all drug concentrations (all p<0.0005). This suggests that the charge-sensitive cluster residues point toward the lipid, whereas the ones that are intolerant tend to be buried in the core of the protein.
Figure 5—figure supplement 2
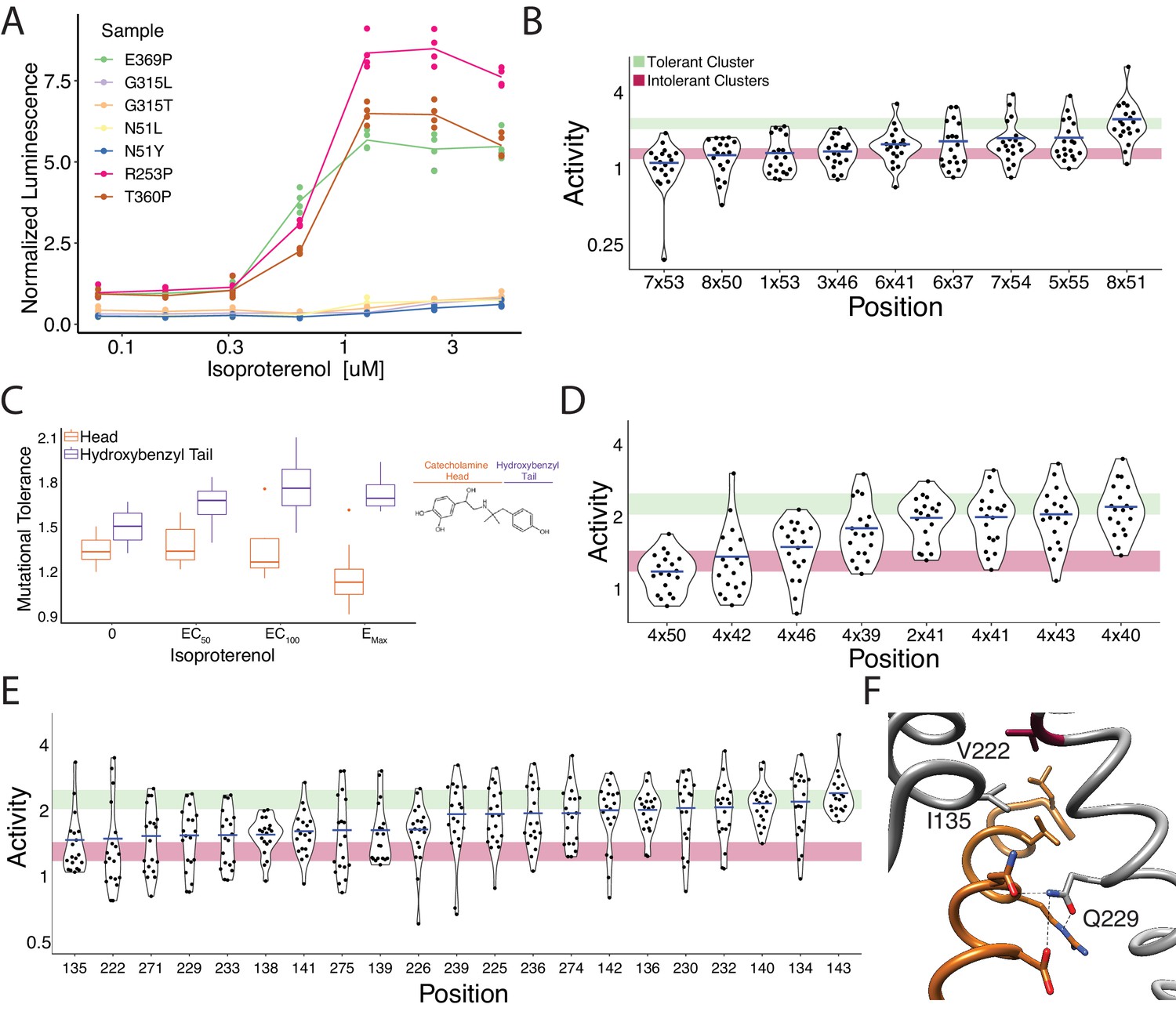
Mutational intolerance of functionally related residues.
(A) Relative activation of an integrated CRE luciferase reporter gene for β2AR missense variants mentioned in the manuscript. (B) Functional consequences of mutation for a set of residues near the G-protein coupling region involved in GPCR activation (Venkatakrishnan et al., 2016). (C) Residues that interact with the catecholamine head (orange) of hydroxybenzyl isoproterenol have significantly lower mutational tolerance than those that interact with the hydroxybenzyl functional group on the tail (purple). In our assay, we screened with isoproterenol lacking the hydroxybenzyl tail. These differences are significantly different at EC50 (p=0.028), EC100 (p=0.016), and saturating agonist concentration (p=0.008). (D) Functional consequences of mutation at predicted contacts of a cholesterol binding pocket determined in the timolol-bound structure of the β2AR inactive state (PDB: 3D4S). As predicted, the highly conserved, across species and class A GPCRs, W1584x50 is the most constrained residue. The shaded bars represent ±1 standard deviation of the mutational tolerance in the tolerant Cluster 6 (green) or the intolerant Clusters 1 and 2 (red). The mean activity of every mutation at a given position (the mutational tolerance) is shown as a blue bar. (E) Effects of mutations at residues in the Gs interface. (F) Three of the four most intolerant β2AR residues at the G-protein interface (brown) from the β2AR-Gs complex crystal structure (PDB: 3SN6), V2225x61, I1353x54, and Q2295x68.
Figure 6 with 1 supplement
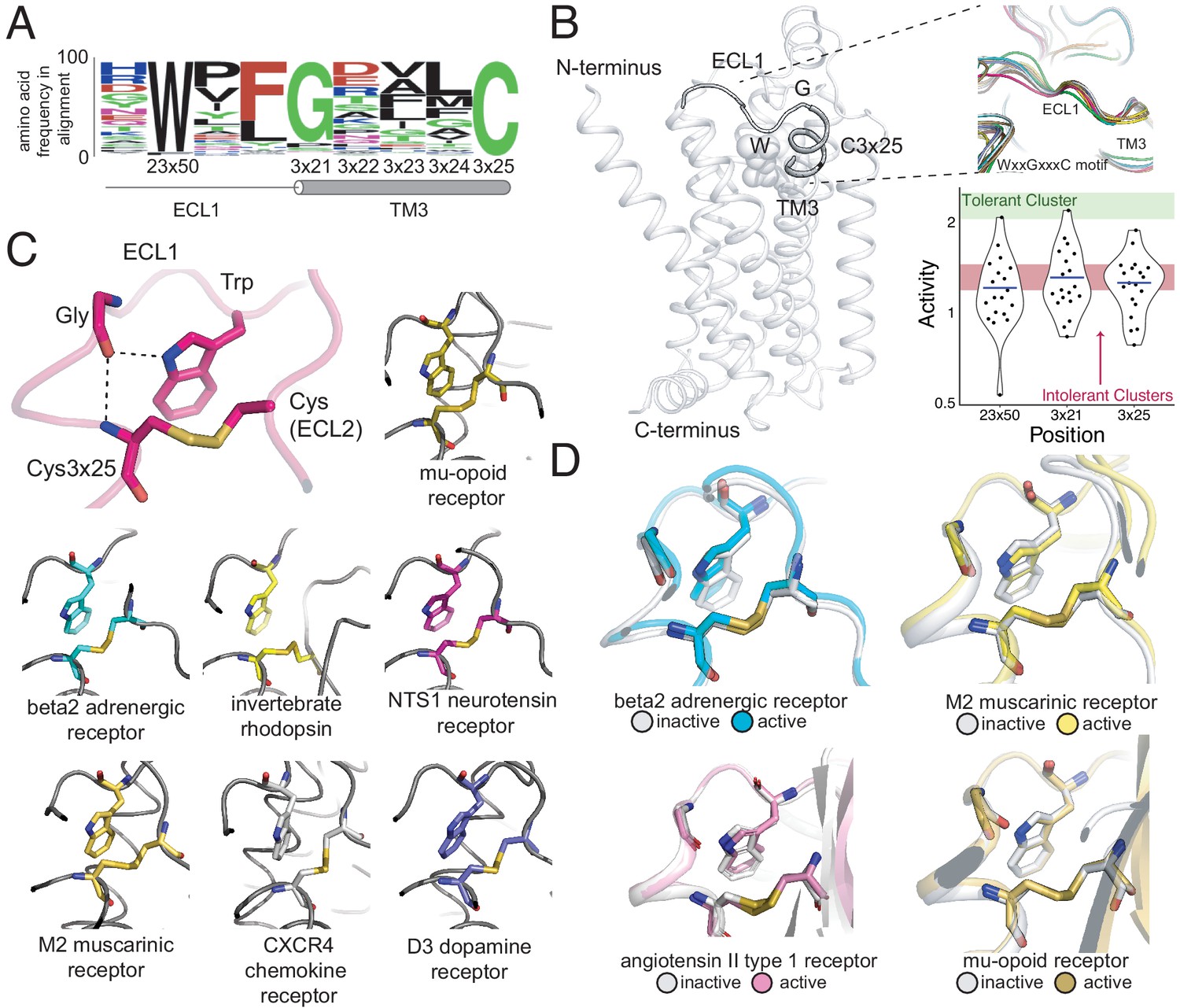
A conserved extracellular tryptophan-disulfide ‘structural latch’ in class A GPCRs is mutationally intolerant and conformation-independent.
(A) Sequence conservation of extracellular loop 1 (ECL1) and the extracellular interface of TM3 (202 Class A GPCRs with a disulfide bridge between TM3 and ECL1). (B) Left: Depiction of the interaction of W9923x50, G1023x21, and C1063x25 in ECL1 of the β2AR. Top Right: Conservation of the structure of the ECL1 region across functionally different class A GPCRs. Bottom Right: Activity of all 19 missense variants assayed for each of the three conserved residues, with the mean activity (mutational tolerance) shown as a blue bar. The shaded bars represent the mean mutational tolerance ± 1 SD of residues in the tolerant Cluster 6 (green) and the intolerant Clusters 1 and 2 (red). (C) A hydrogen bond network between mutationally intolerant positions W9923x50, G1023x21, and C1063x25. Representative examples of the structural latch are shown. (D) This structural latch is maintained in both the inactive and active state structures for the β2AR (inactive: 2RH1, active: 3P0G), the M2 muscarinic receptor (inactive: 3UON, active: 4MQS), the angiotensin II type one receptor (inactive: 4ZUD, active: 6OS1), and the mu-opioid receptor (inactive: 4DKL, active: 5C1M).
Figure 6—figure supplement 1
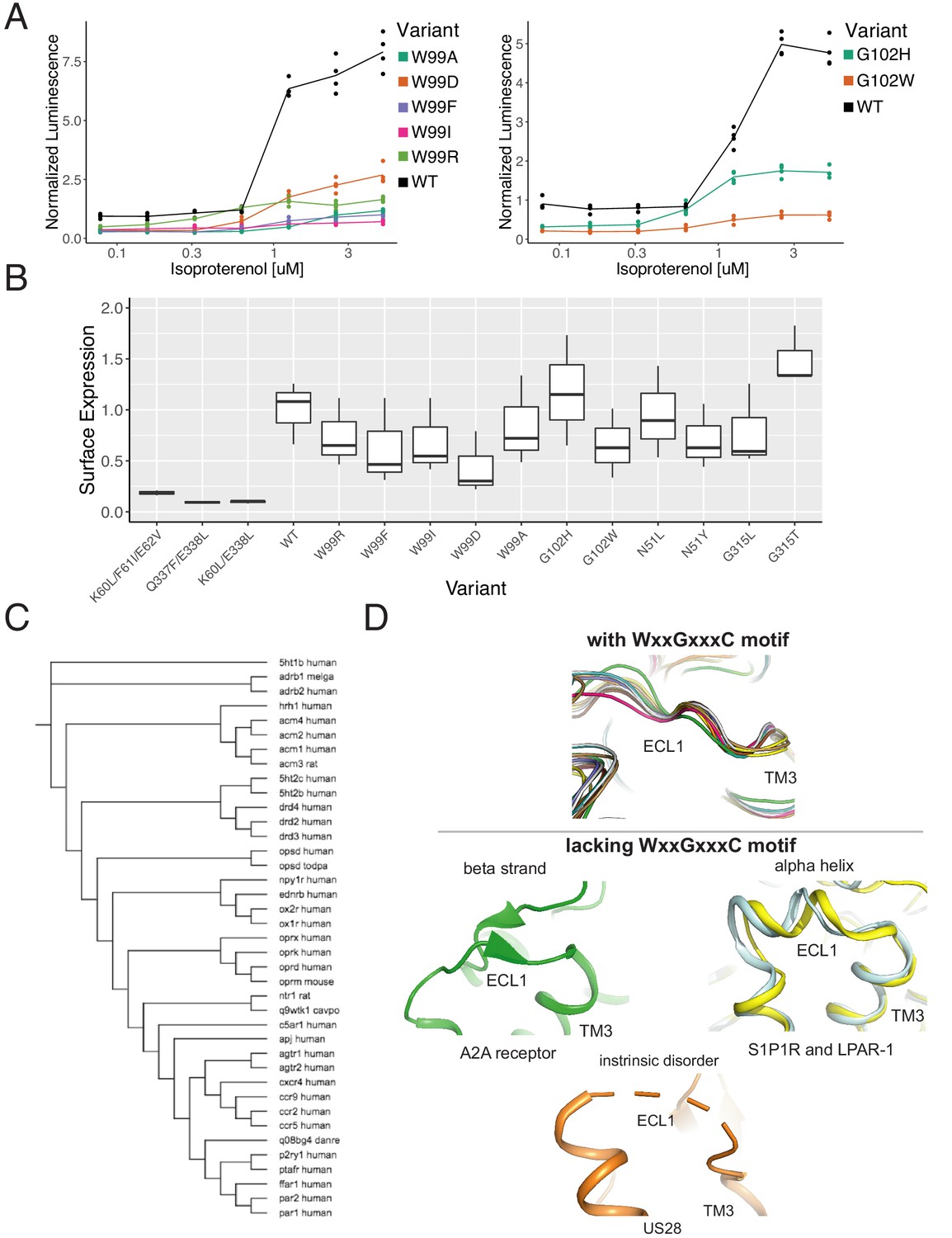
The WxxGxxxC motif is highly conserved across Class A GPCRs.
(A) Individual verification of the mutational intolerance of W9923x50 and G1023x21. Relative activation of an integrated CRE luciferase reporter gene for β2AR missense variants. (B) Surface expression (anti-FLAG immunostaining and flow cytometry) of β2AR missense variants min-max normalized to wild-type and no receptor controls. (C) Functional and evolutionary diversity of Class A GPCRs with the WxxGxxxC motif. (D) ECL1 structures of the four Class A GPCRs lacking the WxxGxxxC Motif, S1P1R, LPAR-1, A2A, and US28.
Author response image 1
Author response image 2
Author response image 3
Author response image 4
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (Homo-sapiens) | HEK293T | ATCC | CRL-3216 | |
| Cell line (Homo-sapiens) | HEK293TΔADRB2 + Landing Pad | This paper | Construction Information found in Endogenous ADRB2 Deletion using CRISPR/Cas9 and Landing Pad Genome Editing Sections | |
| Gene (Homo-sapiens) | ADRB2 | NCBI | Gene ID 154 | |
| Chemical compound, drug | Isoproterenol | Millipore Sigma | I5627 | |
| Chemical compound, drug | Forskolin | Millipore Sigma | F6886 | |
| Commercial assay or kit | Dual Glo Luciferase Assay | Promega | E2920 | |
| Recombinant DNA reagent | TALEN plasmids | Addgene | #51554 #51555 | |
| Recombinant DNA reagent | SpCas9 plasmid | Addgene | pX339 | |
| Sequence-based reagent | Oligonucleotide Microarray | Agilent | Custom Synthesis | |
| Commercial assay or kit | Nextseq Mid Output 300 cycle | Illumina | 20024905 | |
| Commercial assay or kit | Nextseq High Output 75 cycle | Illumina | 20024906 | |
| Strain, strain background (Escherichia coli) | Dh5 alpha | New England Biolabs | C2989K | |
| Antibody | AlexaFluor 488 Anti-Flag rat monoclonal | Thermo Fisher | MA1-142-A488 | (1:100) |
| Transfected construct (Homo-sapiens) | ADRB2 barcoded variant-reporter library | This paper | Reagent Construction Information found in Variant Library Generation and Cloning Section | |
| Commercial assay or kit | RNEasy Miniprep Kit | Qiagen | 74104 | |
| Commercial assay or kit | Plasmid Plus DNA Maxi Kit | Qiagen | 12963 | |
| Commercial assay or kit | Superscript IV | Thermo Fisher | 18091050 | |
| Commercial assay or kit | Lipofectamine 3000 | Thermo Fisher | L3000001 | |
| Commercial assay or kit | D1000 DNA Screen Tape | Agilent | 5067–5582 | |
| Commercial assay or kit | D1000 Reafents | Agilent | 5067–5583 | |
| Commercial assay or kit | SYBR FAST QPCR Master Mix | Roche | 07959362001 | |
| Commercial assay or kit | Zymo Clean Gel DNA Recovery Kit | Zymo Research | D4007 | |
| Commercial assay or kit | Zymo DNA Clean and Concentrator Kit | Zymo Research | D4013 | |
| Chemical compound, drug | CD293 | Thermo Fisher Scientific | 11913019 | |
| Software, algorithm | BBTools | Brian Bushnell | https://jgi.doe.gov/data-and-tools/bbtools/ | |
| Software, algorithm | Jensen-Shannon Conservation | https://doi.org/10.1093/bioinformatics/btm270 | ||
| Software, algorithm | OMA Orthology Database | https://doi.org/10.1093/nar/gkx1019 | ||
| Software, algorithm | FreeSASA | 10.12688/f1000research.7931.1 | ||
| Software, algorithm | EVmutation | doi:10.1038/nbt.3769 | ||
| Software, algorithm | Parasail | http://dx.doi.org/10.1186/s12859-016-0930-z |
Additional files
-
Supplementary file 1
List of species for evolutionary analysis.
A table describing the list of 55 species used for the analysis of evolutionary constraint of residues from the OMA database.
- https://cdn.elifesciences.org/articles/54895/elife-54895-supp1-v2.xls
-
Supplementary file 2
Processed data.
A table with the processed data used in this study. Includes the position, mutation, min activity, max activity, average activity, propagated uncertainty, coefficient of variation, number of barcodes for that mutation, mutation class, and an annotation of where in the β2AR the position is.
- https://cdn.elifesciences.org/articles/54895/elife-54895-supp2-v2.xls
-
Supplementary file 3
Mutational tolerance.
A table containing positions in the β2AR with their mutational tolerance, rank order, and an annotation for known positions in the receptor.
- https://cdn.elifesciences.org/articles/54895/elife-54895-supp3-v2.xls
-
Supplementary file 4
List of primers used in this study.
A table documenting the DNA sequences and application of important primers in this study.
- https://cdn.elifesciences.org/articles/54895/elife-54895-supp4-v2.xls
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/54895/elife-54895-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structural and functional characterization of G protein–coupled receptors with deep mutational scanning
eLife 9:e54895.
https://doi.org/10.7554/eLife.54895




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}