A new protocol for single-cell RNA-seq reveals stochastic gene expression during lag phase in budding yeast
- Laboratory for Systems Biology, VIB-KU Leuven Center for Microbiology, Belgium
- Laboratory of Genetics and Genomics, CMPG, Department M2S, KU Leuven, Belgium
- Laboratory for Translational Genetics, Department of Human Genetics, KU Leuven, Belgium
- VIB Center for Cancer Biology, VIB, Belgium
- Laboratory for Functional Epigenetics, Department of Genetics, KU Leuven, Belgium
Figures
Figure 1 with 2 supplements
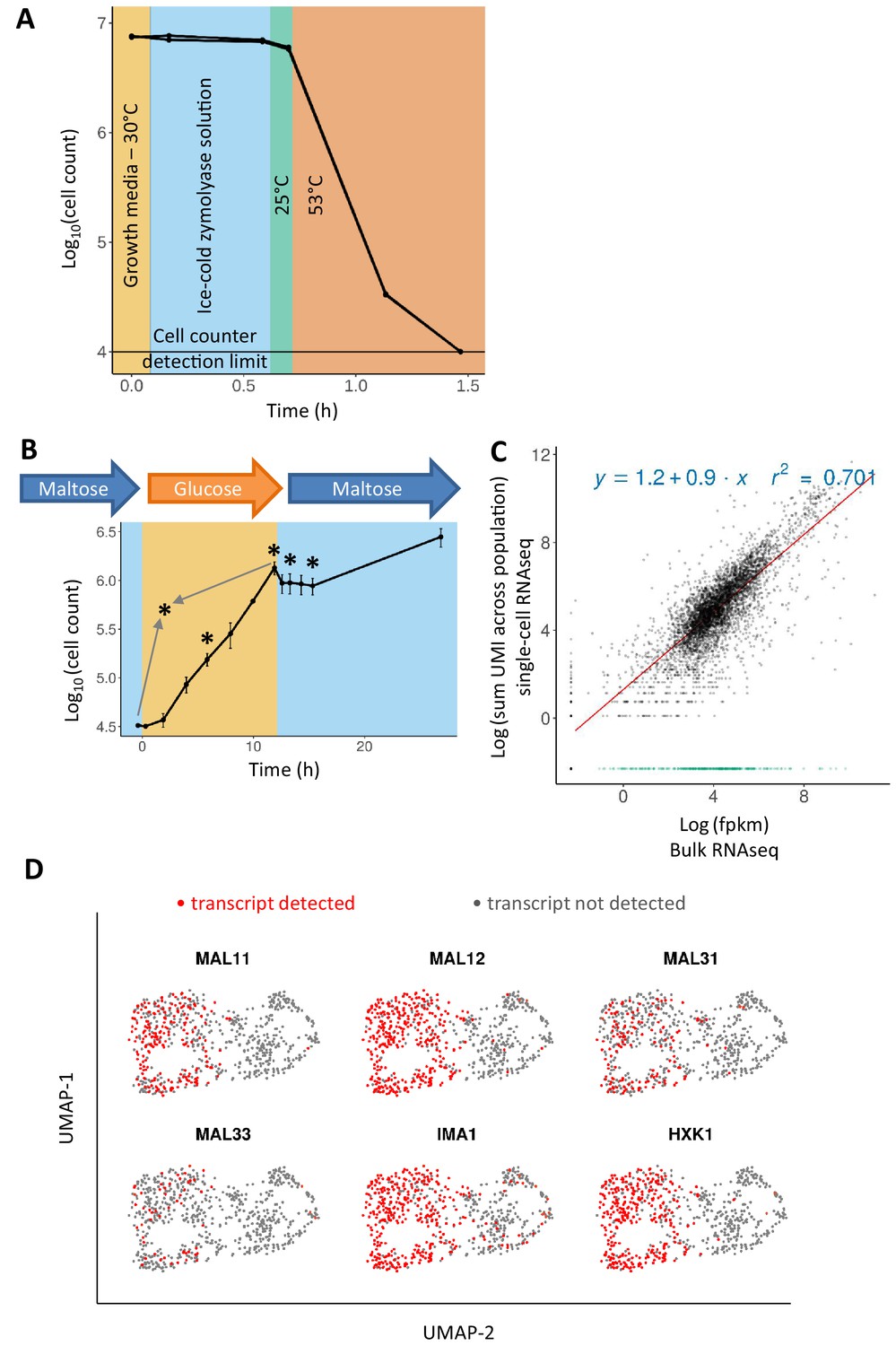
Adapting the 10x Genomics’ single-cell RNA-seq platform for yeast cells.
(A) Zymolyase is required and effective for lysing yeast cells in a temperature treatment regimen similar to the 10x Genomics’ single cell 3’ RNA-seq protocol (see text for details). Cell counts (for biological duplicates) only drop during the incubation at 53°C, showing that cells did not lyse beforehand. (B) Sampling scheme and cell counts for the scRNA-seq experiment. Cells were pre-grown in maltose, transferred to glucose for 12 hr, and finally shifted to maltose where they experience a lag phase. After the initial maltose pre-growth and subsequent wash to glucose, the cells were diluted for inoculation in glucose. Cell count of these two initial time points is scaled to the dilution level. Sampling points for scRNA-seq are represented by an asterisk: after 6 hr in glucose (referred to as ‘glucose-6h’), after 12 hr in glucose (‘glucose-12h’), after 1 hr in lag phase (‘lag-1h’), after 3 hr in lag (‘lag-3h’), and a control sample where cells grown on glucose are mixed in equal parts with cells grown on maltose (grey arrows; ‘mix-glucose-maltose’). (C) Correlation between bulk RNA-seq data and scRNA-seq data from cells in similar conditions, but separate experiments. Bulk RNA-seq data from a similar growth condition (12 hr in glucose) were obtained from a previously published experiment (Cerulus et al., 2018). The expression levels were quantified as fpkm and log-transformed with addition of a pseudo count. UMI counts from single-cell RNA-seq data were summed across the population, a pseudo-count was added, and log-transformed. The R2 is calculated excluding genes that are detected in bulk RNA-seq and not in scRNA-seq (shown in green). (D) Presence or absence of genes expected to be expressed in maltose distinguishes the two subgroups in the mix of cells grown on glucose and cells grown on maltose.
Figure 1—figure supplement 1
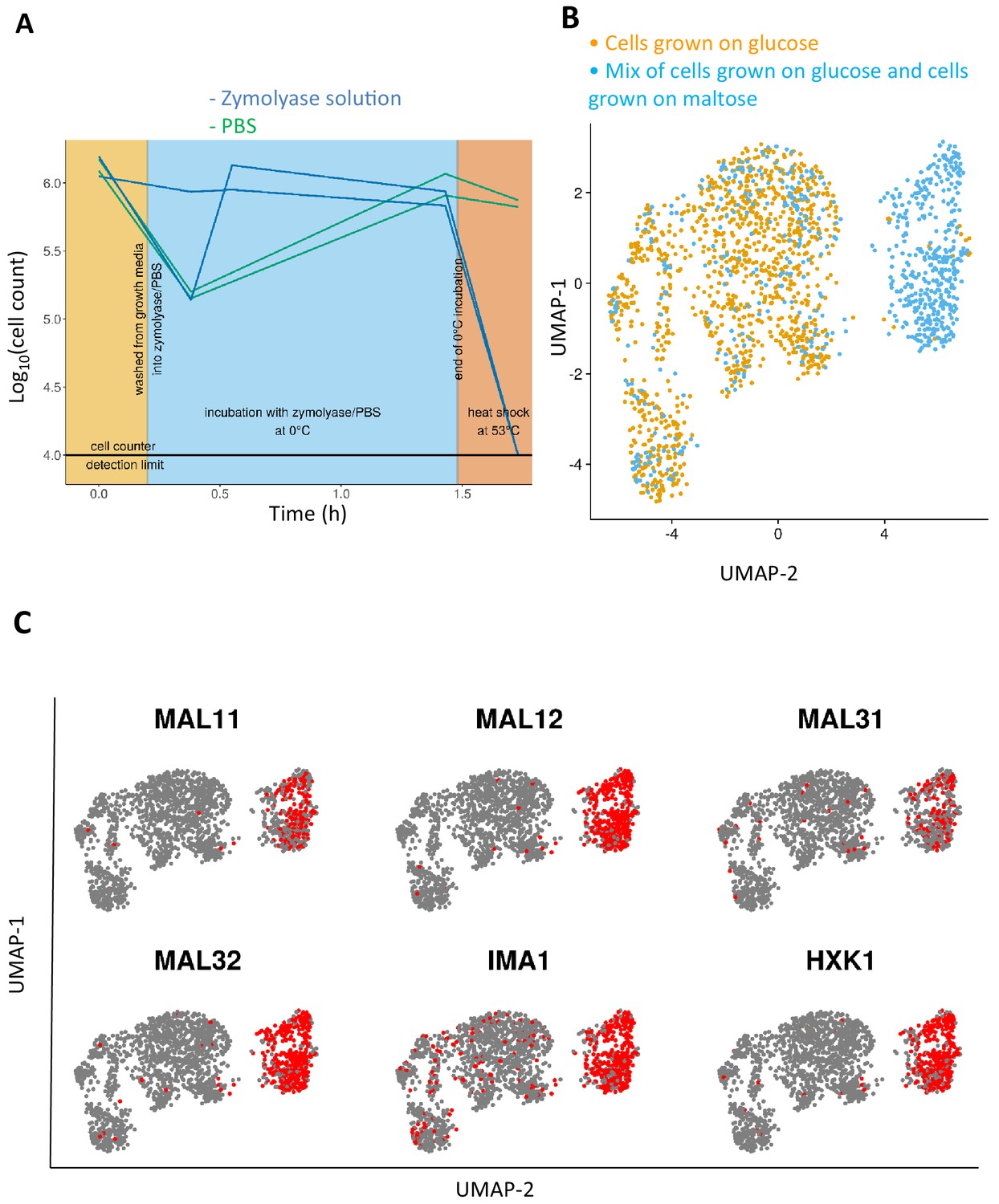
Adapting the 10x Genomics’ single-cell RNA-seq platform for yeast cells.
(A) Comparison of cell lysis efficacy of zymolyase solution to that of PBS buffer without zymolyase enzyme. Zymolyase is required and effective for lysing yeast cells in a temperature treatment regimen similar to the 10x Genomics protocol (see text for details). Cells counts per condition were measured in biological duplicate. (B) Transcriptome projection of two samples sharing cells from a common condition, namely glucose-12h and mix-glucose-maltose, are aggregated. Half of the cells from the mix-glucose-maltose sample are sampled from the same condition as the glucose-12h sample. (C) The group of cells from the mix-glucose-maltose condition that does not overlap with glucose-12h condition, expresses the expected maltose-growth specific genes.
Figure 1—figure supplement 2
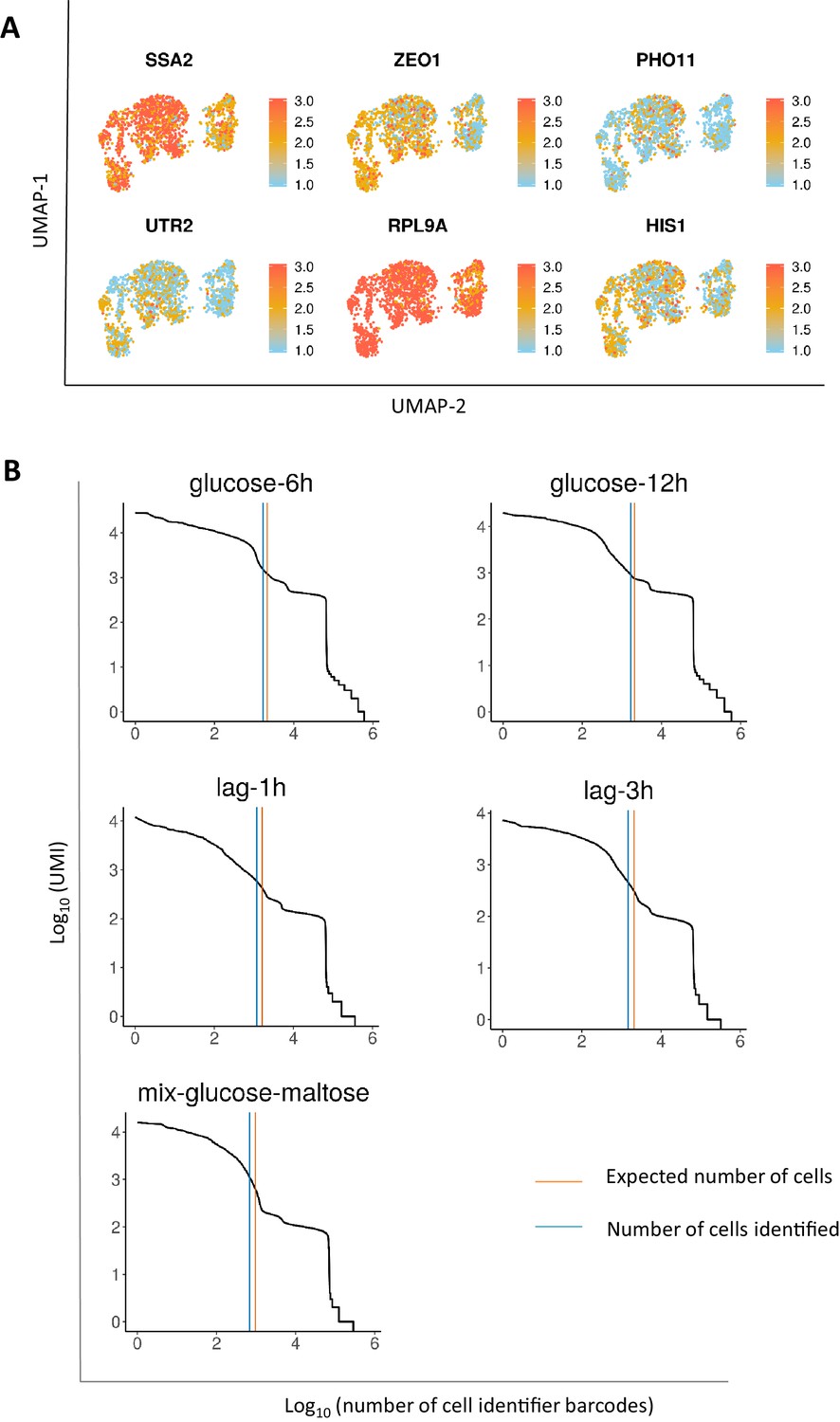
Glucose-growth-specific markers and comparison of expected vs detected number of cells.
(A) UMAP projection of glucose-specific growth markers in the aggregated glucose-12h and mix-glucose-maltose samples. (B) UMI count as a function of per-droplet barcodes. The blue line demonstrates the number of cells identified using the Cell Ranger software. The orange line demonstrates the number of expected cells based on the number of cells loaded. Details on the number of cells loaded and the expected number of cells is provided in the methods section.
Figure 2 with 3 supplements
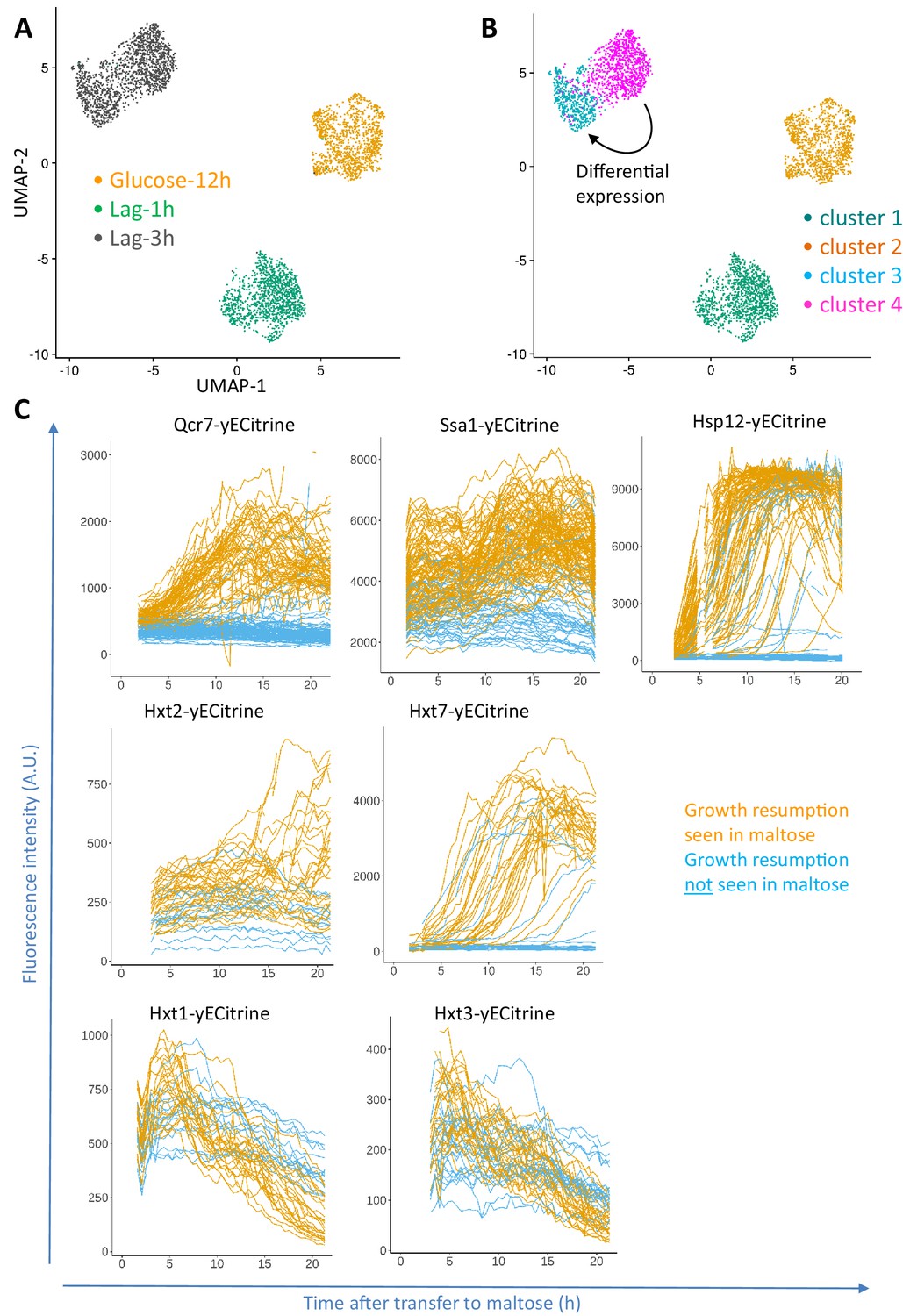
Progression of single-cell transcriptomes during the lag phase and validation of the identified differentially expressed genes.
(A) Samples for scRNA-seq were collected separately at the end of glucose growth and during the lag phase after the glucose-maltose shift. Expression levels of the samples from glucose-12h, lag-1h and lag-3h were aggregated using the Cell Ranger software. The coloring is based on the sample barcode. (B) Clustering based on expression levels to group the glucose-12h, lag-1h and lag-3h samples into four clusters. The number of clusters was determined using the resolution parameter in FindClusters function of Seurat package. The lag-3h sample is grouped into two clusters (clusters 3 and 4). Differential expression analysis between clusters 3 and 4 was performed, of which the list of genes is provided as Supplementary file 1. GO enrichment of the overexpressed genes between these two clusters is provided as Supplementary file 2. (C) Validation of scRNA-seq results using fluorescent protein fusions. Cells carrying fluorescent fusion reporters were pre-grown in maltose, grown in glucose for 6 to 8 hr, then transferred to maltose and prepared for time-lapse microscopy while in maltose medium. The fluorescence intensity was tracked in each cell over time. The plots for cells that eventually managed to resume growth after transfer to maltose are colored orange, whereas plots for cells that failed to escape the lag phase are plotted in blue.
Figure 2—figure supplement 1
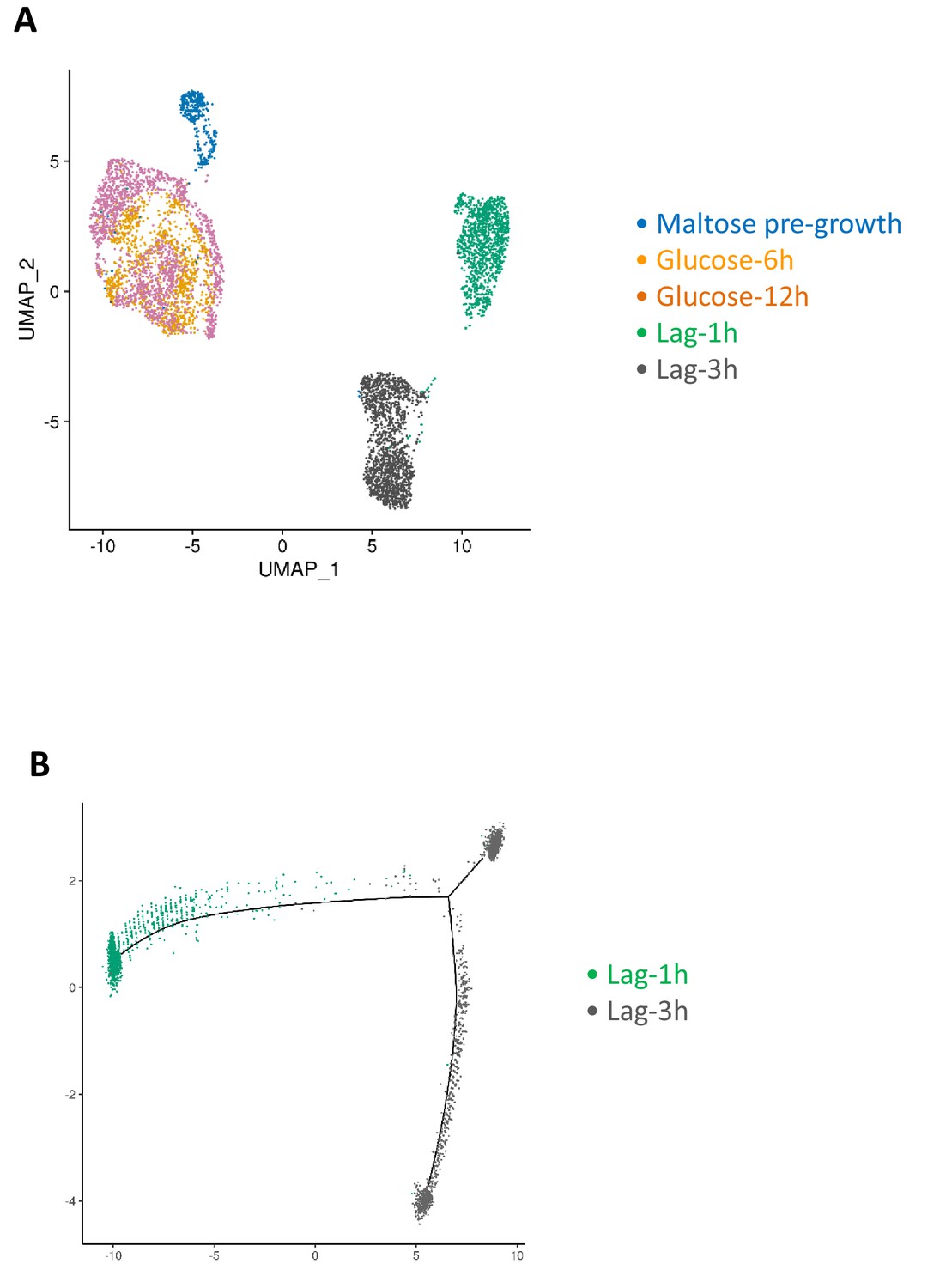
Progression of single-cell transcriptomes during maltose-glucose-maltose shift.
(A) Samples for scRNA-seq were collected separately during glucose growth and during the lag phase after the glucose-maltose shift. The cells labeled as maltose pre-growth in the figure consist of a subset of the mix-glucose-maltose sample, namely those cells that express maltose growth markers. If a cell in mix-glucose-maltose sample had at least a total of two transcripts from MAL11, MAL12, MAL31, MAL32 or HXT1 genes, it was labeled as belonging to the maltose pre-growth condition. (B) Pseudo temporal ordering of the cells in lag phase using Monocle package.
Figure 2—figure supplement 2
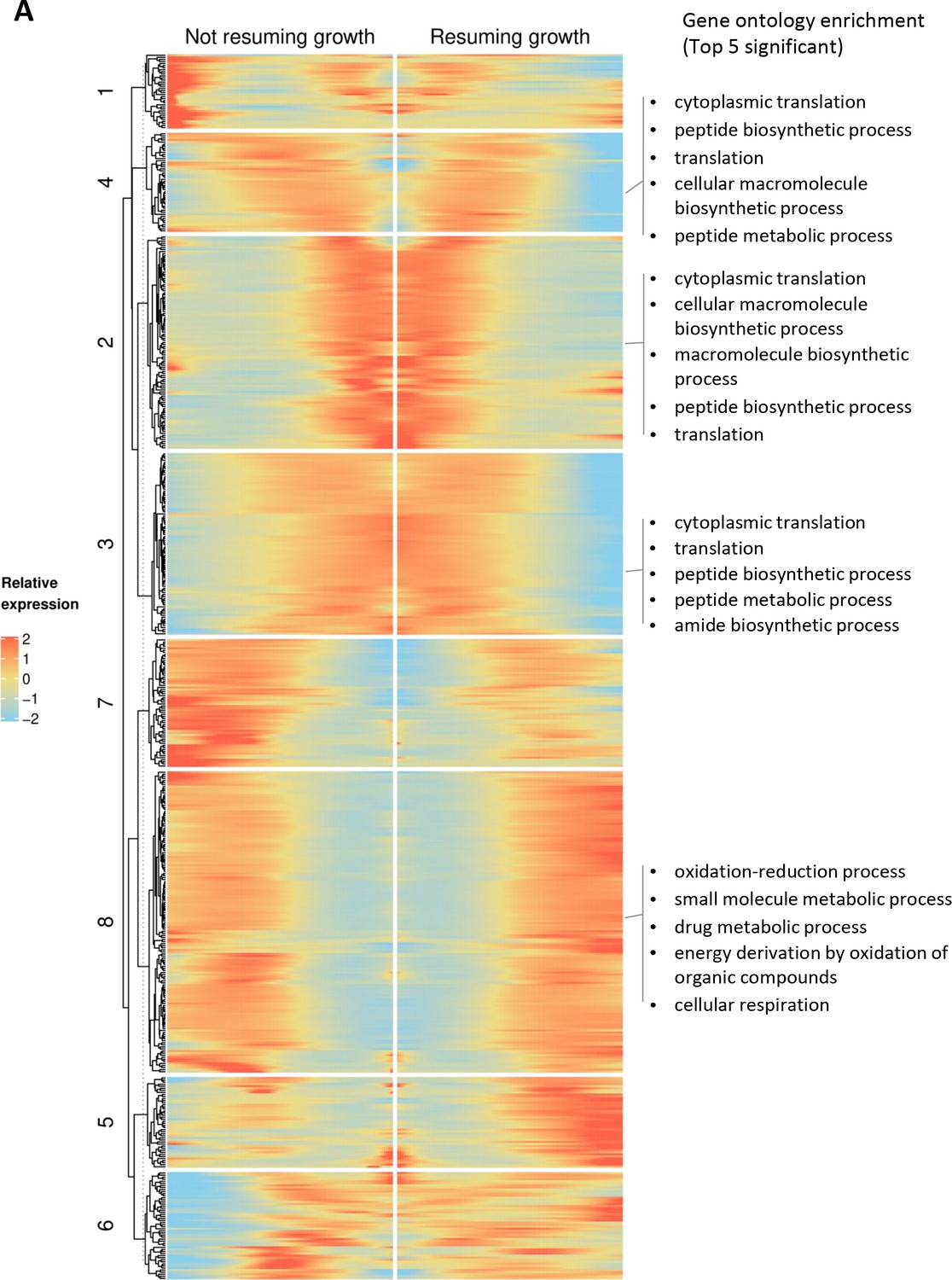
Branched expression analysis modeling (BEAM) using Monocle package showing co-expression patterns in continuously growing cultures.
(A) Rows indicate genes expressed in a branch-dependent manner, and columns represent pseudo-time. The middle of x-axis represents the branching point, and progression from middle to right represents the trajectory of cells that grow in lag phase. Progression from middle to left represents the trajectory of the cells that cannot resume growth in lag phase. The list of the genes in each cluster is provided as Supplementary file 3.
Figure 2—figure supplement 3
Ribosomal gene expression during lag phase.
(A) Expression of the ribosomal genes from clusters 2, 3, and 4 of Figure 2—figure supplement 2A, over time during the lag phase. The blue lines correspond to the genes in clusters 2, 3, and 4 from Figure 2—figure supplement 2A, while the grey lines represent the rest of the genes.
Figure 3 with 1 supplement

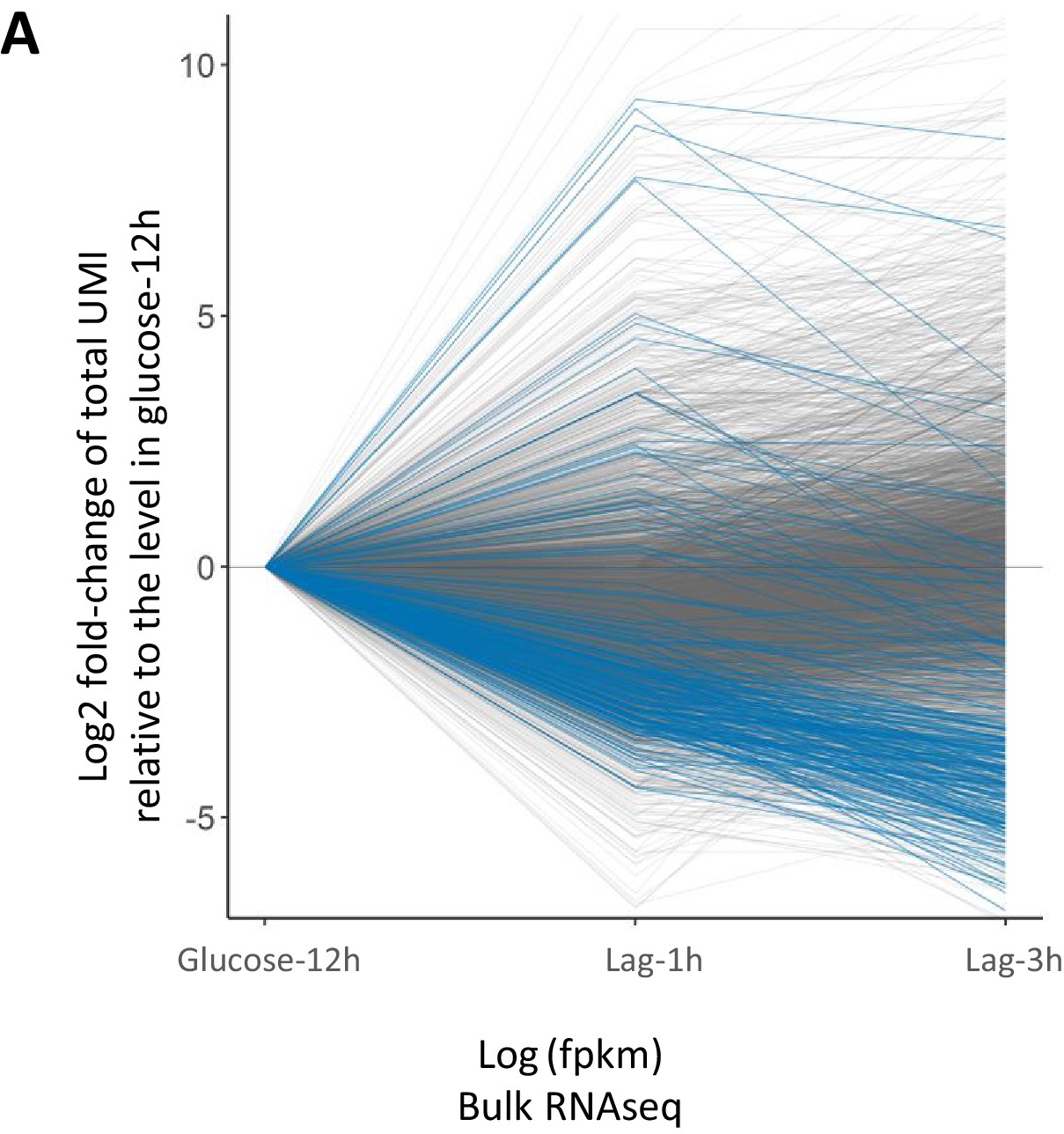
Concurrent analysis of co-expression and expression dynamics during glucose growth (glucose-12h) and lag phase (lag-3h).
(A) Co-expression analysis during glucose growth (glucose-12h) and expression dynamics of genes during glucose-to-maltose shift. This figure is composed of three parts. First, a heat map of expression correlation of the genes (blue-red heat map on the left). Second, a single-column heat map of basal expression level during glucose growth (sky blue-yellow-red heat map in the middle), showing log10 of mean expression level per cell in the population. Third, a two-column heat map of expression change during lag phase (black-green-red on the right) which shows log2 fold change of expression in lag-1h and lag-3h samples relative to glucose-12h sample. The color intensity in the expression correlation heat map (left) shows Pearson correlation coefficient of the expression level of gene pairs in the glucose-12h sample. Genes with less than a total of 10 UMI’s across all cells were excluded from the analysis. Expression levels were normalized by NormalizeData function of Seurat package. For each gene pair, Pearson correlation coefficient between the two vectors of expression levels across the cells was calculated. Genes that were not correlated with at least seven other genes with a minimal absolute correlation coefficient of at least 0.1 and Bonferroni adjusted p-value threshold of 0.05, were filtered out. The identity diagonal elements were set to zero. Genes were grouped together using hierarchical clustering function of ComplexHeatmaps package. The lists of genes in each of the clusters are provided as Supplementary file 4. (B) Co-expression analysis during lag phase (lag-3h) and expression dynamic of genes during glucose-to-maltose shift. Similar to A), but for the lag-3h sample instead of glucose-12h. The lists of genes in each of the clusters are provided as Supplementary file 4.
Figure 3—figure supplement 1

Concurrent analysis of co-expression and expression dynamics during lag phase (lag-1h).
(A) Similar to Figure 3A and B but for lag-1h sample. The lists of genes in each of the clusters are provided as Supplementary file 4.
Figure 4 with 1 supplement

Lag phase is heterogeneous, yet not purely stochastic.
(A) Histogram of single-cell lag times. Colors represent whether the cells resumed growth within the observed time window (orange) or did not resume growth (blue). (B) Same data as in A) but shown as a cumulative fraction rather than a histogram (C) The lag times of genealogically related cells tend to be more similar to each other compared to the mean pair-wise similarity across the population. Each dot represents the mean pair-wise difference of the lag times within a single micro-colony. The red horizontal line represents the mean pair-wise difference of lag times across the population. The median number of cells within each micro-colony is 3. In total 133 single cells were analyzed. (D) Cells expressing a FRET-based ATP sensor (ATeam) were pre-grown in maltose medium, transferred to glucose for 7 hr, and then (at t = 0) loaded into the CellASIC microfluidic chamber with perfusion of glucose-containing medium. After 30 min in glucose the flow was shifted to maltose medium where cells enter the lag phase. Each line represents the FRET signal in a single cell tracked over time. The orange lines represent cells for which growth resumption was seen in 24 hr, while the blue lines represent cells for which growth resumption was not observed. The dark grey circles represent time of growth resumption.
Figure 4—figure supplement 1
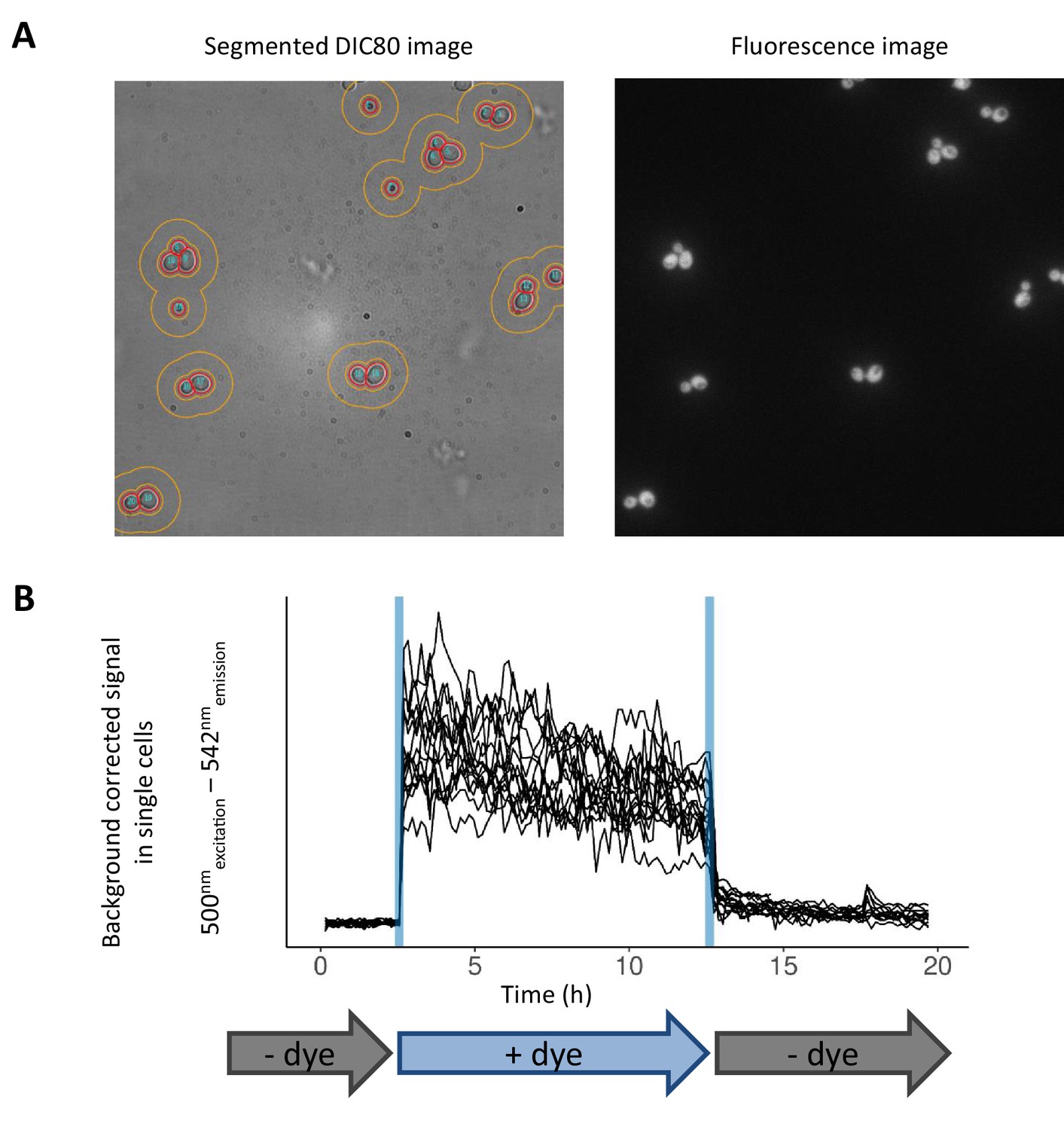
Image analysis pipeline and validation of the microfluidic setup.
(A) Sample microscopy images. DIC80 microscopy image (left) is segmented to separate single cells (red boundary). The donut-shaped boundary around the cells (orange) represents the area used for background fluorescent signal subtraction. The image on the right shows the fluorescence signal of JC10 dye. (B) Demonstration of microfluidic control dynamics and single-cell fluorescence intensity tracking. The cells are trapped in the microfluidic device (CellASIC) and are continuously grown on YP-maltose medium. In the first three hours, cells are growing in absence of fluorescent dye. Afterwards, the medium containing JC10 dye is flown for 10 hr, and finally, the flow is switched back to medium without the dye.
Tables
Table 1
Overview of cell numbers during scRNA-seq experiment
| Sample name | Cell count when sampling | Cell count after thaw and wash in PBS | TARGET conc. of cells | Cell count after extra dilution in PBS | Number of cells expected | Number of cells identified after sequencing |
|---|---|---|---|---|---|---|
| glucose-6h | 1.38E+5 | 2.42E+5 | 2.00E+5 | 2.15E+5 | 2150 | 1695 |
| glucose-12h | 1.17E+6 | 4.19E+5 | 2.00E+5 | 2.07E+5 | 2070 | 1096 |
| lag-1h | 8.13E+5 | 3.19E+5 | 2.00E+5 | 1.62E+5 | 1620 | 1172 |
| lag-3h | 7.34E+5 | 3.72E+5 | 2.00E+5 | 2.09E+5 | 2090 | 1469 |
| mix-glucose-maltose (glucose-12h + pre-growth maltose) | 1.91E+6 (pre-growth maltose) | 3.11E+5 | 1.00E+5 (for glucose-12h and pre-growth maltose) | Pre-growth maltose: 6.06E+4 glucose-12h: 1.32E+5 Together: 1.05E+5 | (606+1320)* 0.5 = 963 | 686 |
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Saccharomyces cerevisiae) | BY4742 | PMID:9483801 | S288c MATalpha; his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | |
| Strain, strain background (Saccharomyces cerevisiae) | KV1156 | PMID:20471265 | BY4742 MAL13:: HygR-MAL63_c9 | |
| Strain, strain background (S. cerevisiae) | AN62 | PMID:24453942 | KV1156 SAL1+ | |
| Strain, strain background (S. cerevisiae) | AN63 | PMID:24453942 | AN62 MATa | |
| Strain, strain background (S. cerevisiae) | AJ78 | This study | AN63 TEFp-ATeam1.03-KanMX inserted in YRO2 intergenic locus | |
| Strain, strain background (S. cerevisiae) | BC73 | This study | AN63 HXT1-yECitrine | |
| Strain, strain background (S. cerevisiae) | BC74 | This study | AN63 HXT2-yECitrine | |
| Strain, strain background (S. cerevisiae) | BC75 | This study | AN63 HXT3-yECitrine | |
| Strain, strain background (S. cerevisiae) | BC79 | This study | AN63 HXT7-yECitrine | |
| Strain, strain background (S. cerevisiae) | MC1 | This study | AN63 HSP12-yECitrine | |
| Strain, strain background (S. cerevisiae) | MC8 | This study | AN63 SSA1-yECitrine | |
| Strain, strain background (S. cerevisiae) | MC23 | This study | AN63 QCR7-yECitrine | |
| Recombinant DNA reagent | pKT140 | RRID:Addgene_8732 | KanMX-yECitrine plasmid | |
| Recombinant DNA reagent | pTEF:ATP | RRID:Addgene_92179 | TEF1p-ATeam1.03-KanMX4 plasmid |
Additional files
-
Supplementary file 1
Differential expression analysis between cells assigned to clusters 3 and 4 in UMAP projection of lag-3h sample.
- https://cdn.elifesciences.org/articles/55320/elife-55320-supp1-v2.csv
-
Supplementary file 2
GO terms corresponding to the differential expression analysis between clusters 3 and 4 in UMAP projection of lag-3h sample.
- https://cdn.elifesciences.org/articles/55320/elife-55320-supp2-v2.xlsx
-
Supplementary file 3
Gene cluster assignment, based on Branched expression analysis modeling in pseudo-temporal analysis.
- https://cdn.elifesciences.org/articles/55320/elife-55320-supp3-v2.csv
-
Supplementary file 4
Cross-correlation matrices of gene expression levels in glucose-12h, lag-1h, and lag-3h samples.
- https://cdn.elifesciences.org/articles/55320/elife-55320-supp4-v2.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/55320/elife-55320-transrepform-v2.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A new protocol for single-cell RNA-seq reveals stochastic gene expression during lag phase in budding yeast
eLife 9:e55320.
https://doi.org/10.7554/eLife.55320




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}