Sequential activation of transcriptional repressors promotes progenitor commitment by silencing stem cell identity genes
- Life Sciences Institute, University of Michigan, United States
- Department of Cell and Developmental Biology, University of Michigan Medical School, United States
- Invertebrate Genetics Laboratory, National Institute of Genetics, Japan
- Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, Sweden
- Riken Center for Brain Science, Japan
- Division of Genetic Medicine, Department of Internal Medicine and Comprehensive Cancer Center, University of Michigan Medical School, United States
Abstract
Stem cells that indirectly generate differentiated cells through intermediate progenitors drives vertebrate brain evolution. Due to a lack of lineage information, how stem cell functionality, including the competency to generate intermediate progenitors, becomes extinguished during progenitor commitment remains unclear. Type II neuroblasts in fly larval brains divide asymmetrically to generate a neuroblast and a progeny that commits to an intermediate progenitor (INP) identity. We identified Tailless (Tll) as a master regulator of type II neuroblast functional identity, including the competency to generate INPs. Successive expression of transcriptional repressors functions through Hdac3 to silence tll during INP commitment. Reducing repressor activity allows re-activation of Notch in INPs to ectopically induce tll expression driving supernumerary neuroblast formation. Knocking-down hdac3 function prevents downregulation of tll during INP commitment. We propose that continual inactivation of stem cell identity genes allows intermediate progenitors to stably commit to generating diverse differentiated cells during indirect neurogenesis.
Introduction
Indirect generation of differentiated cell types through intermediate progenitors allows tissue-specific stem cells to scale their progeny output to accommodate the development of appropriately sized organs in proportion to organism sizes. Indirect neurogenesis by the outer subventricular zone (OSVZ) neural stem cells drives the evolution of lissencephalic brains to gyrencephalic brains in vertebrates (Cárdenas and Borrell, 2020). The competency to generate intermediate progenitors and the maintenance of an undifferentiated state must be coordinately downregulated in stem cell progeny to ensure generation of differentiated cell types. Polycomb-mediated gene repression is thought to inactivate stem-cell-specific genes during differentiation (Tsuboi et al., 2019). However, loss of function of Polycomb Repressive Complex 2 (PRC2) did not lead to intermediate progenitors reverting into neural stem cells (Abdusselamoglu et al., 2019). Thus, the mechanisms that extinguish stem cell functionality during progenitor commitment remains poorly understood.
Two distinct neuroblast lineages function together to generate the number of neurons requisite for the development of adult fly brains (Farnsworth and Doe, 2017; Homem et al., 2015; Janssens and Lee, 2014). Similar to ventricular zone neural stem cells, type I neuroblasts undergo direct neurogenesis. Type I neuroblasts repeatedly undergo asymmetric division to generate one daughter cell that remains a neuroblast and one sibling cell (ganglion mother cell [GMC]) that divides once to generate two neurons. By contrast, type II neuroblasts undergo indirect neurogenesis, similar to OSVZ neural stem cells. Type II neuroblasts continually undergo asymmetric division to self-renew and to generate a sibling cell that commits to an intermediate progenitor (INP) identity (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008). The Sp family transcription factor Buttonhead (Btd) and the ETS-1 transcription factor PointedP1 (PntP1) are specifically expressed in type II neuroblasts (Komori et al., 2014b; Xie et al., 2014; Zhu et al., 2011). Btd and PntP1 expression levels decrease as a newly generated immature INP transitions into a non-Asense-expressing (Ase-) immature INP. Three to four hrs after this transition, an Ase- immature INP upregulates Ase expression as it progresses through INP commitment. Once INP commitment is complete, an Ase+ immature INP transitions into an INP that undergoes six to eight rounds of asymmetric division. These molecularly defined intermediate stages during INP commitment provide critical landmarks to investigate the mechanisms that extinguish the activity of type II neuroblast functional identity genes.
Notch signaling maintains type II neuroblasts in an undifferentiated state, partially by poising transcription of the master regulator of differentiation earmuff (erm) (Janssens et al., 2017). During asymmetric neuroblast division, the TRIM-NHL protein Brain tumor (Brat) segregates into the newly generated immature INP and targets transcripts encoded by Notch downstream effector genes for RNA decay (Bello et al., 2006; Betschinger et al., 2006; Komori et al., 2018; Lee et al., 2006b; Xiao et al., 2012). brat-null brains accumulate thousands of supernumerary type II neuroblasts that originate from the reversion of newly generated immature INPs due to defects in the downregulation of Notch signaling (Komori et al., 2014a). In parallel to Brat, the nuclear protein Insensible (Insb) inhibits the activity of Notch downstream effector proteins during asymmetric neuroblast division, and Insb overexpression efficiently triggers premature differentiation in type II neuroblasts (Komori et al., 2018). This multi-layered gene control allows for the onset of Erm expression, coinciding with the termination of PntP1 expression. Erm expression is maintained in immature INPs but becomes downregulated in INPs (Janssens et al., 2014). Erm belongs to a family of transcription factors that are highly expressed in neural progenitors and can bind histone deacetylase 3 (Hdac3) (Hirata et al., 2006; Koe et al., 2014; Levkowitz et al., 2003; Weng et al., 2010). Erm prevents INP reversion by repressing gene transcription. In erm-null brains, INPs spontaneously revert to type II neuroblasts; however, this phenotype can be suppressed by knocking-down Notch function (Weng et al., 2010). Thus, Erm likely inactivates type II neuroblast functionality genes by promoting histone deacetylation.
By comparing mRNAs enriched in type II neuroblasts or immature INPs, we identified tailless (tll) as a master regulator of type II neuroblast functional identity. Tll is expressed in type II neuroblasts but not in INPs; moreover, Tll is necessary and sufficient for type II neuroblast functionality. Tll overexpression is sufficient to transform type I neuroblasts into type II neuroblasts, as indicated by changes in gene expression and an acquired competence for generating INPs. We identified hamlet (ham) as a new negative regulator of type II neuroblast maintenance. ham is expressed after erm in immature INP during INP commitment, and Erm and Ham function through Hdac3 to continually inactivate tll. Sequential inactivation during INP commitment suppresses tll activation by Notch signaling in INPs. We propose that silencing of the master regulator of stem cell functional identity allows intermediate progenitors to stably commit to the generation of differentiated cell types without reacquiring stem cell functionality.
Results
A novel transient over-expression strategy to identify regulators of type II neuroblast functional identity
Genes that regulate neuroblast functional identity should be expressed in type II neuroblasts and become rapidly downregulated in Ase- and Ase+ immature INPs in wild-type brains (Figure 1A). To enrich for transcripts that fulfill these criteria, we tested if transient Insb overexpression induces supernumerary type II neuroblasts in brat-null brains to synchronously transition into INPs as in wild-type brains. The combination of Wor-Gal4,AseGal80, which is only active in type II neuroblasts, and Tub-Gal80ts, which loses its inhibitory effect on Gal4 activity under non-permissive temperatures, allows for spatial and temporal control of Insb overexpression in all type II neuroblasts. We allowed larvae to grow at 25°C, and induced transient Insb overexpression for 6, 12, 18, or 24 hr by shifting the larvae to a non-permissive temperature of 33°C. Larvae that remained at 25°C for the duration of this experiment served as the control (time 0). We assessed the effect of this transient over-expression strategy on cell identity by quantifying the total type II neuroblasts and INPs per brain lobe based on Deadpan (Dpn) and Ase expression (Figure 1A). Overexpressing Insb for 6 hr did not significantly affect the supernumerary neuroblast phenotype in brat-null brains (Figure 1—figure supplement 1A). By contrast, 12 or more hours of Insb overexpression led to a time-dependent decrease in supernumerary type II neuroblasts and a corresponding increase in INPs. These results demonstrate that transient Insb overexpression can induce supernumerary type II neuroblasts to synchronously transition into INPs in brat-null brains.
Figure 1 with 1 supplement see all
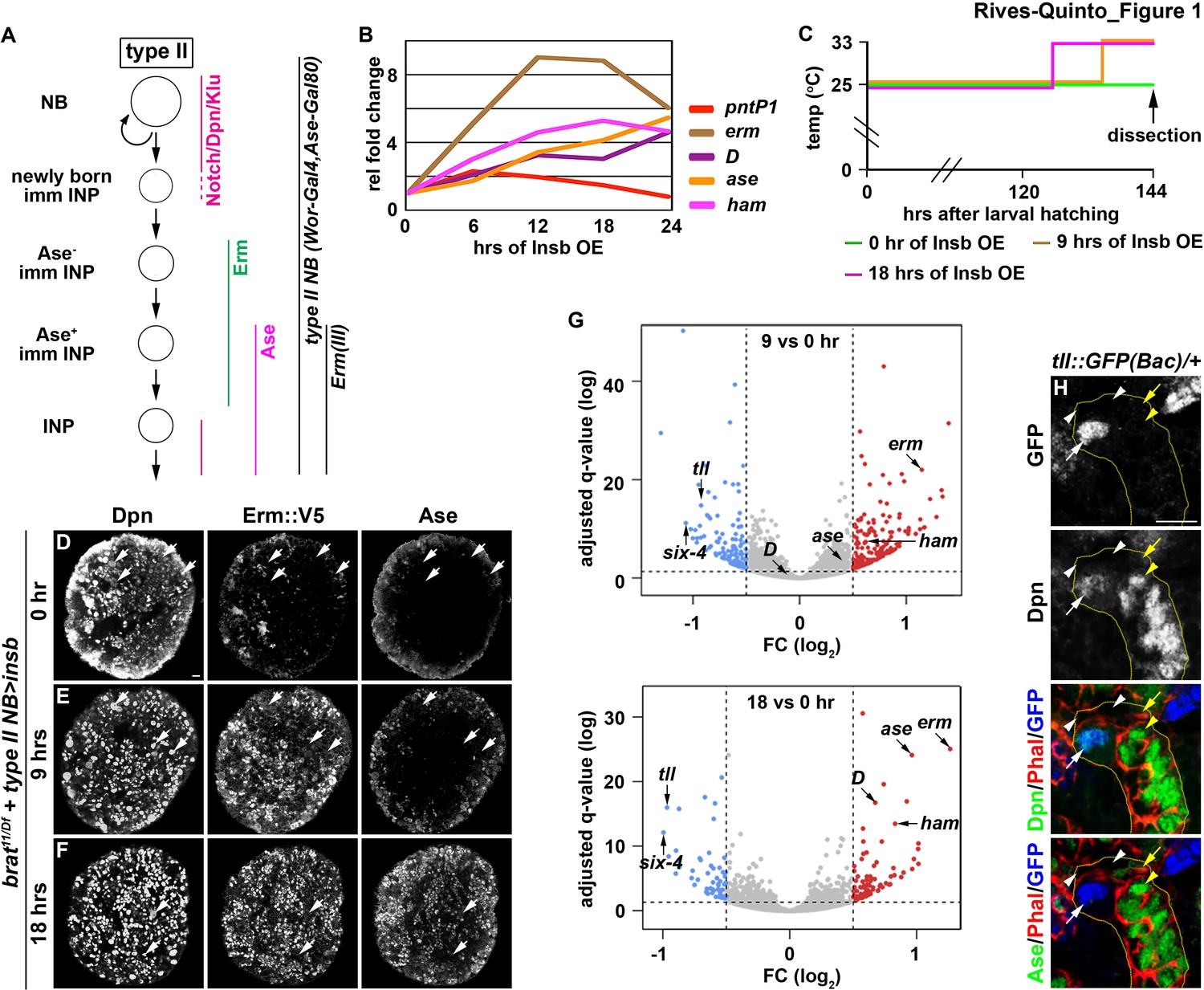
Identification of candidate regulators of type II neuroblast functional identity.
(A) A diagram of the type II neuroblast lineage showing the expression patterns of genes and Gal4 drivers used throughout this study. (B) Gene transcription profiles of brat-null brains transiently overexpressing Insb driven by a type II neuroblast Gal4. Supernumerary type II neuroblasts in brat-null brains transiently overexpressing Insb displayed patterns of gene transcription that are indicative of immature INPs undergoing INP commitment in wild-type brains. (C) A strategy to induce synchronized INP commitment in supernumerary type II neuroblasts in brat-null brains. Larvae carrying a UAS-insb transgene and a type II neuroblast Gal4 were collected and aged at 25°C. One third of larvae were shifted to 33°C at 126 or 135 hr after hatching to induce high levels of transient Insb expression, and the last one-third remained at 25°C serving as the source enriched for type II neuroblast-specific transcripts (time 0). (D–F) Images of brat-null brains transiently overexpressing Insb driven by a type II neuroblast Gal4 for 0, 9, or 18 hr. Transient overexpression of Insb first induced Erm and then Ase expression in supernumerary type II neuroblasts in brat-null brains. (G) Volcano plots showing fold-change of gene expression in brat-null brains transiently overexpressing Insb for 9 or 18 hr. (H) Tll expression pattern in the type II neuroblast lineage. The tll::GFP(Bac) transgene revealed the expression of endogenous Tll in type II neuroblasts but not in immature INPs and INPs. The following labeling applies to all images in this figure: yellow dashed line encircles a type II neuroblast lineage; white arrow: type II neuroblast; white arrowhead: Ase- immature INP; yellow arrow: Ase+ immature INP; yellow arrowhead: INP. Scale bar, 10 μm.
We next assessed if supernumerary type II neuroblasts transiently overexpressing Insb transition through identical intermediate stages to become INPs in brat-null brains as they do in wild-type brains (Figure 1A). We found that pntP1 mRNA levels increased within the first 6 hr of Insb overexpression, and then continually declined (Figure 1B). By contrast, erm transcription rapidly increased in the first 12 hr of Insb overexpression, plateauing between 12 and 18 hr. ase transcription showed little change in the first 6 hr of Insb overexpression, and then steadily increased between 12 and 24 hr. The combined temporal patterns of pntP1, erm, and ase transcription strongly suggest that type II neuroblasts transiently overexpressing Insb for 6 hr in brat-null brains are at an equivalent stage as a newly generated immature INP transitioning to an Ase- immature INP in wild-type brains. brat-null type II neuroblasts are at a stage equivalent to (1) Ase- immature INPs following 6–12 hr of Insb overexpression and (2) Ase+ immature INPs following 12–24 hr of Insb overexpression. We examined the expression of endogenous Erm in brat-null type II neuroblasts transiently overexpressing Insb to confirm their identity. We generated an erm::V5 allele by inserting a V5 epitope at the C-terminus of the erm reading frame, and showed that Erm::V5 recapitulates the published endogenous Erm expression pattern that we previously described (Figure 1—figure supplement 1B; Janssens et al., 2014). Indeed, following 9 hr of Insb overexpression most type II neuroblasts in brat-null brains expressed cell identity markers indicative of Ase- immature INPs (Erm::V5+Ase-), whereas after 18 hr of Insb overexpression they expressed markers indicative of Ase+ immature INPs (Erm::V5+Ase+) (Figure 1C–F). These data indicate that supernumerary type II neuroblasts transiently overexpressing Insb indeed transition through identical intermediate stages during INP commitment in brat-null brains as in wild-type brains.
We predicted that a candidate regulator of type II neuroblast functional identity should become downregulated in brat-null brains following 9 and 18 hr of Insb overexpression. By sequencing mRNA in triplicate following the transient over-expression strategy (Figure 1C), we identified 76 genes that were reproducibly downregulated by 1.5-fold or more in brat-null brains overexpressing Insb for 9 hr (Figure 1G). Of these genes, tll was the most downregulated; similarly, tll was the most downregulated gene in brat-null brains overexpressing Insb for 18 hr. We validated the tll expression pattern in the type II neuroblast lineage using a bacterial artificial chromosome (BAC) transgene [tll::GFP(Bac)] where green fluorescent protein (GFP) is fused in frame with the tll reading frame. Consistent with previous study (Bayraktar and Doe, 2013), we detected Tll::GFP in type II neuroblasts but not in immature or mature INPs (Figure 1H). Tll::GFP expression is also detected in type I neuroblasts in the brain and in the ventral nerve cord, but at a significantly lower level than in type II neuroblasts (Figure 1—figure supplement 1C–E). Thus, tll is an excellent candidate for regulating type II neuroblast functionality.
tll is a master regulator of type II neuroblast functional identity
We defined the type II neuroblast functional identity as the maintenance of an undifferentiated state and the competency to generate INPs. We first tested whether tll is required for maintaining type II neuroblasts in an undifferentiated state by overexpressing a UAS-tllRNAi transgene in the type II neuroblast lineage under the control of the Wor-Gal4,Ase-Gal80 driver (Figure 2A). Whereas control brains always contained eight type II neuroblasts per lobe, brains with tll function knocked down contained 4.3 ± 1.2 type II neuroblasts per lobe (n = 10 brains per genotype) (Figure 2B). A closer examination revealed that the remaining tll mutant neuroblasts showed reduced cell diameters and ectopically expressed Ase, two characteristics typically associated with INPs in the type II neuroblast lineage (Figure 2A, C and D). This result indicated that tll is required for maintaining type II neuroblasts in an undifferentiated state. We next tested whether tll is sufficient to re-establish a type II neuroblast-like undifferentiated state in INPs by misexpressing a UAS-tll transgene under the control of Erm-Gal4(III) (Figure 1A). Brains with tll misexpressed in INPs contained 150 ± 50 type II neuroblasts per lobe, whereas control brains always contained eight type II neuroblasts per lobe (n = 10 brains per genotype) (Figure 2E–G). Thus, Tll misexpression is sufficient to revert INPs to type II neuroblasts. These data together led us to conclude that tll is necessary and sufficient for maintaining type II neuroblasts in an undifferentiated state.
Figure 2
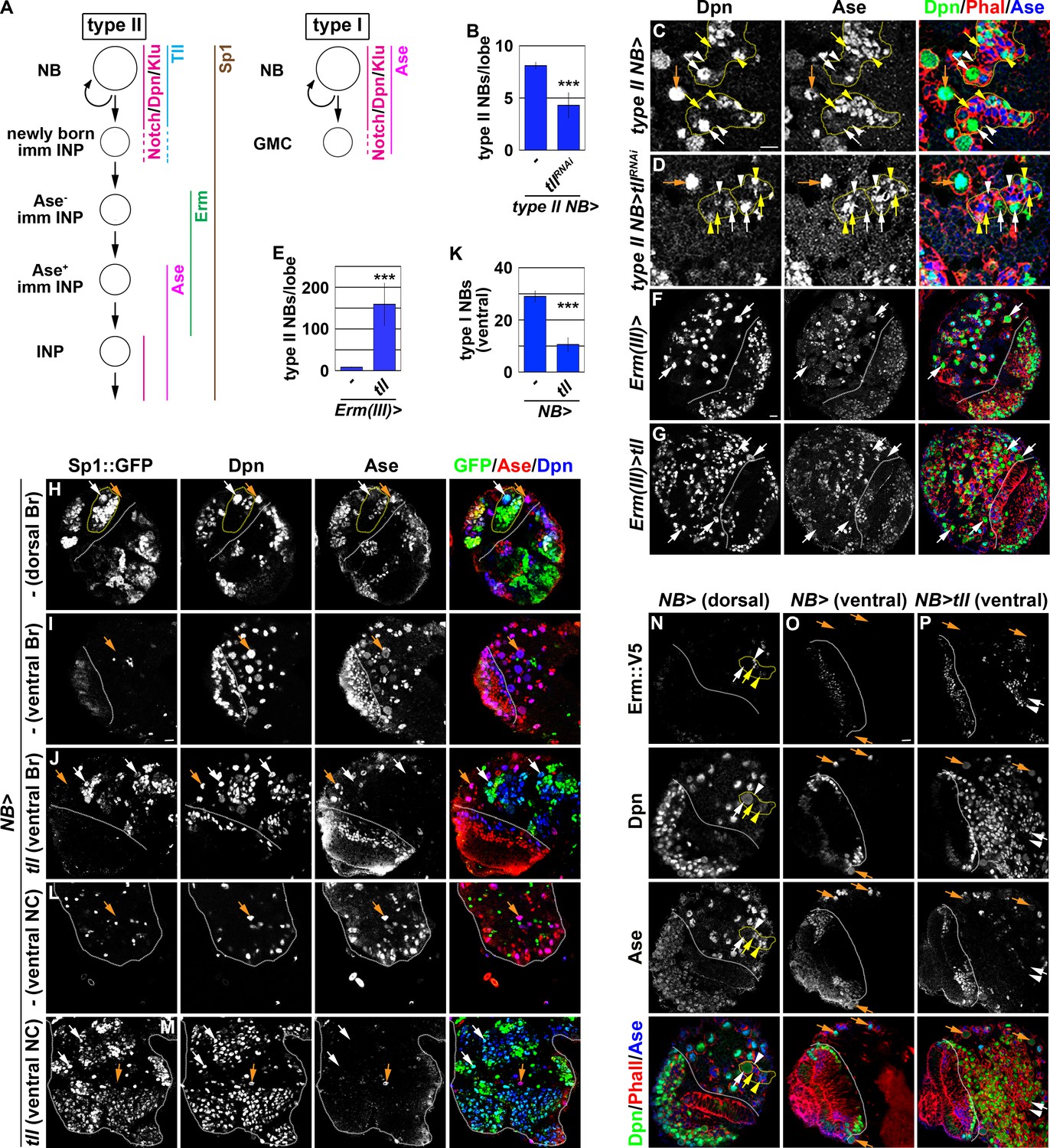
tll is necessary and sufficient for type II neuroblast functional identity.
(A) A diagram showing the expression patterns of genes in the type I and II neuroblast lineages. (B) Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-tllRNAi transgene driven by a type II neuroblast Gal4. Knocking-down tll function reduced the number of type II neuroblasts from 8 to 4 per brain lobe. (C–D) Images of brains that overexpressed a UAS-tllRNAi transgene driven by a type II neuroblast Gal4. Knocking-down tll function led to premature differentiation in type II neuroblast as indicated by reduced cell diameter and ectopic Ase expression but did not affect the maintenance of type I neuroblasts. (E) Quantification of total type II neuroblasts per brain that overexpressed a UAS-tll transgene driven by an INP Gal4. tll overexpression in INPs led to supernumerary type II neuroblast formation. (F–G) Images of brains that overexpressed a UAS-tll transgene driven by an INP Gal4. (H–I) Images of Sp1::GFP(Bac) brains. Sp1::GFP is detected in most cells in all type II neuroblast lineages that are exclusively located in the dorsal brain region, and is detected in few neurons in type I neuroblast lineages in the ventral brain region. (J) Images of the ventral region of Sp1::GFP(Bac) brains that overexpressed a UAS-tll transgene driven by a pan-neuroblast Gal4 (Wor-Gal4). Tll overexpression transforms type I neuroblasts (Ase+Sp1::GFP-) in the ventral brain region into type II neuroblasts (Ase-Sp1::GFP+). (K) Quantification of total ventral type I neuroblasts per brain lobe that overexpressed a UAS-tll transgene driven by a pan-neuroblast Gal4. Tll overexpression transforms 66% of type I neuroblasts in the ventral brain region into type II neuroblasts. (L–M) Images of the thoracic segments on the ventral nerve cord of Sp1::GFP(Bac) larvae that overexpressed a UAS-tll transgene driven by a pan-neuroblast Gal4 (Wor-Gal4). Tll overexpression transforms type I neuroblasts in the thoracic segments into type II neuroblasts. (N–O) Images of dorsal and ventral regions of erm::V5 brains. Erm::V5 is detected in immature INPs in type II neuroblast lineages but is undetectable in type I neuroblast lineages in the ventral region of larval brain. (P) Images of erm::V5 brains that overexpressed a UAS-tll transgene driven by a pan-neuroblast Gal4. Tll overexpression induced type I neuroblasts in the ventral-medial region of the brain to generate supernumerary type II neuroblasts interspersed with Erm::V5+ immature INPs. The following labeling applies to all images in this figure: white dashed line separates the optic lobe from the brain; yellow dashed line encircles a type II neuroblast lineage; white arrow: type II neuroblast; white arrowhead: Ase- immature INP; yellow arrow: Ase+ immature INP; yellow arrowhead: INP; orange arrow: type I neuroblast. Br: brain. NC: nerve cord. Scale bar, 10 μm. Bar graphs are represented as mean ± standard deviation. p-values: ***<0.005.
-
Figure 2—source data 1
Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-tllRNAi transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig2-data1-v2.xlsx
-
Figure 2—source data 2
Quantification of total type II neuroblasts per brain that overexpressed a UAS-tll transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig2-data2-v2.xlsx
-
Figure 2—source data 3
Quantification of total ventral type I neuroblasts per brain lobe that overexpressed a UAS-tll transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig2-data3-v2.xlsx
As tll is required to maintain type II neuroblasts in an undifferentiated state, direct assessment of its role in regulating the competency to generate INPs is not possible. As an alternative, we tested whether Tll overexpression is sufficient to induce ectopic type II neuroblast formation in the ventral brain region and in the ventral nerve cord that exclusively consist of type I neuroblasts. To unambiguously distinguish the two types of neuroblasts, we searched for robust protein markers of type II neuroblasts and their progeny. As Sp1 mRNA is uniquely detected in type II neuroblasts (Yang et al., 2016), we evaluated Sp1 protein expression using an Sp1::GFP(Bac) transgene. We found that Sp1::GFP marks type II neuroblasts and most of their progeny that are found exclusively in the dorsal brain region (Figure 2H). Sp1::GFP is detected in some neurons but never in type I neuroblasts in the ventral brain region and in the ventral nerve cord (Figure 2I and L). Thus, Sp1::GFP is a new marker for the type II neuroblast lineage (Figure 2A). We overexpressed a UAS-tll transgene under the control of a pan-neuroblast driver (Wor-Gal4) in larval brains carrying the Sp1::GFP(Bac) transgene. While the ventral region of control brains contains 29 ± 2.1 type I neuroblasts, the same region of the brains overexpressing Tll contains 10.6 ± 2.5 type I neuroblasts and hundreds of ectopic type II neuroblasts (n = 10 brains per genotype) (Figure 2I–K). This result strongly suggests that Tll overexpression transforms greater than 60% of type I neuroblasts in the ventral brain region into type II neuroblasts. Similarly, Tll overexpression also induced hundreds of supernumerary type II neuroblasts in the ventral nerve cord (n = 10 brains per genotype) (Figure 2L and M). Thus, ectopic tll expression is sufficient to molecularly transform type I neuroblasts into type II neuroblasts.
Next, we tested if type I neuroblasts overexpressing tll can generate immature INPs that are marked by Erm::V5 expression (Figure 2A and N). We reproducibly observed Erm::V5+ immature INPs in the ventral-medial region of the brain overexpressing Tll, but not in the ventral-lateral brain region (Figure 2O and P). This result strongly suggests that progeny of transformed type II neuroblasts in the ventral-medial brain region assume an immature INP identity and then revert into supernumerary type II neuroblasts. By contrast, progeny of transformed type II neuroblasts in the ventral-lateral brain region appeared to rapidly revert into supernumerary neuroblasts instead of assuming an immature INP identity. Thus, tll overexpression is sufficient to molecularly and functionally transform type I neuroblasts into type II neuroblasts, and that tll is a master regulator of type II neuroblast functional identity.
Ham is a new regulator of INP commitment
A previous study found that Suppressor of Hairless, the DNA-binding partner of Notch, binds the tll locus in larval brain neuroblasts, suggesting that tll is a putative Notch target (Zacharioudaki et al., 2016). Thus, mechanisms likely exist to silence tll during INP commitment, preventing re-activation of Notch signaling in INPs from ectopically inducing Tll expression and driving supernumerary type II neuroblast formation (Figure 2A). We predicted that genes required for silencing tll during INP commitment should become upregulated in brat-null brains following 9 and 18 hr of Insb overexpression, and mutations in these genes should lead to supernumerary type II neuroblast formation due to ectopic tll expression in INPs. From our RNA sequencing dataset, we identified genes that encode transcription factors and are upregulated in brat-null brains overexpressing Insb for 9 or 18 hr. These transcription factors include Erm, Dichaete (D), Ase, and Hamlet (Ham) (Figure 1G). Reverse transcriptase PCR confirmed that the transcript levels of these genes indeed become upregulated in brat-null brains overexpressing Insb (Figure 1B). Thus, these genes are candidates for inactivating tll during INP commitment.
We tested if any of the candidate genes is required for inactivating tll expression during INP commitment by knocking-down their function in erm hypomorphic (ermhypo) brains. ermhypo brains display a low frequency of INP reversion to type II neuroblasts, and provides a sensitized genetic background for identifying genes required to prevent INP reversion to type II neuroblasts (Janssens et al., 2017). Because ectopic tll expression in INPs leads to supernumerary type II neuroblast formation, reducing the function of genes that inactivate tll should enhance the supernumerary neuroblast phenotype in ermhypo brains. We found that ermhypo brains alone contained 31.5 ± 6.8 type II neuroblasts per lobe (n = 10 brains) (Figure 3A). Although reducing the function of most candidate genes did not enhance the supernumerary neuroblast phenotype in ermhypo brains, knocking-down ham function with two different UAS-RNAi transgenes reproducibly enhanced the phenotype (n = 10 brains per transgene) (Figure 3A). Consistent with the effect of reducing ham function by RNAi, the heterozygosity of a deficiency that deletes the entire ham locus also enhanced the supernumerary neuroblast phenotype in ermhypo brains (data not presented). Thus, ham likely plays a role in preventing INPs from reverting to supernumerary type II neuroblasts.
ham is the fly homolog of the vertebrate Prdm16 gene that encodes a transcription factor and has been shown to play a key role in regulating cell fate decisions in multiple stem cell lineages (Baizabal et al., 2018; Harms et al., 2015; Moore et al., 2002; Shimada et al., 2017). A previous study concluded that ham regulates INP proliferation but does not play a role in suppressing supernumerary type II neuroblast formation (Eroglu et al., 2014). The discrepancy between our findings and the published observations might be due to the ham1 allele used in the previous study, which encodes a nearly full-length protein and exhibits similar protein stability as wild-type Ham (Figure 3B; Moore et al., 2002). We took two approaches to determine whether ham is required for preventing supernumerary type II neuroblast formation. First, we examined ham deficiency heterozygous brains and reproducibly observed a mild supernumerary type II neuroblast phenotype (9 ± 0.9 per lobe, n = 12 brains) (Figure 3—figure supplement 1A). Second, we knocked down ham function by overexpressing a UAS-RNAi transgene. We found that knocking down ham function also led to a mild but statistically significant increase in type II neuroblasts per lobe (9.8 ± 1.8; n = 20 brains) as compared to control brains (8 ± 0; n = 10 brains) (Figure 3—figure supplement 1B). To confirm that ham is indeed required for suppressing supernumerary type II neuroblast formation, we generated two new ham alleles, hamSK1 and hamSK4, by CRISPR-Cas9 (Figure 3B). We characterized the effect of the hamSK1 or hamSK4 allele on Ham protein expression using a specific antibody. We found that hamSK1 or hamSK4 homozygous brains show undetectable Ham protein expression (Figure 3C and D; data not presented). This result strongly suggests that mutant Ham protein encoded by the hamSK1 or hamSK4 allele is unstable, and that these two new ham alleles are strongly loss-of-function mutants. We then tested if Ham protein is required for suppressing supernumerary type II neuroblast formation. Indeed, hamSK1 homozygous brains contained 40 ± 10 type II neuroblasts per lobe (n = 15 brains) (Figure 3E and F). Because Ham is detectable in Ase+ immature INPs and remains expressed in all INPs (Eroglu et al., 2014), we conclude that ham is a novel regulator of INP commitment.
ham knock-down drastically enhanced the supernumerary neuroblast phenotype in ermhypo brains, leading us to hypothesize that ham functions together with erm to suppress INPs from reverting to supernumerary type II neuroblasts. We tested this hypothesis by examining a potential genetic interaction between erm and ham during INP commitment. Although the heterozygosity of erm did not increase total type II neuroblasts (8.1 ± 0.3 per lobe; n = 11 brains), it enhanced the supernumerary neuroblast phenotype in ham deficiency heterozygous brains and in hamSK1 homozygous brains (14 ± 2.2 vs. 76.2 ± 18.3 type II neuroblasts per lobe; n = 14 brains per genotype) (Figure 3G–I). We conclude that Ham functions synergistically with Erm to suppress supernumerary type II neuroblast formation.
Figure 3 with 1 supplement see all
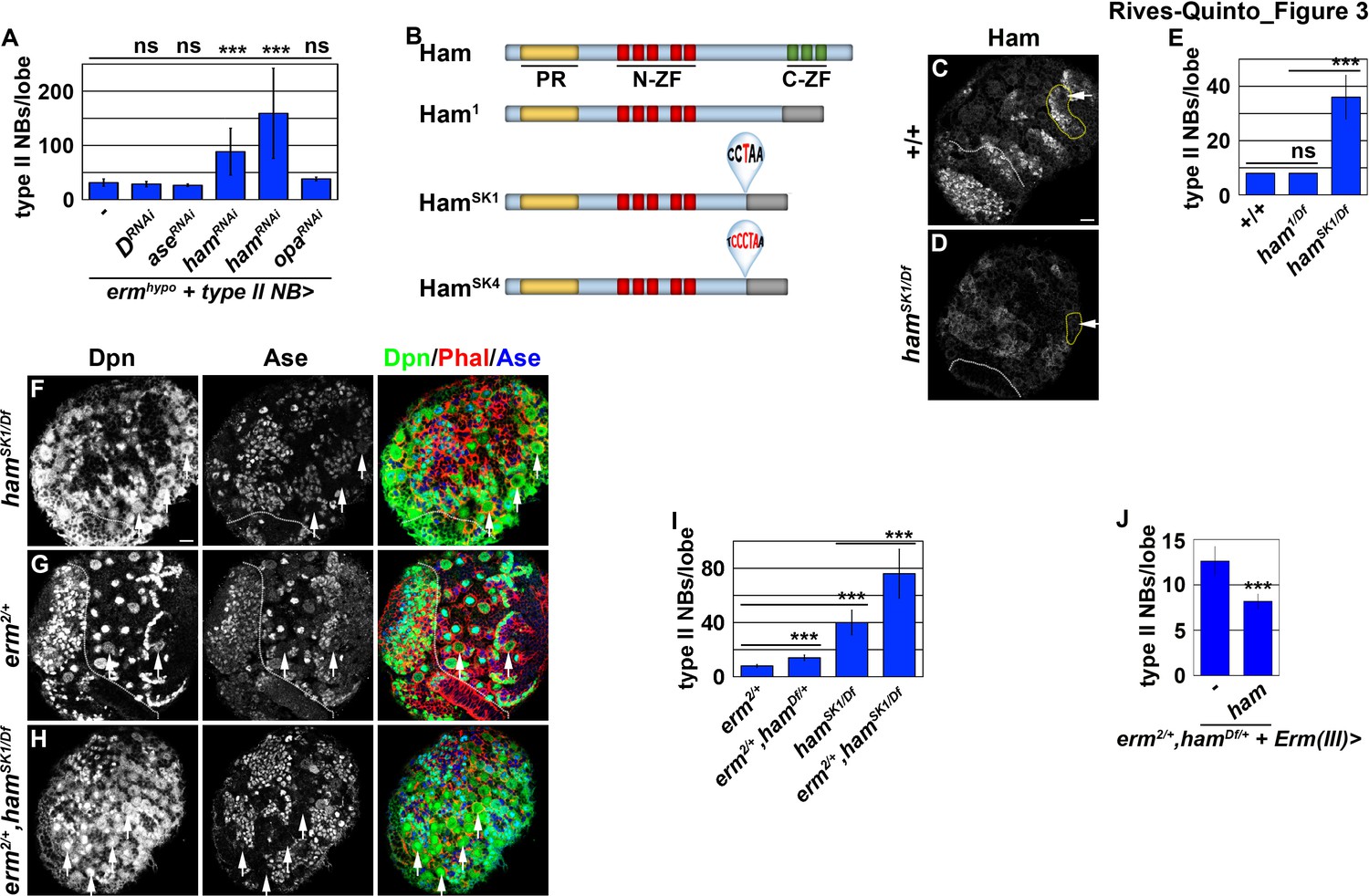
Ham is a novel regulator of INP commitment.
(A) Quantification of total type II neuroblasts per ermhypo brain lobe that overexpressed a UAS-RNAi transgene driven by a type II neuroblast Gal4. Knocking-down ham function consistently enhanced the supernumerary type II neuroblast phenotype in ermhypo brains. (B) A diagram summarizing the lesions in ham alleles. The molecular lesions in hamSk1 and hamSK4 alleles were not independently verified. (C–D) Ham expression in wild-type or hamSK1 homozygous brains. Ham was detected in immature INPs and INPs in wild-type brains, but was undetectable in hamSK1 homozygous brains. (E) Quantification of total type II neuroblasts per ham-mutant brain lobe. hamSK1 homozygous but not ham1 homozygous brains displayed a supernumerary type II neuroblast phenotype. (F–H) Images of ham single mutant or ham,erm double mutant brains. The heterozygosity of erm alone had no effect on type II neuroblasts, but enhanced the supernumerary type II neuroblast phenotype in hamSK1 homozygous brains. (I) Quantification of total type II neuroblasts per brain lobe of the indicated genotypes. erm and ham function synergistically to prevent supernumerary type II neuroblast formation. (J) Quantification of total type II neuroblasts per erm,ham double heterozygous brain lobe that overexpressed a UAS-ham transgene driven by an INP Gal4. Overexpressing Ham in INPs rescued the supernumerary type II neuroblast phenotype in erm,ham double heterozygous brains. The following labeling applies to all images in this figure: white dashed line separates the optic lobe from the brain; yellow dashed line encircles a type II neuroblast lineage; white arrow: type II neuroblast. Scale bar, 10 μm. Bar graphs are represented as mean ± standard deviation. p-values: ***<0.005. ns: not significant.
-
Figure 3—source data 1
Quantification of total type II neuroblasts per ermhypo brain lobe that overexpressed a UAS-RNAi transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig3-data1-v2.xlsx
-
Figure 3—source data 2
Quantification of total type II neuroblasts per ham-mutant brain lobe.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig3-data2-v2.xlsx
-
Figure 3—source data 3
Quantification of total type II neuroblasts per brain lobe of the indicated genotypes.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig3-data3-v2.xlsx
-
Figure 3—source data 4
Quantification of total type II neuroblasts per erm,ham double heterozygous brain lobe that overexpressed a UAS-ham transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig3-data4-v2.xlsx
Ham promotes stable INP commitment by repressing gene transcription
Re-activation of Notch signaling in INPs drives supernumerary type II neuroblast formation in erm-null brains (Weng et al., 2010). Therefore, we hypothesized that supernumerary type II neuroblasts in ham-null brains might also originate from INPs. Whereas erm,ham heterozygous brains alone contained 12.7 ± 1.6 type II neuroblasts per lobe (n = 9 brains), overexpressing ham in INPs driven by Erm-Gal4(III) rescued the supernumerary neuroblast phenotype in erm,ham double heterozygous brains (8.2 ± 0.8 type II neuroblasts per lobe; n = 24 brains) (Figure 3J). This result strongly suggests that supernumerary type II neuroblasts originate from INPs in ham-null brains. We generated GFP-marked mosaic clones derived from single type II neuroblasts to confirm the origin of supernumerary type II neuroblasts in ham-null brains. In wild-type clones (n = 9 clones), the parental type II neuroblast was always surrounded by Ase- and Ase+ immature INPs (Figure 4A). By contrast, supernumerary neuroblasts in hamSK1 homozygous clones (2.5 ± 1.7 type II neuroblasts per clone; n = 11 clones) were always located far away from parental neuroblasts and surrounded by Ase+ cells that were most likely Ase+ immature INPs and ganglion mother cells (Figure 4B and C). It is highly unlikely that supernumerary neuroblasts in hamSK1 homozygous clones originated from symmetric neuroblast division based on their location relative to parental neuroblasts and the cell types that surround them. Thus, we conclude that Ham suppresses INP reversion to supernumerary type II neuroblasts.
Figure 4
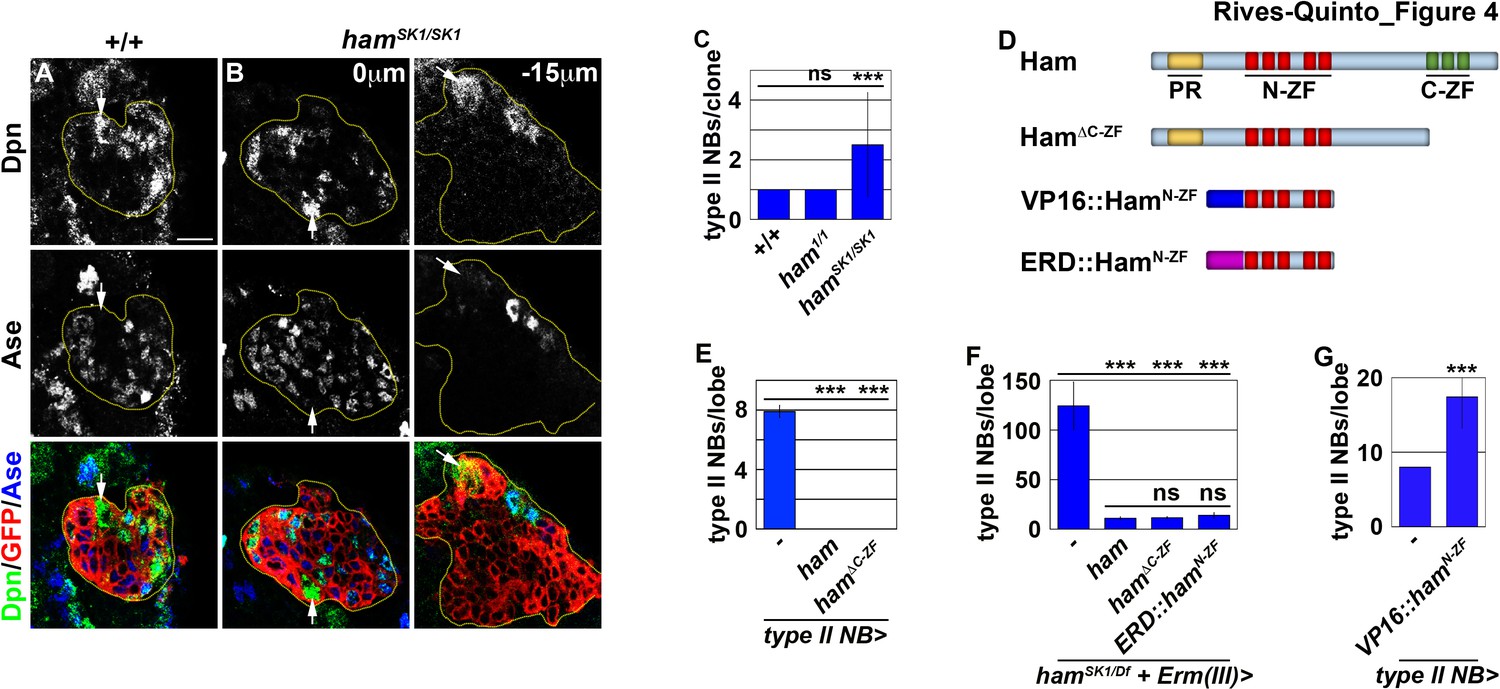
Ham suppresses INP reversion by repressing gene transcription.
(A–B) Images of wild-type or hamSK1 homozygous type II neuroblast mosaic clones. Supernumerary neuroblasts (−15 μm) in hamSK1 homozygous clones were always located far from the parental neuroblast (0 μm), and were surrounded by Ase+ cells. (C) Quantification of total neuroblasts per ham1 or hamSK1 homozygous type II neuroblast clone. hamSK1 homozygous clones contained supernumerary neuroblasts but ham1 homozygous clones did not. (D) A diagram summarizing UAS-ham transgenes used in this study. (E) Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-ham transgene driven by a type II neuroblast Gal4. Overexpressing full-length Ham or HamΔC-ZF led to premature differentiation in type II neuroblasts. (F) Quantification of total type II neuroblasts per hamSK1 homozygous brain lobe that overexpressed various UAS-ham transgenes driven by an INP Gal4. Overexpressed full-length Ham, HamΔC-ZF or ERD::HamN-ZF rescued the supernumerary neuroblast phenotype in hamSK1 homozygous brains. (G) Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-VP16::hamN-ZF transgene driven by a type II neuroblast Gal4. Overexpressing VP16::HamN-ZF led to supernumerary type II neuroblast formation. The following labeling applies to all images in this figure: yellow dashed line encircles a type II neuroblast lineage; white arrow: type II neuroblast. Scale bar, 10 μm. Bar graphs are represented as mean ± standard deviation. Ppvalues: ***<0.005. ns: not significant.
-
Figure 4—source data 1
Quantification of total neuroblasts per ham1 or hamSK1 homozygous type II neuroblast clone.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig4-data1-v2.xlsx
-
Figure 4—source data 2
Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-ham transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig4-data2-v2.xlsx
-
Figure 4—source data 3
Quantification of total type II neuroblasts per hamSK1 homozygous brain lobe that overexpressed various UAS-ham transgenes.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig4-data3-v2.xlsx
-
Figure 4—source data 4
Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-VP16::hamN-ZF transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig4-data4-v2.xlsx
Because Erm suppresses supernumerary type II neuroblast formation by repressing gene transcription, Ham likely prevents INP reversion as a transcriptional repressor. We reasoned that Ham might function through its N-terminal zinc finger to prevent INP reversion based on our finding that hamSK1 but not ham1 homozygous clones displayed a supernumerary neuroblast phenotype (Figure 4C). Consistent with this hypothesis, mis-expressing HamΔC-ZF triggered premature differentiation in type II neuroblasts, identical to mis-expression of full-length Ham (n = 10 brains per genotype) (Figure 4E). We tested if overexpressing HamΔC-ZF in INPs can rescue the supernumerary neuroblast phenotype in hamSK1 homozygous brains by incubating larvae at 33°C for 96 hr. Incubating hamSK1 homozygous larvae at an elevated temperature leads to a severer supernumerary neuroblast phenotype than at 25°C (Figures 3E and 4F). Overexpressing HamΔC-ZF rescued the supernumerary neuroblast phenotype in hamSK1 homozygous brains to a similar extent as overexpressing full-length Ham (11.1 ± 1.4 vs. 11.9 ± 1.3 type II neuroblasts per lobe; n = 11 or 9 brains, respectively) (Figure 4F). Under identical conditions, overexpressing a constitutive transcriptional repressor form of Ham containing only the N-terminal zinc fingers (ERD::HamN-ZF) also rescued the supernumerary neuroblast phenotype in hamSK1 homozygous brains (14 ± 2.3 type II neuroblasts per lobe; n = 9 brains) (Figure 4D and F). These results indicate that Ham functions through the N-terminal zinc finger to repress the transcription of genes that can trigger INP reversion to supernumerary type II neuroblasts.
Finally, we tested if the N-terminal zinc finger of Ham mediates target gene recognition. We mis-expressed a constitutive transcriptional activator form of Ham containing only the N-terminal zinc-finger motif (VP16::HamN-ZF) (Figure 4D). We found that VP16::HamN-ZF misexpression in type II neuroblasts was sufficient to induce supernumerary neuroblast formation (17.1 ± 4.7 type II neuroblasts per lobe; n = 10 brains) (Figure 4G). Because VP16::HamN-ZF can exert a dominant-negative effect, we conclude that Ham suppresses INP reversion by recognizing target genes through its N-terminal zinc-finger motif and repressing their transcription.
Inactivation during INP commitment relinquishes Notch’s ability to activate tll in INPs
Our findings strongly suggest that Erm and Ham inactivate tll during INP commitment, preventing the re-activation of Notch signaling from triggering tll expression in INPs (Figure 5A). Therefore, we tested if Erm and Ham indeed inactivate tll by reducing tll function in erm,ham double heterozygous brains. tll heterozygous brains contained 8.2 ± 0.4 type II neuroblasts per lobe, whereas erm,ham double heterozygous brains contained 11 ± 1.2 type II neuroblasts per lobe (n = 10 or 25 brains, respectively) (Figure 5B). The heterozygosity of tll consistently suppressed the supernumerary neuroblast phenotype in erm,ham double heterozygous brains (8.4 ± 0.6 type II neuroblasts per lobe; n = 22 brains) (Figure 5B). This result strongly supports our hypothesis that Erm and Ham inactivate tll during INP commitment. Consistent with this interpretation, we found that Tll::GFP becomes ectopically expressed in INPs in erm,ham double heterozygous brains (Figure 5C). In these brains, we reproducibly observed small cells expressing Tll::GFP and Deadpan (Dpn) but not Ase that were most likely supernumerary type II neuroblasts newly derived from INP reversion (Figure 5D). Furthermore, we found that one copy of the tll::GFP(BAC) transgene mildly enhanced the supernumerary neuroblast phenotype in erm,ham double heterozygous brains (15.4 ± 1.8 vs. 11.1 ± 1 type II neuroblasts per lobe; n = 12 or 11 brains, respectively) (Figure 5E). These data strongly support our model that Erm and Ham inactivate tll during INP commitment. We tested this model by overexpressing erm or ham in type II neuroblasts that carry one copy of the tll::GFP(Bac) transgene. We found that Tll::GFP expression in type II neuroblasts overexpressing Erm was mildly reduced prior to upregulation of Ase expression, and declined further following upregulation of Ase expression (Figure 5F, G and I). By contrast, Tll::GFP expression in type II neuroblasts overexpressing Ham was unaffected prior to upregulation of Ase expression, but rapidly declined following upregulation of Ase expression (Figure 5H and I). Together, these data strongly support our model that sequential inactivation by Erm and Ham during INP commitment renders tll refractory to activation in INPs.
Figure 5
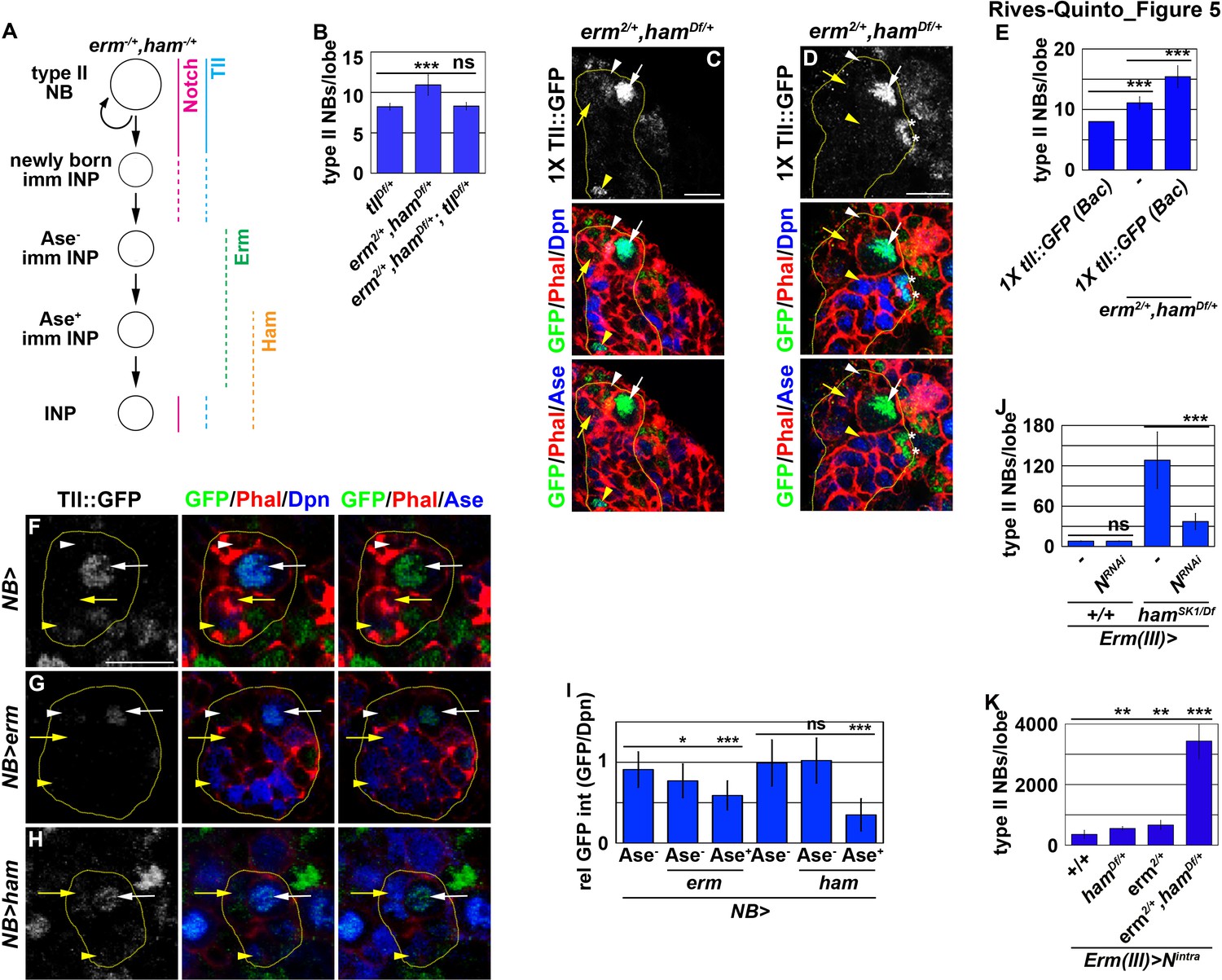
Erm- and Ham-mediated repression renders tll refractory for activation by Notch.
(A) A diagram depicting our hypothesis that ectopic activation of tll in INPs leads to supernumerary type II neuroblasts in erm,ham double heterozygous brains. (B) Quantification of total type II neuroblasts per brain lobe that was erm,ham double heterozygous or erm,ham,tll triple heterozygous. Heterozygosity of tll suppressed the supernumerary neuroblast phenotype in erm,ham double heterozygous brains. (C–D) Images of erm,ham double heterozygous brains that carries a tll::GFP(BAC) transgene. Tll::GFP becomes ectopically expressed in INPs and supernumerary type II neuroblasts (*) in erm,ham double heterozygous brains. (E) Quantification of total type II neuroblasts erm,ham double heterozygous brain lobe that carried one copy of the tll::GFP(BAC) transgene. One copy of the tll::GFP(BAC) transgene was sufficient to enhance the supernumerary type II neuroblast phenotype in erm,ham double heterozygous brains. (F–H) Images of type II neuroblasts that mis-expressed a UAS-erm or UAS-ham transgene driven by a pan-neuroblast Gal4. Erm or Ham mis-expression drastically reduced Tll::GFP expression in type II neuroblasts. (I) Quantification of Tll::GFP expression relative to Dpn expression in type II neuroblasts that mis-expressed a UAS-erm or UAS-ham transgene driven by a pan-neuroblast Gal4. Erm mis-expression reduced Tll::GFP expression in type II neuroblasts before the onset of Ase expression, whereas Ham mis-expression reduced Tll::GFP expression in type II neuroblasts after the onset of Ase expression. (J) Quantification of total type II neuroblasts per wild-type or hamSK1 homozygous brain lobe that overexpressed a UAS-NRNAi transgene driven by an INP Gal4. Knocking-down Notch function in INPs suppressed the supernumerary type II neuroblast phenotype in hamSK1 homozygous brains. (K) Quantification of total type II neuroblasts per erm or ham heterozygous brain lobe that overexpressed a UAS-Nintra transgene driven by an INP Gal4. Overexpressing Nintra in INPs more efficiently induced supernumerary neuroblasts in erm,ham double heterozygous brains than in erm or ham heterozygous brains. The following labeling is applicable to all panels of images in this figure: yellow dashed line encircles a type II neuroblast lineage; white arrow: type II neuroblast; white arrowhead: Ase- immature INP; yellow arrow: Ase+ immature INP; yellow arrowhead: INP; *: supernumerary type II neuroblasts. Scale bar, 10 μm. Bar graphs are represented as mean ± standard deviation. p-values: **<0.05, ***<0.005. ns: not significant.
-
Figure 5—source data 1
Quantification of total type II neuroblasts per brain lobe that was erm,ham double heterozygous or erm,ham,tll triple heterozygous.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig5-data1-v2.xlsx
-
Figure 5—source data 2
Quantification of total type II neuroblasts erm,ham double heterozygous brain lobe that carried one copy of the tll::GFP(BAC) transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig5-data2-v2.xlsx
-
Figure 5—source data 3
Quantification of Tll::GFP expression relative to Dpn expression in type II neuroblasts that mis-expressed a UAS-erm or UAS-ham transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig5-data3-v2.xlsx
-
Figure 5—source data 4
Quantification of total type II neuroblasts per wild-type or hamSK1 homozygous brain lobe that overexpressed a UAS-NRNAi transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig5-data4-v2.xlsx
-
Figure 5—source data 5
Quantification of total type II neuroblasts per erm or ham heterozygous brain lobe that overexpressed a UAS-Nintra transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig5-data5-v2.xlsx
Because tll is a putative target of Notch, we tested if inactivation of tll by Erm and Ham relinquishes the ability of Notch signaling to activate tll in INPs. We knocked-down Notch function in INPs in wild-type or hamSK1 homozygous brains. Knock-down of Notch function in INPs had no effect on type II neuroblasts in wild-type brains (7.9 ± 0.3 type II neuroblasts per lobe; n = 17 brains) (Figure 5J). However, in hamSK1 homozygous brains, knocking-down Notch function in INPs reduced the number of type II neuroblasts per lobe from 128 ± 39.6 to 36.4 ± 11.6 (n = 9 brains per genotype) (Figure 5J). This result indicates that re-activation of Notch signaling triggers INP reversion to supernumerary type II neuroblasts in ham-null brains. We extended our analyses to test if inactivation by Erm and Ham reduces the competency to respond to Notch signaling. Consistently, overexpressing constitutively activated Notch (Notchintra) induced a drastically more severe supernumerary neuroblast phenotype in erm,ham double heterozygous brains (3,437.6 ± 586.8 type II neuroblasts per lobe; n = 9 brains) than in erm or ham single heterozygous brains (687 ± 134.3 vs. 588.6 ± 77.6 type II neuroblasts per lobe; n = 9 or 11 brains, respectively) (Figure 5K). Thus, we propose that inactivation by Erm and Ham during INP commitment renders tll refractory to Notch signaling in INPs (Figure 5A).
Inactivation by Hdac3 relinquishes Notch’s ability to activate tll in INPs
To prevent Notch signaling from activating tll in INPs, Erm and Ham must function through chromatin-modifying proteins. We first tested if Erm and Ham inactivate tll by promoting sequential chromatin changes during INP commitment. We overexpressed full-length Ham or HamΔC-ZF driven by Erm-Gal4(III) in erm-null brains. erm-null brains alone contained 1824.6 ± 520.7 type II neuroblasts per lobe (n = 13 brains) (Figure 6A and Figure 6—figure supplement 1A). Overexpressing full-length Ham or HamΔC-ZF strongly suppressed the supernumerary neuroblast phenotype in erm-null brains (34.5 ± 8.3 vs. 64.6 ± 18.3 neuroblasts per lobe; n = 13 or 7 brains, respectively) (Figure 6A and Figure 6—figure supplement 1B). This result indicates that Ham over-expression can replace endogenous Erm function to suppress INP reversion, suggesting that these two transcriptional repressors function through an identical chromatin-modifying protein to inactivate tll during INP commitment.
Figure 6 with 1 supplement see all
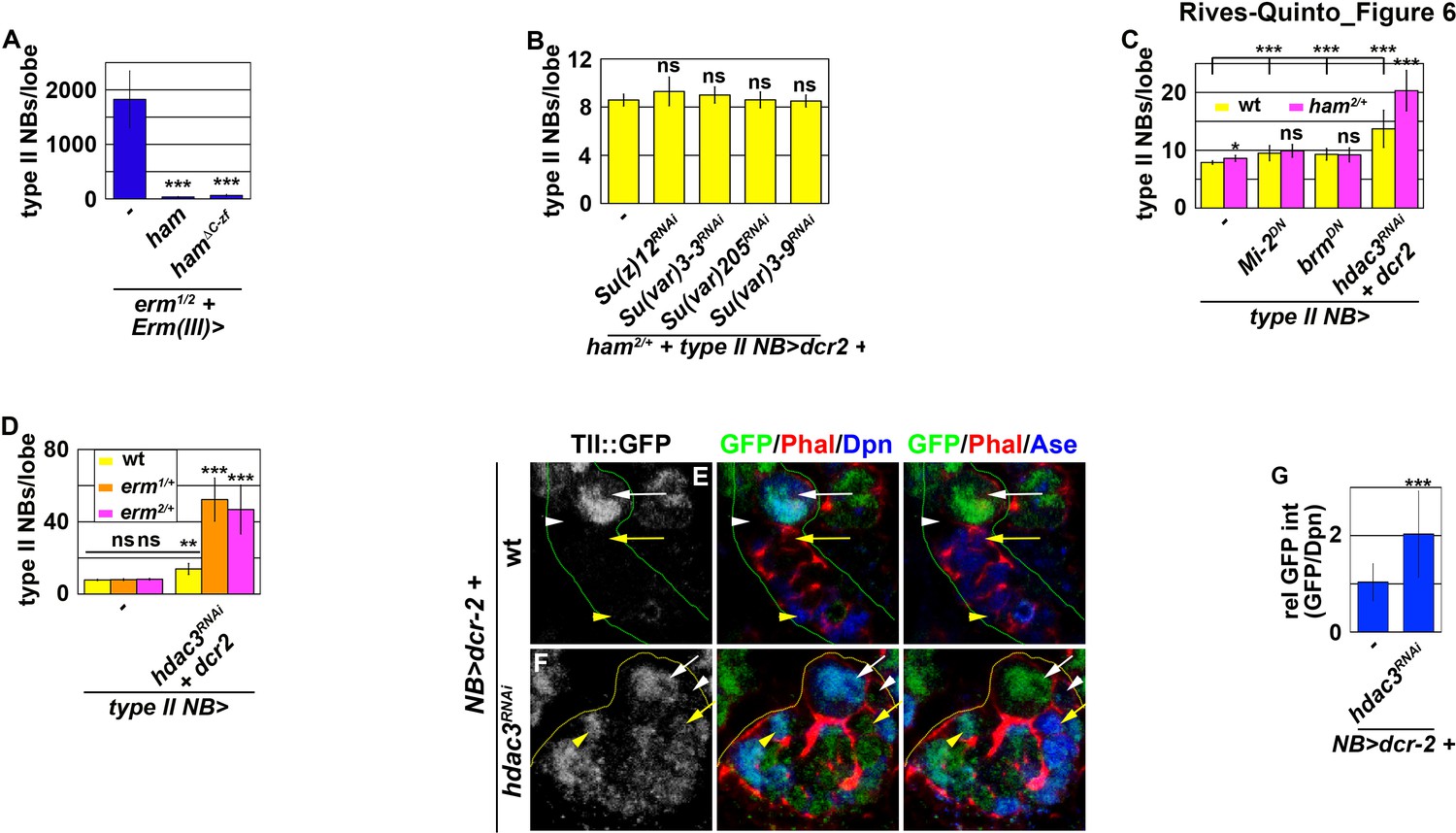
Erm and Ham function through Hdac3 to prevent INPs from reverting to type II neuroblasts.
(A) Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-ham transgene driven by an INP Gal4. Overexpressing full-length Ham or HamΔC-ZF in INPs suppressed the supernumerary type II neuroblast phenotype in erm-null brains. (B) Quantification of total type II neuroblasts per ham heterozygous brain lobe that overexpressed a UAS-RNAi transgene driven by a type II neuroblast Gal4. Reducing activity of the indicated chromatin complex did not increase INP reversion into supernumerary type II neuroblasts in ham heterozygous brains. (C) Quantification of total type II neuroblasts per ham heterozygous brain lobe that overexpressed a UAS transgene driven by a type II neuroblast Gal4. Reducing Hdac3 activity increased INP reversion into supernumerary type II neuroblasts in ham heterozygous brains. (D) Quantification of total type II neuroblasts per erm heterozygous brain lobe that overexpressed a UAS-hdac3RNAi transgene driven by a type II neuroblast Gal4. Reducing Hdac3 activity increased INP reversion into supernumerary type II neuroblasts in erm heterozygous brains. (E–F) Images of tll::GFP(Bac) brains that overexpressed a UAS-hdac3RNAi transgene driven by a pan-neuroblast Gal4. Reducing Hdac3 activity in type II neuroblasts led to ectopic Tll::GFP expression in immature INPs and INPs. (G) Quantification of Tll::GFP expression relative to Dpn expression in INPs derived from type II neuroblasts that overexpressed a UAS-hdac3RNAi transgene. Reducing Hdac3 activity in type II neuroblasts led to ectopic Tll::GFP expression in INPs. The following labeling is applicable to all panels of images in this figure: yellow dashed line encircles a type II neuroblast lineage; white arrow: type II neuroblast; white arrowhead: Ase- immature INP; yellow arrow: Ase+ immature INP; yellow arrowhead: INP. Bar graphs are represented as mean ± standard deviation. p-values: **<0.05, ***<0.005. ns: not significant.
-
Figure 6—source data 1
Quantification of total type II neuroblasts per brain lobe that overexpressed a UAS-ham transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig6-data1-v2.xlsx
-
Figure 6—source data 2
Quantification of total type II neuroblasts per ham heterozygous brain lobe that overexpressed various UAS transgenes.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig6-data2-v2.xlsx
-
Figure 6—source data 3
Quantification of total type II neuroblasts per erm heterozygous brain lobe that overexpressed a UAS-hdac3RNAi transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig6-data3-v2.xlsx
-
Figure 6—source data 4
Quantification of Tll::GFP expression relative to Dpn expression in INPs derived from type II neuroblasts that overexpressed a UAS-hdac3RNAi transgene.
- https://cdn.elifesciences.org/articles/56187/elife-56187-fig6-data4-v2.xlsx
We predicted that decreasing the activity of a chromatin-modifying protein required for Ham-mediated gene inactivation during INP commitment should enhance the supernumerary neuroblast phenotype in ham heterozygous brains. We knocked-down the function of genes known to contribute to the inactivation of gene transcription in ham heterozygous brains. Reducing the activity of PRC2 or heterochromatin protein 1, as well as decreasing the recruitment of Heterochromatin Protein 1, had no effect on the supernumerary neuroblast phenotype in ham heterozygous brains (Figure 6B). While reducing the activity of nucleosome remodelers alone led to a mild supernumerary type II neuroblast phenotype, it did not further enhance the supernumerary neuroblast phenotype in ham heterozygous brains (Figure 6C). By contrast, reducing the activity of histone deacetylase 3 (Hdac3) alone resulted in a mild supernumerary type II neuroblast phenotype and significantly enhanced the supernumerary neuroblast phenotype in ham heterozygous brains (Figure 6C). These results led us to conclude that Ham likely functions through Hdac3 to prevent INP reversion. We next tested if Erm also functions through Hdac3 to prevent INP reversion. erm heterozygous brains alone did not display a supernumerary type II neuroblast phenotype (Figure 6D). Knocking-down hdac3 function in erm heterozygous brains led to a more than three-fold increase in supernumerary type II neuroblasts as compared to wild-type brains (Figure 6D). These data strongly support a model that Hdac3 functions together with Erm and Ham to silence tll during INP commitment.
We tested if hdac3 is indeed required for silencing tll during INP commitment by overexpressing a UAS-hdac3RNAi transgene in type II neuroblasts. Consistent with our hypothesis, knock down of hdac3 function in type II neuroblasts and their progeny led to ectopic Tll::GFP expression in immature INPs and INPs (Figure 6E–G). These results confirmed that Hdac3 is required for inactivating tll during INP commitment. Thus, we conclude that Erm and Ham likely function through Hdac3 to prevent INP from reverting to supernumerary type II neuroblasts by continually inactivating tll.
Discussion
The expansion of OSVZ neural stem cells, which indirectly produce neurons by initially generating intermediate progenitors, drives the evolution of lissencephalic brains to gyrencephalic brains (Cárdenas and Borrell, 2020; Delaunay et al., 2017; Di Lullo and Kriegstein, 2017). Recent studies have revealed important insights into genes and cell biological changes that lead to the formation of OSVZ neural stem cells (Fujita et al., 2020; Namba et al., 2020). However, the mechanisms controlling the functional identity of OSVZ neural stem cells, including the competency to generate intermediate progenitors, remain unknown. In this study, we have provided compelling evidence demonstrating that Tll is necessary and sufficient for the maintenance of an undifferentiated state and the competency to generate intermediate progenitors in type II neuroblasts (Figure 7). We also showed that two sequentially activated transcriptional repressors, Erm and Ham, likely function through Hdac3 to silence tll during INP commitment ensuring normal indirect neurogenesis in larval brains. We propose that continual inactivation of stem cell functional identity genes by histone deacetylation allows intermediate progenitors to stably commit to generating sufficient and diverse differentiated cells during neurogenesis.
Figure 7
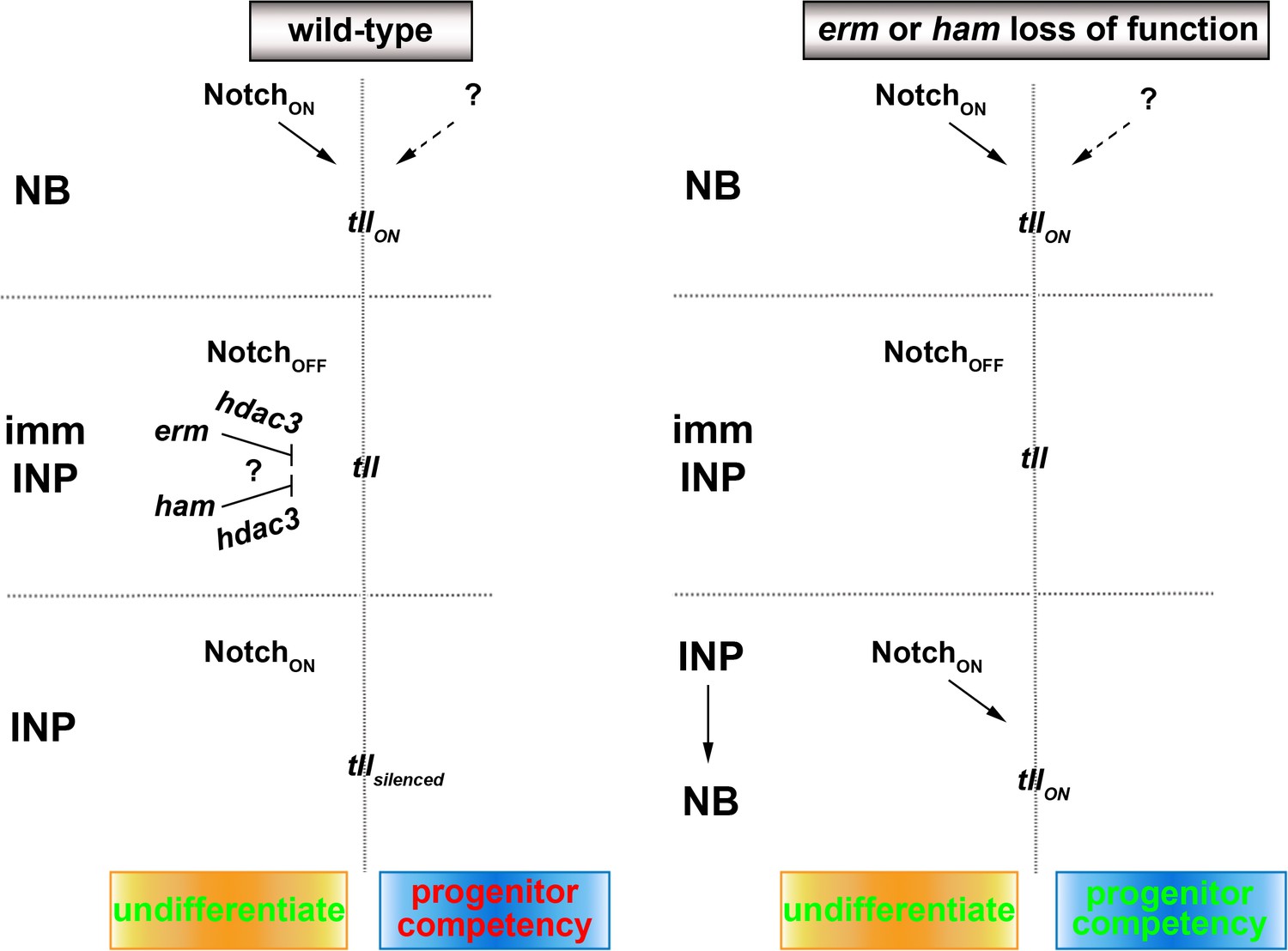
A proposed model for the regulation of type II neuroblast functionality.
We propose that Erm and Ham recruits Hdac3 to silence tll during INP commitment, preventing re-activation of Notch signaling in INPs from triggering Tll expression in wild-type brains. The tll locus remains in an activatable state in erm- or ham-null brains, and re-activation of Notch signaling in INPs triggers aberrant Tll expression driving INP reversion to supernumerary type II neuroblasts.
Regulation of the maintenance of an undifferentiated state and the competency to generate intermediate progenitors in neural stem cells
Stem cell functional identities encompass the maintenance of an undifferentiated state and other unique functional features, such as the competency to generate intermediate progenitors. Because genetic manipulation of Notch signaling perturbs the regulation of differentiation during asymmetric stem cell division (Imayoshi et al., 2010; Kageyama et al., 2008), the role of Notch in regulating other stem cell functions remains poorly understood. In the fly type II neuroblast lineage, overexpressing Notchintra or the downstream transcriptional repressors Dpn, E(spl)mγ, and Klumpfuss (Klu) induces the formation of supernumerary type II neuroblasts at the expense of generating immature INPs via the inhibition of erm activation (Janssens et al., 2017). These results indicate that the Notch-Dpn/E(spl)mγ/Klu axis provides an evolutionarily conserved mechanism to maintain neural stem cells in an undifferentiated state. Although overexpressing Notchintra but not Dpn, E(spl)mγ, and Klu in combination in INPs is sufficient to drive supernumerary type II neuroblast formation, Notchintra overexpression is not sufficient to transform a type I neuroblast into a type II neuroblast (data not presented). Thus, we propose that Notch functions as a general activator of genes expression in type II neuroblasts, and specific regulators of type II neuroblast functional identities must exist.
Our study strongly suggests that tll functions as a master regulator of type II neuroblast functional identities (Figure 7). Identical to Notch, tll is necessary for maintaining type II neuroblasts in an undifferentiated state and is sufficient to induce INP reversion into type II neuroblasts (Figure 2B–G; Hakes and Brand, 2020). Uniquely, high levels of Tll is sufficient to molecularly transform greater than 60% of type I neuroblasts in the ventral brain region into type II neuroblasts (Figure 2H–M). Brain regionalization leads to distinct degrees of sensitivity to Tll overexpression. For example, type I neuroblasts in the dorsal-anterior region of the brain are resistant to Tll-induced lineage transformation. By contrast, Tll overexpression can transform most, if not all, type I neuroblasts in the ventral-lateral and ventral-medial regions of the brain into type II neuroblasts. High levels of Tll expression in ventral-lateral type I neuroblasts leads to accumulation of mostly supernumerary type II neuroblasts and very few Erm::V5+ immature INPs. This result phenocopies Tll overexpression driven by strong drivers in the type II neuroblast lineages, and suggests that neuroblast progeny rapidly re-acquire a neuroblast identity instead of an immature INP identity (data not presented) (Hakes and Brand, 2020). Tll overexpression in ventral-medial type I neuroblasts leads to the formation of supernumerary type II neuroblasts interspersed with Erm::V5+ immature INPs, mimicking Tll overexpression driven by moderate drivers in the type II neuroblast lineages. We speculate that progeny of transformed type II neuroblasts in the ventral-medial region of the brain can assume an immature INP identity and then revert into supernumerary type II neuroblasts. These data strongly support a model that Tll is a potent activator of type II neuroblast functional identities.
A key question regarding the proposed function of Tll in regulating type II neuroblast functional identities is how it mechanistically links to genes previously shown to control these characteristics. ChIP-seq on fly embryonic nuclear extract using the Tll::GFP(Bac) transgenic protein identified hundreds of putative Tll target genes that include all previously characterized regulators of type II neuroblast functional identities (Davis et al., 2018). Tll binds dpn, E(spl)mγ and klu loci in embryos, suggesting that Tll likely regulates their expression. The phenotypic effects of loss- and gain-of-function of tll on type II neuroblasts mimic those of dpn, E(spl)mγ and klu. The vertebrate homolog of Tll, Tlx, has been shown to function as a transcriptional activator during neurogenesis (Sun et al., 2017). Thus, Tll might maintain type II neuroblasts in an undifferentiated state by promoting dpn, E(spl)mγ and klu expression. Tll also binds pntP1 and btd loci in embryos. Similar to Tll overexpression, mis-expression of PntP1 or Btd can transform type I neuroblasts in the ventral brain region into type II neuroblasts (Komori et al., 2014b; Xie et al., 2014; Zhu et al., 2011). Thus, it is plausible that tll functions through pntP1 or btd to regulate the competency to generate INPs in type II neuroblasts. These results strongly support our model that Tll is key component of the regulatory mechanism that endows type II neuroblasts with lineage-specific functional identities. Future experiments to validate the mechanistic links between tll and genes that regulate various lineage-specific functional characteristics will allow for the establishment of gene regulatory circuits that regulate type II neuroblast functional identities.
Successive transcriptional repressor activity inactivates stem cell functional identity genes during progenitor commitment
The identification of ham as a putative regulator of INP commitment was unexpected given that a previously published study concluded that Ham functions to limit INP proliferation (Eroglu et al., 2014). Ham is the fly homolog of Prdm16 in vertebrates and has been shown to play a key role in regulating cell fate decisions in multiple stem cell lineages (Baizabal et al., 2018; Harms et al., 2015; Moore et al., 2002; Shimada et al., 2017). Prdm16 contains two separately defined zinc-finger motifs, with each likely recognizing unique target genes. Prdm16 can also function through a variety of cofactors to activate or repress target gene expression, independent of its DNA-binding capacity. Thus, Ham can potentially inactivate stem cell functionality genes via one of several mechanisms. By using a combination of previously isolated alleles and new protein-null alleles, we demonstrated that the N-terminal zinc-finger motif is required for Ham function in immature INPs. Based on the overexpression of a series of chimeric proteins containing the N-terminal zinc-finger motif, our data indicate that Ham prevents INP reversion to supernumerary type II neuroblasts by recognizing target genes via the N-terminal zinc-finger motif and possibly repressing their transcription. Our results suggest that Ham prevents INPs from reverting to supernumerary type II neuroblasts by possibly repressing target gene transcription.
A key question raised by our study is why two transcriptional repressors that seemingly function in a redundant manner are required to prevent INPs from reverting to supernumerary type II neuroblasts. INP commitment lasts approximately 6–8 hr following the generation of an immature INP (Berger et al., 2012; Homem et al., 2014); after this time, the immature INP transitions into an INP. erm is poised for activation in type II neuroblasts and becomes rapidly activated in the newly generated immature INP less than 90 min after its generation (Janssens et al., 2017). As such, Erm-mediated transcriptional repression allows for the rapid inactivation of type II neuroblast functional identity genes. Because Erm expression rapidly declines in INPs when Notch signaling becomes reactivated, a second transcriptional repressor that becomes activated after Erm and whose expression is maintained throughout the life of an INP is required to continually inactivate type II neuroblast functional identity genes. Ham is an excellent candidate because it becomes expressed in immature INPs 3–4 hr after the onset of Erm expression and is detected in all INPs. Similar to Erm, Ham recognizes target genes and represses their transcription. Furthermore, ham functions synergistically with erm to prevent INP reversion to supernumerary type II neuroblasts, and overexpressed Ham can partially substitute for endogenous Erm. Thus, Erm- and Ham-mediated transcriptional repression renders type II neuroblast functional identity genes refractory to activation by Notch signaling throughout the lifespan of the INP, ensuring the generation of differentiated cell types rather than supernumerary type II neuroblasts instead.
Sustained inactivation of stem cell functional identity genes distinguishes intermediate progenitors from stem cells
Genes that specify stem cell functional identity become refractory to activation during differentiation, but the mechanisms that restrict their expression are poorly understood due to a lack of lineage information. Researchers have proposed several epigenetic regulator complexes that may restrict neural stem-cell-specific gene expression in neurons (Hirabayashi and Gotoh, 2010; Ronan et al., 2013). We knocked-down the function of genes that were implicated in restricting neural stem cell gene expression during differentiation in order to identify chromatin regulators that are required to inactivate type II neuroblast functional identity genes during INP commitment. Surprisingly, we found that only Hdac3 is required for both Erm- and Ham-mediated suppression of INP reversion to type II neuroblasts. Our finding is consistent with a recent study showing that blocking apoptosis in lineage clones derived from PRC2-mutant type II neuroblasts did not lead to supernumerary neuroblast formation (Abdusselamoglu et al., 2019). Our data strongly suggest that genes specifying type II neuroblast functional identity, such as tll, are likely silenced rather than decommissioned in INPs. This result is supported by the finding that overexpressing Notchintra but not Notch downstream transcriptional repressors in INPs can re-establish a type II neuroblast-like undifferentiated state. We speculate that continual histone deacetylation is required to counter the transcriptional activator activity of endogenous Notch and silence tll in INPs (Figure 7). By contrast, the chromatin in the tll locus might be close and inaccessible to the Notch transcriptional activator complex in type I neuroblasts; thus, overexpressing Notchintra cannot transform type I neuroblasts into type II neuroblasts. A key remaining question is what transcription factor is required to maintain the chromatin in the tll loci in an open state. Insights into regulation of the competency of the tll locus to respond to activated Notch signaling might improve our understanding of the molecular determinants of OSVZ neural stem cells.
Materials and methods
Fly genetics and transgenes
Request a detailed protocolFly crosses were carried out in 6-oz plastic bottles, and eggs were collected on apple caps in 8 hr intervals. Newly hatched larvae were genotyped and allowed to grow on corn meal caps. Larvae were shifted to 33°C for 72 hr io induce UAS-transgene expression for overexpression or knock down studies prior to dissection.
Larvae for MARCM analyses were genotyped at hatching and allowed to grow at 25°C for 24 hr. Larvae were then shifted to a 37°C water bath for 90 minto induce clones. Heat-shocked larvae were allowed to recover and grow at 25°C for 72 hr prior to dissection.
Transgenes were inserted into the pUAST-attB M{3xP3-RFP.attP}ZH-86Fb docking site using ΦC31 integrase-mediated transgenesis (Bischof and Basler, 2008). DNA injections were carried out by BestGene Inc or Genetivision Inc, and transgenic flies were identified in the F1 generation based on their red eye color.
Immunofluorescent staining and antibodies
Request a detailed protocolLarval brains were dissected in PBS and fixed in 100 mM PIPES (pH 6.9), 1 mM EGTA, 0.3% Triton X-100, and 1 mM MgSO4 containing 4% formaldehyde for 23 min. Fixed brain samples were washed with PBST (1XPBS and 0.3% Triton X-100). After removing the fix solution, samples were incubated with primary antibodies for 3 hr at room temperature. Three hours later, samples were washed with PBST and then incubated with secondary antibodies overnight at 4°C. On the next day, samples were washed with PBST and then equilibrated in ProLong Gold antifade mountant (ThermoFisher Scientific). The confocal images were acquired on a Leica SP5 scanning confocal microscope (Leica Microsystems, Inc). More than 10 brains per genotype were used to obtain data in each experiment.
RNA extraction and qRT-PCR
Request a detailed protocolTotal RNA was extracted from control or treated brat11/Df(2L)Exel8040 larvae carrying 2xTub-Gal80ts, Wor-Gal4 and UAS-insb transgenes using TRIzol (ThermoFisher Scientific) and mRNA was purified using the RNeasy Micro Kit (Qiagen) according to the manufacturer’s protocol. First strand cDNA was synthesized using a 1st Strand cDNA Synthesis Kit for RT-PCR [AMV] (Roche) according to the manufacturer’s protocol. cDNA is amplified by using gene-specific primers. qRT-PCR was performed using ABsolute QPCR SYBR Green ROX Mix (ThermoFisher Sientific). Data were analyzed by comparative CT method, and the relative mRNA expression was presented.
Sample preparation and RNA sequencing
Request a detailed protocolControl or treated brat11/Df(2L)Exel8040 larvae carrying 2xTub-Gal80ts, Wor-Gal4 and UAS-insb transgenes were cultured in corn meal caps. One third of the larvae (18 hr Insb OE sample) were allowed to grow at 25°C for 102 hr and then transferred to 33°C to induce transient Insb overexpression for 18 hr. One-third of the larvae (9 hr Insb OE sample) were allowed to grow at 25°C for 111 hr and then transferred to 33°C to induce transient Insb overexpression for 9 hr. The remaining third of the larvae (0 hr Insb OE) were allowed to grow at 25°C for 120 hr. Fifty brains from each group were isolated on the same day, and immediately processed for RNA extraction. Biological triplicate brain samples were collected. Paired-end Poly-A-mRNA libraries were generated to be assayed by Illumina HiSeq 4000. Differential genes expression was based on absolute linear fold change >or equal 1.5.
Bioinformatics: RNA-seq analysis
Request a detailed protocolBiological triplicate brain samples for each time point were sequenced using Illumina HiSeq 4000. The samples had around 46, 45 and 44 million reads. Quality of the raw reads for each sample was checked using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). We align the reads to the dm6 reference genome including mRNAs (http://www.genome.ucsu.edu/) with HISAT2 (version 2.1.0) and default parameters. Alignment results were validated through a second round of FASTQ followed by quantification and analysis. For differential expression analysis, Stringtie (version 1.3.4) quantifies expression at the gene and isoforms levels and DESeq2 (version 1.20.0) performs differential expression testing. We compared transcriptional profile between 0 hr vs 9 hr, 0 hr vs 18 hr, and 9 hr vs 18 hr. We identified genes and transcripts as being differentially expressed based on absolute linear fold change >or equal 1.5.
Quantification and statistical analysis
Request a detailed protocolThe Image J software was used to quantify the number of cells of interest. Dpn single-channel confocal images were used to assign the area the type II Neuroblasts or INP nucleus. All biological replicates were independently collected and processed. All statistical analyses were performed using a two-tailed Student's T-test, a p-value<0.05,<0.005, and<0.0005 were indicated by (*), (**) and (***), respectively in figures.
Appendix 1
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | anti-GFP (Chicken polyclonal) | Aves Labs, INC. | Cat#GFP-1020, RRID:AB_2307313 | IF(1:2000) |
| Antibody | anti-V5 (Mouse monoclonal) | ThermoFisher Scientific | Cat#R960-25, RRID:AB_2556564 | IF(1:500) |
| Antibody | anti-Ase (Rabbit polyclonal) | Weng et al., 2010 doi: 10.1016/j.devcel.2009.12.007. | IF(1:400) | |
| Antibody | anti-Hamlet (Rabbit polyclonal) | Eroglu et al., 2014 doi: 10.1016/j.cell.2014.01.053. | IF(1:50) | |
| Antibody | anti-Dpn (Rat monoclonal) | Lee et al., 2006a doi: 10.1038/nature04299. | clone 11D1BC7.14 | IF(1:2) |
| Antibody | Alexa Fluor 488 AffiniPure Anti-Chicken IgY (IgG) (H+L) (Donkey polyclonal) | Jackson Immuno Research Laboratories, INC. | Cat#703-545-155, RRID:AB_2340375 | IF(1:500) |
| Antibody | Alexa Fluor 647 AffiniPure anti-Rat IgG (H+L) (Goat polyclonal) | Jackson Immuno Research Laboratories, INC. | Cat#112-605-167 RRID:AB_2338404 | IF(1:500) |
| Antibody | anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Goat polyclonal) | ThermoFisher Scientific | Cat#A-11029, RRID:AB_2534088 | IF(1:500) |
| Antibody | anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Goat polyclonal) | ThermoFisher Scientific | Cat#A-11034, RRID:AB_2576217 | IF(1:500) |
| Antibody | anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 546 (Goat polyclonal) | ThermoFisher Scientific | Cat#A-11035, RRID:AB_2534093 | IF(1:500) |
| Other | Rhodamine Phalloidin | ThermoFisher Scientific | Cat#R415 | IF(1:100) |
| Genetic reagent (D. melanogaster) | brat11/CyO, Actin-GFP | Lee et al., 2006b doi: 10.1016/j.devcel.2006.01.017. | ||
| Genetic reagent (D. melanogaster) | w1118; Df(2L)Exel8040/CyO | Bloomington Drosophila Stock Center | BDSC: 7847 FlyBase: FBst0007847; RRID:BDSC_7847 | FlyBase symbol: Df(2L)Exel8040/CyO |
| Genetic reagent (D. melanogaster) | y1, M{vas-int.Dm}ZH-2A w*; M{UAS-insbFL-myc}ZH-86Fb | Komori et al., 2018 doi: 10.1101/gad.320333.118. | ||
| Genetic reagent (D. melanogaster) | Wor-Gal4(II) | Lee et al., 2006a doi: 10.1038/nature04299. | ||
| Genetic reagent (D. melanogaster) | Wor-Gal4(III) | Weng et al., 2010 doi: 10.1016/j.devcel.2009.12.007. | ||
| Genetic reagent (D. melanogaster) | y1, w*; P{tubPGAL80} LL10, P{neoFRT}40A/CyO | Bloomington Drosophila Stock Center | BDSC: 5192 FlyBase: FBst0005192; RRID:BDSC_5192 | FlyBase symbol: y1, w*; P{tubPGAL80} LL10, P{neoFRT}40A/CyO |
| Genetic reagent (D. melanogaster) | P{hsFLP}1, P{tubP-GAL80}LL1, w*, P{neoFRT}19A; P{UAS-mCD8::GFP.L}LL5 | Bloomington Drosophila Stock Center | BDSC: 5134 FlyBase: FBst0005134; RRID:BDSC_5134_ | FlyBase symbol: P{hsFLP}1, P{tubP-GAL80}LL1, w*, P{neoFRT}19A; P{UAS-mCD8::GFP.L}LL5 |
| Genetic reagent (D. melanogaster) | y1 w*; PBac{y[+mDint2] w[+mC]=tll EGFP.S}VK00037 | Bloomington Drosophila Stock Center | BDSC: 30874 FlyBase: FBst0030874; RRID:BDSC_30874 | FlyBase symbol: y1 w*; PBac{y[+mDint2] w[+mC]=tll EGFP.S}VK00037 |
| Genetic reagent (D. melanogaster) | Erm-Gal4 (II) | Pfeiffer et al., 2008 doi: 10.1073/pnas.0803697105. | ||
| Genetic reagent (D. melanogaster) | Erm-Gal4 (III) | Pfeiffer et al., 2008 doi: 10.1073/pnas.0803697105. | ||
| Genetic reagent (D. melanogaster) | tllRNAi: y1 sc* v1 sev21; P{TRiP.HMS01316}attP2 | Bloomington Drosophila Stock Center | BDSC: 34329 FlyBase: FBst0034329; RRID:BDSC_34329 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.HMS01316}attP2 |
| Genetic reagent (D. melanogaster) | M{UAS-tll.ORF-VN}ZH-86Fb | FlyORF | F004752 FBst0502964; RRID:FlyORF_ F004752 | FlyBase symbol: M{UAS-tll.ORF-VN}ZH-86Fb |
| Genetic reagent (D. melanogaster) | Ase-Gal80 (II) | Neumüller et al., 2011 doi: 10.1016/j.stem.2011.02.022. | ||
| Genetic reagent (D. melanogaster) | erm1/CyO, Act-GFP | Weng et al., 2010 doi: 10.1016/j.devcel.2009.12.007. | ||
| Genetic reagent (D. melanogaster) | erm2/CyO, Act-GFP | Weng et al., 2010 doi: 10.1016/j.devcel.2009.12.007. | ||
| Genetic reagent (D. melanogaster) | UAS-erm | Weng et al., 2010 doi: 10.1016/j.devcel.2009.12.007. | ||
| Genetic reagent (D. melanogaster) | PBac{erm-flag4C(g)}VK33 | Janssens and Lee, 2014 doi: 10.1242/dev.106534. | ||
| Genetic reagent (D. melanogaster) | DRNAi: y1 v1; P{TRiP.JF02115}attP2 | Bloomington Drosophila Stock Center | BDSC: 26217 FlyBase: FBst0026217; RRID:BDSC_26217 | FlyBase symbol: y1 v1; P{TRiP.JF02115}attP2 |
| Genetic reagent (D. melanogaster) | AseRNAi: y1 sc* v1 sev21; P{TRiP.HMS02847}attP2 | Bloomington Drosophila Stock Center | BDSC: 44552 FlyBase: FBst0044552; RRID:BDSC_44552 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.HMS02847}attP2 |
| Genetic reagent (D. melanogaster) | hamRNAi: y1 v1; P{TRiP.JF02270}attP2 | Bloomington Drosophila Stock Center | BDSC: 26728 FlyBase: FBst0026728; RRID:BDSC_26728 | FlyBase symbol: y1 v1; P{TRiP.JF02270}attP2 |
| Genetic reagent (D. melanogaster) | hamRNAi: y1 sc* v1 sev21; P{y[+t7.7] v[+t1.8]=TRiP.HMS00470}attP2 | Bloomington Drosophila Stock Center | BDSC: 32470 FlyBase: FBst0032470; RRID:BDSC_32470 | FlyBase symbol: y1 sc* v1 sev21; P{y[+t7.7] v[+t1.8]=TRiP.HMS00470}attP2 |
| Genetic reagent (D. melanogaster) | OpaRNAi: y1 sc* v1 sev21; P{TRiP.HMS01185}attP2/TM3, Sb1 | Bloomington Drosophila Stock Center | BDSC: 34706 FlyBase: FBst0034706; RRID:BDSC_34706 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.HMS01185}attP2/TM3, Sb1 |
| Genetic reagent (D. melanogaster) | w1118; Df(2L)Exel7071/CyO | Bloomington Drosophila Stock Center | BDSC: 7843 FlyBase: FBst0007843; RRID:BDSC_7843 | FlyBase symbol: w1118; Df(2L)Exel7071/CyO |
| Genetic reagent (D. melanogaster) | hamSKI, FRT40A/CyO | This paper | A new hamlet mutant fly line | |
| Genetic reagent (D. melanogaster) | P{w[+mW.hs]=GawB}elav[C155], P{w[+mC]=UAS-mCD8::GFP.L}Ptp4E[LL4], P{ry[+t7.2]=hsFLP}1, w[*] | Bloomington Drosophila Stock Center | BDSC: 5146 FlyBase: FBst0005146; RRID:BDSC_5146 | FlyBase symbol: P{w[+mW.hs]=GawB}elav[C155], P{w[+mC]=UAS-mCD8::GFP.L}Ptp4E[LL4], P{ry[+t7.2]=hsFLP}1, w[*] |
| Genetic reagent (D. melanogaster) | w1118; P{w[+mC]=UAS-Dcr-2.D}2 | Bloomington Drosophila Stock Center | BDSC: 24650 FlyBase: FBst00024650; RRID:BDSC_24650 | FlyBase symbol: w1118; P{w[+mC]=UAS-Dcr-2.D}2 |
| Genetic reagent (D. melanogaster) | w*; P{w[+mC]=tubP- GAL8ts}2/TM2 | Bloomington Drosophila Stock Center | BDSC: 7017 FlyBase: FBst00024650; RRID:BDSC_7017 | FlyBase symbol: w*; P{w[+mC]=tubP- GAL8ts}2/TM2 |
| Genetic reagent (D. melanogaster) | ham1,FRT40A/Cyo | Moore et al., 2002 doi: 10.1126/science.1072387. | ||
| Genetic reagent (D. melanogaster) | UAS-ham | Moore et al., 2002 doi: 10.1126/science.1072387. | ||
| Genetic reagent (D. melanogaster) | RNAi of Notch: y1, v1; P{y+t7.7v+t1.8=TRiP.HMS00001}attP2 | Bloomington Drosophila Stock Center | BDSC: 33611 FlyBase: FBst0033611; RRID:BDSC_33611 | FlyBase symbol: y1, v1; P{y+t7.7v+t1.8=TRiP.HMS00001}attP2 |
| Genetic reagent (D. melanogaster) | Oregon-R-C | Bloomington Drosophila Stock Center | BDSC: 5 FlyBase: FBst0000005; RRID:BDSC_5 | FlyBase symbol: Oregon-R-C |
| Genetic reagent (D. melanogaster) | P{hsFLP}1, y1 w*; P{UAS- N.intra.GS}2/CyO; MKRS/TM2 | Bloomington Drosophila Stock Center | BDSC: 52008 FlyBase: FBst0052008; RRID:BDSC_52008 | FlyBase symbol: P{hsFLP}1, y1 w*; P{UAS- N.intra.GS}2/CyO; MKRS/TM2 |
| Genetic reagent (D. melanogaster) | y1; M{vas-int.Dm}ZH-2A w*; M{UAS-ham∆C-ZF-myc}ZH-86Fb | This paper | Transgene expressing Hamlet mutant form of the C-terminal zinc finger deletion version | |
| Genetic reagent (D. melanogaster) | y1; M{vas-int.Dm}ZH-2A w*; M{UAS-ERD::hamN-ZF-myc}ZH-86Fb | This paper | Transgene expressing Hamlet the N-terminal zinc finger fused with ERD transcriptional repression domain | |
| Genetic reagent (D. melanogaster) | y1; M{vas-int.Dm}ZH-2A w*; M{UAS-VP-16::hamN-ZF-myc}ZH-86Fb | This paper | Transgene expressing Hamlet the N-terminal zinc finger fused with VP16 transcriptional activatoin domain | |
| Genetic reagent (D. melanogaster) | cu1, tll49/TM3, P{ftz/lacC}SC1, Sb1, Ser1 | Bloomington Drosophila Stock Center | BDSC: 7093 FlyBase: FBst007093; RRID:BDSC_7093 | FlyBase symbol: cu1, tll49/TM3, P{ftz/lacC}SC1, Sb1, Ser1 |
| Genetic reagent (D. melanogaster) | st1 e1 tll1/TM3, Sb1 | Bloomington Drosophila Stock Center | BDSC: 2729 FlyBase: FBst002729; RRID:BDSC_2729 | FlyBase symbol: st1 e1 tll1/TM3, Sb1 |
| Genetic reagent (D. melanogaster) | hamSK4, FRT40A/CyO | Bloomington Drosophila Stock Center | BDSC: 34329FlyBase: FBst0034329;RRID:BDSC_34329 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.HMS01316}attP2 |
| Genetic reagent (D. melanogaster) | hdac3RNAi: y1 sc* v1 sev21; P{TRiP.HMS00087}attP2 | Bloomington Drosophila Stock Center | BDSC: 34778 FlyBase: FBst0034778; RRID:BDSC_34778 | FlyBase symbol: hdac3RNAi: y1 sc* v1sev21; P{TRiP.HMS00087}attP2 |
| Genetic reagent (D. melanogaster) | Su(z)12RNAi: y1 sc* v1 sev21; P{TRiP.HMS00280}attP2/TM3, Sb1 | Bloomington Drosophila Stock Center | BDSC: 33402 FlyBase: FBst0033402; RRID:BDSC_33402 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.HMS00280}attP2/TM3, Sb1 |
| Genetic reagent (D. melanogaster) | Su(var)3-3RNAi: y1 sc* v1 sev21; P{TRiP.HMS00638}attP2 | Bloomington Drosophila Stock Center | BDSC: 32853 FlyBase: FBst0032853; RRID:BDSC_32853 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.HMS00638}attP2 |
| Genetic reagent (D. melanogaster) | Su(var)205RNAi: y1 sc* v1 sev21; P{TRiP.GL00531}attP40 | Bloomington Drosophila Stock Center | BDSC: 36792 FlyBase: FBti0146447; RRID:BDSC_36792 | FlyBase symbol: y1 sc* v1 sev21; P{TRiP.GL00531}attP40 |
| Genetic reagent (D. melanogaster) | UAS-Mi-2DN | Kovač et al., 2018 doi: 10.1038/s41467-018-04503-2. | ||
| Genetic reagent (D. melanogaster) | UAS-Mi-brmDN | Herr et al., 2010 doi: 10.1016/j.ydbio.2010.04.006. | ||
| Genetic reagent (D. melanogaster) | In(1)wm4; Su(var)3–91/TM3, Sb1 Ser1 | Bloomington Drosophila Stock Center | BDSC: 6209 FlyBase: FBst0006209; RRID:BDSC_6209 | FlyBase symbol: In(1)wm4; Su(var)3–91/TM3, Sb1 Ser1 |
| Genetic reagent (D. melanogaster) | w1118; PBac{Sp1- EGFP.S}VK00033 | Bloomington Drosophila Stock Center | BDSC: 38669 FlyBase: FBst0038669; RRID:BDSC_38669 | FlyBase symbol: w1118; PBac{Sp1- EGFP.S}VK00033 |
| Sequenced-based reagent | Ham_F | Eroglu et al., 2014 doi: 10.1016/j.cell.2014.01.053. | PCR primers | atagatcctttggccagcagac |
| Sequenced-based reagent | Ham_R | Eroglu et al., 2014 doi: 10.1016/j.cell.2014.01.053. | PCR primers | agtactcctccctttcggcaat |
| Sequenced-based reagent | Ase_F | Komori et al., 2014b doi: 10.7554/eLife.03502. | PCR primers | agcccgtgagcttctacgac |
| Sequenced-based reagent | Ase_R | Komori et al., 2014b doi: 10.7554/eLife.03502. | PCR primers | gcatcgatcatgctctcgtc |
| Sequenced-based reagent | D_F | This paper | PCR primers | gcggcggcggtcaacaat |
| Sequenced-based reagent | D_R | This paper | PCR primers | tgcggcgtacagcgaagggt |
| Sequenced-based reagent | Erm_F | Eroglu et al., 2014 doi: 10.1016/j.cell.2014.01.053. | PCR primers | gttacggccaggcatcgggtcaa |
| Sequenced-based reagent | Erm_R | Eroglu et al., 2014 doi: 10.1016/j.cell.2014.01.053. | PCR primers | gggccaggcgggattactcgtctc |
| Sequenced-based reagent | PntP1_F | Komori et al., 2014b doi: 10.7554/eLife.03502. | PCR primers | ggcagtacgggcagcaccac |
| Sequenced-based reagent | PntP1_R | Komori et al., 2014b doi: 10.7554/eLife.03502. | PCR primers | ctcaacgcccccaccagatt |
| Sequenced-based reagent | Dpn_F | Komori et al., 2014b doi: 10.7554/eLife.03502.Komori et al., 2014b | PCR primers | catcatgccgaacacaggtt |
| Sequenced-based reagent | Dpn_R | Komori et al., 2014b | PCR primers | gaagattggccggaactgag |
| Recombinant DNA reagent | pUAST-ham∆C-ZF-myc-attB (plasmid) | This paper | Plasmid DNA of a transgene expressing Hamlet mutant form of the C-terminal zinc finger deletion version | |
| Recombinant DNA reagent | pUAST-ERD::hamN-ZF-myc-attB (plasmid) | This paper | Plasmid DNA of a transgene expressing Hamlet the N-terminal zinc finger fused with ERD transcriptional repression domain | |
| Recombinant DNA reagent | pUAST-VP16::hamN-ZF-myc-attB (plasmid) | This paper | Plasmid DNA of a transgene expressing Hamlet the N-terminal zinc finger fused with VP16 transcriptional activatoin domain | |
| Software, algorithm | LAS AF | Leica Microsystems | RRID:SCR_013673 | |
| Software, algorithm | ImageJ 1.50 g | National Institute of Health | RRID:SCR_003070 |
Data availability
Sequencing data have been deposited in GEO under accession codes GSE152636.
-
NCBI Gene Expression OmnibusID GSE152636. Sequential activation of transcriptional repressors promotes progenitor commitment by silencing stem cell identity genes.
References
-
Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cellsDevelopmental Neurobiology 68:1185–1195.https://doi.org/10.1002/dneu.20648
-
Molecular and cellular evolution of corticogenesis in amniotesCellular and Molecular Life Sciences 77:1435–1460.https://doi.org/10.1007/s00018-019-03315-x
-
The encyclopedia of DNA elements (ENCODE): data portal updateNucleic Acids Research 46:D794–D801.https://doi.org/10.1093/nar/gkx1081
-
Division modes and physical asymmetry in cerebral cortex progenitorsCurrent Opinion in Neurobiology 42:75–83.https://doi.org/10.1016/j.conb.2016.11.009
-
The use of brain organoids to investigate neural development and diseaseNature Reviews Neuroscience 18:573–584.https://doi.org/10.1038/nrn.2017.107
-
Epigenetic control of neural precursor cell fate during developmentNature Reviews Neuroscience 11:377–388.https://doi.org/10.1038/nrn2810
-
Proliferation control in neural stem and progenitor cellsNature Reviews Neuroscience 16:647–659.https://doi.org/10.1038/nrn4021
-
It takes two to tango, a dance between the cells of origin and Cancer stem cells in the Drosophila larval brainSeminars in Cell & Developmental Biology 28:63–69.https://doi.org/10.1016/j.semcdb.2014.03.006
-
Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibitionNature Neuroscience 11:1247–1251.https://doi.org/10.1038/nn.2208
-
Zinc finger protein too few controls the development of monoaminergic neuronsNature Neuroscience 6:28–33.https://doi.org/10.1038/nn979
-
From neural development to cognition: unexpected roles for chromatinNature Reviews Genetics 14:347–359.https://doi.org/10.1038/nrg3413
-
Nuclear receptor TLX in development and diseasesCurrent Topics in Developmental Biology 125:257–273.https://doi.org/10.1016/bs.ctdb.2016.12.003
-
Diverse gene regulatory mechanisms mediated by polycomb group proteins during neural developmentCurrent Opinion in Neurobiology 59:164–173.https://doi.org/10.1016/j.conb.2019.07.003
Article and author information
Author details
Funding
National Institute of Neurological Disorders and Stroke (R01NS107496)
- Hideyuki Komori
- Cheng-Yu Lee
National Institute of Neurological Disorders and Stroke (R01NS111647)
- Hideyuki Komori
- Cheng-Yu Lee
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Drs. E Lai and J Knoblich for providing us with reagents. We thank the Advance Genomics Core and Bioinformatics Core for technical assistance. We thank Dr. Melissa Harrison, Ms. Elizabeth Lawson, and members of the Lee lab for helpful discussions, and Science Editors Network for editing the manuscript. We thank the Bloomington Drosophila Stock Center, FlyORF, and the Vienna Drosophila RNAi Center for fly stocks. We thank BestGene Inc and GenetiVision for generating the transgenic fly lines. This work is supported by NIH grants R01NS107496 and R01NS111647.
Copyright
© 2020, Rives-Quinto et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,776
- views
-
- 260
- downloads
-
- 27
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 27
- citations for umbrella DOI https://doi.org/10.7554/eLife.56187
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Sequential activation of transcriptional repressors promotes progenitor commitment by silencing stem cell identity genes
eLife 9:e56187.
https://doi.org/10.7554/eLife.56187




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}