A new role for histone demethylases in the maintenance of plant genome integrity
- School of Life Science, University of Warwick, United Kingdom
- Université Paris-Saclay, CNRS, INRAE, Univ Evry, Institute of Plant Sciences Paris-Saclay (IPS2), France
- Graduate School of Agriculture, Kyoto University, Kitashirakawa Oiwake-cho, Sakyo-ku, Japan
- Department of Molecular Biology, Max Planck Institute for Developmental Biology, Germany
- Department of Molecular Genetics, The Ohio State University, United States
- Institute of Transformative Bio-Molecules, Nagoya University, Japan
- Division of Biological Science, Graduate School of Science, Nagoya University, Japan
- Donald Danforth Plant Science Center, United States
- Division of Biological Sciences, University of Missouri, United States
- Université de Paris, Institute of Plant Sciences Paris-Saclay (IPS2), F-75006, France
Figures
Figure 1 with 6 supplements
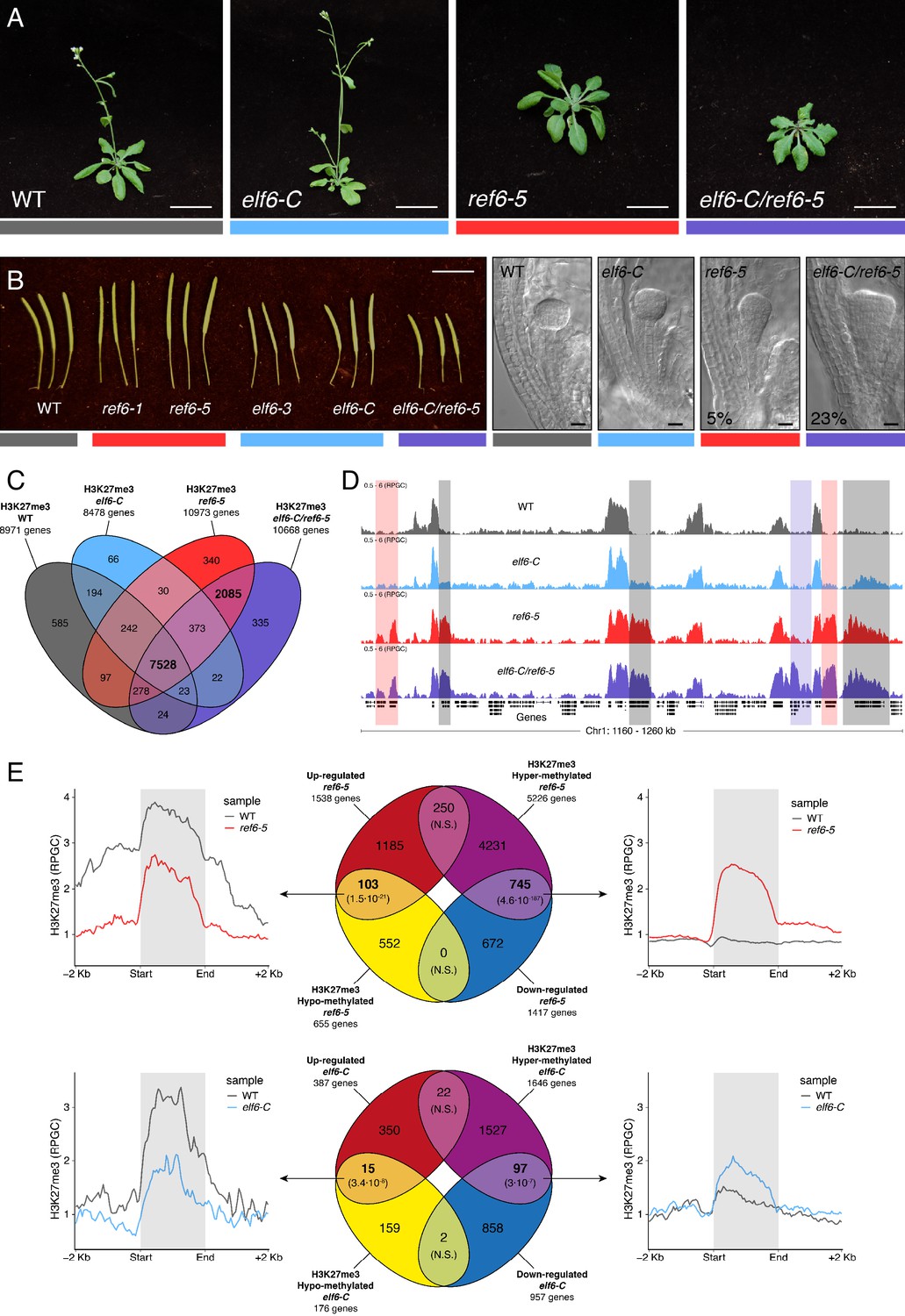
Arabidopsis Histone demethylases ELF6 and REF6 play distinct roles in development and H3K27me3 homeostasis.
(A) Arabidopsis wild-type (WT) and histone demethylase mutants (elf6-C, ref6-5 and elf6-C/ref6-5). Scale bars, 1 cm. (B) Siliques and embryos from Arabidopsis wild-type (WT) and different mutant alleles of histone demethylase ELF6 and REF6. Numbers show the frequency of the abnormal embryos (n = 250). Scale bars 1 cm and 10 μm, respectively. (C) Venn diagram showing the overlap between genes accumulating H3K27me3 in wild-type (WT) and histone demethylase mutants (elf6-C, ref6-5 and elf6-C/ref6-5). (D) Genome browser views of background subtracted ChIP-seq signals as normalized reads per genomic content (RPGC). Shaded red boxes, genes targeted exclusively by REF6. Shaded grey boxes, genes targeted by REF6 and ELF6. Shaded purple boxes, genes targeted by both REF6 and ELF6, and only hyper-methylated in double mutant elf6-C/ref6-5. (E) Venn diagram showing overlap between differentially expressed genes (DEGs) and H3K27me3 differentially methylated genes in histone demethylase mutants. To the left metaplot for H3K27me3 levels for genes both up-regulated and hypo-methylated and to the right metaplot of H3K27me3 levels in genes both down-regulated and hyper-methylated. Top panel, ref6-5; Bottom panel, elf6-C. p-values for Fisher’s exact test are shown in brackets, N.S. Not Significant.
Figure 1—figure supplement 1
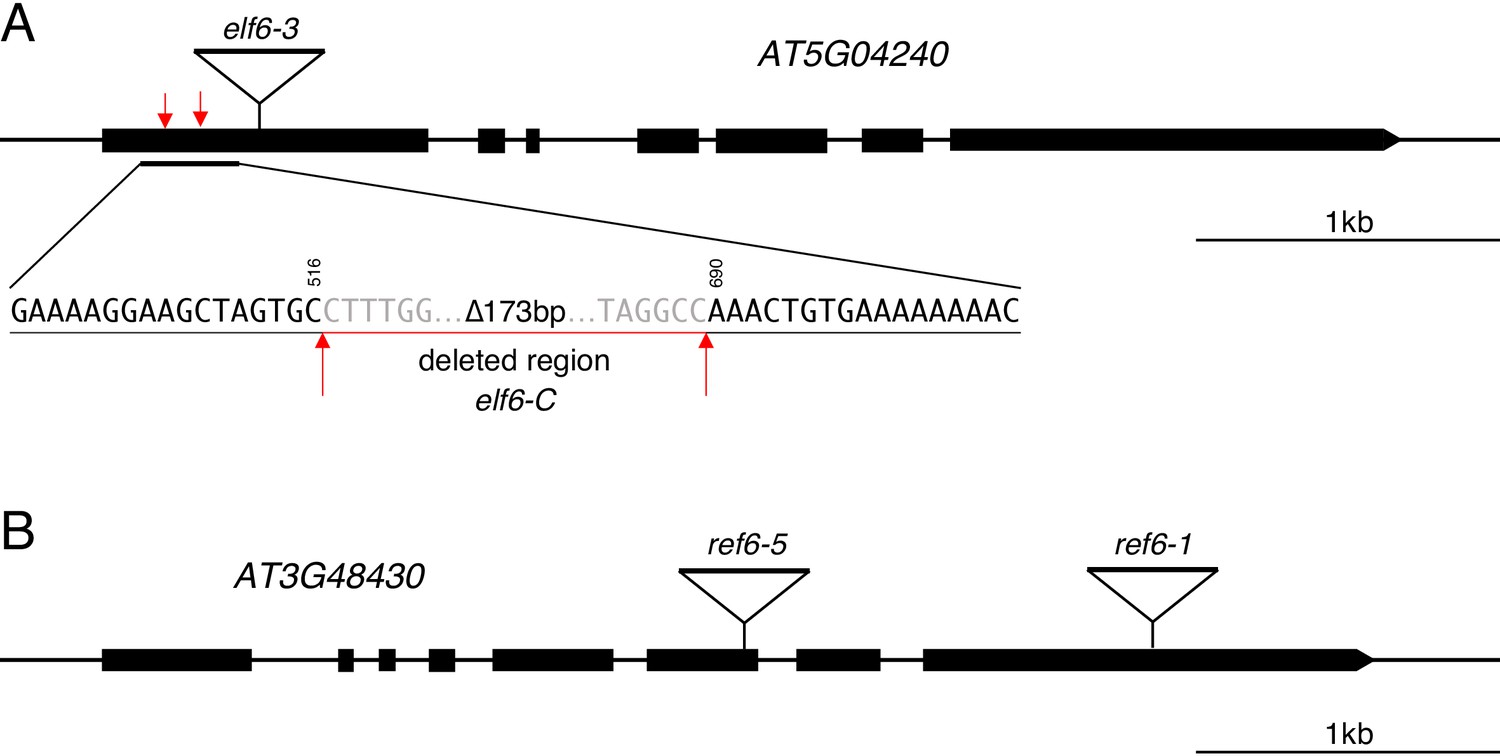
Graphical representation of mutant alleles for ELF6 and REF6 used in this study.
(A) Schematic diagram of ELF6 locus showing the location of different mutant alleles: elf6-3 is a T-DNA insertion (SALK_074694) in the first exon and elf6-C is a CRISPR/Cas9 deletion in the first exon that leads to an early stop codon. Black boxes represent exons, triangle shows the insertion site for elf6-3 and red arrows mark the start and end of the deleted region in elf6-C. Scale bar, 1 kb. (B) Schematic diagram of REF6 locus showing the location of different mutant alleles: ref6-5 (GABI_70E03) and ref6-1 (SALK_001018) are T-DNA insertions in the sixth and last exon, respectively. Black boxes represent exons and triangles shows the T-DNA insertion sites. Scale bar, 1 kb.
Figure 1—figure supplement 2
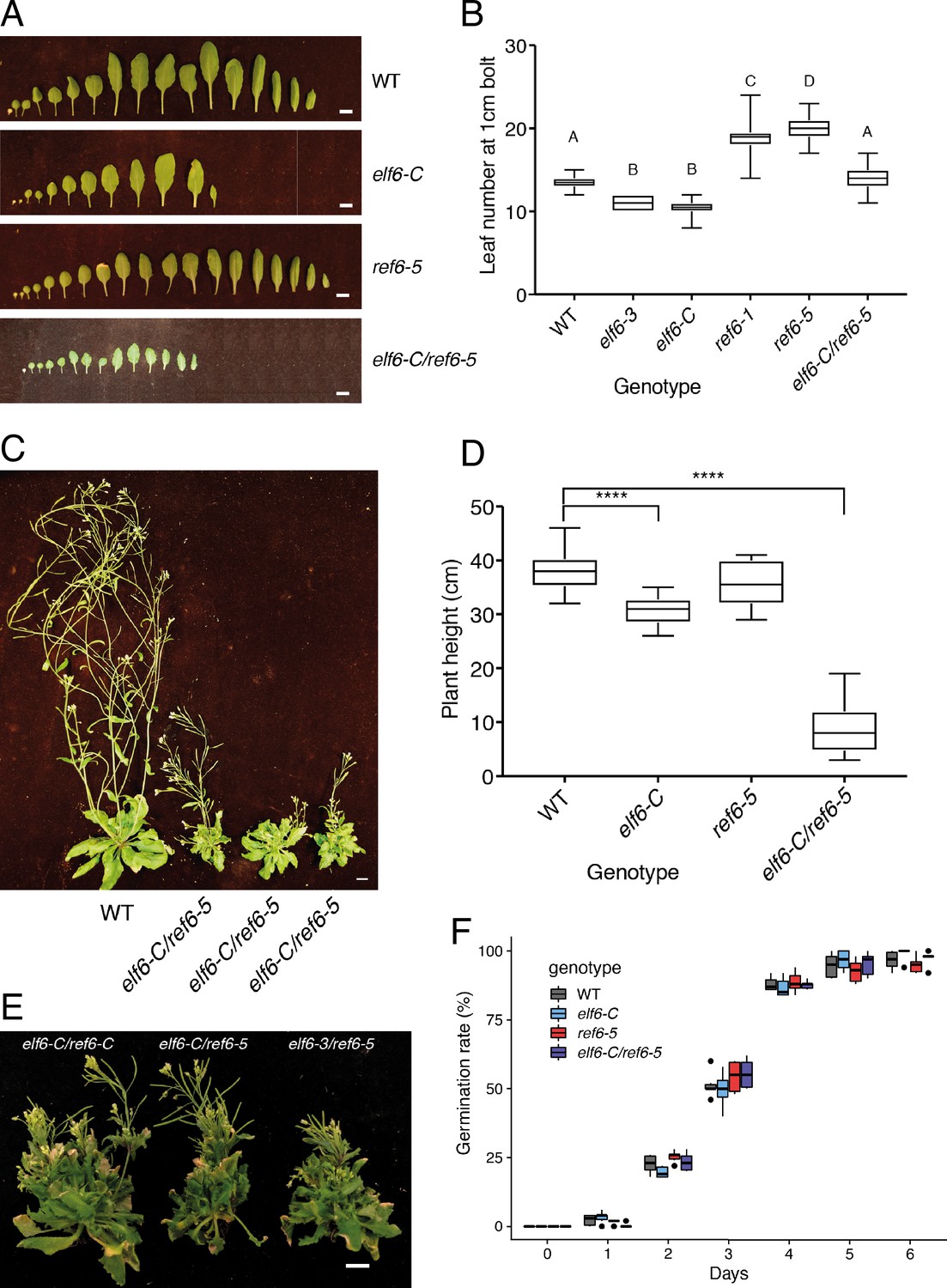
Phenotypic characterization of elf6-C, ref6-5 and double mutants.
(A) Rosette leaves at bolting stage of Arabidopsis wild-type (WT) and histone demethylase mutants (elf6-C, ref6-5 and elf6-C/ref6-5). Scale bars, 1 cm. (B) Boxplot for leaf number at bolting stage for wild-type (WT) and different mutants. Letters represent groups of statistically significantly different samples. Differences between genotypes determined by Students t-test, using a sample size of n = 30. (C) Growth phenotypes of wild-type (WT) and elf6-C/ref6-5 mutant. Scale bar, 1 cm. (D) Boxplot of plant height for wild-type (WT) and histone demethylase mutants. Asterisks represent significant differences between samples. Differences between genotypes determined by Students t-test test, ****p<0.001, sample size of n = 30. (E) Growth phenotypes of elf6-C/ref6-C, elf6-C/ref6-6 and elf6-3/ref6-5 double mutants. Scale bar, 1 cm. (F) Boxplot of seed germination rates in wild-type (WT) and histone demethylase mutants. Germination was scored as radicle protrusion through seed coat. n = 300 from six biological replicates.
Figure 1—figure supplement 3
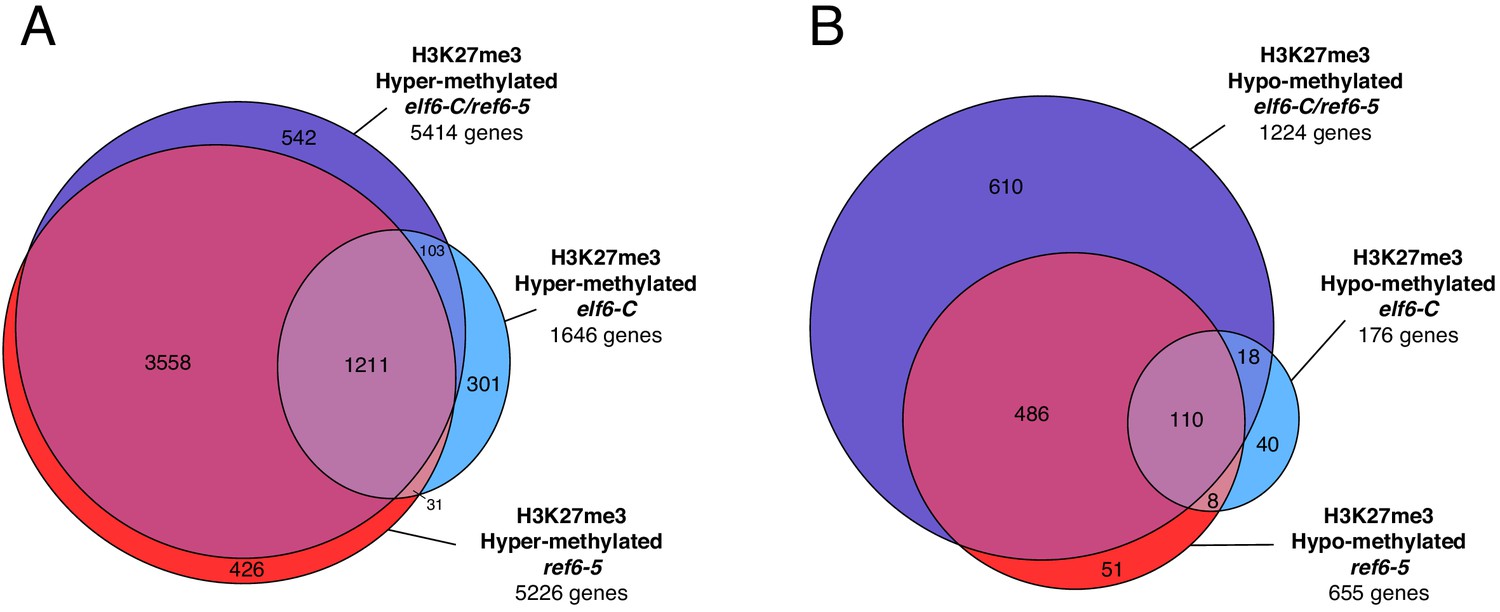
Genes with differential K3K27me3 methylation in histone demethylase mutants.
(A) Euler diagram of H3K27me3 hyper-methylated genes in histone demethylase mutants compared to wild-type (WT). (B) Euler diagram of H3K27me3 hypo-methylated genes in histone demethylase mutants compared to wild-type (WT).
Figure 1—figure supplement 4
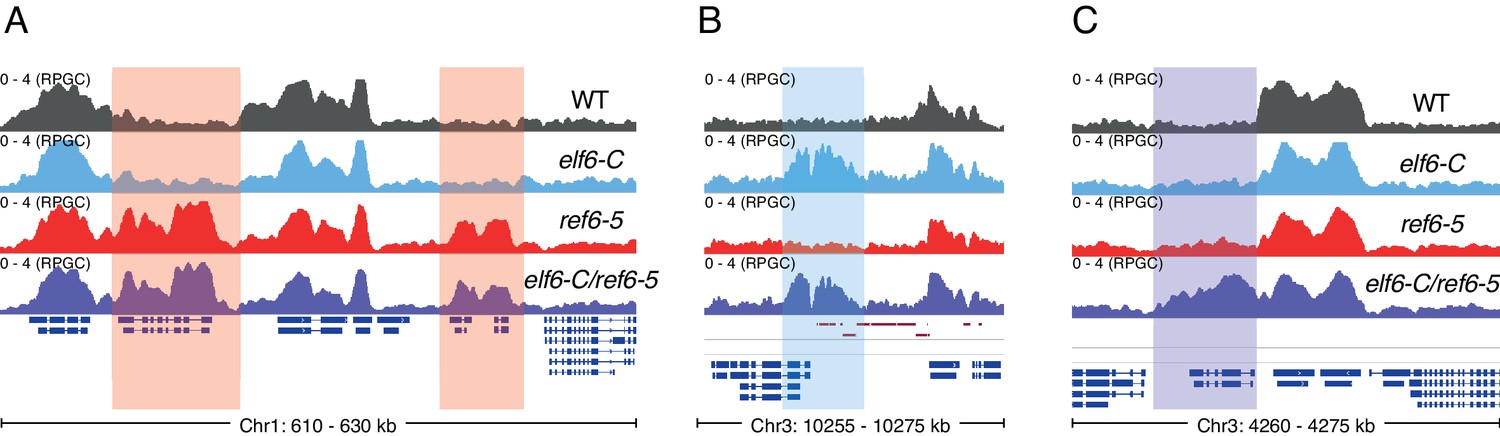
Genomic regions accumulating K3K27me3 in histone demethylase mutants.
(A) Genome browser view of ChIP-seq signal as normalized reads per genomic content (RPGC). Shaded red boxes, genes targeted by REF6. (B) Genome browser view of ChIP-seq signal as normalized reads per genomic content (RPGC). Shaded blue box, gene targeted exclusively by ELF6. (C) Genome browser view of ChIP-seq signal as normalized reads per genomic content (RPGC). Shaded purple boxes, genes targeted by both REF6 and ELF6, and only hyper-methylated in the double mutant elf6-C/ref6-5.
Figure 1—figure supplement 5
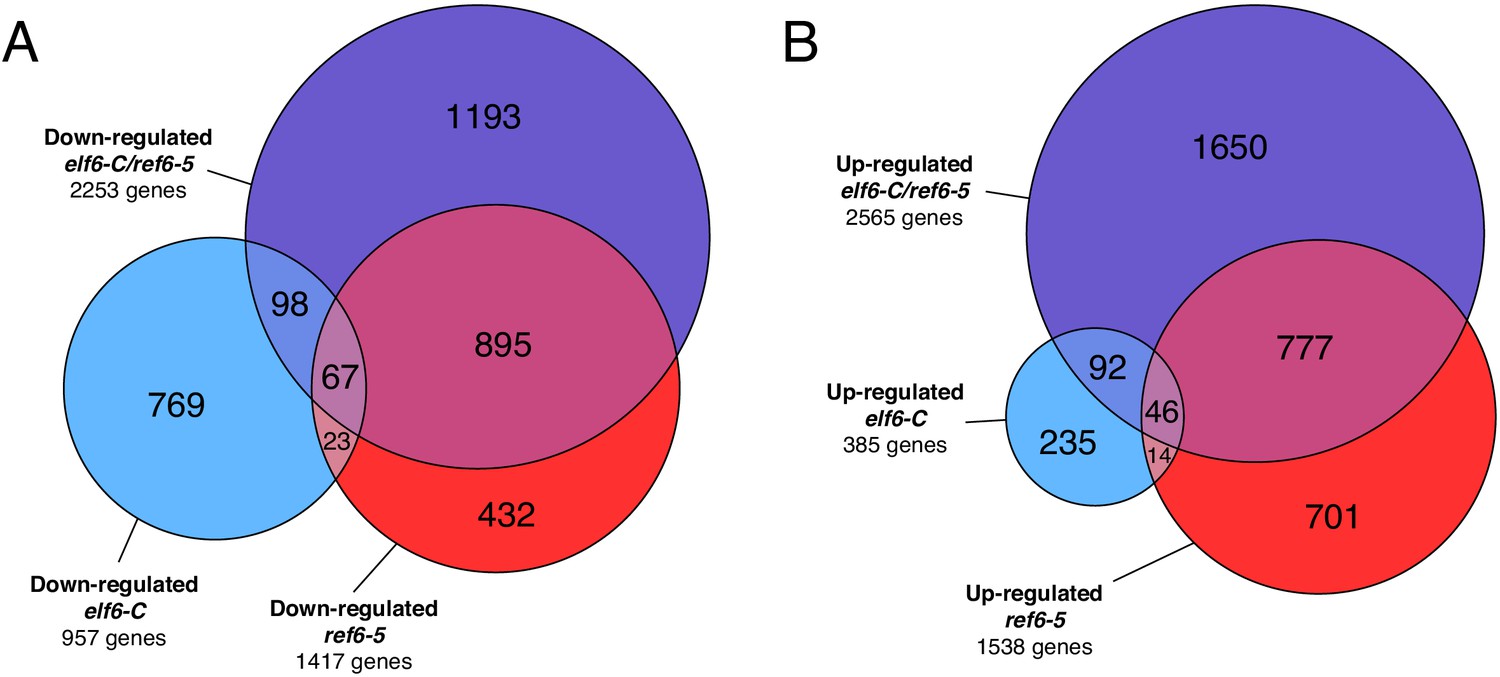
Genes differentially expressed in histone demethylase mutants.
(A) Euler diagram of down-regulated genes in histone demethylase mutants compared to wild-type (WT). (B) Euler diagram of up-regulated genes in histone demethylase mutants compared to wild-type (WT).
Figure 1—figure supplement 6
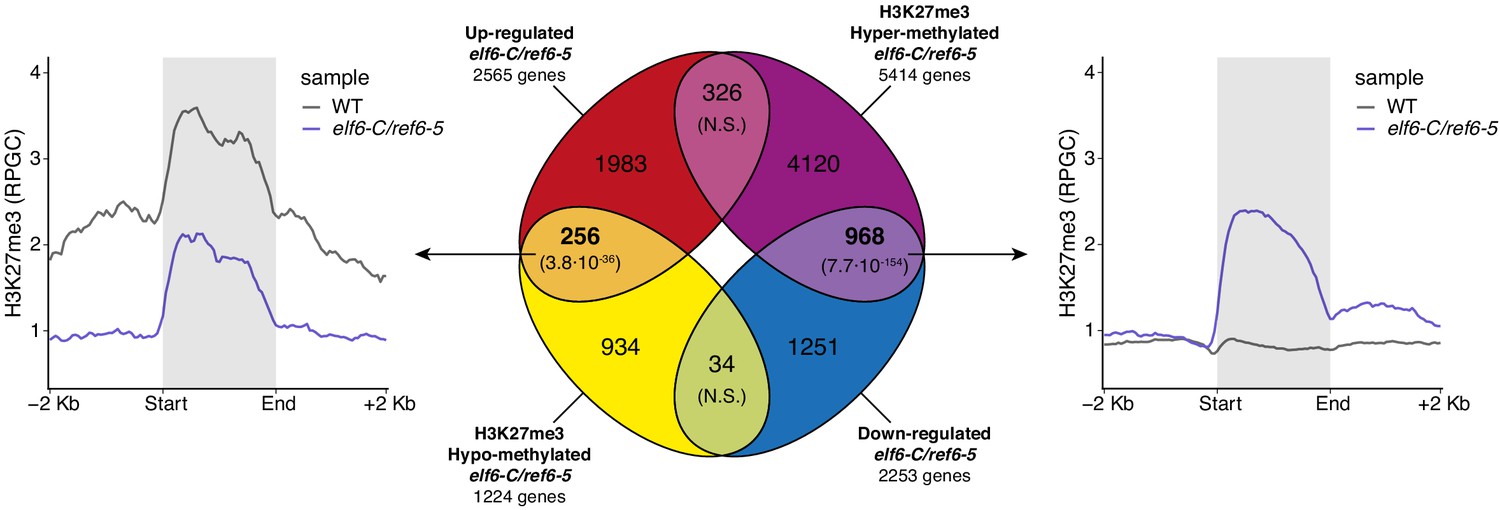
Relation between hypermethylation of H3K27me3 and downregulation of genes in elf6-C/ref6-5.
Venn diagram showing overlap between differentially expressed genes (DEGs) and H3K27me3 differentially methylated genes in elf6-C/ref6-5. Venn diagram showing overlap between differentially expressed genes (DEGs) and H3K27me3 differentially methylated genes in elf6-C/ref6-5. To the left metaplot for H3K27me3 levels for genes both up-regulated and hypo-methylated and to the right metaplot of H3K27me3 levels in genes both down-regulated and hyper-methylated. p-values for Fisher’s exact test are shown in brackets, N.S. Not Significant.
Figure 2 with 5 supplements
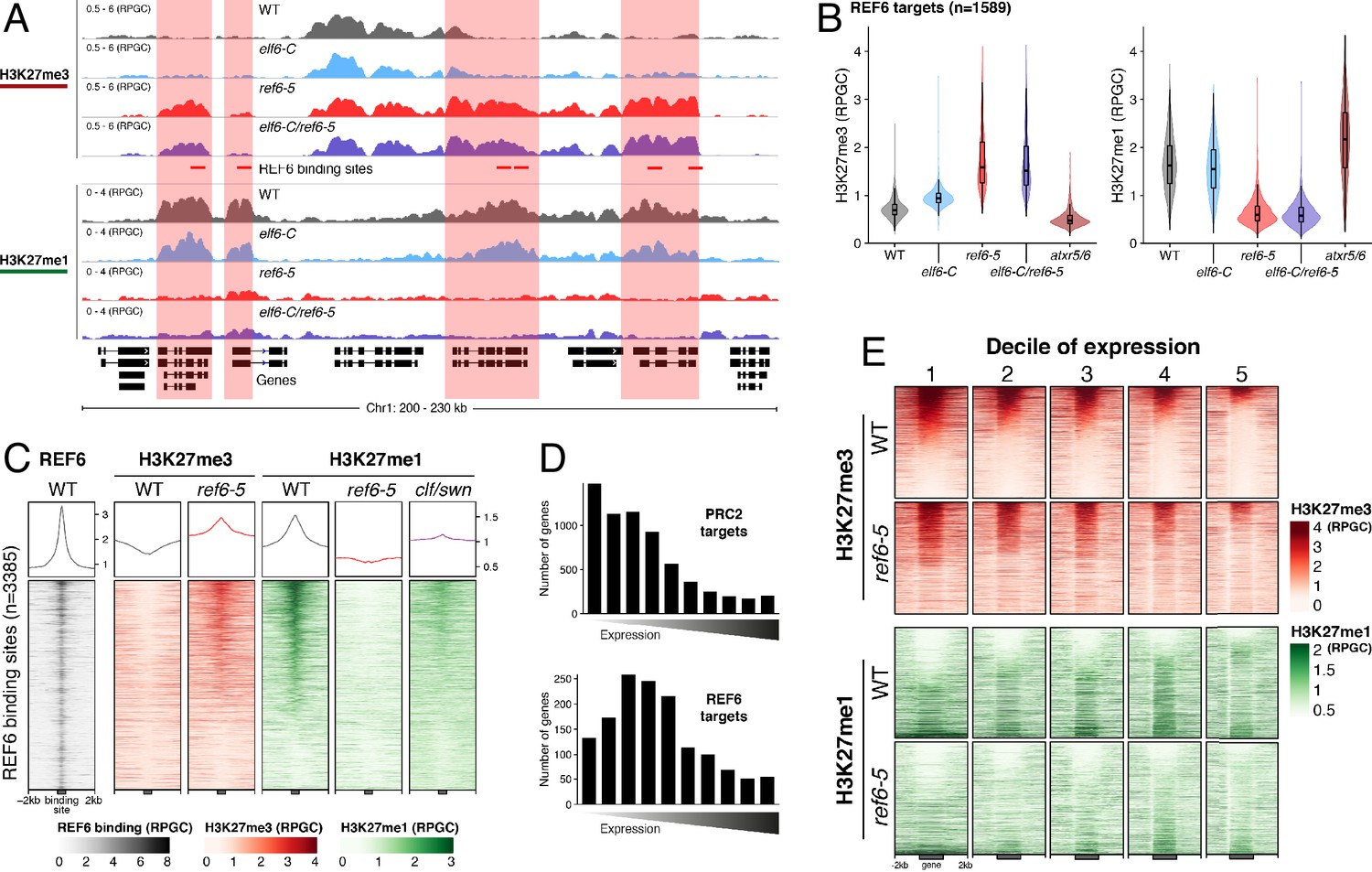
Arabidopsis REF6 plays an essential role in the deposition of H3K27me1 in active chromatin.
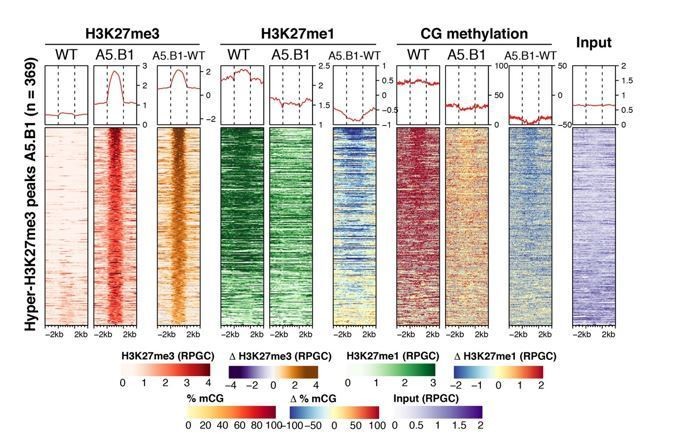
(A) Genome browser views of background subtracted ChIP-seq signals for H3K27me3 and H3K27me1 as normalized reads per genomic content (RPGC) in wild-type (WT) and histone demethylase mutants (elf6-C, ref6-5 and elf6-C/ref6-5). Shaded boxes, genes targeted exclusively by REF6. (B) Violin plots showing the distribution of H3K27me3 and H3K27me1 on genes targeted by REF6. Genes were categorised as targeted if a H3K27me3 peak was annotated on them in ref6-5 and in elf6-C/ref6-5 but not in WT. (C) Heatmap showing the distribution of H3K27me3 and H3K27me1 on genomic sequences targeted by REF6 for wild-type (WT), ref6-5, and clf/swn plants. Sample size n = 3385. (D) Bar charts showing the number of genes for different expression quantiles predicted to be targeted by PRC2 and REF6. (E) Heatmap showing the distribution of H3K27me3 and H3K27me1 present on genes corresponding to low-expression (1-5) quantiles. .
Figure 2—figure supplement 1
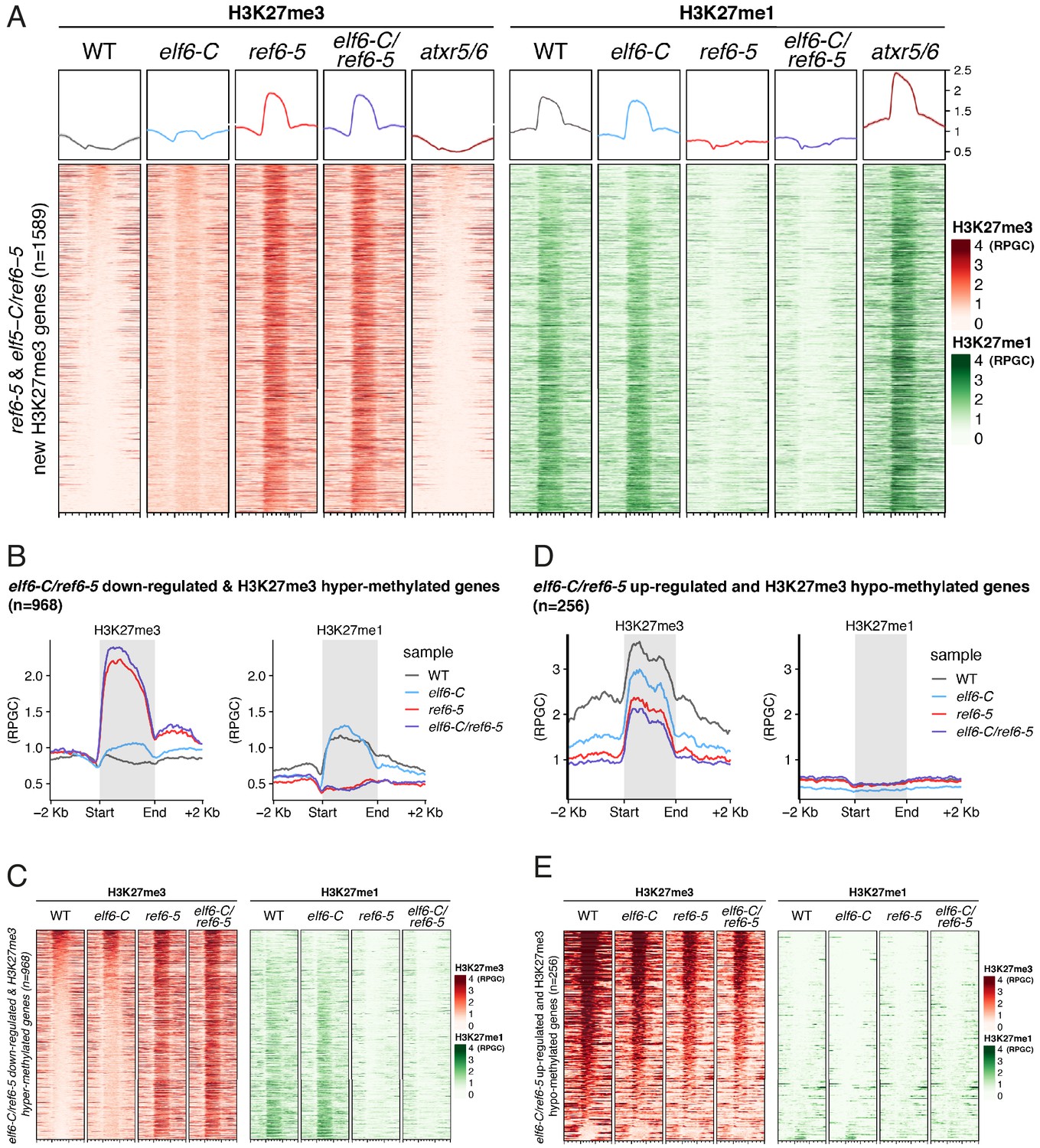
REF6 catalyses H3K27me3 to H3K27me1 conversion in genes causing de-repression.
(A) Heatmap showing the distribution of H3K27me3 (red) and H3K27me1 (green) on genes targeted by REF6. Genes were categorised as targets if a H3K27me3 peak was annotated on them in ref6-5 and in elf6-C/ref6-5 but not in WT. n = 1589. Intensity of the colour represents RPGC of ChIP-seq. Genes are sorted by the amount of H3K27me3 in WT. Boxes at the top represent metaplots of the average signal for all these genes. (B) Metaplot of median of H3K27me3 and H3K27me1 across both downregulated and hypermethylated genes in elf6-C/ref6-5 for wild-type (WT) and histone demethylase mutants, n = 968. (C) Heatmap of the same genes as Fig S 7B. Intensity of the colour represents ChIP-seq signal as RPGC. (D) Metaplot of median of H3K27me3 and H3K27me1 across both upregulated and hypomethylated genes in elf6-C/ref6-5 for wild-type (WT) and histone demethylase mutants, n = 256. (E) Heatmap of the same genes as Fig S 7D. Intensity of the colour represents ChIP-seq signal as RPGC.
Figure 2—figure supplement 2
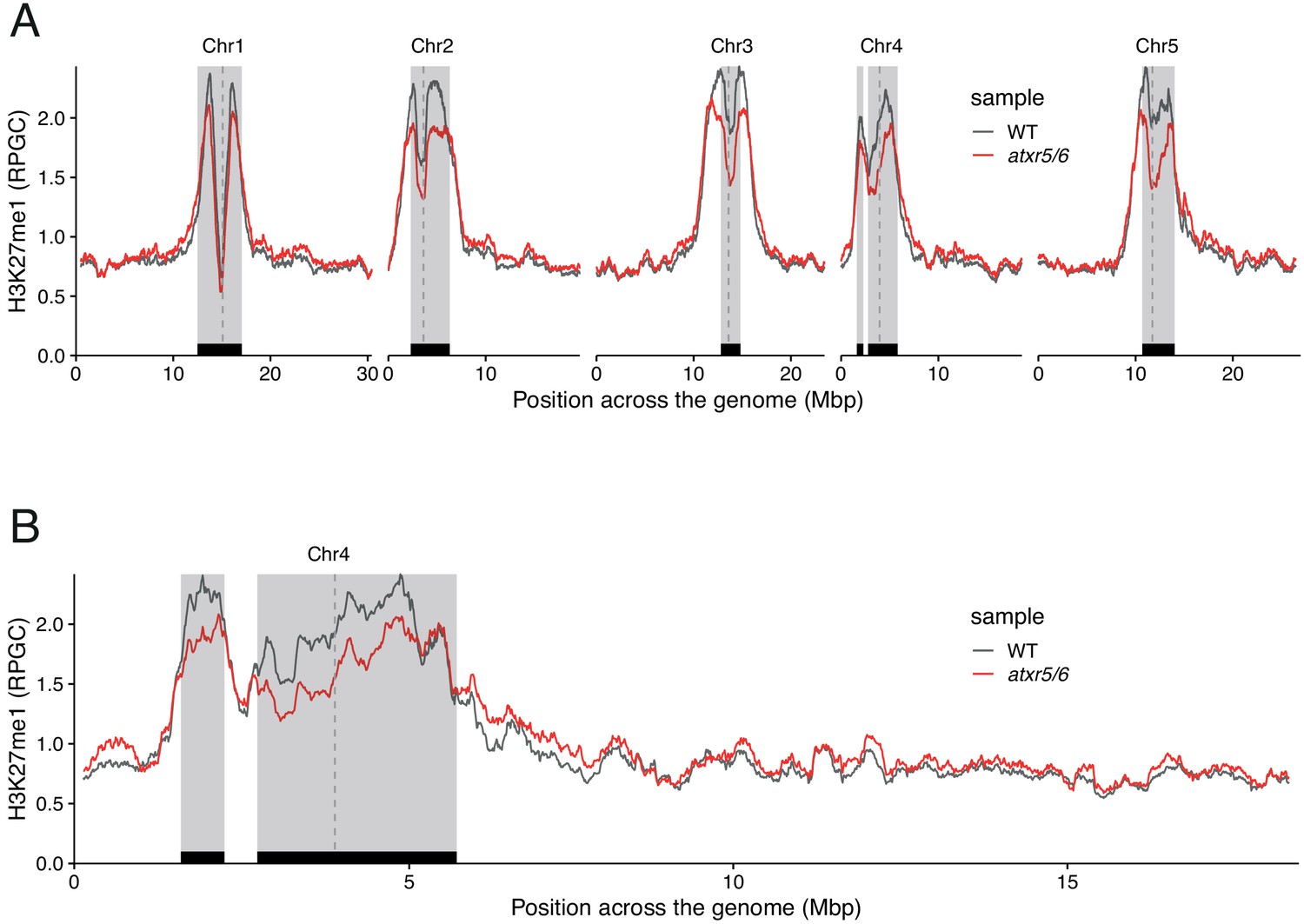
ATXR5/6 contributes to the deposition of H3K27me1 in pericentromeric regions.
(A) Distribution of H3K27me1 across chromosomes of wild-type (WT) and atxr5/6 mutants. Grey shaded boxes, pericentromeric regions. (B) Distribution of H3K27me1 across Chromosome 4 of Arabidopsis genome in wild-type (WT) and atxr5/6 mutants. Grey shaded boxes, pericentromeric regions.
Figure 2—figure supplement 3
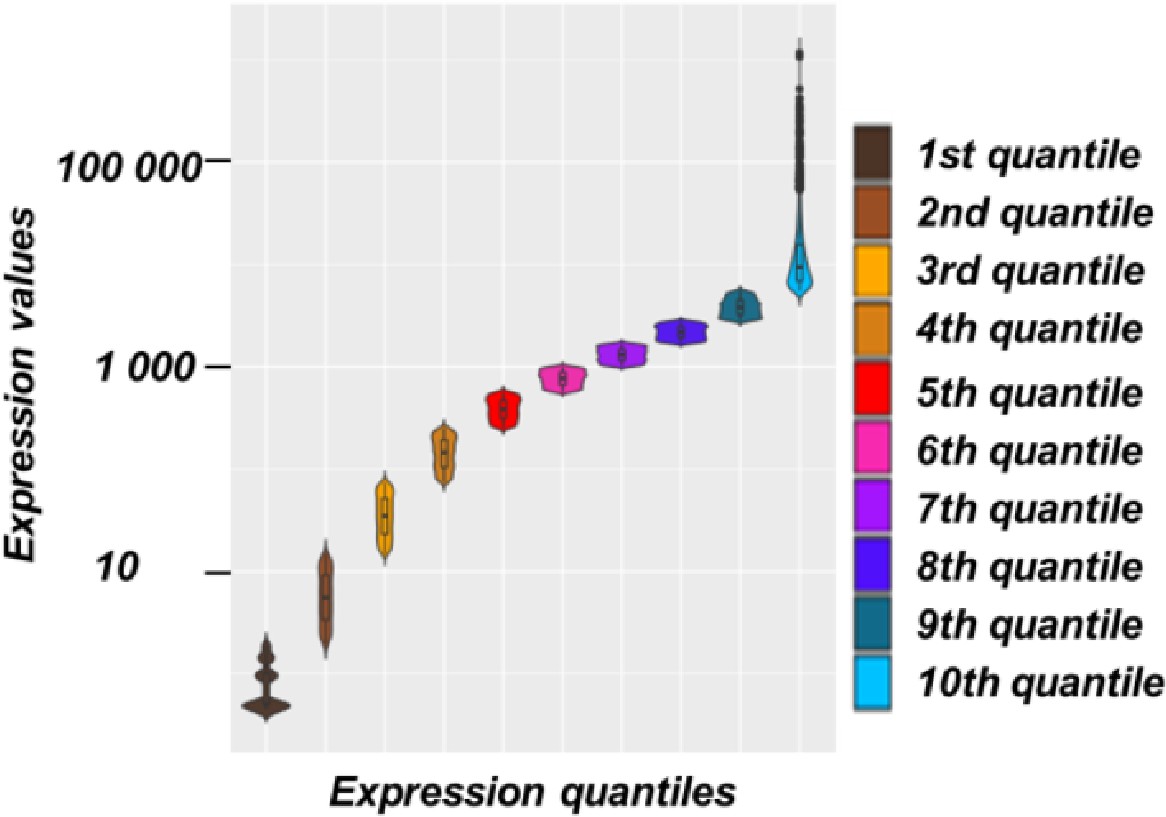
Division of Arabidopsis genes according to their levels of expression.
Violin plot with boxplots representing the expression levels of genes in Arabidopsis divided in 10 quantiles.
Figure 2—figure supplement 4
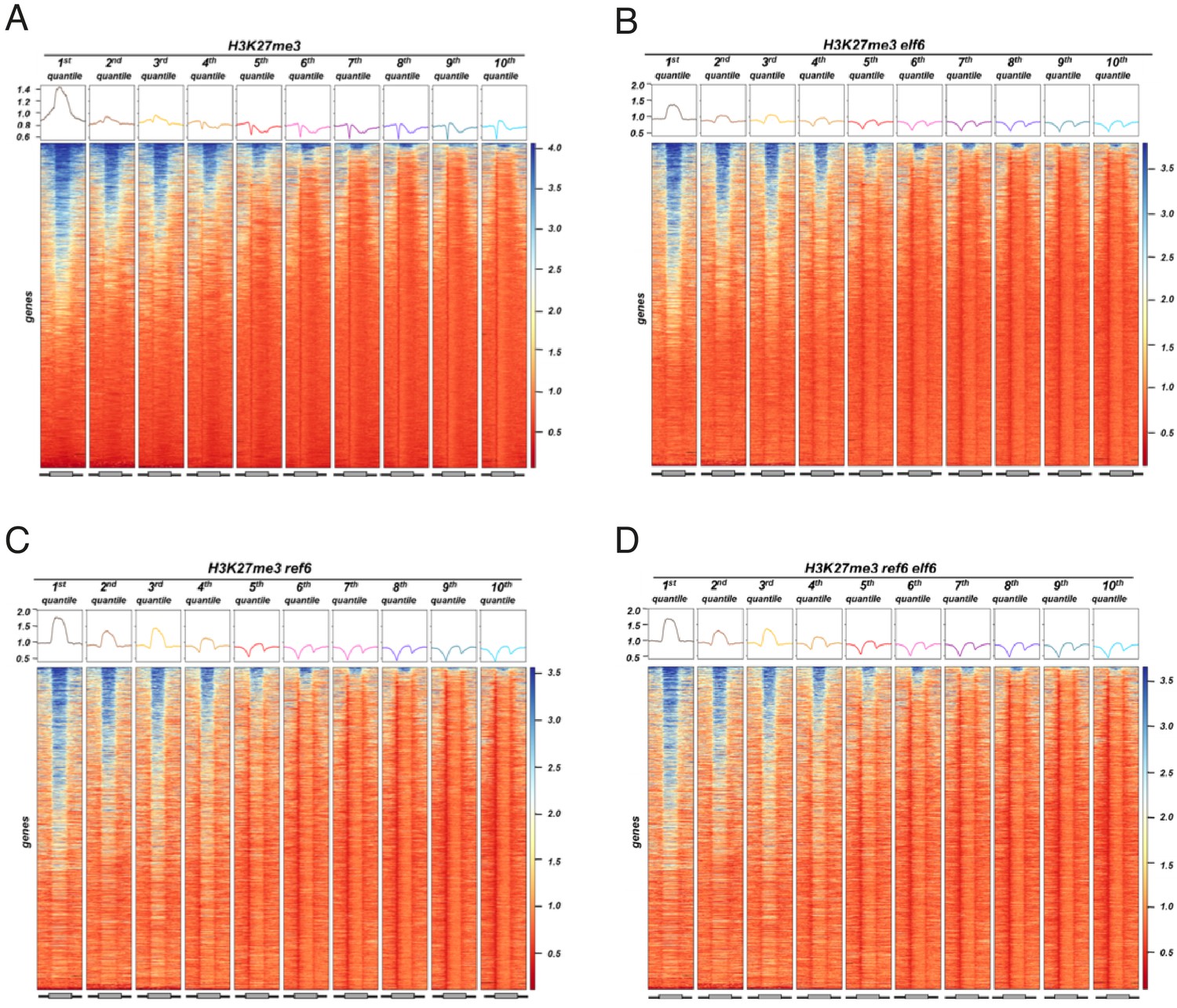
Histone demethylase mutants show an increase in H3K27me3 on genes with intermediate levels of expression Heatmaps showing the H3K27me3.
ChIP-seq signal across genes split by 10 quantiles of expression for wild-type (WT) and histone demethylase mutants. Boxes on top represent metaplots of the median signal in each quantile. Genes are sorted by the amount of H3K27me3 in WT. (A) WT. (B) elf6-C. (C) ref6-5. (D) elf6-C/ref6-5.
Figure 2—figure supplement 5
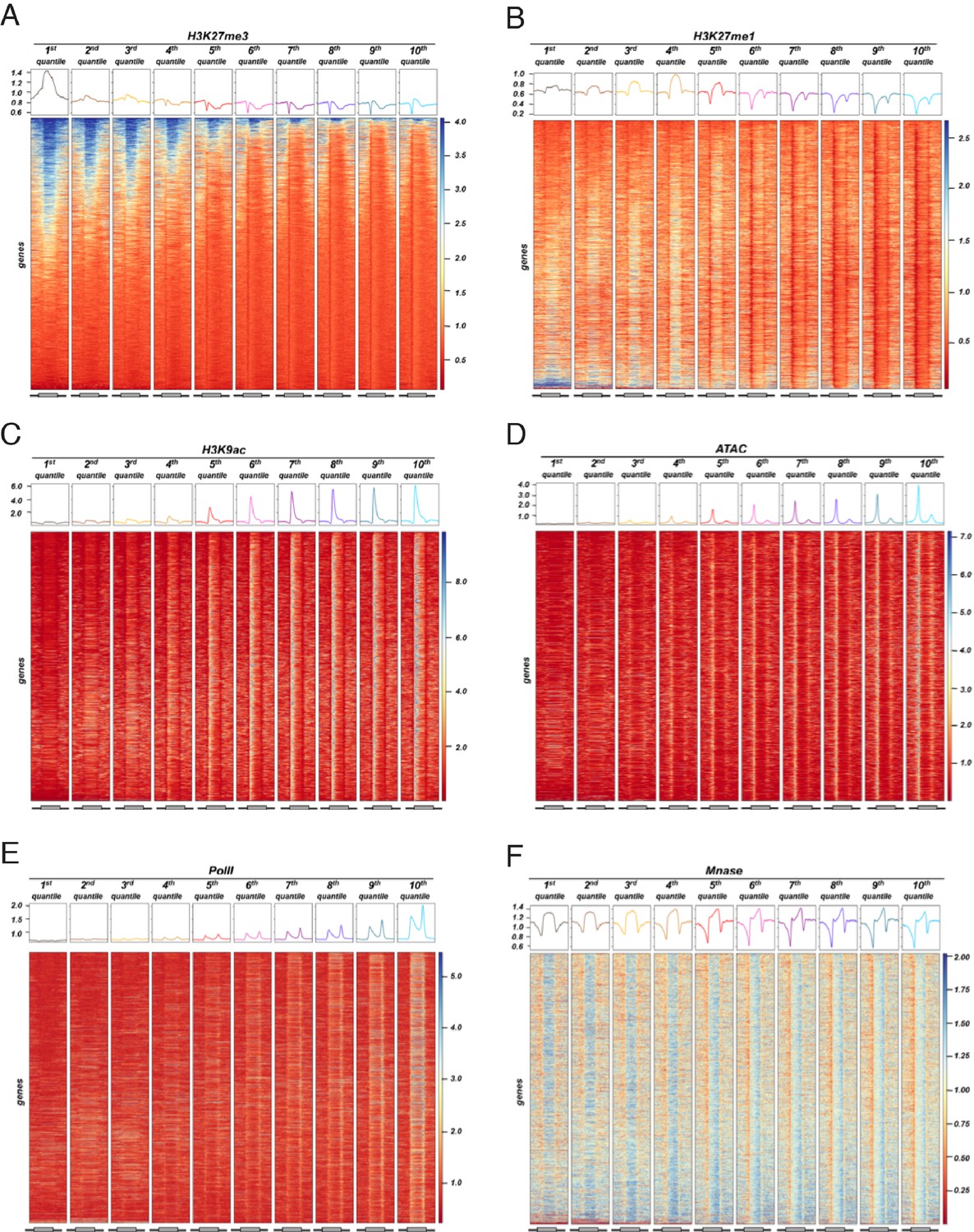
Distribution of different epigenetic features for different expression quantiles.
Heatmaps showing signal for different epigenetic features across 10 expression quantiles for wild-type. Boxes on top represent metaplots of the median signal in each quantile. Genes in all panels are always sorted by the amount of H3K27me3. (A) H3K27me3. (B) H3K27me1. (C) H3K9ac. (D) ATAC-seq. (E) PolII-seq. (F) Mnase-seq.
Figure 3 with 2 supplements
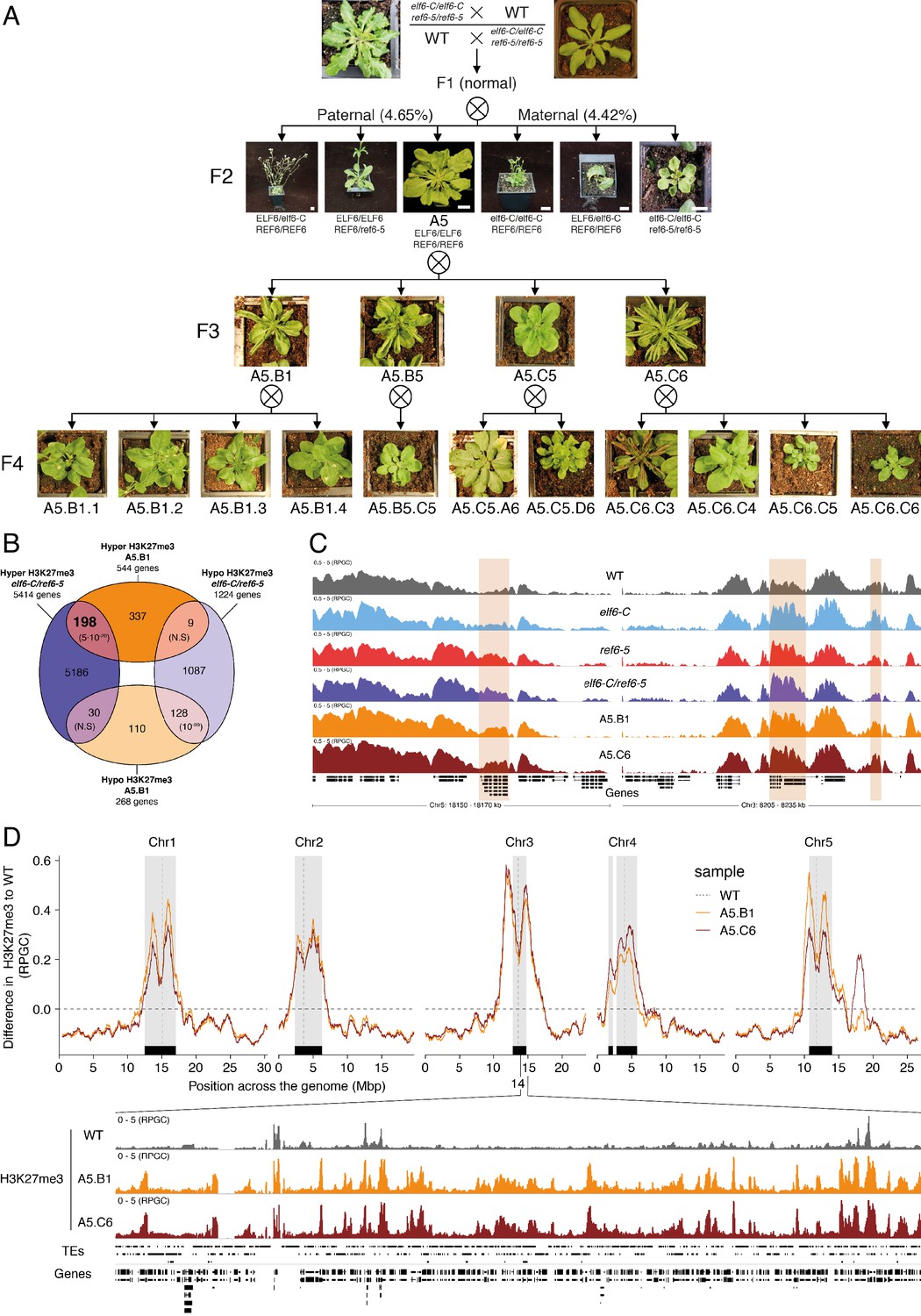
Pleiotropic developmental abnormalities associated with the inheritance of ectopic H3K27me3 imprints in Arabidopsis.
(A) The F2 hybrids from reciprocal crosses between wild-type (WT) and elf6-C/ref6-5 display novel abnormal plant growth phenotypes. Frequency of abnormal phenotypes according to parental transmission of mutant alleles is indicated. Pedigree of an epimutant that was genetically wild-type for ELF6 and REF6 and selected for genomic analysis after propagation by selfing. Scale bars, 1 cm. (B) Venn diagram showing the overlap in genes accumulating H3K27me3 in elf6-C/ref6-5 and F5 progenies from A5.B1. p-values for Fisher’s exact test are shown in brackets, N.S. Not Significant. (C) Genome browser views of background subtracted ChIP-seq signals for H3K27me3 as normalized reads per genomic content (RPGC) in wild-type (WT), elf6-C, ref6-5, elf6-C/ref6-5, and F5 progenies from A5.B1 and A5.C6. Shaded boxes, genes showing transgenerational inheritance of H3K27me3. (D) Top panel: Differences in the chromosomal distribution of H3K27me3 as normalized reads per genomic content (RPGC) between F5 progenies from A5.B1 and A5.C6 and wild-type (WT). Grey shaded boxes, pericentromeric regions. Bottom panel: Genome browser view of ChIP-seq signal for H3K27me3 as normalized reads per genomic content (RPGC) in wild-type (WT), and F5 progenies from A5.B1 and A5.C6 in a pericentromeric region.
Figure 3—figure supplement 1

Phenotypic variation present in epiER progenies.
Representative images of F4 and F5 progenies from epiER line A5 showing a wide range of developmental phenotypes.
Figure 3—figure supplement 2

Inheritance of ectopic H3K27me3 in epiERs.
(A) Venn diagram showing the intersection between genes showing accumulation of H3K27me3 in elf6-C/ref6-5 and epiER A5.C6. p-values for Fisher’s exact test are shown in brackets, N.S. Not Significant. (B) Metaplot of the median of ChIP-seq RPGC across genes hypermethylated in both elf6-C/ref6-5 and epiER A5.C6. n = 198. (C) Euler diagram showing the intersection between the genes hypermethylated in elf6-C/ref6-5 and epiERs A5.B1 and A5.C6.
Figure 4 with 3 supplements
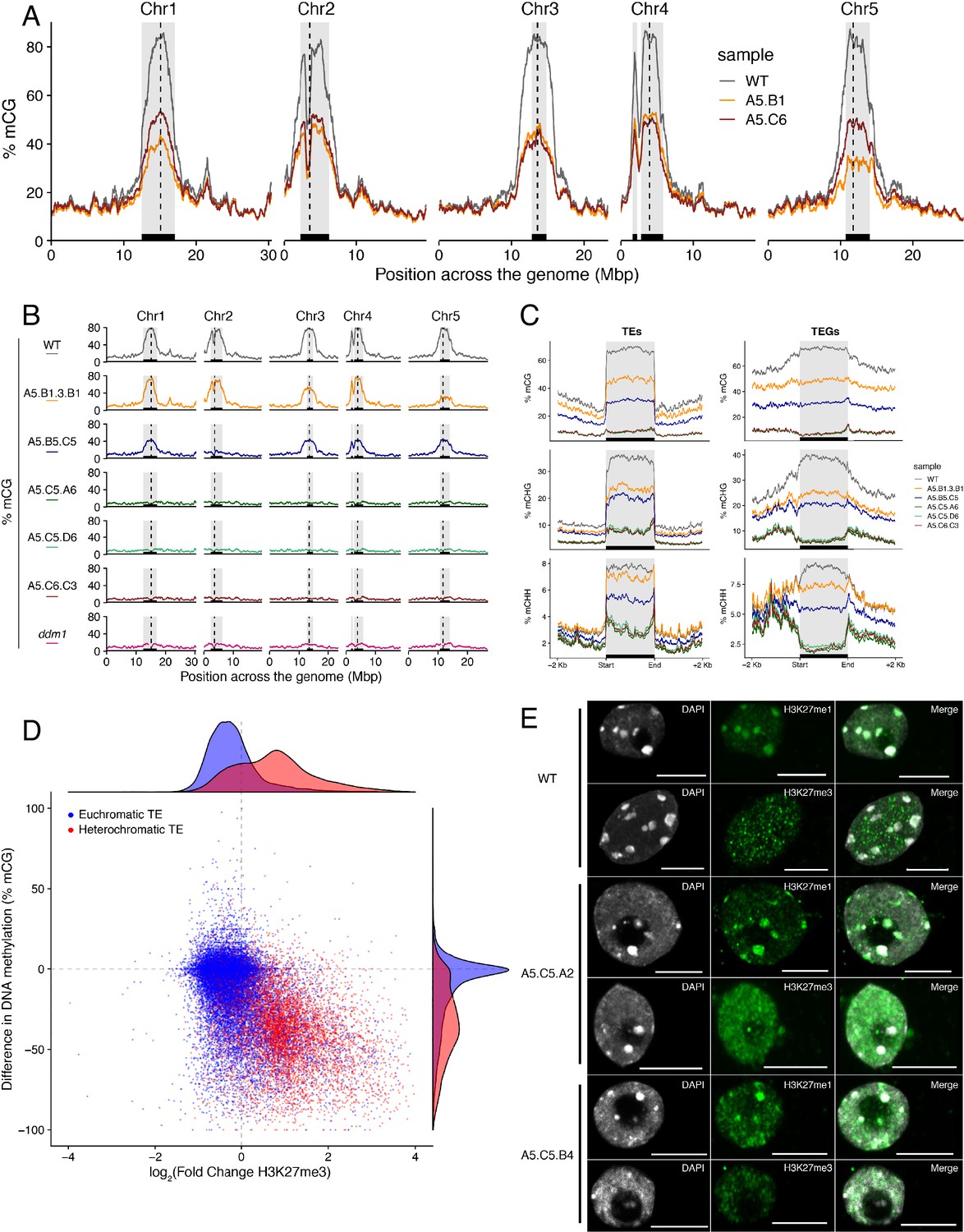
Ectopic accumulation of H3K27me3 is associated with the loss of DNA methylation at pericentromeric heterochromatin and affects chromatin condensation.
(A) Distribution of DNA methylation across chromosomes of wild-type (WT) and progenies from epiERs A5.B1 and A5.C6. Grey shaded boxes, pericentromeric regions. (B) Distribution of DNA methylation across chromosomes of individual plants from wild-type (WT), ddm1, and epiERs A5.B1.3.B1, A5.B5.C5, A5.C5.A6, A5.C5.D6 and A5.C6.C3. Grey shaded boxes, pericentromeric regions. (C) Distribution of DNA methylation across Transposable Elements (TEs) and Transposable Element Genes (TEGs) of individual plants from wild-type (WT) and epiERs A5.B1.3.B1, A5.B5.C5, A5.C5.A6, A5.C5.D6 and A5.C6.C3. Black box, centromeric regions. (D) Correlation between DNA methylation changes and H3K27me3 changes on euchromatic and heterochromatic TEs, in wild-type (WT) and epiER A5.B1. (E) Immunolocalization showing the distribution of H3K27me3 and H3K27me1 in interphase nuclei of wild-type, A5.C5.A2 and A5.C5.B4 plants. Scale bars, 5 μm.
Figure 4—figure supplement 1
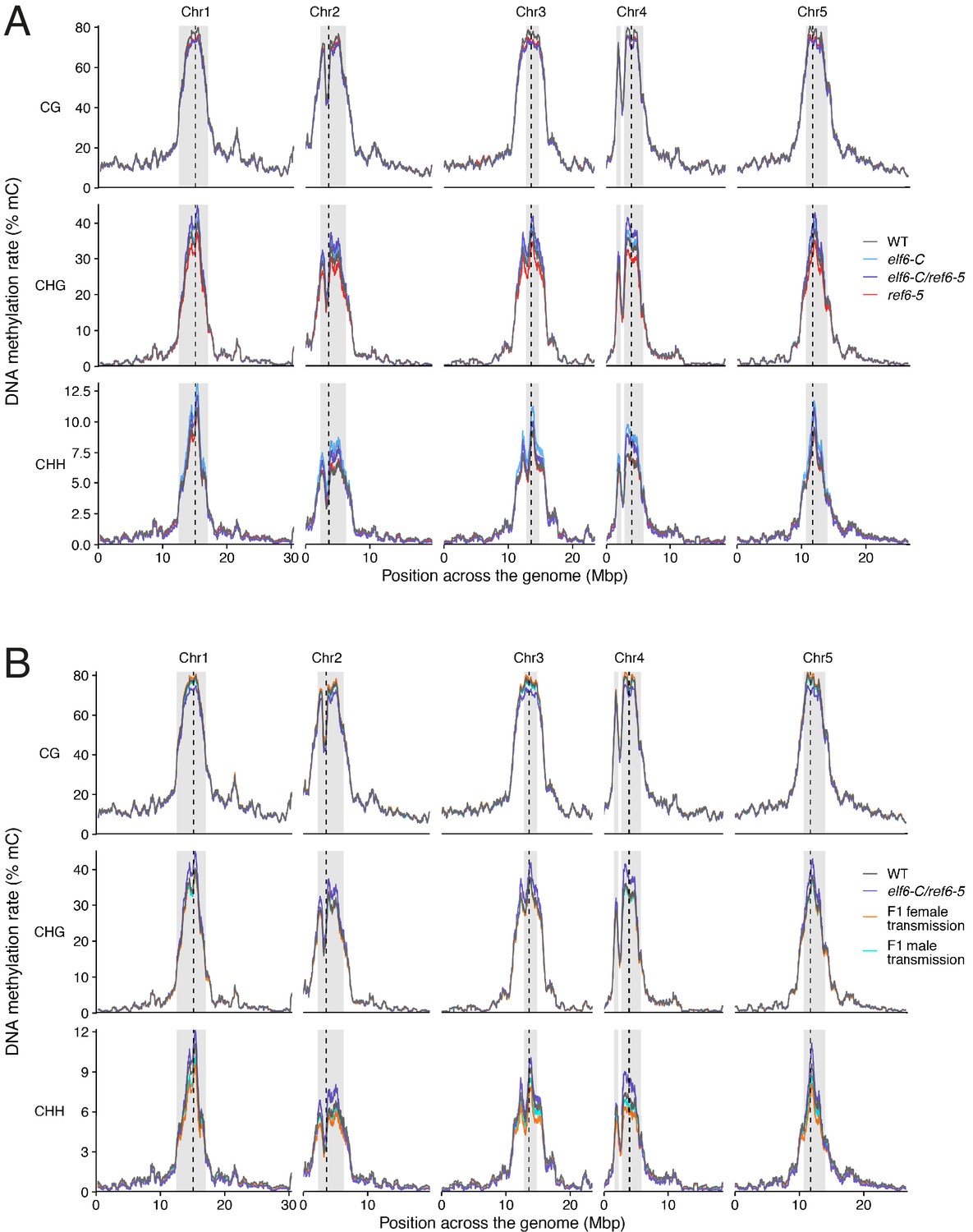
Chromosomal distribution of DNA methylation in histone demethylase mutants and F1 hybrids.
(A) DNA methylation in wild-type (WT), elf6-C, ref6-5 and elf6-C/ref6-5 plants. (B) DNA methylation in wild-type (WT) and F1 hybrids from reciprocal crosses between wild-type and elf6-C/ref6-5. Paternal or maternal transmission of mutant alleles is indicated.
Figure 4—figure supplement 2

DNA hypomethylation of transposable elements in epiERs.
Metaplot showing the proportion of DNA methylation across transposable elements (A) and transposable element genes (B) for wild-type (WT) and progenies from two epiERs.
Figure 4—figure supplement 3
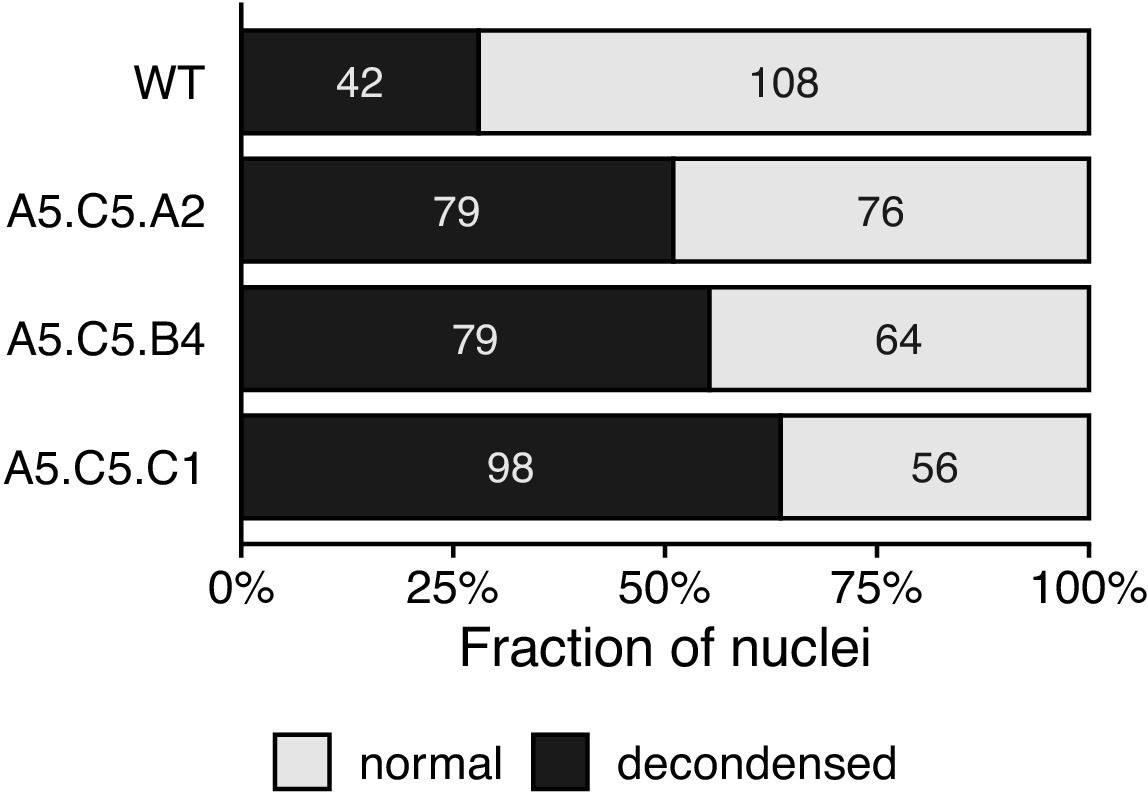
Condensation of chromatin in epiERs.
Bar plot showing the fraction of nuclei categorised as decondensed after DAPI staining in wild-type (WT) and epiERs A5.C5.A2, A5.C5.B4 and A5.C5.C1. Number of nuclei for each category in each line shown inside plot.
Figure 5 with 1 supplement
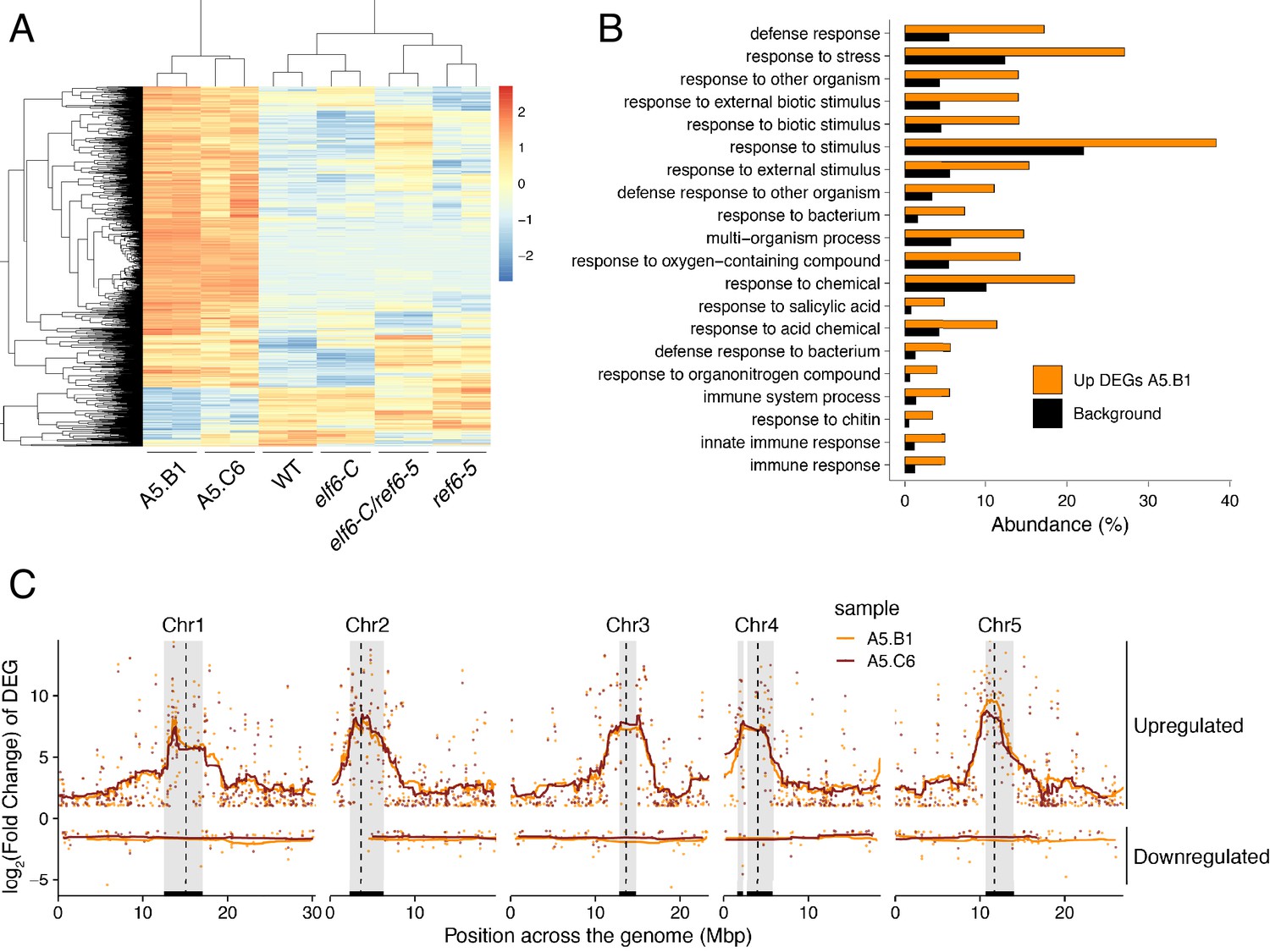
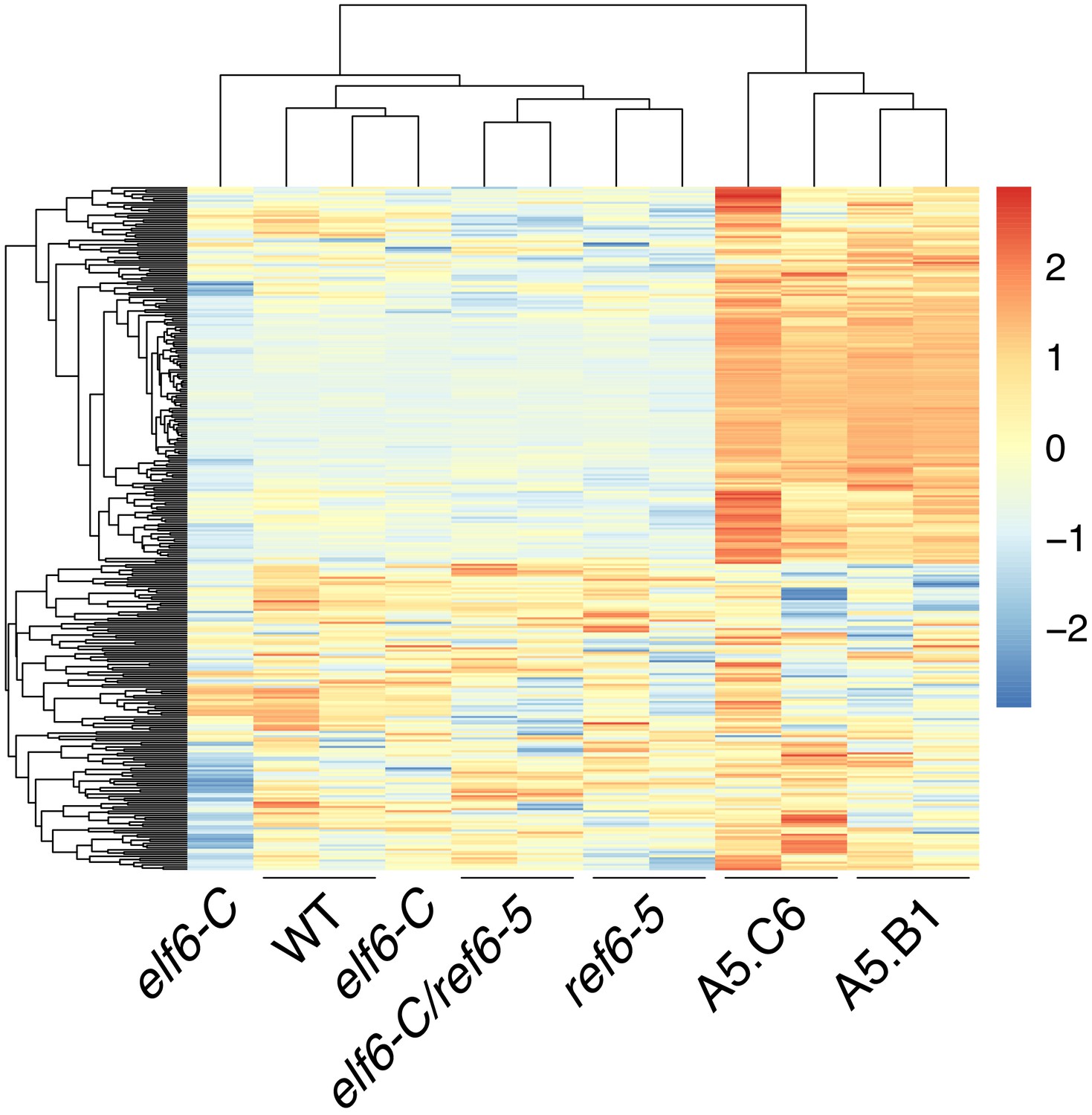
Global upregulation of centromeric gene expression in epiERs.
(A) Heatmap showing scaled expression levels of Differentially Expressed Genes between wild-type and progeny of epiER A5.B1 in wild-type (WT) elf6-C, ref6-5, elf6-C/ref6-5, and progenies of epiERs A5.B1 and A5.C6. (B) Gene Ontology analysis showing the functional categories enriched in genes upregulated in progeny of epiERs A5.B1. (C) Differential gene expression across each Arabidopsis chromosome for genes upregulated and downregulated in progenies of epiERs A5.B1 and A5.C6. Grey shaded boxes, pericentromeric regions.
Figure 5—figure supplement 1
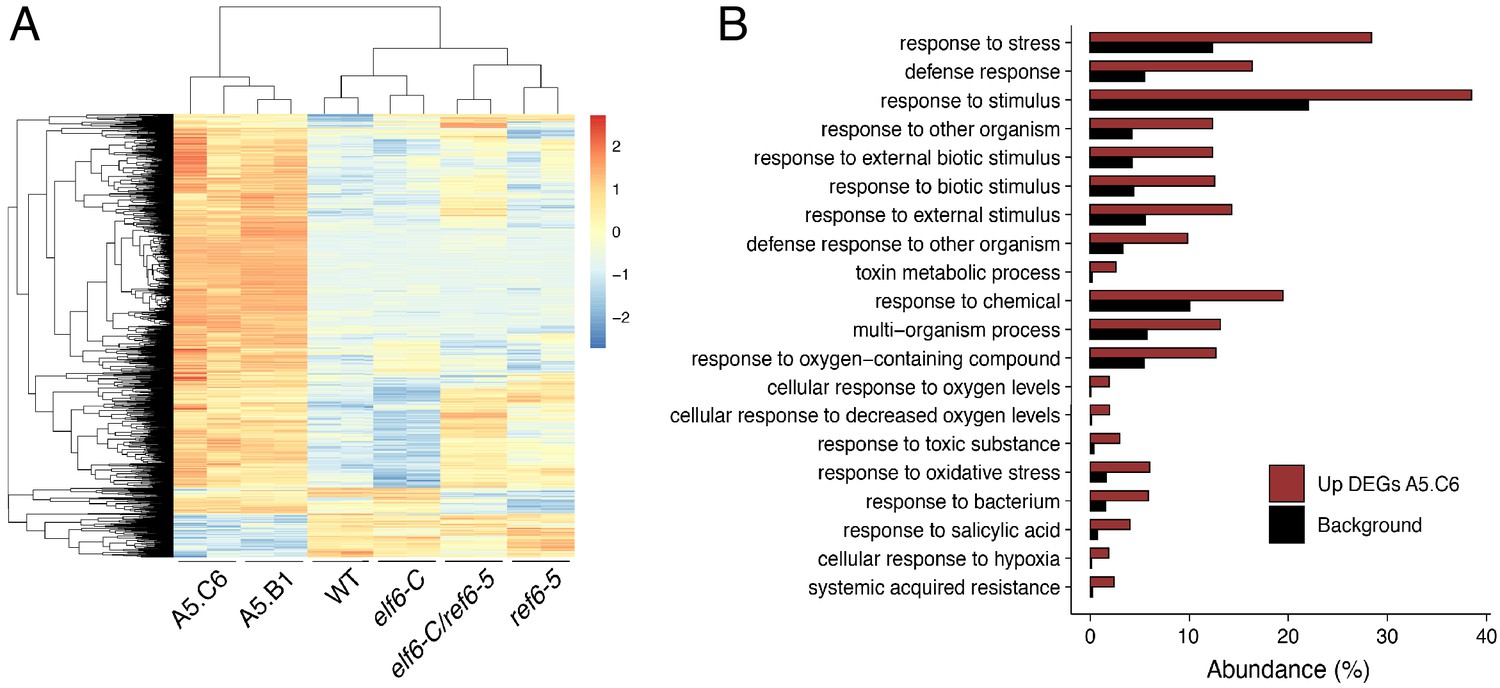
Genes upregulated in epiERs.
(A) Heatmap showing scaled expression levels of Differentially Expressed Genes (DEGs) between wild-type and progeny of epiER A5.C6 in wild-type (WT) elf6-C, ref6-5, elf6-C/ref6-5, and progenies of epiERs A5.B1 and A5.C6. (B) Gene Ontology analysis showing the functional categories enriched in genes upregulated in epiER A5.C6.
Figure 6 with 2 supplements
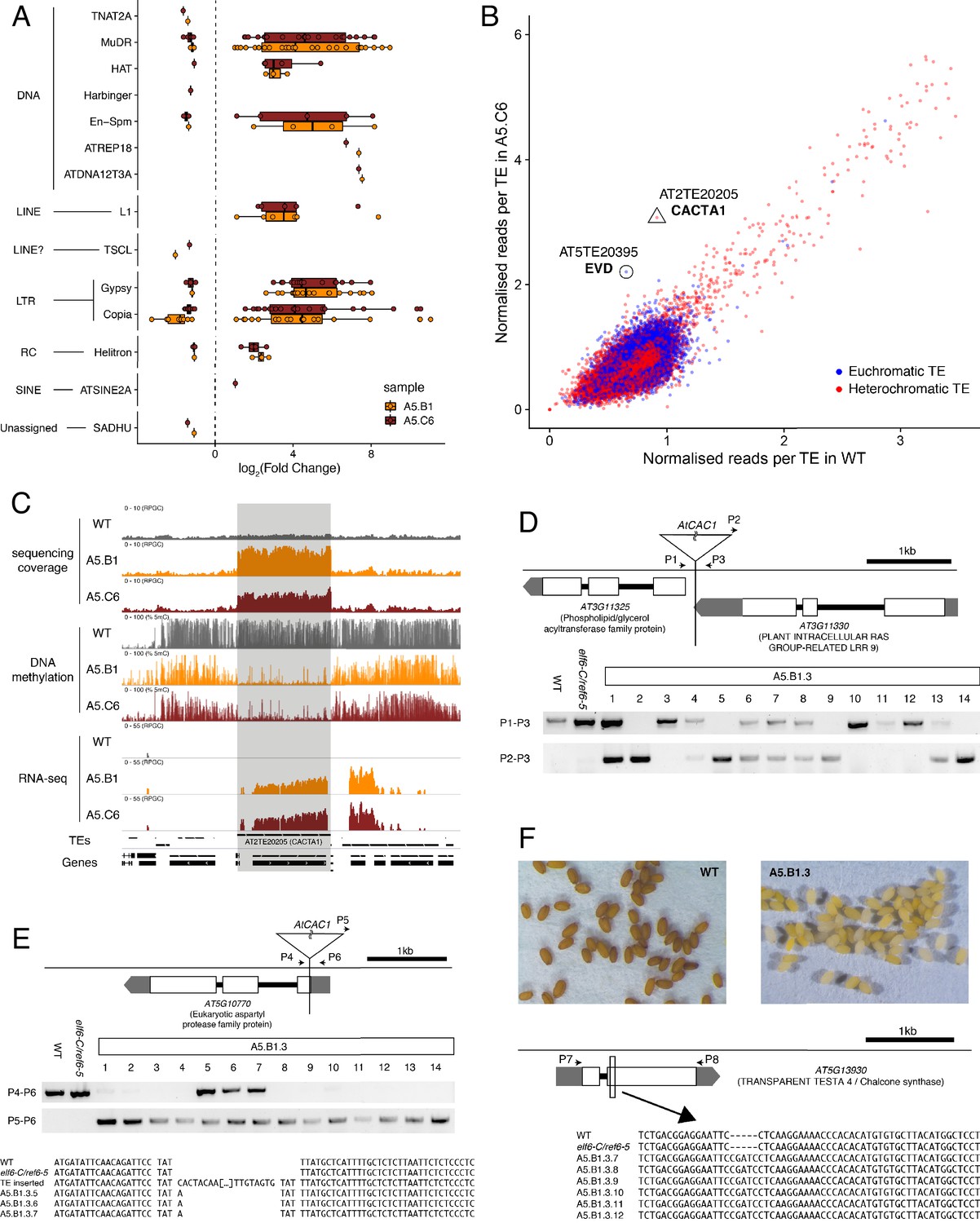
Transposon mobilization in epiERs results in heritable genetic lesions.
(A) Differential expression of DNA and RNA transposon families grouped by superfamily in progenies of epiERs A5.B1 and A5.C6. (B) Copy number variation of transposons in progenies of epiER A5.C6. Blue dots, euchromatic TEs; Red dots, heterochromatic TEs. (C) Genome browser views of normalized sequencing coverage (RPGC), DNA methylation frequency (%) and RNAseq coverage (RPGC) in wild-type (WT) and progenies of epiERs A5.B1 and A5.C6. Grey box, AT2TE20205 (CACTA1). (D) Map of transposon insertion in AT3G11330 and its segregation in epiER A5.B1.3 progenies. P1-3, primers used for PCR amplification and sequencing. (E) Map of transposon insertion in AT5G10770 and sequence footprint resulting from re-mobilization in epiER A5.B1.3 progenies. P4-6, primers used for PCR amplification and sequencing. (F) Seed pigmentation defects caused by a sequence insertion in AT5G13930 (TRANSPARENT TESTA4/CHALCONE SYNTHASE) resulting from transposon re-mobilization in in epiER A5.B1.3 progenies. P7-8, primers used for PCR amplification and sequencing.
Figure 6—figure supplement 1
Transcriptional upregulation of transposons in epiERs.
Heatmap showing scaled logged expression of all the families of transposons or Arabidopsis. Samples represented are wild-type (WT), histone demethylase mutants and epiERs A5.B1 and A5.C6.
Figure 6—figure supplement 2
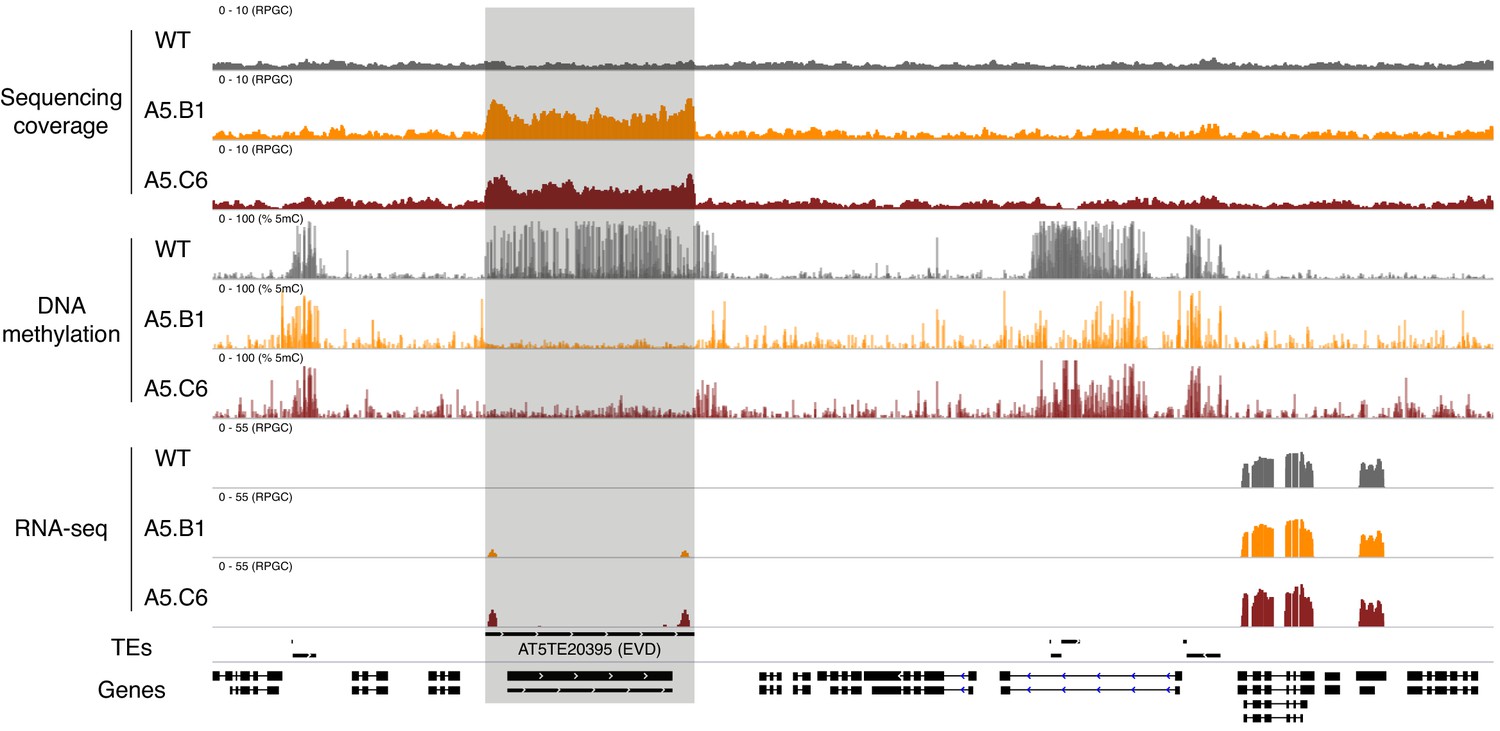
Deregulation of EVD transposon in epiERs.
Genome browser view showing normalised sequencing coverage (RPGC), DNA methylation rate (%) and RNA-seq (RPGC) in wild-type (WT) and progenies from epiERs A5.B1 and A5.C6. Grey box, AT2TE20395 (EVD).
Figure 7
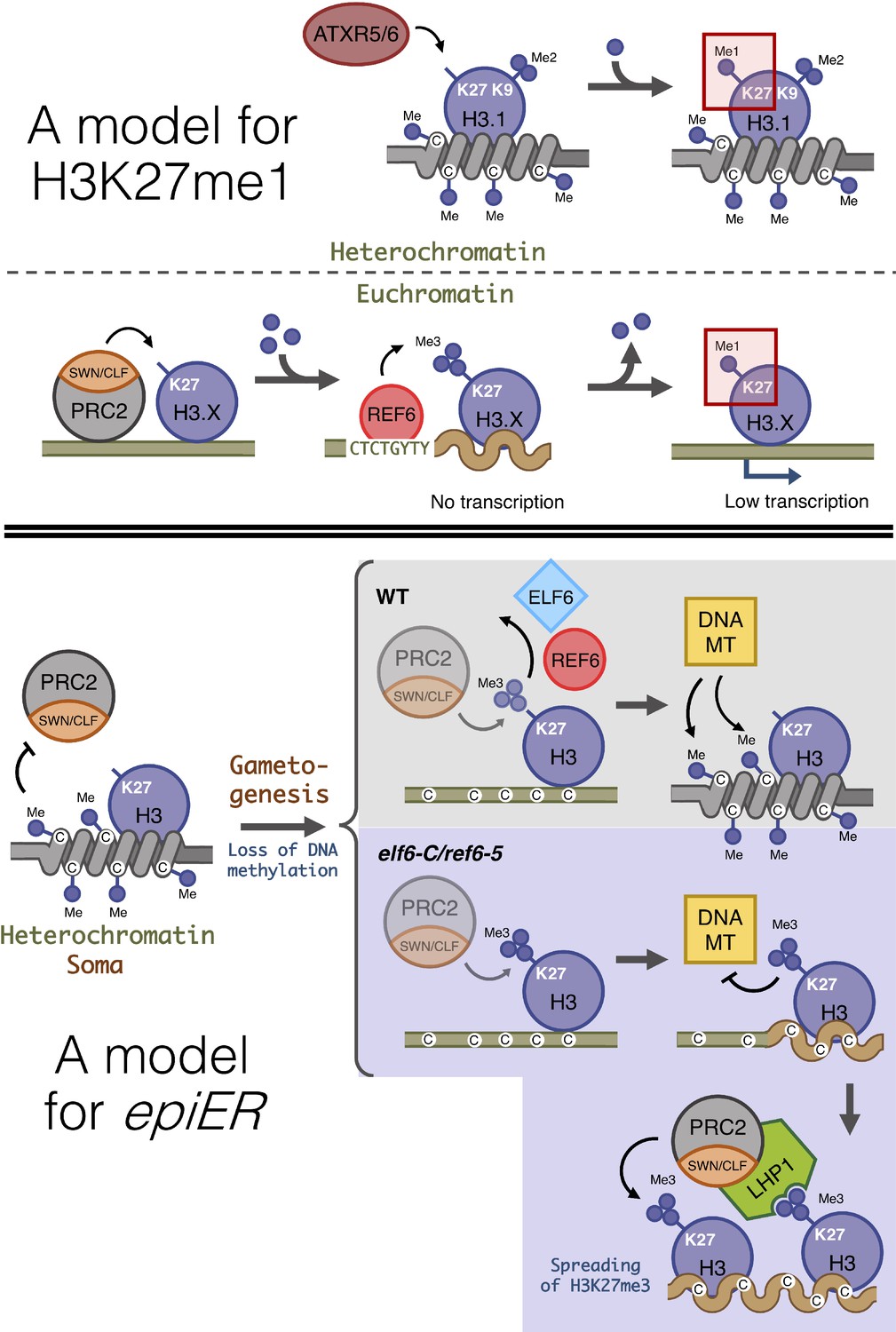
Proposed model for the role of histone demethylases in the accumulation of H3K27me1 and the formation of epimutations arising in ELF6 and REF6 mutants (epiERs).
(Top panel) Model for the mechanisms implicated in the accumulation of H3K27me1. In pericentromeric heterochromatin of somatic cells, ATRX5/6 deposits in a single-step H3K27me1 on histones containing H3K9me2. In euchromatin of somatic cells, the PRC2 complex deposits H3K27me3, which is converted to H3K27me1 by the catalytic activity of REF6. (Bottom panel) Model for the origin of epiERs. In constitutive heterochromatin of wild type somatic cells, DNA methylation obstructs the PRC2 complex from depositing H3K27me3. In gametes of wild-type plants, reprogramming of DNA methylation facilitates the deposition of H3K27me3 by the PRC2 complex, but these imprints are actively removed by histone demethylases, thus permitting DNA methylases to establish normal levels of methylation. In gametes of elf6-C/ref6-5, the H3K27me3 deposited by the PRC2 complex accumulates during the reprogramming of DNA methylation and interfere with the activity of DNA methyltransferases. The ectopic accumulation of H3K27me3 spreads to flanking genomic regions by recruitment of LHP1 and the PRC2 complex. Dark blue circles, methyl groups. C, Cytosines. H3.X, H3 variant that is not H3.1. Coiled lines represent closed and inactive chromatin. Wavy lines represent Polycomb-repressed chromatin. DNA MT: DNA methyltransferases. Faded shapes represent low amount of enzyme.
Author response image 1
Author response image 2
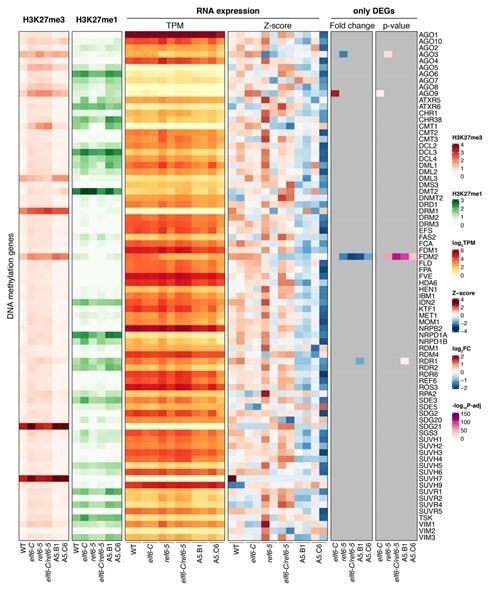
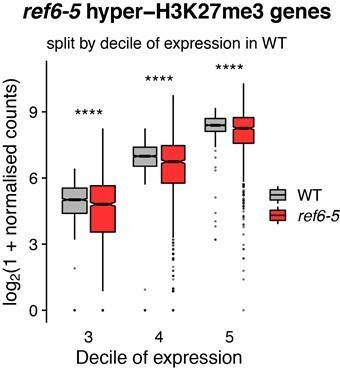
Author response image 3
Additional files
-
Supplementary file 1
Misregulated genes in epiERs.
- https://cdn.elifesciences.org/articles/58533/elife-58533-supp1-v2.xlsx
-
Supplementary file 2
New TE insertion sites predicted by epiTEome in epiERs.
- https://cdn.elifesciences.org/articles/58533/elife-58533-supp2-v2.xlsx
-
Supplementary file 3
Oligonucleotides used to genotype genetic lesions in mutants and epiERs.
- https://cdn.elifesciences.org/articles/58533/elife-58533-supp3-v2.xlsx
-
Supplementary file 4
ChIP peaks and associated genes.
- https://cdn.elifesciences.org/articles/58533/elife-58533-supp4-v2.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/58533/elife-58533-transrepform-v2.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A new role for histone demethylases in the maintenance of plant genome integrity
eLife 9:e58533.
https://doi.org/10.7554/eLife.58533




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}