Structures of diverse poxin cGAMP nucleases reveal a widespread role for cGAS-STING evasion in host–pathogen conflict
- Department of Microbiology, Harvard Medical School, United States
- Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, United States
- Harvard PhD Program in Virology, Division of Medical Sciences, Harvard University, United States
- Parker Institute for Cancer Immunotherapy at Dana-Farber Cancer Institute, United States
Figures
Figure 1 with 1 supplement
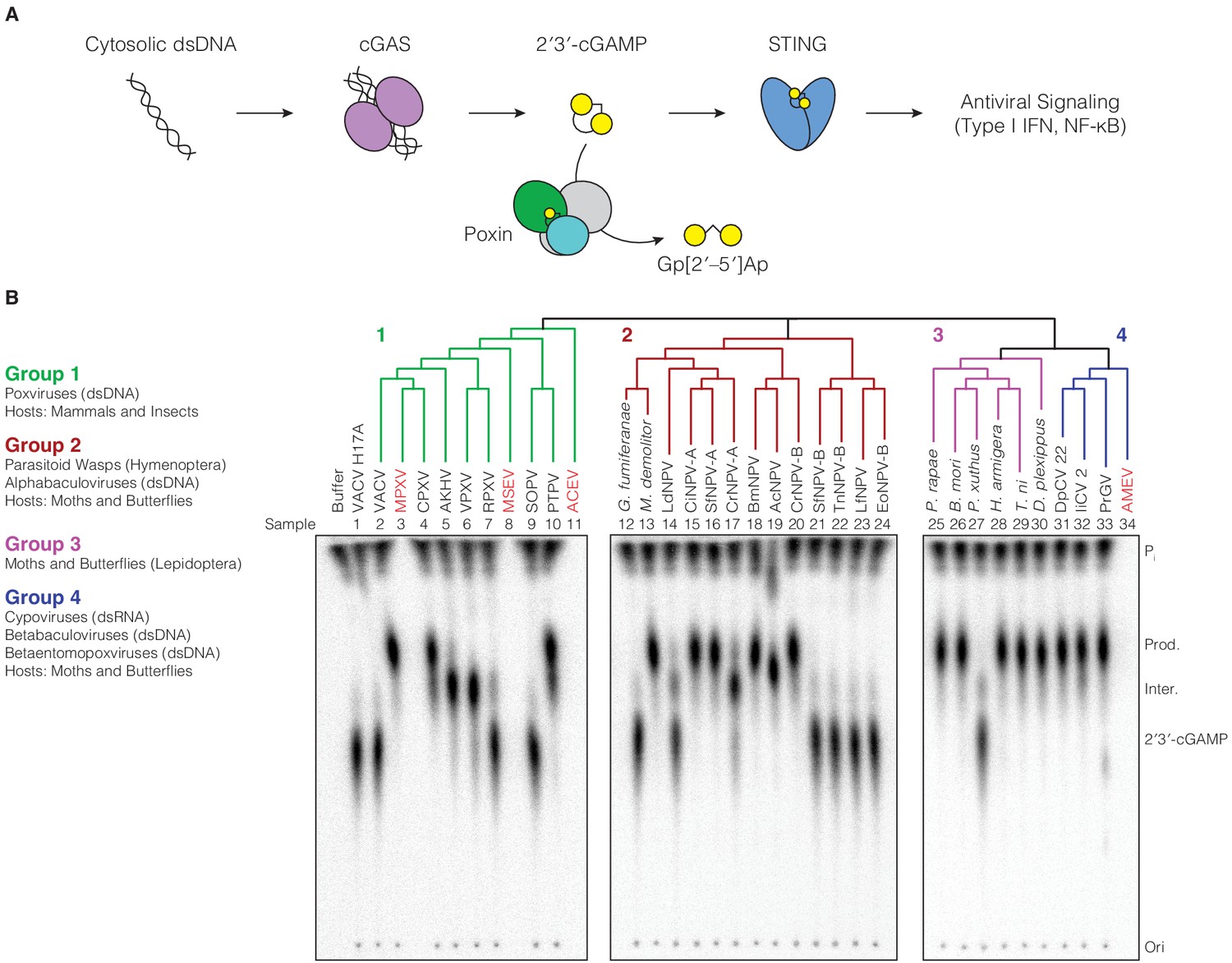
Poxin 2′3′-cGAMP nuclease activity is conserved across three families of viruses and two orders of insects.
(A) Schematic of cGAS-STING signaling. The sensor cyclic GMP–AMP synthase (cGAS) detects cytosolic DNA and synthesizes the second messenger 2′3′-cGAMP from ATP and GTP. 2′3′-cGAMP activates the receptor Stimulator of Interferon Genes (STING) and initiates downstream antiviral signaling. Poxin enzymes inhibit cGAS-STING signaling by degrading 2′3′-cGAMP and blocking activation of STING. (B) Bioinformatic identification (Figure 1—figure supplement 1A) and biochemical verification of diverse poxin enzymes. Poxins can be divided into groups, including enzymes from mammalian and insect poxviruses (Group 1), parasitoid wasps and alphabaculoviruses (Group 2), moths and butterflies (Lepidoptera) (Group 3), and cypoviruses, betabaculoviruses, and betaentomopoxviruses (Group 4). Poxin 2′3′-cGAMP nuclease activity is conserved within each group. Recombinant proteins (Figure 1—figure supplement 1B) were incubated with radioactively labeled 2′3′-cGAMP for 1 hr at 37°C, and degradation products were resolved using thin-layer chromatography (TLC). Four proteins could not be expressed in E. coli (red text, empty lanes on TLC plate). See Materials and methods for protein accession numbers. Data are representative of two independent experiments.
Figure 1—figure supplement 1
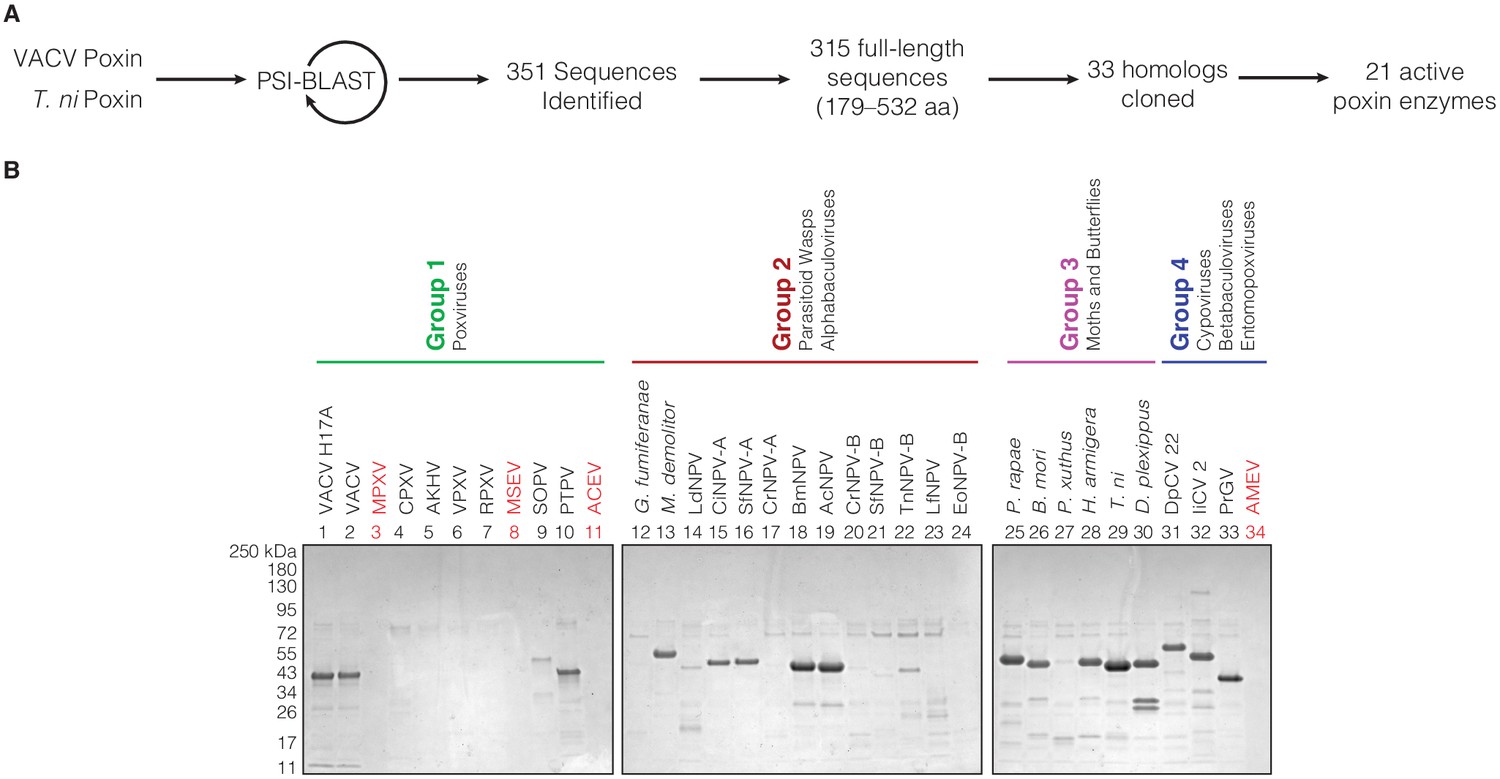
Bioinformatic identification and biochemical characterization of poxin homologs.
(A) Schematic showing process for identifying and selecting poxin homologs for biochemical characterization. (B) SDS-PAGE gels showing level of poxin protein expression for each construct used in the biochemical screen in Figure 1B.
Figure 2 with 2 supplements

Poxin crystal structures define a conserved catalytic mechanism and species-specific active-site adaptations.
(A) X-ray crystal structures of poxin enzymes from an alphabaculovirus (AcNPV), the moth Trichoplusia ni, and a betabaculovirus (PrGV) allow comparison with VACV poxin (PDB: 6EA9) and demonstrate family-wide structural conservation (Figure 2—figure supplement 1A). (B, C) Conservation of the active-site triad between VACV and AcNPV poxin. (D, E) The insect T. ni and betabaculovirus PrGV poxin proteins contain altered active-site triads composed of two histidines, and an arginine or lysine residue. Consistent with alternative active-site residues, lepidopteran poxin enzymes show remarkably different kinetic properties (Figure 2—figure supplement 2). (F) Despite differences in active-site residues (Figure 2—figure supplement 1B,C), 2′3′-cGAMP is contorted within each structure into a similar strained conformation, confirming that poxins operate by the same core metal-independent mechanism established for VACV poxin. (G) Shared poxin active-site features (see Figure 2—figure supplement 1C) include hydrophobic pockets for each base of 2′3′-cGAMP and charged interactions that read out the 2′–5′ phosphodiester linkage.
Figure 2—figure supplement 1
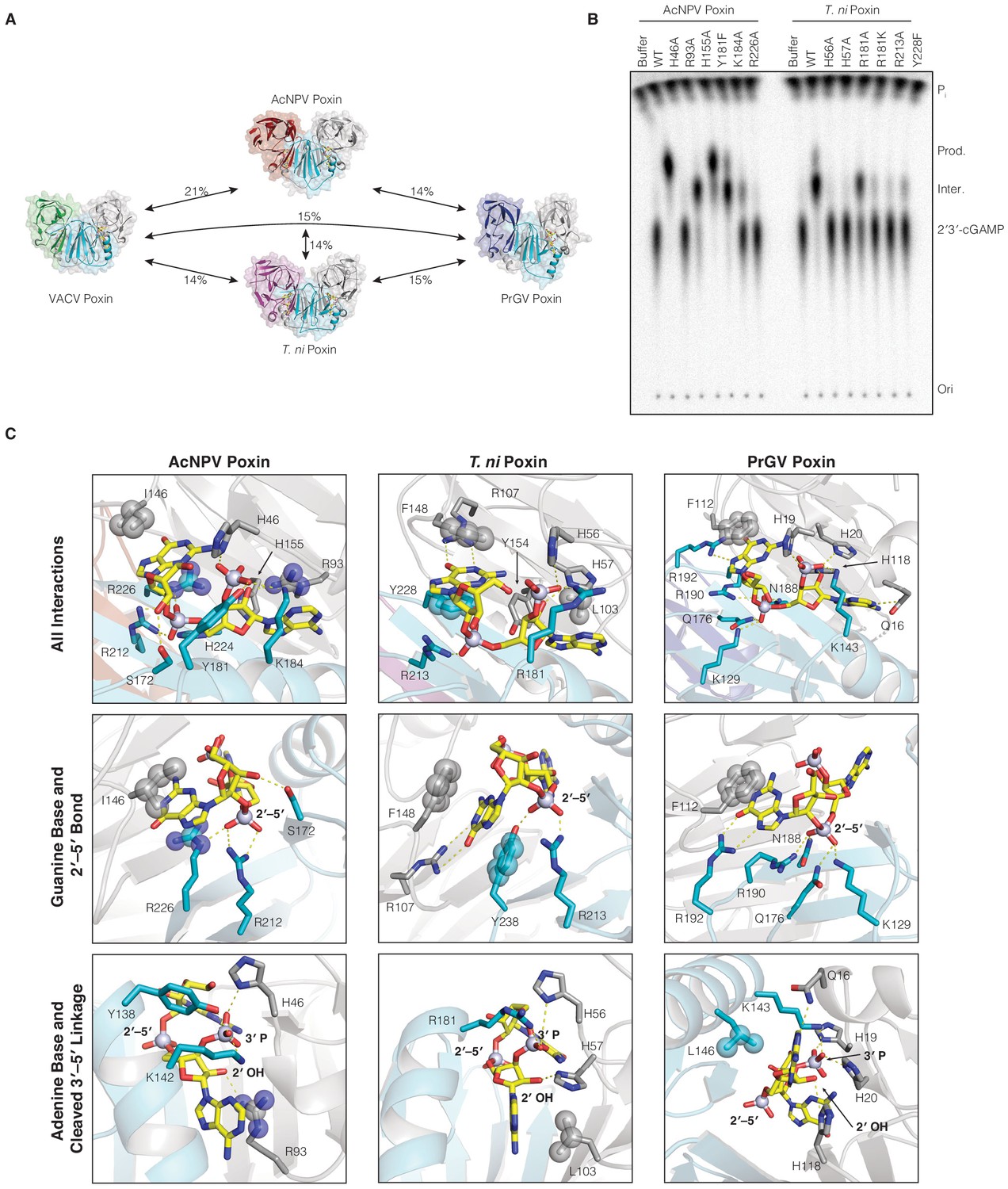
Comparison of 2′3′-cGAMP recognition and degradation in insect virus and host poxin enzymes.
(A) Poxin proteins retain the same core dimeric architecture and protein fold but are extremely divergent at the amino-acid level sharing <21% sequence identity. (B) Mutational analysis of active-site amino acids required for catalysis in AcNPV and T. ni poxin proteins. Data are representative of two biological replicates. (C) Poxin proteins make extensive contacts with 2′3′-cGAMP within the active-site pocket. Despite extensive sequence diversity, each poxin enzyme retains conservation of nucleobase interactions within hydrophobic pockets and basic residues that stabilize the 2′3′-cGAMP phosphodiester backbone. .
Figure 2—figure supplement 2

Host and viral poxins have alternative enzyme kinetics and ligand binding properties.
(A–C) Plots of enzyme kinetics data for poxin enzymes from Groups 1, 2, and 3. Error bars are ± standard error of the mean for n = 3 technical replicates. Each plot is representative of at least two independent experiments. (D) Table showing values computed from plots in (A–C) giving KM, Kcat, and catalytic efficiency for each tested enzyme with 95% confidence interval in parentheses below. Viral poxin homologs from poxviruses and baculoviruses share similar enzyme kinetics and catalytic efficiencies. In contrast, the lepidopteran poxin from T. ni exhibits much higher KM and Kcat values, and a lower catalytic efficiency. (E–G) Gel shift analysis of stable poxin–2′3′-cGAMP complex formation using active-site mutant proteins. No stable 2′3′-cGAMP binding was observed for mutant T. ni poxin using a range of 2′3′-cGAMP concentrations from 0.025 to 20 µM. Each image is representative of three independent experimental replicates. (H) Quantification of VACV H17A and AcNPV H46A gel shifts in E and F, and determination of KD for 2′3′-cGAMP. Note, the histidine inactivating mutation likely also impairs 2’3’-cGAMP resulting in an underestimated KD value. Error bars are ± standard error of the mean for n = 3 independent experimental replicates.
-
Figure 2—figure supplement 2—source data 1
Enzyme Kinetics Source Data.
- https://cdn.elifesciences.org/articles/59753/elife-59753-fig2-figsupp2-data1-v2.xlsx
-
Figure 2—figure supplement 2—source data 2
Enzyme Binding Source Data.
- https://cdn.elifesciences.org/articles/59753/elife-59753-fig2-figsupp2-data2-v2.xlsx
Figure 3
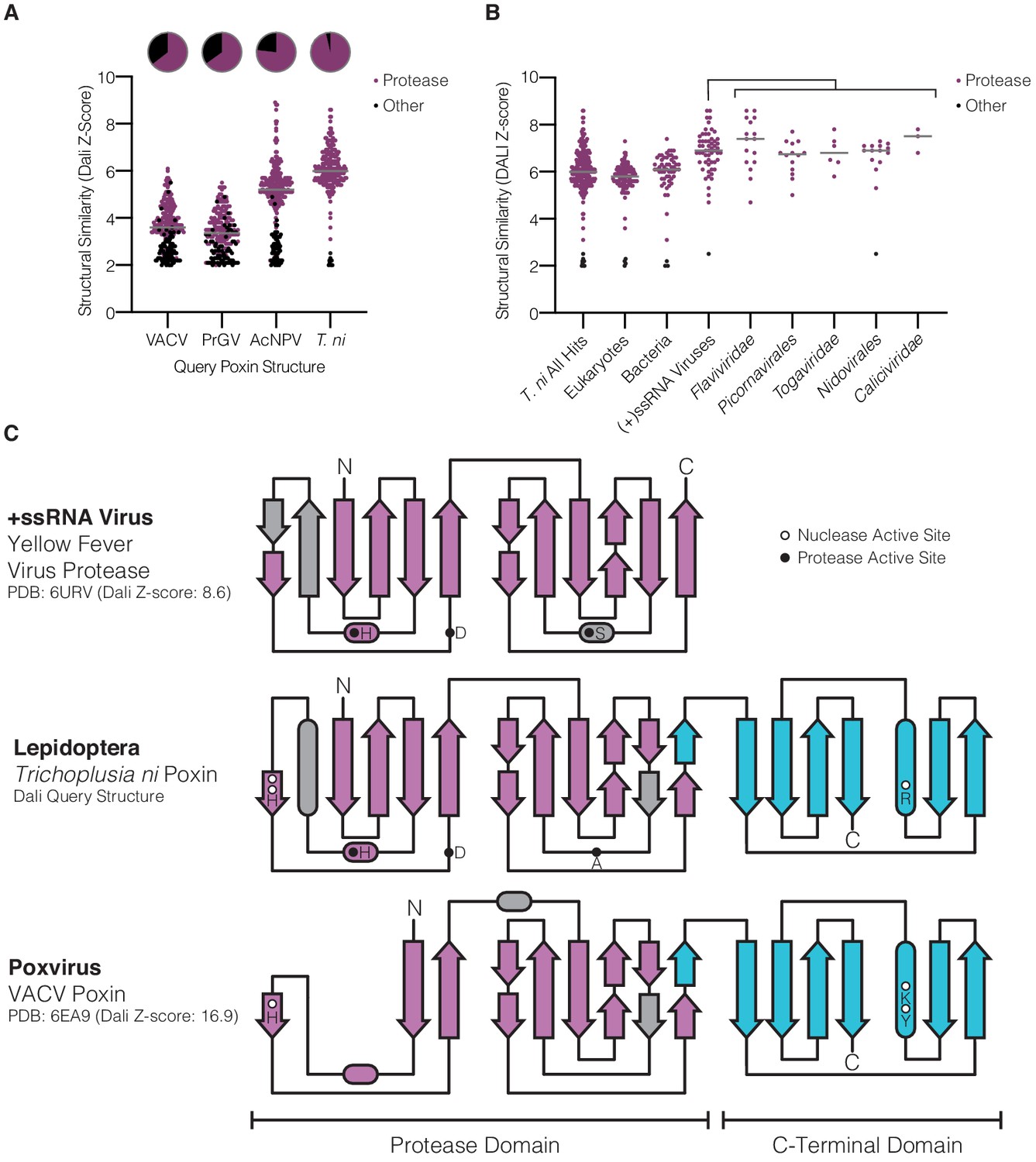
Lepidopteran poxins share structural homology with (+)ssRNA viral proteases.
(A) Dot plot comparing homology of each poxin structure to representatives in the Protein Data Bank. Poxin homologs were identified by DALI and graphed according to Z-score (higher Z-score indicates stronger homology) with a cutoff of 2. Red dots denote homologs that are serine proteases, and the proportion of hits assigned as proteases is indicated in the pie chart above. (B) Dot plot as in A depicting homology of T. ni poxin to enzymes in the Protein Data Bank, broken down by phylogenetic group. T. ni poxin shares the strongest homology with proteases from (+)ssRNA viruses. Gray bars in A and B represent the median Dali Z-score value for each distribution. (C) Topology diagram demonstrating conservation of a dual Greek key β-barrel chymotrypsin-like protease fold in the N-terminal domain of T. ni poxin, compared with the yellow fever virus protease and VACV poxin. Residues corresponding to the protease active site are denoted as filled circles, and poxin nuclease active-site residues are denoted as open circles.
Figure 4 with 1 supplement
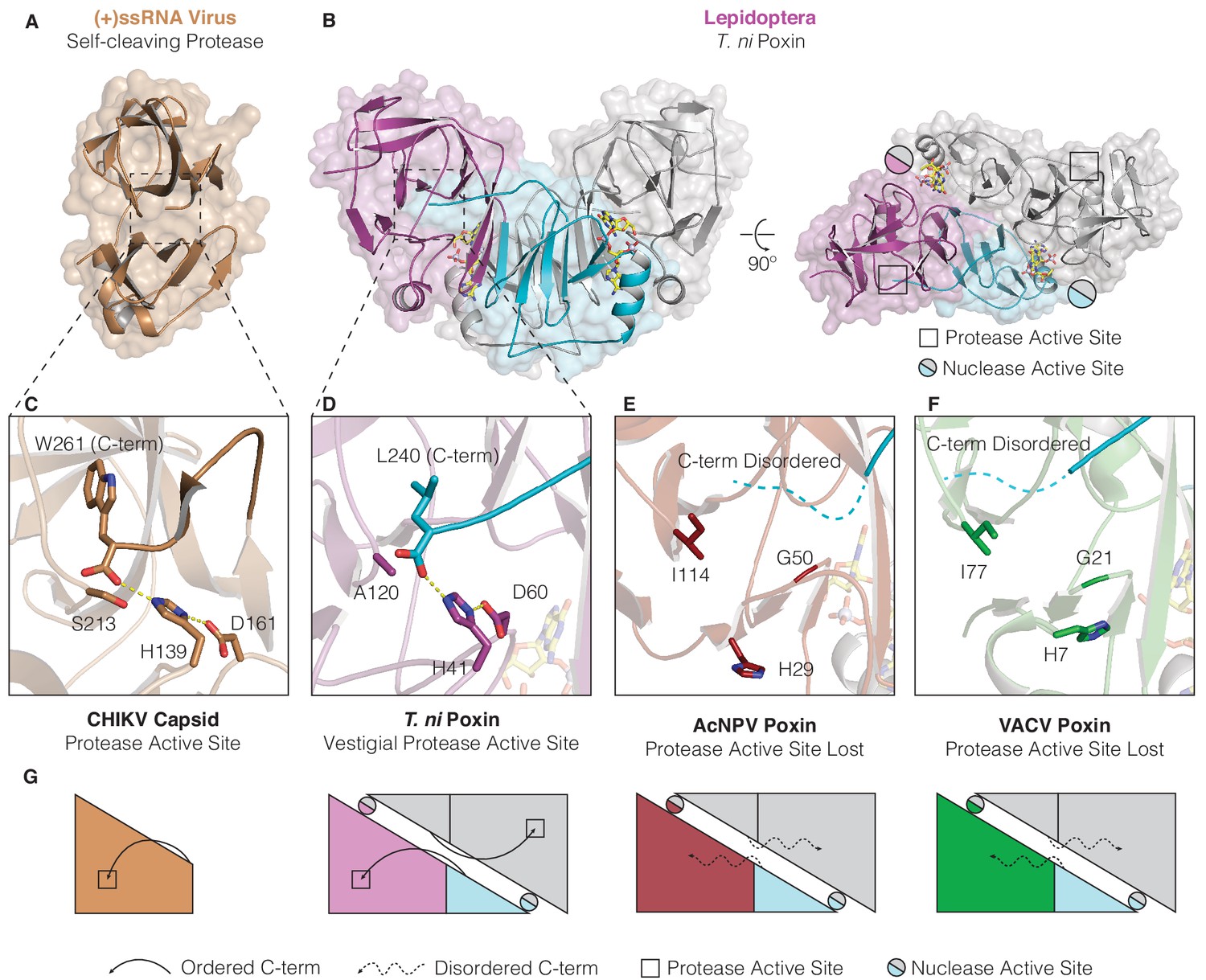
Structures of lepidopteran poxins reveal a vestigial self-cleaving protease active site.
(A) Structure of a (+)ssRNA virus self-cleaving accessory protease, the chikungunya virus capsid protein (PDB: 5H23) (Sharma et al., 2018). (B) The structure of the lepidopteran T. ni poxin reveals a vestigial protease active site (black boxes, at right) that is distinct from the poxin nuclease active site which forms at the dimer interface (circles, at right). Protease-like features are conserved amongst lepidopteran poxins (Figure 4—figure supplement 1). (C, D) Dotted lines show closeup views of the protease active site of a functional (+)ssRNA virus self-cleaving protease, and the vestigial protease active site of T. ni poxin. (E, F) Closeups of the same region of AcNPV and VACV poxin as in D demonstrating degeneration of the protease active site in viral poxins. (G) Schematics indicating how lepidopteran poxins coordinate the C-terminus within a protease active-site pocket similar to the chikungunya virus capsid self-cleaving protease. Although the vestigial protease active site has been lost in AcNPV and VACV poxin, the poxin nuclease active site is preserved at the dimer interface in these enzymes.
Figure 4—figure supplement 1
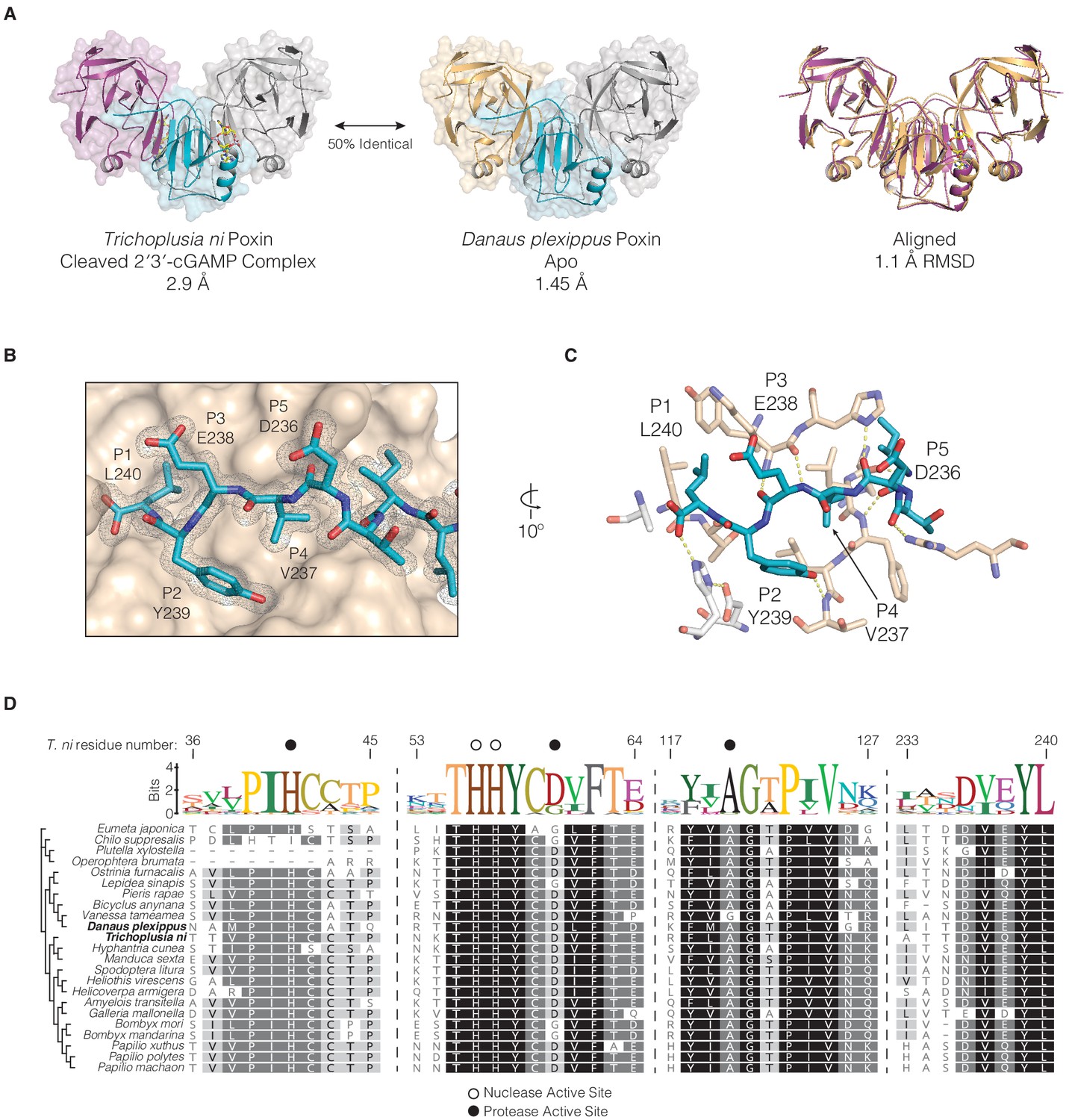
Conservation of protease-like features in lepidopteran poxin enzymes.
(A) Structures of T. ni poxin and the related Danaus plexippus (monarch butterfly) poxin protein. These proteins share 50% amino-acid identity to one another and a similar overall structure with an RMSD of 1.1 Å. (B) The D. plexippus poxin C-terminal peptide is recognized within a groove on the surface of the protease domain. The 2FO−FC electron density map is contoured at 1.0σ and shown in grey mesh. (C) Closeup showing details of interactions between the D. plexippus poxin C-terminal peptide and the surface of the protease domain. The side chains at the P1, P2, and P4 positions are coordinated within hydrophobic pockets demonstrating a mechanism for recognition and binding of the C-terminal peptide in lepidopteran structures. Additional hydrogen bonding interactions between the C-terminal peptide backbone and residues on the surface of the protease domain stabilize and assist in positioning of the C-terminal peptide within the vestigial protease active site (white sticks). (D) Alignment and phylogeny of lepidopteran poxin enzymes. Dotted lines represent breaks in the sequence presented, and residue numbers at top represent the corresponding positions within T. ni poxin. Dark circles denote residues within the vestigial protease active site, and open circles denote nuclease active-site residues.
Figure 5 with 2 supplements
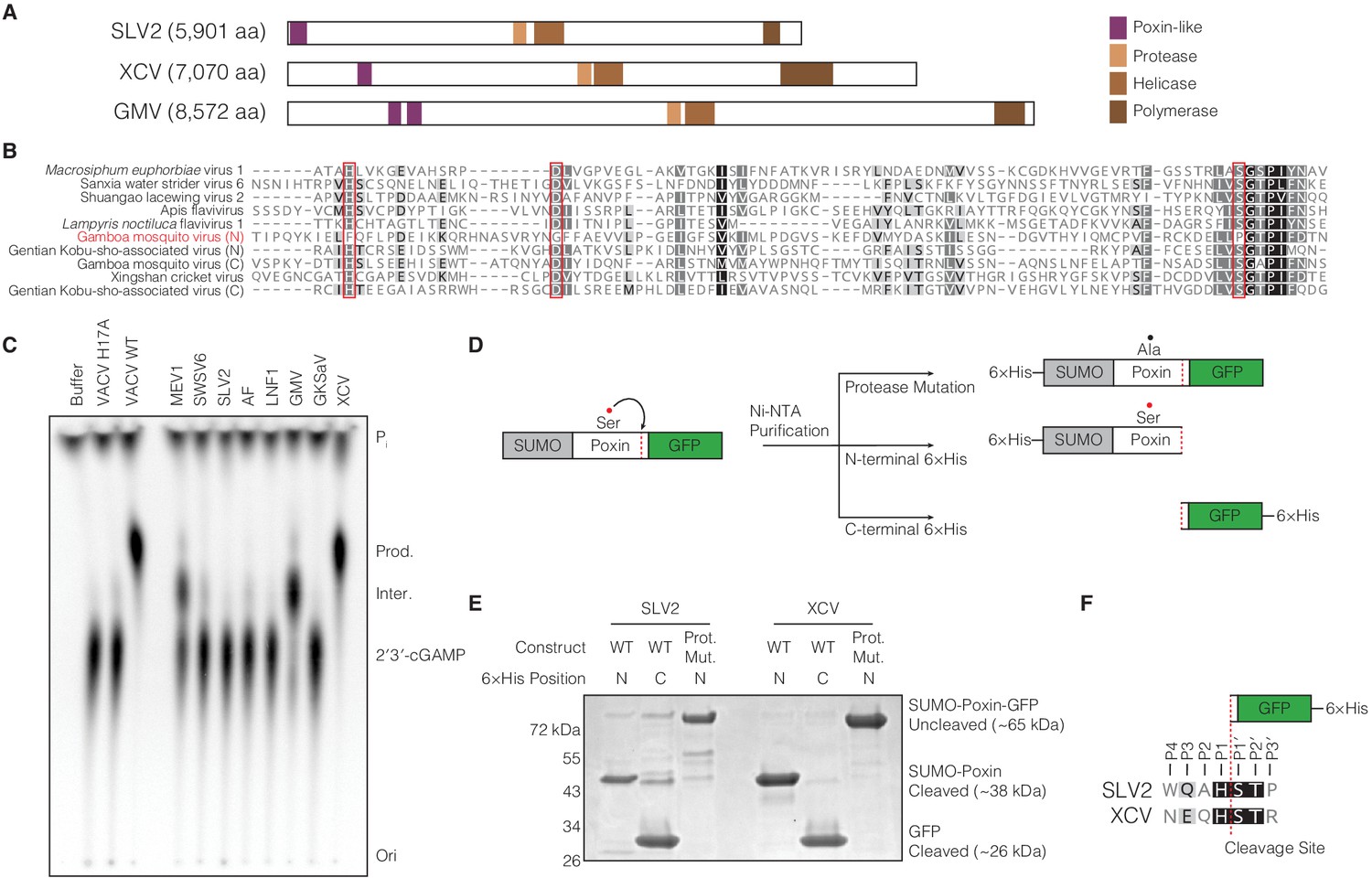
(+)ssRNA viruses encode functional self-cleaving poxin nucleases.
(A) Diagrams of three representative amarillovirus polyproteins with poxin-like regions identified by PSI-BLAST highlighted in purple. Poxin is encoded in (+)ssRNA viral genomes at the extreme N-terminus or up to ~1000 amino acids inside the polyprotein, and is duplicated in some viruses like Gamboa mosquito virus (Figure 5—figure supplement 1). (B) Alignment confirming strong conservation of putative protease catalytic residues (red boxes) within amarillovirus poxins. Duplicated poxin proteins are denoted (N) or (C) indicating their positions in the polyprotein relative to one another. (C) TLC analysis of 2′3′-cGAMP degradation by recombinant amarillovirus poxin proteins (Figure 5—figure supplement 2). Amarillovirus poxin proteins from Macrosiphum euphorbiae virus 1, Gamboa mosquito virus, and XCV retain 2′3′-cGAMP nuclease activity. (D) Schematic showing constructs used to assess autoproteolytic cleavage activity of two amarillovirus poxin homologs from SLV2 and XCV. (E) SLV2 and XCV poxin proteins are active proteases. Self-cleavage during expression and purification allows separation of a SUMO-poxin fragment from a C-terminal GFP tag. Mutation to the amarillovirus poxin predicted protease catalytic site disrupts all detectable cleavage. (F) Edman degradation mapping identifies a conserved cleavage site in the C-terminus of SLV2 poxin and XCV poxin demonstrating strict recognition of an H/ST motif for autoproteolytic processing, despite these enzymes sharing only 21% identity. All data are representative of two independent experiments.
Figure 5—figure supplement 1
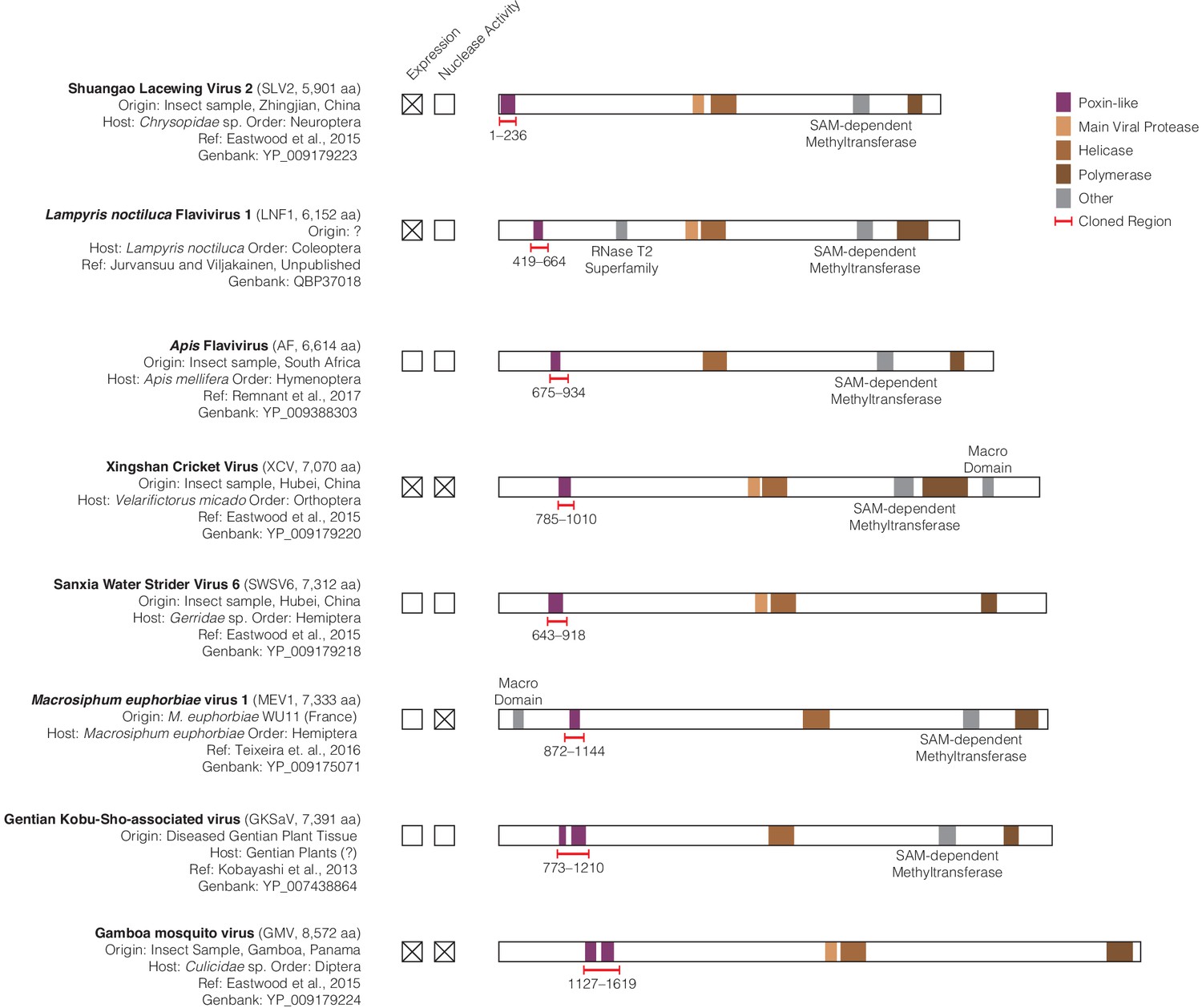
Amarillovirus genome structure and cloning of poxin-like region.
(A) Scale genome polyprotein diagrams providing detailed information on each studied amarillovirus encoding a putative poxin-like protein. These viruses were identified in insect samples from six different orders, collected in diverse locations from China to South America indicating a wide host range and geographic distribution. The poxin-like region identified by PSI-BLAST is shown in purple and resides close to the N-terminus in each polyprotein. Gentian Kobu-sho-associated virus and Gamboa mosquito virus exhibit duplication of the poxin region, with two adjacent poxin-like regions identified by PSI-BLAST. Regions marked with red brackets were selected for recombinant expression in E. coli and tested for 2′3′-cGAMP nuclease activity. All annotations were obtained using the InterProScan plugin (version 1.1.4) for Geneious Prime (Quevillon et al., 2005), with the exception of the poxin-like region, which was identified using PSI-BLAST (Thyssen et al., 2014). Each genome shared four conserved genome annotations: the poxin-like region, the main viral protease, the helicase, and the polymerase. All additional identified domains not shared between all genomes were annotated as ‘other’ and individually labeled.
Figure 5—figure supplement 2
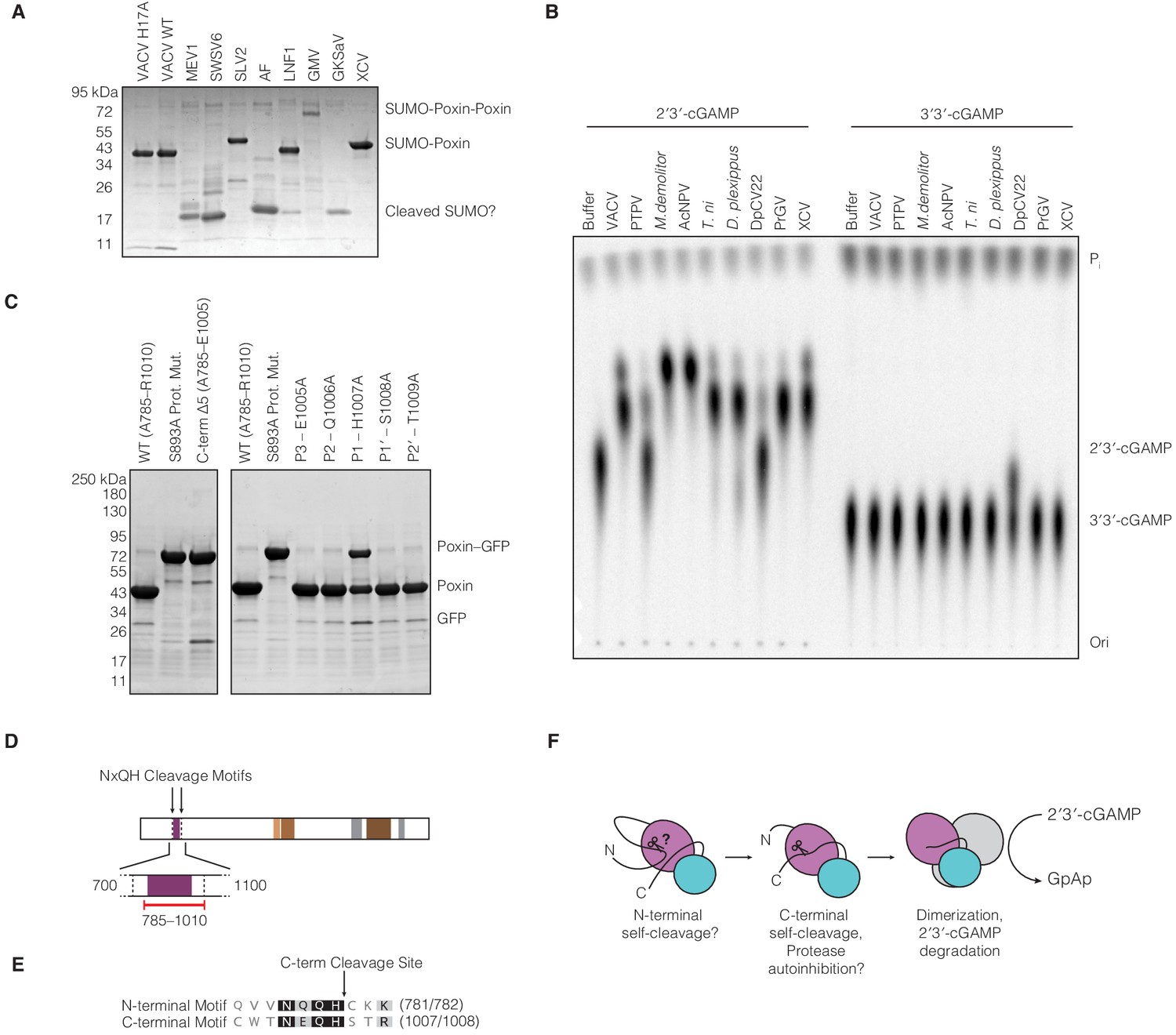
Biochemical analysis of amarillovirus poxin protease and nuclease activity.
(A) SDS-PAGE analysis of purified amarillovirus poxin constructs. All proteins are expected to migrate as a SUMO-poxin fusion ~40–50 kDa, except Gamboa mosquito virus and Gentian Kobu-sho-associated virus which encode two poxin sequences in tandem (SUMO-poxin-poxin). Fragments corresponding in size to SUMO which migrates ~17 kDa appear for most constructs which did not yield a substantial soluble poxin fragment, potentially indicating N-terminal cleavage and self-excision from the N-terminal 6 × His-SUMO2 tag used for purification. (B) TLC analysis of poxin specificity for 2′3′-cGAMP compared to 3′3′-cGAMP. Most poxins, including the amarillovirus XCV poxin, exclusively degrade 2′3′-cGAMP. However, Dendrolimus punctatus cypovirus 22 (DpCV 22) exhibits relaxed specificity and catalyzes some degradation of both 2′3′-cGAMP and 3′3′-cGAMP. Proteins are abbreviated as in Figures 1B and 5C. (C) SDS-PAGE analysis of XCV poxin mutant self-cleavage. Left: Removal of the XCV poxin cleavage site (C-term Δ5) completely abrogates proteolysis. Right: Alanine scanning of the XCV poxin cleavage site motif shows that H1007 at the P1 position, adjacent to the scissile peptide bond, is critical for efficient autoproteolysis. (D) Schematic of the XCV polyprotein showing the two loci at which the cleavage site motif NxQH occurs as dotted vertical lines. The solid black lines indicate a magnified region from amino acid 700–1,100, depicting in red the cloned construct used in this study. This construct included the C-terminal cleavage site but excluded a putative N-terminal cleavage motif. (E) Alignment of the XCV putative N-terminal cleavage motif with the experimentally identified C-terminal cleavage site. (F) Putative model explaining how amarillovirus enzymes like XCV poxin might self-excise from the polyprotein and assemble into functional 2′3′-cGAMP nucleases for immune antagonism.
Figure 6 with 1 supplement
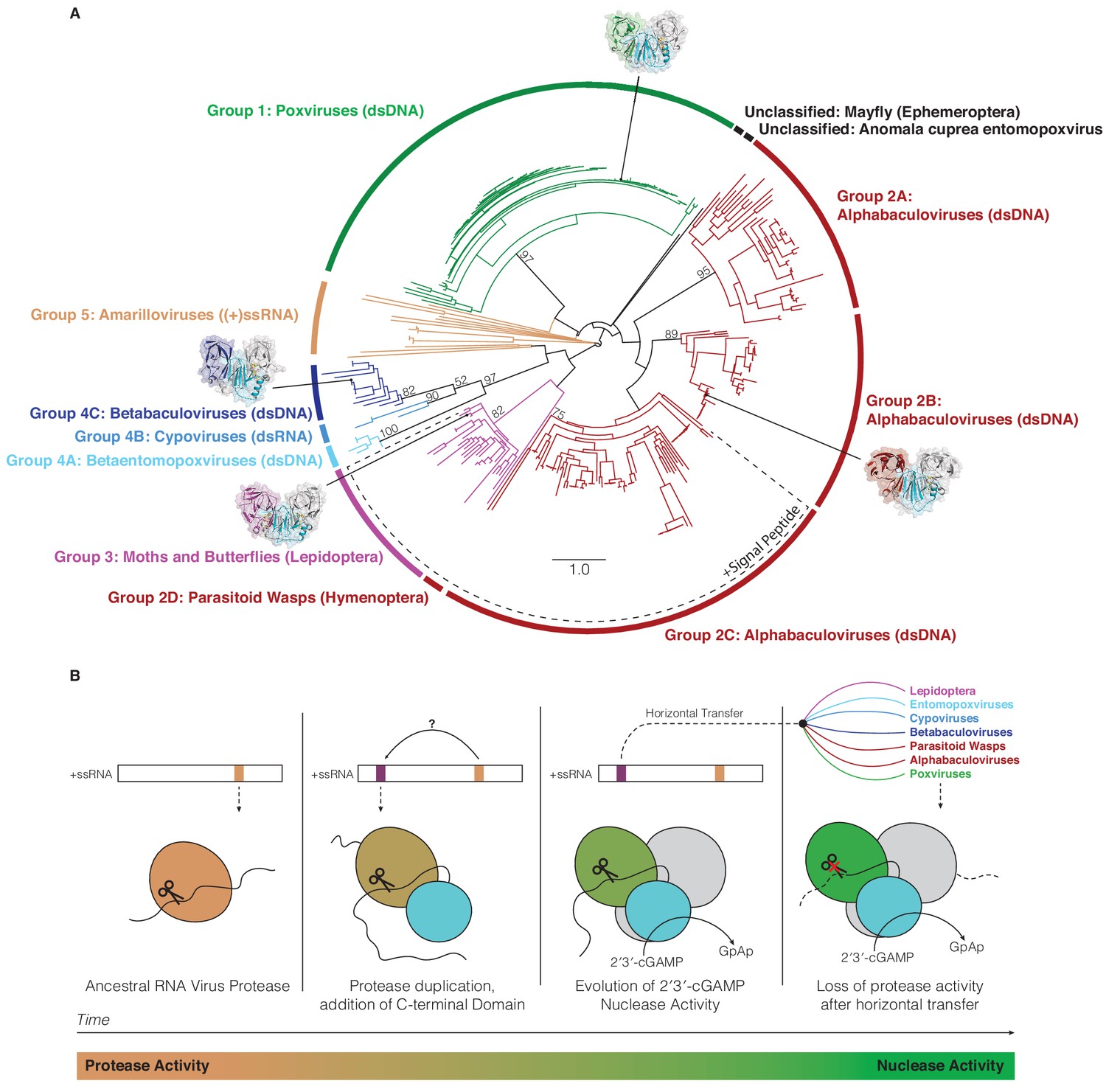
Horizontal transfer and evolution of poxin enzymes from proteases to nucleases.
(A) Structure-guided phylogenetic tree depicting global poxin diversity. 100 bootstrap replicates were performed, and branch support is provided for all poxin groups with support values > 50. Most poxin sequences within groups highlighted with a dotted line encode putative signal peptides, suggesting extracellular or secretory roles of some alphabaculovirus and lepidopteran poxin homologs. Poxin enzymes with crystal structures are highlighted with black dots. Unclassified poxins from the mayfly E. danica and Anomala cuprea entomopoxvirus were grouped together on the tree for simplicity but are not closely related to one another. The tree is visualized here as rooted to emphasize and enumerate global poxin sequence diversity, and the unrooted visualization is available in Figure 6—figure supplement 1, annotated with all bootstrap values for deep branches. (B) Model providing a possible explanation for the evolution of poxin proteins from (+)ssRNA viral proteases. Transition from protease to nuclease activity occurred through evolution of a second active site, and horizontal transfer to other viruses and insect hosts.
Figure 6—figure supplement 1

Unrooted visualization of structure-based maximum-likelihood poxin phylogeny.
A second, unrooted visualization of the tree in Figure 6. All bootstrap scores for deep branches in the tree are provided as numbers out of 100 total bootstrap replicates. Unclassified poxin sequences not belonging to other groups are individually labeled.
Additional files
-
Supplementary file 1
Poxin sequences identified and protein constructs utilized in this study.
- https://cdn.elifesciences.org/articles/59753/elife-59753-supp1-v2.xlsx
-
Supplementary file 2
Crystallographic statistics, related to Figures 2–4 and 6.
This file summarizes data collection, phasing, and refinement statistics for X-ray crystallography data. All crystallography datasets were collected from individual crystals. Values in parentheses are for the highest resolution shell.
- https://cdn.elifesciences.org/articles/59753/elife-59753-supp2-v2.docx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/59753/elife-59753-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structures of diverse poxin cGAMP nucleases reveal a widespread role for cGAS-STING evasion in host–pathogen conflict
eLife 9:e59753.
https://doi.org/10.7554/eLife.59753




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}