Specificity in glycosylation of multiple flagellins by the modular and cell cycle regulated glycosyltransferase FlmG
- Department of Microbiology & Molecular Medicine, Faculty of Medicine / CMU, University of Geneva, Switzerland
Abstract
How specificity is programmed into post-translational modification of proteins by glycosylation is poorly understood, especially for O-linked glycosylation systems. Here we reconstitute and dissect the substrate specificity underpinning the cytoplasmic O-glycosylation pathway that modifies all six flagellins, five structural and one regulatory paralog, in Caulobacter crescentus, a monopolarly flagellated alpha-proteobacterium. We characterize the biosynthetic pathway for the sialic acid-like sugar pseudaminic acid and show its requirement for flagellation, flagellin modification and efficient export. The cognate NeuB enzyme that condenses phosphoenolpyruvate with a hexose into pseudaminic acid is functionally interchangeable with other pseudaminic acid synthases. The previously unknown and cell cycle-regulated FlmG protein, a defining member of a new class of cytoplasmic O-glycosyltransferases, is required and sufficient for flagellin modification. The substrate specificity of FlmG is conferred by its N-terminal flagellin-binding domain. FlmG accumulates before the FlaF secretion chaperone, potentially timing flagellin modification, export, and assembly during the cell division cycle.
Introduction
Post-translational protein modification is essential for various facets in cellular biology, ranging from gene regulation to the organization of cellular structures. In all cases, biological function underlies the capacity to specifically identify and modify the correct target protein. Exquisite control mechanisms must be in place to ensure modification of the designated target, a feat that is more convoluted for proteins that are destined for the cell surface or the exterior, for example, for proteins that are first modified in the cytosol by dedicated glycosyltransferases (Keys and Aebi, 2017; Nothaft and Szymanski, 2010; Valguarnera et al., 2016). Unraveling the determinants underpinning the substrate selection is not only important for understanding the fundamentals and diversity in biological glycosylation systems but also has important translational implications for synthetic biology towards engineering recombinant glycoconjugates as vaccines or glycoproteins in other therapeutic applications (Nothaft and Szymanski, 2010; Vimr et al., 2004; Cuccui and Wren, 2015; Ghaderi et al., 2012).
In bacteria, extracellular proteinaceous surface structures including pili, flagella, and autotransporters as well as toxins are often post-translationally modified by glycosylation (Nothaft and Szymanski, 2010; Valguarnera et al., 2016; Vimr et al., 2004; De Maayer and Cowan, 2016; Miller et al., 2008; Szymanski et al., 2003; Schäffer and Messner, 2017; Goon et al., 2003; Schirm et al., 2003; Shen et al., 2006; Sulzenbacher et al., 2018; Lu et al., 2014). Since pili and flagella may be exposed to immune surveillance systems of eukaryotic cells, glycosylation of the structural subunits of these appendages, the pilin or flagellin, is often linked to virulence and evasion from the host immune system by molecular mimicry (Nothaft and Szymanski, 2010; Schäffer and Messner, 2017; Arora et al., 2005; Logan, 2006). In Salmonella enterica serovar Typhimurium another type of flagellin modification, methylation, was recently shown to promote adhesion to host cells (Horstmann et al., 2020). Flagellin glycosylation may potentially affect flagellar motility in many bacterial lineages since genomic and mass spectrometry data reveal that glycosylation systems are not restricted to pathogens but also occur in non-pathogenic bacteria found in the environment (De Maayer and Cowan, 2016; Schirm et al., 2005). In several polarly flagellated Gram-negative bacteria, flagellin glycosylation is required for assembly of the flagellar filament. In Campylobacter jejuni and Helicobacter pylori, two epsilon-proteobacteria that cause a broad range of human and animal diseases, glycosylation is required for flagellar assembly, motility, and virulence (Schirm et al., 2003; Linton et al., 2000; Guerry et al., 2006; Zebian et al., 2016). H. pylori has a monopolar flagellum, while C. jejuni is bipolarly flagellated (Kostrzynska et al., 1991; Guerry et al., 1991).
In Campylobacter species, the exact chemical nature of glycosylation is variable but generally a nine-carbon sugar related to sialic acids such as a pseudaminic acid or legionaminic acid derivative is appended to the flagellin (Thibault et al., 2001; Logan et al., 2002). Many Campylobacter strains possess three dedicated NeuB-like synthases: one for sialic acid (incorporated into the lipo-oligosaccharide), one for legionaminic acid, and one for pseudaminic acid, both used to modify flagellins (Linton et al., 2000; Sundaram et al., 2004; Chou et al., 2005; McNally et al., 2006; McNally et al., 2007; Schoenhofen et al., 2009). By contrast, Helicobacter species seem to use pseudaminic acid only for flagellin glycosylation (McNally et al., 2006; McNally et al., 2007; Schoenhofen et al., 2006). In both C. jejuni and H. pylori, loss of pseudaminic acid biosynthesis results in non-motile strains lacking flagella. The abundance of intracellular flagellin is severely reduced in these mutants and the flagellins showed increased mobility by SDS-PAGE (polyacrylamide gel electrophoresis), consistent with the loss of glycosylation (Schirm et al., 2003; Linton et al., 2000). Similarly, polar flagellation in the gamma-proteobacteria such as pathogenic Aeromonas spp and the non-pathogenic environmental bacterium Shewanella oneidensis depends on glycosylation of flagellin with pseudaminic acid and another nonulosonic acid derivative, respectively (Sun et al., 2013; Schirm et al., 2005; Wilhelms et al., 2012). Interestingly, pseudaminic acid is also a component of surface polysaccharides such as the O-antigen of lipopolysaccharide (LPS) in Aeromonas caviae or the capsular polysaccharide (K antigen) in the symbiotic alpha-proteobacterium Sinorhizobium fredii NGR234 (Forsberg and Reuhs, 1997; Le Quéré et al., 2006; Margaret et al., 2012). In A. caviae, the genes required for pseudaminic acid biosynthesis are encoded in the O-antigen cluster and their mutation affects both flagellum and LPS O-antigen biosynthesis (Canals et al., 2007; Tabei et al., 2009).
The basis for substrate specificity in protein glycosylation systems is poorly understood and hampers biotechnological exploitation of these protein modification systems for therapeutic purposes. Flagellin glycosylation occurs at serine or threonine residues by O-linking glycosyltransferases (henceforth OGTs) that modify their substrates to various extent for each flagellin system, ranging from modification at a single site for Burkholderia and Listeria species (Shen et al., 2006; Scott et al., 2011; Hanuszkiewicz et al., 2014) to promiscuous modification at 19 serine or threonine residues for the C. jejuni flagellin (Schirm et al., 2005; Thibault et al., 2001). The modification usually occurs at the two surface-exposed central domains of flagellin, ideally positioned to influence the immunogenicity of the filament and the virulence in pathogens (Arora et al., 2005; Verma et al., 2005). Since no consensus sequence determinant in the primary structure of the flagellin acceptor (apart from the serine or threonine modification site) has been identified (Thibault et al., 2001), OGTs likely recognize the tertiary structure of the glycosyl acceptor in a highly specific manner. Evidence has been provided that glycosylation precedes secretion of the flagellin (Parker et al., 2014) via the flagellar export machinery to the tip of the growing flagellar filament (Chevance and Hughes, 2008). Thus, flagellin identification and subsequent glycosylation by the OGT must occur in the cytoplasm, presumably by soluble proteins. During flagellar assembly in Gram-negative (diderm) bacteria, the basal body harboring the export apparatus is assembled first in the cytoplasmic membrane, followed by envelope-spanning structures along with the external hook structure that serves as universal joint between the flagellar filament and the envelope-spanning parts (Chevance and Hughes, 2008). The flagellins are assembled last by polymerization on the hook into the flagellar filament (Figure 1A). They are usually the last proteins to be expressed and secreted during assembly, relying on temporal control mechanisms of gene expression promoting the orderly assembly of the flagellum. A key feature of polarly or bipolarly flagellated bacteria is that they must assemble a new flagellum each cell cycle. Thus, flagellar assembly, including potentially flagellin glycosylation, must be cell cycle regulated, but this remains unexplored.
Figure 1
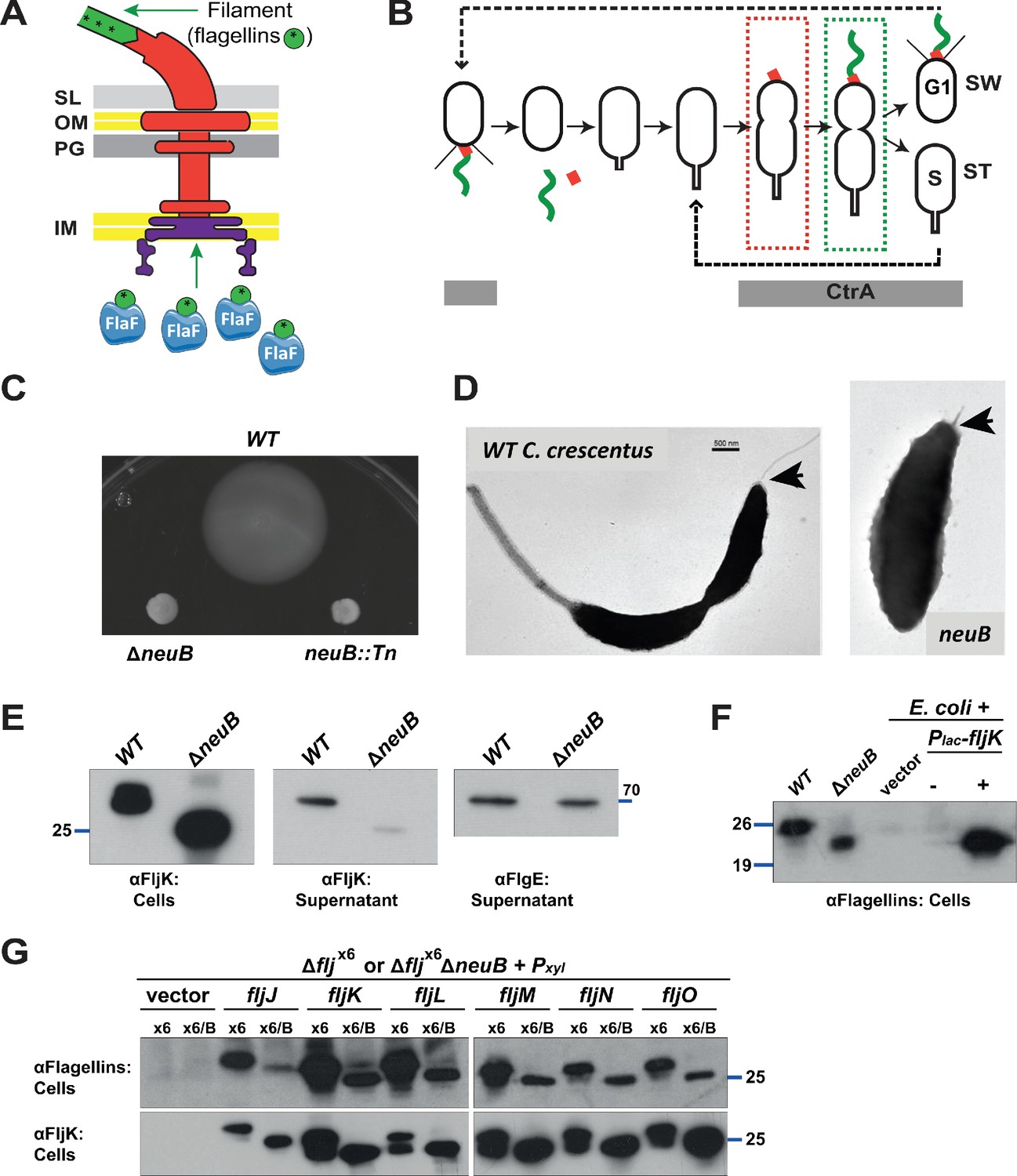
Mutation of neuB affects the assembly of the flagellar filament.
(A) Schematic of the C. crescentus flagellum with the MS- and C-ring structures in the inner membrane (IM), the hook basal body components spanning the periplasm (with the peptidoglycan – PG – layer) and outer membrane (OM), and the filament. Flagellin subunits (in green) are brought to the export machinery by the secretion chaperone FlaF. Purple star on flagellins indicates the post-translational modification by glycosylation. (B) Schematic of the C. crescentus cell cycle. The grey bar represents the time during the cell cycle when CtrA is present and activate transcription of flagellar genes. The hook structure (FlgE, in red) is synthesized in early pre-divisional cells, whereas the flagellar filament (in green) is polymerized from flagellins in late pre-divisional cells. Both,flagellar filament and hook, are shed during the swarmer (SW) to stalked (ST) cell transition. (C) Motility assay of neuB::Tn and ΔneuB mutants compared to WT strain. Overnight cultures were spotted on PYE soft agar plates and incubated for 72 hours at 30°C. Compact swarms indicate that neuB mutant cells are non-motile. (D) WT and neuB::Tn cells analyzed by transmission electron microscopy show that only a short protrusion is visible at the SW pole of neuB::Tn cells, in contrast to the WT strain (black arrow). The images suggest that in neuB mutant cells the flagellar hook is stably assembled, but not the flagellar filament. (E) Immunoblots o performed with anti-FljK (αFljK, raised against FljK produced in E. coli, see methods) and anti-FlgE (Hahnenberger and Shapiro, 1987) antibodies on cell lysates and supernatants of WT and ΔneuB cultures show that flagellins are produced in ΔneuB cells but not efficiently exported, whereas the export of the FlgE hook protein is not affected. The migration of FljK in ΔneuB cells is shifted towards lower molecular mass, suggesting that post-translational modification of flagellin is defective in the ΔneuB mutant. Molecular size standards are indicated by the blue lines with the corresponding value in kDa. (F) Immunoblot performed on extracts from E. coli cells expressing C. crescentus FljK under control of Plac from a plasmid. FljK expressed in E. coli shows the same migration profile as in ΔneuB cells, indicating that E. coli cells cannot post-translationally modify C. crescentus FljK. Molecular size standards are indicated by the blue lines with the corresponding value in kDa. Note that antibodies used in this immunoblot were raised against flagellins purified from C. crescentus (αFlagellins; Hahnenberger and Shapiro, 1987). (G) Immunoblots on extracts from Δfljx6 (x6) and Δfljx6ΔneuB (x6/B) cells expressing each flagellin from a plasmid under Pxyl control. The immunoblots were performed with antibodies raised against purified C. crescentus flagellins (upper panel) or FljK expressed and purified from E. coli (lower panel). In both cases, all six flagellins show a shift to a lower molecular mass in their migration in the absence of neuB, suggesting that all six flagellins are post-translationally modified. Molecular size standards are indicated by the blue lines with the corresponding value in kDa. Both antibodies recognize all six flagellins. Note that antibodies raised against flagellins purified from Caulobacter recognize the glycosylated form of all six flagellins better, whereas the antibodies raised against FljK expressed and purified from E. coli also efficiently recognizes unglycosylated flagellins.
The non-pathogenic and polarized alpha-proteobacterium C. crescentus serves as a model system to study how flagellation is regulated in space and as a function of the cell cycle (Skerker and Laub, 2004; Ardissone and Viollier, 2015). C. crescentus assembles a single flagellum at the newborn cell pole each cell cycle and then divides asymmetrically into a flagellated but non-replicative dispersal (swarmer, SW) cell that resides in a G1-like phase, and a capsulated sessile (stalked, ST) cell that engages in DNA synthesis (S-phase) and harbors the defining stalked appendage at the old cell pole (Figure 1B). Each SW cell undergoes a metamorphosis into a ST cell, replacing the flagellum with a stalk, a cylindrical extension of the cell envelope. In doing so, the flagellar filament and hook are released during the SW to ST cell transition while stalk outgrowth commences. Concurrently, replication competence is acquired and gene expression is reprogrammed toward the production of a SW daughter cell (Laub et al., 2007) that assembles a new polar flagellum in strict coordination with cell cycle progression.
Multiple spatiotemporal cell cycle control mechanisms feed into flagellar assembly in C. crescentus (Ardissone and Viollier, 2015). First, spatial cues that direct assembly of the flagellum to the proper site are deposited during cell division in the preceding cell cycle and inherited by the progeny (Huitema et al., 2006; Lam et al., 2006). Next, in S-phase, the transcriptional cell cycle activator CtrA and the TipF flagellar assembly organizer are expressed (Huitema et al., 2006; Davis et al., 2013; Quon et al., 1996; Holtzendorff et al., 2004; Fioravanti et al., 2013). CtrA induces transcription of early flagellar structural genes and regulators of late flagellar gene expression including the flagellin genes (Quon et al., 1996; Stephens and Shapiro, 1993; Laub et al., 2002; Fumeaux et al., 2014; Fiebig et al., 2014). Once a functional flagellar secretion structure has been assembled, the newly synthesized flagellins are exported by the FlaF secretion chaperone (Ardissone et al., 2020; Llewellyn et al., 2005; Figure 1A).
Here we identify, reconstitute, and dissect the substrate specificity of the O-linked flagellin glycosylation pathway of C. crescentus. A peculiarity of C. crescentus is that it expresses six flagellin paralogs (Nierman et al., 2001; Faulds-Pain et al., 2011): five structural flagellins (FljKLMNO) each of which is sufficient for flagellar filament formation and motility, while the regulatory FljJ flagellin controls translation of the others (Ardissone et al., 2020) but cannot support filament formation and motility in the absence of other flagellins (Faulds-Pain et al., 2011). We show that all six flagellins in C. crescentus are glycosylated in a manner that requires pseudaminic acid as donor in a reaction catalyzed by the newly identified soluble OGT FlmG. Reconstitution of FlmG-dependent flagellin glycosylation in two heterologous systems, S. fredii NGR234 that naturally produces pseudaminic acid to incorporate it into the K-antigen capsule and Escherichia coli K12 cells engineered to produce pseudaminic acid from C. crescentus enzymes, reveals that FlmG is sufficient for flagellin glycosylation. The underlying specificity of glycosylation resides in the modular organization of FlmG: an N-terminal substrate (flagellin) binding domain and a C-terminal glycosyltransferase domain. We show that both domains are required for flagellin glycosylation, formation of the flagellar filament, and motility, but not for flagellin export. Finally, our studies reveal how flagellin glycosylation is tuned with progression of the C. crescentus cell cycle to ensure that glycosylation by FlmG can occur as soon as flagellin is translated, potentially avoiding competition for flagellin binding by the FlaF secretion chaperone (Figure 1A).
Results
NeuB is required for flagellar filament assembly
Our previously assembled library of C. crescentus transposon (Tn) motility mutants (Huitema et al., 2006) included four mutants each harboring a Tn insertion in the uncharacterized gene CCNA_02961, predicted to encode a NeuB-like sialic acid synthase (henceforth neuB). Three Tn mutants harbor a himar1 insertion (NS7, NS44, and NS388) at different locations in neuB, while in the other (NS150) neuB is disrupted by an Ez-Tn5 insertion. All four mutants are non-motile on soft (0.3%) agar plates and do not swim when observed by phase contrast light microscopy. An in-frame deletion of neuB (ΔneuB) recapitulated the motility defect of the Tn insertions (Figure 1C). The expression of NeuB from a plasmid (pMT335 [Thanbichler et al., 2007], see below) corrected the motility defect of ΔneuB cells, indicating that neuB function is required for motility. Transmission electron microscopy (TEM) reveale a flagellar filament on the new pole of WT cells, whereas ΔneuB cells lack a flagellar filament and only harbor a short protrusion corresponding to a hook structure (Figure 1D, see below). The neuB gene is predicted to encode a 38 kDa protein belonging to the NeuB-family of acetylneuraminate synthases (Vimr et al., 2004; Linton et al., 2000; Chou et al., 2005), suggesting that biosynthesis of sugars of the sialic acid family is required for flagellation in C. crescentus.
To gain further insights into the flagellar assembly defect of ΔneuB cells, we investigated whether flagellins are synthesized and exported in the absence of NeuB by immunoblotting using antibodies to the FljK flagellin (that also cross-react with other flagellins, see below). These experiments revealed lower flagellin steady-state levels in the supernatants of ΔneuB cells compared to WT. By contrast, the FlgE hook protein was present in the supernatant of both WT and ΔneuB cells to comparable levels (Figure 1E), in agreement with TEM analyses. Moreover, the increased migration of flagellin through SDS-PAGE suggests that the molecular mass is reduced in the absence of NeuB, consistent with NeuB-dependent post-translational modification of flagellin. In support of this conclusion, FljK expressed in E. coli showed the same mobility as the mobility of FljK in ΔneuB cells (Figure 1F). Next, we asked whether all six flagellins show a NeuB-dependent shift in mobility by immunoblotting and found this to be the case (Figure 1G). In these experiments, we expressed individual flagellins from a plasmid under the control of an inducible promoter (Pxyl) in cells deleted for all six flagellin genes (Δfljx6) and compared the mobility to that of the flagellin expressed in Δfljx6 ΔneuB cells. In all cases, we observed a shift to an apparent lower molecular mass in the absence of NeuB. We conclude that NeuB controls the mobility of all six flagellins.
The pseudaminic acid synthase activity of NeuB is required for flagellation
NeuB family members are phosphoenolpyruvate (PEP)-dependent synthases that catalyze the condensation of PEP with hexoses to form sialic acid or derivatives, such as pseudaminic acid (Vimr et al., 2004; Linton et al., 2000; Sundaram et al., 2004; Chou et al., 2005; Gunawan et al., 2005; Liu et al., 2009). Some bacteria encode more than one NeuB enzyme, for example, the sialic acid synthase NeuB1 and the pseudaminic acid synthase NeuB3 from C. jejuni (Linton et al., 2000; Chou et al., 2005). The Neisseria meningitidis sialic acid synthase NeuB forms a domain-swapped homodimer, in which each monomer consists of an N-terminal TIM barrel domain similar to other PEP-utilizing enzymes and a C-terminal antifreeze-like domain (Gunawan et al., 2005; Liu et al., 2009). The catalytic site is located in the C-terminal end of the TIM barrel domain, but the antifreeze-like domain from the second monomer in the homodimer contributes key residues required for substrate binding (Gunawan et al., 2005). Based on primary structure alignment between NeuB from N. meningitidis and C. crescentus, we identified residues predicted to be involved in catalysis and substrate binding in C. crescentus NeuB (Figure 2A). To confirm the role of these amino acids and the requirement of NeuB catalytic activity for its function in motility and flagellin modification, we engineered single amino acid substitutions in three highly conserved residues, glutamate at position 30 (E30, implicated in the stabilization of the reaction intermediate), histidine at position 245 (H245, involved in the coordination of the Mn2+ cofactor) and arginine at position 322 (R322, one of the residues of the antifreeze-like domain that participate in substrate binding in the active site of the second monomer in the dimer), and tested their ability to correct the motility defect of ΔneuB cells compared to WT NeuB (Figure 2B). The expression of NeuB variants from the vanillate-inducible Pvan promoter on pMT335 (Thanbichler et al., 2007) revealed that none of the variants were functional in the absence of the inducer. Under these conditions all variants, except H245A, were expressed to comparable steady-state levels as indicated by immunoblotting with polyclonal antibodies to C. crescentus NeuB (Figure 2C). In the presence of vanillate, the E30A and R322A variants still showed no activity, whereas H245A exhibited some activity in flagellin modification and motility, albeit far less than WT NeuB, despite accumulating to higher steady-state levels (Figure 2B and C). We conclude that NeuB catalytic activity is required for function in C. crescentus.
Figure 2
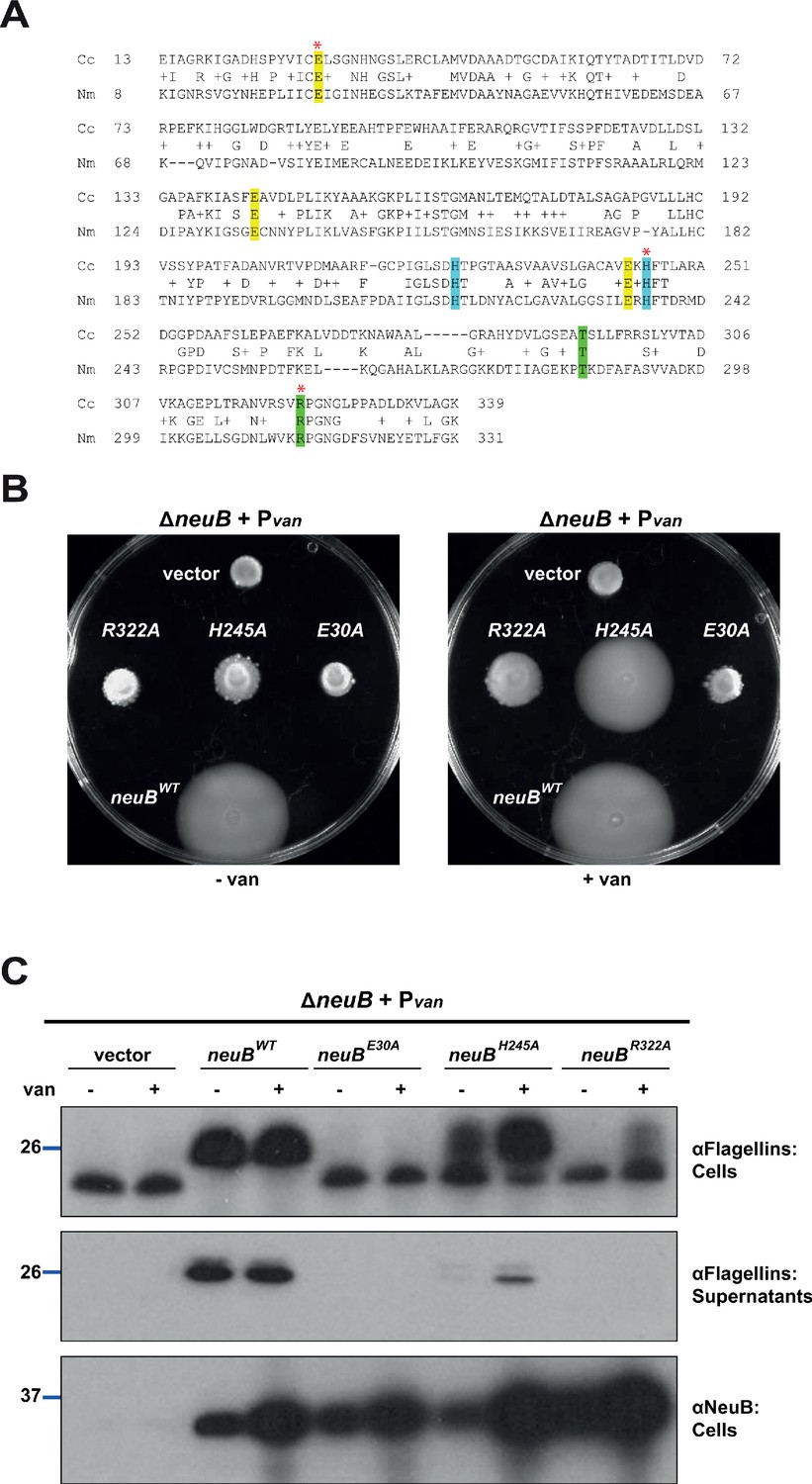
NeuB putative catalytic activity is required for motility and flagellin modification.
(A) Sequence alignment of C. crescentus NeuB (Cc) to N. meningitidis sialic acid synthase (Nm). The three glutamate residues that have been proposed to stabilize the reaction intermediate are highlighted in yellow; the two histidine residues that coordinate the Mn2+ cofactor are highlighted in blue; the threonine and arginine residues highlighted in green are located in the antifreeze-like C-terminal domain and protrude into the active site of the other subunit in the N. meningitidis sialic acid synthase dimer. The residues selected for site-directed mutagenesis in C. crescentus are indicated by a red asterisk. (B) Motility assay of ΔneuB cells complemented with different neuB alleles expressed from Pvan on a plasmid. Only the WT neuB allele can fully complement the motility defect of the ΔneuB strain, whereas the allele encoding NeuB(H245A) complements partially and the NeuB(E30A) and (R322A) versions do not restore motility. (C) Immunoblots showing the levels of flagellins and NeuB in ΔneuB cells complemented with different NeuB versions expressed from Pvan on a plasmid. All the NeuB variants were expressed (lower panel). Immunoblotting for flagellins in whole cell lysates (cells, upper panel) indicates that only the WT NeuB version can restore the migration profile of flagellins, whereas the E30A and R322A variants are inactive and the H245A variant shows an intermediate phenotype. The middle panel shows that only upon induction of the H245A version can the flagellins be detected in the culture supernatant, in agreement with the motility assay shown in panel B. Molecular size standards are indicated by the blue lines, with the corresponding value in kDa. Note that antibodies used in this immunoblot wereraised against flagellins purified from C. crescentus (αFlagellins; Hahnenberger and Shapiro, 1987) and they detect the glycosylated version of the flagellin better than the other flagellin antiserum (αFljK, see Figure 1).
Next, we sought to clarify whether C. crescentus NeuB is a sialic acid or a pseudaminic acid synthase. To resolve this question, we conducted heterologous complementation with the three NeuB variants from C. jejuni, whose enzymatic activities are known: NeuB1 synthesizes sialic acid, NeuB2 produces legionaminic acid, and NeuB3 is a pseudaminic acid synthase (Linton et al., 2000; Sundaram et al., 2004; Chou et al., 2005; McNally et al., 2007; Schoenhofen et al., 2009). Using motility (Figure 3A) and flagellin modification (Figure 3B) as a readout for NeuB activity, we discovered that only NeuB3 can substitute for C. crescentus NeuB, indicating that NeuBCc functions as a pseudaminic acid synthase.
Figure 3
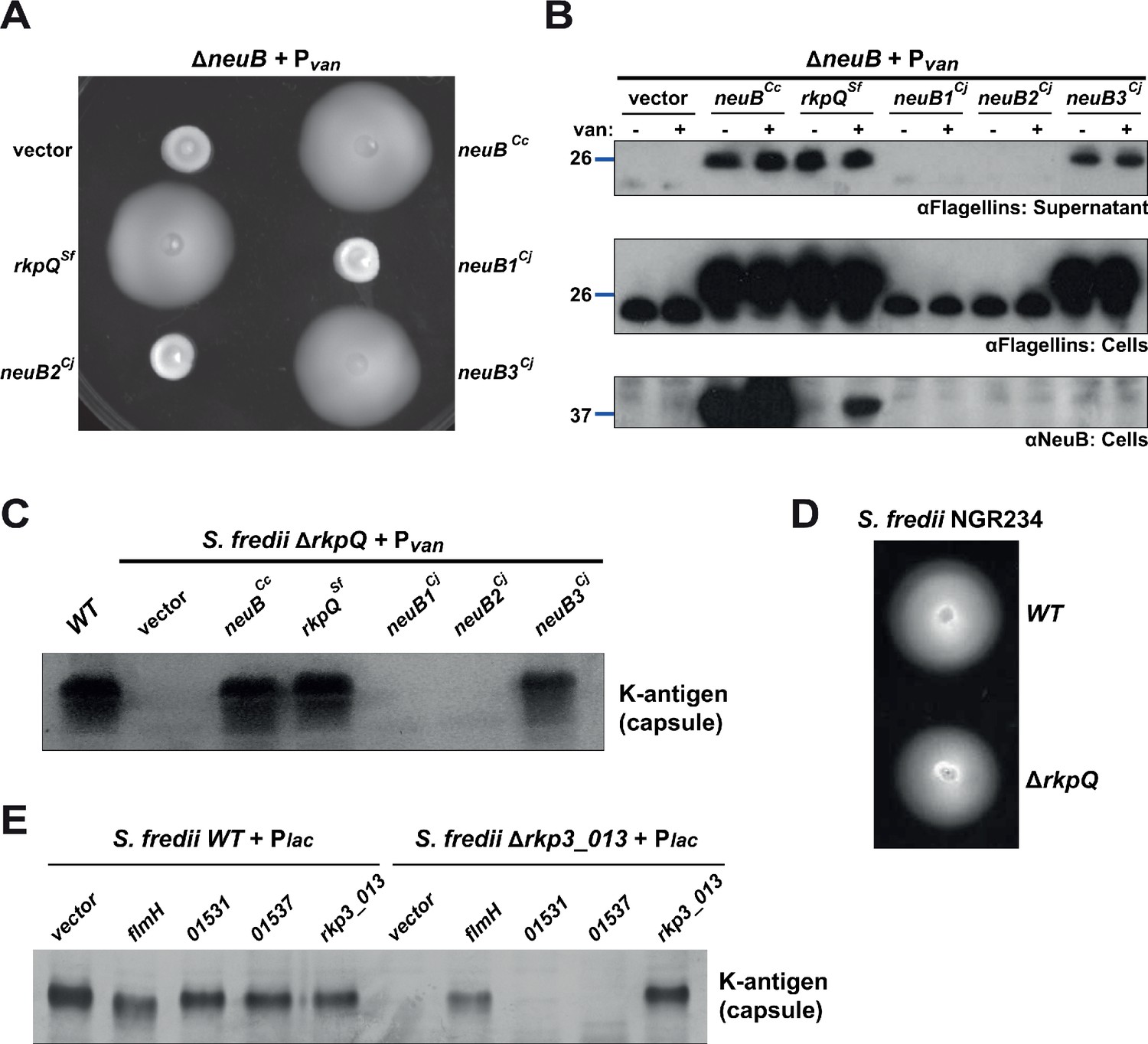
Heterologous complementation of the ΔneuB mutant with pseudaminic acid synthases.
(A) Motility assay of ΔneuB cells complemented with different neuB homologs expressed from Pvan on plasmid. Only NeuB from C. crescentus and the homologs known to be pseudaminic acid synthases (RkpQ from S. fredii and NeuB3 from C. jejuni) can fully complement the motility defect of the ΔneuB strain, whereas NeuB1 and NeuB2 from C. jejuni do not restore motility. (B) Immunoblots showing the intracellular levels of flagellins and NeuB in ΔneuB cells complemented with different NeuB homologs expressed from Pvan on a plasmid. Consistent with the motility assay shown in panel A, only NeuBCc, RkpQSf, and NeuB3Cj can restore the flagellin migration profile (in whole cell lysates, middle panel) and the secretion of flagellin in the supernatant (upper panel) in ΔneuB cells. RkpQSf is the protein that shows the highest similarity to NeuBCc, as shown by the fact that RkpQSf is detected by the antibodies against NeuBCc (lower panel). The blue lines on the left indicate the migration of the molecular size standards, with the corresponding value in kDa. Note that antibodies used in this blot were raised against flagellins purified from C. crescentus (αFlagellins; Hahnenberger and Shapiro, 1987). (C) Capsular polysaccharide profile of S. fredii NGR234 ΔrkpQ mutant cells expressing different NeuB homologs from Pvan on a plasmid. Cells with a ΔrkpQ mutation do not produce capsular polysaccharide, but production of capsular polysaccharide in ΔrkpQ cells can be restored by NeuBCc or NeuB3Cj (but not by NeuB1Cj or NeuB2Cj), which indicates that NeuBCc is a pseudaminic acid synthase. (D) Motility assay of S. fredii NGR234 WT and ΔrkpQ mutant showing that mutation of the pseudaminic acid synthase does not affect motility in S. fredii. (E) Capsular polysaccharide profile of S. fredii WT and Δrkp3_013 expressing putative C. crescentus acetyltransferases from Plac on a plasmid. Production of capsular polysaccharide in Δrkp3_013 cells can be restored only by expression of flmH, which suggests that FlmH can participate in the pseudaminic acid biosynthetic pathway.
Since C. jejuni NeuB3 also functions in the control of motility, we sought to corroborate our conclusion with a pseudaminic acid synthase that does not act in the flagellation pathway and tested whether such an enzyme can also support flagellation in C. crescentus ΔneuB cells. This experiment served to demonstrate that it is the enzymatic activity of NeuBCc in pseudaminic acid synthesis that is required for flagellation in C. crescentus. Conversely, if C. crescentus NeuB is indeed a pseudaminic acid synthase, then it should be able to support pseudaminic acid synthesis in another system. We, therefore, turned to the symbiotic alpha-proteobacterium S. fredii NGR234 that synthetizes a K-antigen capsule composed of pseudaminic acid and glucuronic acid units (Le Quéré et al., 2006; Le Quéré and Ghigo, 2009). S. fredii NeuB (called RkpQ) is encoded in the K-antigen capsular polysaccharide biosynthesis locus (rkp3) on the pNGR234b megaplasmid (Schmeisser et al., 2009). First, we confirmed that S. fredii RkpQ was able to functionally replace NeuB in C. crescentus, restoring motility and flagellin migration to C. crescentus ΔneuB cells (Figure 3A and B), akin to NeuB3 from C. jejuni. To confirm that C. crescentus NeuB is indeed a pseudaminic acid synthase, we constructed an rkpQ deletion mutant (ΔrkpQ) in S. fredii and observed that this mutation blocks synthesis of the K-antigen capsule (Figure 3C), but not motility (Figure 3D). Capsule synthesis was restored by complementation of S. fredii ΔrkpQ cells with a plasmid expressing either RkpQ, C. crescentus NeuB or C. jejuni NeuB3. By contrast, C. jejuni NeuB1 and NeuB2 could not restore capsular polysaccharide production (Figure 3C). Thus, pseudaminic acid synthesis is required for motility and flagellin modification in C. crescentus and pseudaminic acid synthases are interchangeable.
The OGT FlmG is required and sufficient for flagellin modification
Knowing that pseudaminic acid synthesis is required for motility and modification of all six flagellins in C. crescentus, we predicted that our Tn library of motility mutants should also contain Tn insertions in a gene encoding a cognate OGT. Inspection of the Tn insertion sites revealed 10 mutants with a Tn insertion in the CCNA_01524 (henceforth flmG) gene: six bear a himar1 Tn insertion at different positions in flmG (strains NS25, NS55, NS81, NS128, NS157, and NS192), while an Ez-Tn5 insertion disrupts flmG in four other mutants (NS149, NS211, NS322, and NS327). The flmG gene had previously been implicated in motility and flagellin biosynthesis (Leclerc et al., 1998; Schoenlein et al., 1992; Wang et al., 1993; Schoenlein and Ely, 1989; Schoenlein et al., 1989) and is predicted to encode a 596-residue protein of 65 kDa containing an N-terminal domain (NTD) with tetratricopeptide (TPR) repeats, known to be involved in protein-protein interactions, and a C-terminal domain (CTD) resembling glycosyltransferases (GT-B superfamily). We constructed an in-frame deletion in flmG (ΔflmG) and found the resulting mutant cells have a defect in motility (Figure 4A) and flagellin modification (Figure 4B). The motility and flagellin modification defects were corrected by the expression of FlmG in trans from Pvan on pMT335 (Figure 4A and B). Thus, FlmG acts in the same pathway as NeuB as predicted for an OGT responsible for the post-translational O-glycosylation of flagellins in C. crescentus.
Figure 4
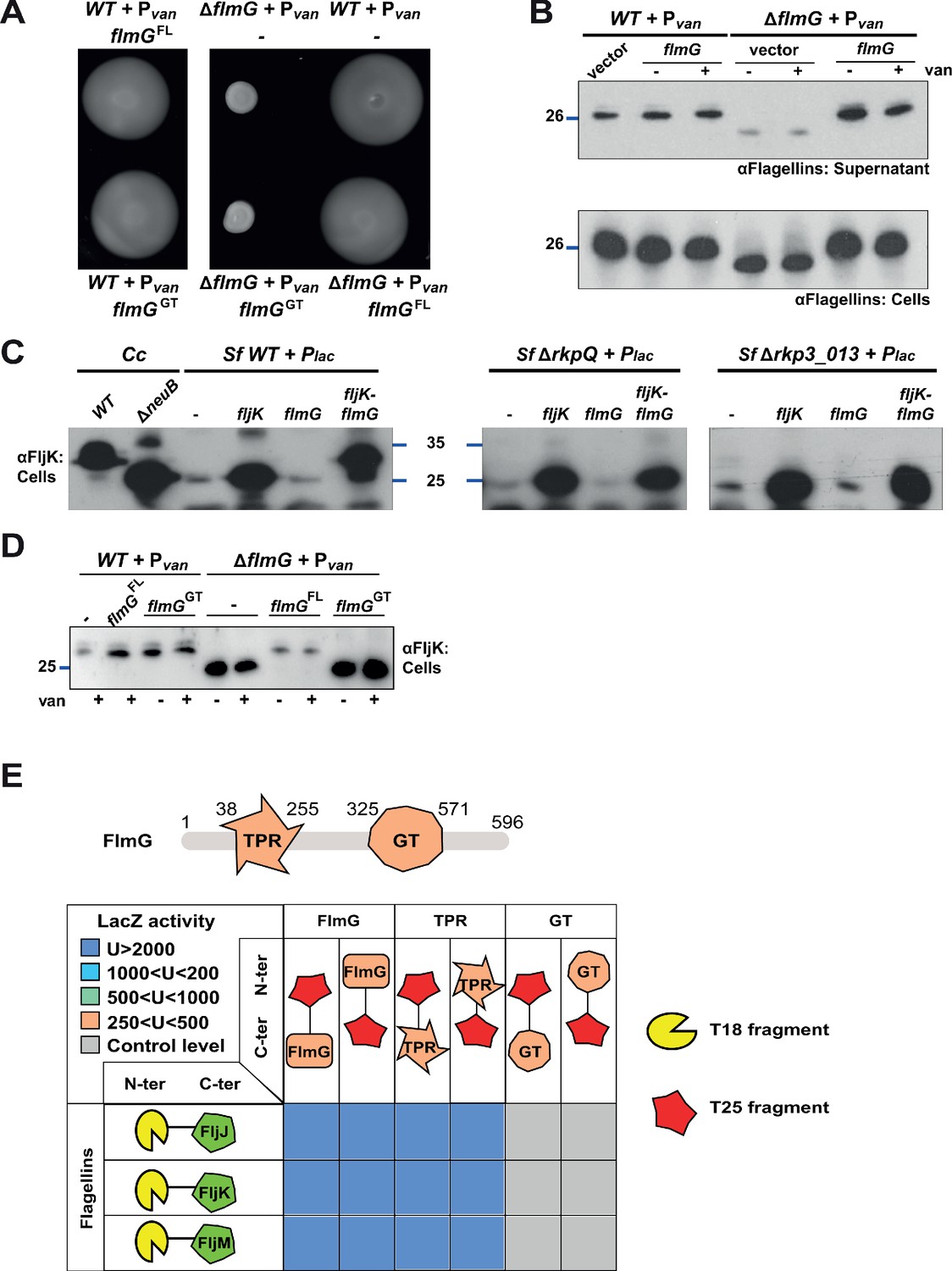
FlmG is the putative glycosyltransferase required to post-translationally modify flagellins.
(A) Motility assay of WT and ΔflmG cells complemented with Pvan-flmG full-length (FL) or glycosyltransferase CTD (GT) on plasmid. ΔflmG cells are non-motile, as indicated by the compact swarming. The expression of FlmG full-length from Pvan restores motility, in contrast to the GT domain alone. (B) Immunoblot showing the levels of flagellins in supernatants and whole cell lysates from WT and ΔflmG cells complemented with Pvan-flmG on a plasmid. Flagellins produced by ΔflmG cells show the same migration profile as those produced by ΔneuB cells, with faster migration and reduced abundance in the supernatant. The blue lines on the left indicate the molecular size standard, with the corresponding value in kDa. Note that antibodies used in this blot were raised against flagellins purified from C. crescentus (αFlagellins; Hahnenberger and Shapiro, 1987). (C) Immunoblot on C. crescentus FljK expressed in S. fredii NGR234 strains. The left panel shows that when expressed alone in WT S. fredii FljK migrates faster, like in the C. crescentus ΔneuB mutant. By contrast, co-expression of FlmG and FljK in S. fredii NGR234 results in a shift in the migration profile of FljK, similar to that observed in C. crescentus WT cells. When FlmG and FljK are co-expressed in S. fredii strains unable to synthesize pseudaminic acid (ΔrkpQ, middle panel; Δrkp3_013, right panel) FljK shows only the fast migrating band, independently from the presence of FlmG. The blue lines indicate the molecular size standards, with the corresponding value in kDa. (D) Immunoblot with anti-FljK antibodies on whole cell lysates from WT and ΔflmG cells expressing FlmG full-length (FL) or the glycosyltransferase CTD (GT) from Pvan on a plasmid. The expression of the GT domain alone does not restore the migration profile of flagellins in ΔflmG cells, in agreement with the motility defect shown in panel A. (E) BACTH assay showing the interaction of FlmG with flagellins. The cartoon represents FlmG, with the N-terminal TPR domain and the C-terminal glycosyltransferase domain. The scheme below shows the β-galactosidase activity, expressed in Miller units (U) of E. coli BTH101 cells containing the pair-wise combinations of the different constructs used for the BACTH assay. T18 and T25 correspond to the two fragments of the adenylate cyclase and are represented as yellow shape and red star, respectively. Hybrids with the proteins of interest (FlmG, FljJ, FljK, and FljM) were created as N-ter or C-ter fusions, as mentioned. In the case of FlmG, fusions were created with the full-length protein, the TPR domain or the glycosyltransferase (GT) domain only. Gray squares correspond to β-galactosidase activity similar to the control (empty plasmid, U < 250), orange squares indicate an activity between 250 and 500 U, light green squares between 500 and 1000 U, light blue between 1000 and 2000 U, and dark blue above 2000 U. The values of β-galactosidase correspond to the mean and standard deviation of three independent experiments and are listed in Supplementary file 1 Table S1.
To prove that FlmG is indeed the OGT in this modification pathway, we probed for sufficiency of flagellin modification by the expression of FlmG in a heterologous system naturally producing pseudaminic acid. We therefore chose to (co-)express FljK with or without FlmG in S. fredii NGR234 and probed for flagellin modification by immunoblotting using antibodies to C. crescentus FljK (Figure 4C). In the absence of FlmG, FljK showed the same mobility on SDS-PAGE as in C. crescentus ΔneuB cells. However, upon co-expression of FlmG, FljK shifted to a species with higher molecular mass and identical apparent migration on SDS-PAGE to that observed for FljK in C. crescentus WT cells. Importantly, this shift was dependent on the presence of pseudaminic acid, since FljK co-expressed with FlmG in S. fredii cells lacking pseudaminic acid (ΔrkpQ or Δrkp3_013, see below) had the same mobility by SDS-PAGE as FljK expressed in C. crescentus ΔneuB or ΔflmG cells, or in WT S. fredii cells without FlmG (Figure 4C). We conclude that FlmG is required and sufficient for flagellin modification in the presence of pseudaminic acid.
A major question in glycosylation is how substrate specificity is programmed into the OGT of the system. Based on the domain organization of FlmG, we reasoned that the NTD might hold the specificity determinant toward the flagellins, perhaps by directly interacting with flagellins. By contrast, the CTD might confer OGT activity, but would not function without the NTD specificity determinant. Indeed, the expression of the CTD alone did not restore motility or flagellin modification to C. crescentus ΔflmG cells (Figure 4A and D). We next probed for a direct interaction of FlmG NTD with flagellins using the bacterial two-hybrid assay (BACTH, Figure 4E). This assay is based on the functional reconstitution of the adenylate cyclase from Bordetella pertussis, composed of two fragments, T25 and T18 (Karimova et al., 1998). When two proteins of interest fused to each fragment interact, adenylate cyclase is reconstituted and produces cyclic AMP, which in turn induces the expression of the lacZ gene. We tested combinations of the FlmG NTD and CTD together with the flagellins FljJ, FljK, and FljM as probes. Notably, a strong interaction was observed between each of the flagellins and the FlmG NTD (TPR), but not FlmG CTD (GT, Figure 4E). These BACTH results along with the domain analysis show the TPR-containing NTD is required and sufficient for a specific interaction of FlmG with multiple flagellins performing structural or regulatory functions, consistent with our finding that all flagellins are modified with pseudaminic acid by FlmG (Figure 1G).
Flagellin glycosylation components are expressed before the FlaF secretion chaperone
We wished to determine if FlmG and the glycosylation pathway components are cell cycle regulated. Toward this goal, we first needed to identify the other pathway components in C. crescentus using a combination of genetics and bioinformatics (Figure 5A; Table 1). The first two enzymes of the pathway elucidated in C. jejuni are PseB (UDP-N-acetylglucosamine 5-inverting 4,6-dehydratase) and PseC (UDP-4-amino-4,6-dideoxy-N-acetyl-beta-L-altrosamine transaminase). Since genes that act in the same pathway in C. crescentus should be required for motility, we scanned our library of Tn mutants for insertions in orthologous genes. Indeed, the gene products of flmA (CCNA_00233) and flmB (CCNA_00234) resemble PseB and PseC, respectively. This scan revealed five mutants with Tn insertions in flmA (NS235, NS246, and NS294 had HyperMu insertions, NS148 harbored an Ez-Tn5 insertion and NS102 a Tn5 insertion) and three mutants with Tn insertions in flmB (Himar1 insertion in NS76 and HyperMu insertions in NS132 and NS255). Importantly, these mutants recapitulate the motility and flagellin modification defect of neuB and flmG mutant cells (Figure 5B–E) and the corresponding orthologs of S. fredii, RkpL and RkpM, can functionally replace C. crescentus FlmA and FlmB (Figure 5B, C and E).
Figure 5 with 1 supplement see all
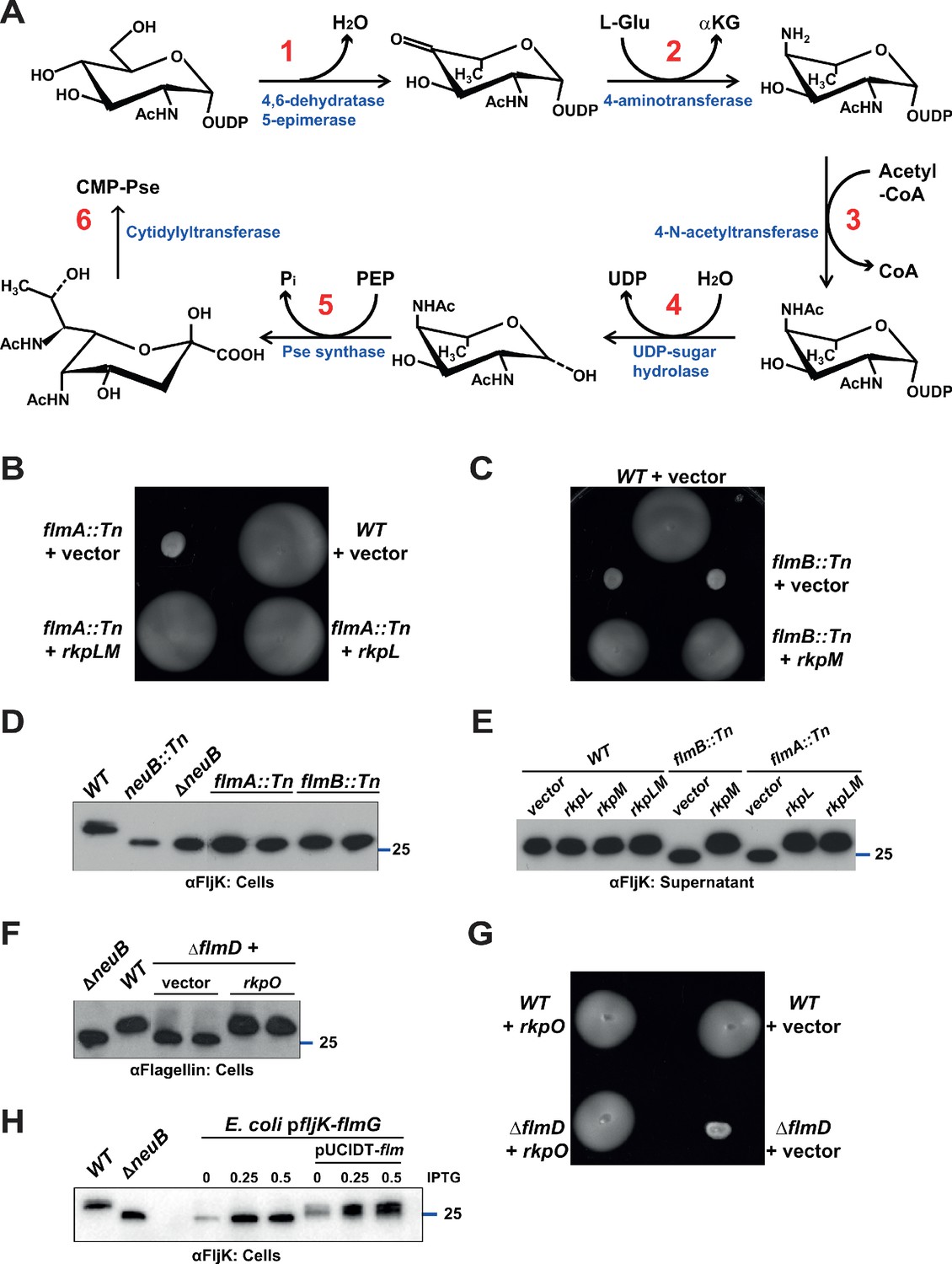
Pseudaminic acid biosynthetic pathway in C. crescentus.
(A) Schematic of the pseudaminic acid biosynthetic pathway as it has been described in C. jejuni and H. pylori (reviewed in Salah Ud-Din and Roujeinikova, 2018). The different steps are catalyzed by PseB (1), PseC (2), PseH (3), PseG (4), PseI (5), and PseF (6). (B) Motility assay of WT and flmA::Tn cells expressing S. fredii NGR234 rkpL and rkpM from Pvan ona plasmid. (C) Motility assay of WT and flmB::Tn cells expressing S. fredii NGR234 rkpM from Pvan on a plasmid. (D) Immunoblots of extracts from WT and mutant cells probed with polyclonal anti-FljK antibodies on whole cell lysates. Flagellins produced by flmA::Tn and flmB::Tn cells show the same migration profile as in ΔneuB mutant cells. The blue line indicates the migration of the molecular size standard, with the corresponding size in kDa. (E) Immunoblots of supernatants from WT, flmA::Tn and flmB::Tn cells probed with polyclonal anti-FljK antibodies. The expression of the S. fredii homologs RkpL and RkpM from Pvan on a plasmid restores the migration profile and secretion of flagellin in flmA::Tn and flmB::Tn cells, respectively. The blue line indicates the migration of the molecular size standard, with the corresponding size in kDa. (F) Immunoblot of extracts from WT and ΔflmD cells. Mutation of flmD impairs post-translational flagellin modification, and the defect is complemented by the expression of the S. fredii NGR234 homologue RkpO from a plasmid. The blue line indicates the migration of the molecular size standard, with the corresponding size in kDa. Note that antibodies used in this blot were raised against flagellins purified from C. crescentus (αFlagellins; Hahnenberger and Shapiro, 1987). (G) Motility assay of WT and ΔflmD cells expressing S. fredii NGR234 RkpO from Pvan on a plasmid. (H) Immunoblot with anti-FljK antibodies on whole cell lysates from E. coli expressing fljK and flmG from Plac on a plasmid, in the presence or absence of a compatible plasmid carrying the complete set of Caulobacter genes for the pseudaminic acid biosynthetic pathway (pUCIDT-flm, see Materials and methods and Supplementary file 1 Table S3). In the absence of pUCIDT-flm, FljK shows the same migration profile as in Caulobacter ΔneuB cells, whereas in the presence of pUCITD-flm FljK migration is shifted toward higher molecular mass, as in Caulobacter WT cells. The values above the panel indicate the concentration of the inducer for Plac-fljK-flmG (mM IPTG). The blue line indicates the migration of the molecular size standard, with the corresponding size in kDa.
Table 1
Homology of the C. crescentus enzymes involved in the pseudaminic acid biosynthesis pathway to S. fredii NGR234 and C. jejuni proteins.
| C. crescentus | S. fredii NGR234 | C. jejuni NCTC 11168 | Enzymatic activity |
|---|---|---|---|
| FlmA (CCNA_00233) | RkpL 52% id; 68% sim | PseB 57% id; 72% sim | Dehydratase |
| FlmB (CCNA_00234) | RkpM 44% id; 58% sim | PseC 34% id; 52% sim | Aminotransferase |
| FlmH (CCNA_01523) | Rkp3_013† 26% id; 37% sim | PseH‡ 26% id; 50% sim | N-acetyltransferase |
| FlmD (CCNA02947) | RkpO 36% id; 49% sim | PseG 22% id; 41% sim | UDP-sugar hydrolase |
| NeuB (CCNA_02961) | RkpQ 56% id; 70% sim | PseI 43% id; 59% sim | Pseudaminic acid synthase |
| FlmC§ (CCNA_02946) | RkpN | PseF | Cytidyltransferase |
| FlmG (CCNA_01524) | - | - | Protein Glycosyltransferase |
-
* Although C. crescentus genomes encodes for several putative acetyltransferases, only CCNA_01531 has significant homology to FlmH (35% identity [id] and 54% similarity [sim]).
† FlmH aligns to Rkp3_013 only for about 50% of its length.
-
‡ FlmH aligns to PseH for about 75% of its length.
§ FlmC shows no homology to RkpN or PseF; RkpN and PseF share 46% identity and 68% similarity.
For the third step of the pathway, enzymatic redundancy or promiscuity exists in C. crescentus as inactivation of the predicted ortholog (flmH, CCNA_01523), even with the inactivation of the paralogous genes CCNA_01531 and CCNA_01537 (i.e. a ΔflmH ΔCCNA_01531 ΔCCNA_01537 triple mutant), did not phenocopy the effects of neuB, flmA, flmB, or flmG single gene disruptions (Figure 5—figure supplement 1). Conversely, however, we demonstrated that inactivation of the flmH ortholog of S. fredii NGR234, rkp3_013, led to a defect in K-antigen capsule synthesis, which could be restored by the expression of C. crescentus flmH in trans (Figure 3E). Thus, FlmH can execute the corresponding acetylating step in pseudaminic acid synthesis, at least in S. fredii.
Bioinformatics predicts that the fourth step in pseudaminic acid biosynthesis is executed by FlmD (CCNA_02947) in C. crescentus and RkpO in S. fredii NGR234. To verify this prediction, we engineered an in-frame deletion in flmD (ΔflmD) and found that the resulting cells are non-motile, consistent with a previous report (Faulds-Pain et al., 2011), and unable to modify flagellins (Figure 5F and G). Importantly, we found that S. fredii RkpO can functionally replace FlmD, restoring motility and flagellin modification to C. crescentus ΔflmD cells (Figure 5F and G). Thus, the FlmD enzyme is also required for pseudaminic acid synthesis. Immediately upstream of and co-encoded with flmD lies flmC whose gene product resembles cytidylyltransferases. Since pseudaminic acid must usually be activated with cytidine 5’-monophosphate (CMP) before being incorporated into a polysaccharide or protein (Salah Ud-Din and Roujeinikova, 2018), FlmC likely executes this last event in C. crescentus. To confirm that these six enzymatic steps are necessary and sufficient for pseudaminic acid synthesis, we reconstituted FlmG-dependent glycosylation in E. coli K12 cells using a plasmid with a synthetic flm operon expressing all six enzymes (FlmA-FlmB-FlmH-FlmD-NeuB-FlmC) from open reading frames that had been codon-optimized for the expression in E. coli. We also introduced a second, compatible plasmid co-expressing FljK and FlmG into these cells and then probed for FljK by immunoblotting using antibodies to FljK. We indeed observed that FljK was modified under these conditions, but not in the absence of the flm-operon plasmid (Figure 5H).
Having identified the components of the flagellin glycosylation pathway, we asked whether the corresponding transcripts are cell cycle regulated. To this end we interrogated a data set of transcripts by RNA-Seq and Ribo-Seq analysis on synchronized WT C. crescentus cells harvested at different stages in the cell cycle. We noted that while the flmD and flmC transcripts do not fluctuate substantially in abundance during the cell cycle (Schrader et al., 2016), the flmGH and flmAB transcripts peak in abundance during the early pre-divisional cell stage (90 min into the cell cycle, Figure 6A), while the neuB transcript is most abundant at the late pre-divisional cell stage (after 120 min, Figure 6A). These findings raise the possibility that the flagellin modification genes are transcribed from a cell cycle regulated promoter. Previous chromatin-immunoprecipitation deep-sequencing (ChIP-Seq) studies revealed that the essential cell cycle regulator CtrA binds to the predicted promoter region of flmG, flmA, and neuB (Fumeaux et al., 2014; Fiebig et al., 2014). To demonstrate that these flagellin modification genes are indeed regulated by CtrA, we conducted promoter probe experiments with transcriptional fusions to a promoterless lacZ gene encoding β-galactosidase. We determined the β-galactosidase activities from these reporters in WT cells and ctrA401 cells that have a missense mutation (encoding the T170I substitution) in ctrA, a partial loss of function mutation that renders cells temperature sensitive for growth at 37°C (Quon et al., 1996). At the permissive temperature (30°C), ctrA401 cells grow well, albeit several CtrA-activated promoters show substantially reduced activity (Quon et al., 1996; Delaby et al., 2019). We observed a similar reduction in activity of the promoter probe reporters of flmG, flmA, and neuB in ctrA401 cells (Figure 6B). Conversely, a gain-of-function mutation in ctrA (ctrA*, encoding the T170A substitution) increases the activity of these reporters compared to the isogenic parent (Figure 6B). In these experiments, the isogenic parent is the ΔmucR1/2 double mutant that has reduced activity of many CtrA-activated promoters. This defect can be ameliorated either by gain-of-function mutations in ctrA (such as ctrA*, Figure 6B) or by loss-of-function mutations in sciP (shown as control in Figure 6B), which encodes a negative regulator of a subset CtrA-activated genes (Fumeaux et al., 2014; Gora et al., 2010; Tan et al., 2010; Figure 6A and B). Thus, transcription of flmG, flmA, and neuB is directly integrated into the cell cycle via CtrA, as is the case for the other flagellar genes that are CtrA-dependent (directly or indirectly), including the FlaF flagellin secretion chaperone that binds all six flagellins (Ardissone et al., 2020) to direct their secretion and that accumulates in dividing cells (Fumeaux et al., 2014; Llewellyn et al., 2005). Immunoblotting of extracts from synchronized cells harvested at different times during the cell cycle probed with antibodies to FlmG and NeuB did not reveal substantial changes in abundance of NeuB during the C. crescentus cell cycle (Figure 6—figure supplement 1A). However, the levels of FlmG increased steadily, peaking at the end of the cell cycle (Figure 6C). Because FlmG is already abundant before cell division, while the FlaF secretion chaperone accumulates in dividing cells (Llewellyn et al., 2005), flagellin modification by FlmG can occur before the FlaF-dependent secretion and potentially avoids competition between FlmG and FlaF for flagellins.
Figure 6 with 1 supplement see all
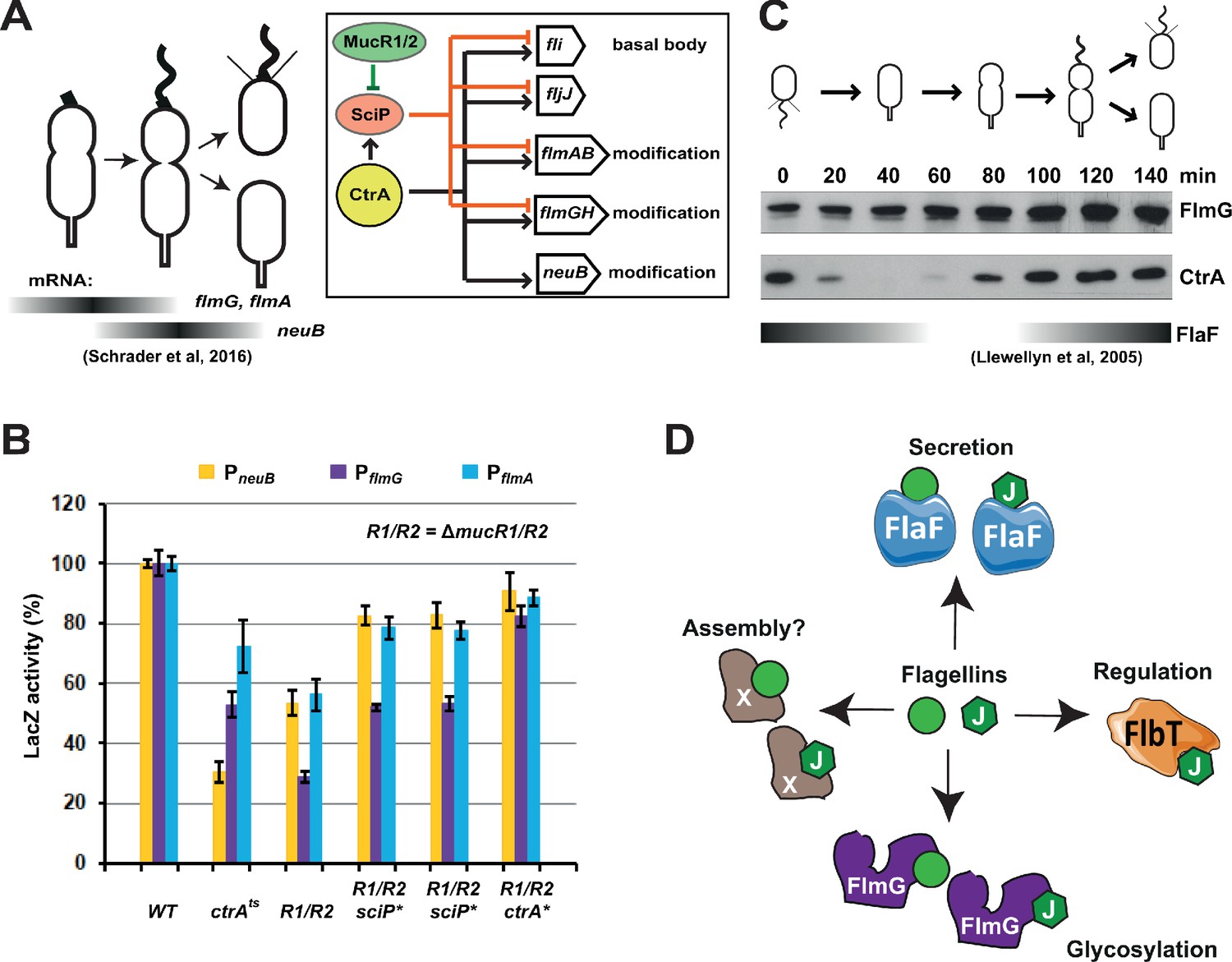
Transcription of genes encoding flagellin-modification proteins is cell cycle regulated.
(A) Schematic of the transcriptional regulatory network controlling the expression of flagellar and pseudaminic acid biosynthetic genes in C. crescentus pre-divisional cells. The gray bars represent the time during the cell cycle when flmA, flmG, and neuB mRNA levels peak (Schrader et al., 2016). The transcriptional regulatory network is represented in the box: CtrA promotes the expression of genes encoding the basal body (fli genes), the regulatory flagellin FljJ and several components of the flagellin glycosylation pathway (flmAB and flmGH operons, neuB). The expression of these same genes is repressed by SciP (with the exception of neuB), whose expression is under control of CtrA (positively) and MucR1/2 (negatively). (B) β-galactosidase activity of PneuB, PflmG, and PflmA transcriptional fusions. Transcription of neuB, flmG and flmAB is significantly reduced in ctrA401 (T170I, temperature sensitive allele ctrAts) and double ΔmucR1ΔmucR2 (R1/R2) strains. β-galactosidase activity of PneuB, PflmG, and PflmA is partially restored in ΔmucR1ΔmucR2 cells carrying ctrA(T170A), sciP(T24I), or sciP(T65A) alleles (ctrA* and sciP*). Values are expressed as percentages (activity in WT NA1000 set at 100%). (C) Immunoblot showing FlmG and CtrA protein levels in synchronized C. crescentus cells. The gray bar represents the abundance of the flagellin chaperone FlaF along the cell cycle, as previously shown (Llewellyn et al., 2005). (D) Model depicting a flagellin-centric view of the events during flagellar assembly in C. crescentus. The FljJ flagellin is illustrated as a green hexagon, whereas the other flagellins (FljKLMNO) are depicted as green circles. The events acting on flagellins are translational regulation (binding of FljJ by FlbT), glycosylation by FlmG, secretion by FlaF and possibly assembly by an unknown factor (X).
Discussion
A new class of flagellin O-glycosyltransferases
There are two principal mechanisms for the transfer of sugar moieties onto an acceptor protein. An oligosaccharide synthesized on a lipid carrier can be transferred onto the acceptor protein by an oligosaccharyl-transferase (OTase)-dependent mechanism, as it occurs in the case of type IV pilin subunits in N. meningitidis and Neisseria gonorrhoeae (Nothaft and Szymanski, 2010; Schäffer and Messner, 2017; Vik et al., 2009; Faridmoayer et al., 2007). By contrast, glycosylation of flagellin subunits is OTase-independent and relies on specific glycosyltransferases that sequentially transfer monosaccharide units on the acceptor protein. Moreover, and importantly, glycosylation of flagellins occurs by a soluble glycosylation donor in the cytoplasm, the same compartment where proteins are synthesized. In the OTase-based systems the acceptor protein synthesized in the cytoplasm is (initially) spatially separated from the glycosylation donor and must be transported to the periplasmic compartment for modification by the OTase, while the lipid-anchored donor must be presented on the periplasmic (extra-cytoplasmic) face of the membrane. Therefore, owing to these advantages, the flagellin glycosylation systems have great engineering potential for custom-designed O-glycosylation of proteins of interest.
The molecular basis underlying the specificity of bacterial protein glycosylation systems, especially for the cytoplasmic O-glycosylation systems, is poorly understood. Recently evidence was provided that the Maf glycosyltransferase from Geobacillus kaustophilus and Clostridium botulinum can modify flagellin with N-acetylneuraminic acid or 3-deoxy-D-manno-octulosonic acid by O-linkage at threonine and serine residues when expressed in E. coli (Khairnar et al., 2020). The Maf orthologs from Aeromonas caviae and Magnetospirillum magneticum magneticum are required for flagellin glycosylation in vivo (Sulzenbacher et al., 2018; Parker et al., 2014). While C. crescentus does not encode a Maf ortholog in its genome, we discovered that it uses FlmG, the defining member of a hitherto unknown class of OGTs, to glycosylate flagellins in hosts that can synthesize pseudaminic acid. The very recent evidence that C. crescentus modifies the FljK flagellin at four threonine residues with a molecule predicted to be an O-linked glycan (Montemayor et al., 2020) aligns well with our finding that pseudaminic acid is required for FlmG to modify FljK. The ability of FlmG to function in a heterologous system when offered pseudaminic acid raises the question whether FlmG can also accept another (activated) nonulosonic acid, such as sialic acid, as donor for the modification reaction, or other sugars that resemble the sialic acids of humans. Our demonstration that FlmG glycosylates flagellin in E. coli expressing a synthetic pseudaminic acid biosynthesis (flm) operon from C. crescentus simplifies future analyses of the donor promiscuity for FlmG-dependent flagellin glycosylation, as it can now be simply achieved by the expression of orthologous sialic acid biosynthesis pathways in E. coli. The NeuB family of nonulosonic acid synthases enzymes may constitute a convenient marker for the identification of genomes encoding biosynthesis pathways for sialic acid-like molecules that could then subsequently be used to probe for glycosylation of flagellin or other (potentially promiscuous) acceptor proteins.
Promiscuous flagellin binding by FlaF and FlmG
Since all six C. crescentus flagellins are glycosylated by FlmG, a certain degree of substrate promiscuity is inherent in the system. While the FljJ flagellin only bears 49% amino acid identity compared to the other flagellins, there are still at least 20 serine and threonine residues that are conserved between FljJ and FljK and that FlmG could potentially modify. However, only one of the four threonines that are modified in FljK is present in all six flagellins (Montemayor et al., 2020). We showed that the FlmG NTD binds the flagellins via the TPR region and that this domain is required for motility and efficient glycosylation (Figure 4A, D and E). A free-floating glycosyltransferase (CTD) in the cytoplasm cannot bind the flagellins alone in BACTH assays (Figure 4E) and can not modify sufficient flagellin to support assembly of the flagellar filament in C. crescentus. At this point, we cannot rule out that the NTD also plays a role in controlling the activity of the glycosyltransferase domain in the CTD.
Recent BACTH assays revealed that the FlaF secretion chaperone binds the six flagellins as well (Ardissone et al., 2020). It is expected that FlaF can bind all flagellins, since it must direct their secretion via the flagellar export apparatus to assemble into the flagellar filament. While all flagellins are assembled into the flagellar filament (Driks et al., 1989), FljJ is the only flagellin that cannot assemble into a flagellar filament on its own (Faulds-Pain et al., 2011). FljJ is the first flagellin to be synthesized and exported during the cell cycle as its location within the filament is the most proximal to the hook (Driks et al., 1989). As an exported flagellin, FljJ is also glycosylated like the other flagellins and it therefore must also be bound by FlmG and FlaF (Figure 1A). Therefore, FlmG and FlaF could potentially compete for the same substrate. However, FlmG is already present well before FlaF accumulates (Llewellyn et al., 2005; Figure 6C). Moreover, flagellin modification does occur in mutants lacking a flagellar hook basal body complex (Figure 6—figure supplement 1B, C) or the flagellin chaperone FlaF (Figure 6—figure supplement 1A), confirming that modification can occur without the secretion apparatus and the secretion chaperone.
It would be interesting to elucidate whether FlaF and FlmG bind the flagellins in the same way, and compare it to the highly specific interaction of the FljJ flagellin with C. crescentus FlbT (Figure 6D), a post-translational regulator of flagellin transcripts that only interacts with FljJ, but not other flagellins as determined by BACTH assay (Ardissone et al., 2020). The preference of FlbT for FljJ underlies the translational repression mechanism used to regulate the expression of transcripts encoding the five (other) structural flagellins. Translational repression by FlbT•FljJ is relieved with the removal of FljJ from the cytoplasm by secretion through the flagellar secretion conduit (hook basal body, HBB; Chevance and Hughes, 2008) once it has been assembled and the FlaF secretion chaperone has accumulated. Since the translational regulation by FlbT requires the presence of FljJ, translational repression is inactivated with the secretion of FljJ.
Roles of pseudaminic acid in cell surface structures
The presence of sialic acid-like sugars on the surface of pathogenic bacteria may serve to evade the host immune system. However, flagellin glycosylation also occurs in environmental (non-pathogenic) bacteria, where the post-translational modification likely serves another purpose. In the case of Campylobacter, Helicobacter, and Aeromonas, the loss of flagellin glycosylation prevents assembly of a functional flagellar filament. By contrast, in Pseudomonas and Burkholderia species, glycosylation of flagellins is not required for filament assembly (Schirm et al., 2003; Linton et al., 2000; Scott et al., 2011; Parker et al., 2014; Taguchi et al., 2008). In C. crescentus, no flagellar filament is assembled in the absence of glycosylation, although flagellins accumulate in the supernatant of ΔneuB or ΔflmG cultures. However, the level of the flagellins in the supernatant is reduced in these mutants compared to the WT, suggesting that non-glycosylated flagellin is less efficiently exported and/or less stable in the supernatant than modified flagellin akin to A. caviae (Parker et al., 2014).
Why pseudaminic acid is essential for flagellation in many polarly flagellated bacteria remains unclear. While the abundance of secreted flagellins is substantially reduced in the absence of glycosylation, secretion is still functional. However, we cannot rule out that the efficiency of secretion is substantially reduced and that the residual level of secreted flagellin does not suffice for assembly of a filament to support motility. However, since only hook structures are observed in glycosylation mutants rather than short flagellar filaments, it appears that filament assembly is defective. Thus, a problem other than flagellin secretion appears to underlie the flagellar assembly defect in glycosylation mutants. It is possible than an assembly factor exists that preferentially binds glycosylated flagellins upon translocation, facilitating their incorporation into the flagellar filament (Figure 6D). Such a factor would likely be encoded in a flagellar assembly gene cluster in polarly flagellated bacteria that glycosylate flagellin, but absent from peritrichous flagellation systems. The loss of such a factor should prevent flagellar filament formation, but neither flagellin secretion, nor glycosylation.
It is possible that glycosylation simply promotes self-assembly of the flagellins into the filament in C. crescentus without the aid of another factor. However, many flagellated bacteria assemble flagellar filaments without the need for glycosylation, indicating that flagellins per se do not depend on glycosylation for self-assembly. It is not evident why specifically the flagellins of polar flagellation systems, such as the C. crescentus flagellins, should have evolved a dependency on glycosylation to (self-) assemble into a filament. Indeed, the glycosylated residues do not appear to reside in the flagellin inter-subunit contact area within the flagellar filament (Montemayor et al., 2020). A more appealing scenario is that glycosylation serves another purpose as it does endow flagellar assembly with an additional regulatory event. Perhaps it permits differentiating flagellins from other secretion substrates present in the cytoplasm (i.e. hook or capping proteins [Chevance and Hughes, 2008]) and/or it permits tuning flagellin assembly with another input such as a cell cycle cue. In monopolar and bipolar flagellation systems, flagellar assembly must be highly coordinated with the cell cycle, since flagellar assembly must occur once per division cycle. Such delicate temporal control is not needed for peritrichous flagellation systems because assembly occurs in an unsynchronized fashion with each machinery being in a different assembly state as the cell elongates. Since pseudaminic acid synthesis by NeuB requires phosphoenolpyruvate (PEP), flagellation in C. crescentus is indirectly PEP-dependent. PEP is also well known for its regulatory functions as a phosphoryl-donor in phosphotransferase signaling (PTS) systems (Tchieu et al., 2001; Pflüger-Grau and Görke, 2010; Postma et al., 1993). Interestingly, a PTS system is required for the production of the alarmone (p)ppGpp (Ronneau et al., 2016; Sanselicio and Viollier, 2015; Hallez et al., 2017) that promotes retention of C. crescentus in the flagellated state. Given this linkage, it is appealing to propose that PEP-dependent glycosylation with pseudaminic acid serves to integrate flagellar assembly with (p)ppGpp-dependent cell cycle control. Interestingly, pseudaminic acid synthesis also branches from a precursor of cell wall (peptidoglycan, PG) and lipopolysaccharide (O-antigen) synthesis, UDP-N-acetyl-glucosamine, indicating that flagellin glycosylation could be linked to cell envelope modifications, for example, to ensure that the formation of the PG-based division septum is linked to the synthesis of the flagellar filament. In other environmental bacteria, such as S. fredii NGR234, pseudaminic acid is not required for motility (Figure 3D) but it is a component of the capsular K-antigen (Le Quéré et al., 2006), where it may still be coordinated with the cell cycle, for example, again with septation. In C. crescentus the capsule is cell cycle regulated (Ardissone et al., 2014), but it is composed of different monosaccharides (Ardissone et al., 2014). Pseudaminic acid or other sialic acid-like sugars synthesized by NeuB paralogs may be generally exploited for metabolic coordination of cell (cycle) processes in bacteria.
Materials and methods
Strains and growth conditions
Request a detailed protocolC. crescentus NA1000 (Evinger and Agabian, 1977) and derivatives were grown at 30°C in PYE (peptone-yeast extract) or M2G (minimal glucose) (Ely, 1991). S. fredii NGR234 (Stanley et al., 1988) was grown at 30°C in TY (tryptone-yeast extract). E. coli S17-1 λpir (Simon et al., 1983), EC100D and Rosetta(DE3)pLysS (Novagen) were grown at 37°C in LB. Motility assays, swarmer cells isolation, electroporations, biparental matings, and bacteriophage φCr30-mediated generalized transductions were performed as described (Ely, 1991; Chen et al., 2005; Viollier and Shapiro, 2003; Viollier and Shapiro, 2004). Antibiotics were used at the following concentrations: nalidixic acid 20 μg/mL, kanamycin 20 μg/mL in solid medium and 5 μg/mL in liquid (50 μg/mL for S. fredii), gentamicin 1 μg/mL (20 μg/mL for E. coli and S. fredii), tetracycline 1 μg/mL (10 μg/mL for E. coli and S. fredii). Plasmids were introduced into S. fredii by bi-parental mating and into C. crescentus by electroporation.
Production of antibodies and immunoblots
Request a detailed protocolFor the production of antibodies, His6-NeuB and His6-SUMO-FlmG(301-500) were expressed in E. coli Rosetta(DE3)pLysS cells and the recombinant proteins were purified using Ni-NTA agarose (Qiagen). Purified His6-NeuB and His6-SUMO-FlmG(301-500) were excised from 12.5% SDS polyacrylamide gels and used to immunize rabbits (Josman LLC, Napa, CA). For immunoblots, protein samples were separated on SDS polyacrylamide gel, transferred to polyvinylidene difluoride (PVDF) Immobilon-P membranes (Merck Millipore) and blocked in TBS (Tris-buffered saline) 0.1% Tween 20% and 5% dry milk. The anti-sera were used at the following dilutions: anti-CtrA (1:10,000; Domian et al., 1997), anti-FlgE (1:50,000; Hahnenberger and Shapiro, 1987), anti-flagellins (raised against purified flagellins from C. crescentus; indicated as αFlagellins in figures; 1:20,000; Hahnenberger and Shapiro, 1987), anti-NeuB (1:10,000), anti-FlmG (1:10,000), anti-FljK (raised against His6-FljK expressed in E. coli; indicated as αFljK in figures; 1:10,000; Ardissone et al., 2020). Protein-primary antibody complexes were visualized using horseradish peroxidase-labelled anti-rabbit antibodies and ECL detection reagents (Merck Millipore).
Site-directed mutagenesis of neuB
Request a detailed protocolTo create neuB alleles mutated in the predicted catalytic residues, the neuB ORF was sub-cloned into pOK12 (Vieira and Messing, 1991) as NdeI/EcoRI fragment from pMT335-neuB. pOK-neuB was used as a template for oligonucleotide site-directed mutagenesis. Two complementary oligonucleotide primers containing the desired mutation were designed for each point mutation (Supplementary file 3 Table S3). PCR reactions were composed of 30 cycles, carried out under the following conditions: denaturation, 94°C for 1 min; annealing, 60°C for 1 min; extension, 68°C for 8 min. The PCR products were treated with DpnI to digest the template DNA and used to transform E. coli EC100D competent cells. The constructions obtained were verified by sequencing and sub-cloned as NdeI/EcoRI fragments into pMT335.
Transmission electron microscopy
Request a detailed protocolTransmission electron microscopy (Skerker and Shapiro, 2000) was performed on a JEOL 1200EX with samples that were fixed in 2.5% gluteraldehyde/25 mM cacodylate-HCl buffer (pH 7.4) and negatively stained with 1% uranyl acetate for 15 min on a 300 mesh nickel formvar-coated grid stabilized with an evaporated carbon film (Huitema et al., 2006; Radhakrishnan et al., 2008).
Capsular polysaccharide analysis by SDS-PAGE and silver staining
Request a detailed protocolStrains were grown in TY supplemented with vanillate 0.5 mM or IPTG 1 mM for 24 hr. Polysaccharide extractions were made from cells collected by centrifuging 4 mL of culture. The cell pellets were resuspended in 30 μL of lysis buffer (1 M Tris-HCl [pH 6.8], 2% [wt/vol] SDS, 4% [vol/vol] β-mercaptoethanol, 10% [vol/vol] glycerol, and 0.03% [wt/vol] bromophenol blue) and boiled for 10 min. Lysed cells were treated with 10 μL of proteinase K (2.5 mg/mL) at 60°C for 3 hr, then the samples were diluted by adding 80 μL of sample buffer (120 mM Tris-HCl [pH 6.8], 3% [wt/vol] SDS, 9% [vol/vol] β-mercaptoethanol, 30% [vol/vol] glycerol, and 0.03% [wt/vol] bromophenol blue). Polysaccharides were separated by SDS-PAGE (18% acrylamide) and stained for KPS as described (Le Quéré et al., 2006).
Bacterial adenylate cyclase two-hybrid (BACTH) assays
Request a detailed protocolProtein-protein interactions were studied through the BACTH system (Euromedex, France). Plasmid constructions and E. coli co-transformation were performed according to the manufacturer’s instructions. This system is based on the reconstitution of the adenylate cyclase (Cya) from Bordetella pertussis which is composed of two different fragments T25 and T18. Heterodimerization of the hybrid-proteins leads to the functional complementation of the adenylate cyclase that results in the production of cAMP leading to lacZ gene expression. The entire open reading frames of fljJ, fljK, fljM, and flmG, as well as the N-terminal (TPR, amino acids 1–313) and C-terminal (GT, amino acids 309–596) parts of FlmG were cloned into plasmids allowing fusion with T18 or T25 fragments either in N-terminal (pKNT25/pUT18) or C-terminal position (pKT25/pUT18C). Different combinations of T18 and T25 fusion plasmids were co-transformed in the E. coli BTH101 Δcya strain. The β-galactosidase activity was measured after an overnight culture of three independent co-transformants in LB medium as recommended by the manufacturer’s instruction. Co-transformation of BTH101 strain with the empty plasmid pK(N)T25 and pUT18(C) served as a control for basal β-galactosidase activity level (around 100 Miller unit).
β-galactosidase activity assays
Request a detailed protocolβ-galactosidase assays were performed at 30°C. Cells (50–200 µL) at OD660nm = 0.1–0.5 were lysed with 30 µL of chloroform and mixed with Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, and 1 mM MgSO4; pH 7) to a final volume of 800 µL. The reaction was started and timed following the addition of 200 µL of ONPG (o-nitrophenyl-β-D-galactopyranoside, 4 mg/mL in 0.1 M potassium phosphate, pH 7). Upon medium-yellow color development, 400 µL of 1 M Na2CO3was added to stop the reaction. OD420nm of the supernatant was recorded and Miller units were calculated as follows: U=(OD420nm*1000)/(OD660nm*t(min)*v(mL)). Error was computed as standard deviation (SD). Data is from three biological replicates.
Strains and plasmids construction
Request a detailed protocolIn-frame deletions were created using pNPTS138 or pK18mobsacB derivatives constructed as follows: pNPTS_ΔneuB: PCR was used to amplify two DNA fragments flanking the neuB ORF, by using primers neuB_ko1/neuB_ko2 and neuB_ko3/neuB_ko4. The PCR fragments were digested with EcoRI/BamHI and BamHI/HindIII, respectively, and ligated into pNPTS138 restricted with EcoRI and HindIII.
pSA37: PCR was used to amplify two DNA fragments flanking the flmG ORF, using primers flmG_ko1/flmG_ko2 and flmG_ko3/flmG_ko4. The PCR fragments were digested with HindIII/BamHI and BamHI/EcoRI, respectively, and ligated into pNPTS138 restricted with EcoRI and HindIII.
pSA252: PCR was used to amplify two DNA fragments flanking the flmH ORF, using primers flmH_ko1/flmH_ko2 and flmH_ko3/flmH_ko4. The PCR fragments were digested with EcoRI/XbaI and XbaI/HindIII, respectively, and ligated into pNPTS138 restricted with EcoRI and HindIII.
pSA265: PCR was used to amplify two DNA fragments flanking the CCNA_01531 ORF, using primers 1531_ko1/1531_ko2 and 1531_ko3/1531_ko4. The PCR fragments were digested with EcoRI/BamHI and BamHI/HindIII, respectively, and ligated into pNPTS138 restricted with EcoRI and HindIII.
pSA617: PCR was used to amplify two DNA fragments flanking the CCNA_01537 ORF, using primers 1537_ko1/1537_ko2 and 1537_ko3/1537_ko4. The PCR fragments were digested with NheI/BamHI and BamHI/HindIII, respectively, and ligated into pNPTS138 restricted with NheI and HindIII.
pSA253: PCR was used to amplify two DNA fragments flanking the flmD ORF, using primers flmD_ko1/flmD_ko2 and flmD_ko3/flmD_ko4. The PCR fragments were digested with EcoRI/BamHI and BamHI/HindIII, respectively, and ligated into pNPTS138 restricted with EcoRI and HindIII.
pSA35: PCR was used to amplify two DNA fragments flanking the rkpQ ORF, using primers rkpQ_ko1/rkpQ_ko2 and rkpQ_ko3/rkpQ_ko4. The PCR fragments were digested with EcoRI/BamHI and BamHI/HindIII, respectively, and ligated into pNPTS138 restricted with EcoRI and HindIII.
pSA326: PCR was used to amplify two DNA fragments flanking the rkp3_013 ORF, using primers 013_ko1/013_ko2 and 013_ko3/013_ko4. The PCR fragments were digested with EcoRI/XbaI and XbaI/HindIII, respectively, and ligated into pK18mobsacB (Schäfer et al., 1994) restricted with EcoRI and HindIII.
Bi-parental matings were used to transfer the resulting constructs into C. crescentus or S. fredii strains. Double recombination was selected by plating bacteria onto PYE (for C. crescentus) or TY (for S. fredii) plates containing 3% sucrose. Putative mutants were confirmed by PCR using primers external to the DNA fragments used for the in-frame deletion constructs.
Plasmids for constitutive expression (from Pvan, Pxyl, or Plac) were constructed as follows: pSA53: neuB ORF was amplified by PCR with primers NeuB_N (with NdeI site overlapping the start codon) and NeuB_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA59: flmG ORF was amplified by PCR with primers FlmG_N (with NdeI site overlapping the start codon) and FlmG_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA645: the flmG sequence encoding the glycosyltransferase domain (residues 309–596) was amplified by PCR with primers FlmG_int_N and FlmG_E and cloned into pMT335, restricted with NdeI and EcoRI.
pSA126: C. jejuni 11168 neuB1 ORF was amplified by PCR with primers NeuB_Cj1_N (with NdeI site overlapping the start codon) and NeuB_Cj1_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA47: C. jejuni 11168 neuB2 ORF was amplified by PCR with primers NeuB_Cj2_N (with NdeI site overlapping the start codon) and NeuB_Cj2_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA48: C. jejuni 11168 neuB3 ORF was amplified by PCR with primers NeuB_Cj3_N (with NdeI site overlapping the start codon) and NeuB_Cj3_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA60: fljK ORF was amplified by PCR with primers FljK_N (with NdeI site overlapping the start codon) and FljK_X (with XbaI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and XbaI.
pSA104: fljK ORF was subcloned from pSA60 into pMT463, using NdeI and XbaI.
pSA42: S. fredii NGR234 rkpQ ORF was amplified by PCR with primers RkpQ_N (with NdeI site overlapping the start codon) and RkpQ_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA263: S. fredii NGR234 rkpO ORF was amplified by PCR with primers RkpO_N (with NdeI site overlapping the start codon) and RkpO_M (with MunI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA569: S. fredii NGR234 rkpL ORF was amplified by PCR with primers RkpL_N (with NdeI site overlapping the start codon) and RkpL_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA568: S. fredii NGR234 rkpM ORF was amplified by PCR with primers RkpM_N (with NdeI site overlapping the start codon) and RkpM_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
pSA570: S. fredii NGR234 rkpLM operon was amplified by PCR with primers RkpL_N (with NdeI site overlapping the rkpL start codon) and RkpM_E (with EcoRI site flanking the rkpM stop codon) and cloned into pMT335, restricted with NdeI and EcoRI.
To create pSA454, a synthetic fragment encoding C. crescentus fljK (sequence optimized for E. coli, Supplementary file 3 Table S3) was ligated into pSRK-Gm (Khan et al., 2008), using NdeI and XbaI.
To create pSA496, the flmG ORF was subcloned from pSA59 into pSRK-Gm, using NdeI and XbaI.
To create the vector for co-expression of fljK and flmG in S. fredii, the synthetic fragment encoding C. crescentus fljK ORF was first ligated into pMT335, using NdeI and EcoRI, creating pSA107. Then flmG ORF was amplified by PCR with primers FlmG_rbs_E (with ribosome binding site and EcoRI site flanking the flmG start codon) and FlmG_X (with XbaI site flanking the flmG stop codon) and cloned into pSA107, restricted with EcoRI and XbaI, creating pSA235. Finally, the fljK and flmG ORFs were subcloned from pSA235 into pSRK-Gm, using NdeI and XbaI, creating pSA236.
pSA571: flmH ORF was amplified by PCR with primers FlmH_N (with NdeI site overlapping the start codon) and FlmH_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI. The flmH ORF was then subcloned from pMT335 into pSRK-Km (Khan et al., 2008), using NdeI and XbaI.
pSA572: CCNA_01531 ORF was amplified by PCR with primers 1531_N (with NdeI site overlapping the start codon) and 1531_E (with EcoRI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI. The CCNA_01531 ORF was then subcloned from pMT335 into pSRK-Km, using NdeI and XbaI.
pSA624: CCNA_01537 ORF was amplified by PCR with primers 1537_N (with NdeI site overlapping the start codon) and 1537_X (with XbaI site flanking the stop codon) and cloned into pSRK-Km, restricted with NdeI and XbaI.
pSA573: S fredii NGR234 rkp3_013 ORF was amplified by PCR with primers rkp3_013_N (with NdeI site overlapping the start codon) and rkp3_013_M (with MunI site flanking the stop codon) and cloned into pMT335, restricted with NdeI and EcoRI. The rkp3_013 ORF was then subcloned from pMT335 into pSRK-Km, using NdeI and XbaI.
For neuB site-directed mutagenesis:
pSA58: neuB ORF was subcloned from pSA53 into pOK12, using NdeI and EcoRI. pSA58 was used as a template for site-directed mutagenesis of neuB.
pSA90: pOK12 carrying neuB E30A allele.
pSA91: pOK12 carrying neuB H245A allele.
pSA92: pOK12 carrying neuB R322A allele.
pSA93: the neuB E30A allele was subcloned from pSA90 into pMT335, using NdeI and EcoRI.
pSA94: the neuB H245A allele was subcloned from pSA91 into pMT335, using NdeI and EcoRI.
pSA95: the neuB R322A allele was subcloned from pSA92 into pMT335, using NdeI and EcoRI.
pIDT-flm: to express the complete C. crescentus pseudaminic acid biosynthesis pathway in E. coli, a synthetic sequence (codon-optimized for E. coli; Supplementary file 3 Table S3) encoding for flmA (CCNA_00233), flmB (CCNA_00234), flmH (CCNA_01523), flmD (CCNA_02947), neuB (CCNA_02961) and flmC (CCNA_02946) was expressed from pUCIDT under control of the T5 promoter.
Plasmid pSA44 is a derivative of pET28a expressing His6-NeuB under control of the T7 promoter. To construct pSA44, neuB ORF was amplified by PCR with primers NeuB_N (with NdeI site overlapping the start codon) and NeuB_E (with EcoRI site flanking the stop codon) and cloned into pET28a, restricted with NdeI and EcoRI.
To express FljK in E. coli without His6 tag, fljK ORF was subcloned from pMT335 into pET47b, using NdeI and SacI, creating pSA106.
Plasmid pSA363 is a derivative of pCWR547 (Radhakrishnan et al., 2010) expressing His6-SUMO-FlmG(301-500) under control of the T7 promoter. To construct pSA363, a fragment encoding residues 301–500 of FlmG was amplified by PCR with primers FlmG_547_N and FlmG_547_S, digested with NdeI and SacI and cloned into pCWR547 restricted with the same enzymes.
Plasmids used for the BACTH experiments were constructed as follows: pNK92: the sequence encompassing the last 288 amino acids of FlmG containing the glycosyltransferase domain (GT) was amplified with primers NK-77 and NK-75 and cloned into pKNT25 restricted with XbaI and KpnI.
pNK93: the sequence encompassing the last 288 amino acids of FlmG containing the glycosyltransferase domain (GT) was amplified with primers NK-77 and NK-79 and cloned into pKT25 restricted with XbaI and KpnI.
pNK95: flmG ORF was amplified with primers NK-74 and NK-75 and cloned into pKNT25 restricted with XbaI and KpnI.
pNK96: flmG ORF was amplified with primers NK-74 and NK-79 and cloned into pKT25 restricted with XbaI and KpnI. pNK98: the sequence encompassing the first 313 amino acids of FlmG containing the TPR domain was amplified with primers NK-74 and NK-76 and cloned into pKNT25 restricted with XbaI and KpnI.
pNK99: the sequence encompassing the first 313 amino acids of FlmG containing the TPR domain was amplified with primers NK-74 and NK-78 and cloned into pKT25 restricted with XbaI and KpnI.
pNK16: fljJ ORF was amplified with primers NK-01 and NK-02 and cloned into pUT18C restricted with XbaI and KpnI.
pNK144: fljK ORF was amplified with primers NK-10 and NK-11 and cloned into pUT18C restricted with XbaI and KpnI.
pNK330: fljM ORF was amplified with primers NK-62 and NK-64 and cloned into pUT18C restricted with XbaI and KpnI.
To create lacZ transcriptional fusions, promoter regions were cloned into pRKlac290 (Gober and Shapiro, 1992) as follows: pRKlac290_PneuB: a 569 bp DNA fragment was amplified by PCR with primers PneuB_E/PneuB_X, digested with EcoRI and XbaI, and ligated into pRKlac290, cut with the same enzymes.
pRKlac290_PflmG: a 579 bp DNA fragment was amplified by PCR with primers PflmG_E/PflmG_X, digested with EcoRI and XbaI, and ligated into pRKlac290, cut with the same enzymes.
pRKlac290_PflmA: a 570 bp DNA fragment was amplified by PCR with primers PflmA_E/PflmA_X, digested with EcoRI and XbaI, and ligated into pRKlac290, cut with the same enzymes.
Appendix 1
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, Strain Background (Caulobacter crescentus NA1000) | Caulobacter crescentus NA1000 | Evinger and Agabian, 1977; PMID:334726 | See Supplementary file 2 Table S2 | |
| Antibody | CtrA Rabbit polyclonal | Domian et al., 1997; PMID:9267022 | Home-made antibodies raised against full-length protein of C. crescentus | Immunoblot: 1/10000 dilution Figure 6, Figure 6—figure supplement 1 |
| Antibody | FlgE Rabbit polyclonal | Hahnenberger and Shapiro, 1987; PMID:3039149 | Home-made antibodies raised against FlgE protein of C. crescentus | Immunoblot: 1/50000 dilution Figure 1 |
| Antibody | Flagellins Rabbit polyclonal | Hahnenberger and Shapiro, 1987; PMID:3039149 | Home-made antibodies raised against purified flagellins from C. crescentus | Immunoblot: 1/20000 dilution Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6—figure supplement 1 |
| Antibody | FljK Rabbit polyclonal | Ardissone et al., 2020 | Home-made antibodies raised against full-length protein of C. crescentus | Immunoblot: 1/10000 dilution Figure 1, Figure 4, Figure 5, Figure 5—figure supplement 1 |
| Antibody | NeuB Rabbit polyclonal | This paper | Home-made antibodies raised against full-length protein of C. crescentus | Immunoblot: 1/10000 dilution Figure 2, Figure 3, Figure 6—figure supplement 1 |
| Antibody | FlmG Rabbit polyclonal | This paper | Home-made antibodies raised against FlmG (amino acids 301–500) of C. crescentus | Immunoblot: 1/10000 dilution Figure 6 |
| Recombinant DNA reagent | Plasmids | This paper | See Supplementary file 2 Table S2 | |
| Sequence-based reagent | PCR primers | This paper | See Supplementary file 3 Table S3 | |
| Sequence-based reagent | FljK synthetic sequence | This paper | FljK coding sequence, codon-optimized for E. coli | See Supplementary file 3 Table S3 |
| Sequence-based reagent | Pseudaminic acid biosynthesis operon synthetic sequence | This paper | FlmA, FlmB, FlmH, FlmD, NeuB and FlmC coding sequences, codon-optimized for E. coli | See Supplementary file 3 Table S3 |
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Interplay between flagellation and cell cycle control in CaulobacterCurrent Opinion in Microbiology 28:83–92.https://doi.org/10.1016/j.mib.2015.08.012
-
Coordinating assembly of a bacterial macromolecular machineNature Reviews Microbiology 6:455–465.https://doi.org/10.1038/nrmicro1887
-
Identification and characterization of NeuB3 from Campylobacter jejuni as a pseudaminic acid synthaseJournal of Biological Chemistry 280:35922–35928.https://doi.org/10.1074/jbc.M507483200
-
Hijacking bacterial glycosylation for the production of glycoconjugates, from vaccines to humanised glycoproteinsThe Journal of Pharmacy and Pharmacology 67:338–350.https://doi.org/10.1111/jphp.12321
-
De- and repolarization mechanism of flagellar morphogenesis during a bacterial cell cycleGenes & Development 27:2049–2062.https://doi.org/10.1101/gad.222679.113
-
Bacterial cell cycle and growth phase switch by the essential transcriptional regulator CtrANucleic Acids Research 47:10628–10644.https://doi.org/10.1093/nar/gkz846
-
The organization of the Caulobacter crescentus flagellar filamentJournal of Molecular Biology 206:627–636.https://doi.org/10.1016/0022-2836(89)90571-8
-
Genetics of caulobacter crescentusMethods in Enzymology 204:372–384.https://doi.org/10.1016/0076-6879(91)04019-k
-
Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cellsJournal of Bacteriology 132:294–301.https://doi.org/10.1128/JB.132.1.294-301.1977
-
Functional characterization of bacterial oligosaccharyltransferases involved in O-linked protein glycosylationJournal of Bacteriology 189:8088–8098.https://doi.org/10.1128/JB.01318-07
-
Flagellin redundancy in Caulobacter crescentus and its implications for flagellar filament assemblyJournal of Bacteriology 193:2695–2707.https://doi.org/10.1128/JB.01172-10
-
Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylationBiotechnology and Genetic Engineering Reviews 28:147–176.https://doi.org/10.5661/bger-28-147
-
A developmentally regulated Caulobacter flagellar promoter is activated by 3' enhancer and IHF binding elementsMolecular Biology of the Cell 3:913–926.https://doi.org/10.1091/mbc.3.8.913
-
Role of two flagellin genes in Campylobacter motilityJournal of Bacteriology 173:4757–4764.https://doi.org/10.1128/JB.173.15.4757-4764.1991
-
Identification of a gene cluster involved in flagellar basal body biogenesis in Caulobacter crescentusJournal of Molecular Biology 194:91–103.https://doi.org/10.1016/0022-2836(87)90718-2
-
Hit the right spots: cell cycle control by phosphorylated guanosines in alphaproteobacteriaNature Reviews Microbiology 15:137–148.https://doi.org/10.1038/nrmicro.2016.183
-
Engineering protein glycosylation in prokaryotesCurrent Opinion in Systems Biology 5:23–31.https://doi.org/10.1016/j.coisb.2017.05.016
-
Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on ti plasmid quorum sensingApplied and Environmental Microbiology 74:5053–5062.https://doi.org/10.1128/AEM.01098-08
-
Systems biology of caulobacterAnnual Review of Genetics 41:429–441.https://doi.org/10.1146/annurev.genet.41.110306.130346
-
Structural characterization of a K-antigen capsular polysaccharide essential for normal symbiotic infection in Rhizobium sp. NGR234: deletion of the rkpMNO locus prevents synthesis of 5,7-diacetamido-3,5,7,9-tetradeoxy-non-2-ulosonic acidThe Journal of Biological Chemistry 281:28981–28992.https://doi.org/10.1074/jbc.M513639200
-
A new class of Caulobacter crescentus flagellar genesJournal of Bacteriology 180:5010–5019.https://doi.org/10.1128/JB.180.19.5010-5019.1998
-
Functional characterization of the flagellar glycosylation locus in Campylobacter jejuni 81-176 using a focused metabolomics approachJournal of Biological Chemistry 281:18489–18498.https://doi.org/10.1074/jbc.M603777200
-
Targeted metabolomics analysis of Campylobacter coli VC167 reveals legionaminic acid derivatives as novel flagellar glycansJournal of Biological Chemistry 282:14463–14475.https://doi.org/10.1074/jbc.M611027200
-
Flagellin glycosylation in Pseudomonas aeruginosa PAK requires the O-antigen biosynthesis enzyme WbpOJournal of Biological Chemistry 283:3507–3518.https://doi.org/10.1074/jbc.M708894200
-
Protein glycosylation in Bacteria: sweeter than everNature Reviews Microbiology 8:765–778.https://doi.org/10.1038/nrmicro2383
-
Regulatory roles of the bacterial nitrogen-related phosphotransferase systemTrends in Microbiology 18:205–214.https://doi.org/10.1016/j.tim.2010.02.003
-
Phosphoenolpyruvate:carbohydrate phosphotransferase systems of BacteriaMicrobiological Reviews 57:543–594.https://doi.org/10.1128/MMBR.57.3.543-594.1993
-
Flagellin glycosylation with pseudaminic acid in Campylobacter and Helicobacter: prospects for development of novel therapeuticsCellular and Molecular Life Sciences : CMLS 75:1163–1178.https://doi.org/10.1007/s00018-017-2696-5
-
Emerging facets of prokaryotic glycosylationFEMS Microbiology Reviews 41:49–91.https://doi.org/10.1093/femsre/fuw036
-
Rhizobium sp. strain NGR234 possesses a remarkable number of secretion systemsApplied and Environmental Microbiology 75:4035–4045.https://doi.org/10.1128/AEM.00515-09
-
Flagellar glycosylation in Burkholderia pseudomallei and Burkholderia thailandensisJournal of Bacteriology 193:3577–3587.https://doi.org/10.1128/JB.01385-10
-
A bifunctional O-GlcNAc transferase governs flagellar motility through anti-repressionGenes & Development 20:3283–3295.https://doi.org/10.1101/gad.1492606
-
Cell-cycle progression and the generation of asymmetry in Caulobacter crescentusNature Reviews Microbiology 2:325–337.https://doi.org/10.1038/nrmicro864
-
Cloning of hemA from Rhizobium sp. NGR234 and symbiotic phenotype of a gene-directed mutant in diverse legume generaMolecular and General Genetics MGG 215:32–37.https://doi.org/10.1007/BF00331299
-
Glycosylate and move! the glycosyltransferase maf is involved in bacterial flagella formationEnvironmental Microbiology 20:228–240.https://doi.org/10.1111/1462-2920.13975
-
Posttranslational modification of flagellin FlaB in Shewanella oneidensisJournal of Bacteriology 195:2550–2561.https://doi.org/10.1128/JB.00015-13
-
Campylobacter--a tale of two protein glycosylation systemsTrends in Microbiology 11:233–238.https://doi.org/10.1016/S0966-842X(03)00079-9
-
The complete phosphotransferase system in Escherichia coliJournal of Molecular Microbiology and Biotechnology 3:329–346.
-
Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellinJournal of Biological Chemistry 276:34862–34870.https://doi.org/10.1074/jbc.M104529200
-
Sugar and spice make Bacteria not nice: protein glycosylation and its influence in pathogenesisJournal of Molecular Biology 428:3206–3220.https://doi.org/10.1016/j.jmb.2016.04.013
-
Diversity of microbial sialic acid metabolismMicrobiology and Molecular Biology Reviews 68:132–153.https://doi.org/10.1128/MMBR.68.1.132-153.2004
-
Spatial complexity of mechanisms controlling a bacterial cell cycleCurrent Opinion in Microbiology 7:572–578.https://doi.org/10.1016/j.mib.2004.10.005
-
Differential glycosylation of polar and lateral flagellins in Aeromonas hydrophila AH-3Journal of Biological Chemistry 287:27851–27862.https://doi.org/10.1074/jbc.M112.376525
Article and author information
Author details
Patrick H Viollier

Funding
Swiss National Science Foundation (31003A_182576)
- Patrick H Viollier
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Laurence Degeorges for excellent technical assistance and acknowledge the Swiss National Science Foundation grant 31003A_182576 for the funding support.
Copyright
© 2020, Ardissone et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,353
- views
-
- 205
- downloads
-
- 18
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 18
- citations for umbrella DOI https://doi.org/10.7554/eLife.60488
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Specificity in glycosylation of multiple flagellins by the modular and cell cycle regulated glycosyltransferase FlmG
eLife 9:e60488.
https://doi.org/10.7554/eLife.60488




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}