Casein kinase 1G2 suppresses necroptosis-promoted testis aging by inhibiting receptor-interacting kinase 3
- National Institute of Biological Sciences, China
- Tsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University, China
- Department of Urology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, China
Abstract
Casein kinases are a large family of intracellular serine/threonine kinases that control a variety of cellular signaling functions. Here we report that a member of casein kinase 1 family, casein kinase 1G2, CSNK1G2, binds and inhibits the activation of receptor-interacting kinase 3, RIPK3, thereby attenuating RIPK3-mediated necroptosis. The binding of CSNK1G2 to RIPK3 is triggered by auto-phosphorylation at serine 211/threonine 215 sites in its C-terminal domain. CSNK1G2-knockout mice showed significantly enhanced necroptosis response and premature aging of their testis, a phenotype that was rescued by either double knockout of the Ripk3 gene or feeding the animal with a RIPK1 kinase inhibitor-containing diet. Moreover, CSNK1G2 is also co-expressed with RIPK3 in human testis, and the necroptosis activation marker phospho-MLKL was observed in the testis of old (>80) but not young men, indicating that the testis-aging program carried out by the RIPK3-mediated and CSNK1G2-attenuated necroptosis is evolutionarily conserved between mice and men.
Introduction
RIPK3 is an intracellular serine/threonine kinase with a key role in necroptosis, a regulated form of necrotic cell death (Christofferson and Yuan, 2010; Vandenabeele et al., 2010; Wallach et al., 2016). During necroptosis, in response of TNF-family of cytokines, toll-like receptors (TLRs), and Z-RNAs, RIPK3 is activated either by a related kinase RIPK1, or adaptor proteins TRIF, or ZBP1/DAI, respectively (Cho et al., 2009; Degterev et al., 2008; He et al., 2009; Zhang et al., 2009; Zhang et al., 2020). Activated RIPK3 phosphorylates the mixed lineage kinase-domain-like protein, MLKL, to trigger its oligomerization and translocation from the cytosol to membranes including plasma membrane for their disruption (Cai et al., 2014; Chen et al., 2014; Sun et al., 2012; Wang et al., 2014). Necroptosis is actively suppressed by caspase-8-mediated cleavage of RIPK1 and RIPK3, whose upstream activation pathway is often shared between caspase-8 and RIPK1 kinase. Embryonic lethality in Caspase-8 knockout mice is rescued by double knockout Ripk3 or Mlkl, and cellular necroptosis induction by TNF-α or TLRs is dramatically enhanced if caspase-8 activity is suppressed (Dannappel et al., 2014; Dillon et al., 2014; Günther et al., 2011; He et al., 2009; Kaiser et al., 2011; Newton et al., 2019; Oberst et al., 2011; Rickard et al., 2014; Takahashi et al., 2014). RIPK3 was also reported to be negatively regulated by the phosphatase Ppm1b but the effect seems minor and the in vivo validation has yet to be obtained (Chen et al., 2015).
One of the important physiological function of necroptosis is to promote the aging of testis in mice (Li et al., 2017). The necroptosis activation marker, the phosphorylated MLKL has been observed in spermatogonium stem cells and Sertoli cells in the seminiferous tubules of old (>18 months) but not young mouse testis. Knockout Ripk3, or Mlkl, or feeding mice with a RIPK1 kinase inhibitor-containing diet, blocks necroptosis from occurring in mouse testis, and allows mice to maintain the youthful morphological features and the function of male reproductive system to advanced ages, when age-matched wild-type mice had lost their reproductive function (Li et al., 2017).
Casein kinases are a large family of intracellular serine/threonine kinases that control a variety of cellular signaling functions that include the circadian clock, Wnt receptor activation, μ opioid receptor modulation, DNA repair, and hypoxia response (Davidson et al., 2005; Elyada et al., 2011; Etchegaray et al., 2009; Goldberg et al., 2017; Pangou et al., 2016). We found several of the casein kinase (CSNK) 1 family members, including CSNK1D1, CSNK1E, and CSNK1G2, bind to and inhibit RIPK3 kinase activity. Interestingly, CSNK1G2 expresses at the highest level in mouse testis, whose expression pattern overlaps with that of RIPK3. Knocking out Csnk1g2 in mouse or multiple cell lines including cell lines derived from spermatocyte and Sertoli cells significantly enhanced necroptosis response and the Csnk1g2 knockout mice showed premature aging of their testis. Our results demonstrate that CSNK1G2 is a major negative regulator of necroptosis.
Results
CSNK1G2 negatively regulates necroptosis by binding to RIPK3
In a course of investigating RIPK3-interacting proteins, we found that several members of the casein kinase 1 family were among the proteins co-precipitated with RIPK3 kinase (Figure 1—figure supplement 1A). The effect of CSNK1 members on RIPK3 kinase activity was then assessed by co-expressing each member with RIPK3 in human embryo kidney 293 T cells, and the RIPK3 kinase activity was measured by probing the serine 227 auto-phosphorylation of RIPK3, an event critical for RIPK3 to recruit its substrate MLKL (Li et al., 2015; Sun et al., 2012). Among the casein kinase family members, CSNK1D1, CSNK1G2, and CSNK1E suppressed serine 227 phosphorylation on RIPK3 (Figure 1—figure supplement 1B). In particular, CSNK1G2, but not its closest family members CSNK1G1 and CSNK1G3, showed the most potent suppression of RIPK3 kinase activity (Figure 1—figure supplement 1C). Two kinase-dead mutants, CSNK1G2(K75A) and CSNK1G2(D165N), failed to suppress RIPK3 kinase activity (Figure 1A), indicating that the kinase activity of CSNK1G2 is required for its function in suppressing RIPK3. Although CSNK1G2 was not among the casein kinases co-precipitated with RIPK3 in 293 T cells, likely due to lack of expression in this cell line, its ability to strongly inhibit RIPK3 kinase activity and its pattern of tissue expression (see below) prompted us to invest this isoform of casein kinase further. Consistent with its ability to inhibit RIPK3 activity when co-expressed in 293 T cells, knockout Csnk1g2 in mouse embryonic fibroblasts, MEFs, significantly accelerated MEF necroptosis induced by the combination of TNF-α (T), a Smac mimetic (S), and a pan-caspase inhibitor Z-VAD-fmk (Z) (Figure 1B). The enhanced necroptosis was mitigated by reintroducing wild-type CSNK1G2 into the Csnk1g2 knockout MEFs, but a similar level of K75A kinase-dead mutant did not restore the necroptosis inhibition activity (Figure 1B). In addition to TSZ, MEFs with their Csnk1g2 knocked out also showed enhanced cell death when treated with death-inducing cytokine TRAIL plus a Smac mimetic and z-VAD (TRAIL/S/Z), or lipopolysaccharide (LPS) plus a Smac mimetic and z-VAD (LPS/S/Z) (Figure 1—figure supplement 2A).
Figure 1 with 3 supplements see all
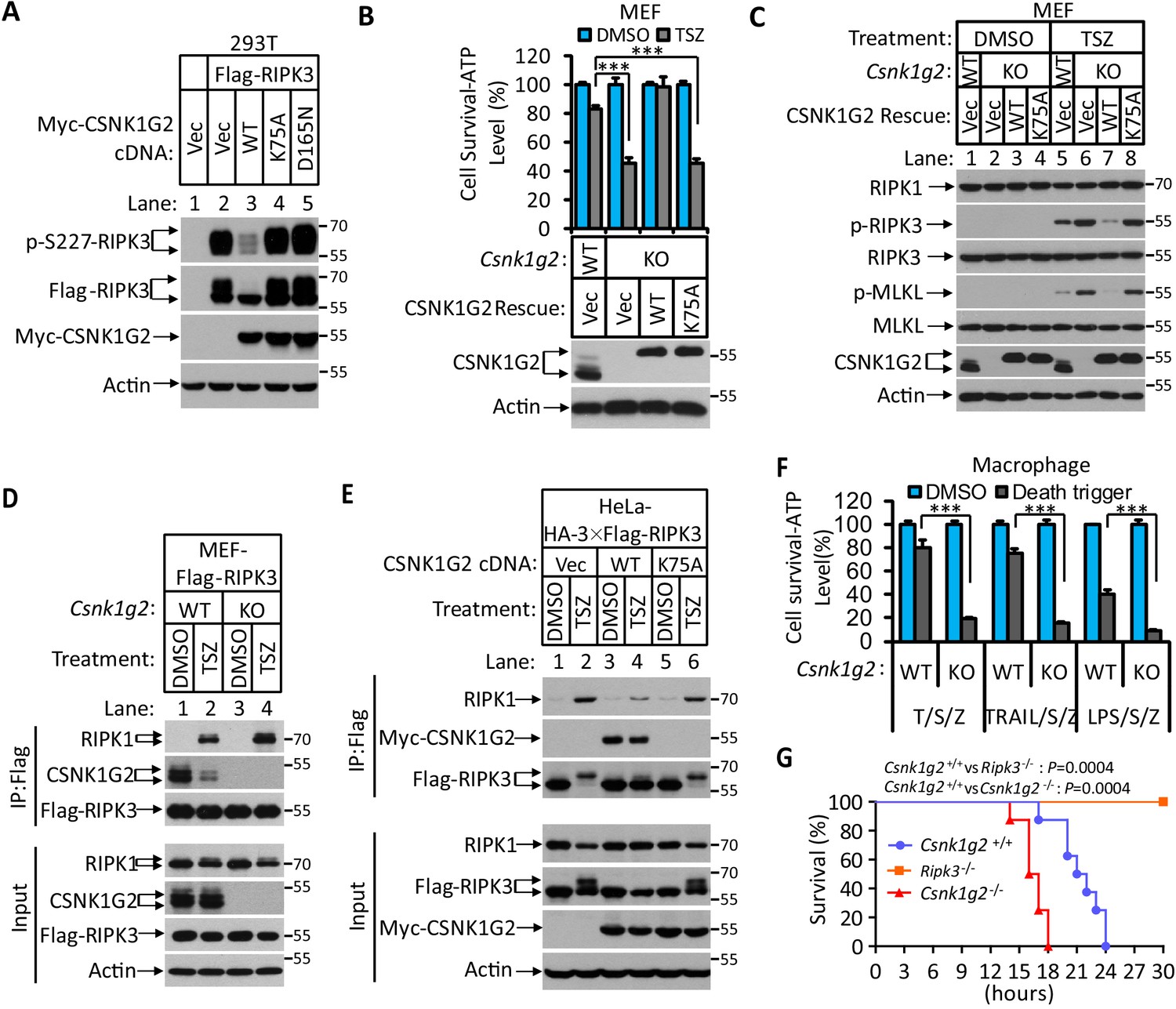
Knockout Csnk1g2 accelerates necroptosis.
(A) Western blotting analysis using antibodies against the indicated proteins. Cultured 293 T cells were transfected with Flag-tagged RIPK3 and the indicated versions of Myc-tagged CSNK1G2, including wild-type (WT) and two kinase-dead point mutants K75A and D165N for 20 hr. Cell extracts were then prepared and used for western blotting analysis. Vec, vector control. Numbers on the right indicate molecular weight markers (kDa). (B) Top: Cell viability as measured by Cell Titer-Glo. Cultured MEF with wild-type Csnk1g2 gene (WT) or with their Csnk1g2 gene knocked out (KO) followed by transfection with vector control (Vec) or indicated wild-type or a kinase-dead (K75A) mutant CSNK1G2 MEF. The cells were then treated with DMSO or TSZ as indicated for 12 hr before the intracellular ATP levels were measured by Cell Titer-Glo. T denotes 20 ng/ml TNF-α; S, denotes 100 nM Smac mimetic; Z denotes 20 μM Z-VAD-FMK. Data are mean ± SD of triplicate wells. ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. Bottom: Aliquots of these treated cells were used for western blotting analysis using an antibody against CSNK1G2. (C) Western blotting of necroptosis activation markers phospho-RIPK3 (p-RIPK3) and phospho-MLKL (p-MLKL). Cultured MEF cells with indicated CSNK1G2 gene as in (B) were treated with indicated stimuli for 4 hr before the cell extracts were prepared and subjected to western blotting analysis as indicated. (D and E) Western blotting analysis of RIPK3-associated RIPK1 and CSNK1G2. Immunoprecipitates using an anti-Flag antibody from extracts of MEF-Flag-RIPK3 and MEF (Csnk1g2−/−)-Flag-RIPK3 cells (D) or HeLa-HA-3×Flag-RIPK3-Myc-CSNK1G2(WT and K75A) cells (E) treated with the indicated stimuli for 6 hr were subjected to western blotting analyzing using antibodies as indicated. (F) Cell viability measurement of bone marrow-derived macrophages from the wild-type (WT) or Csnk1g2 knockout mice. Macrophages were isolated from the WT or Csnk1g2 knockout mice (KO) and treated with the indicated necroptosis stimuli for 12 hr, and the cell viability was measured by Cell-Titer Glo. Trail: TNF-related apoptosis-inducing ligand. LPS: Lipopolysaccharide. Data are mean ± SD of triplicate wells. ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. (G) Kaplan–Meier plot of survival of male Csnk1g2+/+(WT), Csnk1g2−/− (Csnk1g2 knockout littermates), or Ripk3−/− (Ripk3 gene knockout) mice (n = 10 for each genotype, age: 3 months) injected intraperitoneally with one dose of murine TNF-α (300 μg/kg). Body temperature was measured with a lubricated rectal thermometer. Mice with a temperature below 23°C were euthanized for ethical reasons. Generation of Csnk1g2−/− mice.
To further explore the mechanism through which CSNK1G2 suppresses necroptosis, MEFs with their endogenous Csnk1g2 knocked out or rescued with wild-type or kinase-dead CSNK1G2 cDNA were treated with necroptosis-inducing T/TRAIL/LPS+S+Z, and the necroptosis activation markers phospho-RIPK3 (at threonine 231 and serine 232, equivalent to serine 227 of human RIPK3) and phospho-MLKL (at serine 345, equivalent to serine 358 of human MLKL) were analyzed by western blotting. Knocking out Csnk1g2 in MEFs resulted in higher levels of phospho-RIPK3 and phospho-MLKL (Figure 1C, lanes 5 and 6, and Figure 1—figure supplement 2B). Reintroducing wild-type, but not kinase-dead, mutant CSNK1G2, significantly decreased the levels of phospho-RIPK3 and phospho-MLKL when cells were treated with necroptosis inducers (Figure 1C, lanes 7 and 8).
Necroptosis induced by TSZ is initiated by the formation of necrosome, a protein complex containing both RIPK1 and RIPK3 (Cho et al., 2009; He et al., 2009; Zhang et al., 2009). MEFs with their Csnk1g2 knocked out showed more RIPK1 kinase association with RIPK3 (Figure 1D, lanes 2 and 4), indicating that CSNK1G2 suppresses necroptosis by binding to RIPK3 and preventing its recruitment to necrosome.
In addition to MEFs, we investigated the effect of CSNK1G2 in human cells. As shown in Figure 1E, expressing a Myc-tagged human CSNK1G2 reduced RIPK1 and RIPK3 interaction as measured by a co-immunoprecipitation (co-IP) experiment (Figure 1E, lanes 2 and 4). The kinase-dead mutant of CSNK1G2 (K75A) neither co-IPed with RIPK3 nor decreased RIPK1–RIPK3 interaction (Figure 1E, lanes 5 and 6, and Figure 1—figure supplement 2C). Moreover, the wild-type CSNK1G2-blocked necroptosis caused by the FKBP-binding small molecule-induced dimerization of an FKBP–F36V–RIPK3 fusion protein (Li et al., 2020; Orozco et al., 2014), while the kinase-dead CSNK1G2 (K75A) mutants did not, indicating that CSNK1G2 inhibits necroptosis by directly preventing RIPK3 activation (Figure 1—figure supplement 2D).
Csnk1g2 knockout mice showed accelerated TNF-α-induced systematic sepsis
We subsequently knocked out the Csnk1g2 gene in mice using guide RNA specifically targeted to exon-2 of the Csnk1g2 gene (Figure 1—figure supplement 3A). The successful deletion of 56 base pairs of exon-2 in the Csnk1g2 gene caused the deletion of its N-terminus kinase domain and introduced a new premature stop codon in the remaining mRNA (Figure 1—figure supplement 3A,B and D). Knockout of the Csnk1g2 gene resulted in no detection of CSNK1G2 protein in the testis of these animals (Figure 1—figure supplement 3C).
Loss of the Csnk1g2 gene did not affect the development of the knockout mice. However, the bone marrow-derived macrophages (BMDM) from the Csnk1g2 knockout mice showed enhanced cell death when treated with necroptosis-inducing agents, including T/S/Z, TRAIL/S/Z, or LPS/S/Z (Figure 1F) compared to the BMDM from their wild-type littermates. Notably, although the Csnk1g2 knockout mice looked normal, they died within 18 hr after TNF-α administration, much quicker than their wild-type littermates, indicating that TNF-α-induced systematic sepsis was dramatically enhanced (Figure 1G). In contrast, Ripk3 knockout mice were totally resistant to such a treatment as previously reported (Newton et al., 2014; Figure 1G).
CSNK1G2 interaction with RIPK3 requires auto-phosphorylation of its serine 211 and threonine 215 sites
Since the interaction between CSNK1G2 and RIPK3 requires the kinase activity of CSNK1G2, we searched for phosphorylation events on these two proteins that might be required for such an interaction. To this end, we immunoprecipitated CSNK1G2–RIPK3 complex and by mass spectrometry analysis found three clusters of peptides of CSNK1G2 that contained phosphorylated amino acid residues. These residues were: serine 26 and serine 27, serine 211 and threonine 215, and serine 381 (Figure 2A and Figure 2—figure supplement 1A). We subsequently introduced phosphorylation-resistant mutations in these residues and assessed their effect on RIPK3 activity. We found that only the S211A/T215A mutant lost the ability to block RIPK3 S227 phosphorylation, whereas the other two mutants containing serine 26/serine 27 and serine 381 to alanine mutations still blocked RIPK3 S227 phosphorylation as efficiently as the wild type (Figure 2B and Figure 2—figure supplement 1B). Consistently, reintroducing S211A/T215A mutant CSNK1G2 to the Csnk1g2 knockout MEFs did not restored the necroptosis-inhibiting activity of CSNK1G2 (Figure 2C). Furthermore, compared to wild-type CSNK1G2 that efficiently bound to RIPK3 and blocked RIPK1–RIPK3 interaction induced by necroptosis inducer TSZ (Figure 2D, lanes 1–4), S211A/T215A mutant CSNK1G2 lost its ability to bind RIPK3 and did not block RIPK1–RIPK3 interaction in response to TSZ (Figure 2D, lanes 5 and 6). Moreover, single S211A or T215A mutant showed decreased ability to block RIPK3 kinase activity while the double mutant lost all the inhibitory activity, similar to the K75A kinase-dead mutant (Figure 2—figure supplement 1C). These results suggested that the auto-phosphorylation of serine 211 and threonine 215 contributed to the ability of CSNK1G2 to bind and inhibit RIPK3 kinase activity. Not surprisingly, serine 211 and threonine 215 are within the conserved region of CSNK1G2 with amino acid residues between 205 and 240 (human origin) 100% conserved between human, chimpanzee, mouse, and bovine (Figure 2—figure supplement 1D).
Figure 2 with 1 supplement see all
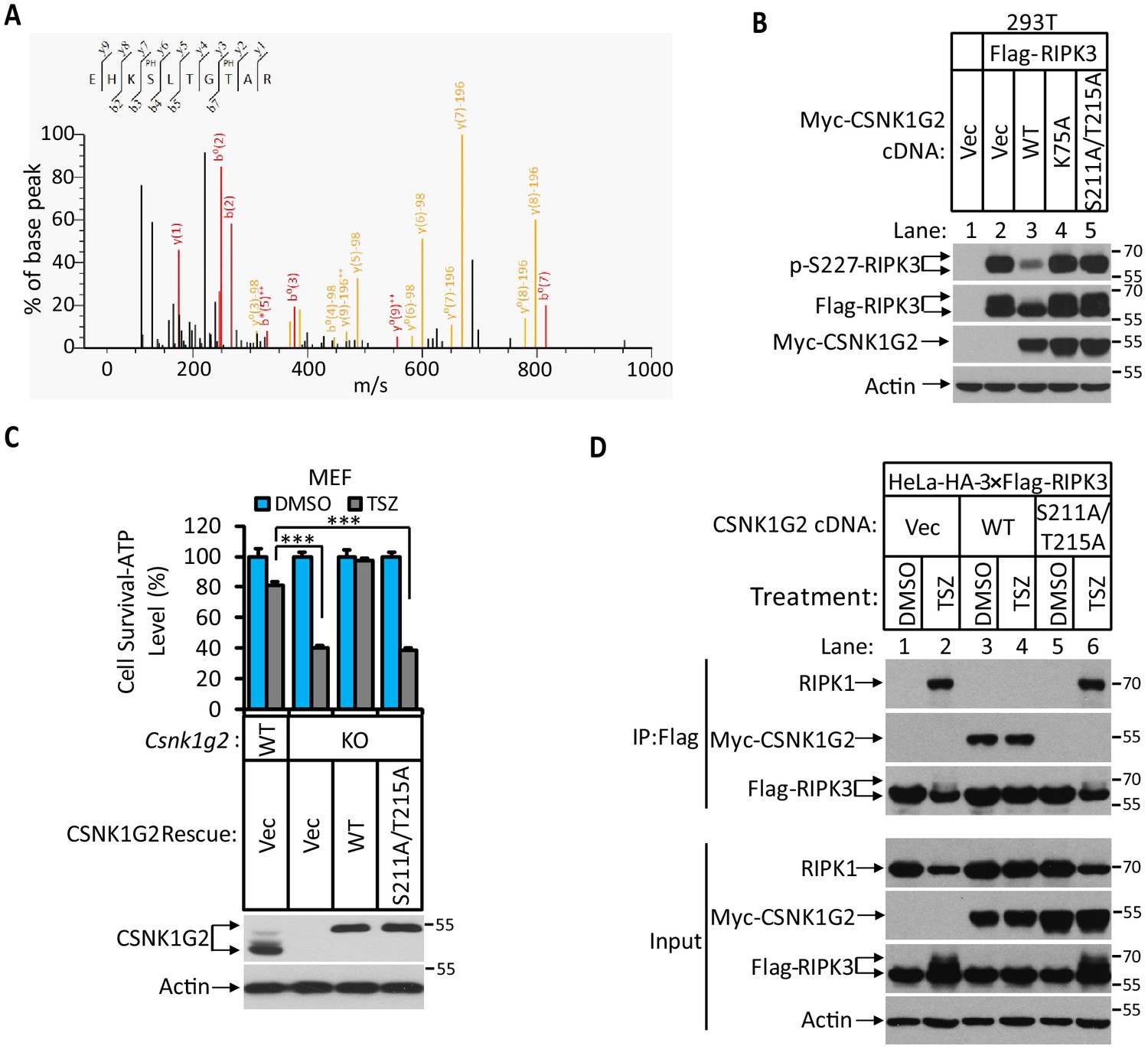
Identification and characterization of CSNK1G2 auto-phosphorylation sites.
(A) MS/MS spectrum of CSNK1G2 phosphorylation sites. The identified phosphorylated peptide to be EHKPSLTGPTAR with S211 and T215 as the phosphorylated amino acid residues. The b- and y-type product ions are indicated in the spectrum. Data are related to those in Figure 2—figure supplement 1. (B) Effect on RIPK3 auto-phosphorylation by co-expression of the indicated version of CSNK1G2. Cultured 293 T cells were transfected with Flag-tagged RIPK3 cDNA together with indicated Myc-tagged wild-type (WT) CSNK1G2, or kinase-dead mutant K75A, or phosphorylation site-resistant mutant S211A/T215A for 20 hr. The cell extracts were then subjected to western blotting analysis using antibodies against phospho-S227-RIPK3, Flag-RIPK3, and Myc-CSNK1G2 as indicated. (C) Top: Cell viability measurement of effect of phosphorylation sites mutant CSNK1G2 on necroptosis. Cultured WT MEF or MEF with their Csnk1g2 gene knocked out (KO) were transfected with either vector control (Vec) or cDNA encoding WT Csnk1g2 or phosphorylation sites mutant (S211A/T215A) followed by treatment of DMSO or necroptotic stimuli TSZ as indicated for 12 hr. The cell viability was measured by Cell-titer Glo. Data are mean ± SD of triplicate wells. ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. Bottom: Aliquots of these treated cells were used to make cell extracts for western blotting analysis using an antibody against CSNK1G2 protein. The effect of CSNK1G2 on RIPK1/RIPK3 interaction as measured by co-IP. Cultured HeLa-HA-3×Flag-RIPK3 cells were transfected with either vector control (Vec) or WT or phosphorylation site mutant Myc-CSNK1G2 (S211A/T215A) as indicated. The cells were then treated with DMSO or necroptosis stimuli TSZ for 6 hr. The cell extracts were prepared and subjected to immunoprecipitation with an anti-Flag antibody. The extracts (Input) and the immunoprecipitates (IP: Flag) were then subjected to western blotting analysis using antibodies as indicated.
Knocking out Csnk1g2 in testis cells significantly enhanced their necroptosis response
To further study the function of Csnk1g2 in vivo, we first measured the expression of this protein in mouse tissues by western blotting. As shown in Figure 3A, CSNK1G expression is low in the brain, heart, liver, ovary, and intestine (Figure 3A, lanes 3, 4, 6, 7, 8, 9, and 10). There was higher CSNK1G2 presence in lung and spleen (Figure 3A, lanes 2 and 5). The highest expression was found in the testis (Figure 3A, lane 1). Immunohistochemical analysis showed that CSNK1G2 was present in the seminiferous tubules of the testis (Figure 3B) and overlapped with that of RIPK3, indicating that, like RIPK3, it is also expressed in the spermatogenic cells and Sertoli cells, two major cell types in the seminiferous tubules that express RIPK3 (Li et al., 2017). When primary cells from the testis of Csnk1g2 knockout mice and their wild-type littermates were treated with necroptosis stimuli TSZ, significantly more cell death was observed in cells from the Csnk1g2 knockout testis (Figure 3C). Additionally, when the endogenous CSNK1G2 was immunoprecipitated from the testis extracts of 3-month-old wild-type mice, the endogenous RIPK3 was co-precipitated, indicating that these two proteins not only co-express in the testis but also interact with each other (Figure 3D). In contrast, the same immunoprecipitation experiment using testis extracts from the CSNK1G2 knockout littermates did not precipitate either CSNK1G2 or RIPK3, confirming the specificity of the anti-CSNK1G2 antibody (Figure 3D). These findings further indicate that the function of CSNK1G2 in testis is to block necroptosis in the RIPK3-expressing spermatogenic cells and Sertoli cells by directly binding to RIPK3.
Figure 3 with 1 supplement see all
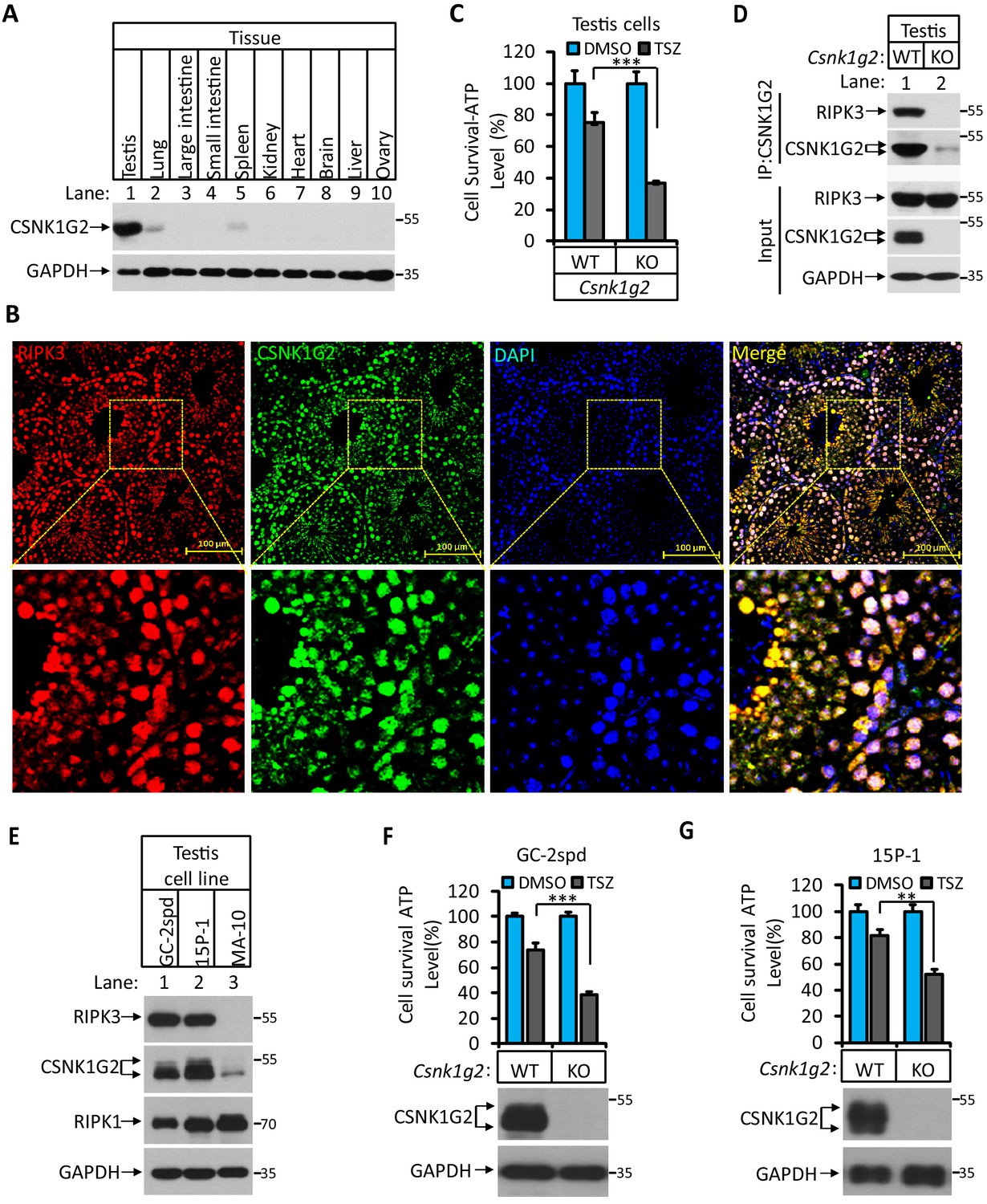
CSNK1G2 and RIPK3 are co-expressed in the seminiferous tubules of the mouse testis.
(A) The expression of CSNK1G2 in mouse testis, lung, large intestine, small intestine, spleen, kidney, heart, brain, liver, and ovary tissues (n = 3, 2 months). The indicated tissue extracts were subjected to western blotting analysis using antibodies against CSNK1G2 and GAPDH as indicated. (B) The expression of RIPK3 and CSNK1G2 in mouse testis. The testis sections of 2-month-old wild-type mice (n = 3) were stained sequentially with antibodies against RIPK3 and CSNK1G2 followed by fluorescent-conjugated secondary antibody. Counterstaining with DAPI, blue. Scale bar on the upper panel is 100 μm. The areas marked by the yellow boxes on the upper panel were shown in the lower panel. (C) The sensitivity of primary cells from the seminiferous tubules of Csnk1g2−/− and Csnk1g2+/+ testis to necroptosis induction. The cells from the seminiferous tubules of 2-month-old littermates with indicated genotype were isolated and cultured in vitro before treated with DMSO or TSZ as indicated for 12 hr. The cell viability was then measured by Cell-Titer Glo. Data are mean ± SD of triplicate wells. ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. (D) Western blotting analysis of CSNK1G2 interaction with RIPK3 in mouse testis. The wild-type and Csnk1g2 knockout testis (3 months) were ground and resuspended in lysis buffer, homogenized for 30 s with a Paddle Blender. The supernatants were collected, and CSNK1G2 was immunoprecipitated using anti-CSNK1G2 antibody. The immunocomplexes and lysates were analyzed by western blotting using antibodies as indicated. Each group was from a pool of three mice. (E) The expression of RIPK3, CSNK1G2, and RIPK1 proteins in GC-2spd, 15 P-1, and MA-10 cells. The extracts from the indicated cultured cells were subjected to western blotting analysis using antibodies against RIPK3, CSNK1G2, RIPK1, and GAPDH as indicated. (F and G) The effect of CSNK1G2 on necroptosis of GC-2spd and 15 P-1 cells. Cultured parental GC-2spd (f) or 15 P-1 cells (g) (WT), and GC-2spd or 15 P-1 cells with their Csnk1g2 gene knocked out (Csnk1g2−/−) were treated with DMSO or TSZ as indicated for 4 hr. The cell viability was measured by Cell-titer Glo. Data are mean ± SD of triplicate wells. **p<0.01, ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. Bottom, immunoblot of CSNK1G2. Cell extracts from aliquots of these cells were also subjected to western blotting analysis using antibodies against CSNK1G2 and GAPDH as indicated, and the results were shown at the bottom.
To further demonstrate necroptosis suppression activity of CSNK1G2 in seminiferous cells in testis, we knocked out the Csnk1g2 gene in cell lines derived from spermatocyte (GC-2spd(ts)), or Sertoli cells (15 p-1) and measured their necroptosis response. Similar to what was seen in testis, GC-2spd and 15 p-1 cells expressed both RIPK3 and CSNK1G2. In contrast, a cell line from the testosterone-producing Leydig cells did not express RIPK3, and CSNK1G2 was present at a much lower level compared to the other two cell lines (Figure 3E and Figure 3—figure supplement 1). Similar to what was observed in primary testis cells, GC-2spd and 15 p-1 with their Csnk1g2 gene knocked out showed significantly more death compared to their respective parental cells when treated with necroptotic stimuli TSZ (Figure 3F and G).
Csnk1g2 knockout mice showed accelerated male reproductive system aging compared to their wild-type littermates
When male mice reach more than one-and-a-half years of age, their body weight increases, their seminal vesicles grow several fold in both size and weight, and their seminiferous tubules become empty as spermatogenic and Sertoli cells undergo necroptosis (Li et al., 2017). We thus measured these aforementioned physiological features of Csnk1g2 knockout mice and their wild-type littermates up to 12 months of age. When these mice were at 2 months of age, their body weight, seminal vesicle size, testis, and appearance of seminiferous tubules were indistinguishable (Figure 4A–F). However, at 12 months of age, the body weight of Csnk1g2 knockout mice became significantly higher than their wild-type littermates, reaching up to 45 g on average compared to ~37 g for the wild type (Figure 4A). Their seminal vesicles weighted approximately 1 g, a 10-fold increase from when these mice were 2 months old, and about twofold higher than their 12-month-old wild-type littermates (Figure 4B and C). The average size and weight of the testis of 12-month-old Csnk1g2 knockout mice were also significantly smaller than their wild-type littermates, and many of their seminiferous tubules were already empty (Figure 4D–F). At 12 months of age, ~31% of Csnk1g2 knockout seminiferous tubules were empty compared to ~2% empty seminiferous tubules of wild-type littermates (Figure 4F).
Figure 4
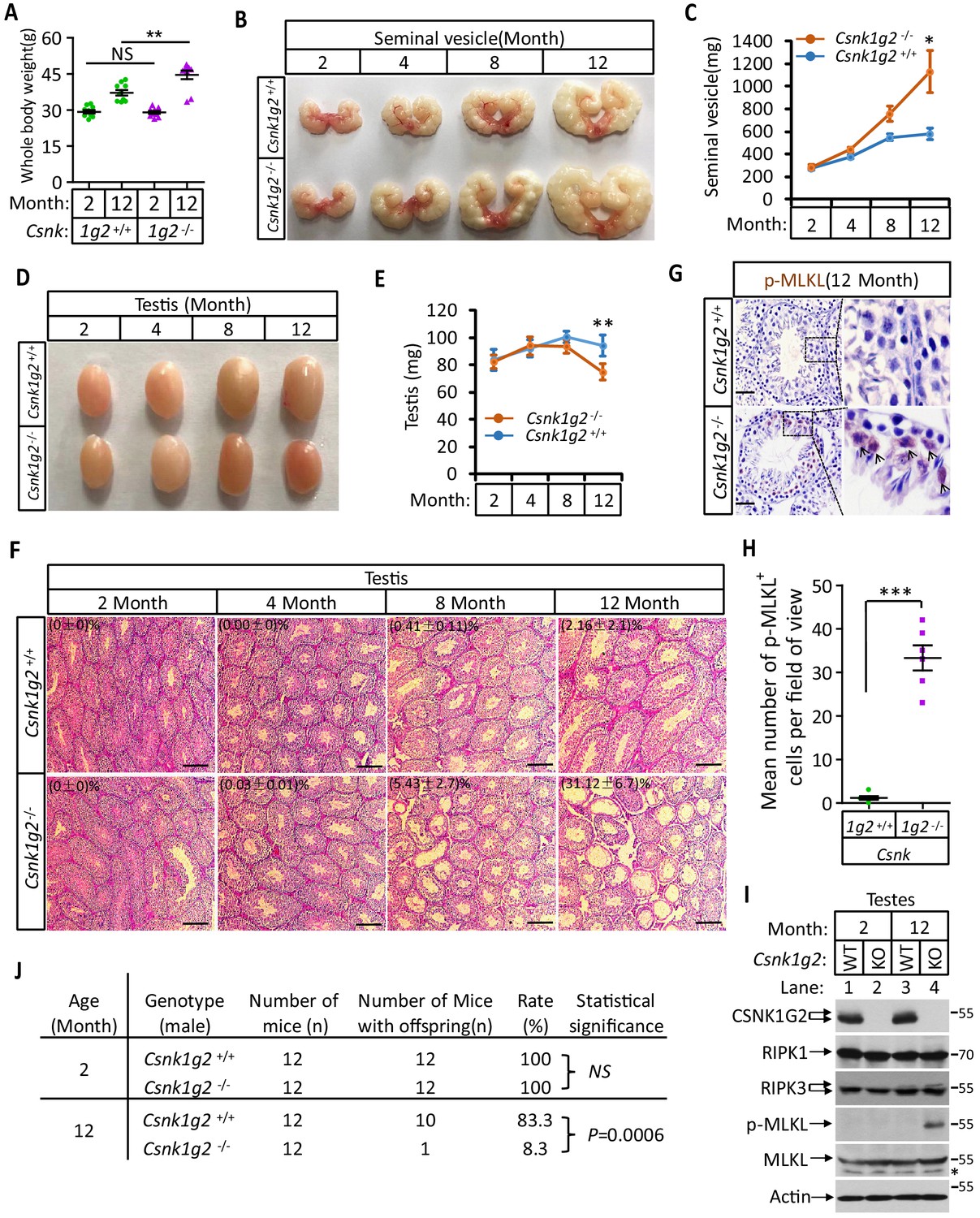
Accelerated reproductive system aging in Csnk1g2−/− male mice.
(A) Body weights of Csnk1g2+/+ and Csnk1g2−/− male littermate mice when they were 2 and 12 months old (n = 10 for each genotype). (B and C) Macroscopic features (B) and weights (C) of seminal vesicles from Csnk1g2+/+ and Csnk1g2−/− male littermate mice (n = 10 for each genotype) at the indicated ages. (D and E) Macroscopic features (D) and weights (E) of testes from Csnk1g2+/+ and Csnk1g2−/− male littermate mice (n = 12 for each genotype) at the indicated ages. (F) H&E staining sections of testis from Csnk1g2+/+ and Csnk1g2−/− male littermate mice (n = 10 for each genotype) at the indicated ages. The number of empty seminiferous tubules was counted based on H&E staining, and the percentage of empty seminiferous tubules of each group is labeled in the upper left corner of the images. Scale bar, 200 μm. (G and H) Immunohistochemical staining (IHC) of testes from Csnk1g2+/+ and Csnk1g2−/− male littermate mice (n = 6 for each genotype) with phospho-MLKL (p-MLKL) antibody in (G). p-MLKL positive cells were counted in five fields per testis and quantified in (H). Scale bar, 100 μm. (I) Western blotting analysis of extracts from phosphate-buffered saline (PBS) perfused testes of Csnk1g2+/+ (WT) and Csnk1g2−/− (KO) male littermate mice of 2 and 12 months of age using antibodies against CSNK1G2, RIPK1, RIPK3, MLKL, and phospho-MLKL (p-MLKL) and β-actin as indicated. The number on the right is markers of molecular weight (kDa). Each group was from a pool of three mice. (J) Summary of fertility rates of Csnk1g2+/+ and Csnk1g2−/− male littermate mice (n = 12). Each male mice of 2 or 12 months of age was housed in the same cage with a pair of 10-week-old wild-type female mice for 2 months; females were replaced every 2 weeks. The number of male mice with reproduction capacity was counted. p-values were determined using Fisher’s exact tests (unpaired, two-tailed). All quantified data in the figure except (J) represent the mean ± SEM. *p<0.05, **p<0.01, ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. NS, not significant.
In addition to the differences in appearance, 12-month-old Csnk1g2 knockout testis had much more necroptosis activation marker phospho-Serine345-MLKL in their seminiferous tubules. As shown in Figure 4G and H, there was prominent staining of phospho-Serine345-MKLK signal in the seminiferous tubules of Csnk1g2 knockout testis, whereas much less signal was seen in that of wild-type littermates. Consistently, the phospho-serine345-MLKL signal was only detected by western blotting in the testis extracts from 12-month-old Csnk1g2 knockout mice (Figure 4I, lane 4). No signal was observed in the extracts of young mice (2 months), nor from the 12-month-old wild-type littermates, although the protein levels of RIPK1, RIPK3, and MLKL were the same (Figure 4I, lanes 1–3).
Finally, we tested the reproductive activity of Csnk1g2 knockout and their wild-type littermates when they were 2 and 12 months of age. As shown in Figure 4J, all twelve 2-month-old male mice, regardless of genotype, impregnated their 10-week-old female partners. In contrast, only one of the twelve 12-month-old Csnk1g2 knockout mice was able to produce pubs when mated with 10-week-old female mice, whereas 10 of the 12 wild-type littermates produced progenies with their 10-week-old partners.
Blocking necroptosis by a RIPK1 inhibitor or Ripk3 gene knockout prevented CSNK1G2-accelerated male reproductive system aging
To test if the accelerated male reproductive system aging in the Csnk1g2 knockout mice was indeed due to enhanced necroptosis in the testis of these animals, the Csnk1g2 knockout animals were either fed a RIPK1 kinase inhibitor RIPA-56-containing diet or crossed with Ripk3 knockout mice to generate Csnk1g2/Ripk3 double knockout mice. As shown in Figure 5A–C, Figure 5—figure supplement 1A and B, the signs of aging, including body weight gain, an increase in the size and weight of seminal vesicles, and a decrease in the size of the testis, were all mitigated when the Csnk1g2 knockout animals were fed with RIPA-56 or their Ripk3 gene was knockout. All three physiological features remained at the same level as when the animals were young (3 months old) (Figure 5—figure supplement 1C).
Figure 5 with 2 supplements see all
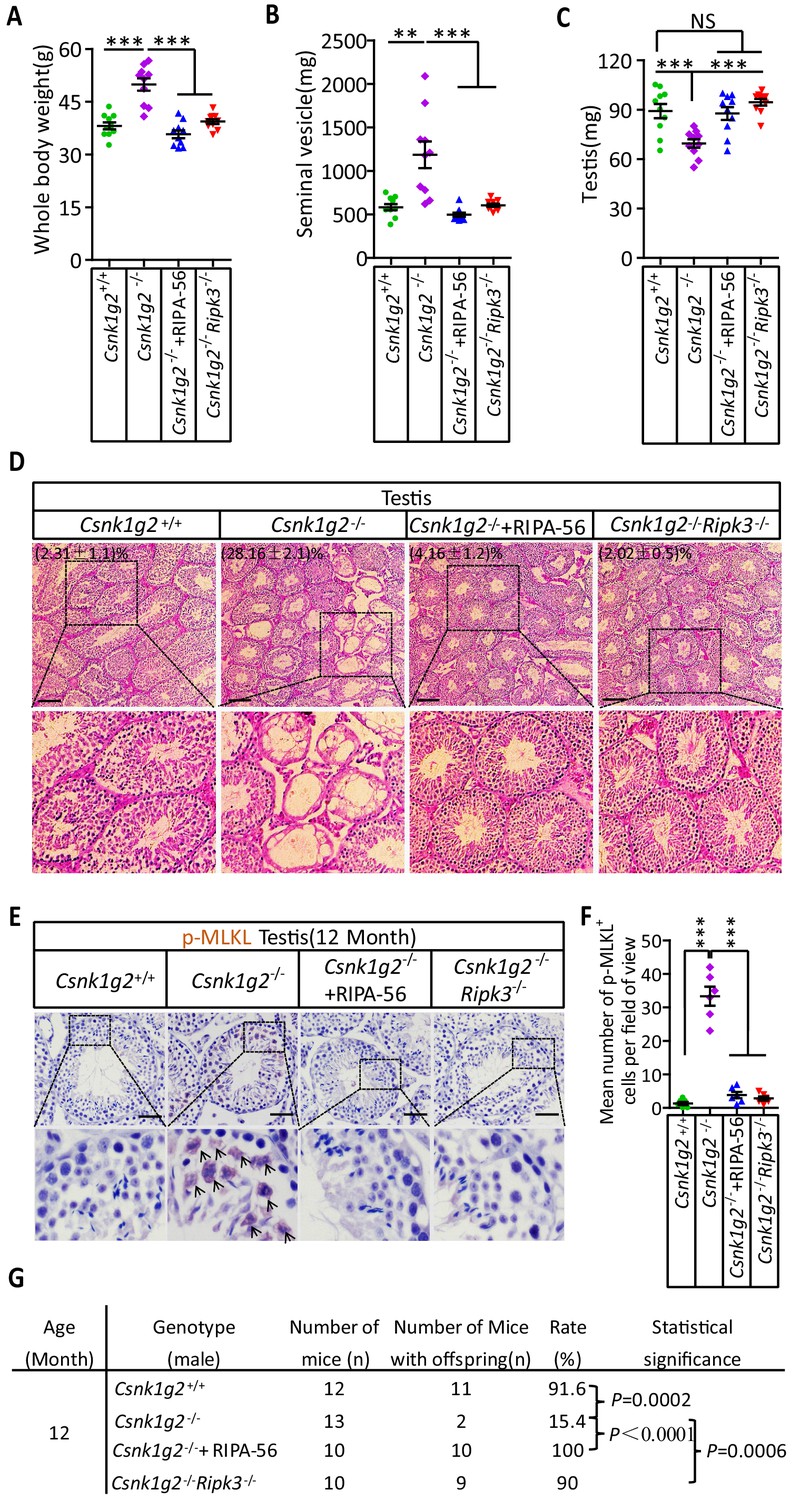
Rescuing the accelerated male reproductive system aging of Csnk1g2 knockout mice with a RIPK1 kinase inhibitor or Ripk3 knockout.
(A) The body weights of 12-month-old Csnk1g2+/+, Csnk1g2−/−, and Csnk1g2−/− fed with a RIPK1 kinase inhibitor (RIPA-56)-containing diet, and Csnk1g2−/−Ripk3−/− male littermate mice (n = 10 for each genotype). (B) The weights of seminal vesicles from 12-month-old Csnk1g2+/+, Csnk1g2−/−, Csnk1g2−/−+RIPA-56, and Csnk1g2−/−Ripk3−/− male littermate mice (n = 10 for each genotype). (C) The weights of testes from 12-month-old Csnk1g2+/+, Csnk1g2−/−, Csnk1g2−/−+RIPA-56, and Csnk1g2−/−Ripk3−/− male littermate mice (n = 10 for each genotype). (D) H&E staining of testis sections from 12-month-old Csnk1g2+/+, Csnk1g2−/−, Csnk1g2−/−+RIPA-56, and Csnk1g2−/−Ripk3−/− male littermate mice (n = 10 for each genotype). The number of empty seminiferous tubules was counted based on H&E staining, and the percentage of empty seminiferous tubules was labeled in the upper left corner of the images: scale bar, 200 μm. (E and F) IHC staining of testes from 12-month-old Csnk1g2+/+, Csnk1g2−/−, Csnk1g2−/−+RIPA-56, and Csnk1g2−/−Ripk3−/− male littermate mice (n = 8 for each genotype) with an anti-phospho-MLKL (p-MLKL) antibody (E). p-MLKL positive cells were counted in five fields per testis and quantified in (F). Scale bar, 100 μm. (G) Summary of the fertility rates of 12-month-old Csnk1g2+/+, Csnk1g2−/−, Csnk1g2−/−+RIPA-56, and Csnk1g2−/−Ripk3−/− male littermate mice (n = 10 for each genotype). Each male mouse was caged with a pair of 10-week-old wild-type female mice for 2 months; females were replaced every 2 weeks. The number of male mice with reproduction capacity was counted. p-values were determined using Fisher’s exact tests (unpaired, two-tailed). Csnk1g2−/−+RIPA-56 mice: Csnk1g2−/− male mice were fed with AIN93G or AING3G containing RIPA-56 (RIPA-56: 300 mg/kg) for 10 months started when they were 2 months old in an SPF facility. All quantified data in the figure except (G) represent the mean ± SEM. **p<0.01, ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests. NS, not significant.
In addition to these gross physical properties, the number of emptied seminiferous tubules in 12-month-old mice was significantly dropped from about 30% in Csnk1g2 knockout mice fed with normal chow diet to about 4% in Csnk1g2 knockout mice fed with RIPA-56-containing diet (Figure 5D). The number decreased to about 2%, similar to that of 2-month-old mice, when the mice also had their RIP3 gene knocked out (Figure 5D, right panel). The necroptosis activation marker phospho-Serine345-MLKL in the seminiferous tubules of Csnk1g2 knockout testis was also dropped to almost non-detectable levels in the littermates fed with RIPA-56 or had their Ripk3 gene knocked out, in contrast to the prominent signal from 12-month-old Csnk1g2 knockout mice on normal chow (Figure 5E and F).
When 12-month-old Csnk1g2 knockout male mice were tested for their reproductive ability, the loss of reproductive function of these mice was almost completely restored when fed with RIPA-56-containing food or had their Ripk3 gene knocked out. All ten 12-month-old male Csnk1g2 knockout mice fed with RIPA-56-containing food produced progenies, and nine of the ten Csnk1g2/Ripk3 double knockout mice generated pubs when paired with young (10-week-old) female partners, whereas only two of the thirteen Csnk1g2 knockout littermates on chow diet still produced progenies under the same condition (Figure 5G).
The accelerated aging of the mouse male reproductive system observed in 12-month-old Csnk1g2 knockout mice also manifested in decreased testosterone levels and increased amount of sex hormone-binding globulin (SHBG), pituitary hormone follicle-stimulating hormone (FSH), and luteinizing hormone (LH) (Figure 5—figure supplement 2E–H). The change in hormonal levels was mitigated when 12-month-old Csnk1g2 knockout animals were fed with RIPA-56 or had their Ripk3 gene knocked out, and all four hormones remained at the same level as when the animals were young (3 months old) (Figure 5—figure supplement 2A–H).
Human testes express CSNK1G2 and showed necroptosis activation marker when old
To test if the observed CSNK1G2 expression in mouse testis also occurred in human testis, we analyzed immunohistochemical sections of human testis surgically removed from young men who suffered from severe testicular torsion. As shown in Figure 6A, CSNK1G2 was expressed in the seminiferous tubules of testis in young men, and the expression of CSNK1G2 completely overlapped with RIPK3. Interestingly, when we examined the testis of both young and older men, we saw that most of the seminiferous tubules in an 82-year-old’s testis were empty, whereas the seminiferous tubules of a 30-year-old young man were full of cells surrounding vacuolar center where the mature sperms were present (Figure 6B and C). The seminiferous tubules of an 80-year-old testis also had high signal for phospho-Serine358-MLKL, a marker of necroptosis activation (Figure 6D and E). No such signal was detected in the seminiferous tubules of a 30-year-old testis (Figure 6D and E). These findings indicated that necroptosis-promoted testis aging observed in mice is also conserved in men.
Figure 6 with 1 supplement see all
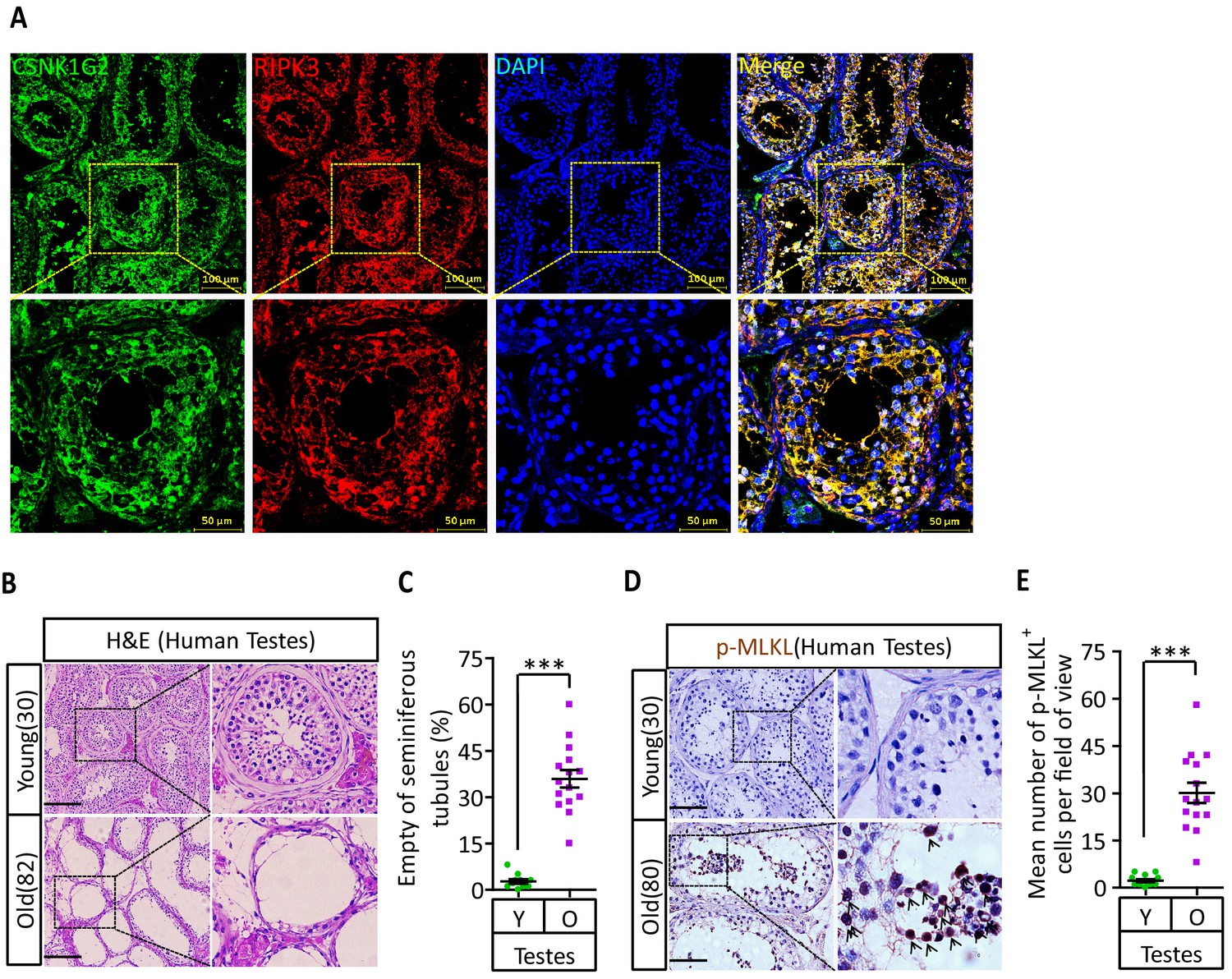
CSNK1G2 expression and necroptosis activation marker phosphor-Serine358-MLKL in human testes.
(A) Expression of CSNK1G2 and RIPK3 in human testes. Sections from a testis sample of a 30-year-old human patient were stained sequentially with antibodies against CSNK1G2 and RIPK3 as indicated followed by green or red fluorescent-conjugated secondary antibodies as indicated. Counterstaining with DAPI, blue. Scale bar, 50/100 μm. Yellow boxes in the upper panels were shown in the lower panels. The experiment was repeated three times with three different patients. (B and C) H&E staining of testes from young and old men. Young man testes (25–30 years, n = 10; from testicular torsion necrosis patients) and old man testes (80–89 years, n = 15; from prostate cancer patients) were sectioned and stained with H&E in (B). The number of empty seminiferous tubules was counted based on H&E staining and quantification in (C); empty seminiferous tubules were counted in five fields per testis. Scale bar, 100 μm. (D and E) IHC of testes from young and old men with phosphor-Serine358-MLKL antibody (p-MLKL). Young man testes (25–30 years, n = 10; from testicular torsion necrosis patients) and old man testes (80–89 years, n = 15; from prostate cancer patients) were sectioned and stained with an antibody against phosphor-Serine358-MLKL antibody (D). p-MLKL+ cells were counted in five fields per testis and quantification in (E). Scale bar, 100 μm. All quantified data in the figure represent the mean ± SEM. ***p<0.001. p-values were determined by two-sided unpaired Student’s t-tests.
Discussion
Necroptosis, although not crucial for normal mammalian development, is known to have critical functions in antimicrobial infection and tissue damage response due to the nature of the inflammation-eliciting necrotic cell death that releases damage pattern recognition (DAMP) signals (Christofferson and Yuan, 2010; Vandenabeele et al., 2010; Wallach et al., 2016). It is thus essential that such a danger signal has to be well controlled to prevent accidental death. So far, the most dominant negative regulator identified is active caspase-8, which cleaves and inactivates RIPK1 and RIPK3 kinases, thereby terminating necroptosis (Günther et al., 2011; Kaiser et al., 2011; Newton et al., 2019; Oberst et al., 2011). Inhibition or genetic inactivation of caspase-8 activates necroptosis both in vitro and in vivo. Additionally, Ppm1b phosphatase has been proposed to directly dephosphorylate threonine 231 and serine 232 sites of RIPK3 to attenuate the recruitment of MLKL, although the effect is modest at best (Chen et al., 2015). Moreover, even after MLKL translocated to the plasma membrane, the MLKL-containing membrane fraction lipid rafts can still be removed from the membrane by flotillin-mediated endocytosis or ESCRT-mediated exocytosis before membrane leakage occurs (Fan et al., 2019; Gong et al., 2017; Yoon et al., 2017). All these regulatory events happen after RIP3 kinase is already activated and thus can only serve as ‘emergency brakes’.
Our current report identified CSNK1G2 as an upstream negative regulator of pre-activated RIPK3. CSNK1G2 physically interacts with RIPK3 and prevents RIPK3 from responding to upstream activating signals, be that TNF-α, TRAIL, or LPS. CSNK1G2’s function appears analogous to cFlip protein for caspase-8 (Chang et al., 2002) or Bcl-2 for Bax/Bak, both of which bind and sequester their pro-death protein targets (Elmore, 2007). The binding of RIPK3 by CSNK1G2 requires its auto-phosphorylation on the serine 211 and threonine 215 sites, possibly a structural requirement to form the CSNK1G2/RIPK3 complex (Figure 6—figure supplement 1). This complex formation prevents RIPK3 from being recruited to the necrosome, thus attenuating necroptosis. Since RIPK3 homo-dimerization is a possible requirement for its activation marked by auto-phosphorylation at serine 227, CSNK1G2, by binding to the monomeric RIPK3, effectively suppresses RIPK3 activation. In addition to CSNK1G2, other members of the casein kinase-1 family, including CSNK1D1 and CSNK1E, seem to be able to bind and suppress RIPK3. These members may regulate necroptosis and other functions of RIPK3 in tissues other than testis, a hypothesis certainly merits future studies.
This CSNK1G2-mediated RIPK3 suppression is particularly important to attenuate necroptosis from occurring in mouse testis, a cell death program that actively promotes male reproductive system aging (Figure 6—figure supplement 1). It is thus not surprising that knockout CSNK1G2 resulted in drastically accelerated aging of the testis. CSNK1G2 is also expressed in human testis, and the necroptosis marker phospho-MLKL was present in the old but not young testis of men. Therefore, the CSNK1G2-accelerated and necroptosis-mediated testis aging phenomenon is evolutionarily conserved between mice and men.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (Homo sapiens) | HEK293T | ATCC | CRL-11268 | Female |
| Cell line (Homo sapiens) | HeLa | ATCC | CCL-2 | Female |
| Cell line (Mus musculus) | GC-2spd(ts) | ATCC | CRL-2196TM | Male |
| Cell line (Mus musculus) | 15 P-1 | ATCC | CRL-2618TM | Male |
| Cell line (Mus musculus) | MA-10 | ATCC | CRL-3050TM | Male |
| Cell line (Homo sapiens) | HeLa-HA-3×Flag-RIPK3 | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Homo sapiens) | HeLa-HA-3×Flag-RIPK3-Myc-CSNK1G2 | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Homo sapiens) | HeLa-HA-3×Flag-RIPK3-Myc-CSNK1G2(K75A) | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Homo sapiens) | HeLa-HA-3×Flag-RIPK3-Myc-CSNK1G2 (S211A/T215A) | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | NIH3T3-Flag-mRIPK3 | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | NIH3T3-Flag-mRIPK3-Myc-mCSNK1G2 | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | NIH3T3-Flag-mRIPK3-Myc-mCSNK1G2(K75A) | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | MEF(Csnk1g2−/−) | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | MEF(Csnk1g2−/−)-Myc-mCSNK1G2 | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | MEF(Csnk1g2−/−)-Myc-mCSNK1G2(K75A) | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Cell line (Mus musculus) | MEF(Csnk1g2−/−)-Myc-mCSNK1G2(S211A/T215A) | Dr. Xiaodong Wang lab at National Institute of Biological Sciences, Beijing | N/A | |
| Antibody | Anti-RIPK3 (rabbit polyclonal) | ProSci | Cat# 2283; RRID:AB_203256 | WB (1:1000) |
| Antibody | Anti-CSNK1G2 (rabbit monoclonal) | Abcam | Cat# ab238121 | First described in this paper;WB (1:1000) |
| Antibody | Anti-GAPDH-HRP (mouse monoclonal) | MBL | Cat# M171-1; RRID:AB_10699462 | WB (1:20,000) |
| Antibody | Anti-β-Actin-HRP (rabbit polyclonal) | MBL | Cat# PM053-7; RRID:AB_10697035 | WB (1:20,000) |
| Antibody | Anti-Flag-HRP (mouse monoclonal) | Sigma-Aldrich | Cat# A8592; RRID:AB_439702 | WB (1:10,000) |
| Antibody | Anti-RIPK (rabbit monoclonal) | Cell Signaling | Cat# 3493S; RRID:AB_2305314 | WB (1:1000) |
| Antibody | Anti-Myc-HRP (mouse monoclonal) | MBL | Cat# M192-7 | WB (1:2000) |
| Antibody | Anti-Mouse-MLKL (rabbit polyclonal) | ABGENT | Cat# AP14272b; RRID:AB_11134649 | WB (1:1000) |
| Antibody | Anti-Mouse-p-MLKL (rabbit monoclonal) | Abcam | Cat# ab196436; RRID:AB_2687465 | WB (1:1000) |
| Antibody | Anti-Human-p-MLKL (rabbit monoclonal) | Abcam | Cat# ab187091; RRID:AB_2619685 | WB (1:1000) |
| Antibody | Anti-p-S227-RIPK3 (rabbit monoclonal) | Abcam | Cat# ab209384; RRID:AB_2714035 | WB (1:1000) |
| Antibody | Anti-p-S232-RIPK3 (rabbit monoclonal) | Abcam | Cat# ab222302 | WB (1:1000) |
| Antibody | Mouse anti-rabbit IgG (mouse monoclonal) | Cell Signaling | Cat# 5127S; RRID:AB_10892860 | WB (1:2000) |
| Antibody | Donkey anti-mouse, Alexa Fluor 488 (mouse polyclonal) | Thermo Fisher | Cat# A-21202; RRID:AB_141607 | IF (1:500) |
| Antibody | Donkey anti-mouse, Alexa Fluor 555 (mouse polyclonal) | Thermo Fisher | Cat# A-31570; RRID:AB_2536180 | IF (1:500) |
| Antibody | Donkey anti-rabbit, Alexa Fluor 488 (rabbit polyclonal) | Thermo Fisher | Cat# A-21206; RRID:AB_141708 | IF (1:500) |
| Antibody | Donkey anti-rabbit, Alexa Fluor 555 (rabbit polyclonal) | Thermo Fisher | Cat# A-31572 RRID:AB_162543 | IF (1:500) |
| Antibody | Anti-Flag M2 affinity gel | Sigma-Aldrich | A2220 | |
| Antibody | Anti-c-Myc Agarose | Sigma-Aldrich | 20168 | |
| Recombinant DNA reagent (plasmid) | pWPI-Flag-RIPK3 | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pWPI-Flag-mRIPK3 | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pCDNA3.1-Myc-CSNK1G2 | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pCDNA3.1-Myc-CSNK1G2(K75A) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pCDNA3.1-Myc-CSNK1G2(D165N) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pCDNA3.1-Myc-CSNK1G2(S211A) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pCDNA3.1-Myc-CSNK1G2(T215A) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pCDNA3.1-Myc-CSNK1G2(S211A/T215A) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pWPI-Myc-CSNK1G2 | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pWPI-Myc-CSNK1G2(K75A) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pWPI-Myc-CSNK1G2(S211A/T215A) | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pWPI-Myc-mCSNK1G2 | This paper | N/A | Described in Materials and methods; available upon request |
| Recombinant DNA reagent (plasmid) | pWPI-Myc-mCSNK1G2(K75A) | This paper | N/A | Described in Materials and methods; available upon request |
| Peptide, recombinant protein | Myc peptide | Sigma-Aldrich | 20170 | 1 mg/ml |
| Peptide, recombinant protein | 3xFlag peptide | ChinaPeptides | DYKDHDGDYKDHDIDYKDDDDK | 1 mg/ml |
| Software, algorithm | ImageJ | NIH | N/A | |
| Software, algorithm | GraphPad | Graphpad Software | N/A | |
| Software, algorithm | Prism | Graphpad Software | N/A | |
| Software, algorithm | Nikon A1-R | Nikon | https://www.nikoninstruments.com/Products/Confocal-Microscopes/A1R-HD |
Mice
The Csnk1g2 knockout mice were generated using the CRISPR-Cas9 system (Figure 1—figure supplement 3). The Ripk3−/− (C57BL/6NCrl strain) were kept in our lab (He et al., 2009). Csnk1g2+/−Ripk3+/− mice were produced by mating Csnk1g2−/− males with Ripk3−/− females. Csnk1g2−/−Ripk3−/− mice were produced by mating Csnk1g2+/−Ripk3+/− males with Csnk1g2+/−Ripk3+/− females. The primers used for genotyping are listed below.
Csnk1g2-KO-F: AGGTTTCGCACTCGGATCTCACG;
Csnk1g2-KO-R: CCCCGAAGTTCCCACAGCCTATC;
Ripk3-KO-F: CAGTGGGACTTCGTGTCCG;
Ripk3-KO-R: CAAGCTGTGTAGGTAGCACATC.
Mice husbandry
Request a detailed protocolMice were group-housed in a 12 hr light/dark (light between 08:00 and 20:00) in a temperature-controlled room (21.1 ± 1°C) at the National Institute of Biological Sciences with free access to water. The ages of mice are indicated in the figure, figure legends, or methods. All animal experiments were conducted following the Ministry of Health national guidelines for the housing and care of laboratory animals and were performed in accordance with institutional regulations after review and approval by the Institutional Animal Care and Use Committee at the National Institute of Biological Sciences, Beijing.
Human tissues
Request a detailed protocolThe research involving human tissue samples dissected from adult human testicular torsion necrosis and prostate cancer patients (n = 10, 25–30 years, testicular torsion necrosis patients; n = 25, 80–89 years, prostate cancer patients) were kindly provided by Shanghai Renji Hospital, China, and were snap-frozen in liquid nitrogen and stored at −80°C. Tissues were cut into appropriately sized pieces and placed in formalin for preservation. After several days of formalin fixation at room temperature, tissue fragments were transferred to 70% ethanol and stored at 4°C.
The medical ethics committee of the National Institute of Biological Sciences, Beijing, China, approved the study.
Ethics
Animal experimentation: Animal care and use followed the institutional guidelines of the National Institute of Biological Sciences (NIBS), Beijing (Approval ID: NIBSLuoM15C) and the Regulations for the Administration of Affairs Concerning Experimental Animals of China.
Constructs psPAX2 and pMD2.G were kept in our lab. Full-length human RIPK3 and mouse Ripk3 cDNA were kept in our lab and subcloned into the pWPI (GFP-tagged) and pCDNA3.1 vector to generate pWPI-Flag-RIPK3, pWPI-Flag-mRIPK3, and pCDNA3.1-HA-3×Flag-RIPK3 constructs. Full-length cDNA for human CSNK1A1, CSNK1A1-L, CSNK1D1, CSNK1D2, CSNK1G1, CSNK1G2, CSNK1G3, CSNK1E, CSNK2A1, CSNK2A2, and CSNK2B were RCR-amplified from cDNA library (Sigma, USA) using KOD polymerase (TOYOBO, China) and subcloned into pcDNA3.1 vector. Quickchange Site-Directed Mutagenesis Kit was used to generate pWPI-Myc-CSNK1G2(K75A)/CSNK1G2(D165N)/CSNK1G2(S211A)/CSNK1G2(T215A)/CSNK1G2(S211A/T215A) and pWPI-Myc-mCSNK1G2/CSNK1G2(K75A)/CSNK1G2(S211A)/CSNK1G2 (T215A)/CSNK1G2 (S211A/T215A) constructs.
The gRNAs for targeting Csnk1g2 (Figure 1—figure supplement 3A) were designed and were cloned into the gRNA-Cas9 expression plasmid pX458-GFP to generate pX458-GFP-mCSNK1G2 construct.
Cells
All cells were cultured at 37°C with 5% CO2. All cell lines were confirmed to be mycoplasma free on a weekly basis using a PCR-based assay with primers: 5′-GGGAGCAAACAGGATTAGATACCCT-3′ and 5′- TGCACCATCTGTCACTCTGTTAACCTC-3′. All cell lines were cultured as follows: HT29 cells were obtained from ATCC and cultured in McCoy’s 5A culture medium(GIBCO, 16600082, USA). HEK293T (293T), HeLa, GC-2spd(ts), and 15 P-1 cells were obtained from ATCC and cultured in DMEM (GIBCO, C11965500BT, USA). NIH3T3-Flag-RIPK3 (NIH3T3 stably transfected with Flag-RIPK3 fused to FKBP-F36V), MEFs, MEF (Csnk1g2−/−), and HeLa-HA-3×Flag-RIPK3 (Sun et al., 2012) cells were cultured in DMEM (GIBCO, C11965500BT, USA). NIH3T3-Flag-RIPK3, HeLa-HA-3×Flag-RIPK3, and MEF (Csnk1g2−/−) cells were infected with virus encoding Myc-CSNK1G2 (WT, K75A, and S211A/T215A) or Myc-mCSNK1G2 (WT, K75A, S211A, T215A, and S211A/T215A), and GFP-positive live cells were sorted to establish the NIH3T3-Flag-RIPk3-Myc-CSNK1G2(WT, K75A), MEF (Csnk1g2−/−)-Myc-CSNK1G2(WT and K75A), and HeLa-HA-3×Flag-RIPK3-Myc-CSNK1G2 (WT and K75A) cell lines. All media were supplemented with 10% FBS (Thermo Fisher, USA) and 100 units/ml penicillin/streptomycin (Thermo Fisher, USA). MA-10 was obtained from ATCC and cultured in DMEM:F12 (GIBCO, USA, additional 20 mM HEPES, horse serum to a final concentration of 15%).
Isolation of cells from testes seminiferous tubules
Request a detailed protocolTestes from 8-week-old mice were collected using a previously reported protocol (Chang et al., 2011; Li et al., 2017). Briefly, a testis was placed in Enriched DMEM:F12 (GIBCO, USA) media and placed on ice. After removal of the tunica albuginea of a testis, the seminiferous tubules were dissociated and transferred immediately into 10 ml of protocol enzymatic solution 1. Tubules were incubated for 15–20 min at 35°C in a shaking water bath at 80 oscillations (osc)/min and were then layered over 40 ml of 5% Percoll/95% 1× Hank’s balanced salt solution in a 50 ml conical tube and allowed to settle for 20 min. Leydig cells were removed from the top 35 ml of the total volume of Percoll. The bottom 5 ml of Percoll was transferred to a fresh 50 ml conical tube containing 10 ml enzymatic solution 2. Tubules were incubated for 20 min at 35°C and 80 osc/min. After incubation, 3 ml of charcoal-stripped FBS was immediately added to halt the digestion. The fraction was immediately centrifuged at 500 × g at 4°C for 10 min. Pellets were resuspended in PBS and washed three times, and then cultured in DMEM:F12 (15% FBS) medium at 37°C.
Cell survival assay
Request a detailed protocolCell survival assay was performed using Cell Titer-Glo Luminescent Cell Viability Assay kit. A Cell Titer-Glo assay (Promega, G7570, USA) was performed according to the manufacturer’s instructions. Luminescence was recorded with a Tecan GENios Pro plate reader.
ELISA
Request a detailed protocolMice were killed and blood was clotted for 2 hr at room temperature before centrifugation at approximately 1000 × g for 20 min. Mice blood sera were collected and assayed immediately or was stored as sample aliquots at −20°C. The testosterone/FSH/LH levels were measured with ELISA kits (BIOMATIK, EKU07605, EKU04284, EKU05693, USA); the SHBG level was measured with an ELISA kit (INSTRUCTION MANUAL, SEA396Mu, USA). The ELISA assays were performed according to the manufacturer’s instructions.
CRISPR/Cas9 knockout cells
Request a detailed protocolFour micrograms of pX458-GFP-mCSNK1G2 plasmid was transfected into 1 × 107 MEF cells using the Transfection Reagent (FuGENEHD, E2311, USA) by following the manufacturer’s instructions. Three days after the transfection, GFP-positive live cells were sorted into single clones by using a BD FACSArial cell sorter. The single clones were cultured into 96-well plates for another 10–14 days or longer, depending upon the cell growth rate. The anti-CSNK1G2 immunoblotting was used to screen for the MEF (Csnk1g2−/−) clones. Genome type of the knockout cells was determined by DNA sequencing.
Western blotting
Request a detailed protocolCell pellet samples were collected and resuspended in lysis buffer (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, Roche complete protease inhibitor set, and Sigma phosphatase inhibitor set), incubated on ice for 30 min, and centrifuged at 20,000 × g for 30 min. The supernatants were collected for western blotting. Testis or other tissues were ground and resuspended in lysis buffer, homogenized for 30 s with a Paddle Blender (Prima, PB100, UK), incubated on ice for 30 min, and centrifuged at 20,000 × g for 30 min. The supernatants were collected for western blotting.
Immunoprecipitation
Request a detailed protocolThe cells were cultured on 15 cm dishes and grown to confluence. Cells at 70% confluence and subjected to indicated treatment for the appropriate time according to different experiments. Then cells were washed once with PBS and harvested by scraping and centrifugation at 800 × g for 5 min. The harvested cells were washed with PBS and lysed for 30 min on ice in the lysis buffer (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, Roche complete protease inhibitor set, and Sigma phosphatase inhibitor set). Cell lysates were then spun down at 12,000 × g for 20 min. The soluble fraction was collected, and the protein concentration was determined by Bradford assay. Cell extracted was mixed with anti-Flag/Myc affinity gel (Sigma-Aldrich, A2220, A7470, USA) in a ratio of 1 mg of extract per 30 μl of agarose. After overnight rocking at 4°C, the beads were pelleted at 2500 × g for 3 min and washed with lysis buffer three times. The beads were then eluted with 0.5 mg/ml of the corresponding antigenic peptide for 6 hr or directly boiled in 1× SDS loading buffer (125 mM Tris, pH 6.8, 2% 2-mercaptoethanol, 3% SDS, 10% glycerol, and 0.01% bromophenol blue).
Harvesting of tissues
Request a detailed protocolAnimals were killed and perfused with PBS, followed by 4% paraformaldehyde. Major organs were removed, cut into appropriately sized pieces, and either flash-frozen in liquid nitrogen and stored at −80°C or placed in 4% paraformaldehyde for preservation. After several days of 4% paraformaldehyde fixation at room temperature, tissue fragments were transferred to 70% ethanol and stored at 4°C. Blood was collected by cardiac puncture and was allowed to coagulate for the preparation of serum.
Immunohistochemistry and immunofluorescence
Request a detailed protocolParaffin-embedded specimens were sectioned to a 5 μm thickness and were then deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E) using standard protocols. For the preparation of the immunohistochemistry samples, sections were dewaxed, incubated in boiling citrate buffer solution for 15 min in plastic dishes, and subsequently allowed to cool down to room temperature over 3 hr. Endogenous peroxidase activity was blocked by immersing the slides in hydrogen peroxide buffer (10%, Sinopharm Chemical Reagent, China) for 15 min at room temperature and were then washed with PBS. Blocking buffer (1% bovine serum albumin in PBS, China) was added, and the slides were incubated for 2 hr at room temperature. Primary antibody against p-mMLKL or p-MLKL was incubated overnight at 4°C in PBS. After three washes with PBS, slides were incubated with secondary antibody (polymer-horseradish-peroxidase-labeled anti-rabbit, Sigma, USA) in PBS. After a further three washes, slides were analyzed using a diaminobutyric acid substrate kit (Thermo Fisher, USA). Slides were counterstained with hematoxylin and mounted in neutral balsam medium (Sinopharm Chemical, China).
Immunohistochemistry analysis for RIPK3 or CSNK1G2 was performed using an antibody against RIPK3 and CSNK1G2. Primary antibody against RIPK3 was incubated overnight at 4°C in PBS. After three washes with PBS, slides were incubated with DyLight-555 conjugated donkey anti-rabbi/mouse secondary antibodies (Life, USA) in PBS for 8 hr at 4°C. After a further three washes, slides were incubated with CSNK1G2 antibody overnight at 4°C in PBS. After a further three washes, slides were incubated with DyLight-488 conjugated donkey anti-mouse/rabbit secondary antibodies (Life, USA) for 2 hr at room temperature in PBS. After a further three washes in PBS, the cell nuclei were then counterstained with DAPI (Invitrogen, USA) in PBS. Fluorescence microscopy was performed using a Nikon A1-R confocal microscope.
Mating and fertility tests
Request a detailed protocolCsnk1g2+/+, Csnk1g2−/−, Csnk1g2−/−+RIPA-56, and Csnk1g2−/−Ripk3−/− male littermate mice were housed in an SPF barrier facility. To score vaginal patency, mice were examined daily from weaning until vaginal opening was observed. The fertility rate of males was determined via a standard method (Cooke and Saunders, 2002; Hofmann et al., 2015; Li et al., 2017) by mating a male with a series of pairs of 10-week-old wild-type females for 2 months; females were replaced every 2 weeks (females were either from our colony or purchased from Vital River Laboratory Co(C57BL/6NCrl)). Each litter was assessed from the date of the birth of pups; when pups were born but did not survive, we counted and recorded the number dead pups; for females that did not produce offspring, the number of pups was recorded as ‘0’ (did not produce a litter with a proven breeder male for a period of 2 months). The number of male mice with reproductive capacity was recorded.
RIPA-56 feeding experiment
Request a detailed protocolRIPA-56 in the AIN93G (LAD3001G, China) at 300 mg/kg was produced based on the Trophic Animal Feed High-tech Co. protocol (Li et al., 2017). Cohorts of 2-month-old Csnk1g2−/− male mice were fed with AIN93G or AING3G containing RIPA-56 (RIPA-56: 300 mg/kg) for 10 months in an SPF facility; each male mouse was then mated with four 10-week-old wild-type female mice successively. The number of male mice with reproductive capacity was recorded.
Mass spectrometry and data analysis
Request a detailed protocolA total of 293 T cells were transfected with Myc-tagged CSNK1G2 (WT and K75) for 24 hr. Then the cell extracts were prepared and used for immunoprecipitation with an anti-Myc antibody. The immunoprecipitates were washed three times with lysis buffer. The beads were then eluted with 0.5 mg/ml of the corresponding antigenic peptide for 6 hr or directly boiled in 1 × SDS loading buffer and subjected to SDS-PAGE. CSNK1G2 bands were excised from SDS-PAGE gel and then dissolved in 2M urea, 50 mM ammonium bicarbonate, pH 8.0, and reduced in 2 mM DTT at 56°C for 30 min followed by alkylation in 10 mM iodoacetamide at dark for 1 hr. Then the protein was digested with sequencing grade modified trypsin (Promega, USA) (1:40 enzyme to total protein) at 37°C overnight. The tryptic peptides were separated by an analytical capillary column (50 μm × 15 cm) packed with 5 μm spherical C18 reversed phase material (YMC, Kyoyo, Japan). A Waters nanoAcquity UPLC system (Waters, Milford, USA) was used to generate the following HPLC gradient: 0–30% B in 40 min and 30–70% B in 15 min (A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile). The eluted peptides were sprayed into a LTQ Orbitrap Velos mass spectrometer (ThermoFisher Scientific, San Jose, CA, USA) equipped with a nano-ESI ion source. The mass spectrometer was operated in data-dependent mode with one MS scan followed by four CID (collision-induced dissociation) and four HCD (high-energy collisional dissociation) MS/MS scans for each cycle. Database searches were performed on an in-house Mascot server (Matrix Science Ltd, London, UK) against human CSNK1G2 protein sequence. The search parameters are: seven ppm mass tolerance for precursor ions; 0.5 Da mass tolerance for product ions; three missed cleavage sites were allowed for trypsin digestion, and the following variable modifications were included: oxidation on methionine, cysteine carbamidomethylation, and serine, threonine, and tyrosine phosphorylation.
Statistical analysis
Request a detailed protocolAll results are representative of three independent experiments. Statistical tests were used for every type of analysis. The data meet the assumptions of the statistical tests described for each figure. Results are expressed as the mean ± SEM or SD. Differences between experimental groups were assessed for significance using a two-tailed unpaired Student’s t-test using GraphPad prism6 and Microsoft Excel 2017. Fertility rate was assessed for significance using Fisher’s exact test (unpaired, two-tailed) using GraphPad prism6 software. The *p < 0.05, **p < 0.01, and ***p < 0.001 levels were considered significant. NS, not significant.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Ppm1b negatively regulates necroptosis through dephosphorylating Rip3Nature Cell Biology 17:434–444.https://doi.org/10.1038/ncb3120
-
Necroptosis as an alternative form of programmed cell deathCurrent Opinion in Cell Biology 22:263–268.https://doi.org/10.1016/j.ceb.2009.12.003
-
Identification of RIP1 kinase as a specific cellular target of necrostatinsNature Chemical Biology 4:313–321.https://doi.org/10.1038/nchembio.83
-
Apoptosis: a review of programmed cell deathToxicologic Pathology 35:495–516.https://doi.org/10.1080/01926230701320337
-
Casein kinase 1 Delta regulates the pace of the mammalian circadian clockMolecular and Cellular Biology 29:3853–3866.https://doi.org/10.1128/MCB.00338-09
-
Casein kinase 1-epsilon deletion increases mu opioid receptor-dependent behaviors and binge eating1Genes, Brain and Behavior 16:725–738.https://doi.org/10.1111/gbb.12397
-
RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosisCell Death & Differentiation 21:1511–1521.https://doi.org/10.1038/cdd.2014.76
-
HIF-2α phosphorylation by CK1δ promotes erythropoietin secretion in liver Cancer cells under hypoxiaJournal of Cell Science 129:4213–4226.https://doi.org/10.1242/jcs.191395
-
Molecular mechanisms of necroptosis: an ordered cellular explosionNature Reviews Molecular Cell Biology 11:700–714.https://doi.org/10.1038/nrm2970
Article and author information
Author details
Funding
Ministry of Science and Technology of the People's Republic of China (Institutional)
- Xiaodong Wang
Beijing Municipal Commission of Education (Institutional)
- Xiaodong Wang
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Mr. Alex Wang for critically reading and editing the manuscript. This work was supported by institutional grants from the Chinese Ministry of Science and Technology and Beijing Municipal Commission of Science and Technology. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Ethics
Human subjects: This work used human surgical samples for immunohistochemical analysis. The protocool was approved by the institutional review board for usage of human samples.
Animal experimentation: This study was peformed in strict accordance with NIBS insitutional guidance of animal use and the protocool was approved by the institutional animal care and use committee. ID: NIBSLuoM15C.
Copyright
© 2020, Li et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,903
- views
-
- 415
- downloads
-
- 27
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 27
- citations for umbrella DOI https://doi.org/10.7554/eLife.61564
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Casein kinase 1G2 suppresses necroptosis-promoted testis aging by inhibiting receptor-interacting kinase 3
eLife 9:e61564.
https://doi.org/10.7554/eLife.61564




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}