Combinations of maternal-specific repressive epigenetic marks in the endosperm control seed dormancy
- Department of Plant Biology, Uppsala BioCenter, Swedish University of Agricultural Sciences and Linnean Centre for Plant Biology, Sweden
- Max Planck Institute of Molecular Plant Physiology, Germany
Abstract
Polycomb Repressive Complex 2 (PRC2)-mediated trimethylation of histone H3 on lysine 27 (H3K27me3) and methylation of histone 3 on lysine 9 (H3K9me) are two repressive epigenetic modifications that are typically localized in distinct regions of the genome. For reasons unknown, however, they co-occur in some organisms and special tissue types. In this study, we show that maternal alleles marked by H3K27me3 in the Arabidopsis endosperm were targeted by the H3K27me3 demethylase REF6 and became activated during germination. In contrast, maternal alleles marked by H3K27me3, H3K9me2, and CHG methylation (CHGm) are likely to be protected from REF6 targeting and remained silenced. Our study unveils that combinations of different repressive epigenetic modifications time a key adaptive trait by modulating access of REF6.
Introduction
Animals, fungi, and plants make use of two major epigenetic repressive modifications on histones that are destined to silence genes and transposable elements (TEs). The Polycomb repressive system applies the repressive trimethylation on lysine 27 of histone H3 (H3K27me3), which mainly serves to silence genes during defined stages of development (Mozgova and Hennig, 2015; Schuettengruber et al., 2017). In contrast, the repressive di- or trimethylation on lysine 9 of histone H3 (H3K9me2/3) is mainly associated with constitutively silenced heterochromatin, present in repeat-rich regions and TEs (Saksouk et al., 2015; Wendte and Schmitz, 2018). Repression mediated by the Polycomb system is transient and dynamically regulated, while repression mediated by H3K9me2/3 is generally stable during development (Mozgova and Hennig, 2015; Saksouk et al., 2015; Schuettengruber et al., 2017; Wendte and Schmitz, 2018). The establishment of H3K27me3 is mediated by the evolutionary conserved Polycomb repressive complex 2 (PRC2). In plants, there are distinct PRC2 complexes that are active during different stages of plant development (Mozgova and Hennig, 2015). In Arabidopsis, the EMBRYONIC FLOWER (EMF) and VERNALIZATION (VRN) PRC2 regulate sporophyte development, while the FERTILIZATION-INDEPENDENT SEED (FIS) PRC2 is active in the central cell of the female gametophyte and the descendent endosperm (Mozgova and Hennig, 2015). The establishment of H3K9me2 in Arabidopsis depends on the SU(VAR)3–9 homologous proteins (SUVHs), KRYPTONITE (or SUVH4), SUVH5 and SUVH6 that act in a feedback loop with CHG methylation (CHGm) established by the CHROMOMETHYLTRANSFERASE 3 (CMT3) (Kenchanmane Raju et al., 2019; Zhang et al., 2018). In animals and plants, H3K27me3 and H3K9me2 are generally exclusive repressive marks (Wiles and Selker, 2017); however, they co-occur in certain tissue types like the plant endosperm (Klosinska et al., 2016; Moreno-Romero et al., 2016; Weinhofer et al., 2010), as well as in specific organisms as filamentous fungi and bryophytes (Carlier et al., 2020; Montgomery et al., 2020). Furthermore, redistribution of H3K27me3 to heterochromatic regions occurs when constitutive heterochromatin is disrupted, indicating that specific features of heterochromatin prevent PRC2 recruitment (Deleris et al., 2012; Jamieson et al., 2016; Peters et al., 2003; Wiles and Selker, 2017).
Erasure of H3K27me3 and H3K9me2 is mediated by different types of JUMONJI lysine demethylases (KDMs) that belong to the KDM4 and KDM3 type, respectively (Cheng et al., 2020). RELATIVE OF EARLY FLOWERING 6 (REF6) and EARLY FLOWERING 6 (ELF6) function redundantly as major erasers of H3K27me3 during Arabidopsis development (Lu et al., 2011; Yan et al., 2018). The zinc-finger (ZnF) domains of REF6 recognize the sequence motif CTCTGYTY (Y = T or C) (Cui et al., 2016; Li et al., 2016). Binding of REF6 to this motif is negatively affected by non-CG methylation (Qiu et al., 2019), revealing cross-talk between different epigenetic modifications.
Seed development in flowering plants is initiated by a double fertilization event, where one of the sperm cells fertilizes the haploid egg, giving rise to the diploid embryo and the other sperm cell fertilizes the diploid central cell, initiating development of the triploid endosperm (Zhou and Dresselhaus, 2019). The endosperm regulates the transfer of nutrients to the developing embryo, similar as the placenta in mammals. (Costa et al., 2012; Lee et al., 2010; Linkies et al., 2009). The endosperm furthermore regulates seed dormancy, a process preventing germination of seeds under favourable conditions (Nonogaki, 2019). Both processes have been linked to genomic imprinting, an epigenetic phenomenon causing parent-of-origin specific gene expression (Batista and Kohler, 2020; Gehring and Satyaki, 2017; Piskurewicz et al., 2016). Imprinted genes are epigenetically modified during gametogenesis, establishing parental-specific expression patterns that are maintained after fertilization. Genomic imprinting leading to maternally biased expression is predominantly connected to DNA demethylation of maternal alleles mediated by the DNA glycosylase DEMETER (Gehring et al., 2009; Hsieh et al., 2011; Park et al., 2016). In contrast, paternally biased expression is predominantly connected to the presence of H3K27me3, H3K9me2, and CHGm on maternal alleles (Klosinska et al., 2016; Moreno-Romero et al., 2019; Moreno-Romero et al., 2016; Moreno-Romero et al., 2017). The FIS-PRC2 establishes H3K27me3 in the central cell, which is maintained on the maternal alleles after fertilization. Loss of FIS-PRC2 function impairs CHGm establishment, indicating that H3K27me3 is the primary imprinting mark and CHGm and H3K9me2 are recruited after H3K27me3 establishment (Moreno-Romero et al., 2019). The biological meaning of the co-occurance of H3K27me3, H3K9me2, and CHGm on the maternal alleles of paternally expressed genes (PEGs) is unclear. In this study, we reveal that genes marked by H3K27me3 on their maternal alleles are targeted by REF6 and become reactivated during germination. In contrast, REF6 fails to activate genes marked by H3K27me3, H3K9me2, and CHGm, consistent with the finding that DNA methylation prevents REF6 binding (Qiu et al., 2019). Among those genes with triple repressive marks was the ABA INSENSITIVE 3 (ABI3) gene, a known positive regulator of seed dormancy (Liu et al., 2013; Shu et al., 2016; Tian et al., 2020). Genes activated by REF6 are enriched for ethylene signaling, a process activated during germination. Consistently, loss of REF6 causes increased dormancy, as recently reported (Chen et al., 2020). Together, our study unveils a mechanism that employs the combination of different repressive epigenetic modifications to time a crucial developmental process.
Results
Genes marked by single H3K27me3 are expressed during germination
To understand the functional relevance of different combinations of repressive epigenetic modifications in the endosperm, we analyzed gene ontologies (GOs) of genes marked with single H3K27me3, single H3K9me2, and both modifications (referred to as H3K27me3/H3K9me2) using AgriGO v2.0 (http://systemsbiology.cau.edu.cn/agriGOv2/; Tian et al., 2017; Figure 1—figure supplement 1). Genes marked by single H3K9m2 were not enriched for any GO category and genes marked by H3K27me3/H3K9me2 only for one; in contrast, genes marked with single H3K27me3 were significantly enriched for many GOs (Figure 1—figure supplement 1). Interestingly, among those GOs was an enrichment of genes related to ethylene signaling (Figure 1—figure supplement 1; Dubois et al., 2018), a pathway which is activated in the endosperm tissue during germination to allow root protrusion through cell loosening (Linkies and Leubner-Metzger, 2012; Linkies et al., 2009) and known to release seed dormancy (Beaudoin et al., 2000; Corbineau et al., 2014). Supporting the GO enrichment, we found that genes with single H3K27me3 significantly overlapped with ethylene-inducible genes identified in a previous study (Das et al., 2016), while genes with single H3K9me2 and H3K27me3/H3K9me2 did not (Figure 1a). We analyzed the expression of ethylene-inducible genes marked by single H3K27me3 during germination in the endosperm using previously published data (Dekkers et al., 2013). We found that ethylene-inducible genes with single H3K27me3 were induced during germination, especially around the time point of endosperm rupture (p<0.05, Wilcoxon test) (Figure 1b), while there were no significant expression differences between different time points for genes with single H3K9me2 and genes with both H3K27me3/H3K9me2. Increased gene expression in the endosperm during germination was not restricted to ethylene inducible genes marked by H3K27me3, but also detectable for all H3K27me3 marked genes (Figure 1—figure supplement 2). This data reveal that genes with single H3K27me3 are initially suppressed during early endosperm development and become reactivated during germination, while genes with single H3K9me2 and genes with H3K27me3/H3K9me2 remain continuously suppressed throughout endosperm development and germination. This implies the presence of a molecular mechanism that allows to differentially regulate the expression of the three groups of epigenetically marked genes during germination.
Figure 1 with 2 supplements see all
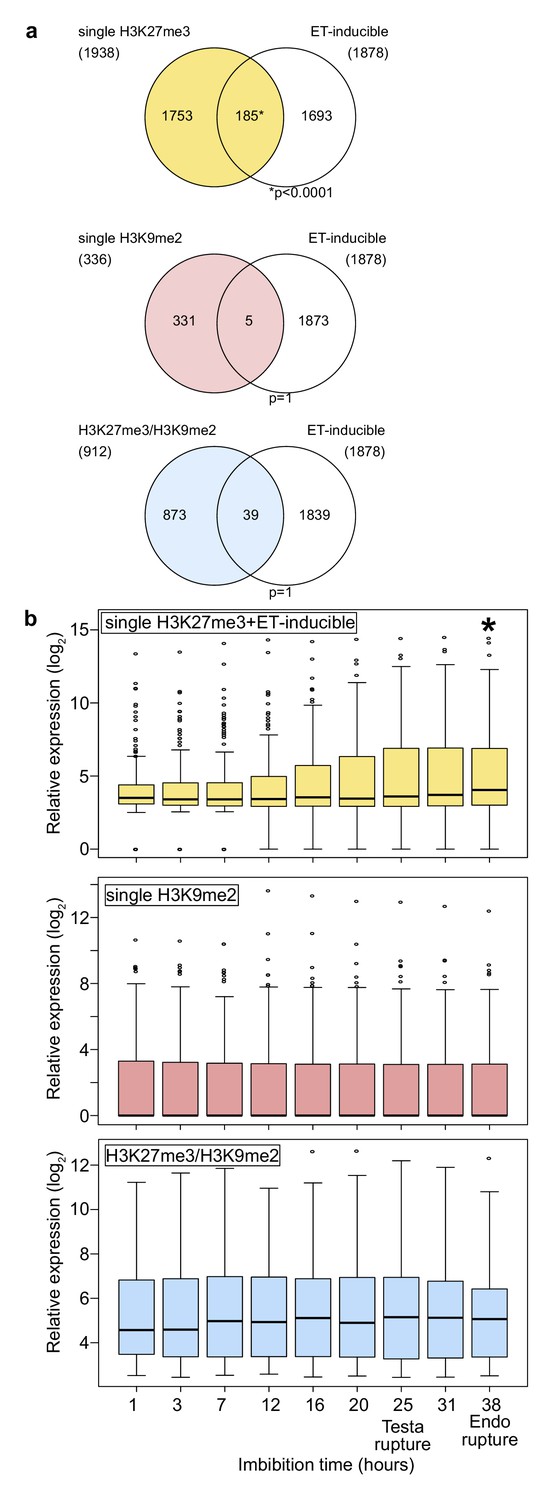
Genes marked by H3K27me3 become activated in the endosperm during germination.
(a) Venn diagrams show overlaps between three groups of genes with different patterns of histone modifications and ethylene (ET)-inducible genes in seedlings. ET-inducible genes identified in a previous study (Das et al., 2016) were compared with genes marked by the indicated histone modifications in the developing endosperm (Moreno-Romero et al., 2016). Asterisks indicate significant overlap (p<0.0001, Fisher’s exact test). (b) Box plots show relative expression profiles of genes marked by the indicated histone modifications in the endosperm during germination. Time-course expression profiles of ethylene inducible genes with single H3K27me3 (top; 185 genes), single H3K9me2 (middle; 336 genes), and H3K27me3, H3K9me2 and CHG methylation (bottom; 227 genes). Expression data are endosperm-specific transcriptome data during germination (Dekkers et al., 2013). The time points of testa rupture and endosperm (Endo) rupture are shown. Asterisks indicate significant differences compared to the time point at 1 hr of imbibition (p<0.01, Wilcoxon test).
Genes with double H3K27me3/H3K9me2 modifications are suppressed through CHG methylation on REF6-binding sites during germination
Recent work revealed that the presence of CHG methylation (referred to as CHGm) prevents binding of the H3K27me3 histone demethylase RELATIVE OF EARLY FLOWERING 6 (REF6/JMJ12) to its target motif CTCTGYTY (Y = T or C) (Qiu et al., 2019). We recently showed that genes with dual H3K27me3/H3K9me2 modifications are marked by CHGm in the endosperm and that CHGm is established in the central cell (Moreno-Romero et al., 2019). We thus hypothesized that the presence of CHGm may prevent REF6 from targeting and activating those genes. To test this hypothesis, we identified genes with REF6 binding sites in their gene bodies and associated the presence of the REF6 motif, CHGm status, and expression during germination. We found that REF6 binding sites within genes that were marked either by single H3K9me2 or by H3K27me3/H3K9me2 were more frequently CHG methylated in the central cell (Figure 2a) and endosperm (Figure 2—figure supplement 1) compared to single H3K27me3 marked genes (Moreno-Romero et al., 2016; Park et al., 2016). Importantly, genes marked by H3K27me3/H3K9me2 were frequently marked by CHGm at position five within the REF6-binding site (Figure 2b). Methylation at position five was found to be sufficient to abolish the affinity of REF6 for this target site, while methylation at position three was less effective (Qiu et al., 2019). Consistent with the hypothesis that CHGm on REF6 binding sites prevents REF6 targeting and thus gene activation, we found that genes with non-methylated REF6 binding sites were significantly more frequently activated during germination than genes with methylated REF6 binding sites (Figure 2c). Together, this data supports the idea that the methylation status of REF6-binding sites affects the reactivation of genes in the endosperm during germination. While H3K27me3 marked genes can be activated during germination, genes with H3K27me3, H3K9me2, and CHGm fail to be activated since REF6 (or homologous proteins) recruitment is blocked by DNA methylation.
Figure 2 with 1 supplement see all
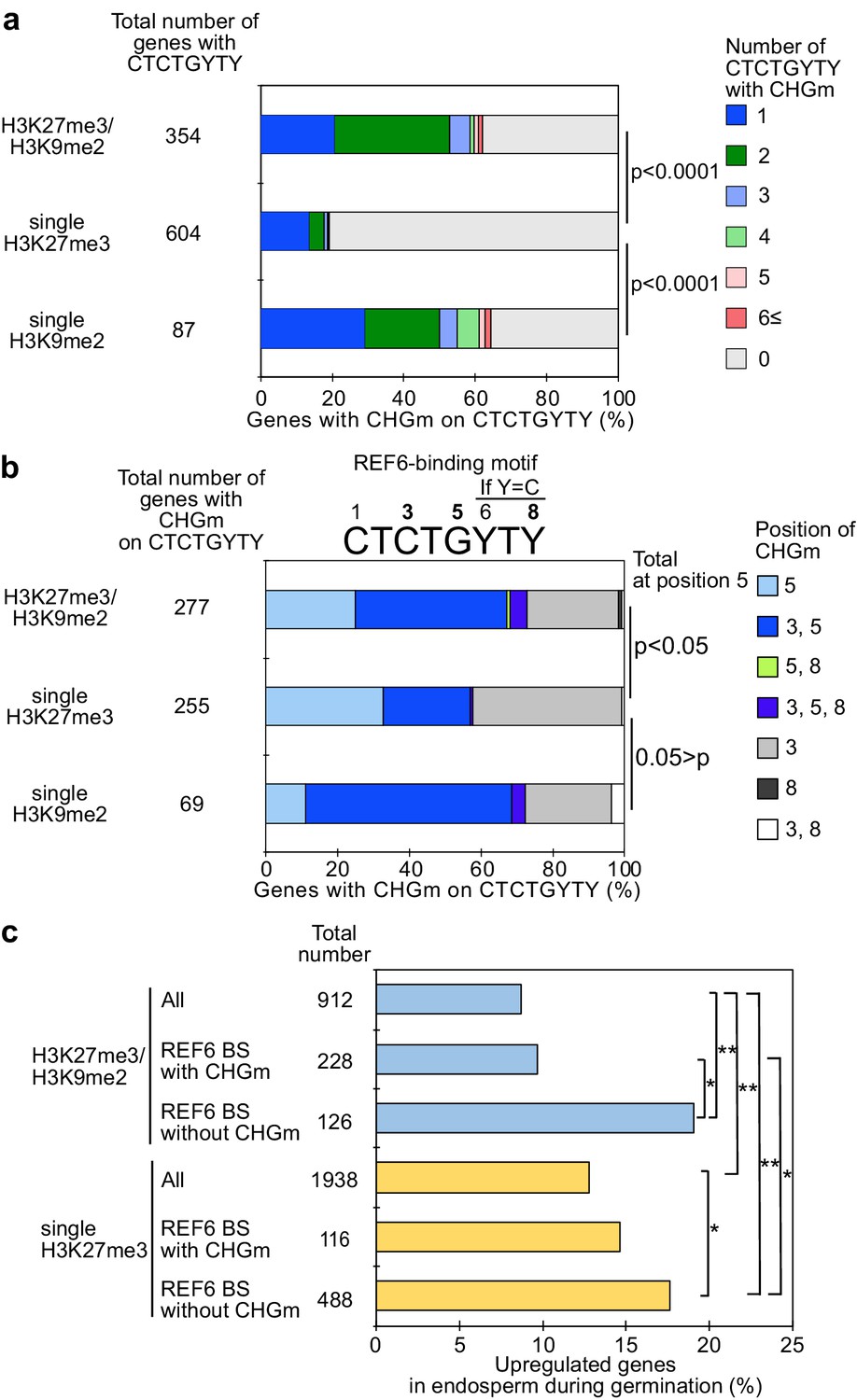
Gene activation during germination depends on CHG methylation (CHGm) on REF6 binding sites (REF6-BS).
(a) Plot shows the percentage of REF6-BS with CHGm in the three groups of genes with different combinations of histone modifications. CHGm on REF6-BS in gene bodies was determined based on published DNA methylome data (Park et al., 2016). p values were calculated using pairwise Fisher’s exact test and Benjamini–Hochberg correction. (b) Plot shows the percentage of CHGm on different cytosine positions of REF6-BS in the three groups of genes with different combinations of histone modifications. CHGm on REF6-BS in gene bodies was determined based on published DNA methylome data (Park et al., 2016). Numbers above the REF6-BS indicate possible positions of DNA methylation, of which positions 3, 5, and 8 can be targeted by CHGm. p values were calculated using pairwise Fisher’s exact test and Benjamini–Hochberg correction and indicate significance for CHGm on position five in REF6-BS. (c) Plot shows the percentage of upregulated genes with double H3K27m3/H3K9me2 and single H3K27me3 in the endosperm during germination depending on CHGm on REF6-BS. Gene expression is based on previously published endosperm-specific transcriptome data during germination (Dekkers et al., 2013). Compared were expression levels between 1 and 38 hours of imbibition (1≦Log2FC, p<0.05). Asterisks indicate significant differences between two categories (*p<0.05; **p<0.01; pairwise Fisher’s exact test with Benjamini–Hochberg correction).
REF6 is required to promote germination
Loss of REF6 was recently reported to cause increased dormancy (Chen et al., 2020). There are two REF6 orthologs in Arabidopsis thaliana, EARLY FLOWERING 6 (ELF6), and JUMONJI 13 (JMJ13). The DNA-binding domains of REF6 and ELF6 share amino acids that are required for binding to REF6-sequence motifs (Qiu et al., 2019) contrasting with lack of conservation of those sites in JMJ13 (Figure 3—figure supplement 1). This suggests that ELF6 acts redundantly with REF6 to promote germination. We tested two independent mutant alleles of ref6 and elf6 that all showed late germination phenotypes, but the ref6 mutants were more strongly delayed than elf6 (Figure 3—figure supplement 2). To test whether there are genetic redundancies between REF6, ELF6, and JMJ13 in controlling germination, we analyzed double elf6 ref6 and triple elf6 ref6 jmj13 mutants (Yan et al., 2018) for their ability to germinate. The elf6 ref6 double was slightly, but significantly, more delayed compared to the ref6 single mutant, while the elf6 ref6 jmj13 triple mutant behaved like the efl6 ref6 double mutant (Figure 3a). This data reveal that REF6 is the major factor releasing seed dormancy and promoting germination and that ELF6 is partially redundant with REF6, consistent with the conserved DNA binding domain (Figure 3—figure supplement 1). Prolonged after-ripening or stratification of ref6 seeds decreased primary dormancy and promoted germination (Figure 3—figure supplement 3), similar to other dormancy-related mutants (MacGregor et al., 2019; Piskurewicz et al., 2016; Zheng et al., 2012). Loss of maternal, but not paternal REF6 caused a significant delay in germination (Figure 3b), revealing that REF6 acts in maternal tissues. Nevertheless, combined loss of maternal and paternal REF6 had a stronger effect than loss of the maternal REF6 (Figure 3b), indicating that REF6 also acts after fertilization. To test whether the maternal effect of ref6 is caused by a functional role of REF6 in maternal sporophytic tissues or in the female gametophyte, we analyzed the phenotypes of F1 seeds generated by crossing maternal heterozygous ref6-1 mutant plants with paternal wild-type plants. The F1 seeds showed significantly delayed germination compared to wild-type seeds, with germination frequencies being in between that of wild-type seeds and F1 seeds of homozygous ref6 mutants crossed with wild-type pollen (Figure 3c). Since REF6 is not imprinted (Hsieh et al., 2011; Pignatta et al., 2014), this data strongly suggests that REF6 acts before fertilization in the female gametophyte.
Figure 3 with 3 supplements see all
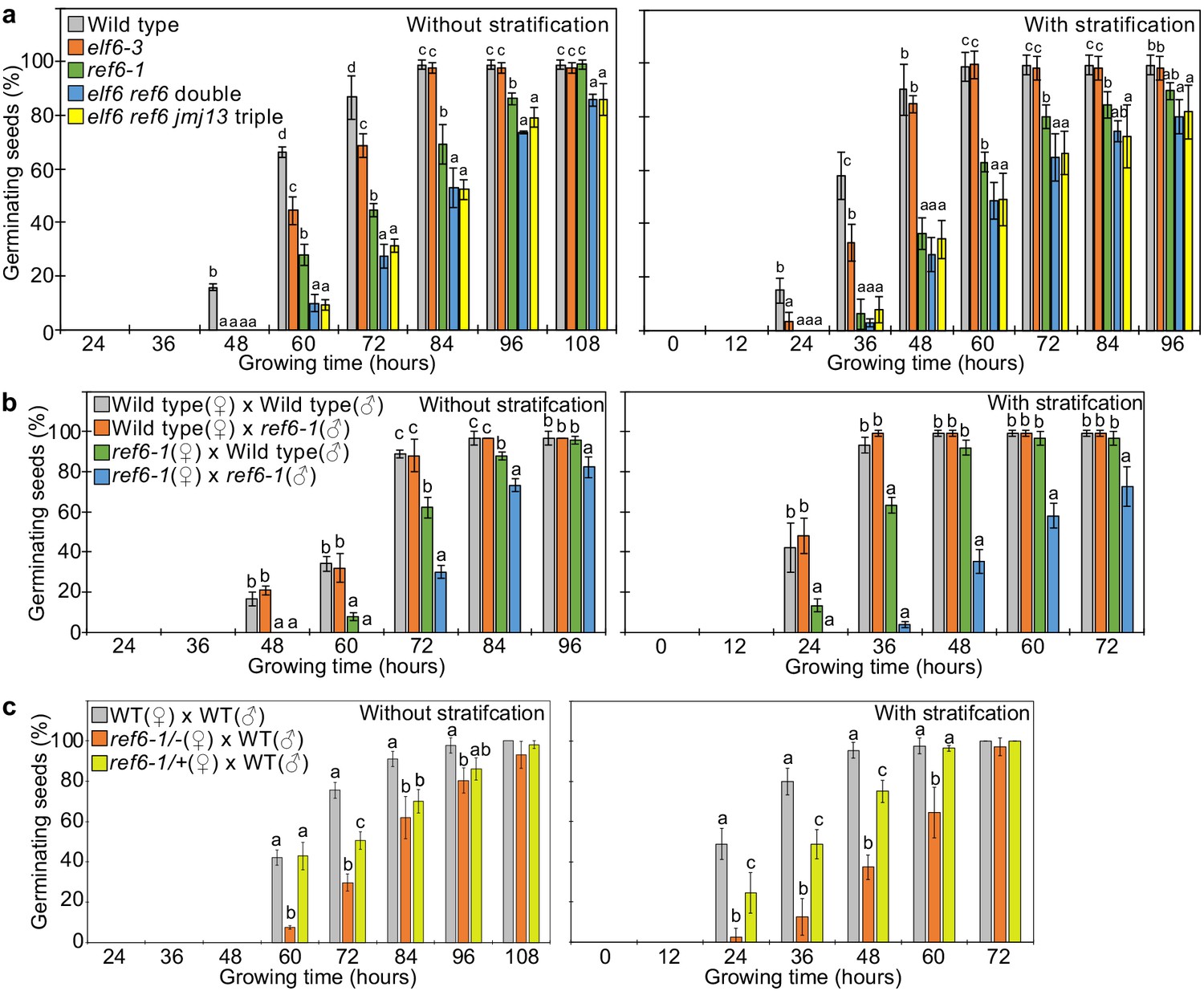
REF6 is the major H3K27me3 demethylase controlling seed dormancy.
(a) Plots show the percentage of germinating seeds of wild type, elf6-3 single, ref6-1 single, elf6-3 ref6C double, and elf6-3 ref6C jmj13G triple mutants with and without stratification. Error bars indicate SD from three biological replicates (n = 120 total). Letters above bars indicate significant differences among genotypes (p<0.05, Tukey’s multiple range test). (b) Germination phenotypes of seeds generated by reciprocal crosses between wild type and ref6-1. Plots show the percentage of germinating seeds with and without stratification. Error bars indicate SD from three biological replicates (n = 120 total). Letters above bars indicate significant differences among genotypes (p<0.05, Tukey’s multiple range test). (c) Plots show the percentage of germinating seeds with and without stratification of wild type (WT), F1 seeds generated by crossing maternal heterozygous or homozygous ref6-1 and paternal wild type. Error bars indicate SD from three biological replicates (n = 90 total). Letters above bars indicate significant differences among genotypes (p<0.05, Tukey’s multiple range test).
Endosperm-specific expression of REF6 is important for germination
Our data suggests a major role of REF6 in the endosperm to promote the expression of genes required for releasing dormancy. To further challenge this model, we tested whether expression of REF6 in the micropylar endosperm was sufficient to suppress the germination delay of ref6 mutants. We expressed REF6 in the ref6 mutant under control of the EARLY-PHYTOCHROME-RESPONSIVE1 (EPR1) promoter that is specifically expressed in the micropylar endosperm during germination (Dubreucq et al., 2000; Figure 4—figure supplement 1). Two independent transgenic EPR1::REF6; ref6 lines showed a significant suppression of the ref6 germination delay (Figure 4a), indicating that expression of REF6 in the micropylar endosperm is functionally required to release dormancy and promote germination. We also expressed REF6 under control of the TREHALOSE-6-PHOSPHATE SYNTHASE 1 (TPS1) promoter that is specifically active in the embryo during germination (Bae et al., 2009; Figure 4—figure supplement 1). However, TPS1::REF6 did not suppress the germination delay of ref6 (Figure 4b), indicating that REF6 has no functional role in the embryo during germination. In support of this notion, dissected ref6 embryos did not show any delay in growth compared to dissected wild-type embryos (Figure 4c,d). Together, this data supports our model that REF6 is required in the endosperm to promote the expression of genes required for releasing dormancy.
Figure 4 with 1 supplement see all
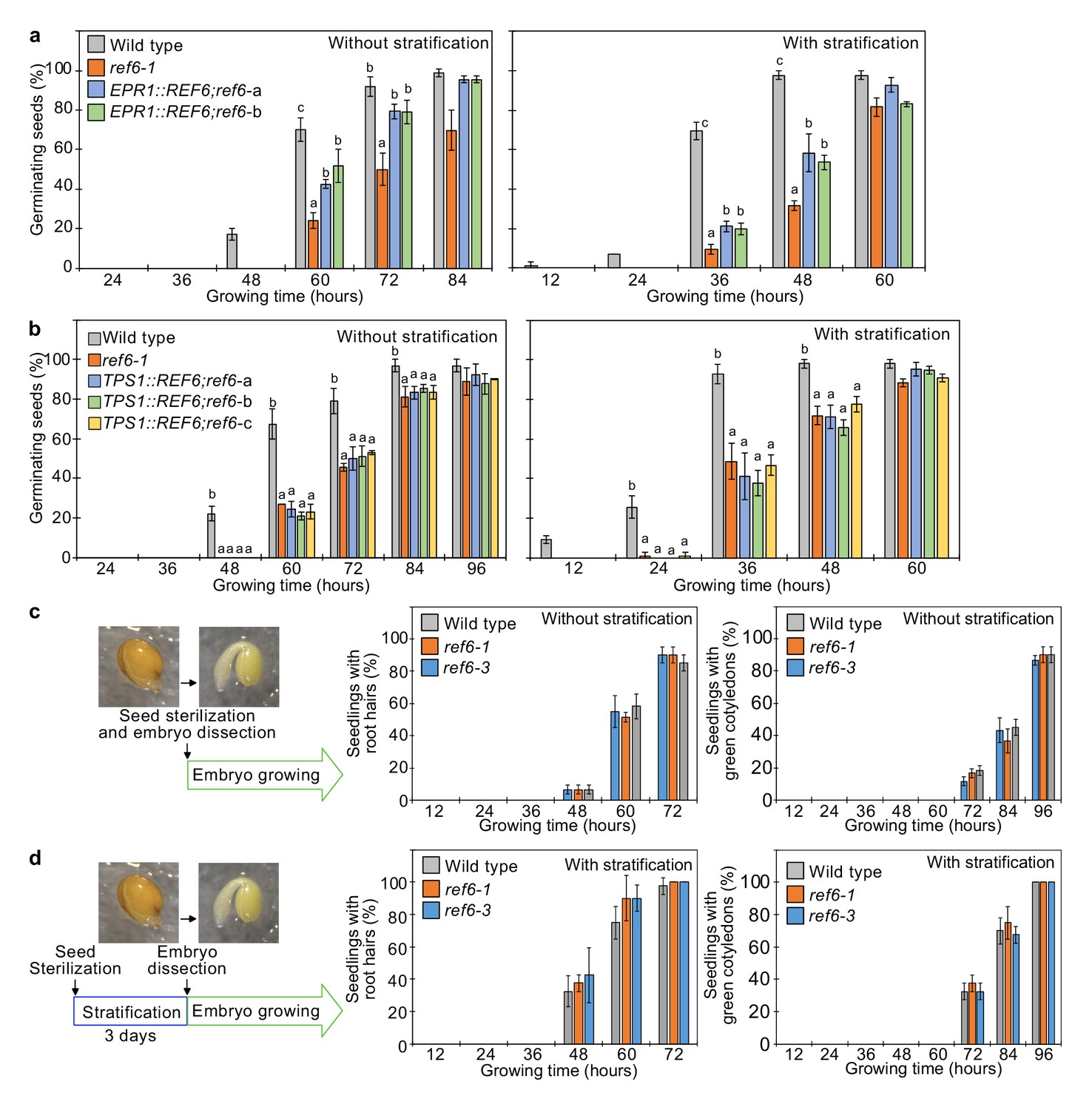
REF6 expression in the endosperm promotes germination.
(a) Germination phenotypes of the ref6-1 mutant and transgenic lines in the ref6-1 background expressing REF6 under control of the micropylar endosperm-specific EPR1 promoter. Plots show the percentage of germinating seeds of wild-type, ref6-1, and complemented lines (EPR1::REF6;ref6-1-a and b) with and without stratification. Error bars indicate SD from three biological replicates (n = 120 total). The letters above the bars indicate significant differences among plant lines (p<0.05, Tukey’s multiple range test). (b) Germination phenotypes of the ref6-1 mutant and transgenic lines in the ref6-1 background expressing REF6 under control of the embryo-specific TPS1 promoter during germination. Plots show the percentage of germinating seeds of wild-type, ref6-1, and complemented lines (TPS1::REF6;ref6-1 a, b, and c) with and without stratification. Details are shown in the legend of (a). (c) and (d) Germination phenotypes of ref6-1 mutant embryos dissected from seeds without (c) and with (d) stratification. Embryos of wild-type, ref6-1, and ref6-3 plants were manually dissected from seeds. Plots show the percentage of seedlings with root hairs and green cotyledons. Error bars indicate SD from three biological replicates (n = 90 total). The data at each time point were evaluated using one-way ANOVA, no significant differences were detected (p>0.05).
REF6 activates genes marked by single H3K27me3 during germination
To identify the genes controlled by REF6 during germination, we profiled the transcriptome of ref6 mutant endosperm during germination. We found more than twice as many significantly downregulated than upregulated genes in ref6 compared to wild type (1471 downregulated [Log2FC≦–1, p<0.05] vs 655 upregulated [Log2FC ≥ 1, p<0.05];Figure 5—source data 1), consistent with the proposed function of REF6 to activate genes during germination. Genes with single H3K27me3 and unmethylated REF6-binding sites were significantly enriched among downregulated but not upregulated genes (Figure 5a,b), supporting the model that REF6 activates genes with single H3K27me3 through the unmethylated REF6-binding sites. Furthermore, there was no significant enrichment of genes with single H3K9me2 among deregulated genes in ref6 (Figure 5—figure supplement 1). Additionally, while genes with double H3K27me3/H3K9me2 and CHG methylated REF6-binding sites were not significantly enriched among deregulated genes, genes with double H3K27me3/H3K9me2 and unmethylated REF6-binding sites were slightly enriched among downregulated genes, although the enrichment was not significant (p=0.0568, pairwise Fisher’s exact test) (Figure 5—figure supplement 2). The 59 genes with single H3K27me3 and REF6-binding sites that were downregulated in ref6 (Figure 5—figure supplement 3) were significantly enriched for germination- and ethylene-inducible genes, but not for gibberellin-inducible genes according to previous transcriptome analyses (Cao et al., 2006; Das et al., 2016; Dekkers et al., 2013; Figure 5c). These findings are consistent with the idea that REF6 promotes germination by activating germination-inducible genes acting in the ethylene pathway. Among the candidate REF6 target genes (Figure 5—figure supplement 3) was ETHYLENE RESPONSE DNA BINDING FACTOR 4 (EDF4) that was previously reported to mediate ethylene responses (Alonso et al., 2003; Chen et al., 2015). We confirmed significant suppression of EDF4 in the ref6 mutant endosperm (Figure 5d), supporting the proposed endosperm-specific function of REF6 during germination (Figure 4a,b). We furthermore found three genes encoding for ethylene biosynthesis enzymes converting the ethylene precursor 1-aminocyclopropane-1-carboxylic acid (ACC) to biologically active ethylene (ACC oxidases, ACOs) significantly downregulated in ref6 (Figure 5—figure supplement 3). ACO genes were not marked by H3K27me3 in the 4 days after pollination (DAP) endosperm, suggesting that REF6 activates these genes at an earlier timepoint. REF6 was shown to target the ABA catabolism genes CYP707A1 and CYP707A3, but not CYP707A2 during seed development (Chen et al., 2020). We also found CYP707A1 and CYP707A3 slightly, but significantly suppressed in ref6 endosperm (Figure 5—figure supplement 3), consistent with the presence of REF6-BS in CYP707A1 and CYP707A3, but not CYP707A2. Like for ACO genes, we did not detect H3K27me3 on all three genes in 4 DAP endosperm (Moreno-Romero et al., 2016), suggesting that REF6 acts on these genes before this timepoint. To further test the contribution of the ethylene and ABA pathways to the delayed germination phenotypes in ref6 mutants, we analyzed germination of ref6 in the presence of the ethylene precursor 1-aminocyclopropane-1-carboxylic acid (ACC) and the ABA biosynthesis inhibitor fluridone, which both were reported to induce seed germination (Linkies et al., 2009; Martinez-Andujar et al., 2011). Treatment with only ACC or fluridone partially suppressed the ref6 delayed germination phenotype, but the germination ratios were significantly lower than non-treated wild type after stratification (Figure 5—figure supplement 4). In contrast, treatment of cyp707a2 mutants with fluridone resulted in comparable germination frequencies as for wild-type seeds without treatment (Figure 5—figure supplement 4), suggesting that induction of the ABA pathway can be effectively suppressed by fluridone, in line with previous data (Martinez-Andujar et al., 2011). The failure of fluridone to suppress the ref6 mutant phenotype is also consistent with previous findings showing that a mutation in the ABA biosynthesis gene ABA2 does not completely rescue the delayed germination phenotype of ref6-1 after stratification (Chen et al., 2020). Importantly however, treatment with both ACC and fluridone could completely suppress the delayed germination of ref6-1 after stratification (Figure 5—figure supplement 4). This data support the idea that loss of REF6 causes failure to activate the ethylene pathway (Figure 5c) and at the same time causes induction of the ABA pathway as previously shown (Chen et al., 2020).
Figure 5 with 4 supplements see all
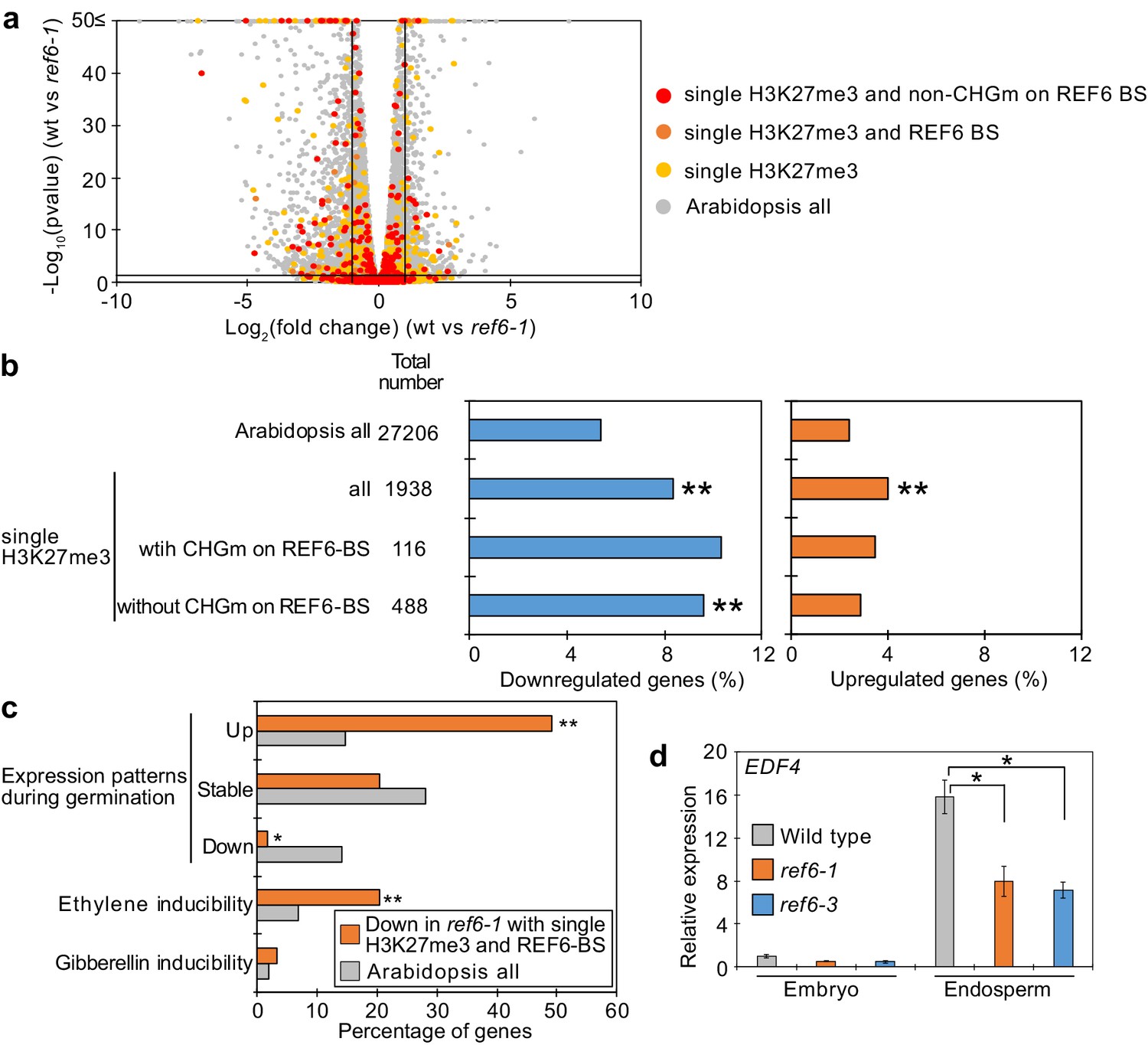
Genes marked by single H3K27me3 are activated by REF6 during germination.
(a) Volcano plot shows differentially expressed genes with single H3K27me3 and REF6-binding sites (REF6-BS) in ref6-1. The color of dots corresponds to the following gene categories: red dots represent genes with single H3K27me3 and non-CHG methylation (CHGm) on REF6-BS, orange dots represent genes with single H3K27me3 and REF6-BS (excluding non-CHGm REF6-BS), yellow dots represent genes with single H3K27me3 (excluding REF6-BS), and gray dots represent Arabidopsis all genes (excluding genes with single H3K27me3). Vertical and horizontal lines mark thresholds of up- and downregulated genes (1≦Log2FC, Log2FC≦–1 and p<0.05). (b) Plots show the percentage of up- (1≦Log2FC, p<0.05) and downregulated (Log2FC≦–1, p<0.05) genes in the endosperm of ref6-1 during germination. Compared are genes with single H3K27me3 and presence or absence of (CHGm on REF6-BS. CHGm was determined based on published data Park et al., 2016). Asterisks indicate significant differences compared to Arabidopsis all genes (*p<0.05, **p<0.01, pairwise Fisher’s exact test with Benjamini–Hochberg correction). (c) Plot shows the expression of genes with single H3K27me3 and REF6-BS that were downregulated in endosperm of ref6-1. Gene expression was calculated based on tissue-specific transcriptome data in the endosperm during germination (Dekkers et al., 2013). Expression levels were compared between 1 and 38 hours after imbibition. Expression changes were defined as follows: upregulated genes (1≦Log2FC and p<0.05), downregulated genes (Log2FC≦–1 and p<0.05), stably expressed genes (–1≦Log2FC≦1 and p<0.05) and non-expressed genes. Percentage of ethylene and gibberellin inducible genes was calculated based on transcriptome data from seedlings (Cao et al., 2006; Das et al., 2016). Asterisks indicate significant differences compared to Arabidopsis all genes (*p<0.01, **p<0.001, Fisher’s exact test). (d) Plot shows expression of EDF4 in the endosperm of ref6-1 during germination as determined by RT-qPCR. Embryos of wild-type, ref6-1, and ref6-3 mutants were dissected after 72 hr of stratification and 24 hr of incubation under normal conditions. Error bars indicate SD from technical triplicates. Asterisks indicate significant differences compared to wild type (p<0.01, Bonferroni-corrected Student’s t test).
-
Figure 5—source data 1
RNA-seq data in the ref6 mutant endosperm during germination.
- https://cdn.elifesciences.org/articles/64593/elife-64593-fig5-data1-v2.xlsx
Together, our data support a model where REF6 is required to activate genes with single H3K27me3 and unmethylated REF6-binding sites in the endosperm during germination, among them genes in the ethylene and ABA catabolism pathway.
SUVH4/5/6 and CMT3 are required to promote germination
Our data indicate that REF6 is able to activate genes marked by single H3K27me3, but fails to access genes marked by H3K27me3/H3K9me2/CHGm due to the block of REF6 targeting by CHGm. The H3K9me2 modification is established by the redundantly acting SUVH4/5/6 histone methyltransferases and recruits CMT3 to establish DNA methylation in CHG context (Du et al., 2012; Ebbs and Bender, 2006; Jackson et al., 2002). Since the suvh456 mutant is strongly depleted for CHGm (Stroud et al., 2014), we speculated that in suvh456 REF6 targets should become accessible and activated. Previous work revealed that the suvh45 double mutant has increased dormancy (Zheng et al., 2012). Similarly, we found that suvh456 required prolonged after-ripening time and had reduced germination rates at 10 days after harvesting or required stratification when seeds developed under low temperature (Figure 6—figure supplement 1). Interestingly, the mutations in SUVH4/5/6 only caused a delay in germination when maternally, but not when paternally inherited (Figure 6a), revealing a maternal effect. Since SUVH4/5/6 genes are not regulated by genomic imprinting (Moreno-Romero et al., 2019; Pignatta et al., 2014), one possible explanation is that activity of SUVH4/5/6 in the central cell of the female gametophyte has a lasting effect until germination. Based on published expression studies, SUVH4 and SUVH6 are expressed in the central cell (Belmonte et al., 2013; Hsieh et al., 2011; Wuest et al., 2010), supporting this notion. Furthermore, we found a similar enrichment of methylated REF6 binding sites in genes marked with H3K9me2 and H3K27me3/H3K9me2 in central cells and developing endosperm (Figure 2a and Figure 2—figure supplement 1), supporting the idea that H3K9me2 and CHGm are established in the central cell. We tested this hypothesis by analyzing germination of F1 seeds generated from heterozygous suvh4/5/6 (suvh4/+ suvh5/+ suvh6/+) mutants pollinated with wild-type plants. Germination of these F1 seeds was clearly delayed (Figure 6b), supporting the idea that SUVH4/5/6 act in the female gametophyte. We tested whether indeed CHGm has a functional role in controlling seed dormancy by testing the effect of mutations in CMT3. Consistently, loss of CMT3 caused a delay in germination (Figure 6c) and prolonged after-ripening of cmt3 seeds decreased primary dormancy (Figure 6—figure supplement 2). Interestingly, contrary to the maternal effect of suvh4/5/6, loss of maternal and paternal CMT3 alleles was required to cause a delay in germination (Figure 6d). Based on DNA methylome data CHGm is established in the central cell before fertilization (Figure 2a; Park et al., 2016); nevertheless, the paternal CMT3 allele was sufficient to suppress delay of germination in cmt3. Previous work revealed that loss of SUVH4/5/6 causes severe depletion of both H3K9me2 and CHGm (Stroud et al., 2014; Underwood et al., 2018), while loss of CMT3 causes depletion of CHGm but only intermediate reductions of H3K9me2 (Stroud et al., 2014; Inagaki et al., 2010). Together, this suggests that H3K9me2 is established first and recruits CHGm and that CHGm establishment after fertilization is sufficient to control seed dormancy.
Figure 6 with 2 supplements see all
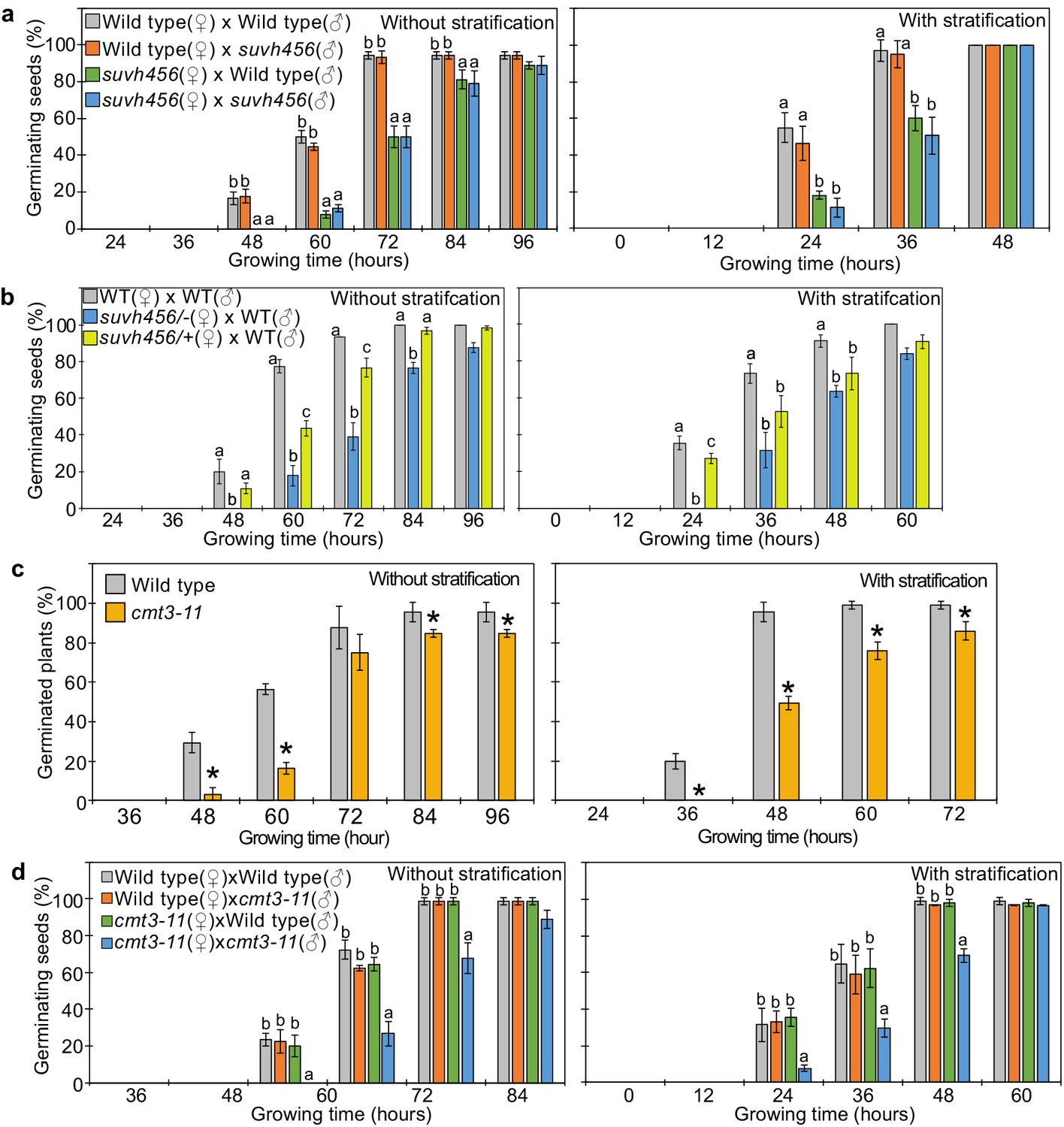
SUVH4/5/6 and CMT3 are necessary to promoter germination.
(a) Germination phenotypes of seeds generated by reciprocal crosses between wild-type and suvh456 triple mutants. Plots show the percentage of germinating seeds with and without stratification. Error bars indicate SD from three biological replicates (n = 120 total). The letters above the bars indicate significant differences between genotypes (p<0.05, Tukey’s multiple range test). (b) Plots show the percentage of germinating seeds with and without stratification of wild-type (WT) and F1 seeds generated by crossing maternal heterozygous or homozygous suvh456 and paternal wild type. Error bars indicate SD from three biological replicates (n = 90 total). Letters above bars indicate significant differences among genotypes (p<0.05, Tukey’s multiple range test). (c) Plots show percentage of germinating seeds of wild-type and cmt3-11 mutant with and without stratification. Error bars indicate SD from three biological replicates (n = 120 total). Asterisks indicate significant differences compared to wild type (p<0.05, Student’s t test). (d) Germination phenotypes of seeds generated by reciprocal crosses between wild-type and cmt3-11 mutant. Details are shown in the legend of (a).
SUVH4/5/6 suppress genes with H3K27me3/H3K9me2 and CHGm on REF6-binding sites
To further test the hypothesis that H3K9me2/CHGm block the access of REF6, we analyzed transcriptome data of suvh456 endosperm during germination. There were more than twice as many significantly up- than downregulated genes in suvh456 compared to wild type (3,203 upregulated [Log2FC ≥ 1, p<0.05] vs 1,502 downregulated [Log2FC ≦ –1, p<0.05]; Figure 7—source data 1). Importantly, genes marked with H3K27me3/H3K9me2 and CHGm on REF6 binding sites were significantly enriched among upregulated, but not downregulated genes (Figure 7a,b), supporting the idea that REF6 targeting is blocked by H3K9me2/CHGm in the endosperm. Consistently, REF6-binding sites were significantly enriched in the gene bodies of upregulated genes marked with H3K27me3/H3K9me2/CHGm (Figure 7c). REF6-binding sites were not significantly enriched among the downregulated genes in suvh456 (Figure 7—figure supplement 1). We also did not find significant differences among the up- and downregulated genes with single H3K9me2 (Figure 7—figure supplement 2), revealing that the suppressive effect of SUVH4/5/6 on gene repression connects to the presence of H3K27me3. Together, our data show that SUVH4/5/6 suppress genes with H3K27me3/H3K9me2/CHGm and that loss of SUVH4/5/6 causes upregulation of genes through REF6-binding sites. The ABA INSENSITIVE 3 (ABI3) gene was previously shown to be upregulated in suvh45 mutants (Zheng et al., 2012) and was also present among the 26 upregulated genes in suvh456 marked with H3K27me3/H3K9me2/CHGm and having REF6-binding sites (Figure 7—figure supplement 3). ABI3 is a B3 type transcription factor and positively regulates seed dormancy (Liu et al., 2013; Shu et al., 2016; Tian et al., 2020). HEAT SHOCK TRANSCRIPTION FACTOR C1 (HSFC1) was also reported to be induced in developing seeds by heat stress and suggested to activate HEAT SHOCK PROTEIN (HSP) genes that are abundant in seed maturation (Chiu et al., 2012). The upregulation of the two genes in the endosperm of suvh456 were also confirmed by RT-qPCR (Figure 7d). We found that genes with single H3K27me3 were significantly enriched for downregulated genes in the suvh456 endosperm (Figure 7—figure supplement 4), suggesting an indirect effect. Interestingly, we found RY binding motifs for B3 type transcription factors to be significantly enriched among downregulated genes in suvh456 containing single H3K27me3 (Figure 7—figure supplement 4). This raised the hypothesis that upregulated ABI3 in suvh456 suppressed genes with single H3K27me3. In support of this idea, we found that genes with single H3K27me3 significantly overlapped with previously identified direct target genes of ABI3 (Tian et al., 2020), while genes with single H3K9me2 and double H3K27me3/H3K9me2 did not significantly overlap (Figure 7—figure supplement 4).
Figure 7 with 5 supplements see all
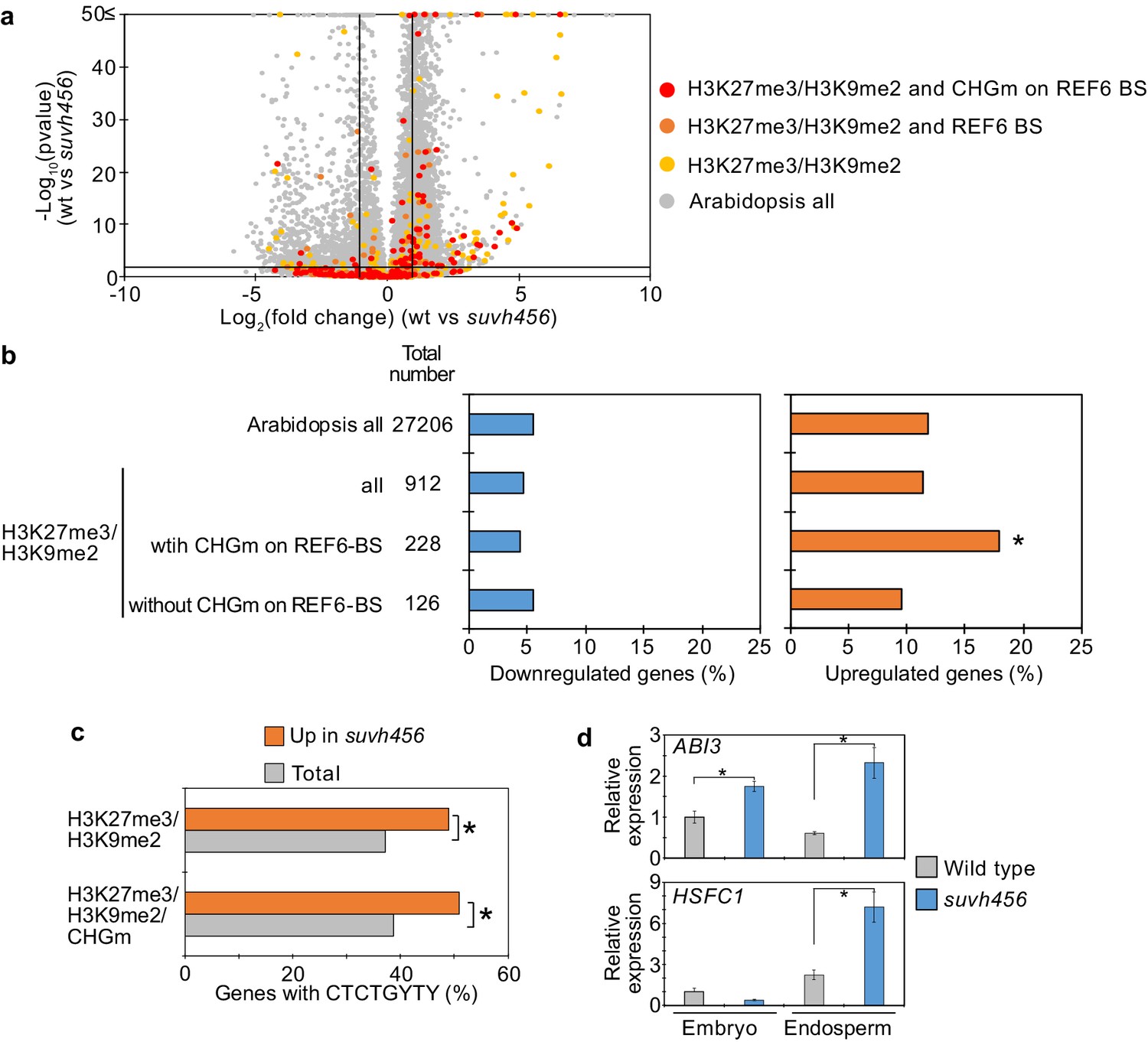
SUVH4/5/6 are required for the suppression of genes with H3K27me3/H3K9me2 and CHG methylation (CHGm) on REF6-binding sites (REF6-BS).
(a) Volcano plot shows differentially expressed genes with double H3K27me3/H3K9me2 and CHGm on REF6-BS in suvh456. The color of dots corresponds to the following gene categories: red dots represent genes with double H3K27me3/H3K9me2 and CHGm on REF6-BS, orange dots represent genes with double H3K27me3/H3K9me2 and REF6-BS (excluding CHGm REF6-BS), yellow dots represent genes with double H3K27me3/H3K9me2 (excluding REF6-BS), and gray dots represent Arabidopsis all genes (excluding genes with H3K27me3/H3K9me2). Vertical and horizontal lines mark thresholds of up- and downregulated genes are shown (1≦Log2FC, Log2FC≦–1, p<0.05). (b) Plots show the percentage of up-(1≦Log2FC, p<0.05) and downregulated (Log2FC≦–1, p<0.05) genes in the endosperm of suvh456 during germination. Compared are genes with double H3K27me3/H3K9me2 and presence or absence of CHGm on REF6-BS. CHGm was determined based on published data (Park et al., 2016). Asterisks indicate significant differences compared to Arabidopsis all genes (p<0.05, pairwise Fisher’s exact test with Benjamini–Hochberg correction). (c) Plot shows the enrichment of REF6-BS among upregulated (1≦Log2FC, p<0.05) genes in the endosperm of suvh456 during germination. Compared are genes with the indicated combinations of histone modifications and presence (>0.04) or absence (≤0.04) of CHGm. CHGm levels were determined based on published data (Park et al., 2016). Asterisks indicate significant differences between two categories (p<0.05, Fisher’s exact test). (d) Plots show the expression of ABI3 and HSFC1 in the endosperm of suvh456 during germination as determined by RT-qPCR. Embryos of wild type and the suvh456 triple mutant were dissected after 72 hr of stratification and 24 hr of incubation under normal conditions. Error bars indicate SD from technical triplicates. Asterisks indicate significant differences between wild type and suvh456 (p<0.01, Student’s t test).
-
Figure 7—source data 1
RNA-seq data in the suvh456 mutant endosperm during germination.
- https://cdn.elifesciences.org/articles/64593/elife-64593-fig7-data1-v2.xlsx
Parentally biased expression during germination is regulated by REF6 and SUVH456
Our data obtained in this (Figure 2) and previous studies (Moreno-Romero et al., 2019; Moreno-Romero et al., 2016) indicate that H3K27me3, H3K9me2, and CHGm are established in the central cell and control imprinted expression in the endosperm after fertilization. We next addressed the question whether maternal-specific epigenetic marks also control parental-specific gene expression in the endosperm during germination. Using previously published parental-specific transcriptome data of the endosperm during germination (F1 seeds of maternal Col and paternal Cvi accessions) (Piskurewicz et al., 2016), we found that genes with triple repressive marks H3K27me3/H3K9m2/CHGm on REF6-binding sites were significantly paternally biased compared to Arabidopsis all genes in both dormant and non-dormant seeds (Figure 8a, upper blue boxes), consistent with our data showing the relationship between triple repressive marks and expression during germination (Figure 2c). This data suggests that triple repressive marks are associated with paternally-biased gene expression in germinating endosperm, similar as in the developing endosperm (Moreno-Romero et al., 2019; Moreno-Romero et al., 2016). In contrast, genes with single H3K27me3 and unmethylated REF6-binding sites were significantly maternally biased in dormant seeds (Figure 8a, lower orange boxes, left panel), but not in non-dormant seeds (Figure 8a, lower gray boxes, right panel). The maternally-biased expression in dormant seeds supports the idea that genes with single H3K27me3 are reactivated during germination. We observed a weak maternal bias of genes marked with single H3K27me3 already in the developing endosperm at 4 DAP (Figure 8—figure supplement 1), suggesting that gene activation by removing single H3K27me3 starts before germination, consistent with the observed maternal effect of ref6 (Figure 3b,c). In non-dormant seeds, however, unbiased expression of genes with single H3K27me3 suggests that the paternal alleles of these genes are also activated. Indeed, this expression pattern has been previously reported for the germination-related CP1 gene, which is maternally-biased in dormant seeds, while biallelically expressed in non-dormant seeds (Piskurewicz et al., 2016). We did not observe a significant parental bias of genes with single H3K9me2 (Figure 8—figure supplement 1).
Figure 8 with 5 supplements see all
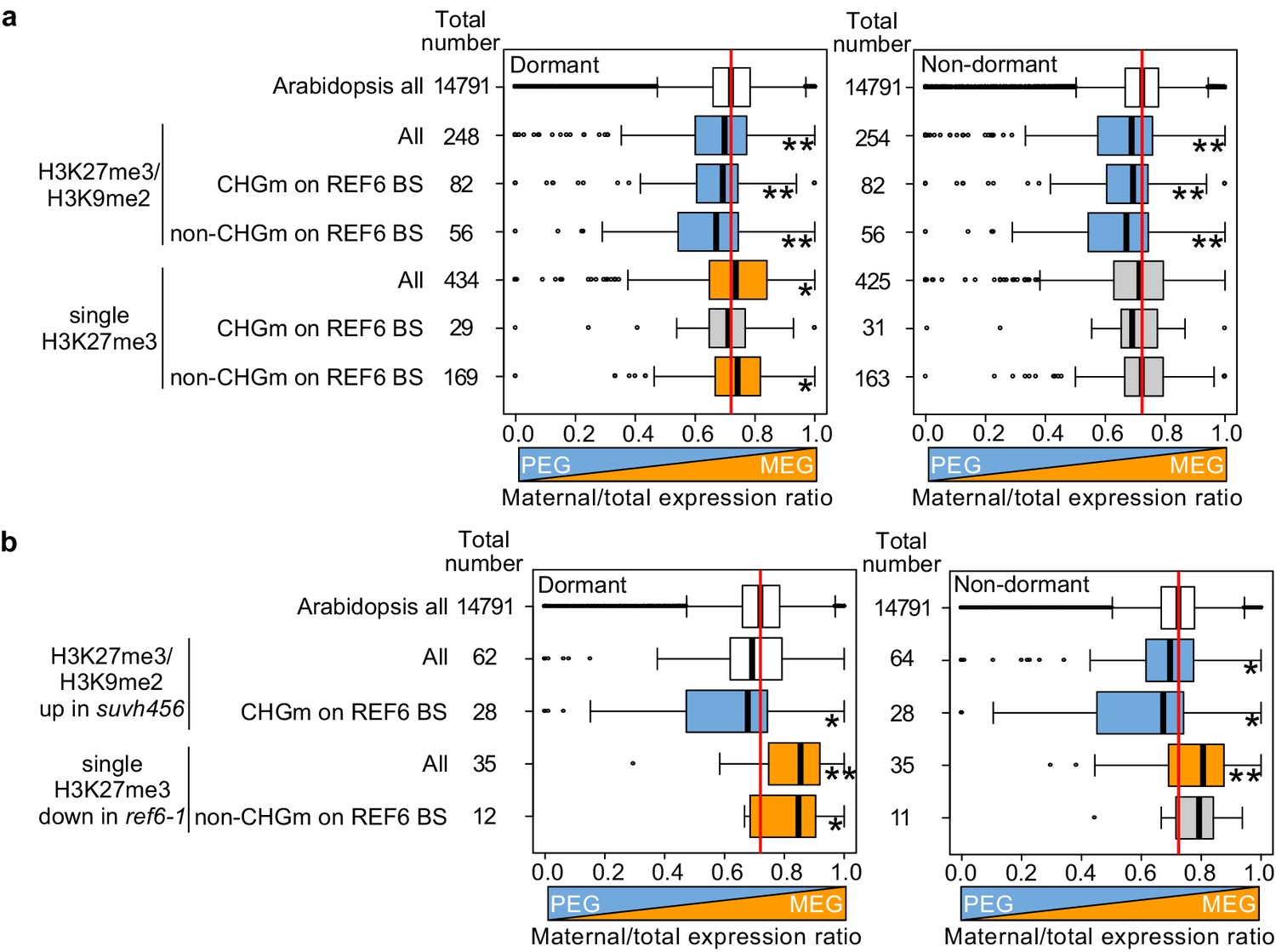
Maternal- and paternal-specific epigenetic marks regulate parental-specific gene expression in the endosperm during germination.
(a) Box plots show the mean values of maternal to total reads of genes expressed in the endosperm in dormant and non-dormant seeds. Compared are Arabidopsis all genes, all genes with double H3K27me3/H3K9me2 and single H3K27me3 depending on the presence or absence of CHG methylation (CHGm) on REF6-binding sites (REF6-BS) based on published data (Park et al., 2016). Parental-specific transcriptome data in the endosperm during germination from F1 seeds (maternal Col × paternal Cvi) were previously published (Piskurewicz et al., 2016). Boxes show medians and the interquartile range, and error bars show full-range excluding outliers. Numbers of genes in each category are shown beside the boxes. Asterisks indicates significant differences compared to Arabidopsis all genes (*p<0.05, **p<0.01, pairwise Wilcoxon test with Benjamini–Hochberg correction). Significantly paternally and maternally biased groups are shown in blue and orange, respectively. (b) Box plots show mean values of maternal to total reads in the endosperm of dormant and non-dormant seeds of Arabidopsis all genes, upregulated genes with double H3K27me3/H3K9me2 in suvh456, and downregulated genes with single H3K27me3 in ref6-1. Details are shown in the legend of (a).
Based on our data showing that ref6 mainly affected genes with single H3K27me3 while suvh4/5/6 mainly affected genes with triple repressive marks on REF6-binding sites (Figure 5a, b and Figure 7a,b), we selected genes that were likely to be regulated by REF6 and SUVH4/5/6 and analyzed the maternal expression ratios. Using the same published parental-specific transcriptome data (Piskurewicz et al., 2016), we found that genes with single H3K27me3 that were downregulated in ref6 were significantly maternally biased in both dormant and non-dormant seeds (Figure 8b, orange boxes). Conversely, upregulated genes in suvh456 that were marked with triple repressive marks were significantly paternally biased (Figure 8b, blue boxes). Nevertheless, the number of genes in each category was relatively small and further experiments should validate these conclusions. Together, this data suggests that REF6 and SUVH4/5/6 regulate genomic imprinting during germination.
The maternally biased expression of genes with single H3K27me3 suggests suppression of the paternal alleles by an epigenetic mechanism. Maternally biased gene expression during germination was previously reported to be mediated by the non-canonical RNA-directed DNA methylation (RdDM) pathway targeting the paternal alleles (Iwasaki et al., 2019). We therefore analyzed DNA methylation levels in genic as well as 2 kb up- and downstream regions on the paternal alleles of genes with single H3K27me3 on the maternal alleles using previously published data (Ibarra et al., 2012; Moreno-Romero et al., 2016). We found that paternal CHHm levels in downstream regions of genes with maternal H3K27me3 and in gene bodies of genes with maternal H3K27me3 and unmethylated REF6-binding sites were significantly higher compared to Arabidopsis all genes in 7–8DAP endosperm (Figure 8—figure supplement 2). CHGm levels in downstream regions were significantly higher in 4DAP endosperm, but this trend was not detected at 7–8DAP (Figure 8—figure supplement 3). Levels of CG methylation (CGm) were marginally increased in gene bodies of genes with maternal H3K27me3 and unmethylated REF6-binding sites in 4DAP and 7–8DAP endosperm (Figure 8—figure supplement 4). This data suggests that CHHm of genes with maternal H3K27me3 and unmethylated REF6-binding sites can be one possible factor to establish maternally biased expression of genes with single H3K27me3 in dormant seeds. However, detailed parental-specific DNA methylome data in dormant and non-dormant seeds are necessary to validate this conclusion.
Discussion
In this study, we uncovered the molecular function and physiological role of the endosperm-specific triple modification of the maternal genome with H3K27me3/H3K9me2 and CHGm. We found that genes marked by only H3K27me3 became reactivated during seed germination, a process that depends on the H3K27me3 demethylase REF6. In contrast, genes marked by triple repressive modifications remained silenced during germination because REF6 binding is repelled by H3K9me2/CHGm, consistent with previous work (Qiu et al., 2019). Supporting the functional requirement of REF6 to initiate germination, loss of REF6 caused higher seed dormancy (Figure 3, Figure 3—figure supplement 2; Chen et al., 2020), connected with decreased expression of many germination-inducible genes. Loss of SUVH4/5/6 also increased dormancy (Figure 6—figure supplement 1; Zheng et al., 2012), which was associated with increased expression of potential REF6 target genes. Since H3K9me2 acts in a feedback loop with CHGm (Du et al., 2012; Jackson et al., 2002; Lindroth et al., 2001; Malagnac et al., 2002), loss of SUVH4/5/6 is expected to expose unmethylated binding sites for REF6 that can then be activated by REF6. Consistent with this model, loss of CMT3 also caused increased dormancy (Figure 6b, Figure 6—figure supplement 2). Interestingly, among those upregulated SUVH4/5/6 targets was the positive regulator of seed dormancy ABI3 (Liu et al., 2013), providing a possible explanation for the increased dormancy in suvh4/5/6. Consistent with this notion we found that downregulated genes with single H3K27me3 were enriched for ABI3 binding motifs. Nevertheless, the small fraction of germination-inducible genes with triple repressive marks suggests possible additional mechanisms removing triple epigenetic marks.
Our work furthermore revealed the connection between maternal-specific repressive marks, gene imprinting, and seed dormancy. Imprinted genes were previously shown to play a role in regulating seed germination and were connected to the maternal inheritance of seed dormancy levels (Iwasaki et al., 2019; Piskurewicz et al., 2016). The non-canonical RdDM pathway was found to target and silence the paternal alleles of the dormancy-related gene ALN1 (Iwasaki et al., 2019). Our data suggests that in addition to RdDM, maternally biased expression of dormancy-related genes also requires REF6, which activates maternal H3K27me3 marked genes. We propose that both mechanisms together ensure the maternal control of seed dormancy; while REF6 regulates the time of activation of certain genes, RdDM-mediated CHHm on the paternal alleles prevents their activation. Interestingly, the timing of REF6 action probably differs for distinct genes. While some genes, like ALN1, CYP707A1, and CYP707A3 (Chen et al., 2020) are likely activated in the central cell of the female gametophyte and therefore lack detactable H3K27me3 in the endosperm, other genes are activated during endoperm development. To the latter category belongs the previously reported gene CP1, which is maternally biased in dormant seeds and biallelically expressed in non-dormant seeds (Piskurewicz et al., 2016) and marked with single H3K27me3 on the maternal alleles (Moreno-Romero et al., 2016). In contrast to genes with single H3K27me3, genes with triple repressive marks on maternal alleles are not targeted by REF6, causing the maternal alleles to be silenced and the paternal alleles to be active. Genes with triple repressive marks on REF6-binding sites were upregulated in suvh4/5/6 and were paternally biased during germination. Although more detailed parental-specific DNA methylome analyses in germinating endosperm are required to precisely determine the molecular mechanism repressing the paternal alleles of genes with maternal H3K27me3 during germination, this data supports the model that the presence of single H3K27me3 and triple repressive marks determines whether genes are maternally or paternally biased during germination, respectively (Figure 8—figure supplement 5).
The SUVH4/5/6 genes are not imprinted (Hsieh et al., 2011; Moreno-Romero et al., 2019; Pignatta et al., 2014); however, the delay of germination in suvh4/5/6 was under maternal control (Figure 6a). This indicates that maternal-specific triple repressive marks are established before fertilization in the central cell and maintained after fertilization, consistent with previous data (Moreno-Romero et al., 2019). This suggests that maternal-specific triple epigenetic marks of H3K27me3/H3K9me2 and CHGm contribute to maternally controlled seed dormancy (Iwasaki et al., 2019; Piskurewicz et al., 2016). Likewise, also REF6 is not imprinted (Hsieh et al., 2011; Pignatta et al., 2014), but ref6 had a maternal effect (Figure 3b), supporting the notion that derepression of some dormancy-controlling genes starts in the central cell of the female gametophyte. Nevertheless, the fact that some REF6 targets have detectable levels of H3K27me3 in the endosperm and since we could partially complement the ref6 mutant by expressing REF6 in the micropylar domain of the endosperm (Figure 5a), strongly suggests that REF6 also acts after fertilization. In support of this view, homozygous ref6 mutants had a stronger delay in germination than maternal ref6 mutants (Figure 3b).
Our data furthermore uncovered a relationship among REF6, ethylene, and ABA. Consistent with previous work showing that loss of ABA2 does not completely suppress the delayed germination of ref6 after stratification (Chen et al., 2020), we found that the ABA biosynthesis inhibitor fluridone was not sufficient to suppress delayed germination phenotypes of ref6 (Figure 5—figure supplement 4). Similarly, the ethylene precursor ACC caused only a partial suppression of the germination delay. However, the combined treatment with fluridone and ACC completely suppressed the ref6 germination phenotype (Figure 5—figure supplement 4), supporting our and previous data revealing that REF6 is required to activate ABA catabolism and ethylene signaling through removal of H3K27me3. Since the activation of the ABA catabolism genes CYP707A1 and CYC707A3 likely occurs in the central cell while activation of ethylene signaling genes occurs during endosperm development, we propose that there is a two-step maternal control of germination by REF6. The first step occurs in the central cell and leads to the suppression of ABA signaling, while the second step occurs in the endosperm and leads to the activation of ethylene signaling. This two-step control of germination may ensure the best timing of germination. Understanding by which mechanism the timing of REF6 action is regulated will be subject of future investigations.
Previous data showed that loss of FIS-PRC2 function causes upregulation of genes with both H3K27me3 and H3K9me2 (Hsieh et al., 2011; Moreno-Romero et al., 2016), suggesting that establishment of H3K9me2 depends on H3K27me3. This idea was substantiated by data showing that deficiency of FIS-PRC2 in the central cell causes depletion of CHGm (Moreno-Romero et al., 2019), pointing that H3K9me2/CHGm depend on FIS-PRC2. Also data shown in this study are consistent with this scenario; we demonstrate that loss of H3K9me2/CHGm only caused an effect if H3K27me3 was removed by REF6, revealing that H3K27me3 is the primary repressive mark. Additionally, we found that maternal loss of SUVH4/5/6 was sufficient to cause delayed gemination (Figure 6a), while that of CMT3 was not (Figure 6c). One possible explanation is that the effects caused by loss of SUVH4/5/6 and CMT3 function differ. While H3K9me2 and CHGm are severely depleted in suvh4/5/6; loss of CMT3 mainly affects CHGm with only intermediate effects on H3K9me2 (Stroud et al., 2014; Underwood et al., 2018). Thus, genes with H3K27me3/H3K9me2 can likely be suppressed by CMT3 after fertilization.
A possible mechanism is that the FIS2-PRC2 complex or H3K27me3 recruits SUVH4/5/6 and that H3K9me2 recruits CMT3, suggesting the presence of a molecular mechanism that distinguishes target genes to be marked by single H3K27me3 or triple marks of H3K27me3/H3K9me2 and CHGm. To identify this distinguishing mechanism remains an important future endeavor.
Our data bear striking similarities to maternal H3K27me3-dependent non-canonical imprinting reported in mammals (Chen et al., 2019; Hanna et al., 2019; Inoue et al., 2017a; Inoue et al., 2017b). In non-canonical imprinting, maternal-specific H3K27me3 inherited from oocytes serves as the primary epigenetic mark for imprinted paternal-specific expression. Most of this non-canonical imprinting is transient and restricted to preimplantation embryos, with only some genes important for placental development remain imprinted in extraembryonic cells (Inoue et al., 2017a). Maintenance of non-canonical imprinting is mediated by DNA methylation, which is specifically applied on the maternal alleles of previously H3K27me3 marked regions (Chen et al., 2019; Hanna et al., 2019). Thus, similar to the findings reported in this study, genes regulated by non-canonical imprinting in mammals are transiently imprinted by inheriting maternal-specific H3K27me3 and only few genes remain imprinting by establishing DNA methylation as secondary imprint. Genomic imprinting has independently evolved in flowering plants and mammals; however, rather remarkably, similar molecular mechanisms have been coopted to regulate imprinted genes in the accessory, nutrient-transferring tissues.
The combined presence of H3K27me3 with the heterochromatic modification H3K9me3 was also reported to occur on TEs and repeat-rich regions in the protozoa Tetrahymena and Paramecium (Frapporti et al., 2019; Liu et al., 2007; Zhao et al., 2019) and the fungus Podospora anserine (Carlier et al., 2020). In P. anserine, the presence of double repressive modifications is restricted to TEs and repeat-rich regions, while H3K27me3 alone is located in genic regions, indicating a similar molecular distinction of constitutively silenced regions marked by double modifications from regions that require activation during specific developmental stages marked by H3K27me3 alone. It is tempting to speculate that the co-occurrence of H3K9me3 and H3K27me3 may prevent H3K27me3 demethylases to target and activate those regions, similar to the findings reported in this study.
In summary, in this study, we uncovered a new molecular mechanism leading to maternal-specific dormancy control that distinguishes genes marked by either H3K27me3 alone from genes marked by the generally antagonistic repressive epigenetic modifications, H3K27me3, H3K9me2, and CHGm. We uncovered how the H3K27me3 demethylase REF6 controls release of dormancy and revealed the underlying mechanism for the H3K9me2 methylases SUVH4/5/6 in controlling dormancy and propose that these regulators form a regulatory network through ABA and ethylene pathways. Our data provide new insights into how maternal-specific epigenetic modifications control dormancy and thus ensure timely germination.
Materials and methods
Plant material and growing conditions
Request a detailed protocolAll mutants used in this study are in the Col background. The T-DNA insertion lines of ref6-1 (SALK_001018) (Cui et al., 2016), ref6-3 (SAIL_747_A07) (Cui et al., 2016), elf6-3 (SALK_074694) (Tao et al., 2017), elf6-4 (SAIL_371_D08) (Tao et al., 2017), and an ethylmethane sulfonate-mutated line of cmt3-11 (Lindroth et al., 2001) were previous described. The T-DNA insertions were confirmed by amplifying the left border-flanking regions of the genomic DNA using the primer set given in Supplementary file 1. The suvh456 mutant was kindly provided by Judith Bender. The elf6-3 ref6C double and elf6-3 ref6C jmj13G triple mutants were kindly provided by Kerstin Kaufmann (Yan et al., 2018). The cyp707a2-1 and cyp707a2-2 mutants were kindly provided by Eiji Nambara (Kushiro et al., 2004). All plants were grown on half-strength Murashige and Skoog agar medium containing 1% sucrose or on soil under long-day conditions (16 hr light/8 hr darkness) at 21°C and a light intensity of 150 µE. Seeds were surface sterilized (5% sodium hypochlorite) before sawing on medium and 14-day-old plants were transferred into soil. For germinating assays, seeds from dry siliques were harvested on the same day 3 weeks after pollination and after-ripened at room temperature for 10 days unless otherwise indicated. Transgenic plants were generated through the floral dip method by using Agrogacterium tumefaciens GM3101 as described previously (Clough and Bent, 1998).
Germinating assays
Request a detailed protocolFor germinating assays, seeds from 20 siliques were sterilized and grown on half-strength Murashige and Skoog medium with or without stratification at 4°C for 3 days. To analyze the effects of chemicals on germination, 1-aminocyclopropanecarboxylic acid (ACC) (Sigma-Aldrich) and fluridone (Merck Chemicals and Life Science AB) were added to half-strength Murashige and Skoog medium. For seed dormancy assays, plants were grown under normal conditions or under lower temperature (10°C). The sterilized seeds were sown on minimal media (0.9% agar) and grown under normal conditions above with or without stratification. For growth analysis of dissected embryos, seeds were sterilized and manually dissected after treatment of stratification as previously described (Piskurewicz et al., 2016). Root protrusion, root hair formation, and cotyledon greening were observed under a microscope (Leica, EZ4) at different times during germination, and photographs were taken by a high-resolution digital camera (Leica, M205 FA). Experiments were done in biological replicates, as indicated.
RNA preparation and qRT-PCR
Request a detailed protocolFor gene expression analysis, embryos and endosperm were manually dissected from seeds that were imbibed for 24 hr as previously described (Endo et al., 2012; Piskurewicz et al., 2016). Total RNA from embryos and endosperm was extracted using MagMAX Plant RNA Isolation Kit (Thermo Fisher Scientific) according to the supplier’s instructions for RNA extraction from plant seeds. For quantitative RT-PCR, cDNA was synthesized from total RNA using the Thermo Scientific RevertAid First-Strand cDNA Synthesis Kit according to the supplier’s instructions. Quantitative RT-PCR analyses were performed using a BIO RAD CFX Connect Real-Time PCR Detection System. HOT FIREPol EvaGreen qPCR Mix Plus (ROX) (Solis BioDyne) was used for the reactions. The primers used for quantitative RT-PCR are listed in Supplementary file 1. Triplicate measurements were generated for each cDNA sample, and the obtained values were normalized to ACT2.
RNA sequencing and data analysis mRNA was isolated from total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Kit (NEB) and cDNA libraries were constructed using the NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB). Libraries were sequenced on Illumina NovaSeq platforms as paired-end through Novogene mRNA sequencing services. For each replicate, the 150 bp long reads were trimmed by removing the 20 bp from the 5’ end and 30 bp from the 3’ and mapped in single-end mode to the Arabidopsis (TAIR10) genome, masked for rRNA genes, using TopHat v2.1.0 (Trapnell et al., 2009) (parameters adjusted as -g 1 -a 10 -i 40 -I 5000 F 0 r 130). Mapped reads were counted and normalized to reads per kilobase per million mapped reads for chromosomal protein coding genes using GFOLD (Feng et al., 2012). Differentially regulated genes across the three replicates of each condition were detected using DESeq (v. 1.12.0) (Anders and Huber, 2010) and in R (v. 3.3.2) (R Development Core Team, 2016). Only changes in expression with a FDR of <0.05 were considered statistically significant. Overrepresentation analysis of hexamers on the promoters of sets of genes by RNA-sequencing data were performed as previously described (Maruyama et al., 2012) using 1 kb upstream sequences from the translational start sites obtained through Phytozome (version 12; https://phytozome.jgi.doe.gov/pz/portal.html).
Data analysis
Request a detailed protocolPublished data of maternal-specific histone modifications of H3K27me3 and H3K9me2 in the developing endosperm derived from crosses of maternal Col and paternal Ler accessions (Moreno-Romero et al., 2019) were used to identify three groups of genes with H3K27me3, H3K9me2, and H3K27me3/H3K9me2. We used CHG methylation scores on gene bodies in central cells (Park et al., 2016) as defined in a previous study (Moreno-Romero et al., 2019). The time-course of expression profiles in the endosperm during germination was determined using previously published data of tissues containing micropylar and chalazal endosperm (Dekkers et al., 2013). To analyze the genomic loci containing CHG, CHH, and CG methylation, published DNA methylation data of central cells (Park et al., 2016) and the developing endosperm at 4 days (Moreno-Romero et al., 2016) and 7–8 days (Ibarra et al., 2012) after pollination were reanalyzed. For the analyses of methylated sites on REF6-binding sites, methylated CHG sites were defined as those containing one or more methylated cytosines among at least five reads in both replicates. For the analyses of parental-specific methylation, numbers of methylated and unmethylated cytosines of two replicates were pooled and methylated sites were defined as those containing one or more methylated cytosines among at least five reads. To analyze parental-specific gene expression patterns in the endosperm during germination, published data of endosperm-specific transcriptomes in F1 seeds generated by reciprocal crosses of Col and Cvi (Piskurewicz et al., 2016) was used. Maternal expression ratios were calculated as the maternal read counts to total read counts (maternal-specific read counts/total read counts).
Protein sequence alignment and gene ontology analysis
Request a detailed protocolThe peptide sequences of REF6, ELF6, and JMJ13 were obtained from Phytozome (Phytozome v12, https://phytozome.jgi.doe.gov/pz/portal.html). Alignment of the proteins was performed using SnapGene and ClustalW and the result of the alignment was manually adjusted to detect conserved amino acids. Gene Ontology (GO) analyses were performed using the public GO database agriGO (Version 2.0) (Tian et al., 2017) and processed through REVIGO using p values (Supek et al., 2011).
Plasmid constructions
Request a detailed protocolTo generate the constructs for complementation assays, the pB7WG vector (https://gatewayvectors.vib.be/) was digested with Eco32I and ligated to remove the cassette of the LR reaction. The promoter regions of EPR1 or TPS1 were inserted into the KpnI and XbaI sites of the vector, and the REF6 coding sequence was inserted into the AatII and Eco32I sites using the In-Fusion HD Cloning Kit (TaKaRa).
Data availability
All data generated in this study were deposited to the Gene Expression Omnibus and are available under the accession number GSE158907.
-
NCBI Gene Expression OmnibusID GSE158907. Combinations of maternal-specific repressive epigenetic marks in the endosperm control seed dormancy.
-
NCBI Gene Expression OmnibusID GSE66585. Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm.
-
NCBI Gene Expression OmnibusID GSE89789. DNA demethylation is initiated in the central cells of Arabidopsis and rice.
-
NCBI Gene Expression OmnibusID GSE41212. Arabidopsis thaliana seed germination timecourse.
-
NCBI Gene Expression OmnibusID GSE38935. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes.
References
-
In situ expression of trehalose synthesizing genes, TPS1 and TPPB, in Arabidopsis thaliana using the GUS reporter genePlant Cell, Tissue and Organ Culture (PCTOC 98:311–319.https://doi.org/10.1007/s11240-009-9565-3
-
Genomic imprinting in plants-revisiting existing modelsGenes & Development 34:24–36.https://doi.org/10.1101/gad.332924.119
-
Interactions between abscisic acid and ethylene signaling cascadesThe Plant Cell 12:1103–1115.https://doi.org/10.1105/tpc.12.7.1103
-
Histone tales: Lysine methylation, a protagonist in arabidopsis developmentJournal of Experimental Botany 71:793–807.https://doi.org/10.1093/jxb/erz435
-
Ethylene, a key factor in the regulation of seed dormancyFrontiers in Plant Science 5:539.https://doi.org/10.3389/fpls.2014.00539
-
The pivotal role of ethylene in plant growthTrends in Plant Science 23:311–323.https://doi.org/10.1016/j.tplants.2018.01.003
-
Endosperm and Imprinting, Inextricably LinkedPlant Physiology 173:143–154.https://doi.org/10.1104/pp.16.01353
-
Genomic imprinting of xist by maternal h3k27me3Genes & Development 31:1927–1932.https://doi.org/10.1101/gad.304113.117
-
Establishment, maintenance, and biological roles of non-CG methylation in plantsEssays in Biochemistry 63:743–755.https://doi.org/10.1042/EBC20190032
-
Arabidopsis ref6 is a histone h3 lysine 27 demethylaseNature Genetics 43:715–719.https://doi.org/10.1038/ng.854
-
The Polycomb Group Protein Regulatory NetworkAnnual Review of Plant Biology 66:269–296.https://doi.org/10.1146/annurev-arplant-043014-115627
-
Seed germination and dormancy: The classic story, new puzzles, and evolutionJournal of Integrative Plant Biology 61:541–563.https://doi.org/10.1111/jipb.12762
-
DNA methylation repels targeting of Arabidopsis REF6Nature Communications 10:2063.https://doi.org/10.1038/s41467-019-10026-1
-
SoftwareR: A language and environment for statistical computing, version 3.3. 2R Foundation for Statistical Computing, Vienna, Austria.
-
Constitutive heterochromatin formation and transcription in mammalsEpigenetics & Chromatin 8:3.https://doi.org/10.1186/1756-8935-8-3
-
Non-CG methylation patterns shape the epigenetic landscape in ArabidopsisNature Structural & Molecular Biology 21:64–72.https://doi.org/10.1038/nsmb.2735
-
Agrigo v2.0: A GO analysis toolkit for the agricultural community, 2017 updateNucleic Acids Research 45:W122–W129.https://doi.org/10.1093/nar/gkx382
-
TopHat: discovering splice junctions with RNA-SeqBioinformatics 25:1105–1111.https://doi.org/10.1093/bioinformatics/btp120
-
H3K27 methylation: A promiscuous repressive chromatin markCurrent Opinion in Genetics & Development 43:31–37.https://doi.org/10.1016/j.gde.2016.11.001
-
Dynamics and function of DNA methylation in plantsNature Reviews. Molecular Cell Biology 19:489–506.https://doi.org/10.1038/s41580-018-0016-z
-
Rnai-dependent polycomb repression controls transposable elements in tetrahymenaGenes & Development 33:348–364.https://doi.org/10.1101/gad.320796.118
-
Friend or foe: Signaling mechanisms during double fertilization in flowering seed plantsCurrent Topics in Developmental Biology 131:453–496.https://doi.org/10.1016/bs.ctdb.2018.11.013
Article and author information
Author details
Juan Santos-González

Funding
Vetenskapsrådet (2017-04119)
- Claudia Köhler
Knut och Alice Wallenbergs Stiftelse (2018-0206)
- Claudia Köhler
Goran Gustafsson Foundation for Research in Natural Sciences and Medicine
- Claudia Köhler
Human Frontier Science Program (LT000162/2018-L)
- Hikaru Sato
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Cecilia Wärdig for technical support and Katarzyna Dziasek for helpful comments on the manuscript. This research was supported by grants from the Swedish Research Council VR (2017–04119, to CK), a grant from the Knut and Alice Wallenberg Foundation (2018–0206, to CK), a grant from the Göran Gustafsson Foundation for Research in Natural Sciences and Medicine (to CK), and a fellowship grant from the HFSP (to HS).
Copyright
© 2021, Sato et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,913
- views
-
- 386
- downloads
-
- 27
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 27
- citations for umbrella DOI https://doi.org/10.7554/eLife.64593
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Combinations of maternal-specific repressive epigenetic marks in the endosperm control seed dormancy
eLife 10:e64593.
https://doi.org/10.7554/eLife.64593




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}