Highly contiguous assemblies of 101 drosophilid genomes
- Department of Biology, Stanford University, United States
- Department of Genetics, University of North Carolina, United States
- Department of Pediatrics, Division of Genetic Medicine, University of Washington and Seattle Children’s Hospital, United States
- Department of Evolution and Ecology, University of California Davis, United States
- School of Natural Sciences, Bangor University, United Kingdom
- Biology Department, University of North Carolina, United States
- Department of Integrative Biology, University of California, Berkeley, United States
- Molecular and Cellular Biology Program, University of Washington, United States
- Department of Biological Sciences, Tokyo Metropolitan University, Japan
- Faculty of Biology, University of Belgrade, Serbia
- University of Belgrade, Institute for Biological Research "Siniša Stanković", National Institute of Republic of Serbia, Serbia
- School of Ecology and Environmental Science, Yunnan University, China
- Hokkaido University Museum, Hokkaido University, Japan
- Biological Laboratory, Sapporo College, Hokkaido University of Education, Japan
- Graduate School of Science and Engineering, Ehime University, Japan
- Department of Biology, University of Kentucky, United States
- Department of Biology, Indiana University, United States
- Neurobiology and Genetics, Theodor Boveri Institute, Biocentre, University of Würzburg, Germany
- Institute of Entomology, Biology Centre, Academy of Sciences of the Czech Republic, Czech Republic
- Department of Molecular and Integrative Physiology, University of Kansas Medical Center, Stowers Institute for Medical Research, United States
- School of Life Science, University of Nevada, United States
Figures
Figure 1 with 4 supplements
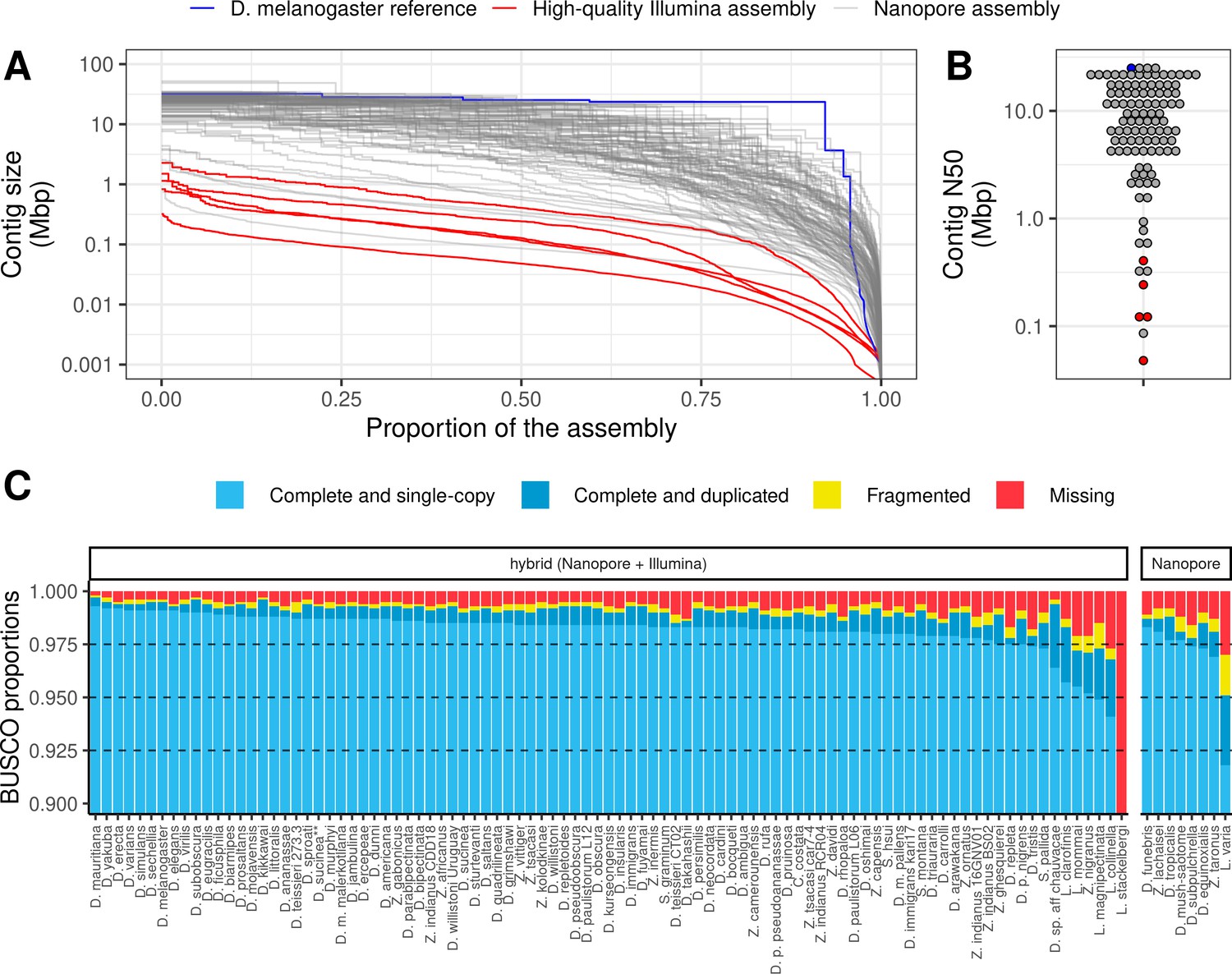
Nanopore-based assemblies are highly contiguous and complete.
(A,B) Assembly contiguity is compared to the D. melanogaster v6.22 reference genome (blue) as well as five recently published, highly contiguous Illumina assemblies (red lines, D. birchii, D. bocki, D. bunnanda, D. kanapiae, D. truncata; Bronski et al., 2020). (A) Nx curves, or the (y-axis) size of each contig when contigs are sorted in descending size order, in relation to the (x-axis) cumulative proportion of the genome assembly that is covered. (B) The distribution of contig N50, the size of the contig at which 50% of the assembly is covered. (C) Assembly completeness assessed by BUSCO v4.0.6 (Seppey et al., 2019). Note, D. equinoxialis was evaluated with BUSCO v4.1.4 due to an issue with v4.0.6. L. stackelbergi has >10% missing BUSCOs. Individual assembly summary statistics are provided in Supplementary file 2.
Figure 1—figure supplement 1
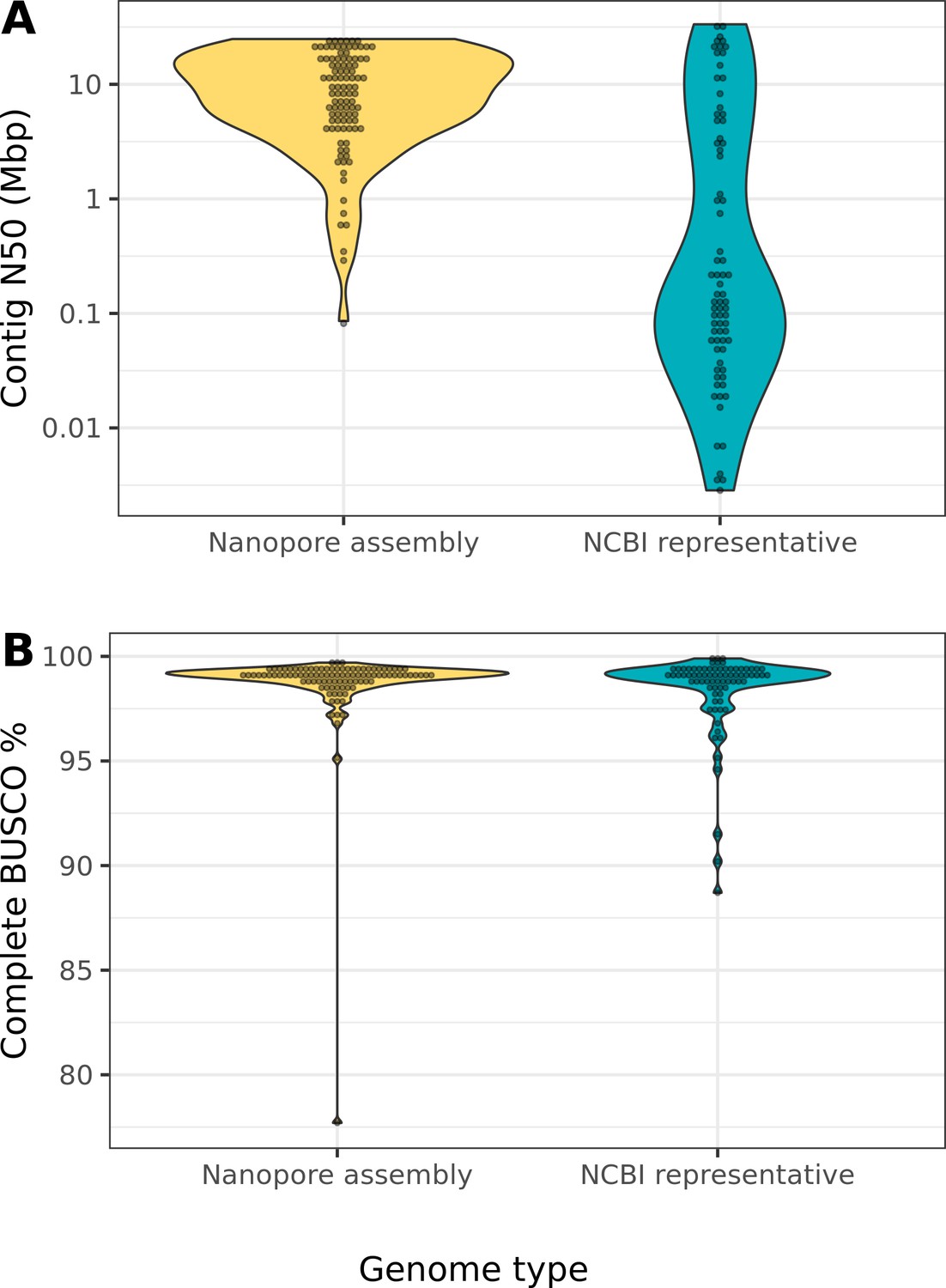
Nanopore-based assemblies compare favorably to representative genomes on NCBI.
(A) The contig N50 of the representative genome assembly for 75 different species on NCBI (right) is compared to the contig N50s of our assemblies (left). (B) The BUSCO (Simão et al., 2015) completeness (sum of complete single-copy and complete duplicated) of the NCBI assemblies of our assemblies is compared to the BUSCO completeness of our assemblies. The list of drosophilid genomes, contig N50s, and BUSCO completeness statistics were obtained from Hotaling et al., 2021. Note, BUSCO v4 was used for both genome assessments, but the OrthoDB v10 (Kriventseva et al., 2019) Diptera gene set was used to evaluate our assemblies while the OrthoDB v10 Insecta set was used to evaluate the NCBI assemblies.
Figure 1—figure supplement 2
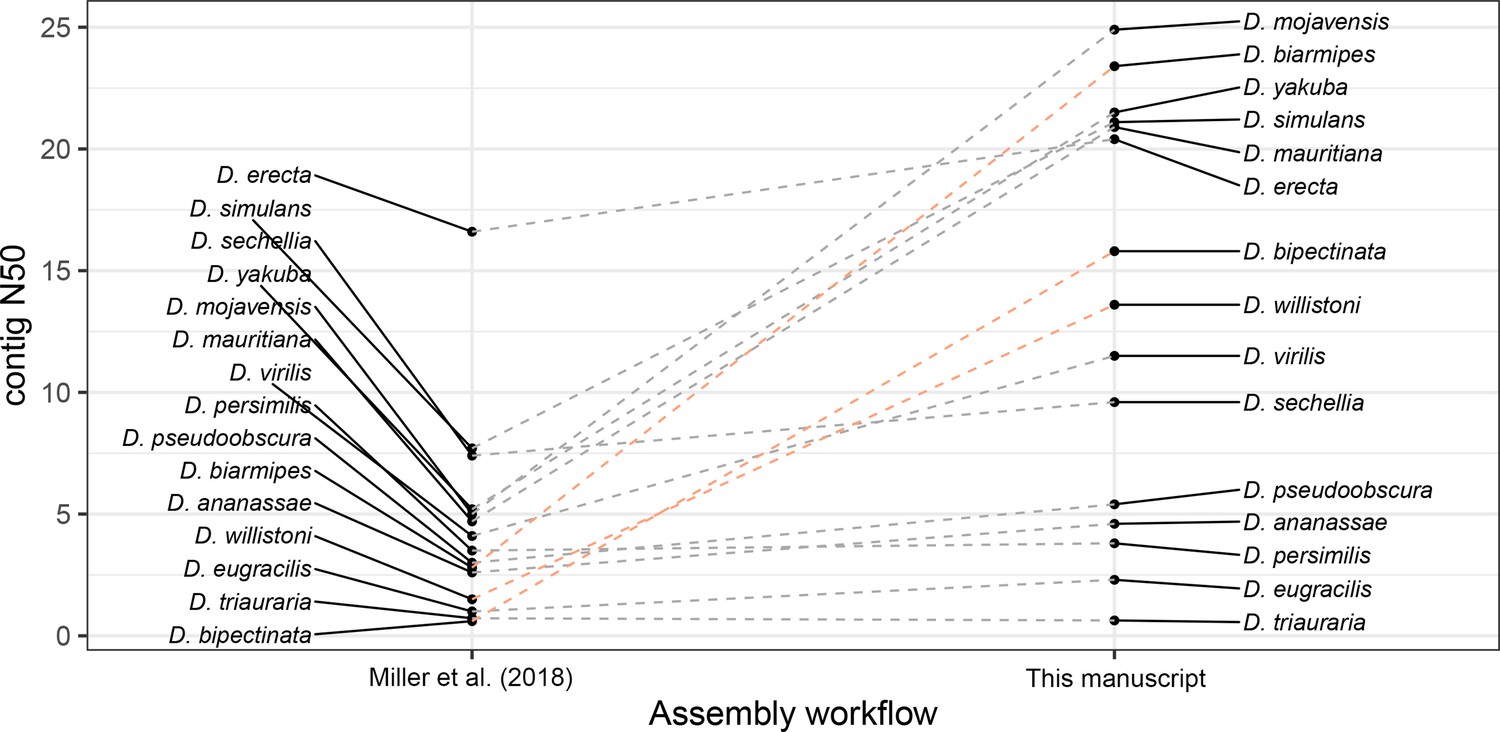
Large improvements in assembly contiguity from an updated assembly workflow.
Points on the left depict contig N50s from Miller et al., 2018. Points on the right depict contig N50s with our updated assembly workflow. In the updated workflow, ONT raw data are basecalled with Guppy in high-accuracy mode and assembled with Flye v2.6. For D. bipectinata, D. biarmipes, and D. willistoni (depicted with the light orange lines), new ONT sequencing optimized for longer reads and of a different strain than Miller et al., 2018 was performed. For all other species, the same raw data was used for both assembly workflows.
Figure 1—figure supplement 3
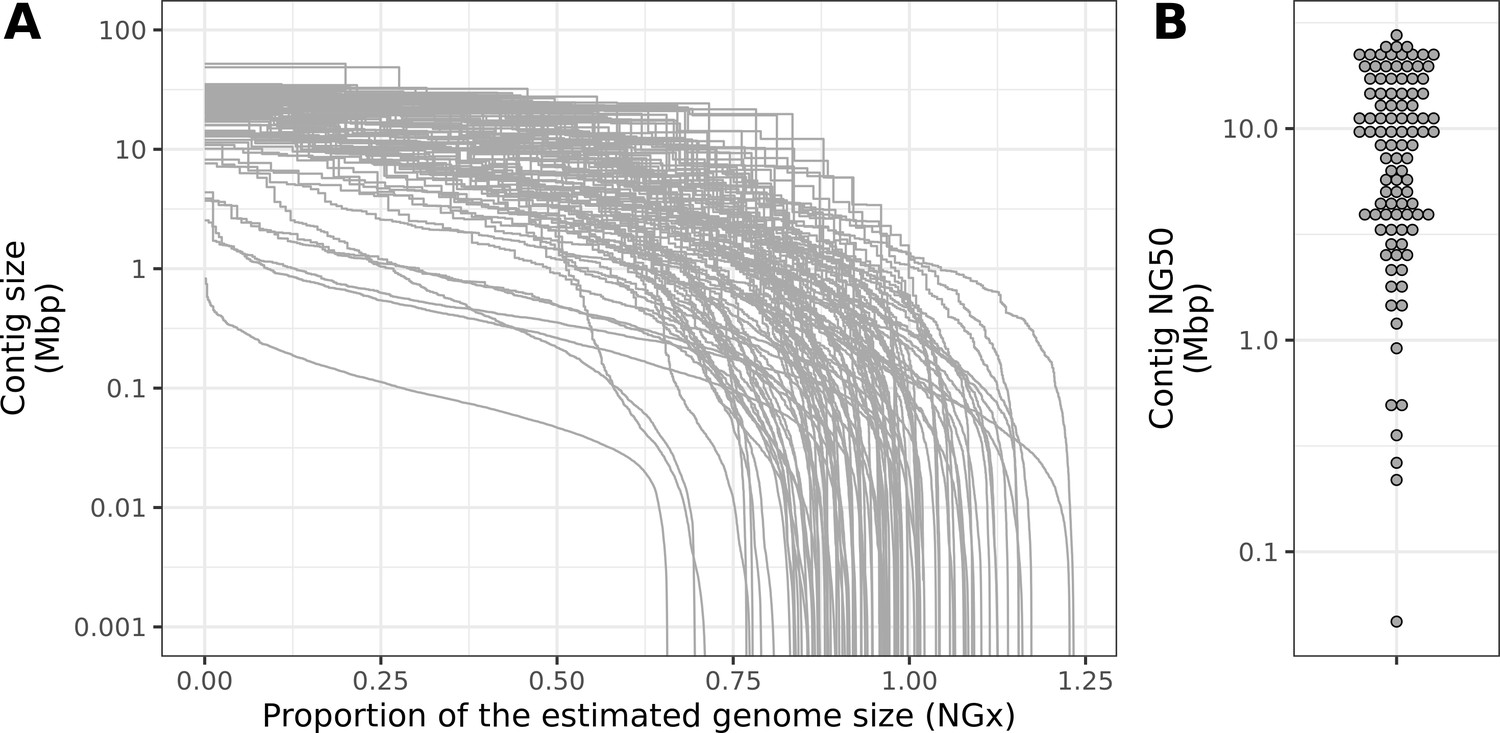
Contiguity metrics standardized by the estimated genome size.
(A) NGx curves, or the (y-axis) size of each contig when contigs are sorted in descending size order, in relation to the (x-axis) cumulative proportion of the estimated genome size that is covered. (B) The distribution of contig NG50, the size of the contig at which 50% of the estimated genome is accounted for.
Figure 1—figure supplement 4
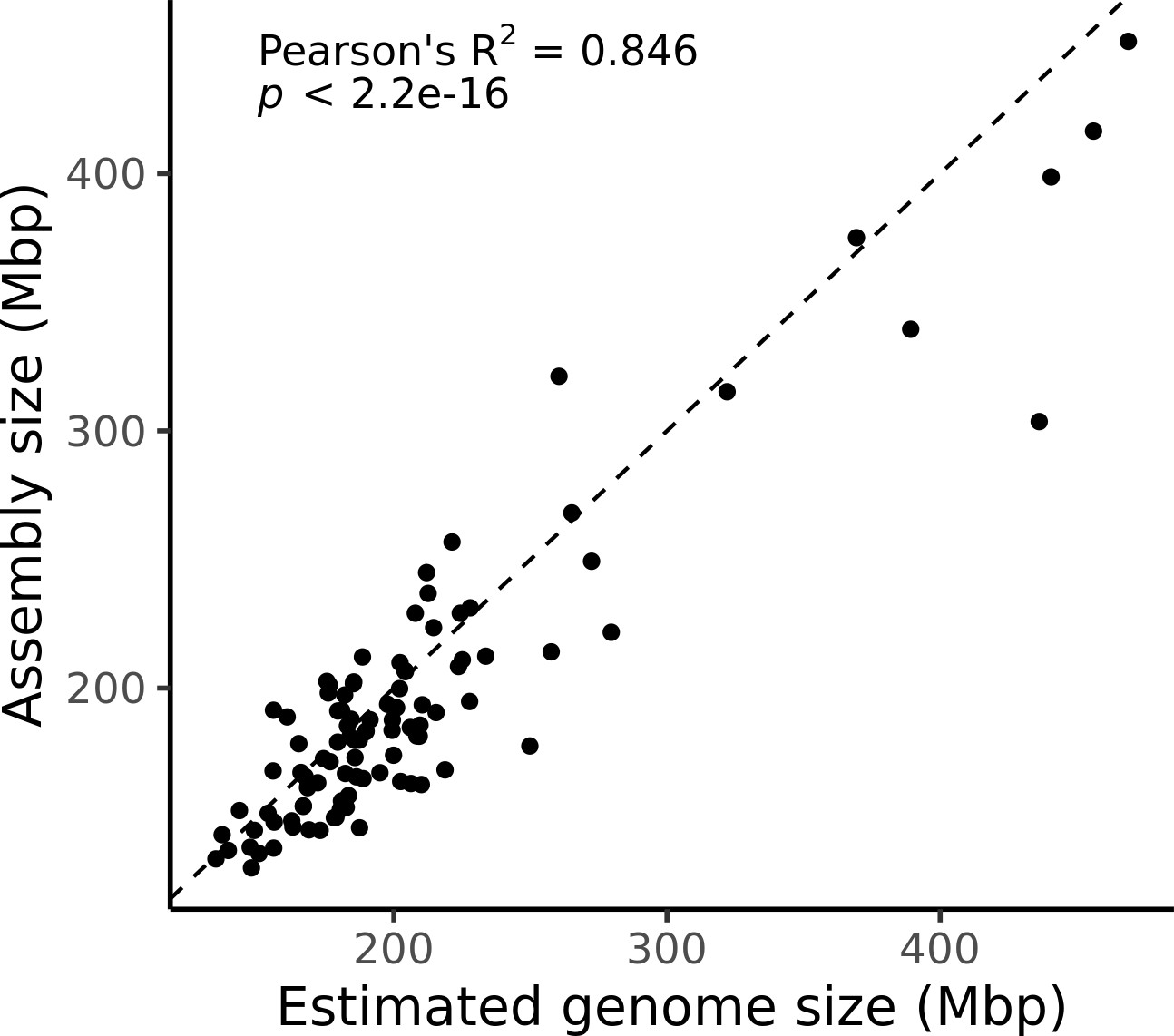
Estimated genome size is similar to assembly size.
The genome size estimated from read coverage over known single-copy genes in each assembly (x-axis) is compared to the length of each final assembly (y-axis). The dotted line is the 1:1 line.
Figure 2 with 1 supplement
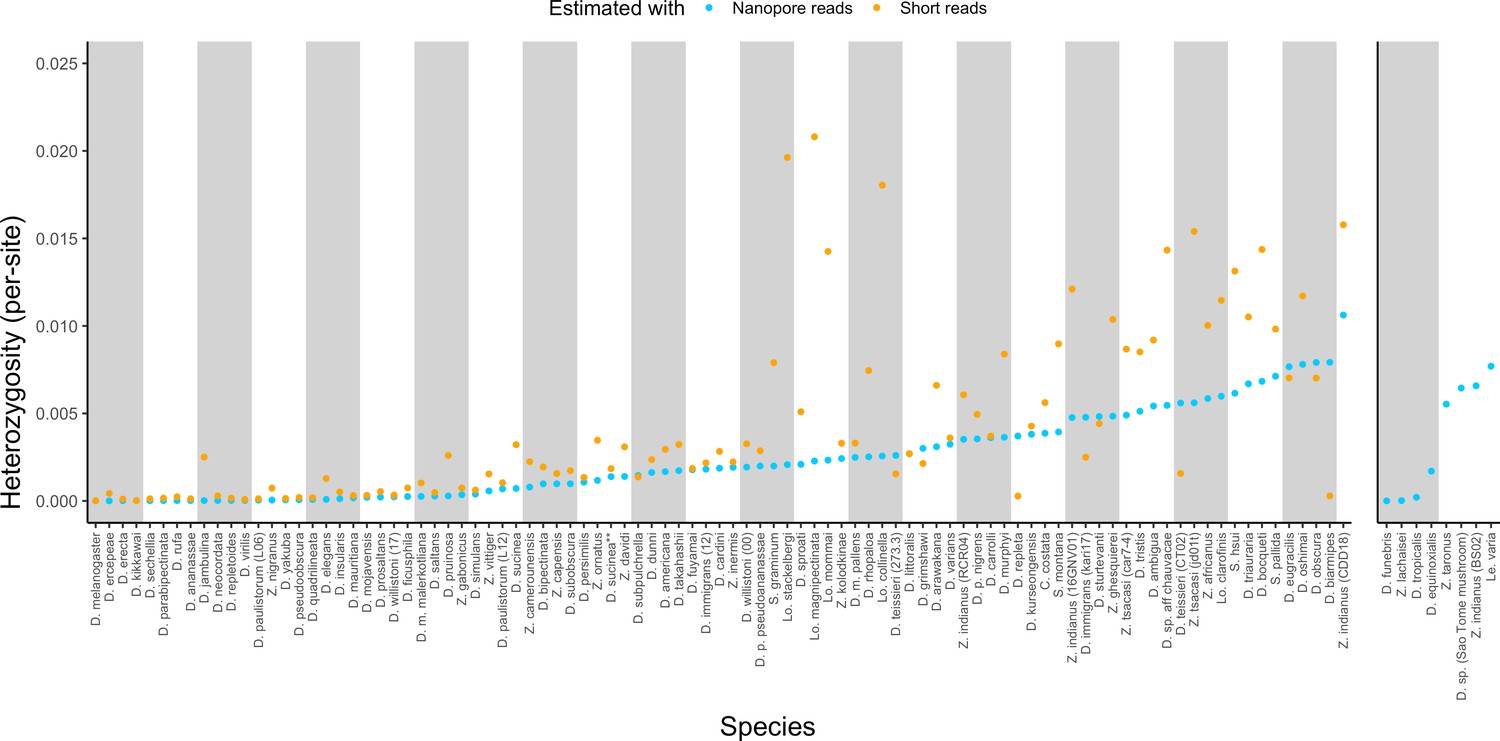
Estimated heterozygosity in the data used for genome assembly.
Per-site SNP heterozygosity (number of heterozygous SNPs/number of callable sites) is plotted for each of the 101 assembled lines. Blue dots represent heterozygosity estimates from Nanopore reads with PEPPER-Margin-DeepVariant (Shafin et al., 2021). Orange dots represent heterozygosity estimates from short reads with BCFtools (Li, 2011). The genomes on the right are for species that did not have available short-read data. Numerical values for these estimates are provided in Supplementary file 4.
Figure 2—figure supplement 1
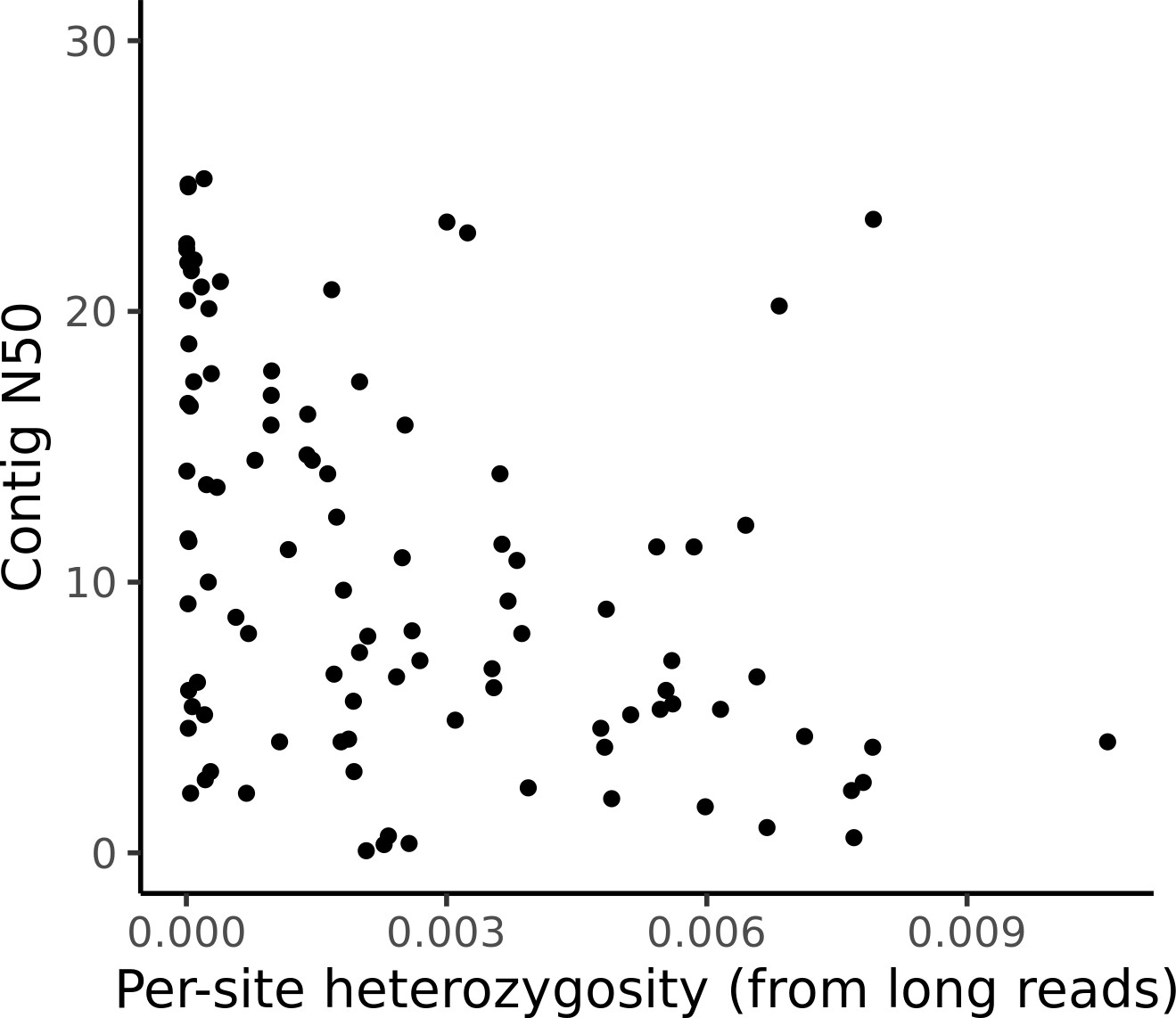
Assembly contiguity is not related to sample heterozygosity.
Per-site estimates of heterozygosity are plotted against the contig N50 for all assemblies. No significant correlation (Pearson’s correlation p=0.30) was observed.
Figure 3 with 2 supplements
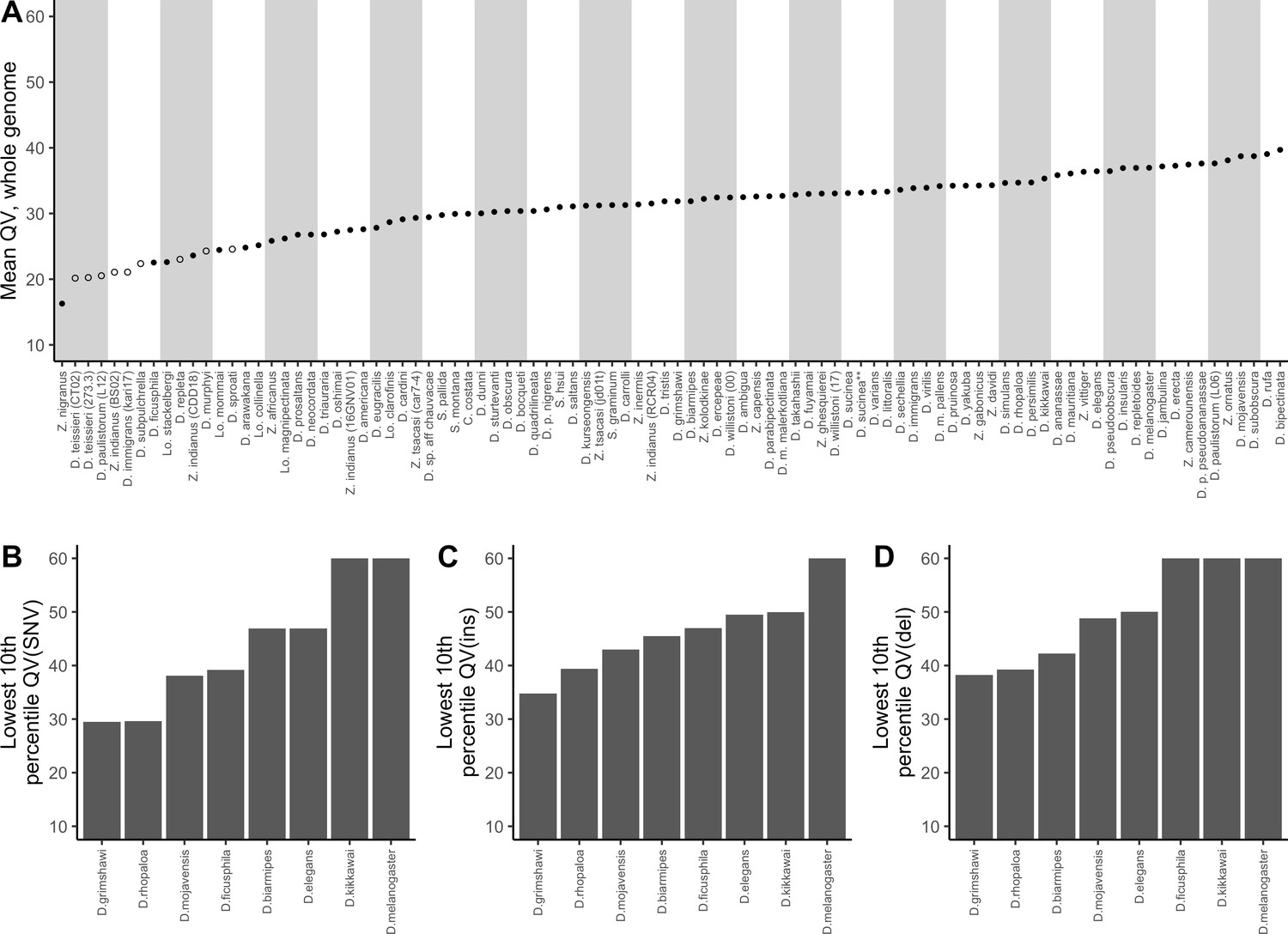
Nanopore-based Drosophila assemblies are accurate, particularly in coding regions.
(A) Genome-wide, Phred quality scores estimated with the reference-free, k-mer based approach implemented in Merqury (Rhie et al., 2020). Merqury requires a short-read dataset to perform the evaluation. Filled circles represent QV estimates with short-read data from the same strain used for Nanopore sequencing, and empty circles denote estimates using short-read data from a different strain than used for Nanopore sequencing. (B, C, D) Phred quality score cutoffs for the bottom 10th percentile of 100 kb genomic windows, as evaluated with a reference-based approach, in coding sequences only. Quality scores are capped at 60 for visualization purposes. At least 90% of 100 kb windows are this accurate. Only Nanopore assemblies with an NCBI RefSeq genome counterpart of the same strain were evaluated. Accuracy is shown for SNVs (B), insertions (C), and deletions (D) separately. Additional details on quality score estimates are provided in Figure 3—figure supplement 1 and Supplementary file 4.
Figure 3—figure supplement 1
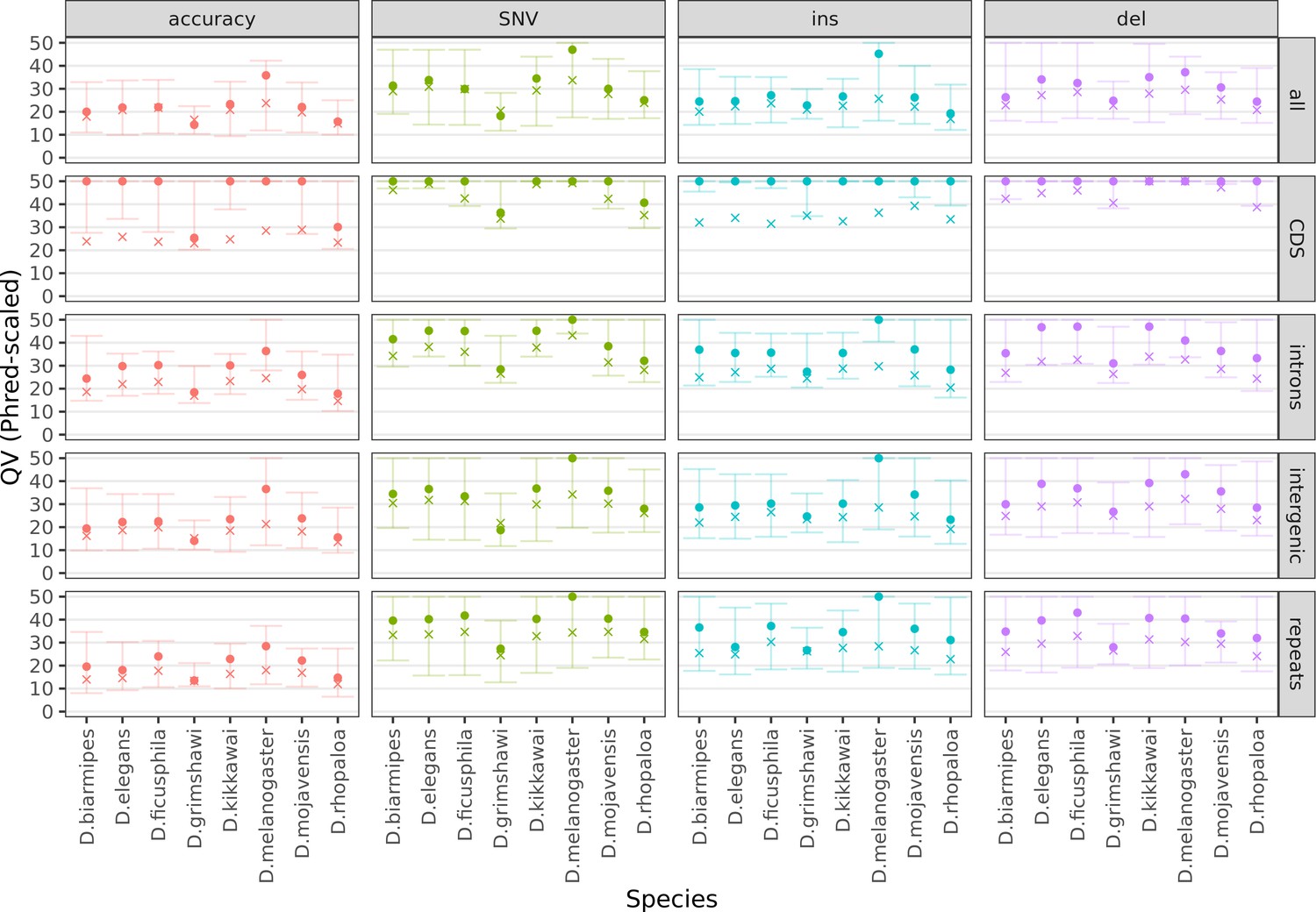
Variation in sequence accuracy within the genome assemblies.
Phred-scaled quality scores were computed by a reference-based comparison in non-overlapping 100 kb windows. All variants were considered together (accuracy), then SNVs, insertions, and deletions separately. All sequences in each window were considered together (all) then coding sequences, introns, intergenic regions, and repeats separately. All scores above QV50 were set to QV50 for visualization purposes. The cross denotes the mean score, weighted by the bases considered for each window. The dot and both whiskers denote the median, 10th percentile, and 90th percentile scores across all windows, respectively. Only Nanopore assemblies with an NCBI RefSeq genome counterpart of the same strain were evaluated.
Figure 3—figure supplement 2
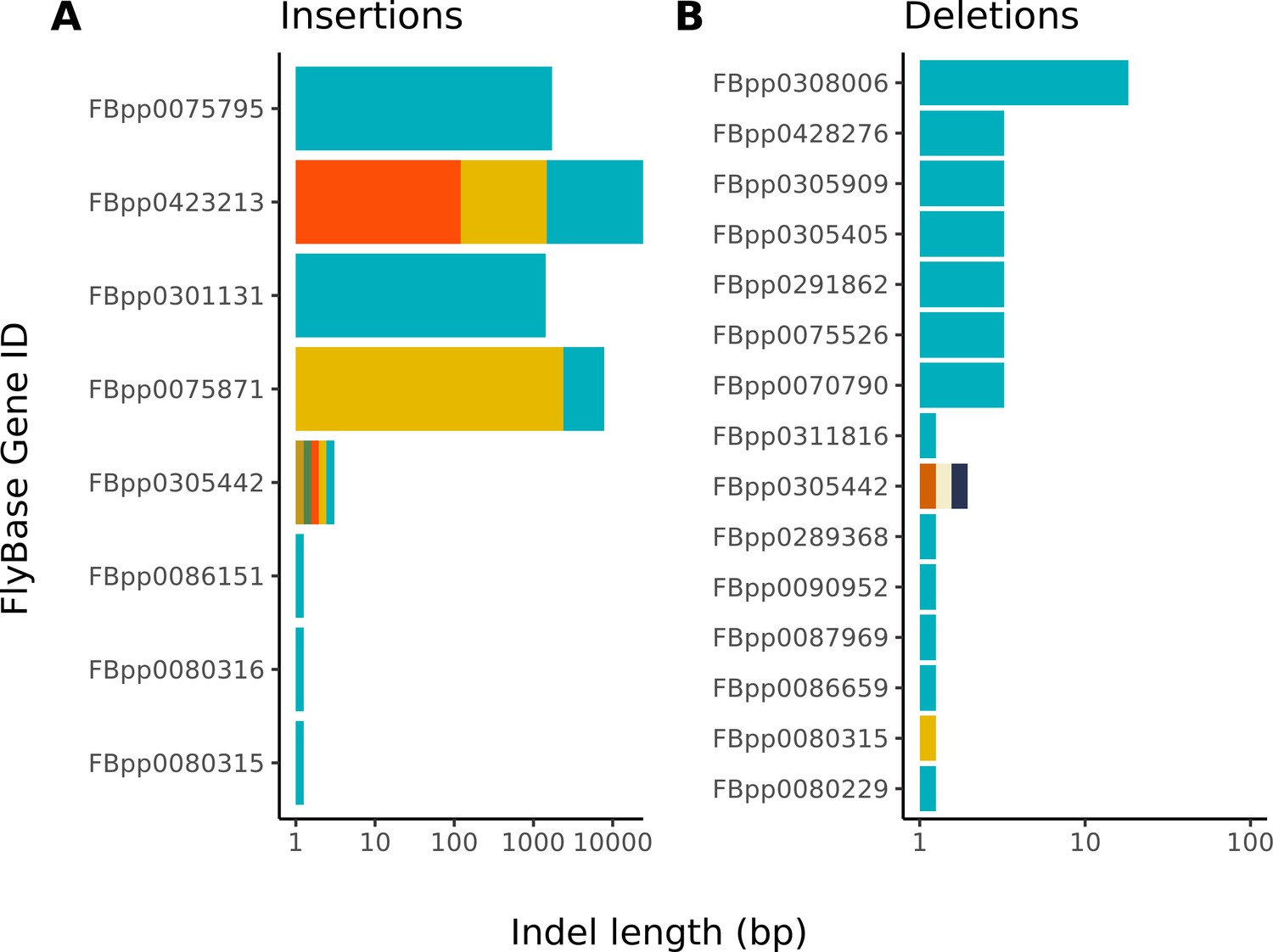
Large insertions account for nearly all differences between the Nanopore-based and reference D. melanogaster assembly.
The distribution of indel differences between our Nanopore-based assembly and the reference are shown. Each color represents a unique indel per FlyBase protein-coding gene. Note, the x-axis scale of insertions is much larger than that of deletions. Additional details on each indel are provided in Table S5.
Figure 4
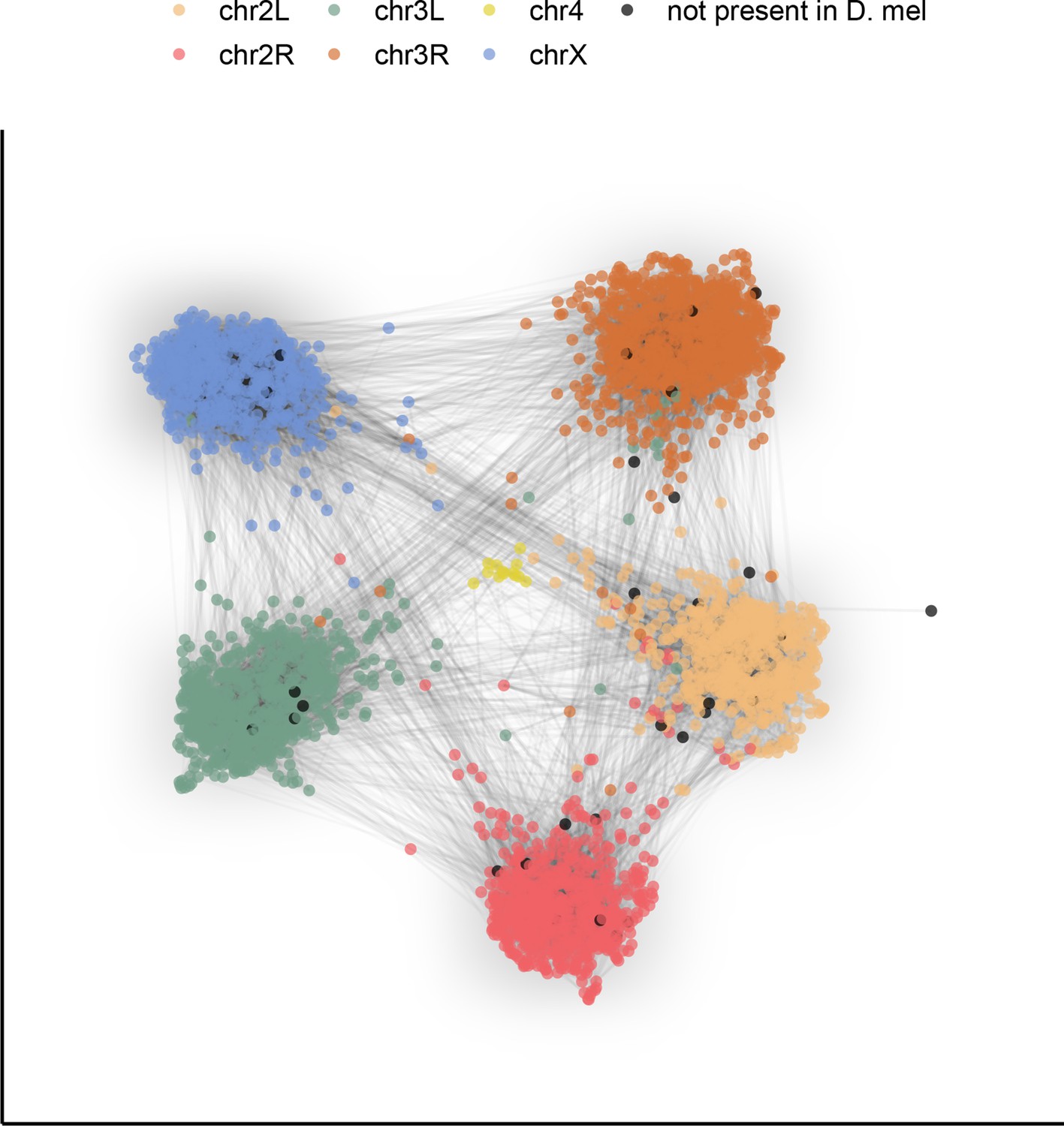
Gene content of Muller elements is conserved across drosophilids while gene order changes.
Each node in this graph represents an orthologous marker corresponding to single-copy orthologs annotated by BUSCOv4 (Seppey et al., 2019). An edge between two nodes represents the number of times that BUSCO pair is directly connected within an assembly. Each BUSCO is colored by the chromosome arm in D. melanogaster that it is found on. The ForceAtlas2 (Jacomy et al., 2014) graph layout algorithm was used for visualization.
Figure 5 with 2 supplements
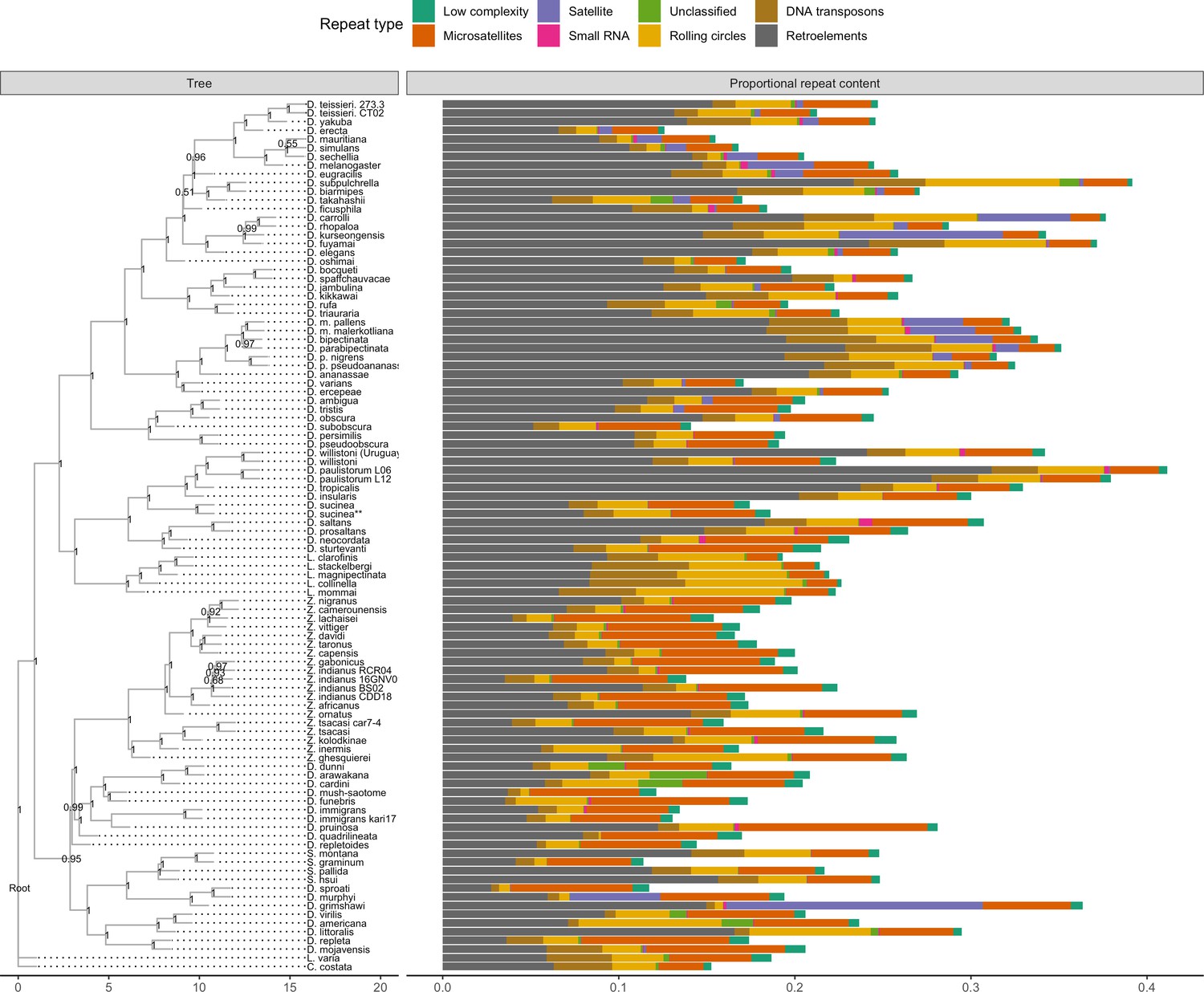
Repeat content varies greatly between drosophilid groups.
For each species, the proportion of each genome annotated with a particular repeat type is depicted. Species relationships were inferred by randomly selecting 250 of the set of BUSCOs (Seppey et al., 2019) that were complete and single-copy in all assemblies. RAxML-NG (Kozlov et al., 2019) was used to build gene trees for each BUSCO then ASTRAL-MP (Yin et al., 2019) to infer a species tree. Repeat annotation was performed with RepeatMasker (Smit et al., 2013) using the Dfam 3.1 (Hubley et al., 2016) and RepBase RepeatMasker edition (Bao et al., 2015) databases. ASTRAL local posterior probabilities are reported at each node.
Figure 5—figure supplement 1
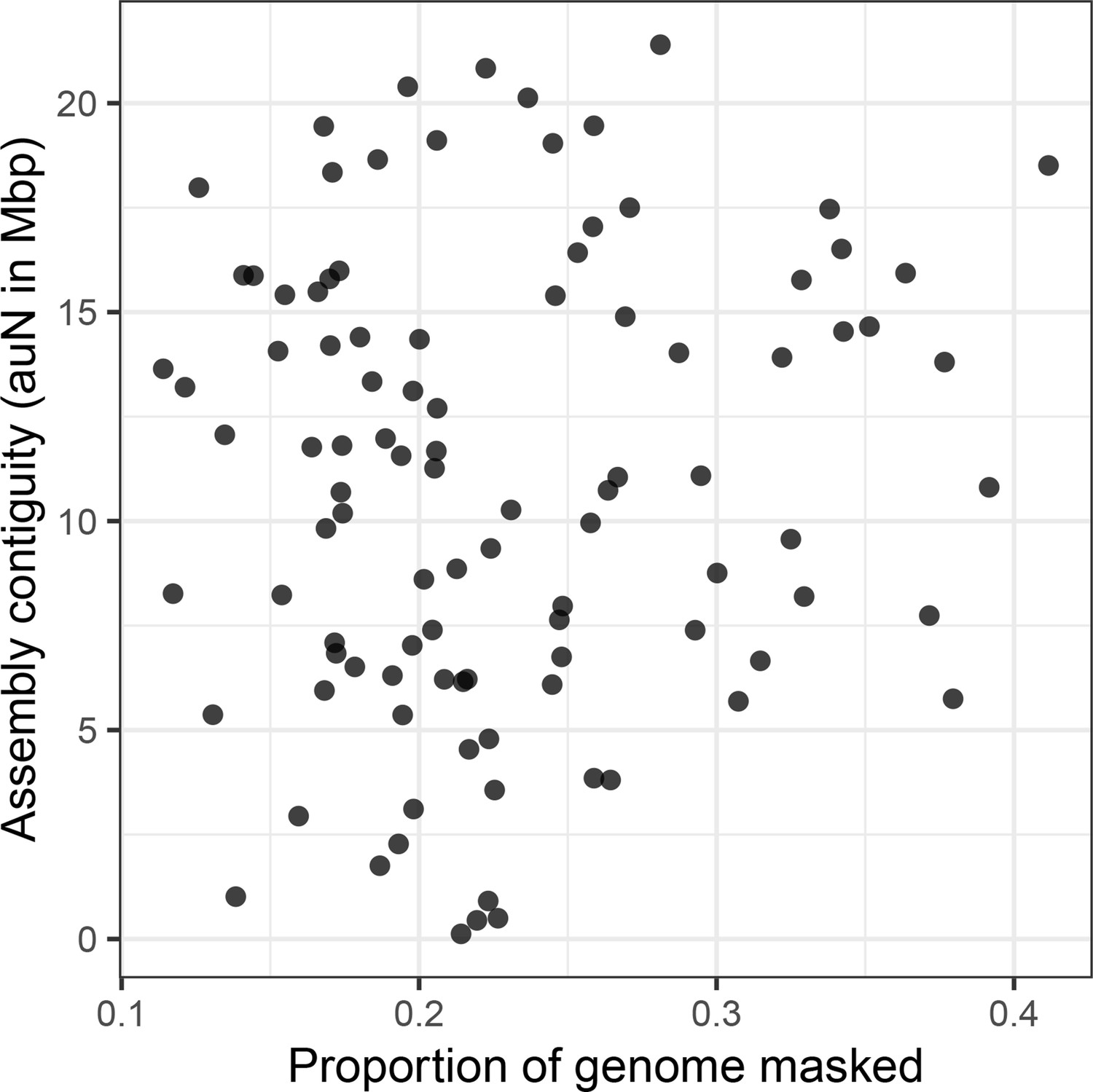
Assembly contiguity is not determined by repeat content.
There is no relationship (Spearman’s ρ=0.036, p=0.725) between repeat content (as annotated by RepeatMasker) in a genome and the contiguity of the resulting assembly.
Figure 5—figure supplement 2
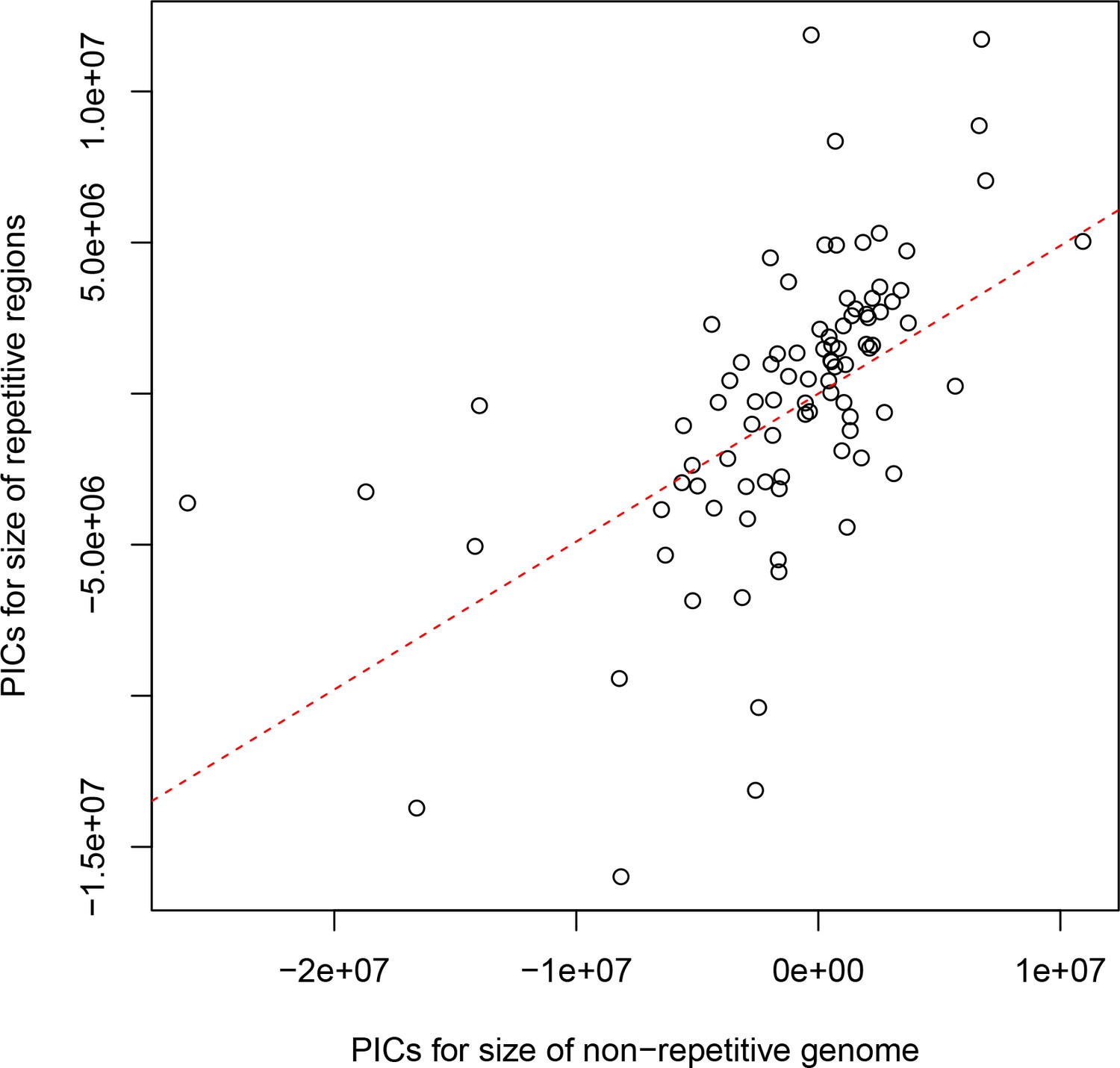
The non-repetitive and repetitive portions of the genome both contribute to genome size differences between drosophilids.
Phylogenetically independent contrasts (PICs) are shown for the number of bases in each genome not annotated as repetitive sequence (x-axis) and the number annotated as repeat by RepeatMasker (y-axis). The red dotted line is the best-fitting line through the origin. A positive relationship between the non-repetitive and repetitive portions of the genome is observed (Spearman’s ρ=0.679, p<2.2e-16), suggesting that both play a role in determining the genome size of drosophilids.
Figure 6
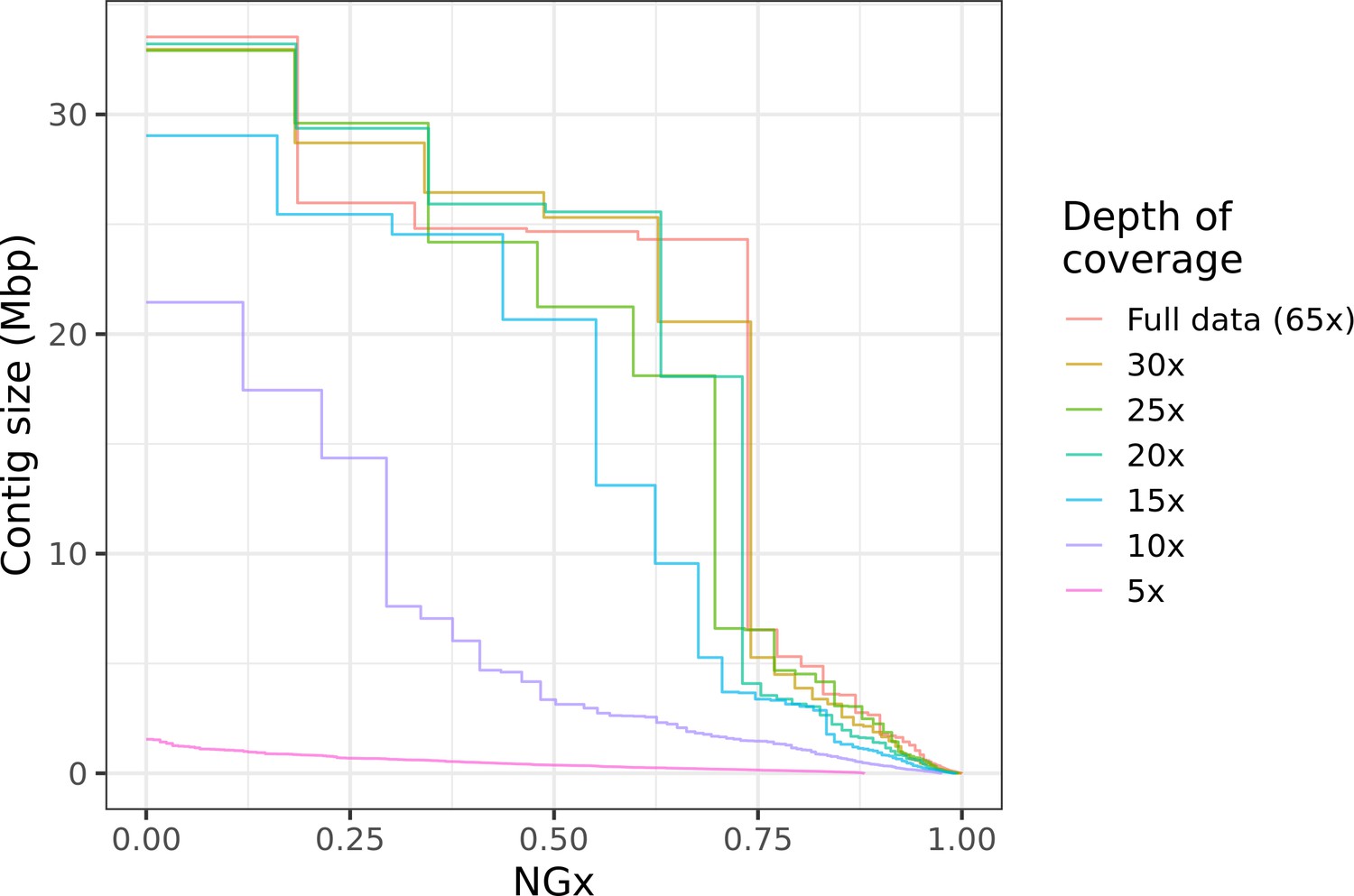
Highly contiguous assemblies can be obtained with lower coverage of ultra-long reads.
The NGx curve is shown for Drosophila jambulina assemblies at varying levels of coverage. The length of the assembly with the full data is assumed to be the genome size. Read sets used for each assembly were obtained by randomly downsampling the basecalled reads (read N50 ~27.5 kb) to varying (5× to 30×) depth of coverage. Proportionally, these read sets contain ~55% of total sequenced bases in reads longer than 25 kb, ~25% of bases in reads longer than 50 kb, and ~7% of bases in reads longer than 100 kb. Near chromosome scale assemblies (N50>20Mb) were achievable even at 15× to 20× depth with this read length distribution. This corresponds to approximately 8× to 10× depth in reads longer than 25 kb.
Figure 7
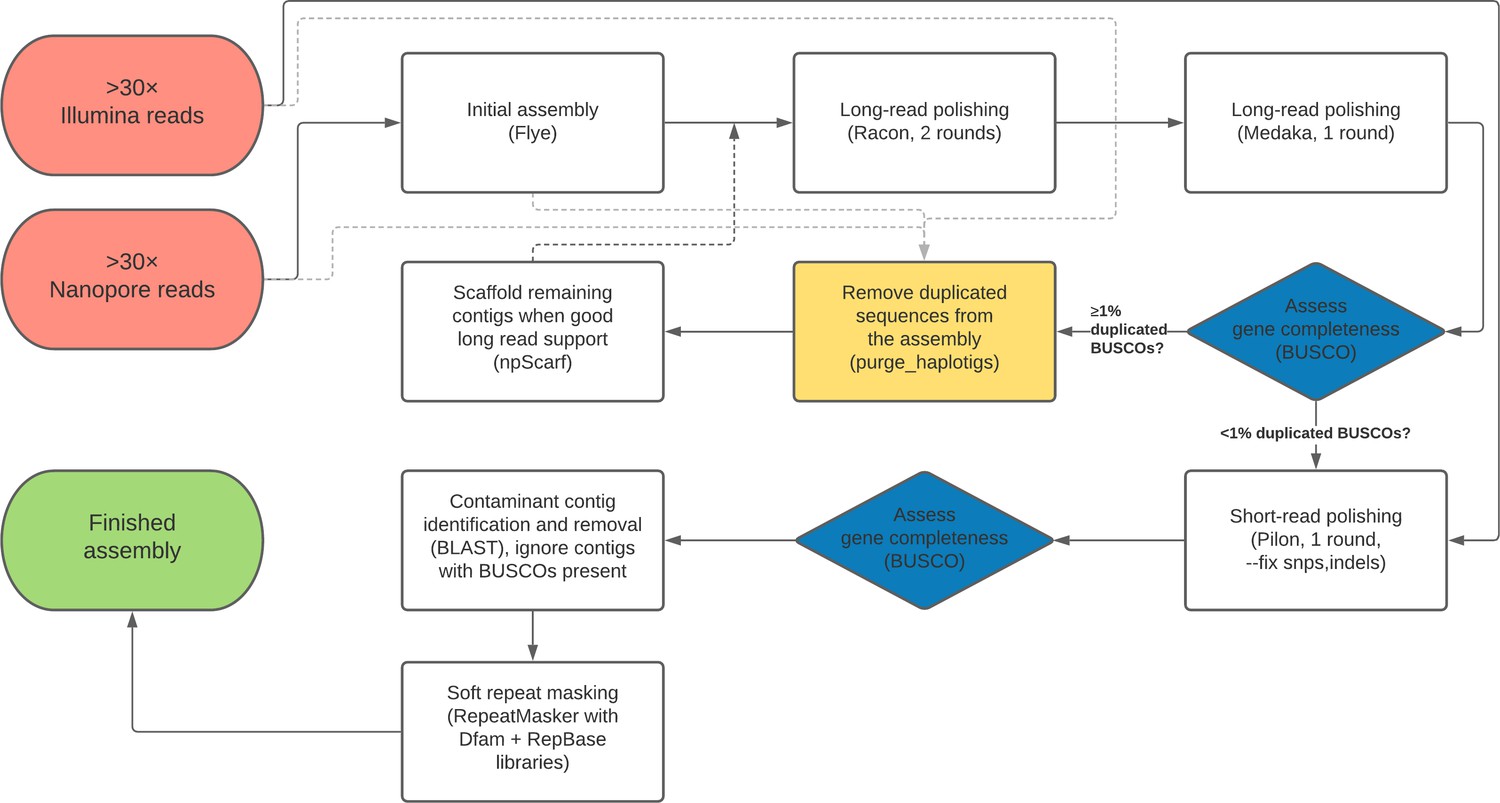
Flow chart depiction of the assembly pipeline.
Tables
Table 1
Species and strain information for all samples assembled for this work.
Note: Species group and subgroup information is taken from the NCBI Taxonomy Browser with slight modifications following O'Grady and DeSalle, 2018. Strain names along with corresponding NDSSC and Kyoto DGRC stock center numbers are provided to the best of our knowledge. See Supplementary file 1 and Supplementary file 6 for detailed information on samples and data. When multiple lines of a species are listed, * denotes the preferred assembly.
| Subgenus | Group | Subgroup | Species | Sex | Strain name | NDSSC | Kyoto DGRC/ Ehime | Additional notes |
|---|---|---|---|---|---|---|---|---|
| Sophophora | melanogaster | melanogaster | D. melanogaster | MF | ISO-1 GENOME | 14021-0231.36 | NA | BDGP reference strain |
| D. mauritiana | F | NA | 14021-0241.01 | NA | Miller et al., 2018 | |||
| D. simulans | F | NA | 14021-0251.006 | NA | Miller et al., 2018 | |||
| D. sechellia | F | NA | 14021-0248.01 | NA | Miller et al., 2018 | |||
| D. teissieri * | M | 273.3 | NA | NA | ||||
| D. teissieri | M | CT02 | NA | NA | ||||
| D. yakuba | F | NA | 14021-0261.01 | NA | Miller et al., 2018 | |||
| D. erecta | F | NA | 14021-0224.01 | NA | Miller et al., 2018 | |||
| eugracilis | D. eugracilis | F | NA | 14026-0451.02 | NA | Miller et al., 2018 | ||
| suzukii | D. subpulchrella | M | L1 | NA | NA | |||
| D. biarmipes | MF | 361.0 iso1 l-11 GENOME strain 1 | 14023-0361.10 | NA | modENCODE strain | |||
| takahashii | D. takahashii | F | IR98-3 E-12201 | NA | E-912201 | inbred derivative of Ehime stock IR98-3 | ||
| ficusphila | D. ficusphila | F | 631.0-iso1 l-10 GENOME | 14025-0441.05 | NA | modENCODE strain | ||
| rhopaloa | D. carrolli | MF | KB866 | NA | NA | |||
| D. rhopaloa | MF | BaVi067 GENOME | 14029-0021.01 | E-24701 | modENCODE strain | |||
| D. kurseongensis | F | SaPa58 | NA | NA | ||||
| D. fuyamai | F | KB-1217 | 14029-0011.01 | NA | ||||
| elegans | D. elegans | F | HK0461.03 GENOME | 14027-0461.03 | NA | modENCODE strain | ||
| suzukii | D. oshimai | M | MT-04 | NA | NA | |||
| montium | D. bocqueti | M | YAK3_mont-66 | NA | NA | |||
| D. sp aff chauvacae | M | mont_up-71 | NA | NA | ||||
| D. jambulina | MF | st-2 | 14028-0671.01 | NA | ||||
| D. kikkawai | F | 561.0-iso4 l-10 GENOME | 14028-0561.14 | NA | modENCODE strain | |||
| D. rufa | F | EH091 iso-C L_3 | NA | 914802 | inbred derivative of Ehime stock EH091 | |||
| D. triauraria | F | NA | 14028-0691.9 | NA | Miller et al., 2018; previously mis-identified as D. kikkawai | |||
| ananassae | D. malerkotliana pallens | F | palQ-isoG | NA | NA | |||
| D. malerkotliana malerkotliana | MF | mal0-isoC | 14024-0391.00 | NA | inbred derivative of strain 14024-0391.00 | |||
| D. bipectinata | MF | 4-4-2-3-1-1-1-1-1 BackUp | 14024-0381.04 | NA | Inbred derivative of NDSSC strain | |||
| D. parabipectinata | MF | par2-isoB | 14024-0401.02 | NA | inbred derivative of strain 14024-0401.02 (now extinct) | |||
| D. pseudoananassae pseudoananassae | F | Wau 125 | NA | NA | ||||
| D. pseudoananassae nigrens | F | VT04-31 | NA | NA | ||||
| D. ananassae | F | 14024-0371.13 | NA | NA | Miller et al., 2018 | |||
| D. varians | MF | CKM15-L1 | NA | NA | ||||
| D. ercepeace | MF | 164-14 | 14024-0432.00 | NA | ||||
| obscura | obscura | D. ambigua | M | R42 | NA | NA | isofemale strain from the wild | |
| D. tristis | M | D2 | NA | NA | isofemale strain from the wild | |||
| D. obscura | M | BZ-5 | NA | NA | isofemale strain from the wild | |||
| D. subobscura | M | Küsnacht | NA | NA | standard laboratory strain | |||
| pseudoobscura | D. persimilis | F | NA | 14011-0111.01 | NA | Miller et al., 2018 | ||
| D. pseudoobscura | F | NA | 14011-0121.94 | NA | Miller et al., 2018 | |||
| willistoni | willistoni | D. willistoni (Uruguay) * | M | L-G3 | 14030-0811.17 | NA | ||
| D. willistoni | F | NA | 14030-0811.00 | NA | Miller et al., 2018 | |||
| D. paulistorum L06 * | M | (Heed) H66.1C | 14030-0771.06 | NA | ||||
| D. paulistorum L12 | M | L12 | 14030-0771.12 | NA | ||||
| D. tropicalis | M | (Heed) H65.2 | 14030-0801.00 | NA | ||||
| D. insularis | M | jp01i | NA | NA | isofemale line from J. Powell | |||
| bocainensis | D. sucinea | M | 49.15 | 14030-0791.01 | NA | |||
| D. sucinea** | M | H176.10 | 14030-0761.01 | NA | NDSSC strain is misidentified as D. nebulosa | |||
| saltans | saltans | D. saltans | M | (Heed) H180.40 | 14045-0911.00 | NA | ||
| D. prosaltans | M | (Heed) H29.6 | 14045-0901.02 | NA | ||||
| neocordata | D. neocordata | M | 2536.7 | 14041-0831.00 | NA | |||
| sturtevanti | D. sturtevanti | F | H191.23 | 14043-0871.01 | NA | |||
| Lordiphosa | miki | L. clarofinis | MF | Guizhou062018LC | NA | NA | Line inbred for 2 generations in the lab before sequencing | |
| L. stackelbergi | MF | UCILTSSapporo052019LS | NA | NA | Pool of 50 wild-caught flies | |||
| L. magnipectinata | MF | UCKTSapporo052019LM | NA | NA | Pool of 50 wild-caught flies | |||
| fenestrarum | L. collinella | MF | UCKTSapporo052019LC | NA | NA | Pool of 30 wild-caught flies | ||
| L. mommai | MF | MMSapporo052014LM | NA | NA | ||||
| Drosophila | Zaprionus | vittiger | Z. nigranus | M | st01n | NA | NA | line derived from wild collection |
| Z. camerounensis | M | jd01cam | NA | NA | isofemale line from J. David | |||
| Z. lachaisei | M | jd01l | NA | NA | line derived from wild collection | |||
| Z. vittiger | M | jd01v | NA | NA | isofemale line from J. David | |||
| Z. davidi | M | jd01d | NA | NA | isofemale line from J. David | |||
| Z. taronus | M | st01t | NA | NA | line derived from wild collection | |||
| Z. capensis | M | jd01cap | NA | NA | isofemale line from J. David | |||
| Z. gabonicus | M | jd01gab | NA | NA | isofemale line from J. David | |||
| Z. indianus RCR04 | M | RCR04 | NA | NA | ||||
| Z. indianus 16GNV01 | M | 16GNV01 | NA | NA | ||||
| Z. indianus BS02 * | M | BS02 | NA | NA | ||||
| Z. indianus CDD18 | M | CDD18 | NA | NA | ||||
| Z. africanus | M | BS06 | NA | NA | ||||
| Z ornatus | M | jd01o | NA | NA | isofemale line from J. David | |||
| tuberculatus | Z. tsacasi | M | car7-4 | NA | NA | |||
| Z. tsacasi * | M | jd01t | NA | NA | isofemale line from J. David | |||
| inermis | Z. kolodkinae | M | jd01k | NA | NA | isofemale line from J. David | ||
| Z. inermis | M | 18BSZ10 | NA | NA | ||||
| Z. ghesquierei | M | jd01ghe | NA | NA | isofemale line from J. David | |||
| cardini | dunni | D. dunni | M | H254.21 | 15182-2291.00 | NA | ||
| D. arawakana | M | MONHI050227(B)-104 | 15182-2261.03 | NA | ||||
| cardini | D. cardini | M | NA | 15181-2181.03 | 917701 | |||
| funebris | funebris? | undescribed (Sao Tome mushroom) | M | st01m | NA | NA | undescribed species collected on mushroom, Sao Tome | |
| funebris | D. funebris | M | fst01 | NA | NA | line derived from wild collection | ||
| immigrans | immigrans | D. immigrans * | F | FK05-19 | 15111.1731.12 | NA | ||
| D. immigrans kari17 | M | kari17 | NA | NA | ||||
| (incertae sedis) | D. pruinosa | M | iso-A1 l-9 | NA | NA | |||
| quadrilineata | D. quadrilineata | M | quad-TMU | NA | 914402 | |||
| tumiditarsus | D. repletoides | M | ISZ-isoB I-10 | NA | NA | |||
| Scaptomyza | Scaptomyza | S. montana | MF | iso-CA-L1 | NA | NA | ||
| S. graminum | F | TMU-2019 | NA | NA | 30 wild-caught females | |||
| Parascaptomyza | S. pallida | MF | iso-CA-L1 | NA | NA | |||
| Hemiscaptomyza | S. hsui | MF | iso-CA-L1 | NA | NA | |||
| HawaiianDrosophila | orphnopeza | D. sproati | MF | DKPTOMS02 | NA | NA | Pool of wild-caught flies | |
| D. murphyi | MF | DKPHETFM01 | NA | NA | Flies from recently established but not inbred lab line | |||
| grimshawi | D. grimshawi | F | NA | 15287-2541.00 | NA | Same line as caf1 genome | ||
| virilis | virilis | D. virilis | F | NA | 15010-1051.87 | NA | Miller et al., 2018 | |
| D. americana | M | 3367.1 | 15010-0951.00 | NA | Also called Anderson strain | |||
| D. littoralis | M | Kilpisjärvi 1 | NA | NA | Originally misidentified as D. ezoana (Lankinen 1986, J Comp Physiol A 159: 123-142) | |||
| repleta | repleta | D. repleta | M | kari30 | NA | NA | ||
| mulleri | D. mojavensis | F | 15081-1352.22 | NA | NA | Miller et al., 2018 | ||
| genus: Leucophenga | L. varia | M | nc01v | NA | NA | Sequenced single wild-caught fly, no amplification | ||
| genus: Chymomyza | C. costata | M | Sapporo | NA | NA | |||
-
* denotes the genome of best quality when multiple assemblies are available for a species.
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Drosophila spp. and relatives) | See Table 1 and Supplementary files 1–6 for sample information, strain designations, stock center line identifiers (when applicable), biomaterial provider, and NCBI accession numbers. | |||
| Commercial assay or kit | Blood and Cell Culture DNA Mini Kit | Qiagen | cat # 13323 | |
| Commercial assay or kit | Ligation Sequencing Kit | Oxford Nanopore | SQK-LSK109 | Superseded by SQK-LSK110 |
| Commercial assay or kit | Flow cell wash kit | Oxford Nanopore | EXP-WSH003 | Superseded by EXP-WSH004 |
| Commercial assay or kit | Short Read Eliminator kit | Circulomics | SKU # SS-100-101-01 | |
| Commercial assay or kit | Companion Module for ONT Ligation Sequencing | NEBNext | cat # E7180S | |
| Commercial assay or kit | Nextera XT DNA Library Preparation Kit | Illumina | cat # FC-131–1002 | Superseded by version 2 |
| Commercial assay or kit | Kapa HyperPrep Kit | Roche | cat # KK8502 | |
| Software, algorithm | Flye | Kolmogorov et al., 2019 | 2.6 | |
| Software, algorithm | Canu | Koren et al., 2017 | 1.8 | |
| Software, algorithm | Miniasm | Li, 2016 | 0.3 | |
| Software, algorithm | Guppy | Oxford Nanopore | 3.2.4 | |
| Software, algorithm | Medaka | Oxford Nanopore | 0.9.1 | |
| Software, algorithm | Minimap2 | Li, 2016 | 2.17 | |
| Software, algorithm | SAMtools | Li et al., 2009 | 1.12 | |
| Software, algorithm | Racon | Vaser et al., 2017 | 1.4.3 | |
| Software, algorithm | BUSCO | Simão et al., 2015 | 3.0.2 | |
| Software, algorithm | BUSCO | Seppey et al., 2019 | 4.0.6 | |
| Software, algorithm | Purge_haplotigs | Roach et al., 2018 | 1.1.1 | |
| Software, algorithm | npScarf | Cao et al., 2017 | 1.9-2b | |
| Software, algorithm | Pilon | Walker et al., 2014 | 1.23 | |
| Software, algorithm | BLAST | Altschul et al., 1990 | 2.10.0 | |
| Software, algorithm | SPAdes | Bankevich et al., 2012 | 3.11.1 | |
| Software, algorithm | FMLRC | Wang et al., 2018 | 1.0.0 | |
| Software, algorithm | LINKS | Warren et al., 2015 | 1.8.7 | |
| Software, algorithm | RepeatMasker | Smit et al., 2013 | 4.1.0 | |
| Software, algorithm | Dfam repeat databse | Hubley et al., 2016 | 3.1 | Library for RepeatMasker |
| Software, algorithm | RepBase RepeatMasker edition | Bao et al., 2015 | 20181026 | Library for RepeatMasker |
| Software, algorithm | cross_match | Green, 2009 | 1.090518 | |
| Software, algorithm | Tandem Repeat Finder | Benson, 1999 | 4.0.9 | |
| Software, algorithm | Bioawk | Li, 2017 | 1.0 | |
| Software, algorithm | GenomeScope | Vurture et al., 2017 | 1.0.0 | |
| Software, algorithm | Jellyfish | Marçais and Kingsford, 2011 | 2.2.3 | |
| Software, algorithm | Sambamba | Tarasov et al., 2015 | 0.8.0 | |
| Software, algorithm | PEPPER-Margin-Deepvariant | Shafin et al., 2021 | 0.4 | |
| Software, algorithm | BCFtools | Li, 2011 | 1.12 | |
| Software, algorithm | Merqury | Rhie et al., 2020 | 1.3 | |
| Software, algorithm | Pomoxis | Oxford Nanopore | 0.3.7 | |
| Software, algorithm | bedtools | Quinlan and Hall, 2010 | 2.30.0 | |
| Software, algorithm | HALtools | Hickey et al., 2013 | 2.1 | |
| Software, algorithm | Integrative Genomics Viewer | Robinson et al., 2011b | 2.9.4 | |
| Software, algorithm | MAFFT | Katoh and Standley, 2013 | 7.453 | |
| Software, algorithm | RAxML-NG | Kozlov et al., 2019 | 0.9.0 | |
| Software, algorithm | ASTRAL-MP | Yin et al., 2019 | 5.14.7 | |
| Software, algorithm | ForceAtlas2 | Jacomy et al., 2014 | Implemented in R package https://github.com/analyxcompany/ForceAtlas2 | |
| Software, algorithm | ape | Paradis and Schliep, 2019 | 5.4.1 | R package |
| Software, algorithm | Docker | docker.com | ||
| Software, algorithm | Singularity | sylabs.io | ||
Additional files
-
Supplementary file 1
Detailed information on both long-read and short-read data used for this project, including accession numbers if publicly available data were used for assembly.
- https://cdn.elifesciences.org/articles/66405/elife-66405-supp1-v3.xlsx
-
Supplementary file 2
Assembly summary statistics and genome size estimates.
- https://cdn.elifesciences.org/articles/66405/elife-66405-supp2-v3.xlsx
-
Supplementary file 3
Counts of SNPs, indels, and per-site heterozygosity estimated from both long reads and short reads.
- https://cdn.elifesciences.org/articles/66405/elife-66405-supp3-v3.xlsx
-
Supplementary file 4
Consensus quality scores estimated with reference-free and reference-based methods.
- https://cdn.elifesciences.org/articles/66405/elife-66405-supp4-v3.xlsx
-
Supplementary file 5
Characterization of all coding sequence indel differences between Nanopore and Release six reference D. melanogaster assemblies.
- https://cdn.elifesciences.org/articles/66405/elife-66405-supp5-v3.xlsx
-
Supplementary file 6
Detailed sample information.
- https://cdn.elifesciences.org/articles/66405/elife-66405-supp6-v3.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/66405/elife-66405-transrepform-v3.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Highly contiguous assemblies of 101 drosophilid genomes
eLife 10:e66405.
https://doi.org/10.7554/eLife.66405




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}