De novo macrocyclic peptides dissect energy coupling of a heterodimeric ABC transporter by multimode allosteric inhibition
- Institute of Biochemistry, Biocenter, Goethe University Frankfurt, Germany
- Department of Chemistry, Graduate School of Science, The University of Tokyo, Japan
- Institute of Physical and Theoretical Chemistry, Goethe University Frankfurt, Germany
Figures
Figure 1 with 1 supplement
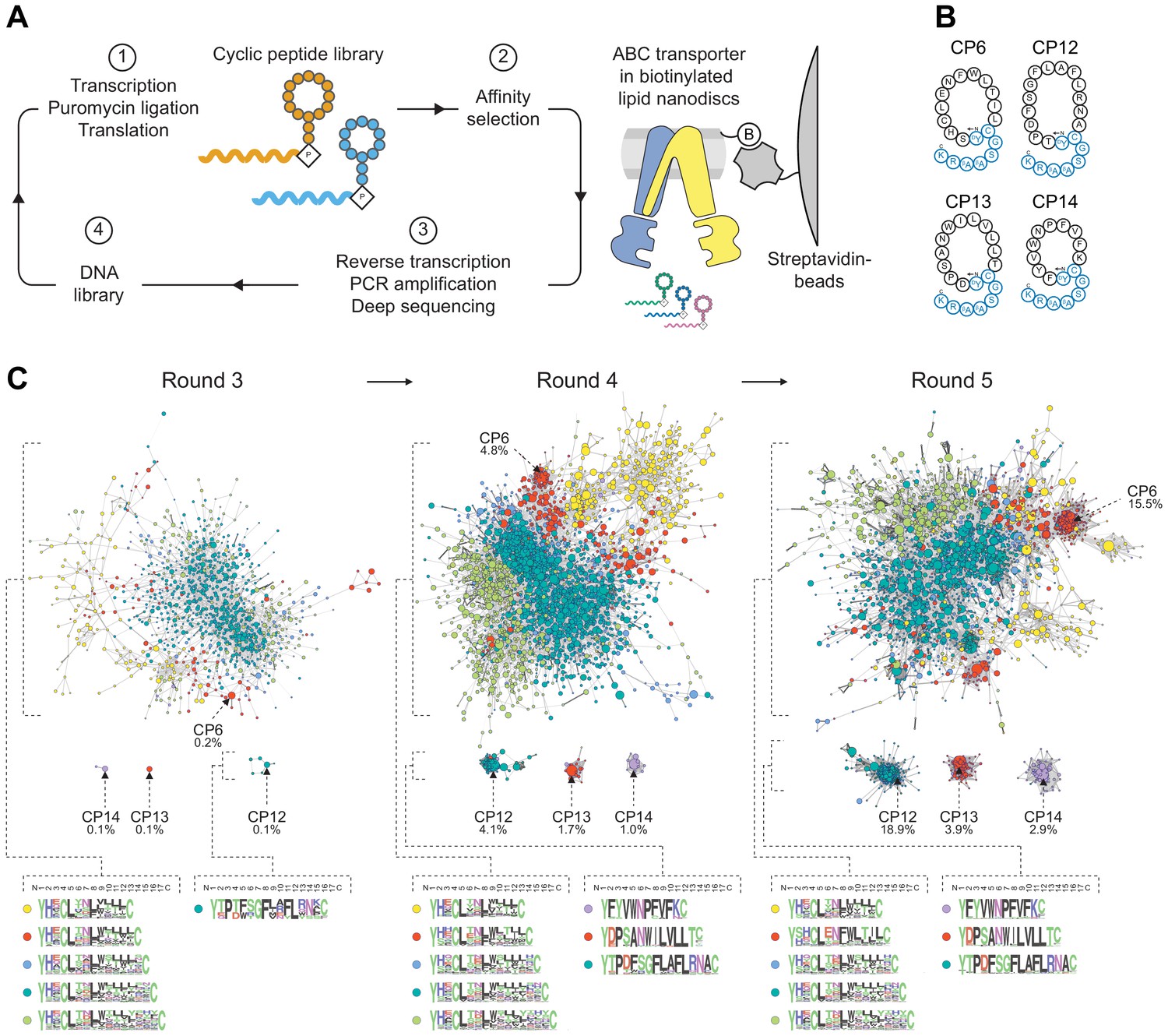
Selection of macrocyclic peptides (CPs) by random nonstandard peptide integrated discovery (RaPID).
(A) RaPID selection of CPs. Starting from a DNA library, macrocyclic peptides were generated through transcription and ribosomal translation using the Flexible In-vitro Translation (FIT) system. Cognate mRNA was covalently attached to the nascent peptide through incorporation of mRNA-linked puromycin. ABC transporter TmrAEQB reconstituted in lipid nanodiscs (Nds) was immobilized on streptavidin matrices and used as bait during affinity selections. After several iterative rounds of selection, high-affinity binders were isolated and identified by deep sequencing. (B) CPs targeting TmrAB. Enriched CPs were produced by solid-phase synthesis and conjugated with a short linear extension (colored in blue). Head-to-side chain thioether cyclization was mediated between the N-terminal N-chloroacetyl-D-tyrosine and a cysteine residue. The C-terminal lysine was used for site-directed labeling of fluorescein (F) or biotin (B). (C) Enrichment of CPs targeting TmrAB. The 5000 most abundant macrocyclic peptides per selection round based on deep sequencing were utilized to generate sequence similarity networks (Gerlt et al., 2015). Nodes represent unique peptide sequences, node sizes depict peptide frequency, and node colors exhibit peptide length. Sequence alignments of peptide clusters were generated using WebLogo (Crooks et al., 2004).
-
Figure 1—source data 1
Sequencing Data for Section of CPs.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig1-data1-v2.zip
Figure 1—figure supplement 1
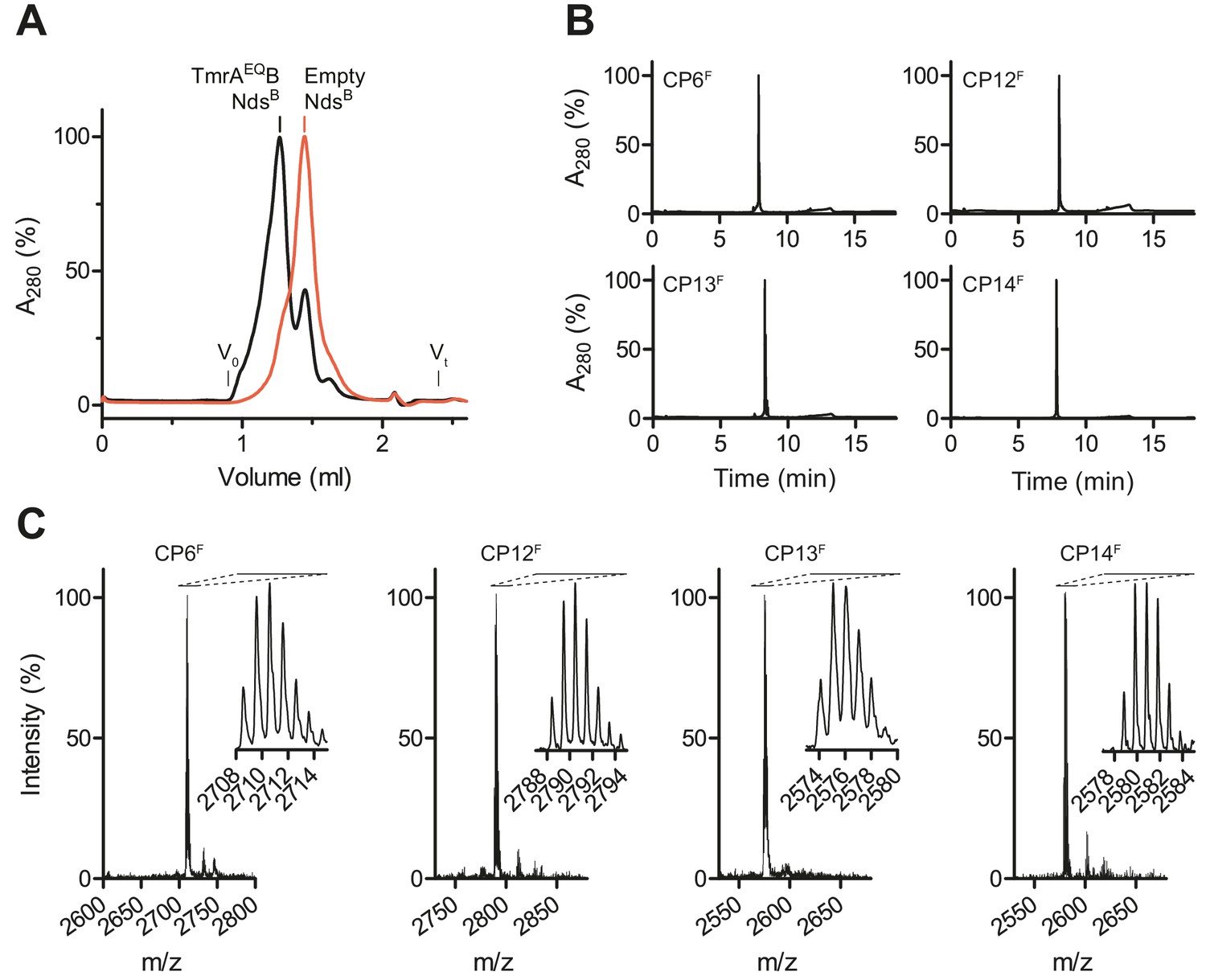
Selection and synthesis of CPs.
(A) TmrAEQB was reconstituted in lipid nanodiscs composed of biotinylated (B) MSP. Reconstituted nanodiscs were isolated by size-exclusion chromatography (SEC). Empty nanodiscs without TmrAEQB were isolated by SEC and served as bait for negative selections. (B) Purification of macrocyclic peptides (CPs). Selected CPs were synthesized on solid support followed by on-resin labeling and cyclization. Macrocycles were purified by C18 reversed-phase HPLC. (C) Identity of CPs. After synthesis, cyclization, and purification, molecular masses of macrocycles were confirmed by MALDI-TOF mass spectrometry. Derived masses, CP6F, calculated: 2708.18 Da, observed: 2707.85 Da; CP12F, calculated: 2788.24 Da, observed: 2788.19 Da; CP13F, calculated: 2574.19 Da, observed: 2574.61 Da; CP14F, calculated: 2579.14 Da, observed: 2579.41 Da.
Figure 2 with 2 supplements
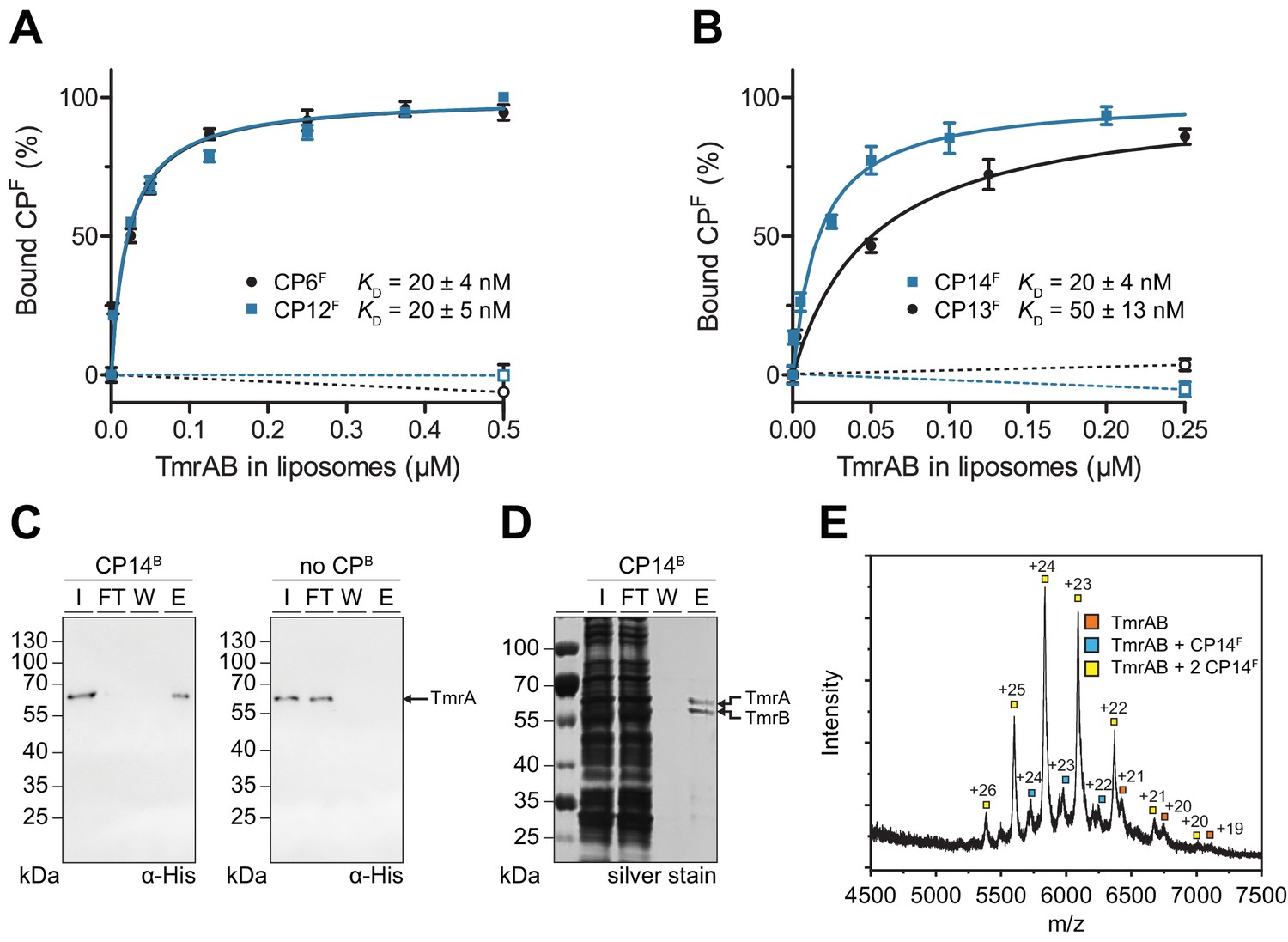
CPFs specifically interact with TmrAB displaying nanomolar binding affinities.
(A, B) Equilibrium binding analysis. Fluorescence anisotropy of CPFs (50 nM) was determined at λex/em = 485/520 nm with increasing concentrations of TmrAB which was reconstituted in liposomes. TmrAB concentrations were calculated based on the random (50/50%) orientation in proteoliposomes (Stefan et al., 2020). As a control, equal amounts of empty liposomes were added to CPFs (dashed lines, open symbols). The difference in fluorescence polarization was normalized to free (0%) and 100% bound CPF. Mean values ± SD (n = 3) are shown, and data were analyzed by a one-site binding model. (C) TmrAB binds specifically to CP14B-loaded matrices. Streptavidin beads loaded with CP14B (1 µM) were mixed with purified TmrAB (60 nM) for 1 hr at 4°C. Beads were washed and bound TmrAB was eluted by adding SDS loading buffer for 10 min at 95°C. Amounts of TmrA in input (I), flow-through (FT), wash fraction (W), and eluate (E) were analyzed by SDS-PAGE and immunoblotting (α-His). Beads without CPB served as negative control (right panel). (D) One-step purification of TmrAB via immobilized CP14B matrices. Streptavidin-agarose beads were loaded with CP14B (1 µM) and mixed with DDM-solubilized membranes of E. coli containing TmrAB as described in (C). Bound TmrAB was eluted in SDS loading buffer and analyzed by SDS-PAGE (silver stain). (E) Native mass spectrometry. TmrAB (4 µM) was buffer exchanged to ESI buffer and incubated with a 2-fold molar excess of CP14F for 10 min on ice. Protein complexes were investigated by ESI-TOF-mass spectrometry. Derived masses, TmrAB: 134.9 kDa, TmrAB +CP14F: 137.4 kDa, TmrAB +2 CP14F:140.0 kDa.
-
Figure 2—source data 1
Source data for Figure 2.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig2-data1-v2.docx
Figure 2—figure supplement 1

CPs specifically bind to TmrAB.
(A) CPBs-mediated isolation of solubilized TmrAB. Streptavidin matrices were loaded with CPBs (1 µM) and incubated with purified TmrAB (60 nM) for 1 hr at 4°C. Beads without CPBs served as control (see Figure 2C). Bound TmrAB complexes were eluted by the addition of SDS loading buffer for 10 min at 95°C. TmrAB in input (I), flow-through (FT), washing fraction (W), and eluate (E) was analyzed by SDS-PAGE and immunoblotting (α-His). (B) Isolation of TmrAB using CP13B. Streptavidin matrices were loaded with CP13B (1 µM) and mixed with DDM-solubilized membranes of E. coli expressing TmrAB as described in (A). Bound TmrAB complexes were eluted in SDS loading buffer and analyzed by SDS-PAGE (silver stain). (C) Formation of CPF-TmrAB complexes. DDM-solubilized TmrAB (1 µM) was incubated with CP12F, CP13F, CP14F (2 µM each), or CP6F (5 µM) for 10 min on ice. CPF-TmrAB complexes were analyzed by gel filtration. Non-specific peptide aggregation was not observed. (D) Conformational epitopes. CPFs (50 nM) were incubated with TmrAB (0.6 µM for CP6F and CP13F, 0.7 µM for CP12F, 0.5 µM for CP14F) in the presence of 1% (v/v) SDS. Fluorescence anisotropy was determined at λex/em = 485/520 nm. Mean values ± SD (n = 3) are displayed.
-
Figure 2—figure supplement 1—source data 1
Source Data for Figure 2—figure supplement 1.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig2-figsupp1-data1-v2.docx
Figure 2—figure supplement 2
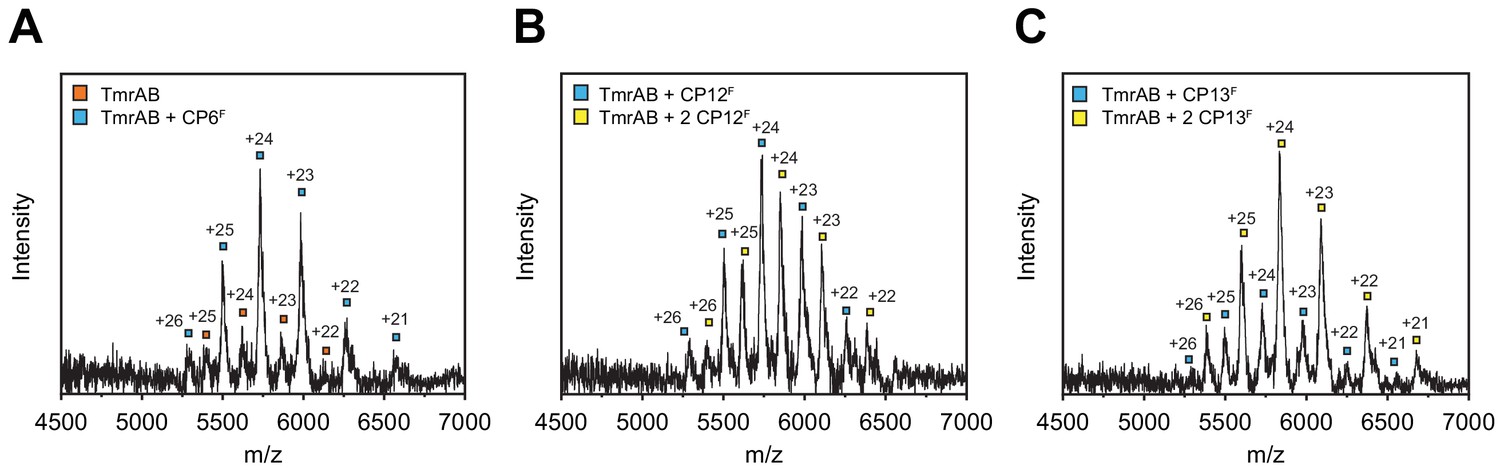
Analysis of CPF-TmrAB complexes by native mass spectrometry.
TmrAB (4 µM) was buffer exchanged to ESI buffer and incubated with a 2-fold molar excess of CPFs for 10 min on ice. Protein complexes were investigated by ESI-TOF-mass spectrometry. (A) CP6F-TmrAB complexes. Derived masses, TmrAB: 135.1 kDa, TmrAB +CP6F: 137.7 kDa. (B) CP12F-TmrAB complexes. Derived masses, TmrAB +CP12F: 137.6 kDa, TmrAB +2 CP12F: 140.5 kDa. (C) CP13F-TmrAB complexes. Derived masses, TmrAB +CP13F: 137.4 kDa, TmrAB +2 CP13F: 140.0 kDa.
Figure 3
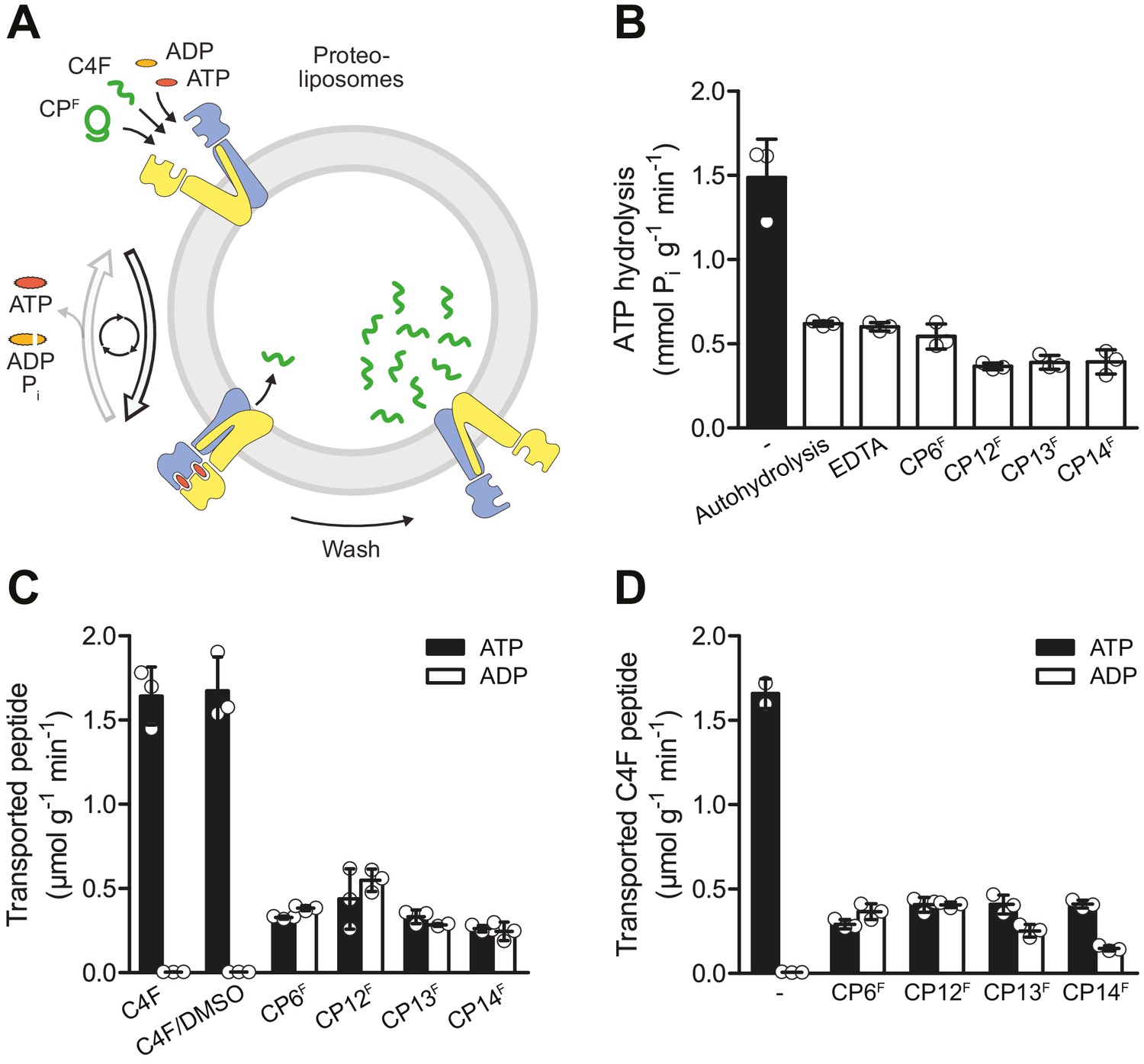
CPFs are potent inhibitors of ATP turnover and substrate transport.
(A) Experimental scheme of ATP hydrolysis and peptide transport by TmrAB. (B) CPFs inhibit ATP hydrolysis. TmrAB in liposomes (100 nM) was incubated with ATP (2 mM, traced with [γ32P]-ATP), and MgCl2 (5 mM) in the presence and absence of CPFs (1 µM) for 10 min at 45°C. Autohydrolysis was determined in the absence of TmrAB, and background turnover was conducted in the presence of EDTA (10 mM). Release of [γ32P] was quantified by thin layer chromatography. (C) CPFs are not transported by TmrAB. TmrAB (0.4 µM) reconstituted in liposomes was incubated with C4F peptide (3 µM) or CPFs (1 µM) in the presence of ATP/ADP (3 mM) and MgCl2 (5 mM) for 5 min at 45°C. Since CPFs were dissolved in maximal 0.5% (v/v) of DMSO, C4F plus 0.5% DMSO served as control. Proteoliposomes were washed on filter plates, and transported peptides were quantified at λex/em = 485/520 nm. (D) Inhibition of substrate translocation. TmrAB in liposomes (0.4 µM) were incubated with C4F peptide (3 µM), ATP/ADP (3 mM), and MgCl2 (5 mM) in the presence and absence of CPFs (1 µM) for 15 min at 45°C. Transported peptides were quantified as described in (C). In (B–C), mean values ± SD (n = 3) are shown.
-
Figure 3—source data 1
Source data for Figure 3.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig3-data1-v2.docx
Figure 4
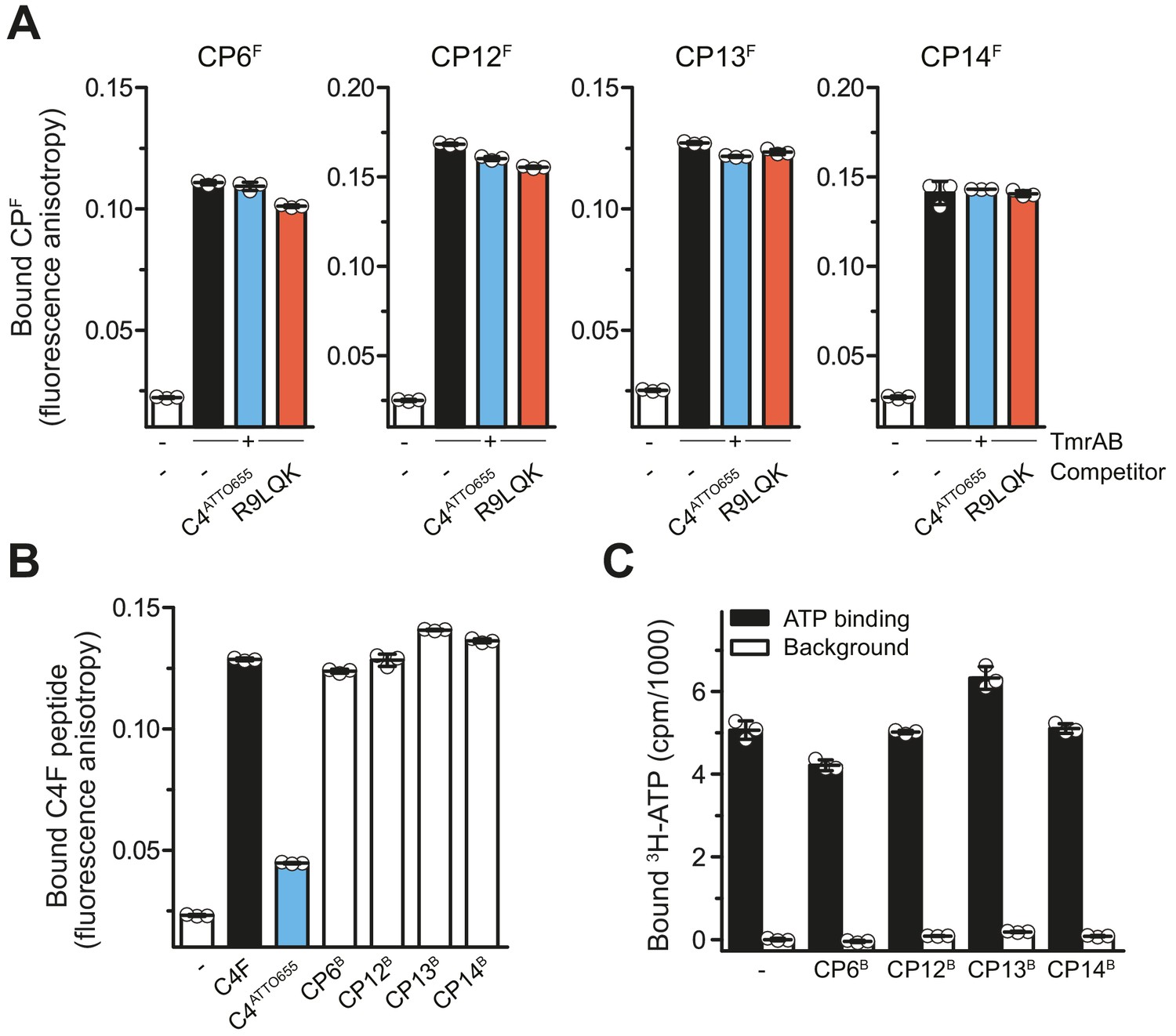
CPs do not affect peptide or ATP binding of TmrAB.
(A) CPFs binding TmrAB is not affected by substrate peptide binding. After the addition of TmrAB (0.6 µM for CP6F and CP13F, 0.7 µM for CP12F, 0.5 µM for CP14F), fluorescence anisotropy of CPFs (50 nM) were monitored at λex/em = 485/520 nm. For competition, C4ATTO655 peptide (2 µM) or R9LQK peptide (200 µM) were added. (B) Binding of C4F peptide is not affected by CPBs. C4F peptides (50 nM) were mixed with TmrAB (4 µM), and fluorescence anisotropy was monitored as described in (A). For competition, C4ATTO655 (10 µM) or CPBs (6 µM) were added. (C) CPBs do not largely affect ATP binding. TmrAB (0.2 µM) were immobilized on SPA beads and incubated with ATP (3 μM, traced with 3H-ATP) in the presence and absence of CPBs (1 µM each) for 30 min on ice. ATP binding was monitored by SPA. Background values were determined after releasing TmrAB complexes from the beads by adding imidazole (200 mM). The background signal in the absence of CPBs was set to 0 cpm. In the case of CP13, the value of bound ATP exceeds the control without CPs. As this is an equilibrium experiment, this can be rationalized by the fact that CP13 stabilizes the ATP-bound OF conformer.
-
Figure 4—source data 1
Source data for Figure 4.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig4-data1-v2.docx
Figure 5 with 1 supplement
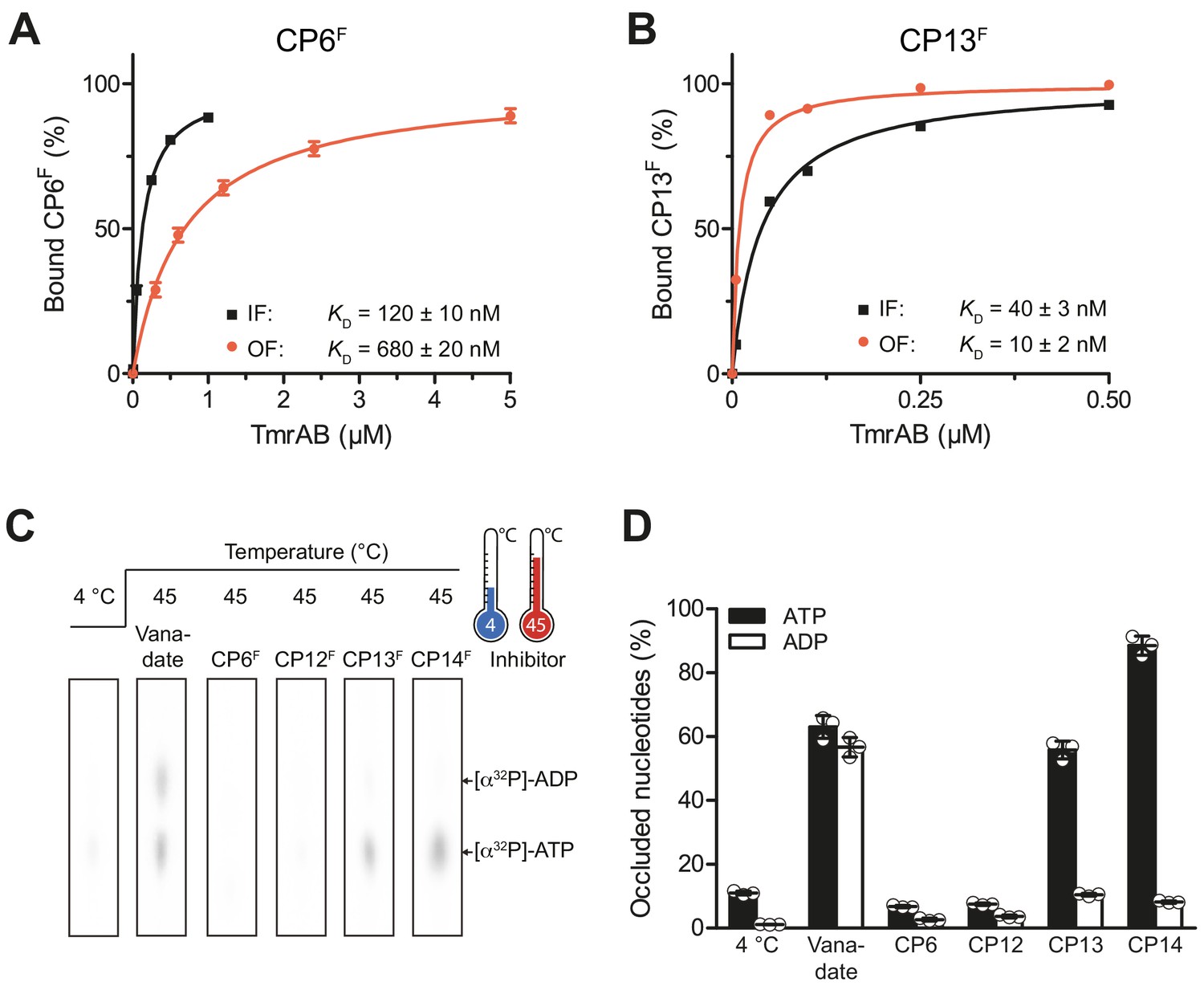
CPs bind preferentially to IF- und OF conformation and stabilize nucleotide occlusion.
Conformation-specific preference of CPs. (A, B) CP6F (A) or CP13F (B, 50 nM each) were incubated with increasing concentrations of detergent-solubilized inward-facing TmrAB (in the absence of Mg-ATP) or outward-facing TmrAEQB, which was trapped with Mg-ATP (1 mM) for 5 min at 45°C as described (Hofmann et al., 2019; Stefan et al., 2020). Immediately after, the fluorescence anisotropy was assayed at λex/em = 485/520 nm. The difference in fluorescence polarization was normalized to free and fully bound CPF. Data were fitted by a one-site binding model. (C) Nucleotide occlusion promoted by CPs. TmrAB (2 µM each) were mixed with CPFs (4 µM), ATP (1 mM, traced with [α32P]-ATP), and MgCl2 (5 mM) for 5 min at 4°C or 45°C. Cold ATP (10 mM) was added, and freely exchangeable nucleotides were removed by rapid gel filtration. Occluded nucleotides were analyzed by thin layer chromatography and autoradiography. Representative radiograms for independent triplicates are shown. (D) Stably occluded nucleotides, [α32P]-ATP and [α32P]-ADP, were quantified by autoradiography. Data were normalized to the vanadate-trapped state. In (A, B, D), mean values ± SD (n = 3) are shown.
-
Figure 5—source data 1
Source Data for Figure 5.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig5-data1-v2.docx
Figure 5—figure supplement 1
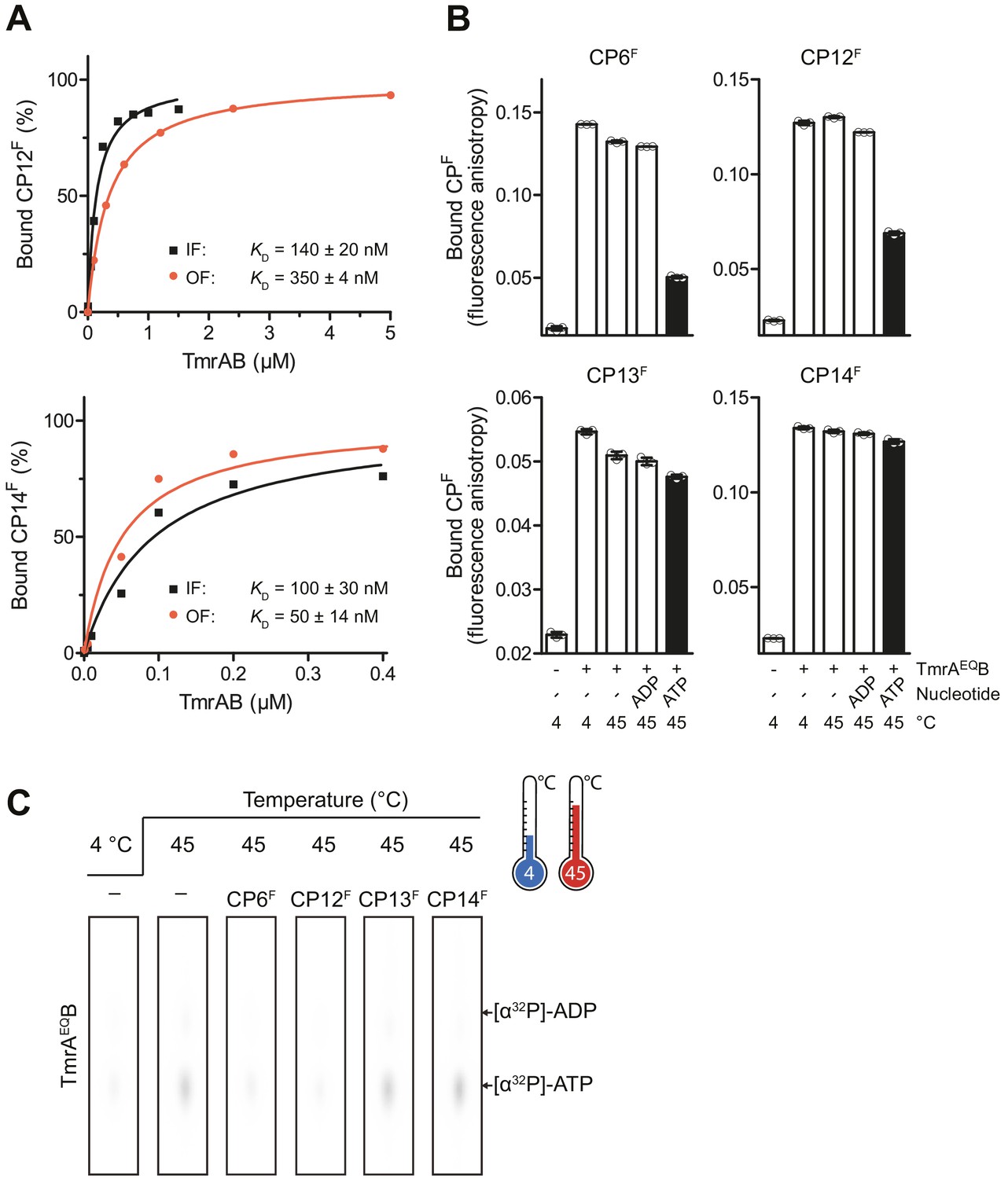
Conformational arrest of TmrAB by CPFs.
(A) CPFs (50 nM each) were incubated with increasing concentrations of detergent-solubilized IF TmrAB in the inward-facing (IF) conformation (absence of Mg-ATP) or TmrAEQB populated in the outward-facing (OF) conformation, which was trapped in the OF conformation by Mg-ATP (1 mM) after 5 min incubation at 45°C. The difference in fluorescence polarization was normalized to unbound (0%) and 100% bound CPF. Data were fitted by a one-site binding model. Fluorescence anisotropy was recorded at λex/em = 485/520 nm. (B) CPFs (50 nM each) were incubated with detergent-solubilized IF or OF TmrAEQB (150 nM for CP6F, 170 nM for CP12F, 50 nM for CP13F, and 120 nM for CP14F). (C) Nucleotide occlusion of the catalytically reduced TmrAB variant. TmrAEQB (2 µM) were incubated in the absence and presence of CPFs (4 µM each), ATP (1 mM, traced with [α32P]-ATP), and MgCl2 (5 mM) for 5 min at 4°C or 45°C. Cold ATP (10 mM) was added, and free nucleotides were removed by rapid gel filtration. Occluded nucleotides were analyzed by thin layer chromatography and autoradiography. Representative radiograms for independent triplicates are shown. In (A, B), mean values ± SD (n = 3) are displayed.
-
Figure 5—figure supplement 1—source data 1
Source Data for Figure 5—figure supplement 1.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig5-figsupp1-data1-v2.docx
Figure 6 with 1 supplement
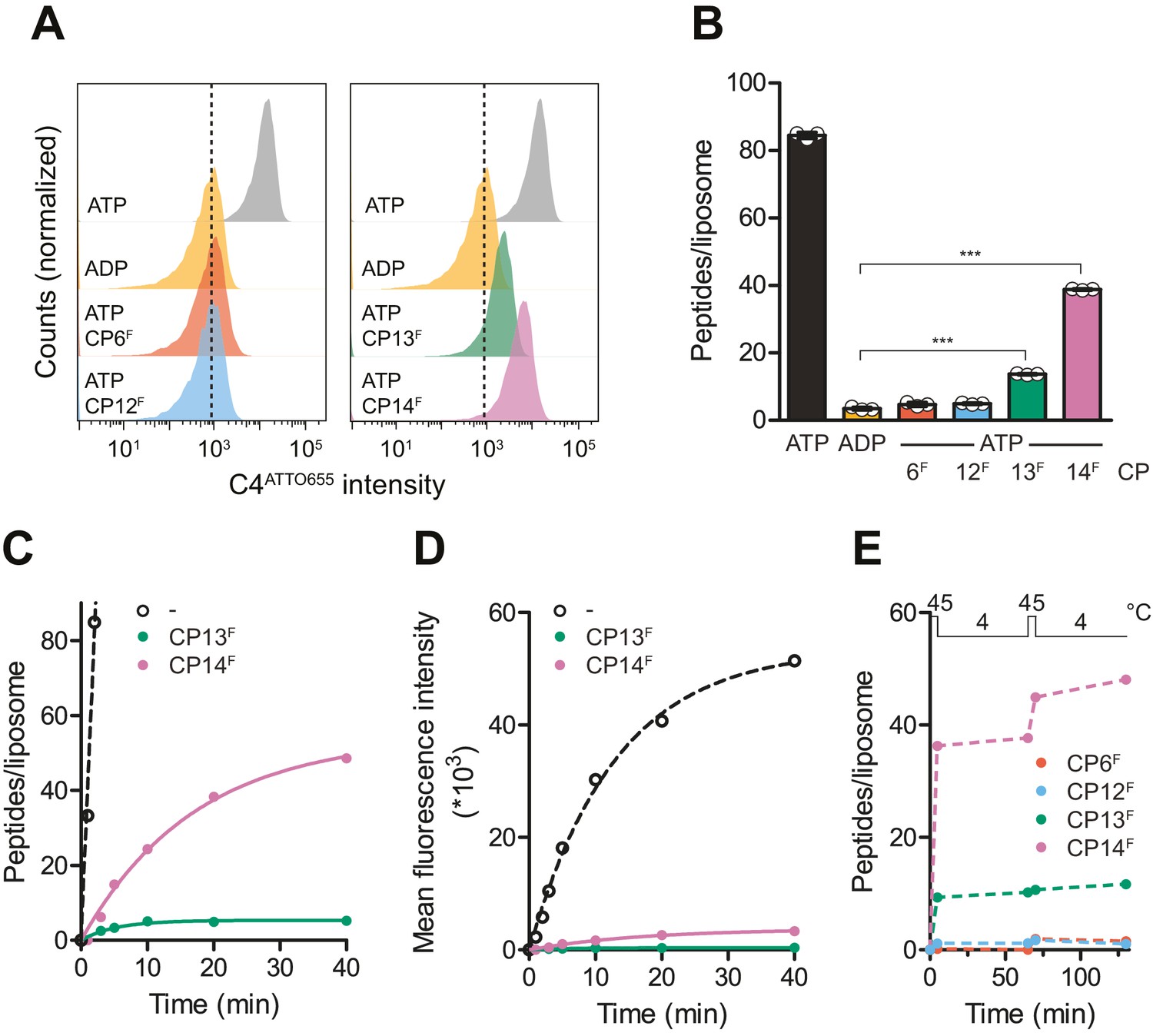
CPs block multiple-turnover transport monitored by quantitative flow cytometry.
(A) Single turnover by IF-to-OF switch monitored by single-liposome flow cytometry. TmrAB in liposomes (0.4 µM) were incubated with C4ATTO655 peptide (1 µM), ATP/ADP (3 mM), MgCl2 (5 mM), and CPFs (1 µM) for 5 min at 45°C. Transport reactions were stopped by the addition of EDTA (10 mM). 100,000 proteoliposomes were analyzed by flow cytometry monitoring fluorescein and ATTO655 intensities. (B) Mean fluorescence intensities of C4ATTO655 were converted into the number of peptides per liposome using the regression analysis described above (two-tailed T-test, ***p<0.0001). (C, D) Slowdown of multiple-turnover substrate transport. TmrAB reconstituted in liposomes (0.4 µM) was incubated with C4ATTO655 peptide (1 µM), ATP/ADP (3 mM), MgCl2 (5 mM), and CPFs (1 µM) for various periods of time at 45°C. Transported peptides per liposomes were evaluated and corrected by ADP background levels as described in (B). Transport kinetics were fitted monoexponentially. In (C), mean fluorescence intensities of transported C4ATTO655 were converted into the number of peptides per liposome as described above. (E) Two consecutive transport cycles. TmrAB reconstituted in liposomes was incubated with C4ATTO655 peptide, ATP/ADP, MgCl2, and CPFs as described in (A) for 5 min at 45°C, 60 min at 4°C, 5 min at 45°C, and 60 min at 4°C. Transported peptides per liposome were evaluated as described in (B) and corrected by background levels in the presence of ADP. In (B–E), mean values ± SD (n = 3) are displayed.
-
Figure 6—source data 1
Source data for Figure 6.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig6-data1-v2.docx
Figure 6—figure supplement 1
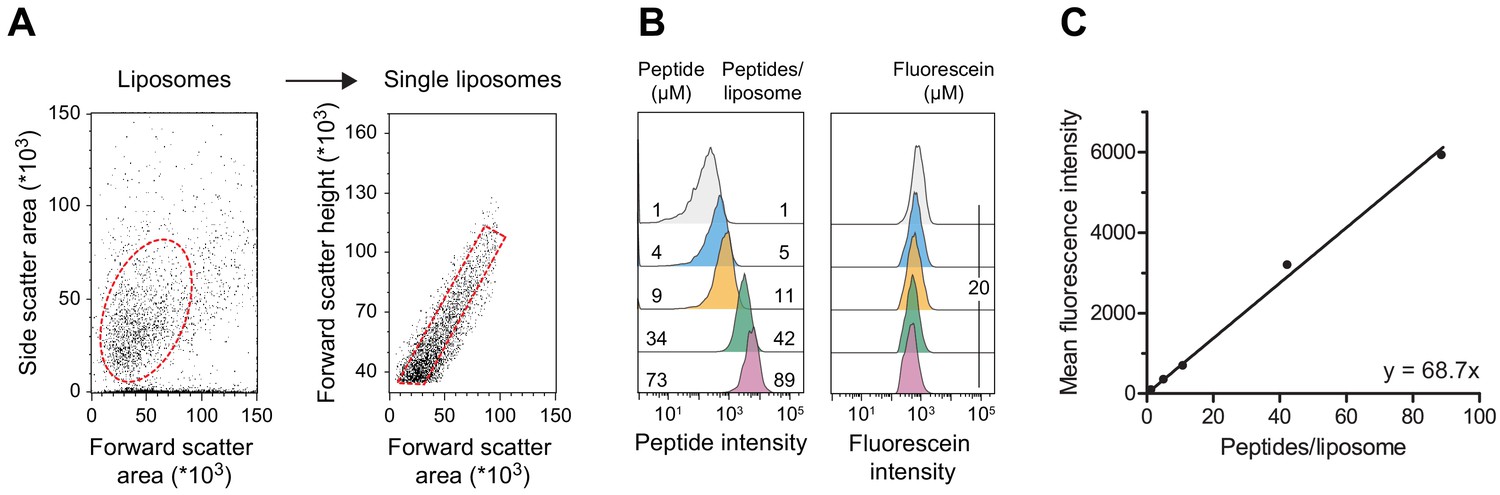
Linear regression of quantitative flow cytometry analysis.
(A) Gating strategy. Liposomes were selected according to side and forward scatter areas. Single liposomes were gated based on the height of forward scatter correlated to the area of forward scatter. Fluorescence intensities of 20,000 to 100,000 single liposomes were evaluated. (B) Liposomes without reconstituted TmrAB were destabilized by Triton X-100 and incubated with defined amounts of C4ATTO655 peptide (left). Detergent was removed, and liposomes were extensively washed by centrifugation. Mean fluorescence intensities of 100,000 liposomes were analyzed by flow cytometry. Equal concentrations of fluorescein served as loading control (right). (C) Linear regression. Encapsulated C4ATTO655 peptide intensities were correlated with the number of peptides per liposome. Data were analyzed by linear regression yielding y = 68.7 x±1.7 x and R2 = 0.99. Mean values ± SD (n = 3) are shown.
-
Figure 6—figure supplement 1—source data 1
Source Data for Figure 6—figure supplement 1.
- https://cdn.elifesciences.org/articles/67732/elife-67732-fig6-figsupp1-data1-v2.docx
Figure 7
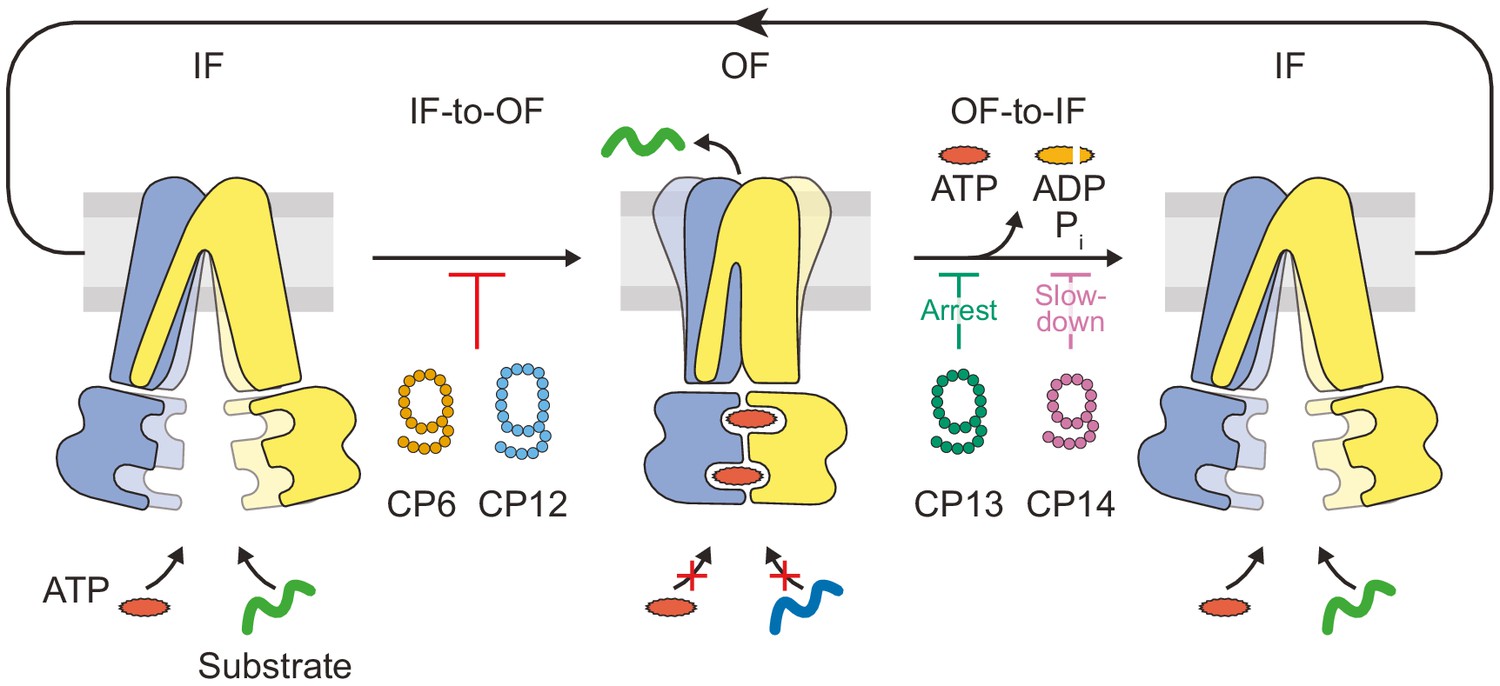
Substrate translocation precedes ATP hydrolysis in a heterodimeric ABC transporter.
In the resting IF state, TmrAB binds nucleotides and substrates independently, which is not affected by CPs. ATP binding induces an IF-to-OF switch, which drives unidirectional substrate translocation. CP6F and CP12F bind preferentially to the IF state and block the transition to the OF state, preventing ATP occlusion and ATP hydrolysis. CP13F and CP14F favor and stabilize a pre-hydrolysis OF state after the IF-to-OF conformation switch and peptide translocation. CPs block ATP hydrolysis at different steps of the transport cycle. In the absence of CPs, ATP hydrolysis and phosphate release initiate the OF-to-IF return restoring transporter function.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | Monoclonal α-His antibody | Sigma-Aldrich | SAB1305538 | Mouse origin. Final dilution: 1/2,000 (v/v) |
| Antibody | α-Mouse-HRP conjugate | Sigma-Aldrich | AP130P | Goat origin. Final dilution: 1/20,000 (v/v) |
| Chemical compound, drug | β-n-Dodecyl β-D-maltoside (DDM) | Carl Roth | CN26.5 | |
| Chemical compound, drug | Bovine brain lipid extract | Sigma-Aldrich | B1502 | |
| Chemical compound, drug | [2,5’,8-3H(N)]-ATP (3H-ATP) | PerkinElmer | NET118900 | |
| Chemical compound, drug | [α32P]-ATP | Hartmann Analytic | FP-207 | |
| Chemical compound, drug | Copper-chelated PVT SPA beads | PerkinElmer | RPNQ0095 | |
| Chemical compound, drug | ClAc-D-Tyr-CME | Synthesized according to DOI: 10.1021/cb200388k | ||
| Chemical compound, drug | Fmoc-protected amino acids | Merck Millipore/Watanabe Chemical Industries | various | |
| Chemical compound, drug | HBTU | Watanabe Chemical Industries | A00149 | |
| Chemical compound, drug | HOBt | Watanabe Chemical Industries | A00014 | |
| Chemical compound, drug | NovaPEG Rink Amide resin | Merck Millipore | 855047 | |
| Chemical compound, drug | N,N-Diisopropylethylamine | Nacalai Tesque | 14014–55 | |
| Chemical compound, drug | N,N-Dimethylformamide | Nacalai Tesque | 13016–23 | |
| Chemical compound, drug | 5/6-Carboxyfluorescein succinimidyl ester | Thermo Fisher Scientific | 46410 | |
| Chemical compound, drug | Acetonitrile | Wako Chemicals | 015–08633 | |
| Chemical compound, drug | Trifluoroacetic acid | Nacalai Tesque | 3483305 | |
| Chemical compound, drug | D-Biotin | Nacalai Tesque | 04822–91 | |
| Chemical compound, drug | Pluronic F127 | Sigma-Aldrich | P2443 | |
| Chemical compound, drug | Albumin, Bovine, Acetylated | Nacalai Tesque | 01278–44 | |
| Chemical compound, drug | NTPs | Jena Bioscience | NU-1010 NU-1011 NU-1012 NU-1013 | |
| Chemical compound, drug | Dynabeads M-280 Streptavidin | Thermo Fisher Scientific | 11206 | |
| Sequence-based reagent | T7g10M.F46 | Eurofins Genomics K.K. (Japan) | PCR primer | TAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATA |
| Sequence-based reagent | NNK(n).R(3n + 45)n = 10–15 | Eurofins Genomics K.K. (Japan) | PCR primer for DNA library | GCTGCCGCTGCCGCTGCCGCA(MNN)nCATATGTATATCTCCTTCTTAAAG |
| Sequence-based reagent | CGS3an13.R36 | Eurofins Genomics K.K. (Japan) | PCR primer | TTTCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCC |
| Sequence-based reagent | Ini-3'.R20-Me | Gene Design Inc (Japan) | PCR primer for tRNAfMetCAU assembly | TGmGTTGCGGGGGCCGGATTT (Gm = 2'-Methoxylated G) |
| Sequence-based reagent | Ini-3'.R38 | Eurofins Genomics K.K. (Japan) | PCR primer for tRNAfMetCAU assembly | TGGTTGCGGGGGCCGGATTTGAACCGACGATCTTCGGG |
| Sequence-based reagent | Ini1-1G-5'.F49 | Eurofins Genomics K.K. (Japan) | PCR primer for tRNAfMetCAU assembly | GTAATACGACTCACTATAGGCGGGGTGGAGCAGCCTGGTAGCTCGTCGG |
| Sequence-based reagent | Ini cat.R44 | Eurofins Genomics K.K. (Japan) | PCR primer for tRNAfMetCAU assembly | GAACCGACGATCTTCGGGTTATGAGCCCGACGAGCTACCAGGCT |
| Sequence-based reagent | Fx5'.F36 | Eurofins Genomics K.K. (Japan) | PCR primer for eFx assembly | GTAATACGACTCACTATAGGATCGAAAGATTTCCGC |
| Sequence-based reagent | eFx.R45 | Eurofins Genomics K.K. (Japan) | PCR primer for eFx assembly | ACCTAACGCTAATCCCCTTTCGGGGCCGCGGAAATCTTTCGATCC |
| Sequence-based reagent | eFx.R18 | Eurofins Genomics K.K. (Japan) | PCR primer for eFx assembly | ACCTAACGCTAATCCCCT |
| Sequence-based reagent | T7e × 5 .F22 | Eurofins Genomics K.K. (Japan) | PCR primer for eFx assembly | GGCGTAATACGACTCACTATAG |
| Sequence-based reagent | DNA-PEG-puromycin | Gene Design Inc, Osaka, Japan | linker for mRNA display | CTCCCGCCCCCCGTCC-(PEG18)5-CC-Pu |
| Gene | TmrA | Q72J05 | TTC0976 | Species: Thermus thermophilus |
| Gene | TmrB | Q72J04 | TTC0977 | Species: Thermus thermophilus |
| Peptide, recombinant protein | RRY-C*-KSTEL | This study (methods and material) | C* denotes fluorescein-labeled Cys | |
| Peptide, recombinant protein | Macrocyclic peptides CP6, CP12, C13 and CP14 | This study (methods and material) | ||
| Peptide, recombinant protein | KOD DNA Polymerase | Prepared in house (methods and material) | ||
| Peptide, recombinant protein | T7 RNA polymerase | Prepared in house (methods and material) | ||
| Peptide, recombinant protein | T4 RNA ligase | Prepared in house (methods and material) | ||
| Peptide, recombinant protein | FIT system | Prepared in house according to DOI: 10.1038/nprot.2015.082 | 50 mM HEPES-KOH (pH 7.6), 12 mM magnesium acetate, 100 mM potassium acetate, 2 mM spermidine, 20 mM creatine phosphate, 2 mM DTT,2 mM ATP, 2 mM GTP, 1 mM CTP,1 mM UTP, 0.5 mM 19 proteinogenic amino acids other than Met, 1.5 mg/ml E. coli total tRNA, 0.73 µM AlaRS, 0.03 µM ArgRS, 0.38 µM AsnRS, 0.13 µM AspRS,0.02 µM CysRS, 0.06 µM GlnRS, 0.23 µM GluRS, 0.09 µM GlyRS, 0.02 µM HisRS,0.4 µM IleRS, 0.04 µM LeuRS, 0.11 µM LysRS, 0.03 µM MetRS, 0.68 µM PheRS,0.16 µM ProRS, 0.04 µM SerRS, 0.09 µM ThrRS, 0.03 µM TrpRS, 0.02 µM TyrRS,0.02 µM ValRS, 0.6 µM MTF, 2.7 µM IF1,0.4 µM IF2, 1.5 µM IF3,0.26 µM EF-G, 10 µM EF-Tu,10 µM EF-Ts, 0.25 µM RF2,0.17 µM RF3, 0.5 µM RRF,0.1 µM T7 RNA polymerase, 4 µg/ml creatine kinase, 3 µg/ml myokinase,0.1 µM pyrophosphatase, 0.1 µM nucleotide-diphosphatase kinase, 1.2 µM ribosome | |
| Recombinant DNA reagent | pET-22b | Merck Millipore | 69744 | Vector for protein expression in E. coli |
| Strain, strain background (Escherichia coli) | BL21(DE3) | Thermo Fisher | C600003 | Chemically competent cells |
| Software, Algorithm | Prism 5 | GraphPad | ||
| Software, algorithm | Cytoscape | Shannon P et al. Genome Research 2003 13(11) 2498–504 | ||
| Software, algorithm | EFI-EST | Gerlt JA et al. Biochim Biophys Acta 1854: 1019-37 | ||
| Software, algorithm | WebLogo | Crooks GE et al. Genome 561 Res 14: 1188–90 |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
De novo macrocyclic peptides dissect energy coupling of a heterodimeric ABC transporter by multimode allosteric inhibition
eLife 10:e67732.
https://doi.org/10.7554/eLife.67732




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}