Novel mechanistic insights into the role of Mer2 as the keystone of meiotic DNA break formation
- Friedrich Miescher Laboratory of the Max Planck Society, Germany
- Department of Mechanistic Cell Biology, Max Planck Institute of Molecular Physiology, Germany
- Columbia University Medical Center, United States
- Department of Chemical Biology, Max Planck Institute of Molecular Physiology, Germany
- Section of Oncogenetics, Department of Human Genetics, Cancer Centre Amsterdam and Amsterdam Reproduction and Development Research Institute, Netherlands
Figures
Figure 1 with 4 supplements
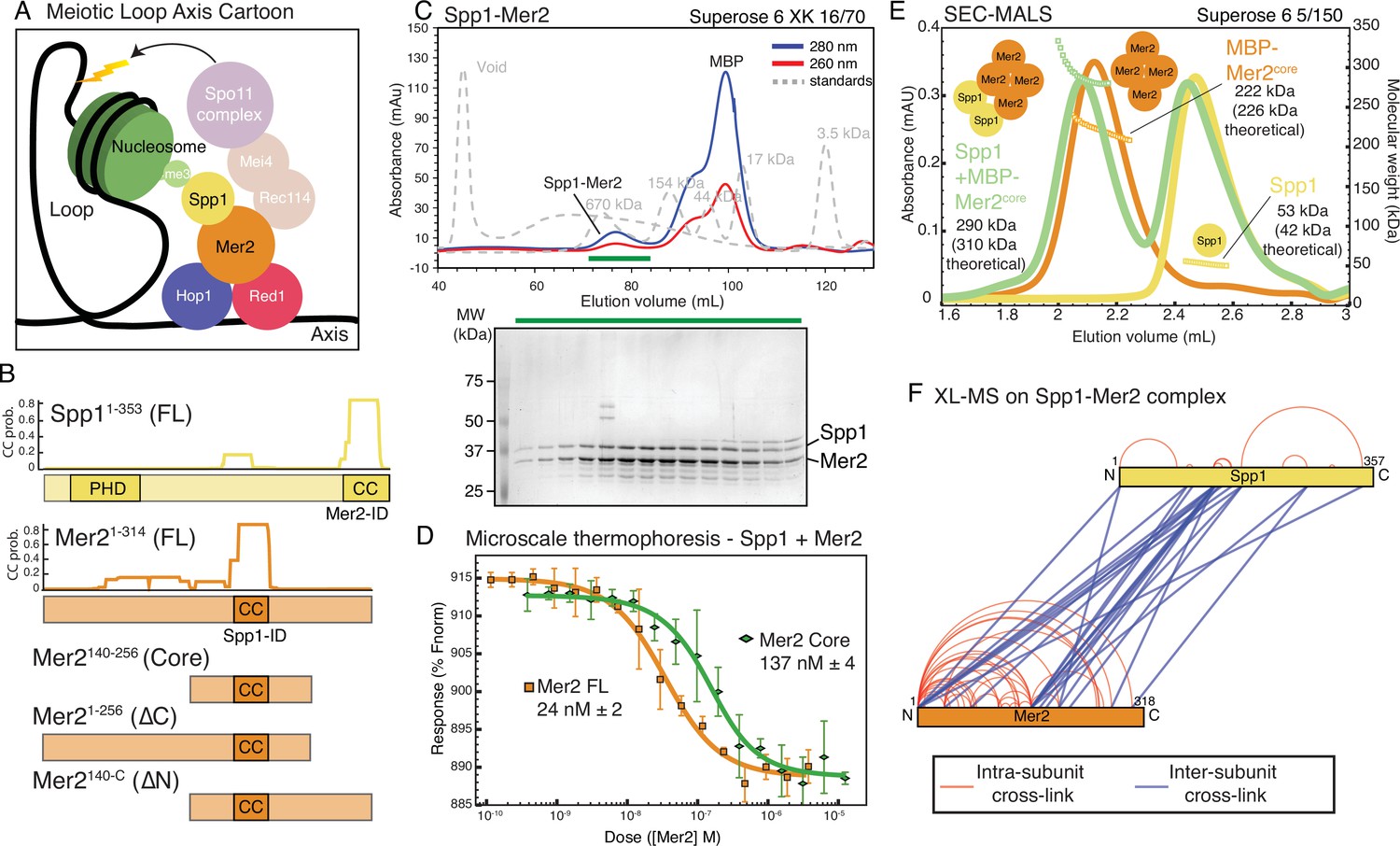
Spp1 binds to Mer2 tetramerisation domain with a 2:4 stoichiometry.
(A) Cartoon of meiotic loop axis architecture and the role of Mer2. During meiosis proteins Red1 and Hop1 form a protein:DNA (coloured black) axis together with cohesin (not shown for clarity). Loops of chromatin are extruded from the axis, where DNA breaks are made by the Spo11 complex (pale magenta). Breaks are directed to the proximity of H3K4me3 nucleosomes (green) through the combined activity of Mer2 and Spp1. Mer2 (orange) is also thought to interact with additional Spo11 accessory proteins (Rec114 and Mei4, pale orange). (B) Domain diagram of Mer2 and Spp1. The four principal Mer2 constructs used throughout this study are shown. The only clear feature of Mer2 (and its orthologs IHO1 in mammals, Rec15 in fission yeast, and Prd3 in plants) is the central coiled-coil motif. Spp1 is shown for comparison with its N-terminal PHD domain and C-terminal Mer2 interaction domain which is predicted to contain a coiled-coil. Predicted coiled-coil probability (CC prob.) is shown for Spp1 full-length and Mer2 and is based on a 28 amino acid window using PCOILS (Gruber et al., 2006). (C) Purification of Mer2-Spp1 complex. A complex of Mer2 and Spp1 was purified to homogeneity. The MBP tags on Spp1 and on Mer2 were cleaved prior to loading. Degradation products of Mer2 full-length protein are also visible. Molecular weight markers are shown in grey. The relative absorbance of the complex at 280 and 260 nm shows that it is free of any significant nucleic acid contamination. The selected fractions (green line) were loaded onto an SDS-PAGE gel and stained with InstantBlue. (D) Microscale thermophoresis of Mer2-Spp1. Two different Mer2 constructs (Mer2FL, orange squares; Mer2core green diamonds) were titrated against Red-NHS labelled untagged Spp1 (20 nM constant concentration), and the change in thermophoresis was measured. Experiments were carried out in triplicate and the KD was determined from the fitting curve. (E) Size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) of different Mer2-Spp1 samples. Measured molecular masses are indicated Three illustrative SEC-MALS experiments are shown for Spp1 (yellow) (theoretical mass of monomer = 42 kDa), MBP-Mer2core (orange) (theoretical mass of a tetramer = 226 kDa) and a complex of Spp1 bound to MBP-Mer2core (green) (theoretical mass of 2:4 complex = 310 kDa). (F) XL-MS of Mer2-Spp1 complex. Full-length Spp1 and Mer2 were used, and in both cases the overhang remaining after cleaving the N-terminal fusion proteins was present. Samples were cross-linked with disuccinimidyl dibutyric urea (DSBU), and data analysis carried out according to the Materials and methods. Cross-links were filtered so as to give a 1% false discovery rate. Figure was prepared using XVis (Grimm et al., 2015). Intramolecular cross-links shown in red, intermolecular cross-links in blue.
-
Figure 1—source data 1
Raw gel data for Figure 1 size exclusion chromatography (SEC) profile.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig1-data1-v2.zip
Figure 1—figure supplement 1
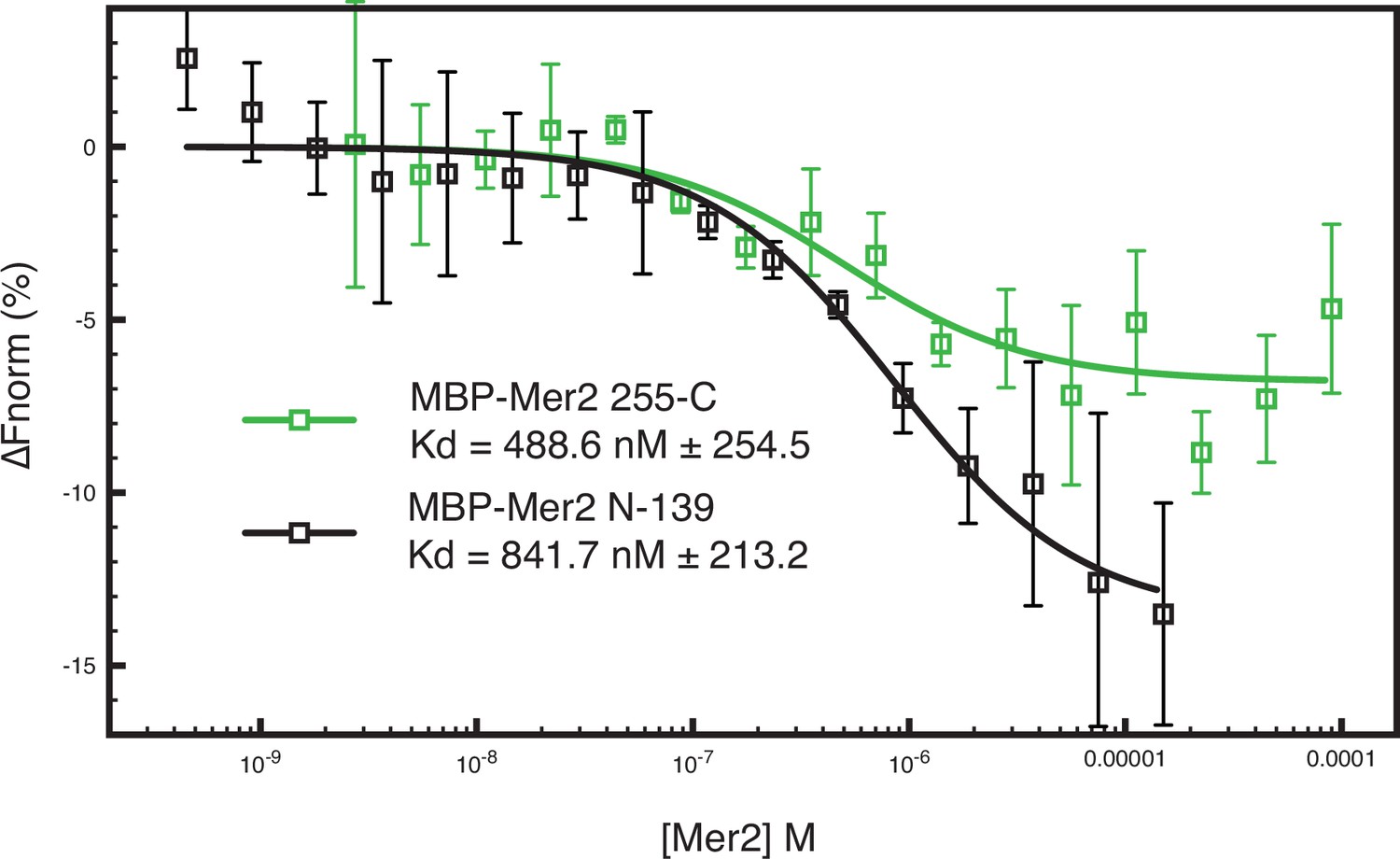
Microscale thermophoresis (MST) measurements on additional Mer2 constructs.
Two MBP-tagged Mer2 constructs, MBP-Mer2 255C (green trace) and MBP-Mer2 N-139 (black trace), were titrated against labelled Spp1 (as in Figure 1). Error bars are the SD from three independent experiments.
Figure 1—figure supplement 2
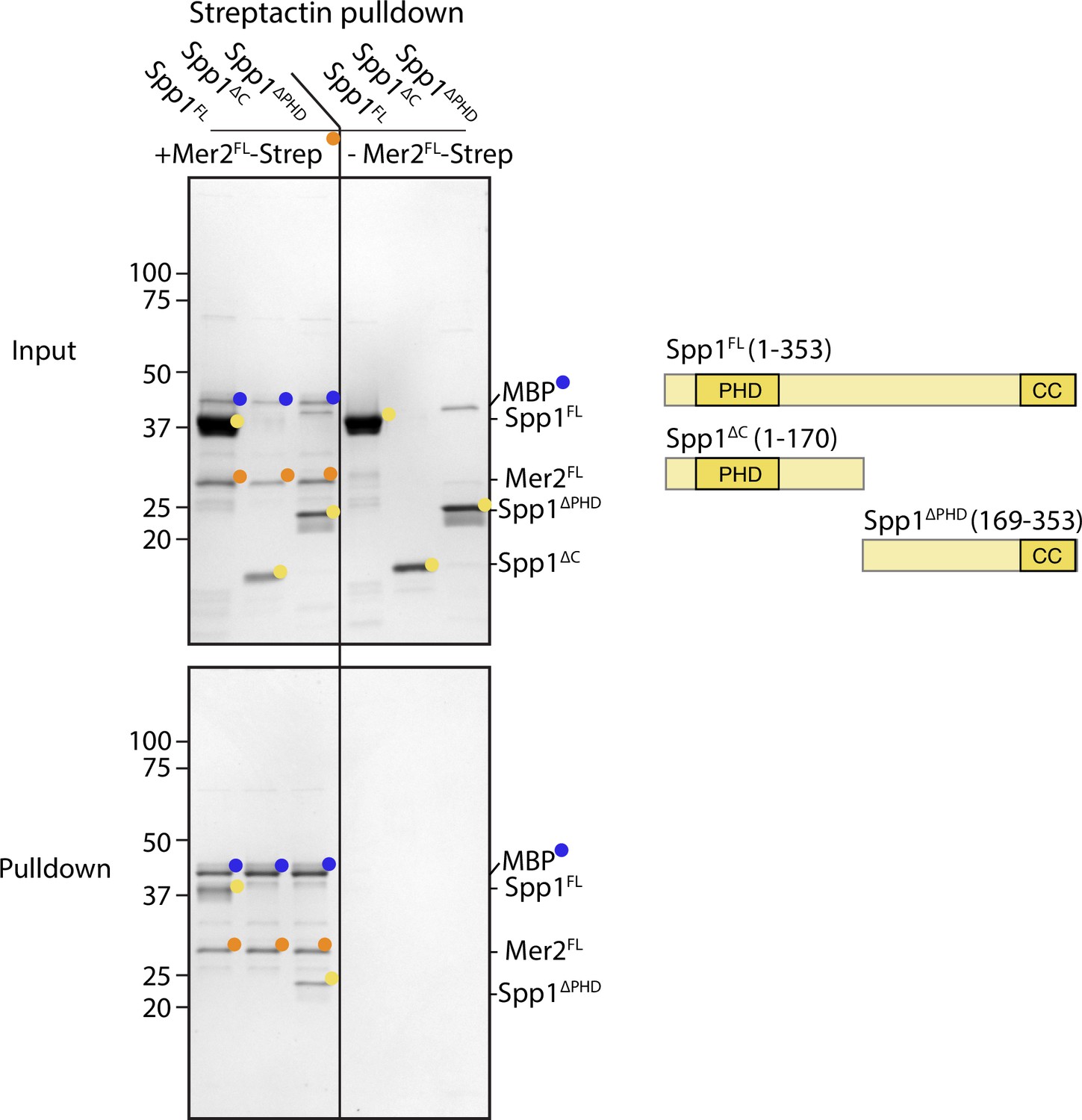
Interaction regions of Spp1 with Mer2.
Full-length Spp1 (Spp1FL), Spp1 1–170 (containing only the PHD domain; Spp1∆C) and Spp1 169–353 (removal of the PHD domain; Spp1∆PHD) were incubated in the presence (left half of the gel) or absence (right half of the gel) of C-terminally 2xStrepII tagged Mer2 1–314 (Mer2∆FL). Samples were then incubated in Streptactin beads and washed as described in the Materials and methods. Pulldown samples were eluted in 1× Laemmli buffer, run on a 12% SDS-PAGE, and stained with coomassie. Mer2FL-Strep was expressed with an N-terminal MBP tag to improve solubility which was then cleaved during the purification. Nonetheless, residual MBP remains in the Strep-Mer2FL sample and is indicated by blue circles.
-
Figure 1—figure supplement 2—source data 1
Raw gel data for Figure 1—figure supplement 2 pulldowns.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig1-figsupp2-data1-v2.zip
Figure 1—figure supplement 3
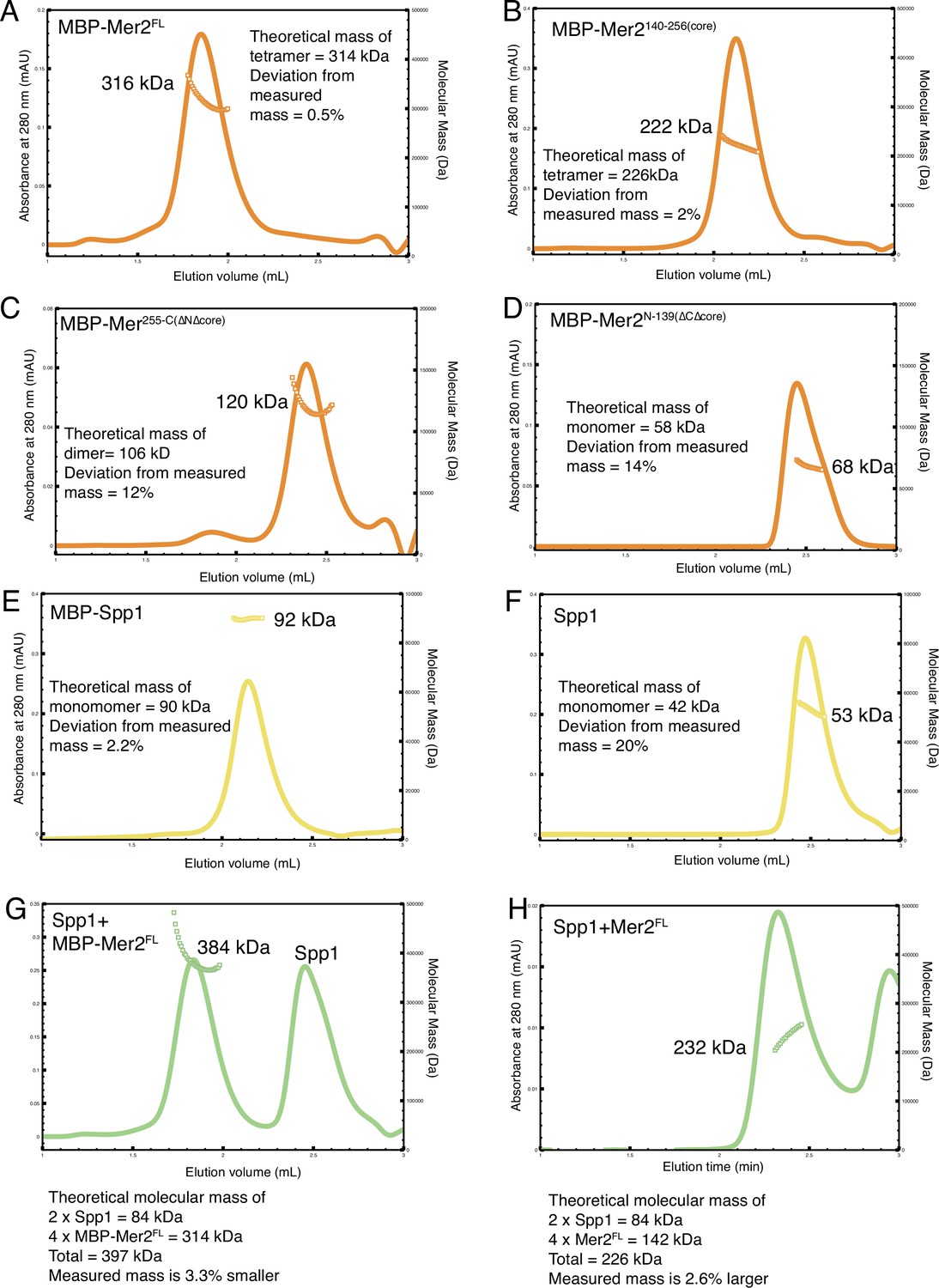
Additional size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) chromatographs and summary.
SEC-MALS experiments were carried out as described in Materials and methods. (A) N-terminally tagged MBP-Mer2FL; (B) N-terminally tagged MBP-Mer2core; (C) N-terminally tagged MBP-Mer2 255C; (D) N-terminally tagged MBP-Mer2 1–139; (E) N-terminally tagged MBP-Spp1; (F) untagged Spp1; (G) a mixture of untagged Spp1 with N-terminally tagged MBP-Mer2FL the excess of Spp1 is in the right hand peak; (H) a mixture of untagged Spp1 with N-terminally tagged MBP-Mer2core the excess of Spp1 is in the right hand peak.
Figure 1—figure supplement 4
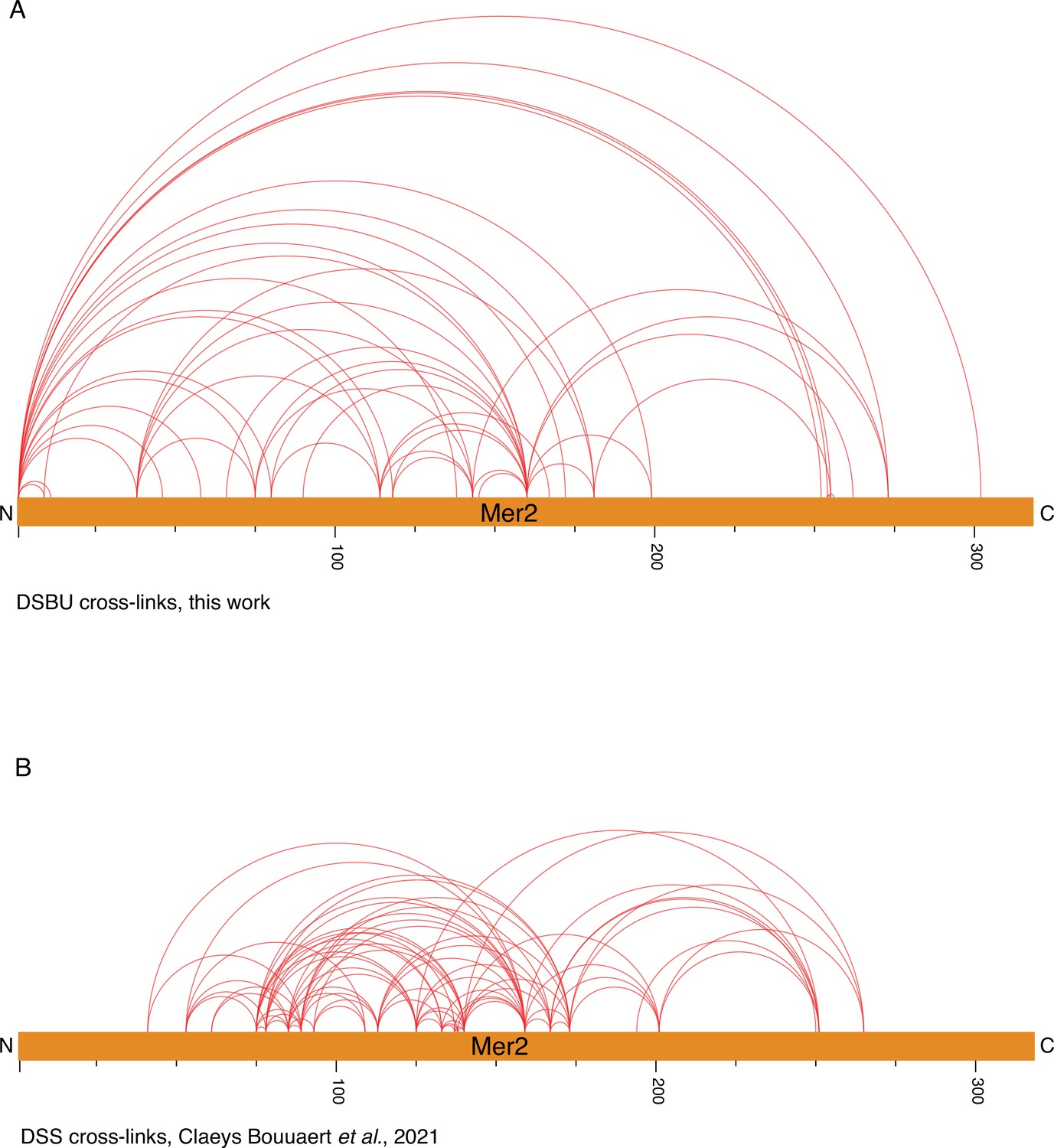
Cross-linking coupled to mass spectrometry (XL-MS) on Mer2 alone.
(A) Visualisation of disuccinimidyl dibutyric urea (DSBU) cross-linked Mer2FL alone. (B) Visualisation of the DSS cross-linked Mer2 sample (Claeys Bouuaert et al., 2021). Both images prepared using XVis (Grimm et al., 2015).
Figure 2
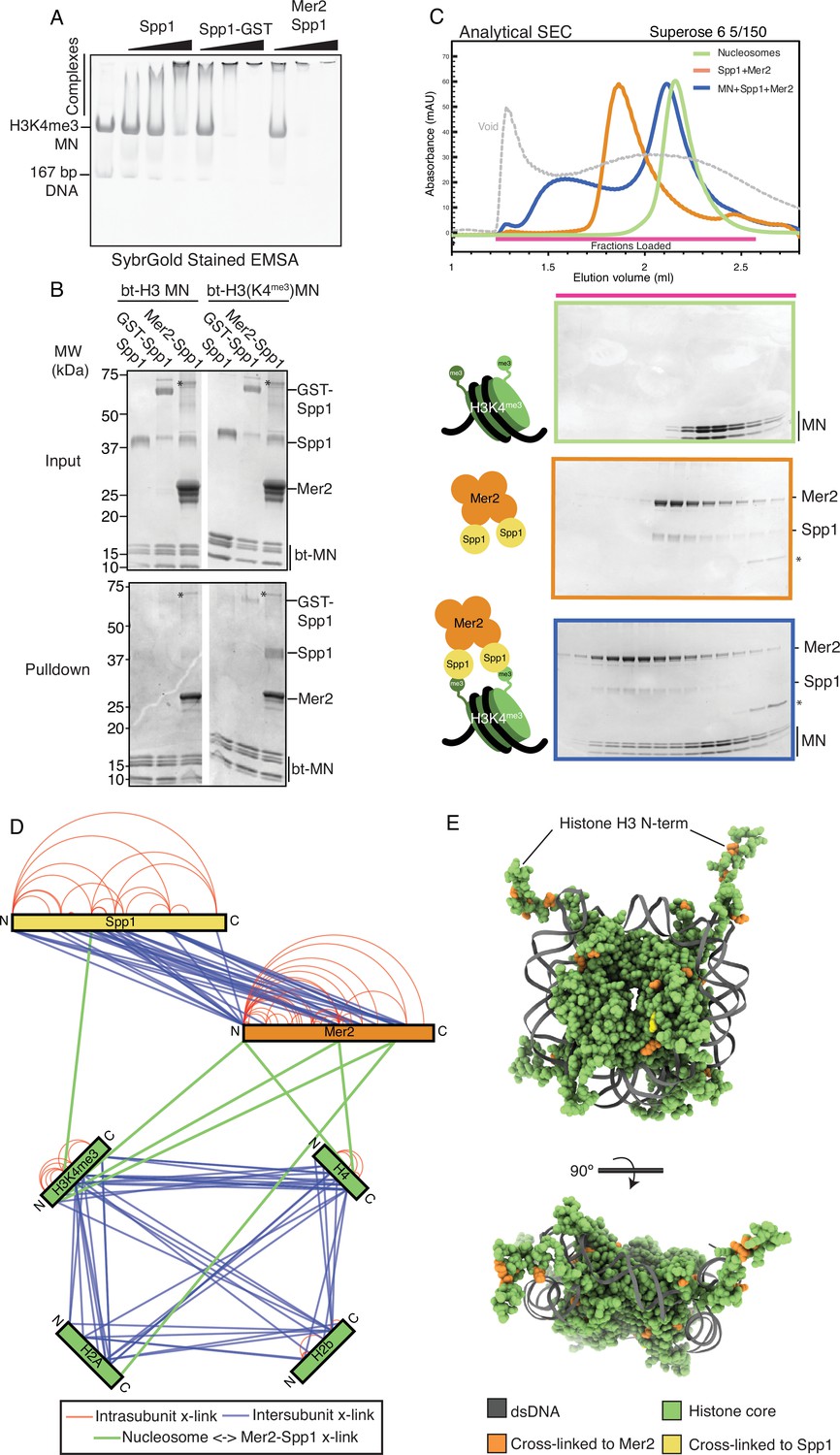
Mer2-Spp1 complex binding to H3K4me3 mononucleosomes.
(A) Electrophoretic mobility shift assay (EMSA) of different Spp1 variants on H3K4me3 nucleosomes. 0.2 μM of H3K4me3 nucleosomes were incubated with 0.33, 1, and 3 μM protein (Spp1, Spp1 with an N-terminal GST fusion, and Mer2-Spp1). Gel was post-stained with SYBRGold. (B) Biotinylated mononucleosome pulldown of different Spp1 variants. 0.5 µM nucleosomes wrapped with 167 bp of biotinylated DNA (either with or without the H3K4me3 modification) were incubated with 1.5 µM protein (same proteins as in A). Samples were taken for the input before incubation with streptavidin beads. The beads were then washed and eluted with 1× Laemmli buffer. Input and elution samples were run on a 10–20% SDS-PAGE gel and stained with InstantBlue. Asterisk marks residual uncleaved MBP-Mer2 in the Mer2-Spp1 lanes. (C) Size exclusion chromatography coupled to multi-angle light scattering (SEC) analysis of Mer2-Spp1-MN complex. 50 µL of 5 µM H3K4me3 mononucleosomes (green), 50 µL of 5 µM Mer2-Spp1 complex (orange), and 50 µL of a 1:1 (5 µM of each) mixture (blue) were run on a Superose 6 5/150 column. The same fractions were loaded in each case (magenta line) onto an SDS-PAGE gel and stained with InstantBlue. (D) Cross-linking coupled to mass spectrometry (XL-MS) analysis of the Mer2-Spp1-mononucleosome complex. Samples were cross-linked with disuccinimidyl dibutyric urea (DSBU), and data analysis carried out according to the Materials and methods. Cross-links were filtered so as to give a 1% false discovery rate. Figure was prepared using XVis (Grimm et al., 2015). Intramolecular cross-links are shown in red, intermolecular cross-links shown in blue. Cross-links between Mer2 and nucleosomes, and Spp1 and nucleosomes shown as thick green lines. (E) Model of nucleosome cross-links. The nucleosomes proximal cross-links from (D) were modelled onto a crystal structure of a Xenopus laevis nucleosome (PDB ID 1K × 5; Davey et al., 2002). Those histone residues that cross-linked to Mer2 are coloured in orange, those that cross-link to Spp1 in yellow. A side and top-down view of the nucleosome are provided. DNA is coloured in dark grey, and histone residues that did not cross-link to Spp1 or Mer2 are coloured green.
-
Figure 2—source data 1
Raw gel data for electrophoretic mobility shift assays (EMSAs) (triplicate), pulldowns, and size exclusion chromatography (SEC) experiments.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig2-data1-v2.zip
Figure 3 with 2 supplements
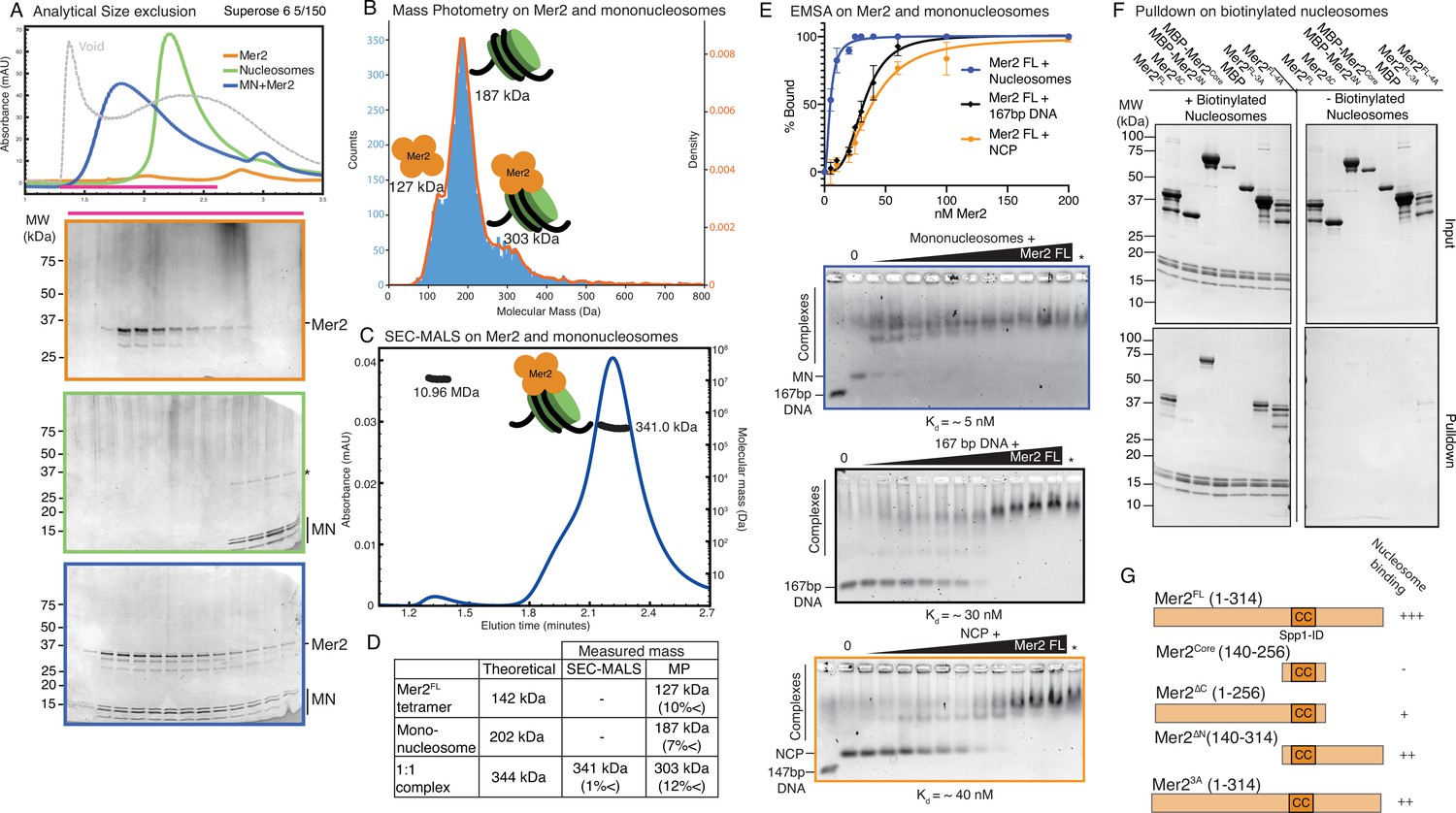
Mer2 binds directly to nucleosomes.
(A) Size exclusion chromatography (SEC) analysis of Mer2-MN complex. 50 µL sample of 5 µM mononucleosomes (green), 50 µL of 5 µM full-length untagged Mer2 (orange), and a mixture of Mer2 and mononucleosomes (blue) were run on a Superose 6 5/150 column. The same fractions were loaded in each case (magenta line) onto an SDS-PAGE gel and stained with InstantBlue. (B) Mass photometry of Mer2 and mononucleosomes. 60 nM of Mer2 and mononucleosomes were mixed and analysed using a Refeyn One mass photometer. Three separate species were identified and the molecular mass determined using a molecular mass standard curve created under identical buffer conditions. Negative data points (i.e. unbinding events) were excluded. (C) SEC coupled to multi-angle light scattering (SEC-MALS) on Mer2-MN complex. Full-length, untagged Mer2 (Mer2FL) was incubated with mononucleosomes and subject to SEC-MALS. Absorbance at 280 nm was constantly monitored (blue trace). Two distinct species were observed, one at 341 kDa, and another at 10.96 MDa (black trace). (D) Summary of molecular mass values for Mer2 FL, mononucleosomes (with 167 bp DNA), and a 1:1 complex. (E) Electrophoretic mobility shift assays (EMSAs) of untagged Mer2 FL. Mer2 was titrated against a constant 5 nM concentration of mononucleosomes (blue), 167 bp ‘601’ DNA (black), or nucleosome core particle (orange). Binding curves were derived based on the Mer2-dependent depletion of free nucleosomes, DNA or NCP, and based on four independent experiments with error bars indicating the SD. Asterisk denotes the background SYBRGold staining from the highest protein concentration alone. The Hill coefficient was added to improve the goodness of fit (h = 1.9 for nucleosomes, 3.3 for DNA and 2.6 for NCP). (F) Pulldowns of Mer2 constructs with nucleosomes. Biotinylated nucleosomes (left panel) were incubated with different Mer2 constructs (as indicated), and samples taken for the input gel. The complexes were captured using streptavidin beads, washed, and eluted in 1× Laemmelli buffer for the pulldown gel. A control experiment (right panel) was conducted without biotinylated nucleosomes to measure non-specific Mer2 interaction with the streptavidin beads. (G) Domain cartoons of Mer2 summarising the different apparent affinities that each Mer2 construct has for mononucleosomes. Based on E and F and Figure 3—figure supplement 2.
-
Figure 3—source data 1
Raw gel data for electrophoretic mobility shift assays (EMSAs) (triplicate), pulldowns, and size exclusion chromatography (SEC) experiments.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig3-data1-v2.zip
Figure 3—figure supplement 1
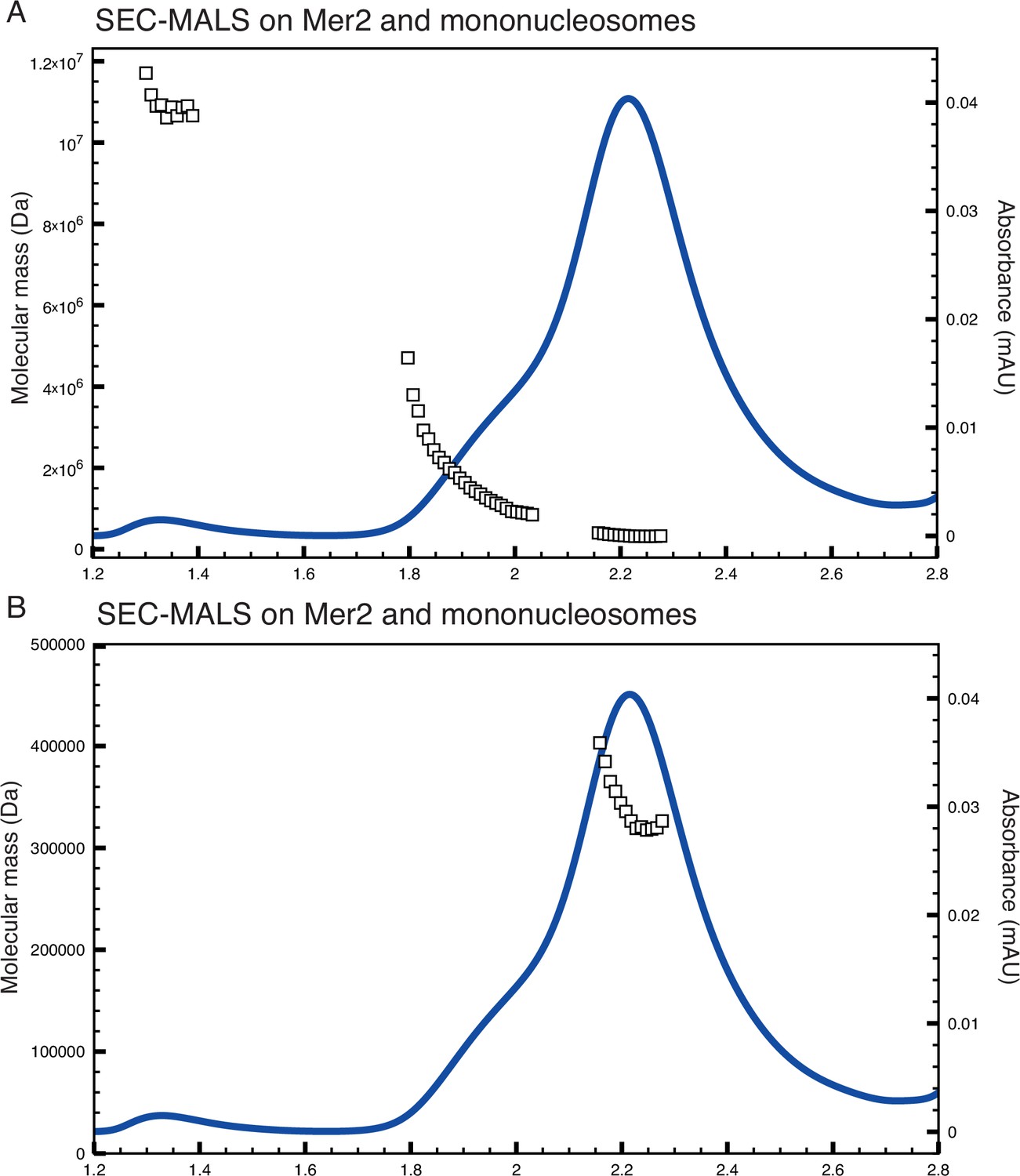
Size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) on Mer2-nucleosome complexes.
(A) SEC-MALS trace as shown in Figure 3C, with the molecular mass plotted on a linear (rather than logarithmic) scale. Also included is the ‘shoulder’ on the complex peak between 1.8 and 2 mL. (B) Trace as in (A), but with the higher molecular mass complexes excluded and a linear x-axis to show the molecular mass distribution of the complex peak.
Figure 3—figure supplement 2
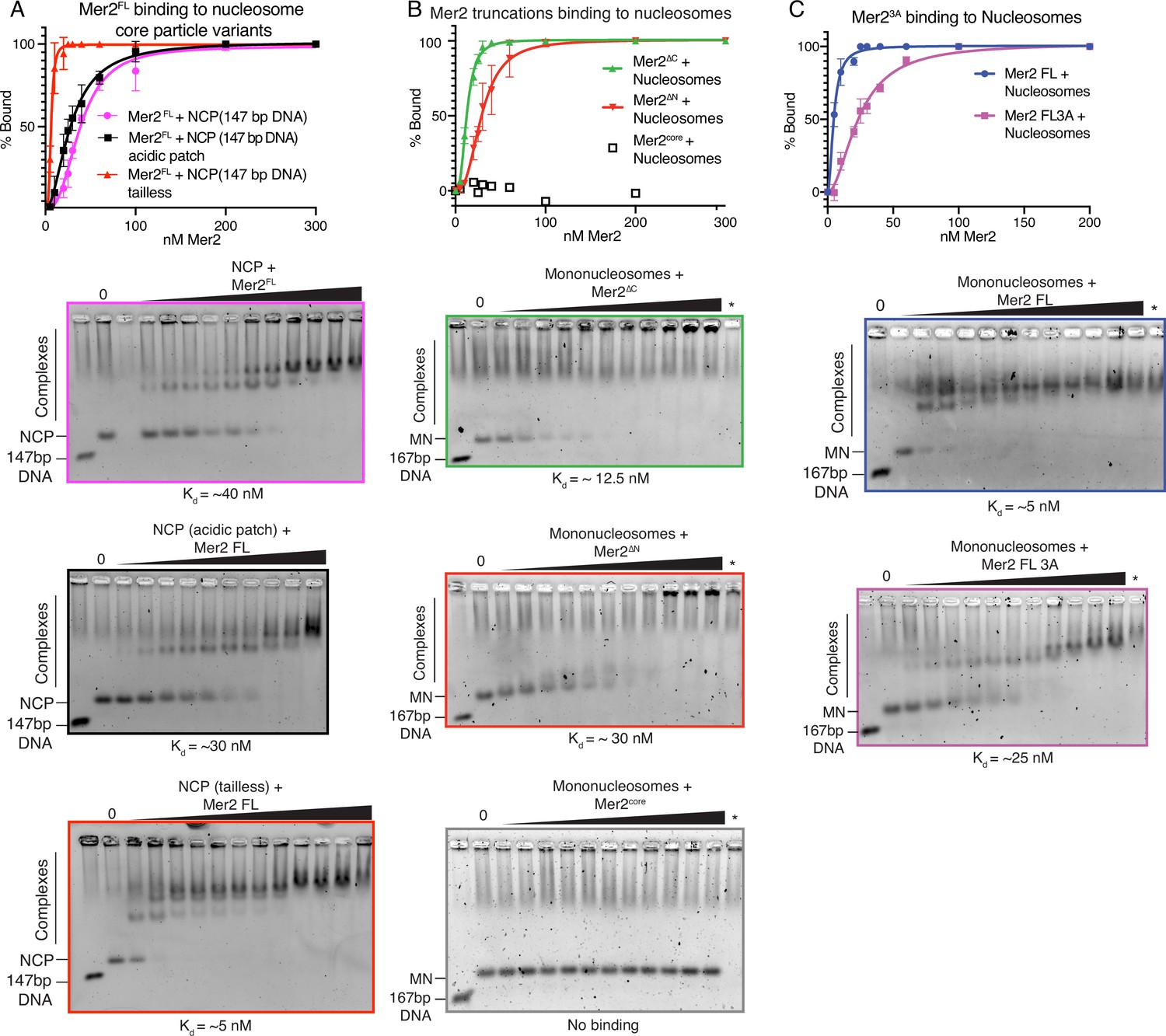
Electrophoretic mobility shift assays (EMSAs) on Mer2 binding to nucleosomes.
(A) Mer2 binding to nucleosome core particle (NCP) variants. Mer2FL was titrated against different variants of the NCP. Either wildtype (WT) NCPs (pink), acidic patch (56T-E61T-E64T-D90S-E91T-E92T) NCPs (black), or tailless NCPs (red) were used at a constant concentration of 10 nM. Error bars indicate the SD of three independent experiments. Hill coefficient for fitting in each case was 2.6 for NCP, 1.9 for NCP acidic patch mutant, and 3.4 for NCP tailless. (B) Mer2 truncation binding to mononucleosomes. Different Mer2 constructs were titrated against mononucleosomes at a constant concentration of 10 nM. Mer2∆C (green), Mer2∆N (red), or Mer2core were used. Error bars indicate the SD of three independent experiments. Hill coefficient for fitting in each case was 2.6 for Mer2∆C and 2.8 Mer2∆N. (C) Mer23A binding to mononucleosomes. Either Mer2FL (blue trace) or Mer23A (purple trace) titrated against mononucleosomes. Error bars indicate the SD of three independent experiments. Hill coefficient for fitting both Mer2FL and Mer23A in each case was 1.9.
-
Figure 3—figure supplement 2—source data 1
Raw gel data for EMSAs (triplicate).
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig3-figsupp2-data1-v2.zip
Figure 4 with 1 supplement

Mer2 binds to the axis via Hop1.
(A) Quantification of Streptactin pulldowns, or amylose pulldowns on either Strep-Hop1 (black) or the Strep-Hop1/Red1I743R-MBP (red) complex against Mer2 (from gels shown in Figure 4—figure supplement 1). In all cases the intensity of Mer2 was quantified as a factor of Hop1 intensity. Error bars are based on the SD from three independent experiments. (B) Amylose pulldown of the co-expressed Red1I743R-MBP/Hop1 complex and Red1I743R-MBP alone against Mer2. Pulldown was carried out as described in Materials and methods. In the rightmost lane Mer2 alone was used as a negative control on the beads. (C) Streptactin pulldown of different 2xStrepII-Hop1 constructs (as indicated) against untagged Mer2FL. N-terminally 2xStrep-II tagged Hop1 constructs were purified as indicated and used as a bait against Mer2 full length. Pulldown was carried out as described in the Materials and methods. (D) Quantification of experiments equivalent to those shown in C. Intensity of Mer2 was quantified as a factor of Strep-Hop1 intensity. Error bars are based on the SD from four independent experiments. (E) Streptactin pulldown of Strep-Hop1K593A as bait to capture different Mer2 constructs as indicated. The rightmost pulldown is the control showing the non-specific binding of different Mer2 constructs to the Streptactin beads. An anti-Mer2 Western was carried out (lower panel) to confirm the specific interactions due to the weakness of the Mer2 bands in a coomassie stained gel. (F) Representation of cross-linking mass-spectrometry (XL-MS) analysis of the Hop1K593A-Mer2 complex. Cross-links were filtered so as to give a <1% false discovery rate. Figure was prepared using XVis (Grimm et al., 2015). Intramolecular cross-links are shown in red, intermolecular cross-links shown in blue. Thick blue lines represent those intermolecular cross-links that were observed five or more times. The HORMA domain of Hop1, the safety belt (SB), and the closure motif (CM) are highlighted. (G) Homology model of the Hop1 HORMA domain (residues 16–247) using open-Mad2 as a template (PDB 1duj; Luo et al., 2000) is shown in two orientations. The safety belt is highlighted in pink. Those residues that cross-link to Mer2 are highlighted in orange.
-
Figure 4—source data 1
Raw gel data for pulldowns (triplicate and singular), anti-Mer2 Western blot raw blot data.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig4-data1-v2.zip
Figure 4—figure supplement 1
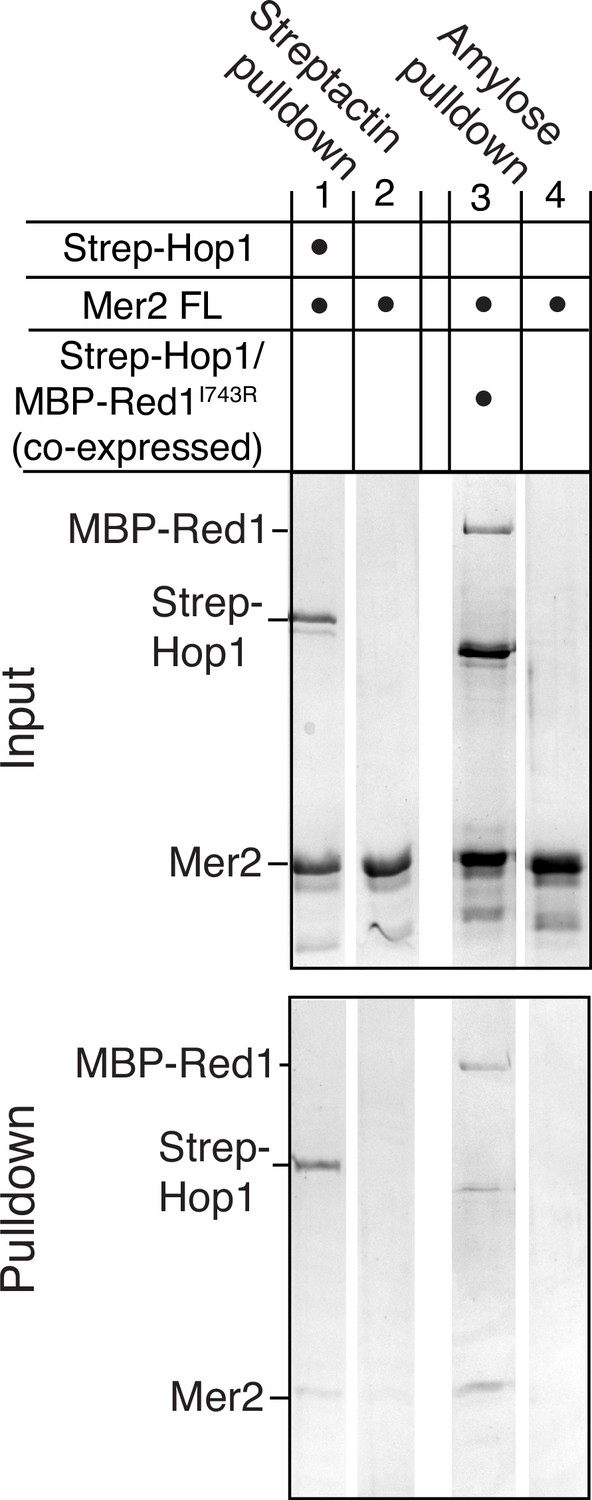
Mer2 pulldowns on Hop1-Red1 complexes.
Mer2FL was incubated either with or without N-terminally tagged 2xStrep-II-Hop1. Samples were then incubated with Streptactin beads to either capture the complex (lane 1) or to determine background binding of Mer2 (lane 2). Mer2FL was also incubated with or without a co-expressed and purified complex of 2xStrep-II-Hop1 and Red1I743R-MBP. To exclude the excess Hop1 in the complex, amylose beads were used to pull down Red1 (lane 3) or to determine background binding of Mer2 (lane 4).
-
Figure 4—figure supplement 1—source data 1
Raw gel data for pulldowns.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig4-figsupp1-data1-v2.zip
Figure 5 with 2 supplements
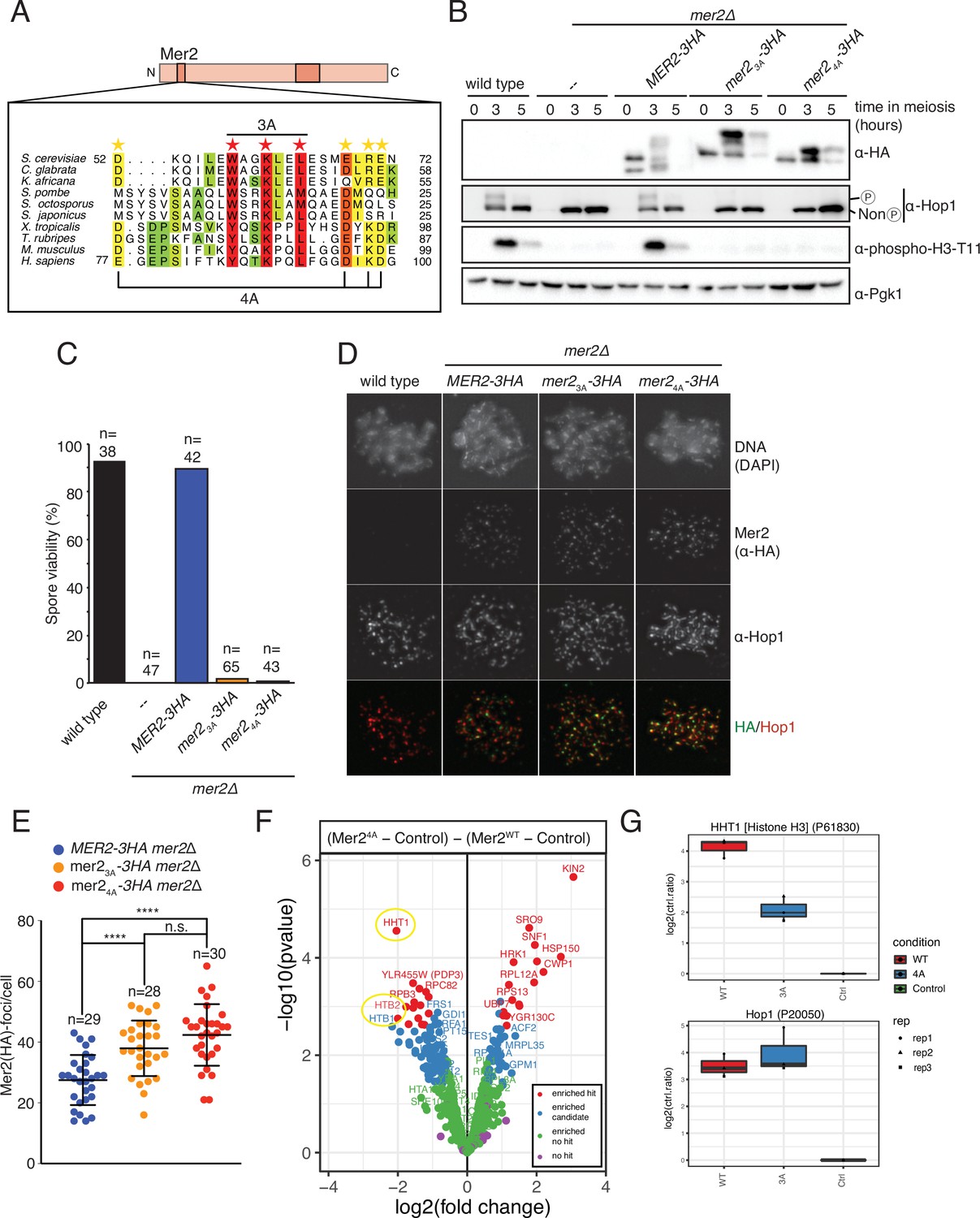
Mer2 3A and 4A mutations lead to inviable spores.
(A) Sequence alignments of Mer2 orthologs from multiple clades revealed a previously undescribed conservation in the N-terminal region. We created two mutants, the ‘3A’ mutant (W58A, K61A, L64A; red stars) based on three universally conserved residues and the ‘4A’ mutant (D52A, E68A, R70A, E71A; yellow stars). The periodicity of the red star conserved residues, combined with some secondary structure predictions, suggest that these residues might be on the same face of an alpha-helix. (B) Western blot analysis of meiotic yeast cultures of wildtype, mer2Δ, MER2-3HA mer2Δ, mer2-3a-3HA mer2Δ, and mer2-4a-3HA mer2Δ strains. Time of induction into the meiotic program indicated. See Yeast strains used in this study for strain information. Pgk1 was used as a loading control. Please note the indicated phospho-Hop1, anti-phospho histone H3 was used to measure the activation of Mek1 kinase in response to DSB formation. (C) Quantification of spore viabilities of wildtype, mer2Δ, MER2-3HA mer2Δ, mer2-3a-3HA mer2Δ, and mer2-4a-3HA mer2Δ strains. Number of dissected tetrads is indicated. See Yeast strains used in this study for strain information. (D) Representative images of meiotic chromosome spreads stained for Mer2 (α-HA [green], Hop1, and DNA [blue]) using 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI) from wildtype, MER2-3HA mer2Δ, mer2-3a-3HA mer2Δ, and mer2-4a-3HA mer2Δ strains at t = 3 hr after induction into the meiotic time course. (E) Quantification of the number of Mer2 foci on chromosome spreads of strains used in (D). Mean and standard deviation are indicated. *p ≤ 0.05, **p ≤ 0.01, and ****p ≤ 0.0001; n.s. (non-significant) >0.05. Mann-Whitney U test. Number of analysed cells is indicated. (F) Volcano plots of quantitative immunoprecipitation (IP) mass spectrometry of Mer2WT vs. Mer24A. Proteins are grouped into hits (red) and candidates (blue), according to the fold change and p-value. Proteins appearing on the left show reduced association with Mer23A when compared with Mer2WT, whereas proteins on the right show an increased association with Mer24A when compared with Mer2WT. Circled in yellow are the histone proteins Hht1 (histone H3) and Htb1/Htb2 (histone H2B). (G) Detail of Mer24A vs. Mer2WT in IP quantitative mass spectrometry analysis for Hht1 (histone H3); top panel, and Hop1; lower panel. Association with Hht1 is reduced ~2-fold in Mer24A whereas it is essentially unchanged for Hop1.
-
Figure 5—source data 1
Raw Western blot data plus explanatory data.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig5-data1-v2.zip
Figure 5—figure supplement 1
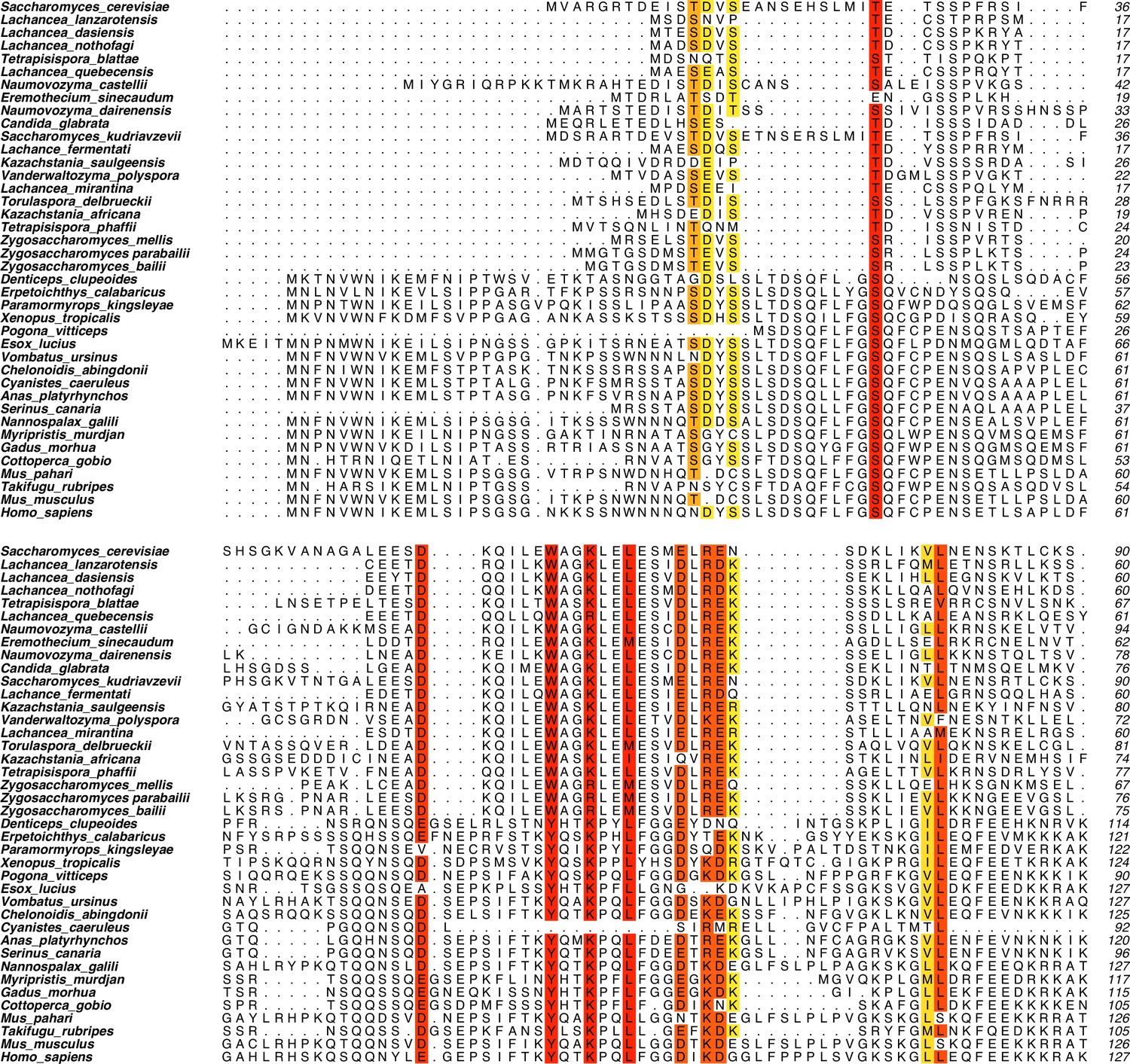
Mer2 sequence alignment.
An iterative round of BLAST searches and MUSCLE alignments using Mer2 and IHO1 as starting points were used to generate an alignment of Mer2 orthologs. Residues were coloured from red to yellow according to degree of similarity within set similarity groups (DE, FWY, HKR, ILMV, NQ, ST).
Figure 5—figure supplement 2
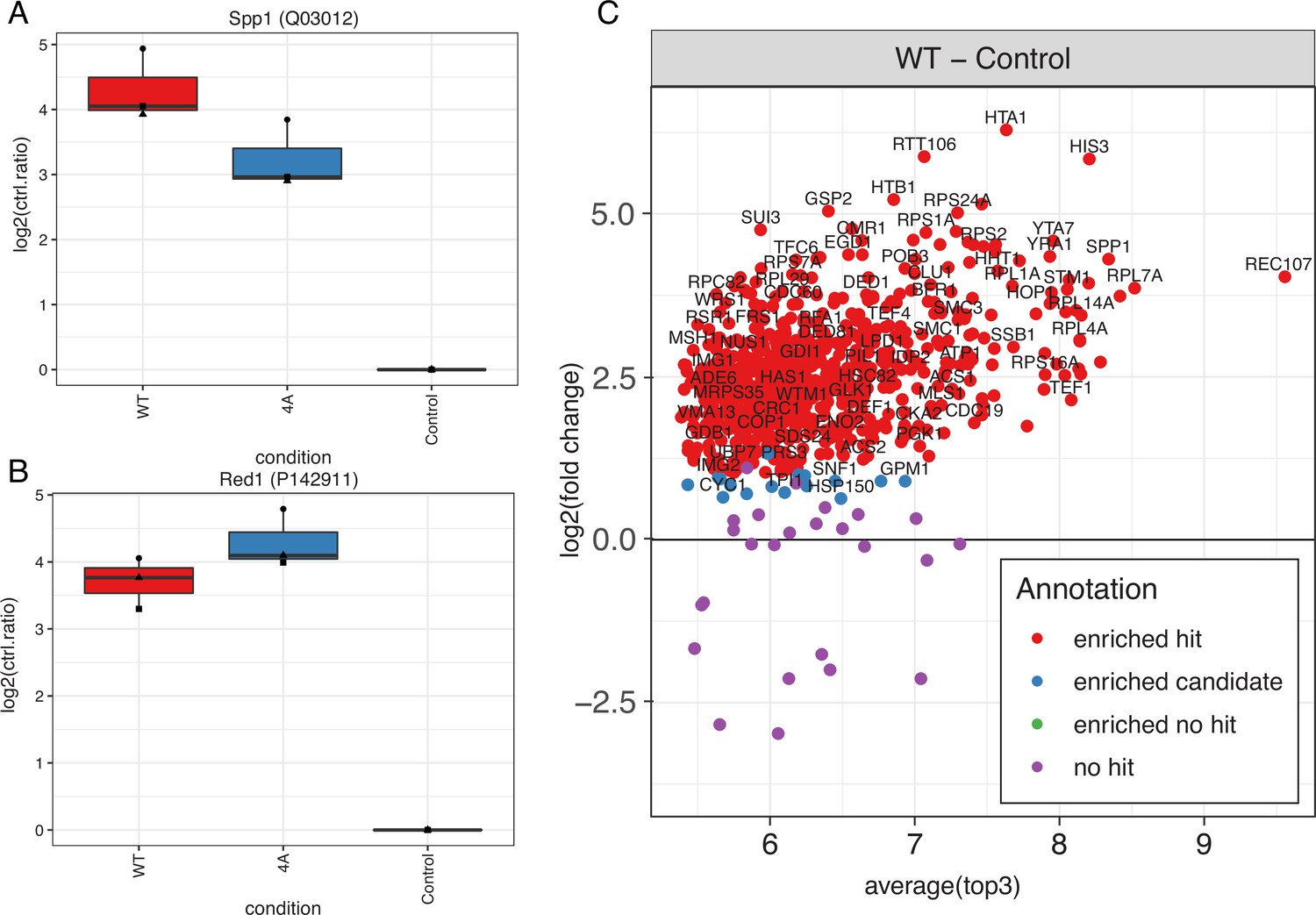
Immunoprecipitation mass spectrometry (IP-MS) of Mer2WT vs. Mer24A (nucleosomes, Hop1, Red1).
Quantitative analysis revealed that in the Mer24A mutant association with both Spp1 (A) and Red1 (B) was unperturbed (Red1 presumably via Mer2 binding to Hop1). (C) Summary of the extent of protein enrichment in the Mer2WT IP (Rec107 = Mer2).
Figure 6 with 4 supplements
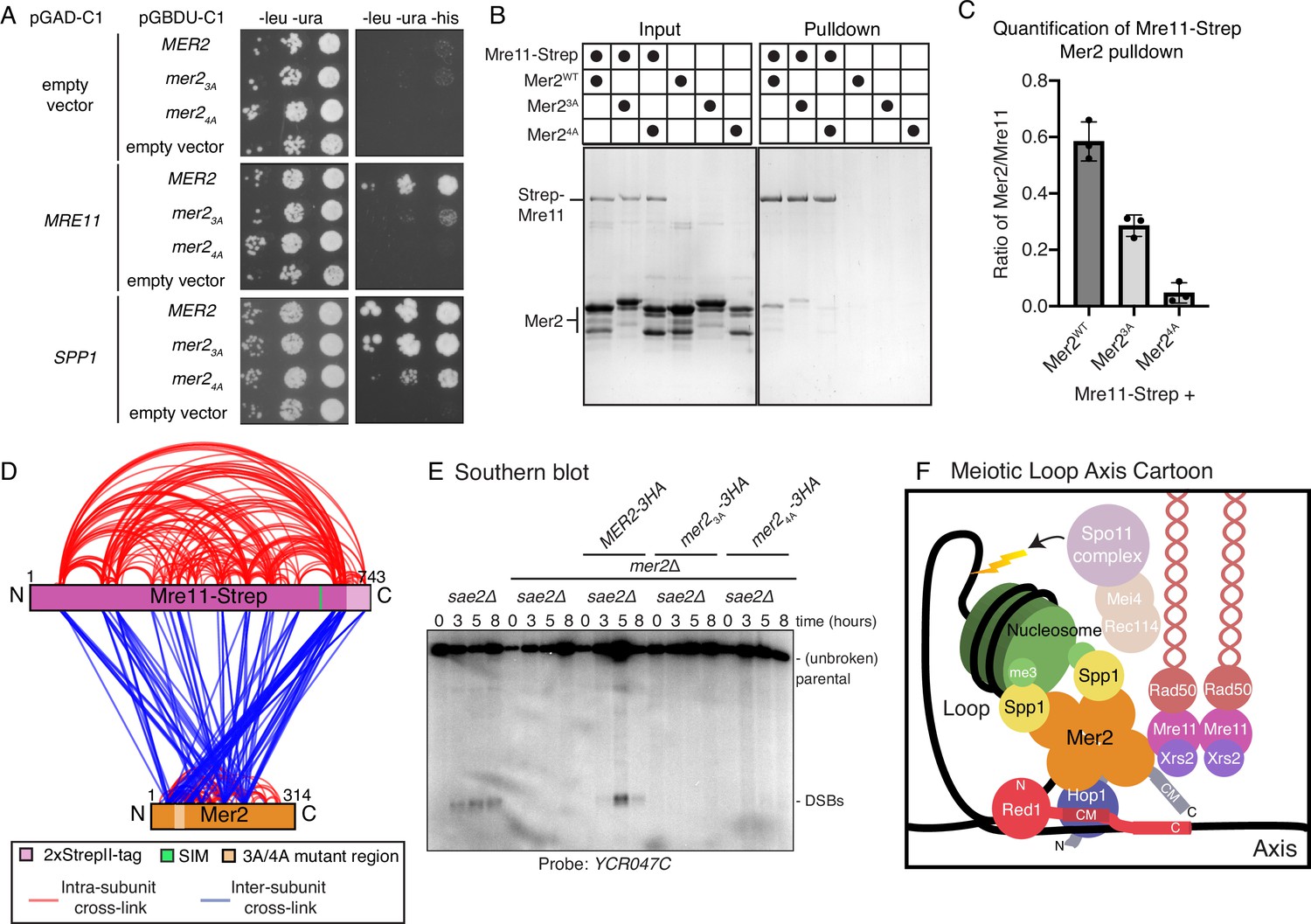
Mer2 binds directly to Mre11.
(A) Yeast-2-hybrid experiments with Mer2WT, Mer23A, and Mer24A mutants. Yeast were transformed with the pGAD-C1 (activation domain) and pGBDU (DNA binding domain) plasmids as indicated. Cells were grown and pipetted onto non-selective (left) or selective plates (right) at three serial concentrations. (B) In vitro pulldowns between Strep-Mre11 and Mer2. C-terminally 2xStrepII-tagged Mre11 was used to capture Mer2WT, Mer23A, and Mer24A on Streptactin beads. Simultaneously Mer2WT, Mer23A, and Mer24A in the absence of Mre11-Strep were used to determine non-specific binding to the beads. (C) Quantification of Strep-Mre11 pulldowns against Mer2WT, Mer23A, and Mer24A. Intensity of Mer2 was quantified as a factor of Strep-Mre11 intensity. Error bars are based on the SD from three independent experiments. (D) Representation of cross-linking mass spectrometry (XL-MS) analysis of the Mre11-Mer2 complex. Cross-links were filtered so as to give a <1% false discovery rate. Figure was prepared using XVis (Grimm et al., 2015). Intramolecular cross-links are shown in red, intermolecular cross-links shown in blue. The location of the SIM is the highest scoring motif (score = 64.397) in Mre11 as determined by the GPS-SUMO prediction tool (Zhao et al., 2014). (E) Southern blot analysis comparing double-strand break (DSB) levels in sae2Δ, mer2Δsae2Δ, MER2-3HA mer2Δsae2Δ, mer2-3a-3HA mer2Δsae2Δ, and mer2-4a-3HA mer2Δsae2Δ strains. Cells were harvested at indicated time points post induction of meiosis and faster migrating DNA species (indicative of DSBs) were detected using a probe for the YCR047C locus. sae2Δ cells are resection-deficient and cannot repair DSBs. (F) Final meiotic loop axis cartoon. Summarises the novel interactions shown in the previous figures while highlighting the integration of Mre11 via its direct binding to Mer2, into the loop axis model. Briefly, a tetrameric Mer2 binds to two copies of Spp1, while also binding to nucleosomes. Mer2 is also able to interact directly with Hop1, in a manner that is enhanced when Hop1 is bound to Red1. Finally Mer2 also interacts with Mre11 via a conserved N-terminal motif.
-
Figure 6—source data 1
Raw gel data for pulldowns.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig6-data1-v2.zip
-
Figure 6—source data 2
Raw gel data for Southern blots.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig6-data2-v2.zip
Figure 6—figure supplement 1

Yeast-2-hybrid (Y2H) with Mer2 and Spo11 complex components.
Y2H experiments with Mer2WT, Mer23A, and Mer24A mutants were done similarly as in Figure 6A.
Figure 6—figure supplement 2
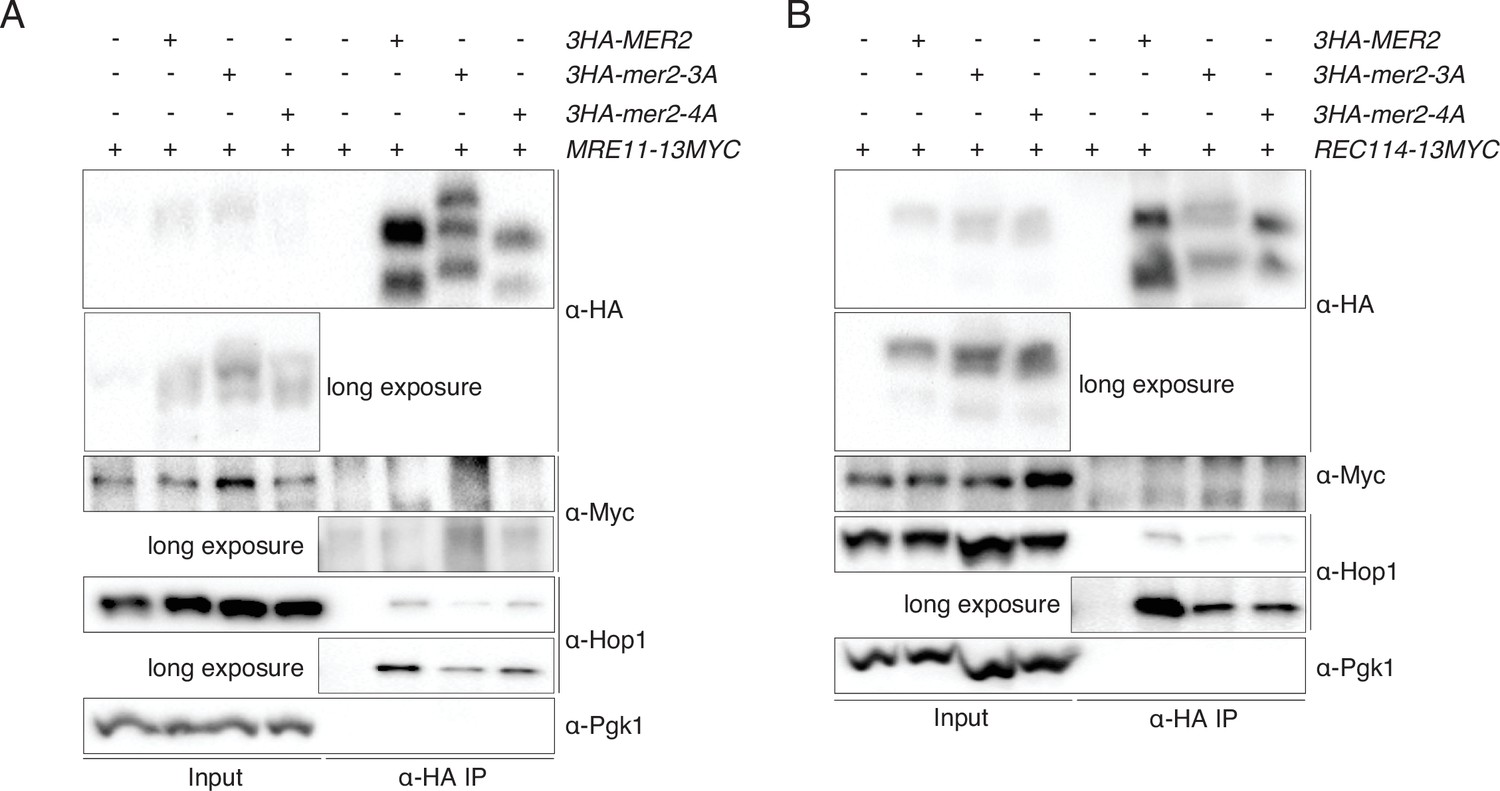
Immunoprecipitations (IPs) of Mer2WT, Mer23A, and Mer24A against Rec114 and Mre11.
(A) IP of Mer2WT-3HA, Mer23A-3HA, and Mer24A-3HA to query potential interaction between Mre11-13myc and Mer2WT-3HA, Mer23A-3HA, and Mer24A-3HA. Cells were harvested at 4 hr post induction of meiosis. Pgk1 was probed as negative control, and Hop1 was probed as a positive control. (B) IP of Mer2WT-3HA, Mer23A-3HA, and Mer24A-3HA to query potential interaction between Rec114-13myc and Mer2WT-3HA, Mer23A-3HA, and Mer24A-3HA. Cells were harvested at 4 hr post induction of meiosis. Pgk1 was probed as negative control, and Hop1 was probed as a positive control.
-
Figure 6—figure supplement 2—source data 1
Raw gel data for western blots, plus explanatory data.
- https://cdn.elifesciences.org/articles/72330/elife-72330-fig6-figsupp2-data1-v2.zip
Figure 6—figure supplement 3
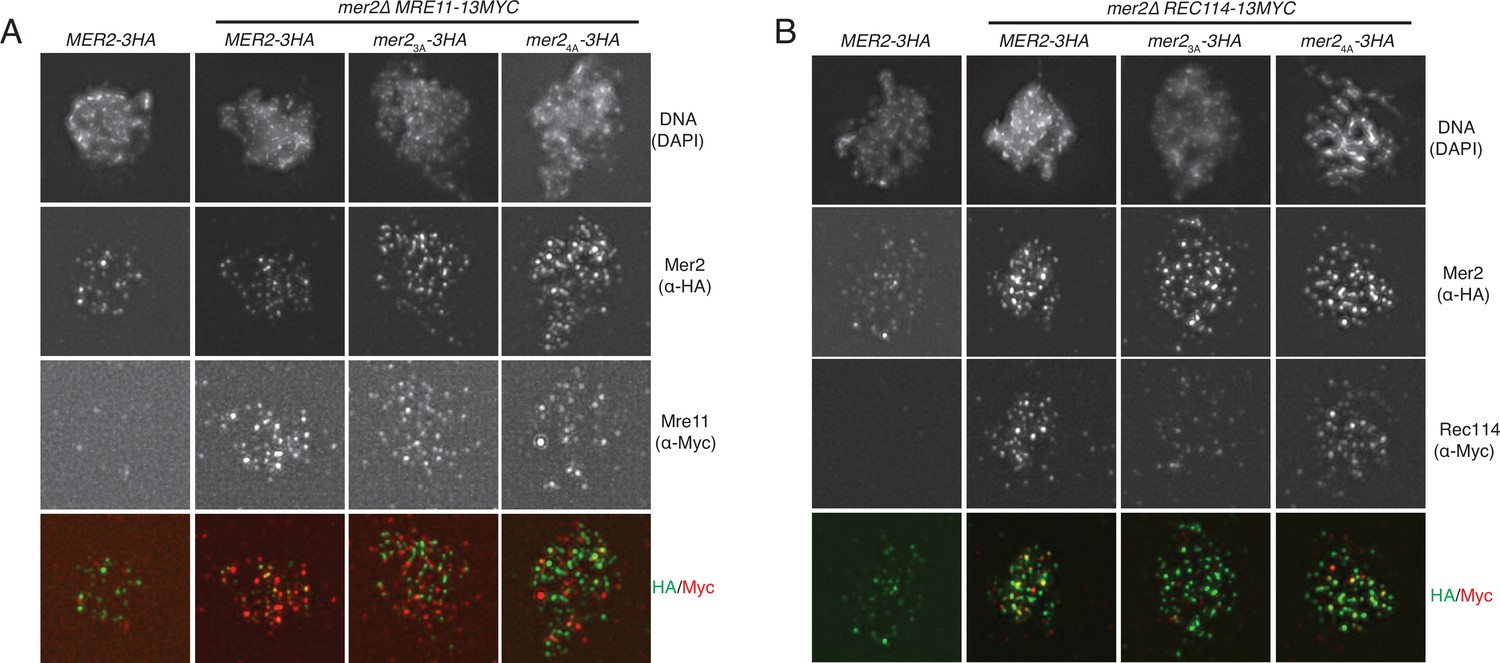
IFs of Mer2WT, Mer23A, and Mer24A against Rec114 and Mre11.
(A) Representative images of meiotic chromosome spreads stained for Mer2 (α-HA [green]), and Mre11 (α-Myc [red]), and DNA (blue) using 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI) from MER2-3HA mer2Δ, MRE11-13MYC MER2-3HA mer2Δ, MRE11-13MYC mer2Δmer2-3a-3HA mer2Δ, and MRE11-13MYC mer2-4a-3HA mer2Δ strains at t = 3 hr after induction into the meiotic time course. (B) Representative images of meiotic chromosome spreads stained for Mer2 α-HA [green], and Rec114 (α-Myc [red]), and DNA (blue) using DAPI from MER2-3HA mer2Δ, REC114-13MYC MER2-3HA mer2Δ, REC114-13MYC mer2Δmer2-3a-3HA mer2Δ, and REC114-13MYC mer2-4a-3HA mer2Δ strains at t = 3 hr after induction into the meiotic time course.
Figure 6—figure supplement 4
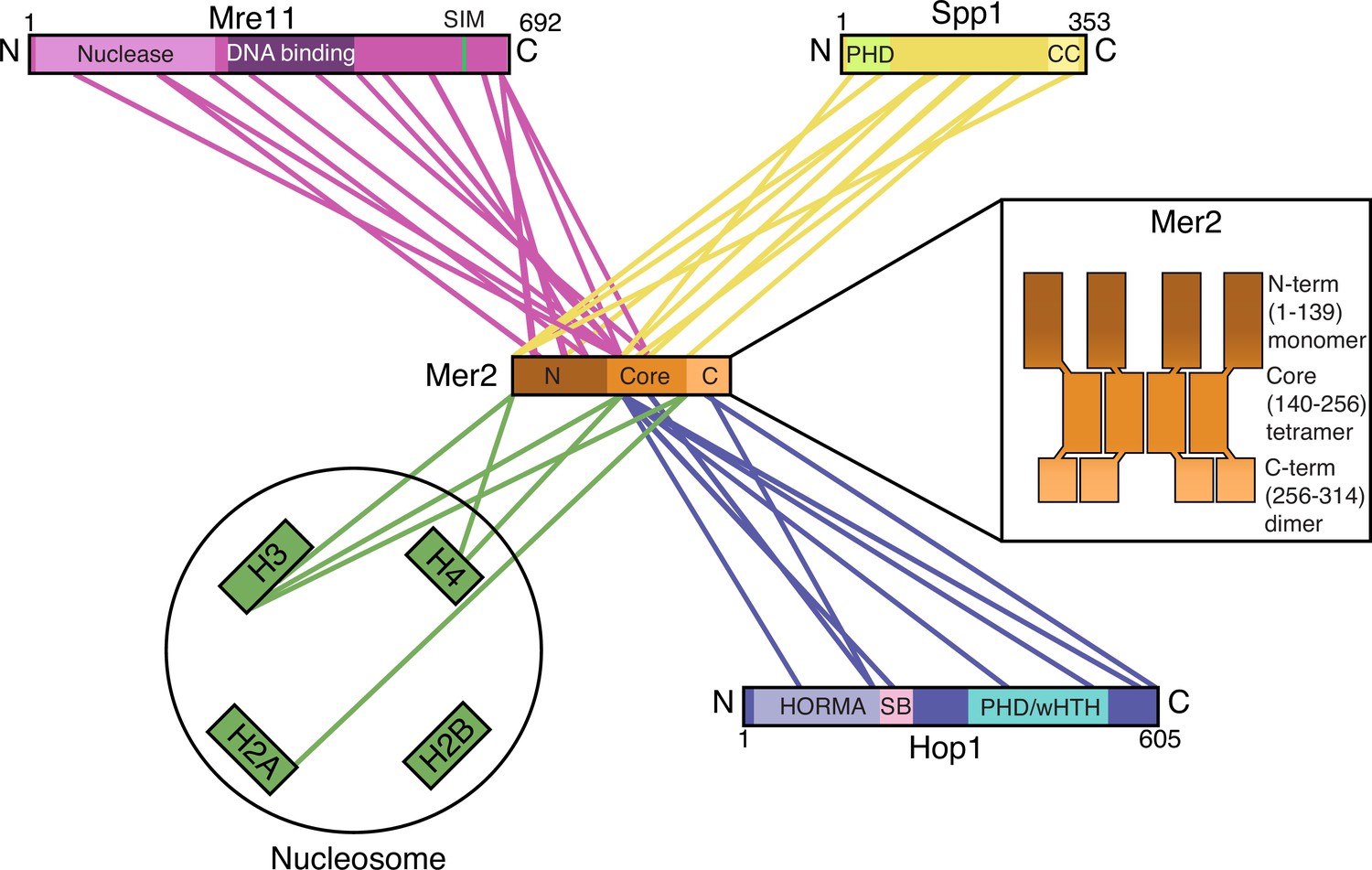
Summary of Mer2 interactions and organisation.
Simplified cartoon representation of four cross-linking coupled to mass spectrometry (XL-MS) experiments (Mer2-Spp1, Figure 1F; Mer2-nucleosomes, Figure 2E; Mer2-Hop1 Figure 4F; Mer2-Mre11 Figure 6D) highlighting those regions of Mer2 that cross-link with different regions of direct binding partners. Only a minimum number of cross-links were retained for representative purposes. Domains are annotated according to Interpro (Mre11 nuclease and DNA binding, Spp1 PHD domain), Pcoils (CC – coiled-coil domain of Spp1), GPS-SUMO (SIM – SUMO interaction motif on Mre11), this work (Mer2 N-terminal domain, core domain, C-terminal domain), or Ur and Corbett, 2021 (Hop1 HORMA domain, safety belt, PHD, winged helix-turn-helix). The internal organisation of Mer2 is based on the size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) experiments shown in Figure 1—figure supplement 3B, C, D. The organisation of the Mer2 domains is not supposed to indicate a parallel arrangement of coiled-coils, but is indicative of stoichiometry. Please note that according to our analyses, we believe that the Mer2-Spp1 complex exists in a 4:2 stoichiometry. Due to the fact that we currently do not know the stoichiometries of the Mer2-Mre11 and Mer2-Hop1 assemblies, we have chosen to not represent the known stoichiometry of Mer2-Spp1 in this schematic representation.
Additional files
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/72330/elife-72330-transrepform1-v2.pdf
-
Source data 1
Data files for XL-MS used to make the XL-MS maps in various figures.
- https://cdn.elifesciences.org/articles/72330/elife-72330-data1-v2.zip
-
Source data 2
All IP-MS source data, used for quantitative mass-spec.
- https://cdn.elifesciences.org/articles/72330/elife-72330-data2-v2.zip
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Novel mechanistic insights into the role of Mer2 as the keystone of meiotic DNA break formation
eLife 10:e72330.
https://doi.org/10.7554/eLife.72330




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}