Dynamic proteomic and phosphoproteomic atlas of corticostriatal axons in neurodevelopment
- Department of Neurobiology, Northwestern University, United States
- The Chemistry of Life Processes Institute, Northwestern University, United States
- Department of Psychiatry, University of Pittsburgh, United States
Figures
Figure 1 with 1 supplement
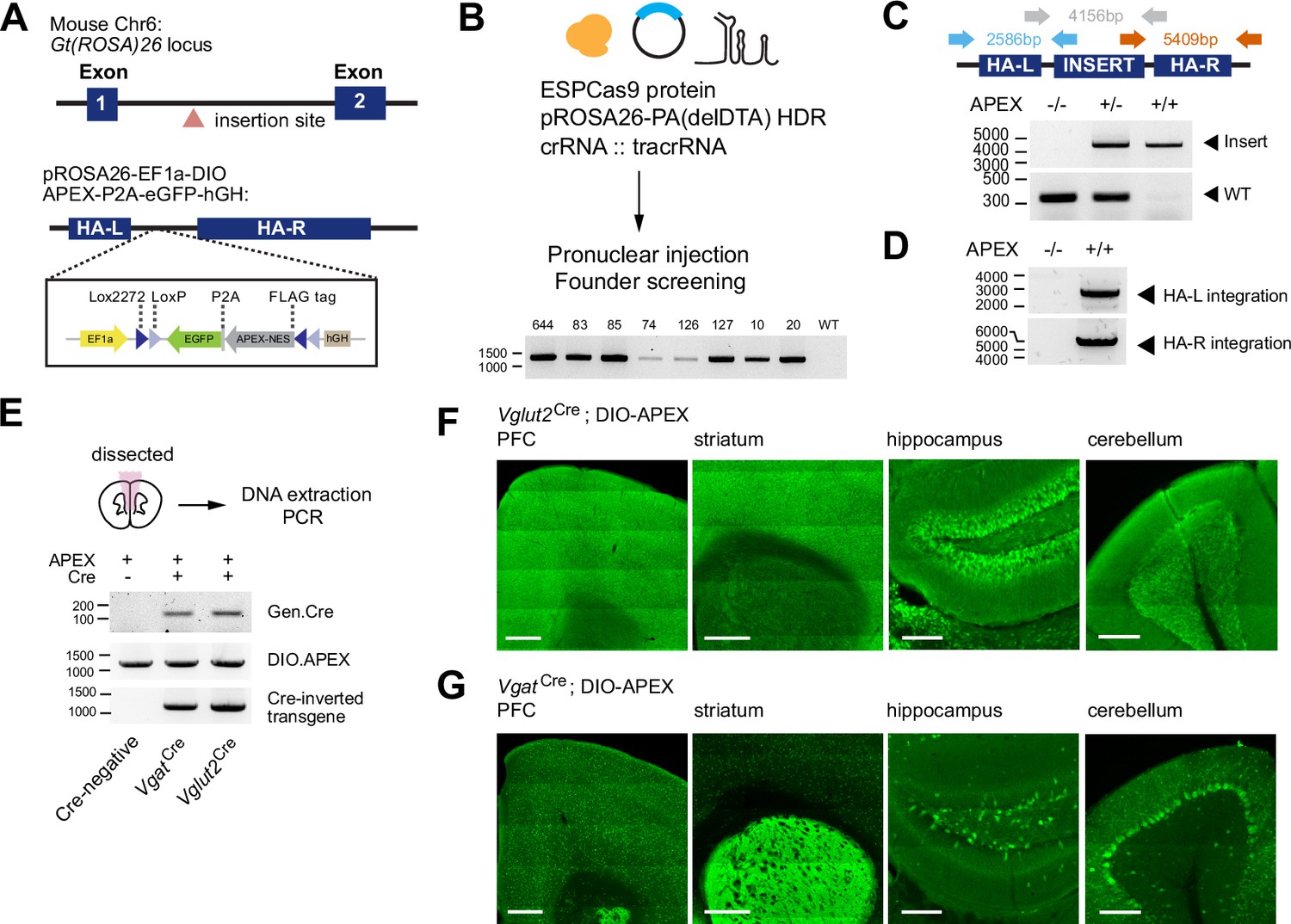
Generation of a Cre-dependent APEX2 reporter mouse line.
(A) Design of a Cre-dependent APEX reporter mouse line. A Cre-dependent APEX transgene under the control of EF1a promoter is targeted for knock-in at the Gt(ROSA)26 locus in the mouse genome. Inverted APEX-NES (nuclear exporting sequence) is flanked by lox sites in a double floxed inverted orientation (DIO). (B) CRISPR/Cas9 strategy for mouse line generation. Enhanced specificity Cas9 (ESPCas9), targeting vector, and crRNA::tracrRNA duplex were used in pronuclear injection to generate founders. Agarose gel electrophoresis shows founders containing APEX transgene. (C) PCR amplification across the APEX transgene. Gray arrows: 317 bp WT loci and ~4 kb APEX transgene. (D) PCR amplification across the homology arms evaluating genomic integration. Blue arrow, left homology arm (HA-L), 2586 bp. Red arrow, right homology arm (HA-R), 5409 bp. (E) PCR amplification of genomic DNA extracted from the brain for the presence of Cre, the APEX transgene before and after Cre-mediated recombination in Cre-/-, VgatCre+/- and Vglut2Cre+/- animals. (F) Cre-dependent expression of EGFP reporter in the prefrontal cortex (PFC), striatum (STR), hippocampus (HC), and cerebellum (CB) of Vglut2Cre mice crossed to APEX reporter line (immunostaining using anti-GFP antibody). Scale bars, 500 µm for PFC and STR, and 200 µm for HC and CB. (G) same as (F) but for VgatCre.
-
Figure 1—source data 1
Raw and uncropped gel images for Figure 1b, c, d and e.
- https://cdn.elifesciences.org/articles/78847/elife-78847-fig1-data1-v2.zip
Figure 1—figure supplement 1
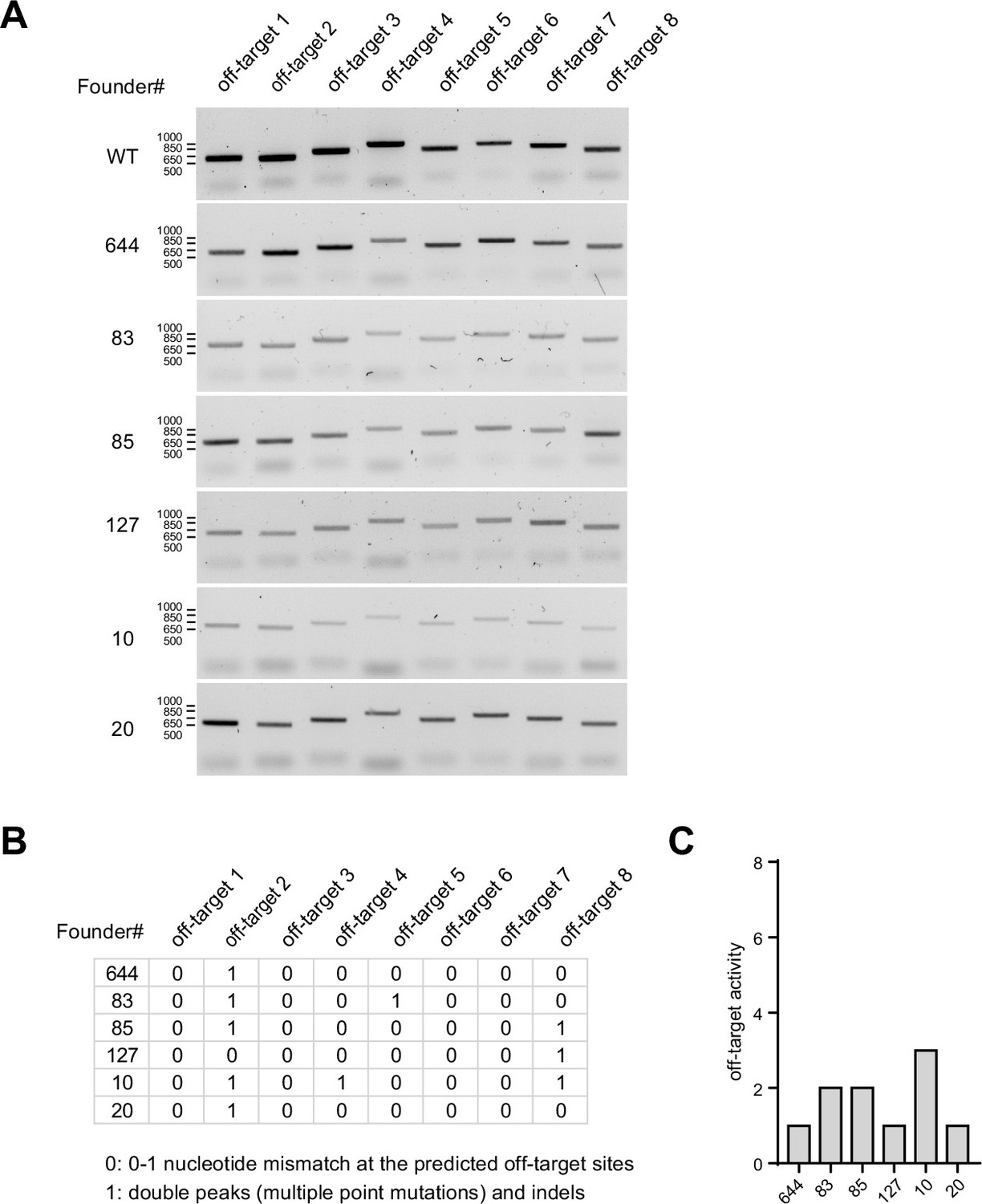
Cas9 off-target analyses in founder animals.
(A) PCR amplification of genomic regions containing off-target sites predicted by Benchling algorithm across six founder animals including C57BL/6 wild type control. (B) Tabulated off-target activity based on Sanger sequencing of PCR products (0 indicates up to 1 nucleotide mismatch compared to the reference genome, and 1 indicates double peak results or indels). (C) Total number of off-target activity events detected in each founder.
-
Figure 1—figure supplement 1—source data 1
Raw and uncropped gel images for Figure 1—figure supplement 1a.
- https://cdn.elifesciences.org/articles/78847/elife-78847-fig1-figsupp1-data1-v2.zip
Figure 2 with 1 supplement
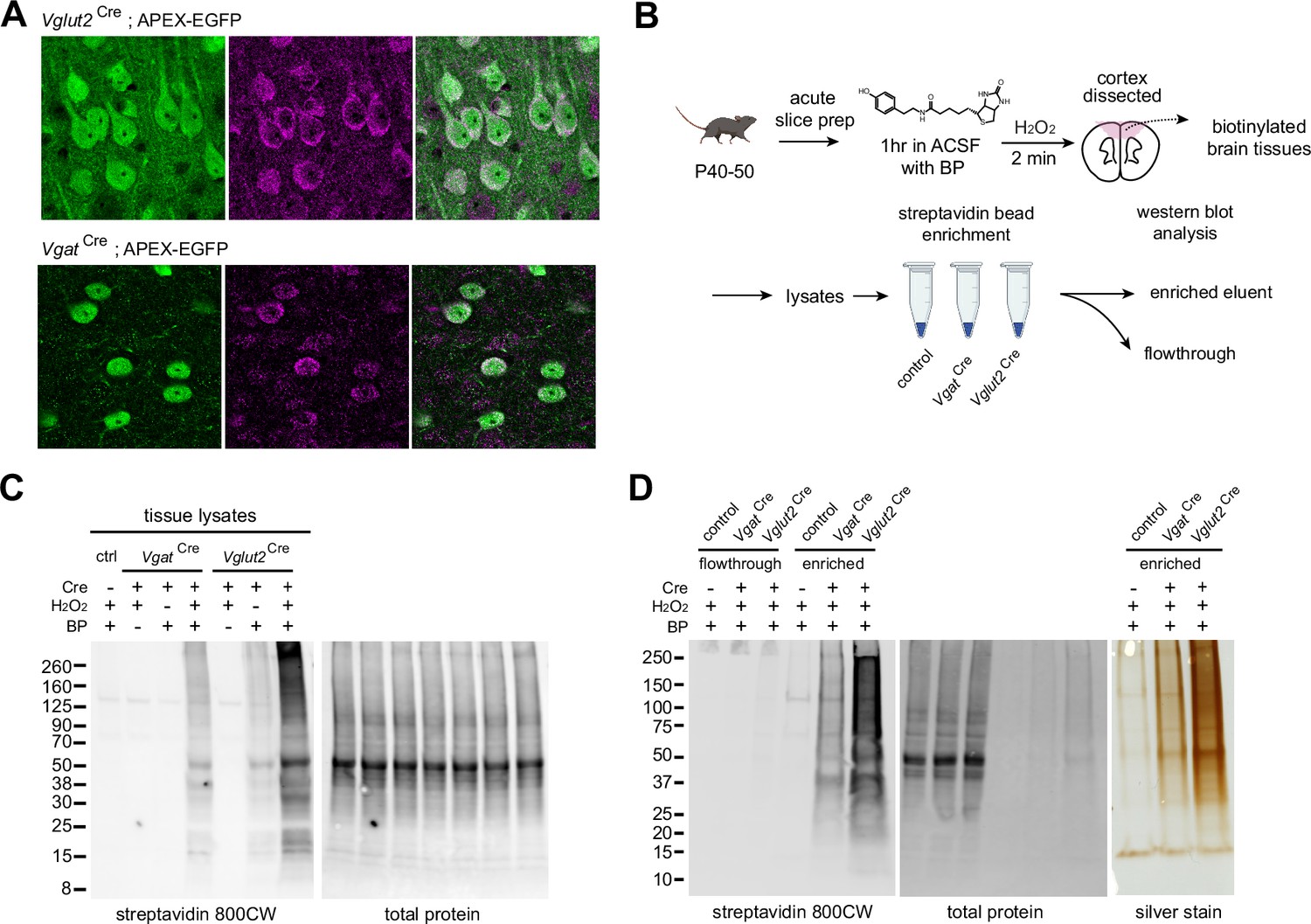
APEX-mediated biotinylation in brain tissue.
(A) APEX subcellular localization in the mPFC of Vglut2Cre;APEX and VgatCre;APEX (immunostained EGFP 488, anti-FLAG 647 for APEX, scale bars 20 µm). (B) Sample preparation schematic for protein analysis. Acute slices were incubated in carbogenated artificial cerebrospinal fluid (ACSF) supplemented with 500 µM biotin phenol for 1 hr. Sections were briefly rinsed in ACSF. APEX was activated by 0.03% H2O2 ACSF for 2 min and quenched in Na ascorbate ACSF solution. Protein lysates were prepared from dissected tissues. Streptavidin beads were used to enrich tagged proteins for analysis. (C) Cell-type-specific biotinylation in Cre-negative, VgatCre, and Vglut2Cre crosses. Western blot analysis of cortical protein lysates. Biotinylated proteins and total protein loading control were detected by streptavidin-CW 800 and REVERT 700 stain, respectively. (D) Validation of streptavidin bead enrichment. Left, detection of biotinylated proteins in flow-through and enriched fractions. Right, silver stain gel of enrichment output proteins. Differential enrichment outputs reflect differences in GABAergic and glutamatergic cell proportions in the cortex.
-
Figure 2—source data 1
Raw and uncropped blots for Figure 2c and d.
- https://cdn.elifesciences.org/articles/78847/elife-78847-fig2-data1-v2.zip
Figure 2—figure supplement 1
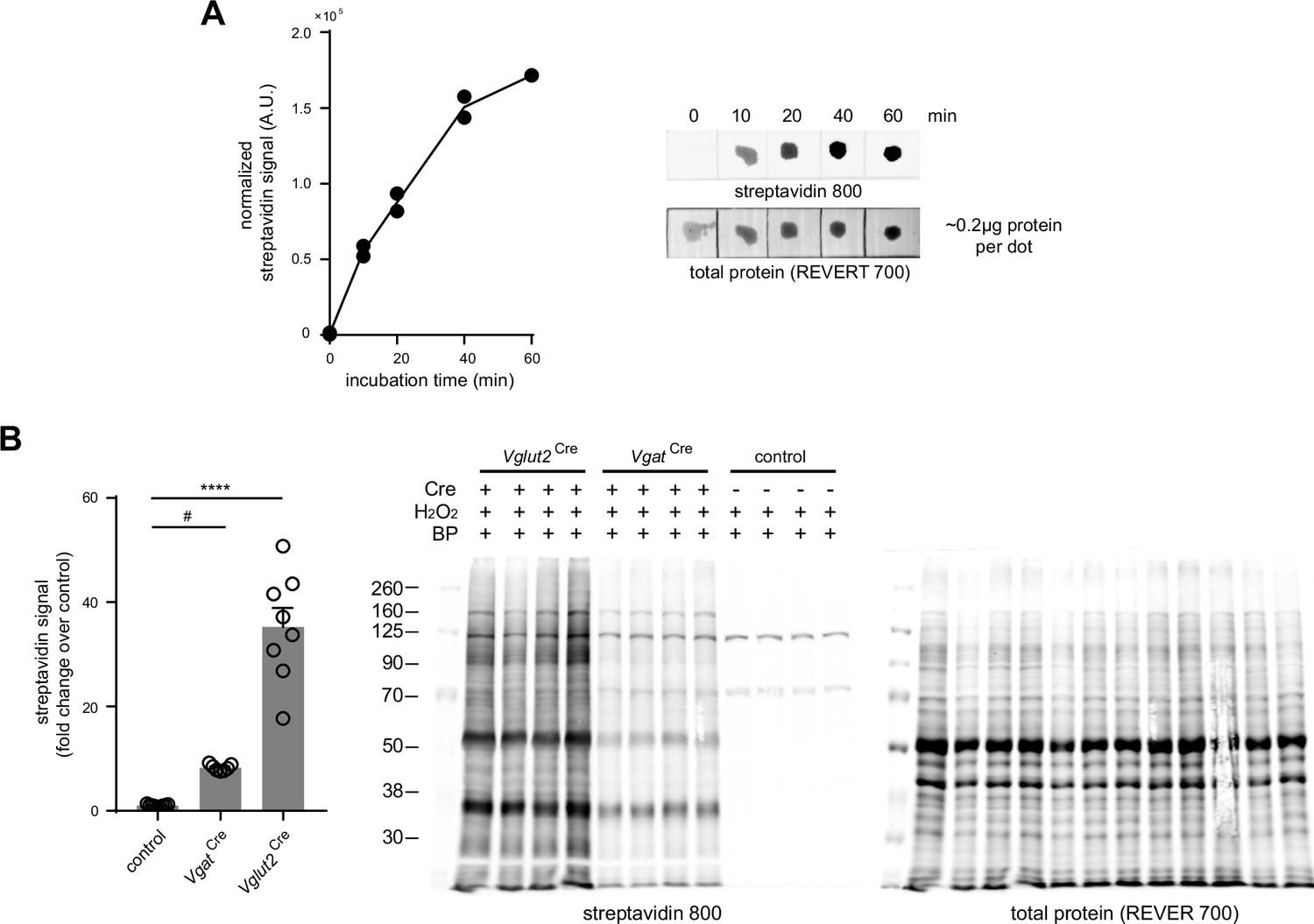
Optimization of biotin phenol ACSF incubation.
(A) Dot blot analysis of cortical lysates with increasing BP incubation time. Left, normalized streptavidin signal plot at time points 0 min, 10 min, 20 min, 40 min, and 60 min (n=2 biological replicates). Right, representative dot blot. (B) Differential biotinylation in mPFC tissues at 1 hr BP incubation. Left, normalized streptavidin signal (one-way ANOVA, p<0.0001, F (2, 21)=74.68, Holm-Sidak’s multiple comparison test, - Vglut2Cre vs control, p<0.0001, and VgatCre vs control, P=0.0254) (n=7 control, 8 VgatCre 8 Vglut2Cre animals). Error bars reflect SEM. Right, representative streptavidin blot used in quantification.
-
Figure 2—figure supplement 1—source data 1
Raw and uncropped blots for Figure 2—figure supplement 1a, b.
- https://cdn.elifesciences.org/articles/78847/elife-78847-fig2-figsupp1-data1-v2.zip
Figure 3 with 3 supplements
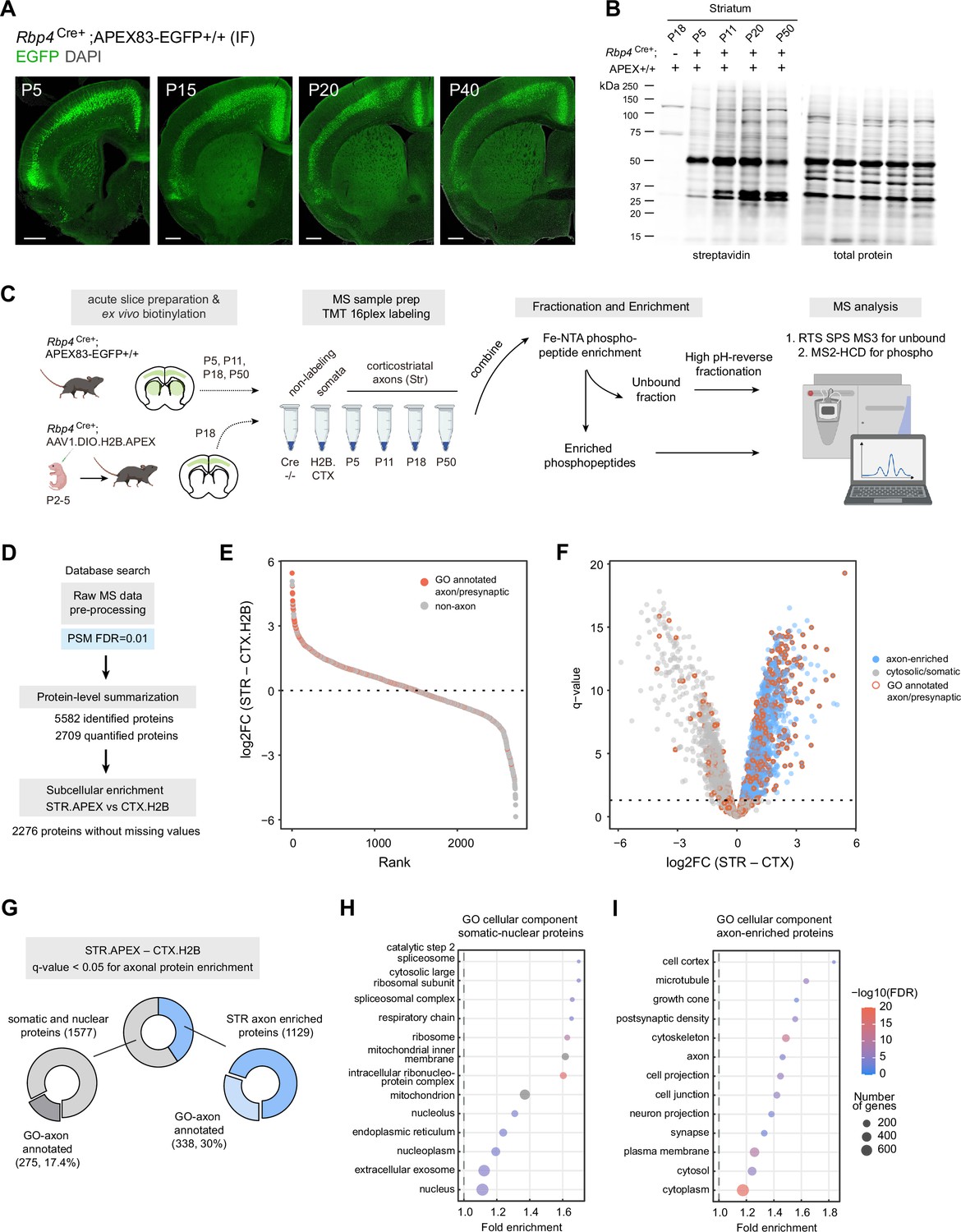
Genetic targeting approach for proteomics analysis of corticostriatal axonal compartment.
(A) Cre-dependent APEX.EGFP expression in excitatory cortical inputs to the striatum. Expression pattern of APEX.EGFP (immunostained EGFP) in Rbp4Cre+;APEX-EGFP+/+animals across postnatal days 5, 15, 20, and 40. Coronal brain section, scale bar: 1 mm. (B) Biotinylation of proteins in corticostriatal axons across development. Western blot analysis of striatal lysates prepared from biotinylated acute brain slices. Acute slices were prepared and biotinylated from Rpb4Cre+;APEX animals at the indicated age. Cre-negative APEX reporter line at P18 was used as non-labeling control. Left, streptavidin blot. Right, total protein loading control. Lower amount of protein biotinylation at early age (P5) is consistent with developing innervation by axonal projections (see (A) ). (C) Sample preparation workflow for proteomics analysis. Acute slices were prepared and biotinylated from Rbp4Cre+;APEX or Rbp4Cre+ injected with H2B.APEX at the indicated time points. Streptavidin beads were used to enrich biotinylated proteins for the bottom-up proteomics workflow. Samples were labeled with tandem mass tags (TMT) and combined. TMT mixtures were enriched for phosphopeptide analysis using Fe-NTA tips. The unbound fractions were fractionated using high pH-reverse phase resin for protein abundance analysis. Synchronous precursor selection (SPS) MS3 method was used for proteome analysis. MS2 with high energy collisional dissociation (HCD) method was used for phosphopeptide analysis. (D) Data analysis flow chart. Raw MS data were analyzed by Proteome Discoverer with false discovery rate (FDR) set at 1% for peptide spectrum matches (PSMs). Protein-level summarization was performed using MSstatsTMT using unique peptides. Striatal (STR) and cortical (CTX.H2B) samples prepared from P18 were compared to determine the axon-enriched proteome. Proteins with missing values were removed prior to this comparison. (E) Log2-fold change (FC) plot for STR–CTX comparison. Proteins were ranked by their log2FC. Red and gray dots, proteins with or without presynaptic or axon gene ontology annotations, respectively. (F) Volcano plot for the STR–CTX comparison. Blue dots, axon-enriched proteins with log2FC >0 and q-value <0.05 (horizontal dotted line). Gray dots, somatic or nuclear proteins that did not pass axon enrichment cutoff. Red border, proteins with presynaptic or axon gene ontology annotations. (G) Proportion of proteins with GO axon/presynaptic annotation for somatic/nuclear and axon protein lists. Using STR–CTX comparison, the proteome is classified into somatic/nuclear and axon-enriched proteins. (H) Over-representation analysis using gene ontology (GO) cellular component database for somatic and nuclear enriched protein list. Dot plot ranked by fold enrichment. Dot size and color correspond to the number of genes, and -log10(FDR), respectively. (I) Same as (H), but for the axon-enriched protein list.
Figure 3—figure supplement 1
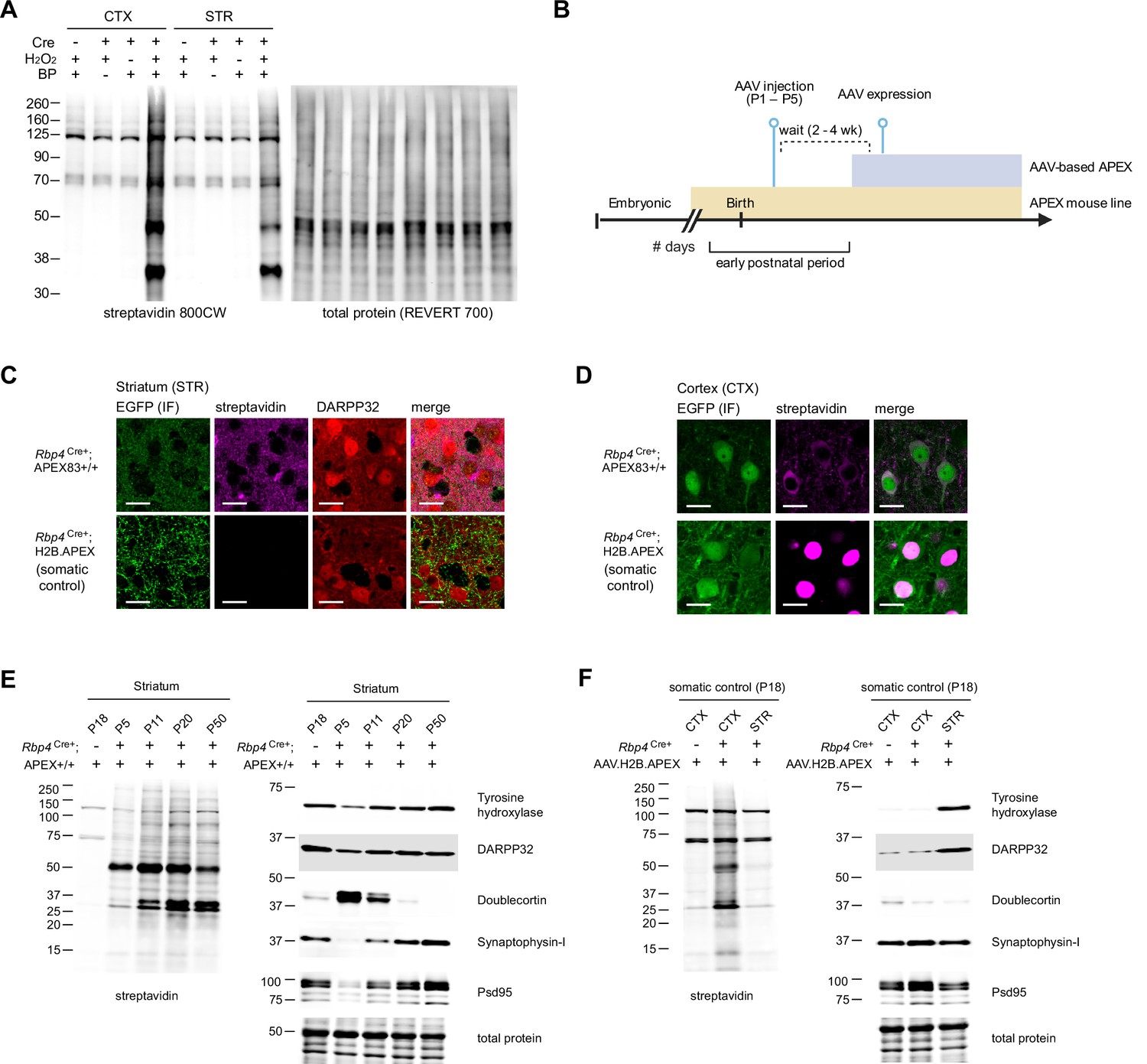
Cre-dependent biotinylation of Rbp4Cre+ corticostriatal projecting neurons.
(A) Western blot analysis of Rbp4Cre+;APEX tissue lysates after biotinylation. Left, streptavidin blot. Right, total protein loading control. Protein labeling by APEX in the cortex (CTX) and striatum (STR) is dependent on Cre, H2O2, and biotin phenol. (B) APEX expression timeline comparison between AAV and transgenic reporter APEX delivery. Neonatal AAV transduction is usually performed around postnatal days 1–5. Typical wait time before AAV-based expression is approximately 2–4 weeks. Expression from a transgenic reporter usually begins in the embryonic stage, enabling experiments during the early postnatal time window. (C) APEX expression in corticostriatal axons in the dorsal striatum (STR). Rbp4Cre+;APEX mouse line expresses APEX.NES in both somata and axons. For APEX-compartment control, Cre-dependent AAV1-H2B.APEX-P2A-EGFP was injected in the cortex of Rbp4Cre+ animals to express nucleus-localized H2B.APEX. Confocal images show immunofluorescence for EGFP, streptavidin, and DARPP32 (scale bar: 20 µm). P2A-linked EGFP broadly distributes in axons, while streptavidin labeling confirms proximity labeling of axonal proteins in Rbp4Cre+;APEX mouse line but not in H2B.APEX-expressing animals. (D) same as (C) for APEX expression in the cortex (CTX). Confocal images show immunofluorescence for EGFP and streptavidin (scale bar: 20 µm). P2A-linked EGFP broadly distributes in somata, while streptavidin labeling confirms proximity labeling of cytosolic and nuclear proteins in Rbp4Cre+;APEX mouse line and H2B.APEX-expressing animals, respectively. (E) Western blot analysis of striatal lysates across postnatal development. Acute slices were prepared and biotinylated from Rpb4Cre+;APEX animals at the indicated age. Cre-negative APEX reporter line at P18 was used as pull-down (non-labeling) control. Left, streptavidin blot (same as Figure 3b). Right, developmental protein markers (Tyrosine hydroxylase, DARPP32, Doublecortin, Synaptophysin-I, and Psd95) and total protein loading control. APEX was expressed in the corticostriatal projections across development confirmed by protein biotinylation. (F) Western blot analysis of striatal and cortical lysates from H2B.APEX-injected Rbp4Cre+ animals. Acute slices were prepared and biotinylated from Rpb4Cre+;APEX animals at the indicated age. Cre-negative H2B.APEX-injected animal was used as non-labeling control. Left, streptavidin blot. Right, protein markers (Tyrosine hydroxylase, DARPP32, Doublecortin, Synaptophysin-I, and PSD95) and total protein loading control. H2B.APEX primarily labels proteins in the cortex (CTX), with minimal biotinylation in the striatum (STR), serving as somatic protein compartment control.
-
Figure 3—figure supplement 1—source data 1
Raw and uncropped blots for Figure 3b, and Figure 3—figure supplement 1a, e, f.
- https://cdn.elifesciences.org/articles/78847/elife-78847-fig3-figsupp1-data1-v2.zip
Figure 3—figure supplement 2
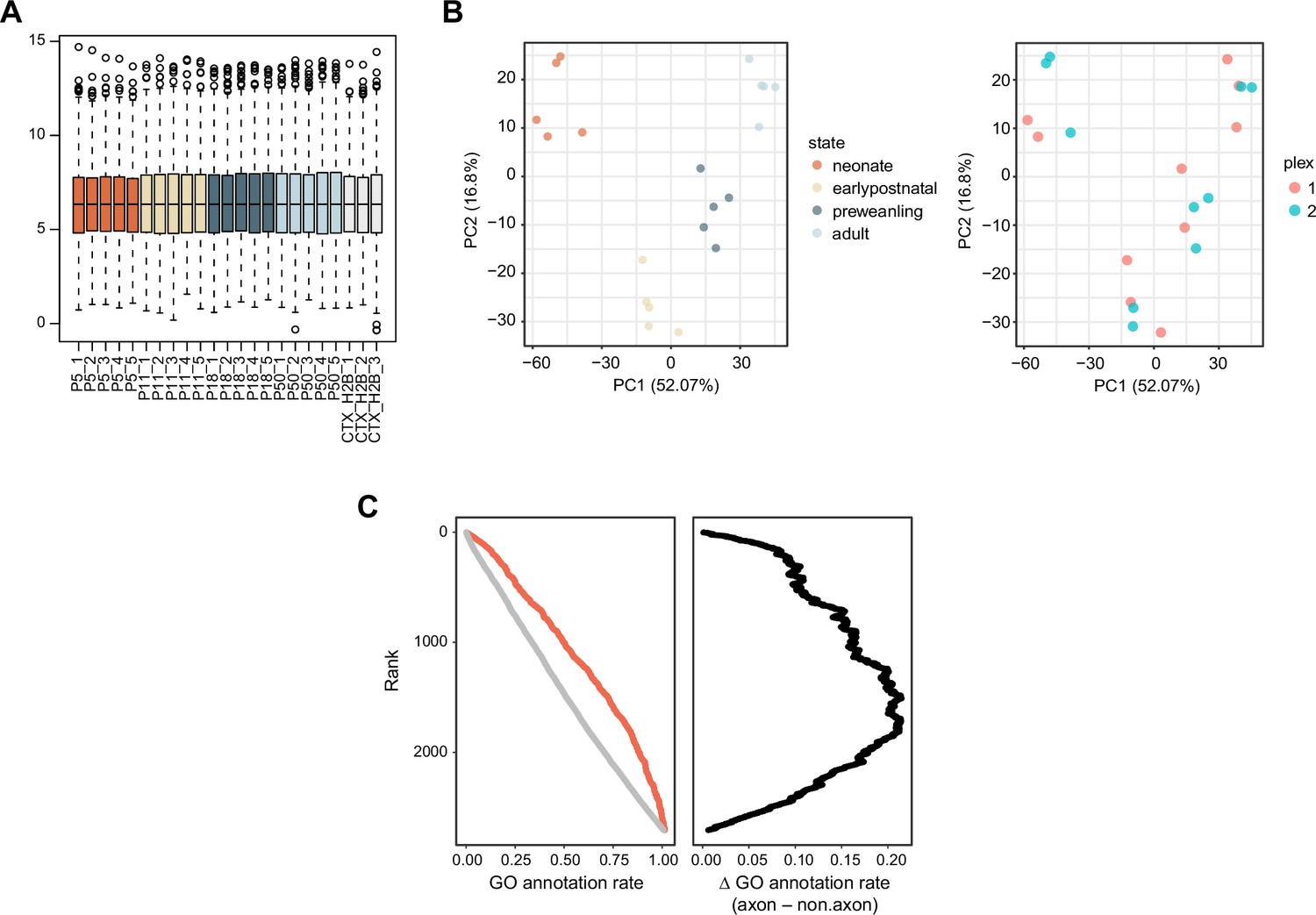
Quality control plots for the proteomic data.
(A) Boxplot of normalized log2 protein abundance. (B) Principal component analysis of striatal samples. (C) Gene ontology (GO) annotation rate. Proteins were ranked by their log2FC (STR–CTX.H2B). Each protein was marked by GO term, either as axon/presynaptic or not. Cumulative proportion of GO annotation as a function of rank. Red, axon/presynaptic GO terms.
Figure 3—figure supplement 3
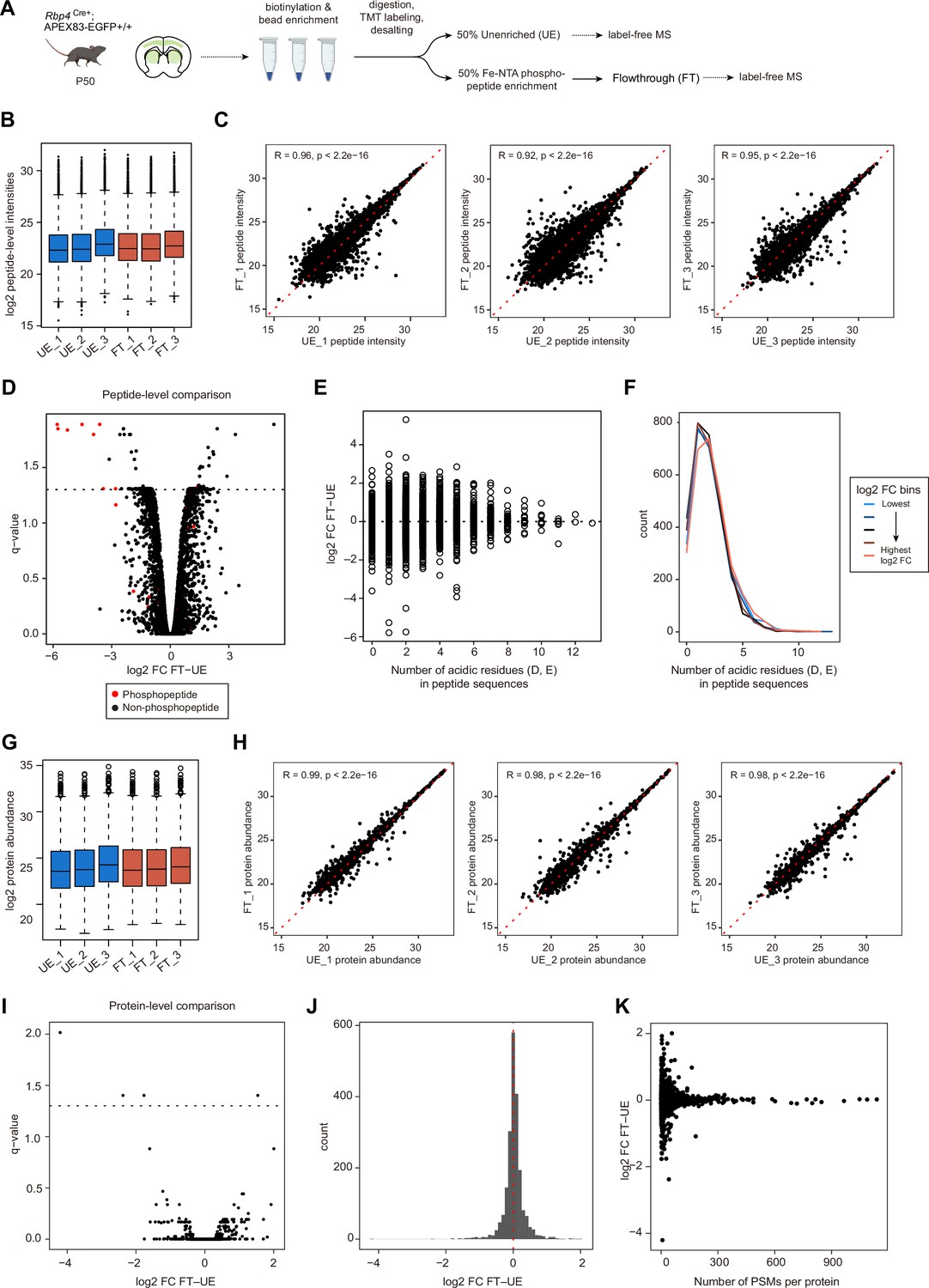
Evaluation of the flowthrough fraction for protein abundance.
(A) Sample preparation schematic for analysis of flowthrough fractions. Biotinylated proteins were enriched and digested from lysates prepared from Rbp4Cre;APEX acute brain slices. Digested peptides were labeled with TMTPro-zero, desalted, and split into 2 equal volumes: unenriched (UE) and phosphoenriched fractions (PE). PE samples were enriched for phosphopeptides and the flowthrough (FT) was collected. UE and FT were dried down and equal amount of each sample was analyzed with label-free quantification method. (B) Box plot of log2-transformed intensities of TMT-modified peptide groups. Unnormalized intensities of 13905 peptide groups. The distribution of peptide intensities was comparable across all replicates in UE and FT fractions. (C) Correlation of peptide intensities between UE and FT fractions. Spearman rank correlation coefficients for each UE and FT pair (R=0.96, R=0.92, and R=0.95, p<2.2e–16 for UE_1 vs. FT_1, UE_2 vs. FT_2, and UE_3 vs. FT_3, respectively). (D) Differential abundance analysis of peptides in UE and FT fractions. Paired LIMMA analyses were used and peptides with BH corrected p values, q-value <0.05 were considered significant (252 out of 13905 peptide groups, 1.8%). Red dots, TMT-modified phosphopeptides. (E) Distribution of log2 foldchange (FC) for peptides with at least one acidic residue (D, aspartic acid, E, glutamic acid). Gain or loss of peptide intensities in FT was centered around logFC = 0, irrespective of the number of acidic residues in the peptide sequence. (F) Histogram of the number of acidic residues by log2 FC bins. Peptides were divided into 5 equal bins by ranks of log2 FC (bin size = 2781 peptides). Histograms retain similar distribution across log2 FC bins. (G) Box plot of log2-transformed protein abundance. Unnormalized abundance of 2088 proteins. The distribution of protein abundance was comparable across all replicates in UE and FT fractions. (H) Correlation of protein abundance between UE and FT fractions. Spearman rank correlation coefficients for each UE and FT pair (R=0.99, R=0.98, and R=0.98, p<2.2e-16 for UE_1 vs. FT_1, UE_2 vs. FT_2, and UE_3 vs. FT_3, respectively). (I) Differential abundance analysis of proteins in UE and FT fractions. Paired LIMMA analyses were used and proteins with q-value <0.05 were considered significant (4 out of 2088 proteins, 0.2%). (J) Histogram of protein log2 FC between unenriched and flowthrough fractions. Histrogram peak centers around log2 FC = 0, suggesting that phosphoenrichment does not significantly alter protein abundance in the flowthrough. (K) Distribution of protein log2 FC by the number of PSMs detected per protein.
Figure 4 with 2 supplements
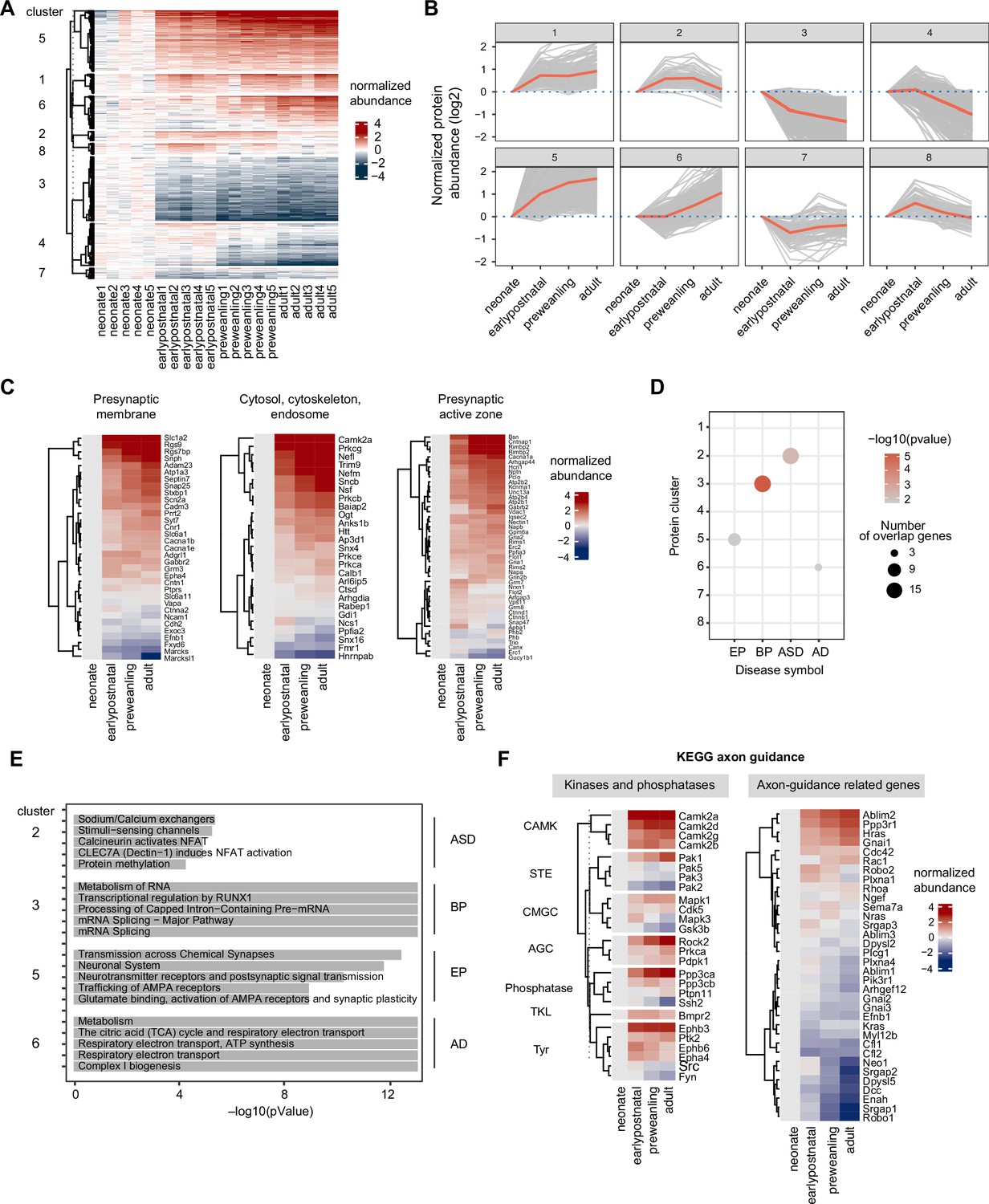
Temporal trajectories of the corticostriatal axonal proteome.
(A) Heat map showing temporal expression across development. Protein clusters were defined using a time course regression analysis implemented in maSigPro R package. The row dendrogram shows hierarchical clustering of proteins in each protein cluster. Log2 protein abundance was normalized to the mean of the neonatal time point. All biological replicates are shown in columns. (B) Temporal trajectories of protein expression in each cluster. Log2 protein abundance was normalized to the mean of the neonatal time point. Red lines represent the average expression of all proteins in a cluster. Gray lines represent individual protein trajectories. Neonate (P5), early postnatal (P11), preweanling (P18–20), and adult (P50). (C) SynGO analysis of presynaptic protein maturation, annotated by gene names. Proteins mapped to SynGO presynapse ontology term were grouped into seven categories: presynapse, cytosol-cytoskeleton-endosome, presynaptic membrane, presynaptic active zone, presynaptic endocytic zone, presynaptic ribosome and ER, and neuronal dense core vesicle. Heatmap shows log2 protein abundance normalized to neonatal time point. Three categories, presynaptic membrane, presynaptic active zone, and presynaptic endocytic zone are shown. Remaining categories are in Figure 4—figure supplement 2. (D) Over-representation analysis of protein clusters for central nervous system (CNS) traits and disorders. Dot size indicates the number of genes enriched for a particular disease. Color density indicates the degree of significance, –log(pvalue). Only significantly enriched diseases are shown. AD (Alzheimer’s disease), EP (Epilepsy), BP (bipolar disorders), and ASD (Autism spectrum disorders). (E) Reactome pathway enrichment analysis for protein clusters that are associated with CNS traits and disorders (clusters 2, 4, 7, and 8). Top 5 Reactome pathway terms are plotted. Gray bar indicates –log(p value). (F) KEGG axon guidance pathway analysis, annotated by gene names. Heatmap of normalized Log2 protein abundance for proteins mapped to the KEGG axon guidance pathway (mmu04360). Left, protein abundance for kinases and phosphatases clustered by family. Right, other proteins in the pathway.
Figure 4—figure supplement 1
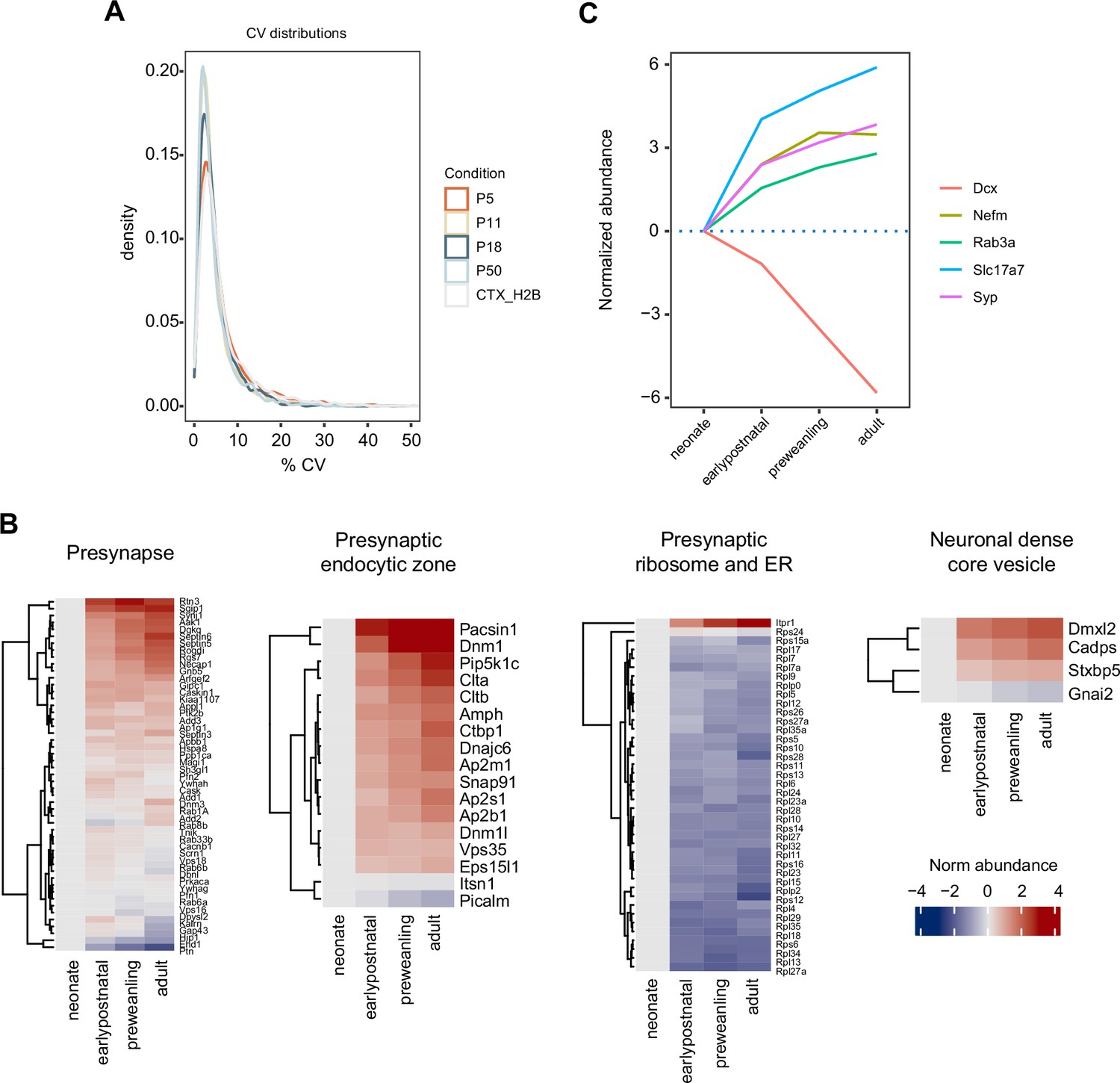
Sample coefficients of variation and expression of selected developmental markers.
(A) Density histogram for sample coefficients of variation (CVs). (B) Proteins mapped to SynGO presynapse ontology term, annotated by gene name, were grouped into seven categories: presynapse, cytosol-cytoskeleton-endosome, presynaptic membrane, presynaptic active zone, presynaptic endocytic zone, presynaptic ribosome and ER, and neuronal dense core vesicle. Heatmap shows log2 protein abundance normalized to neonatal time point. Four categories are shown here—presynapse, presynaptic endocytic zone, presynaptic ribosome and ER, and neuronal dense core vesicle—while the remaining three are in Figure 4b. (C) Developmental trajectories of immature and mature neuronal markers. Doublecortin (DCX), Synaptophysin (SYP), vesicular glutamate transporter VGLUT1, intermediate neurofilament (NEFM), and vesicle marker RAB3A. Line color, gene names.
Figure 4—figure supplement 2
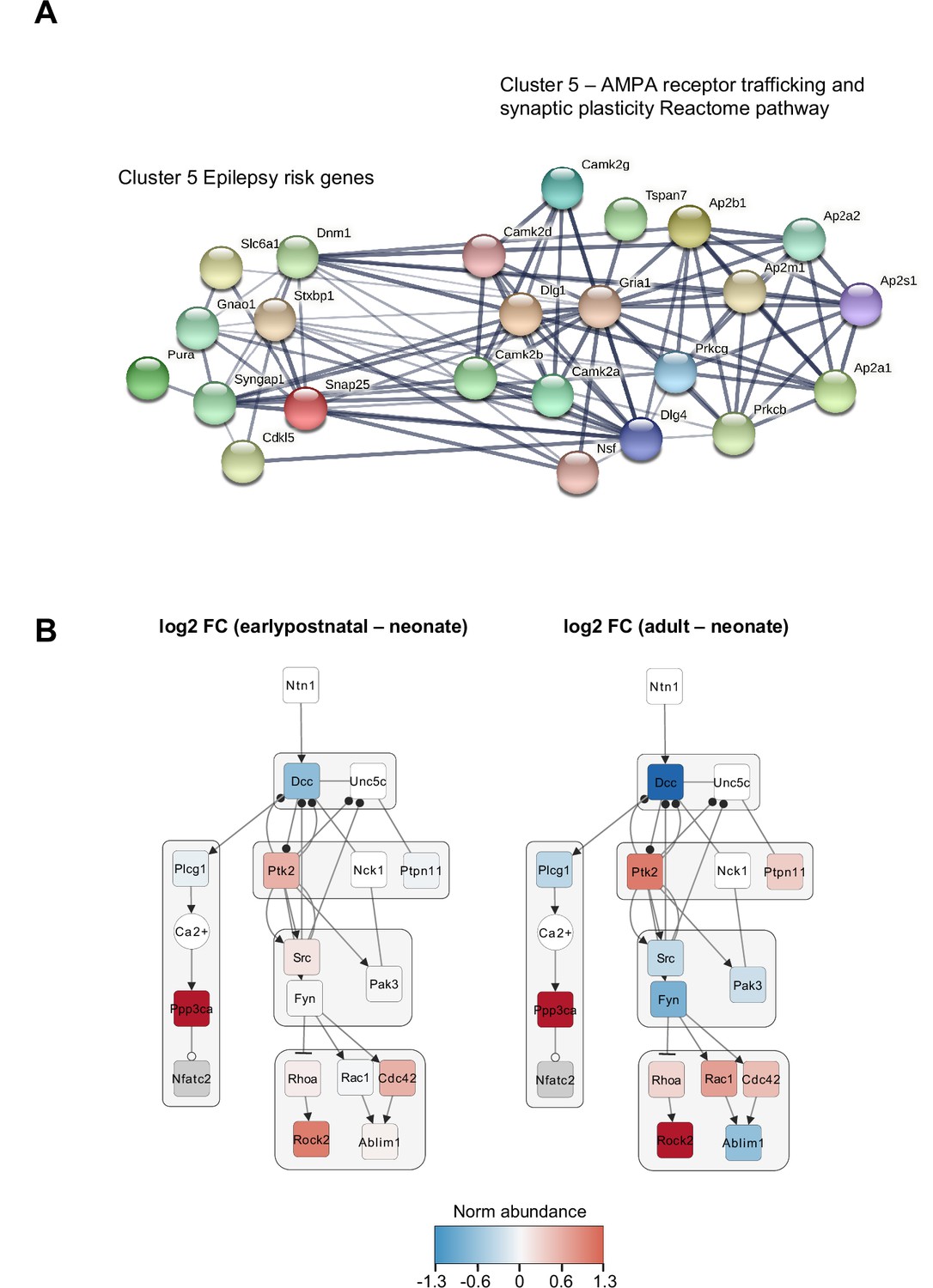
Protein network and Netrin-DCC pathway analysis.
(A) Proposed relationship between epilepsy (EP) risk genes and cluster 5 biological functions. STRING-DB analysis of EP risk genes in cluster 5 and proteins found in the Reactome pathway for AMPA receptor trafficking and plasticity. (B) Temporal changes in protein abundance in the NTN1-DCC signaling subnetwork between early postnatal and adult, annotated by gene name. Node color, Log2 fold change normalized to the neonate condition. Edges, solid and blunt arrows for activation and inhibition, closed and opened circles for phosphorylation and dephosphorylation, respectively.
Figure 5 with 1 supplement
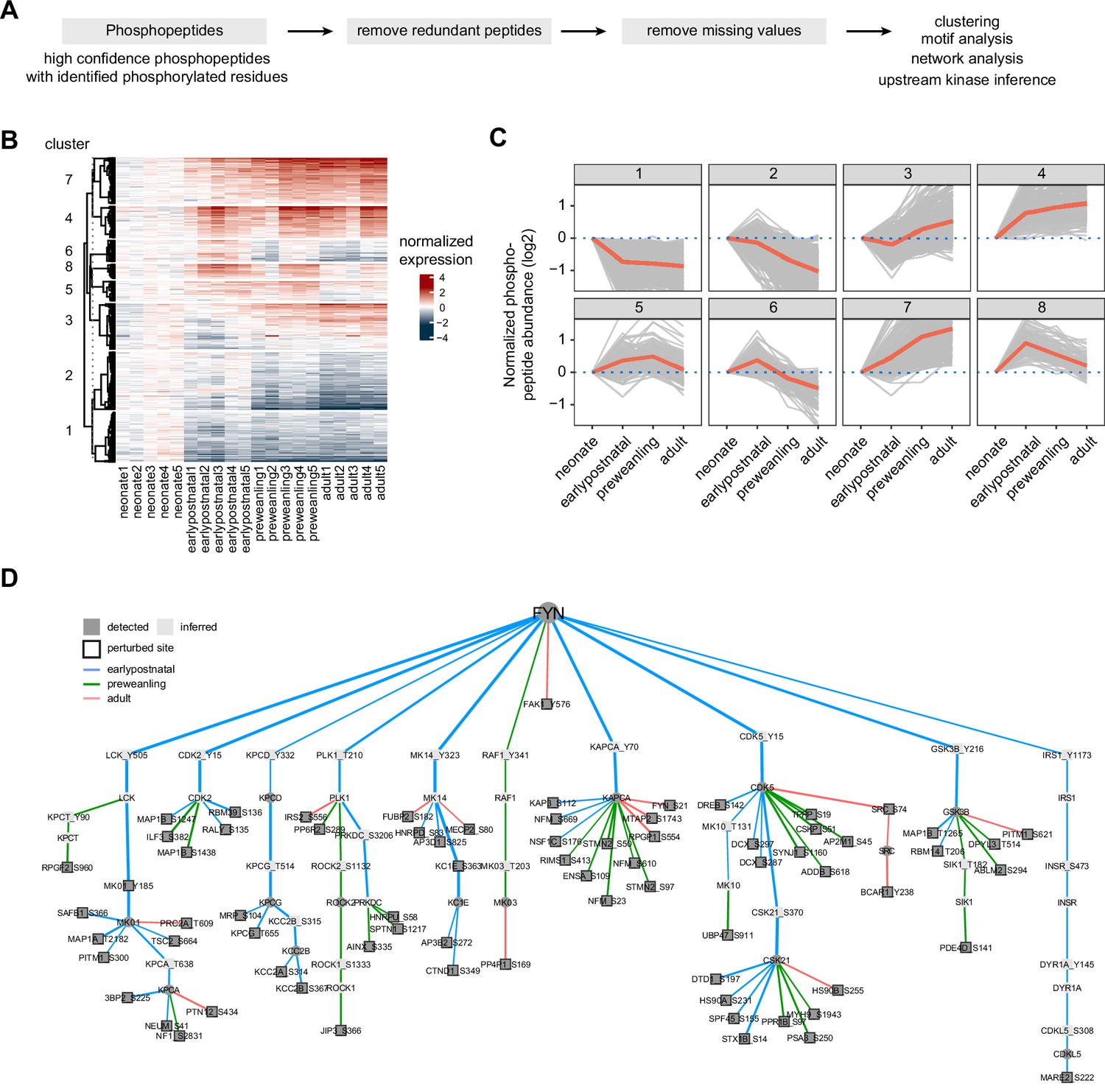
Temporal trajectories of phosphopeptide abundance and kinase-substrate interaction network.
(A) Phosphopeptide analysis workflow. Raw MS data were analyzed by Proteome Discoverer with false discovery rate (FDR) set at 1% for peptide spectrum matches (PSMs). Only phosphopeptide groups with identified phosphorylated residues were used in the analysis. A unique phosphopeptide is defined by a combination of phosphopeptide and its corresponding protein. Entries without missing abundance values across replicates were used in the timecourse and clustering analysis. (B) Heat map showing temporal expression of phosphorylation across development. Peptide clusters were defined using a time-course regression analysis implemented in maSigPro R package. The row dendrogram shows hierarchical clustering of peptide in each cluster. Log2 peptide abundance was normalized to the mean of the neonatal time point. All biological replicates are shown in columns. (C) Temporal trajectories of phosphorylation in each peptide cluster. Log2 peptide abundance was normalized to the mean of the neonatal time point. Red lines represent the average expression of all proteins in a cluster. Gray lines represent individual protein trajectories. Neonate (P5), early postnatal (P11), preweanling (P18–20), and adult (P5). (D) Kinase-substrate interaction network downstream of FYN kinase. PHOsphorylation Networks for Mass Spectrometry (PHONEMeS) was used to model phosphosites using known prior knowledge network from PhosphoSitePlus database. Nodes, kinase or phosphosite. Node color: dark gray, detected, light gray, inferred by the model. Edges, kinase-substrate interactions. Edge thickness shows the degree of importance inferred for each kinase-substrate interaction. Edge color: blue, green, red for earlypostnatal–neonate, preweanling–neonate, and adult–neonate comparison, respectively.
Figure 5—figure supplement 1
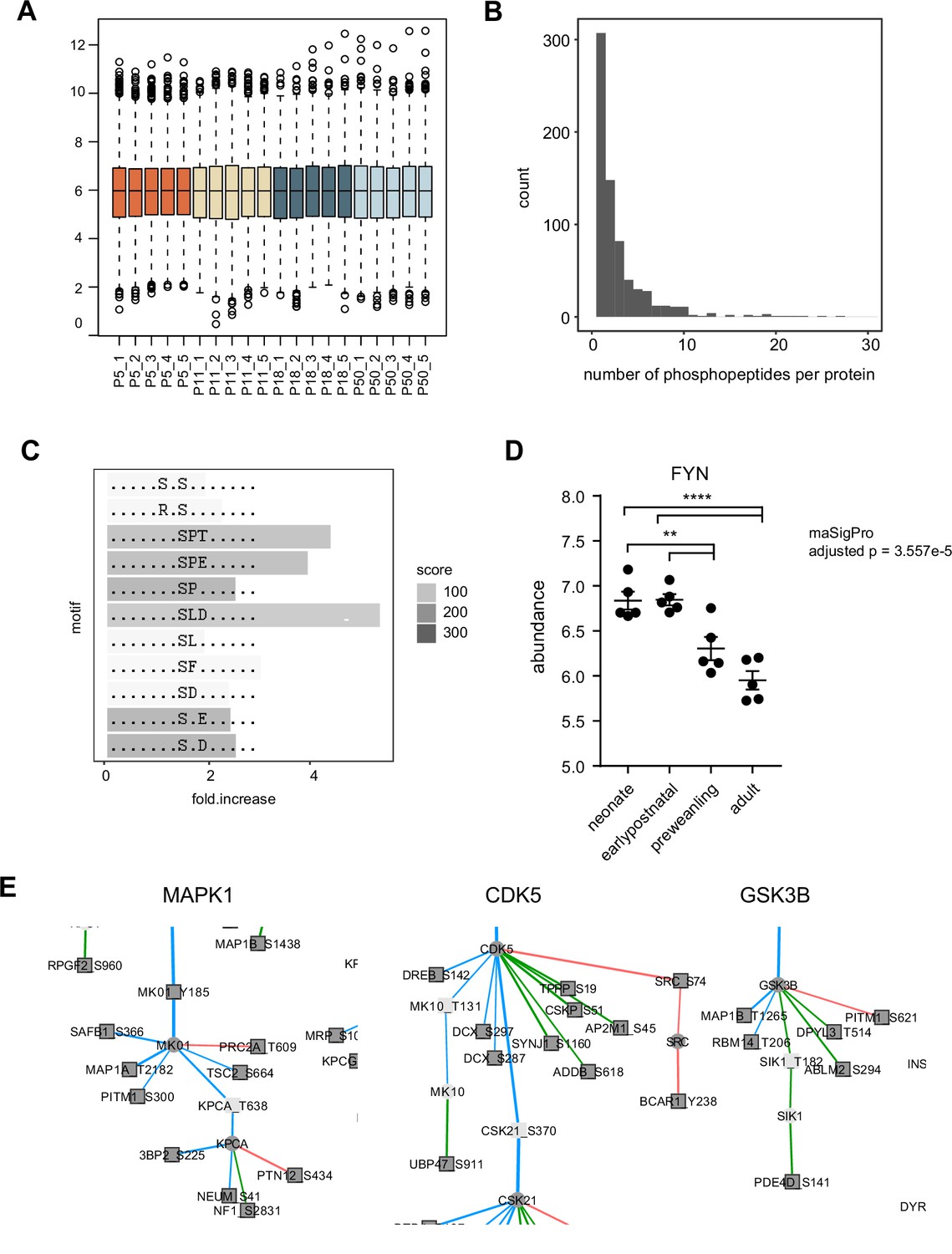
Phosphopeptide abundance normalization and motif analysis.
(A) Boxplot of normalized log2 phosphopeptide abundance.(B) Number of phosphopeptides per protein. Histogram bin width = 1.(C) Motif-x enrichment analysis.(D) FYN protein log2 abundance. maSigPro adjusted P=3.557e-5. n=5 biological replicates each. One-way ANOVA, p<0.0001, F (3, 16)=18.95, Holm-Tukey’s multiple comparison test. Early postnatal vs preweanling, adjusted p=0.0078. Earlypostnatal vs adult, adjusted p<0.0001; neonate vs preweanling, adjusted P=0.0088; neonate vs adult, adjusted p<0.0001. Error bars reflect SEM. (E) Proline-directed kinases in FYN phosphoregulatory network. Close-up for MAPK1, CDK5, and GSK3B subnetworks in Figure 5d.
Figure 6
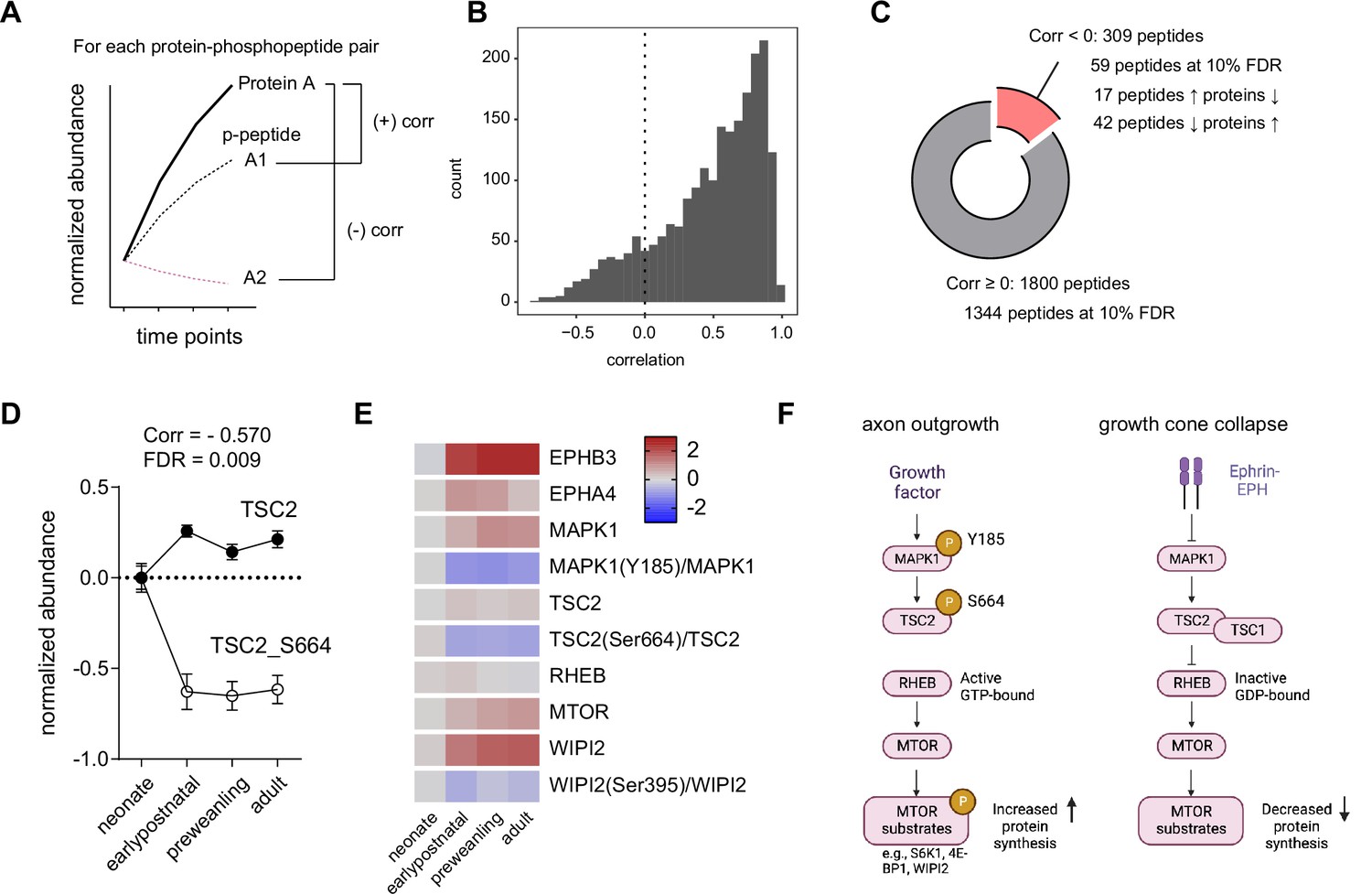
Correlation analysis of phosphorylated peptides and proteins implicates Tsc2 Ser664 in MTOR regulation in corticostriatal axon development.
(A) Correlation analysis workflow. Spearman rank correlation is calculated for each protein and phosphopeptide pair across time points. One protein may have multiple phosphopeptide-protein pairs, yielding multiple correlation values. False-discovery rate was controlled by the Benjamini-Hochberg approach.(B) Distribution of protein-phosphopeptide correlations. Left-skewed histogram (bin width = 1). Most phosphopeptides correlate with their respective protein expression across time. (C) Classification of protein-phosphopeptide correlations. Cutoff for correlated peptides, Spearman correlation ≥0. Of 1800 phosphopeptides, 1344 are positively correlated with proteins by their temporal expression at 10% false discovery rate (FDR). A small proportion of peptides (59 of 309 phosphopeptides) are decorrelated (inversely correlated) at 10% FDR. 42 phosphopeptides decrease in abundance as protein levels increases, while 17 phosphopeptides show the opposite pattern. (D) TSC2 and TSC2_Ser664 expression. Log2 abundance for TSC2 (open circles) and TSC2_Ser664 (solid circles) were normalized to the neonatal time point (Spearman rank correlation: –0.570, FDR = 0.009. n=5 for each time point. Error bars represent SEM).(E) Expression heatmap for proteins and phosphosites in the TSC2-RHEB-MTOR signaling pathway. Log2 protein expression was normalized to the neonate time point. Log2 phosphopeptide signal was normalized to the corresponding protein abundance and to the neonate time point. (F) Model for MTOR-mediated growth cone collapse.
Author response image 1
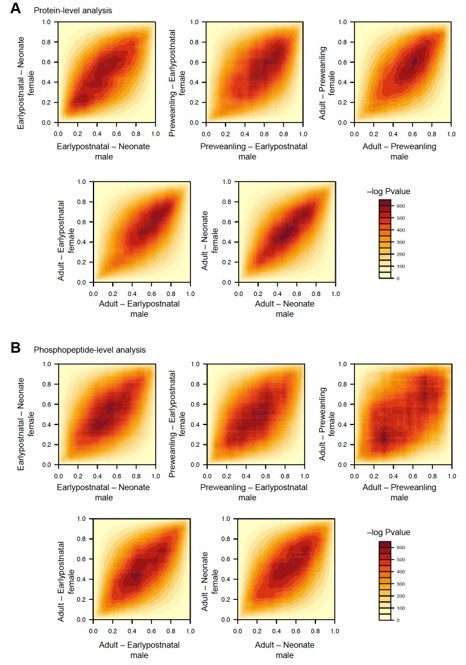
Rank-rank hypergeometric overlap analysis (RRHO) for sex differences in protein and phosphopeptide levels across development.
(A) RRHO analysis for protein abundance across any two time points with male and female as subcategories. Heatmap of –log Pvalue, where higher values indicate greater statistically significant overlapping proteins between male and female. X- and Y axes are normalized rank from highest to lowest log2FC (0 – 1 rank, 0 highest log2FC). Sets of proteins are regulated in a similar manner between male and female if hotspots are in quadrants Q1 and Q3 for both upregulated, or both downregulated, respectively. In contrast, if hotspots are in Q2 and Q4, proteins are differentially regulated between males and females. (B) Same as (A) for phosphopeptides, showing less clustering around the diagonal compared to protein level data.
Author response image 2

Preliminary test for perfusion-based delivery of biotin phenol (BP).
Rbp4Cre;APEX+ animals were transcardially perfused with 15 ml ACSF +/- 500 μM BP. Following perfusion, acute slices were prepared and treated with 0.03% H2O2. No difference in streptavidin staining was detected between BP and no-labeling control, while samples that underwent ex vivo biotinylation show robust signal. Total protein loading control was visualized by REVERT stain.
Additional files
-
Supplementary file 1
Primers and PCR protocol for APEX reporter line genotyping.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp1-v2.xlsx
-
Supplementary file 2
Primers and PCR protocol for off-target analysis.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp2-v2.xlsx
-
Supplementary file 3
Sample information and method validation for flowthrough vs. unenriched samples for protein abundance measurement.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp3-v2.xlsx
-
Supplementary file 4
Protein identification and quantification.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp4-v2.xlsx
-
Supplementary file 5
Statistical analysis for axon vs. soma comparison.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp5-v2.xlsx
-
Supplementary file 6
Gene ontology analysis results for axon vs. soma comparison.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp6-v2.xlsx
-
Supplementary file 7
Protein temporal trajectory analysis and SynGO gene ontology.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp7-v2.xlsx
-
Supplementary file 8
List of risk genes for neurodevelopment, neurodegenerative, and neuropsychiatric disorders.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp8-v2.xlsx
-
Supplementary file 9
Disease and Reactome pathway enrichment analysis for different protein clusters.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp9-v2.xlsx
-
Supplementary file 10
Phosphoproteome temporal trajectory analysis.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp10-v2.xlsx
-
Supplementary file 11
Phosphopeptide vs. protein correlation analysis.
- https://cdn.elifesciences.org/articles/78847/elife-78847-supp11-v2.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/78847/elife-78847-mdarchecklist1-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Dynamic proteomic and phosphoproteomic atlas of corticostriatal axons in neurodevelopment
eLife 11:e78847.
https://doi.org/10.7554/eLife.78847




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}