Searching for molecular hypoxia sensors among oxygen-dependent enzymes
- Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, United States
- Department of Psychiatry, University of California, San Francisco, United States
- Hypoxia Research Laboratory, University of California San Francisco, San Francisco, United States
- Center for Health Equity in Surgery and Anesthesia, University of California San Francisco, San Francisco, United States
- Anesthesia and Perioperative Care, University of California San Francisco, San Francisco, United States
Abstract
The ability to sense and respond to changes in cellular oxygen levels is critical for aerobic organisms and requires a molecular oxygen sensor. The prototypical sensor is the oxygen-dependent enzyme PHD: hypoxia inhibits its ability to hydroxylate the transcription factor HIF, causing HIF to accumulate and trigger the classic HIF-dependent hypoxia response. A small handful of other oxygen sensors are known, all of which are oxygen-dependent enzymes. However, hundreds of oxygen-dependent enzymes exist among aerobic organisms, raising the possibility that additional sensors remain to be discovered. This review summarizes known and potential hypoxia sensors among human O2-dependent enzymes and highlights their possible roles in hypoxia-related adaptation and diseases.
Introduction
In aerobic organisms, the dioxygen molecule (O2) is essential for many biochemical pathways, particularly as the final electron acceptor for bioenergetics. Hypoxia—conditions of decreased O2 availability—is both an essential stimulus for normal development and a pathological trigger of cellular dysfunction and eventual cell death for humans and other mammals (Bickler and Buck, 2007). To maintain O2 homeostasis, aerobic organisms have developed diverse cellular mechanisms for sensing and responding to alterations in O2 level. For multiorgan organisms, the term ‘hypoxia’ is often loosely used to describe decreased O2 levels. More precisely, the term ‘tissue hypoxia’ is meaningful when used in comparison to the baseline for the tissue. Physiological tissue O2 (physoxia, the typical range of function), physiological hypoxia (reduction or fluctuation of pO2 into a range at which adaptation is possible), and hypoxia with pathological impact (pO2 at which cellular injury and death occur) are often cited as ~5, 2, and 1%, respectively, for humans (McKeown, 2014). However, these values can vary widely across tissues and even within a tissue and are affected by tissue-level regulation (e.g., blood flow) and cellular effects (e.g., changes in metabolic state) (Table 1; McKeown, 2014; Ortiz-Prado et al., 2019; Carreau et al., 2011; Jagannathan et al., 2016; Cigognini et al., 2016; Donovan et al., 2010; Mas-Bargues et al., 2019). Here, we focus on O2 sensing in humans, using the term ‘hypoxia’ to denote decreased O2 level relative to physoxia, that is, encompassing both physiological hypoxia and hypoxia with pathological impact.
Table 1
Physiological O2 distribution in different organs/tissues*.
| Organ/tissue | %O2 | pO2 (mmHg) | Concentration(μM) |
|---|---|---|---|
| Ambient air | 21 | 160 | 206 |
| Alveoli | 14 | 104 | 134 |
| Arterial blood | 13 | 100 | 129 |
| Kidney | 4–9.5 | 30–73 | 39–94 |
| Liver | 4–7 | 30–54 | 39–69 |
| Heart | 2–6 | 15–46 | 19–59 |
| Brain | 3–5 | 23–39 | 29–50 |
| Small intestine | 2–9 | 15–69 | 19–89 |
| Large intestine | 0–6 | 0–46 | 0–59 |
| Bone marrow | 1.5–7 | 11–54 | 14–69 |
-
*
The O2 levels in different organs are adjusted from references Burmester and Hankeln, 2014; Lecomte et al., 2005; Hatefi, 1985; Zaccara et al., 2019; Ball et al., 2014 and the partial pressure and concentration are calculated according to references Ortiz-Prado et al., 2019; Carreau et al., 2011; Jagannathan et al., 2016; Cigognini et al., 2016; Donovan et al., 2010; Mas-Bargues et al., 2019; Place et al., 2017.
Discovery of the PHD-HIF-pVHL pathway was pivotal to understanding hypoxia responses and has been reviewed extensively (Majmundar et al., 2010; Kaelin and Ratcliffe, 2008; Ivan and Kaelin, 2017; Schofield and Ratcliffe, 2004). Briefly, in normoxia, prolyl hydroxylase domain proteins (PHDs) use O2 as a substrate to hydroxylate prolines on the transcription factor hypoxia-inducible factor α subunit (HIFα, i.e., HIF1α or HIF2α). The hydroxylated form of HIFα is recognized by the E3 ubiquitin ligase pVHL (von Hippel-Lindau protein), which promotes degradation of HIFα. By contrast, in hypoxia, the decreased catalytic activity of PHDs results in decreased hydroxylation and hence decreased pVHL recognition of HIFα, promoting the accumulation of HIFα. HIFα then translocates to the nucleus and, as a heterodimer with HIF1β, regulates transcription of a broad range of target genes. Thus, PHDs directly sense a decrease in the availability of molecular O2 and transduce this signal to downstream effectors.
What defines a molecular hypoxia sensor? In engineering, a sensor is a device that detects changes to a physical property and transmits this information so that a system can respond to this change. Here, by analogy to human-engineered sensors, we define biological hypoxia sensors as proteins that (1) directly interact with O2 molecules, (2) have activities that are strongly affected by physiological hypoxia, and (3) are coupled to downstream responses that depend on changes of their activities. Of note, many proteins respond to hypoxia by acting downstream of a sensor (e.g., HIF acting downstream of PHD) or by responding to changes in the cellular redox state. In this review, we specifically exclude these as they are not direct sensors of molecular O2 —that is, their response to changes in O2 levels does not involve direct interaction with O2 (Bickler and Donohoe, 2002).
Strong candidates for hypoxia sensors include O2-dependent enzymes, which by definition meet criterion 1. These enzymes constitute a mechanistically, structurally, and biologically diverse group of proteins. There are a number of reviews on the enzymology (Islam et al., 2018; Palfey and McDonald, 2010; Decker and Solomon, 2005; Jasniewski and Que, 2018; Biringer, 2020; Ferguson-Miller and Babcock, 1996; Finney et al., 2014; Bassan et al., 2003; Guengerich, 2007; Ponnaluri et al., 2013; Ivanov et al., 2010; Daff, 2010; Wikström et al., 2018; Huang and Groves, 2018; Romero et al., 2018; Sono et al., 1996; Martinez and Hausinger, 2015; Roberts and Fitzpatrick, 2013; Itoh, 2006; Solomon et al., 2001; Bugg, 2001; Abu-Omar et al., 2005), biological function (Schofield and Ratcliffe, 2004; Islam et al., 2018; Paton and Ntambi, 2009; Shmakova et al., 2014; Danielson, 2002; Donkó et al., 2005; Bundred et al., 2018; Fletcher and Coleman, 2020; Kuhn et al., 2015; Zhuang et al., 2015; Kooistra and Helin, 2012; Mashima and Okuyama, 2015; Wu and Zhang, 2017; Johansson et al., 2014; Daubner et al., 2011; Fong and Takeda, 2008; Markolovic et al., 2015), and evolution (Danielson, 2002; Taylor and McElwain, 2010; Chandrasekharan and Simmons, 2004; Wilks, 2002) of individual subclasses of O2-dependent enzymes. Here, we provide a global map of human O2-dependent enzymes in potential hypoxia sensing. We first survey the broad categories and then discuss specific members that are known or speculated hypoxia sensors. Finally, we investigate the links between O2-dependent enzymes and hypoxia-related evolutionary adaptations and diseases.
O2-dependent enzymes as hypoxia sensor candidates
We start by providing background and taxonomies for considering the three basic ‘sensor’ requirements discussed above.
First, O2-dependent enzymes directly interact with O2 molecules as one of the substrates. Non-enzymatic proteins that directly interact with O2, for example, globins, have been reviewed elsewhere (Burmester and Hankeln, 2014; Lecomte et al., 2005). In humans, 221 enzymes are known or likely to be O2-dependent (Supplementary file 1), that is, utilizing O2 as an electron acceptor for the oxidation of other substrates. Based on their catalyzed reactions, O2-dependent enzymes can be divided into three subclasses: dioxygenases, which catalyze the insertion of both oxygen atoms of the O2 molecule into substrates; monooxygenases, which catalyze the insertion of one oxygen atom of the O2 molecule into a substrate and the reduction of the other oxygen atom to H2O; and oxidases, which catalyze the reduction of O2 molecules to 2H2O or H2O2 (Figure 1).
Figure 1
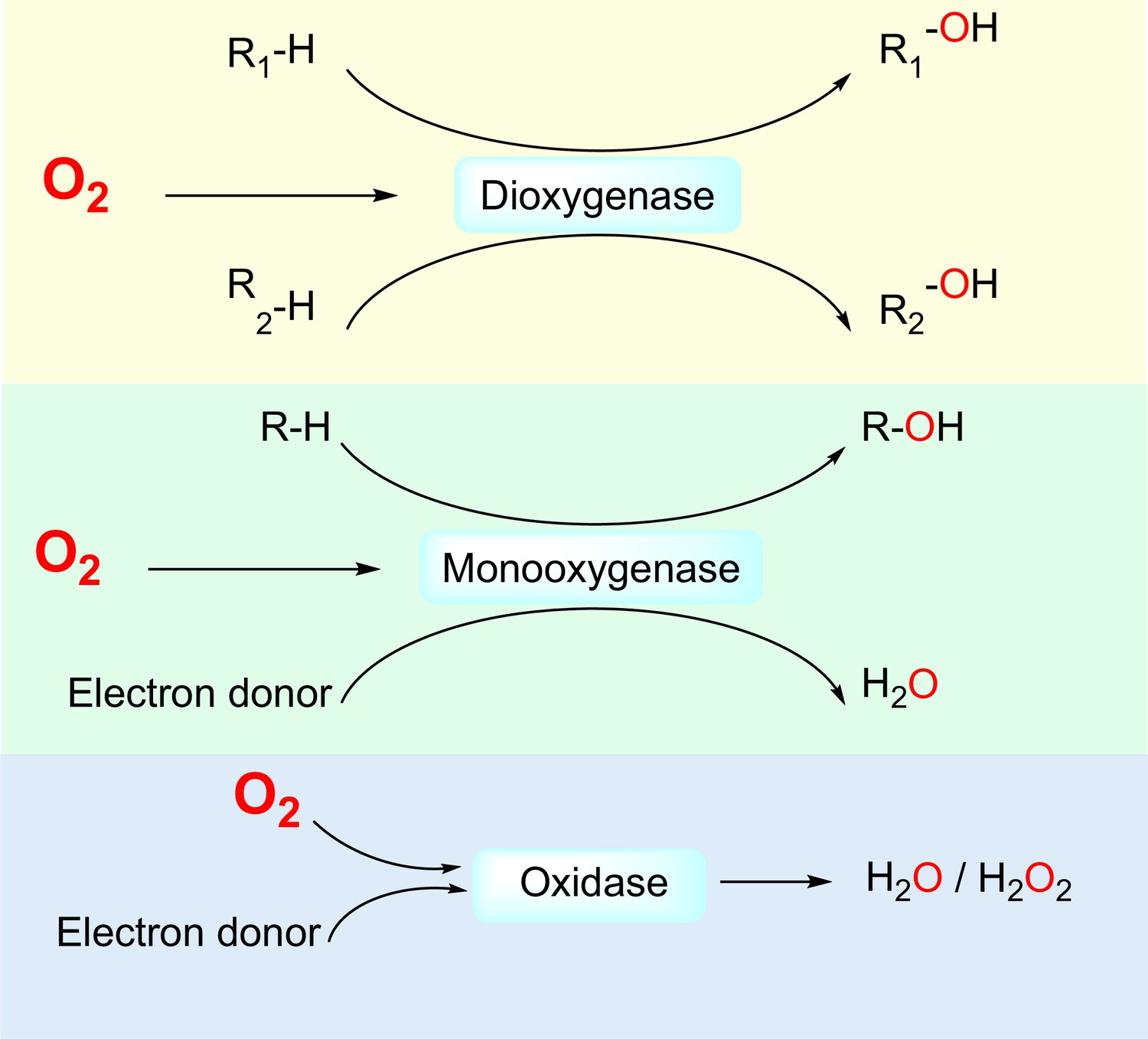
Three classes of by O2-dependent enzymes (dioxygenases, monooxygenases, and oxidases) and the reactions they catalyze.
Dioxygenases catalyze the insertion of both oxygen atoms of the dioxygen molecule into substrates. Monooxygenases catalyze the insertion of one oxygen atom of the dioxygen molecule into a substrate and the other oxygen atom is reduced to H2O. Oxidases catalyze the reduction of dioxygen to H2O or H2O2.
Second, O2-dependent enzymes have diverse mechanisms for utilizing O2 as a substrate, resulting in different sensitivities to O2 concentrations. Sensitivity is determined, in part, by the binding affinity of O2 with the enzyme’s catalytic center. Most O2-dependent enzymes (177/221) use, or are speculated to use, O2-binding metal ions at their catalytic centers. Factors affecting the O2-binding affinity include the metal center (iron or copper in humans), ligands (enzyme residues and other substrates) for the metal center, and the environment of the catalytic pocket. The other O2-dependent enzymes with known non-metal catalytic centers (37/221) utilize flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN) to activate O2. For these enzymes, the accessibility of O2 to FAD or FMN at the catalytic center affects the binding affinity. Dioxygenases, monooxygenases, and oxidases can be further subdivided by their catalytic centers (Table 2). Ultimately, these mechanisms affect the threshold at which enzymatic activities are saturated with O2, thus determining whether the enzyme’s activities are strongly affected by physiological-range hypoxia.
Table 2
Categories of O2-dependent enzymes.
| Category | Subcategory by catalytic center | Metal species at catalytic center | Ligands for the metal species at catalytic center (cofactor/substrate and enzyme residues) | Number of enzymes |
|---|---|---|---|---|
| Dioxygenase | 2-OG-dependent dioxygenase | Fe | 2-OG, His, His, Asp/Glu | 59 |
| Heme-dependent dioxygenase | Fe | Heme, His | 5 | |
| Lipoxygenase | Fe | His, His, His, Ile, His/Asa/Asn/none | 6 | |
| Others | Fe | His, His, His/Asp/Glu* | 10 | |
| Monooxygenase | Heme-dependent monooxygenase | Fe | Heme, Cys/His/Glu | 61 |
| Non-Heme Fe-dependent monooxygenase | Fe | His, His, His/Asp/Glu* | 9 | |
| Cu-dependent monooxygenase | Cu | His, His, Met | 5 | |
| Flavin-dependent monooxygenase | None (uses flavin) | N/A | 12 | |
| Others† | N/A | N/A | 2 | |
| Oxidase | Heme-copper | Fe and Cu | His, His, His for Cu; Heme and His for Fe | 1 |
| Fe-dependent oxidase | Fe | Varies | 14 | |
| Cu-dependent oxidase | Cu | Varies | 7 | |
| Flavin-dependent oxidase | None (uses flavin) | N/A | 25 | |
| Others† | N/A | N/A | 5 |
-
*
Substrates/cofactor ligands for this category varies for each member depending on the reaction it catalyzes.
-
†
Members in this category are not fully studied.
Third, O2-dependent enzymes regulate diverse cellular processes: (1) oxidative phosphorylation is responsible for mitochondrial ATP production and cellular survival (Hatefi, 1985); (2) post-translational modifications (hydroxylation, demethylation, or thiol oxidation) of proteins can regulate protein conformation, stability, and activity (Schofield and Ratcliffe, 2004; Bundred et al., 2018; Fletcher and Coleman, 2020; Kooistra and Helin, 2012; Johansson et al., 2014); (3) hydroxylation and demethylation of DNA/RNA can regulate DNA damage repair, epigenetic modifications, and transcription/translation (Wu and Zhang, 2017; Zaccara et al., 2019); (4) metabolism of amino acids and lipids can maintain cellular hemostasis and regulate cellular pathways through signaling molecules (Paton and Ntambi, 2009; Kuhn et al., 2015; Mashima and Okuyama, 2015; Daubner et al., 2011; Chandrasekharan and Simmons, 2004; Ball et al., 2014); and (5) metabolism of xenobiotics can regulate drug clearance and detoxification (Danielson, 2002; Poulos and Johnson, 2005). Typically, dioxygenases have macromolecules as substrates and regulate cellular processes at a transcriptional or translational level (Schofield and Ratcliffe, 2004; Islam et al., 2018; Bundred et al., 2018; Fletcher and Coleman, 2020; Kooistra and Helin, 2012; Wu and Zhang, 2017; Johansson et al., 2014; Hancock et al., 2015), while monooxygenases and oxidases often have small molecules as substrates and function in metabolism (Romero et al., 2018; Paton and Ntambi, 2009; Danielson, 2002; Daubner et al., 2011). Together, O2-dependent enzymes are integral to a plethora of physiological processes in aerobic animals.
Candidate hypoxia sensors can be identified among the O2-dependent enzymes, in part by the binding affinity between O2 and the enzyme as quantified by the O2 Km value, which suggests the level at which the enzyme is most sensitive to changes in O2 (Kaelin and Ratcliffe, 2008; Schofield and Ratcliffe, 2004; Shmakova et al., 2014; Hancock et al., 2015; Wilson et al., 2020; Holdsworth and Gibbs, 2020; Baik and Jain, 2020). (The Km value, also known as the Michaelis constant, is the concentration of a substrate at which an enzymatic reaction rate is 50% of the maximum reaction rate. A larger Km value reflects lower O2 affinity.) Importantly, the measured Km value is affected by the measurement method, for example, mass spectrometry vs. isotope assays. Besides the Km value, other cellular factors such as the concentration and conformation of the enzyme, as well as concentrations of other substrates or products, also affect the net enzymatic activity and hence the downstream effects of the enzyme. Beyond cellular-level effects, whether an O2-dependent enzyme functions as a hypoxia sensor in vivo can depend on the tissue pO2 context (Table 1). Taken together, whether an O2-dependent enzyme functions as a hypoxia sensor in vivo depends not only on the O2 Km value but also on multiple other factors.
O2-dependent enzymes that are known or potential hypoxia sensors
Below, we classify dioxygenases, monooxygenases, and oxidases into different subgroups based on their catalytic centers and discuss known (Figure 2A) and potential (Figure 2B) hypoxia sensors in each subgroup.
Figure 2
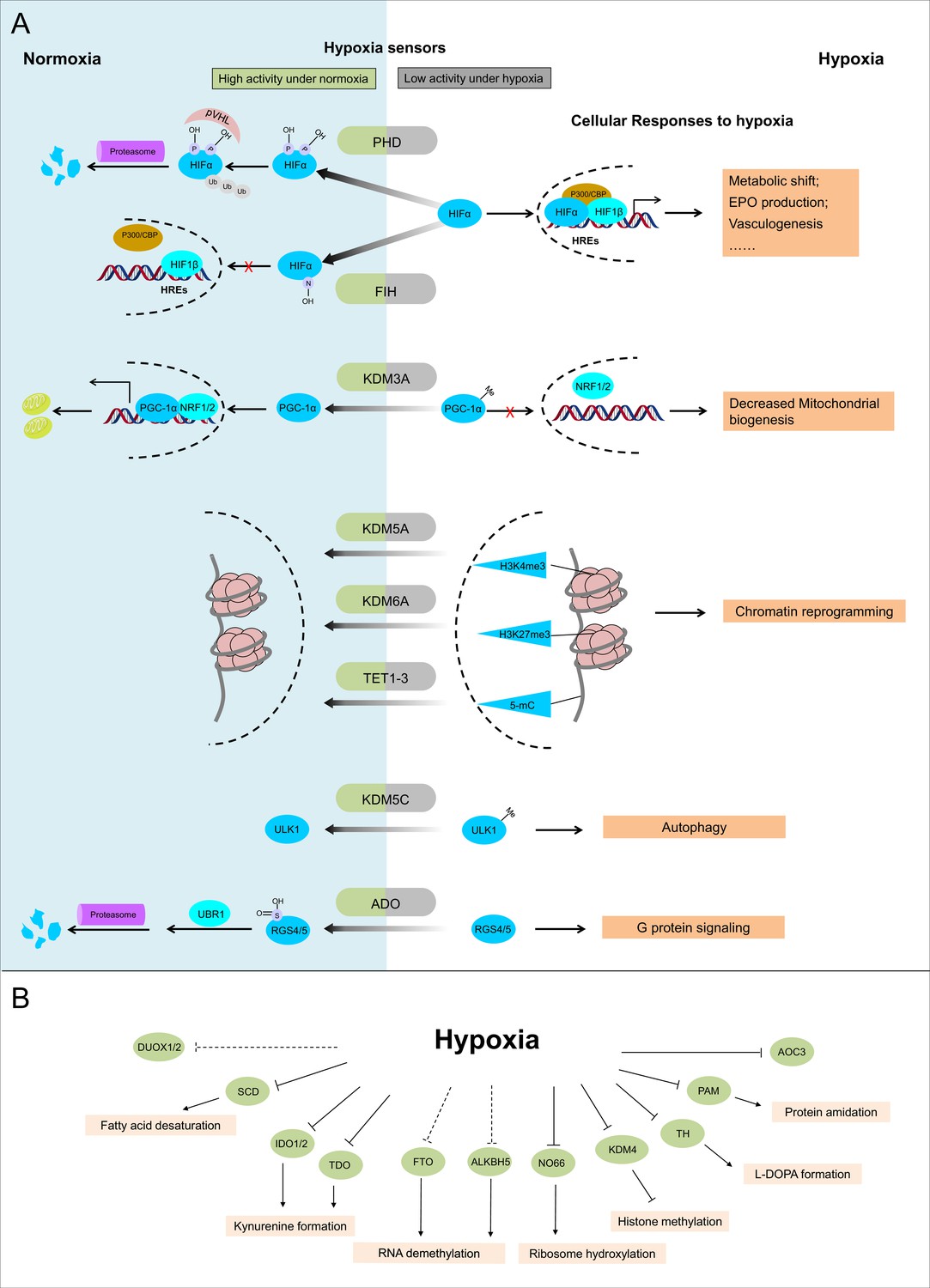
Known and candidate sensors for hypoxia inside O2-dependent enzymes.
(A) Known hypoxia sensors and their corresponding cellular responses to hypoxia. Decreased O2 concentration inhibits activities of hypoxia sensors in O2-dependent enzyme category and results in changes downstream signaling pathway as the cellular response to hypoxia. PHD catalyzes the hydroxylation at two prolyl residues of HIFα, and then the hydroxylated HIFα is recognized and ubiquitylated by pVHL. Following ubiquitilation, HIFα is degraded by proteasome. During hypoxia, activity of PHD is diminished and HIFα is stabilized. Accumulated HIFα translocates to the nucleus, and in dimerization with HIF1β, recruits other transcriptional coactivators (p300, CBP), binds with the hypoxia response elements (HREs) and activates the transcription of HIF target genes. The products of these genes participate in adaptation to hypoxia including metabolic shift, EPO production, vasculogenesis, etc. FIH catalyzes the asparaginyl hydroxylation of HIFα, and this hydroxylation inhibits HIFα from recruiting transcriptional coactivators. Compared with PHD, FIH is inhibited by more severe hypoxia. KDM3A catalyzes the demethylation of K244 monomethylation of PGC-1α, which is a transcriptional coactivator and regulates mitochondrial biogenesis. Under normoxia, PGC-1α binds with transcriptional factor NRF1/2 and activates the transcription of nucleus-encoded mitochondrial genes. Under hypoxia, the inhibited activity of KDM3A leads to accumulation of K224 monomethylation at PGC-1α. The maintained monomethylation at K224 of PGC-1α reduces its binding ability with NRF1/2 and results in decreased mitochondrial biogenesis. KDM5A catalyzes the demethylation at Lys4 of histone H3 (H3K4). Hypoxia inhibits its activity and results in the hypermethylation at H3K4, which is responsible for the gene activation. Similarly, hypoxia also inhibits KDM6A, and results in the hypermethylation at its target site H3K27 and gene repression. TET methylcytosine dioxygenases (TET1, TET2, and TET3) catalyze conversion of DNA 5-methylcytosine (5-mC) to the 5-hydroxymethylcytosine (5hmC) and mediates DNA demethylation. Hypoxia reduces TET activity and causes DNA hypermethylation. Together, these proteins sense hypoxia and lead to transcription alteration by chromatin reprogramming. KDM5C catalyzes the demethylation of ULK1 R170me2s, which regulates ULK1 activity. Under normoxia, R170me2s of ULK1 is removed by KDM5C and ULK1 remains inactive. Under hypoxia, the inhibited activity of KDM5C leads to accumulation of ULK1 R170me2s, and results in ULK1 activation and autophagy induction. ADO catalyzes the thiol oxidation at the N terminal Cys of a protein, which then triggers its degradation through N-degron pathway. Hypoxia inhibits the activity of ADO and leads to the stabilization of its substrates. One of the identified ADO substrates is RSG4/5, regulators of the G protein signaling. Stabilization of RGS4/5 results in the modulation of G-protein-coupled calcium ion signaling. (B) Candidate O2 sensors with reduced enzymatic activities in hypoxia. Hypoxia leads to: inhibition of KDM4A and KDM4B and accumulated hypermethylation at H3K9; inhibition of SCD and increased cellular fatty acid saturation; inhibition of IDO and changes of immunoregulation; inhibition of PAM and reduced protein amidation; in vitro inhibition of RIOX1 and RIOX2 which are responsible for ribosome hydroxylation; in vitro inhibition of AOC3; RNA hypermethylation possibly through inhibition of FTO/ALKBH5; potential inhibition of DUOX1 and DUOX2. PHD: prolyl hydroxylase domain-containing protein; HIF: hypoxia-inducible factor; pVHL: von Hippel-Lindau protein E3 ligase; CBP, cyclic-AMP response element binding protein binding protein; EPO: erythropoietin; FIH: factor inhibiting HIF1; KDM: JmjC (Jumonji C) domain lysine demethylase; PGC: peroxisome proliferator-activated receptor gamma coactivator; NRF: nuclear respiratory factor; TET: ten-eleven translocation methylcytosine dioxygenases; ADO: cysteamine (2-aminoethanethiol) dioxygenase; RGS: regulators of G protein signalling; SCD: stearoyl-CoA desaturases; IDO: indoleamine 2,3-dioxygenase; AOC: amine oxidase, copper containing; PAM: peptidylglycine α-amidating monooxygenase; RIOX: ribosomal oxygenase, FTO: fat mass and obesity-associated protein; ALKBH: AlkB homolog; DUOX: dual oxidase.
Dioxygenases
Based on their catalytic centers, the dioxygenase family members can be further classified into 2-OG-dependent dioxygenase, heme-dependent dioxygenases, lipoxygenases, and other dioxygenases (Table 2).
2-Oxyglutarate (2-OG)-dependent dioxygenases
In humans, there are ~60 identified or postulated dioxygenases (Table 2, Supplementary file 1) that use 2-OG as the co-substrate to catalyze the hydroxylation of their primary substrates, which include proteins, nucleic acids, and lipids (Figure 3A, Figure 3—figure supplement 1; Islam et al., 2018; Fletcher and Coleman, 2020; Rose et al., 2011). We note that when hydroxylation occurs on the carbon of an N-methyl group, this can lead to demethylation, which occurs through spontaneous fragmentation to formaldehyde and the demethylated product (Figure 3B; Islam et al., 2018; Fletcher and Coleman, 2020; Rose et al., 2011).
Figure 3 with 3 supplements see all
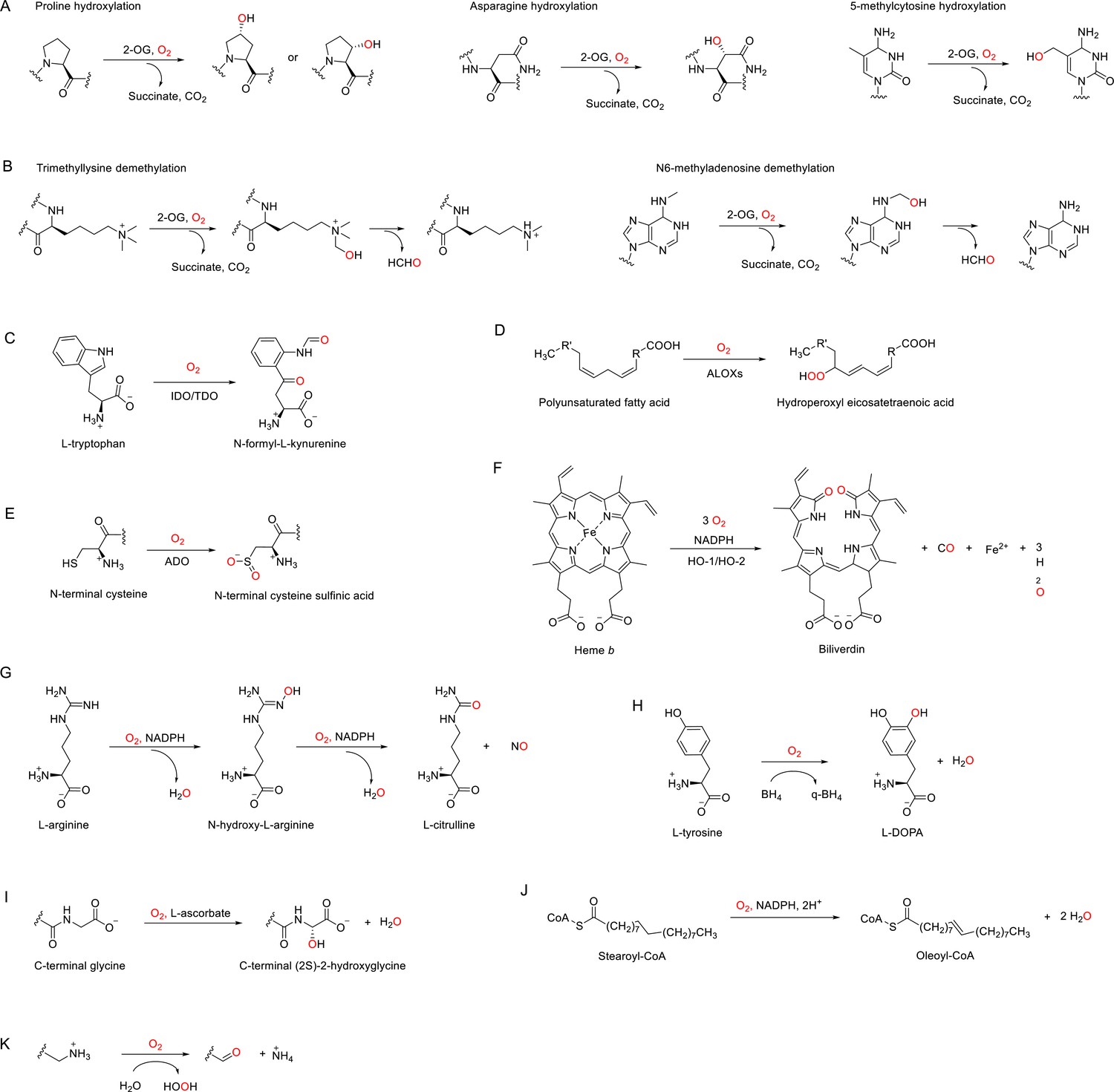
Enzymatic reactions catalyzed by discussed O2-dependent enzymes.
(A) Examples of hydroxylation reactions catalyzed by 2-OG-dependent dioxygenases. (B) Examples of demethylation reactions catalyzed by 2-OG-dependent dioxygenases. (C–K) Reactions catalyzed by indoleamine 2,3-dioxygenase (IDO)/tryptophan 2,3-dioxygenase (TDO) (C), arachidonate lipoxygenases (ALOXs) (D), (2-aminoethanethiol) dioxygenase (ADO) (E), heme oxygenases (HOs) (F), nitric oxide synthases (NOSs) (G), tyrosine 3-hydroxylase (TH) (H), peptidylglycine α-amidating monooxygenase (PAM) (I), stearoyl-CoA desaturase 1 (SCD1), (J) and copper amine oxidases (CAOs) (K).
Among O2-dependent enzymes, 2-OG-dependent dioxygenases are relatively well studied. A majority of members in this subgroup catalyze hydroxylation or demethylation on proteins, DNA, and RNA, and are involved in the regulation of transcription and translation (Islam et al., 2018; Fletcher and Coleman, 2020; Rose et al., 2011). We focus on three subgroups relevant to hypoxia biology: direct HIF modulators, epigenetic modulators, and translational modulators. These subgroups encompass most known hypoxia sensors, including PHDs, factor inhibiting HIF (FIH1), lysine demethylases (KDMs), and ten-eleven translocation methylcytosine dioxygenases (TET1-3), as well as potential sensors that have impaired activities during hypoxia. For each subgroup, we highlight the most well-known sensors and propose additional, potential sensors.
Direct HIF modulators
These include PHDs (which catalyze prolyl hydroxylation of HIFα) and FIH (which catalyzes asparaginyl hydroxylation of HIFα) (Table 3).
Table 3
Direct HIF modulator in 2-OG-dependent dioxygenases.
| Gene symbol | Protein name | Type of reaction | Hydroxylation sites in HIFα | Non-HIF substrate examples |
|---|---|---|---|---|
| EGLN1 | PHD2 | Prolyl hydroxylation | HIF1α Pro402, Pro564; HIF2α Pro405, Pro531; HIF3α Pro492 | FLNA, Akt |
| EGLN2 | PHD1 | Prolyl hydroxylation | HIF1α Pro402, Pro564; HIF2α Pro405, Pro531; HIF3α Pro492 | FOXO3, Cep192, TP53 |
| EGLN3 | PHD3 | Prolyl hydroxylation | HIF1α Pro564; HIF2α Pro405, Pro531; HIF3α Pro492 | ATF-4, ADRB2, TP53 |
| HIF1AN | FIH1 | Asparaginyl hydroxylation | HIF1α Asn803, HIF2α Asn847 | IκBα, Notch, OTUB1, RIPK4 |
The PHD enzymes and their critical role in regulating the PHD–HIF-pVHL signaling pathway are a paradigm for cellular sensing and response to hypoxia (Figure 2A; Majmundar et al., 2010; Kaelin and Ratcliffe, 2008; Ivan and Kaelin, 2017; Schofield and Ratcliffe, 2004). In humans, HIF is composed of an α subunit (HIF1α, HIF2α, or HIF3α) and invariant β subunit (HIF1β), and there are three PHD isoforms, namely PHD1 (EGLN2), PHD2 (EGLN1), and PHD3 (EGLN3). These PHDs are canonical sensors that illustrate our criteria for O2 sensors.
First, PHDs directly interact with O2, utilizing O2 to hydroxylate prolines in the O2-dependent degradation domain (ODD) of HIFα (Epstein et al., 2001; Hirsilä et al., 2003).
Second, the enzymatic activities of PHDs are sensitive to cellular/tissue hypoxia. The O2-binding affinities of all three PHDs, represented by O2 Km values, have been measured in vitro with HIF1α peptides as substrates. Using a short 19-residue HIF1α fragment as the substrate, the reported O2 Km values for PHD1-3 are in the range of 229–1746 μM (Table 4; Hirsilä et al., 2003; Dao et al., 2009; Tarhonskaya et al., 2014). However, recent measurements using longer HIF1α fragments estimate O2 Km values for PHD2 in the range of 67–85 μM (Table 4; Ehrismann et al., 2007), corresponding to pO2 values in the (physoxia) range of 6–8% (Table 1), consistent with the sensitivities of PHDs to changes in physiological O2 concentrations.
Table 4
Reported Km values of O2-dependent enzymes.
Km values vary based on the assay method and tested substrate. Some enzymes have multiple Km values listed, reflecting measurements from different studies.
| Category | Enzyme* | Km for O2 | Assay details | Reference |
|---|---|---|---|---|
| Dioxygenase | 250 μM | In vitro radioactivity 2-OG turnover assay with HIF1α (556–574) peptide as substrate | Hirsilä et al., 2003 | |
| 1746 μM | In vitro time-resolved fluorescence resonance energy transfer assay with P564-HIF1α peptide (DLEMLAPYIPMDDDFQL) as substrate | Dao et al., 2009 | ||
| 67 μM | In vitro O2 consumption assay with HIF1α (502–697) peptide as substrate | Ehrismann et al., 2007 | ||
| 81 μM | In vitro O2 consumption assay with HIF1α (530–698) peptide as substrate | Ehrismann et al., 2007 | ||
| 230 μM | In vitro radioactivity 2-OG turnover assay with HIF1α (556–574) peptide as substrate | Hirsilä et al., 2003 | ||
| PHD3 | 230 μM | In vitro radioactivity 2-OG turnover assay with HIF1α (556–574) peptide as substrate | Hirsilä et al., 2003 | |
| KDM4E | 197 μM | In vitro O2 consumption assay with ARK(me3)STGGK peptide as substrate | Cascella and Mirica, 2012 | |
| 173 μM | In vitro MALDI-TOF-MS assay with H31−15K9me3 peptide as substrate | Hancock et al., 2017 | ||
| 57 μM | In vitro O2 consumption assay with ARK(me3)STGGK peptide substrate | Cascella and Mirica, 2012 | ||
| 60 μM | In vitro radioactivity 2-OG turnover assay with histone H3(1–19)K9me3 as substrate | Chakraborty et al., 2019 | ||
| KDM6A | 180 μM | In vitro radioactivity 2-OG turnover assay with histone H3(21–44)K27(me3) as substrate | Chakraborty et al., 2019 | |
| KDM4C | 158 μM | In vitro O2 consumption assay with ARK(me3)STGGK peptide substrate | Cascella and Mirica, 2012 | |
| KDM4B | 150 μM | In vitro radioactivity 2-OG turnover assay with histone H3(1–19)K9me3 as substrate | Chakraborty et al., 2019 | |
| 90 μM | In vitro radioactivity 2-OG turnover assay with HIF1α (788–822) peptide as substrate | Koivunen et al., 2004 | ||
| 90 μM | In vitro radioactivity 2-OG turnover assay with histone H3(1–21)K4me3 as substrate | Chakraborty et al., 2019 | ||
| KDM3A | 75 μM (7.6% O2) † | In vitro demethylation-formaldehyde dehydrogenase-coupled reaction assay with K224-monomethylated PGC-1α peptide as substrate | Qian et al., 2019 | |
| KDM5B | 40 μM | In vitro radioactivity 2-OG turnover assay with histone H3(1–21)K4me3 as substrate | Chakraborty et al., 2019 | |
| P4HA1 | 40 μM | Standard P4H activity assay with (Pro-Pro-Gly)10 (Peptide Institute) as a substrate | Hirsilä et al., 2003 | |
| KDM5C | 35 μM | In vitro radioactivity 2-OG turnover assay with histone H3(1–21)K4me3 as substrate | Chakraborty et al., 2019 | |
| TET1 | 30 μM | In vitro radioactivity 2-OG turnover assay with oligonucleotides containing a 5-mC as substrate | Laukka et al., 2016 | |
| 3.0 μM (0.31% O2) † | In vitro DNA hydroxymethylation assay with genomic DNA as substrate | Thienpont et al., 2016 | ||
| TET2 | 30 μM | In vitro radioactivity 2-OG turnover assay with oligonucleotides containing a 5-mC as substrate | Laukka et al., 2016 | |
| 5.2 μM (0.53% O2) * | In vitro DNA hydroxymethylation assay with genomic DNA as substrate | Thienpont et al., 2016 | ||
| KDM5D | 25 μM | In vitro radioactivity 2-OG turnover assay with histone H3(1–21)K4me3 as substrate | Chakraborty et al., 2019 | |
| KDM6B | 20 μM | In vitro radioactivity 2-OG turnover assay with histone H3(21–44)K27(me3) as substrate | Chakraborty et al., 2019 | |
| IDO1 | 11.5–24 μM | In vitro O2 consumption assay with L-Trp as substrate | Kolawole et al., 2015 | |
| PTGS1 | 10 μM (sheep) | In vitro radioactivity label assay with [1-14C]arachidonic acid as substrate | Juránek et al., 1999 | |
| PTGS2 | 13 μM (mouse) | In vitro radioactivity label assay with [1-14C]arachidonic acid as substrate | Juránek et al., 1999 | |
| ALOX5 | 13 μM (porcine) | In vitro radioactivity label assay with [1-14C]arachidonic acid as substrate | Juránek et al., 1999 | |
| ALOX12 | 13 μM | In vitro radioactivity label assay with [1-14C]arachidonic acid as substrate | Juránek et al., 1999 | |
| ALOX15 | 26 μM (porcrine) | In vitro radioactivity label assay with [1-14C]arachidonic acid as substrate | Juránek et al., 1999 | |
| ALOX15 | 26 μM (rabbit) | In vitro radioactivity label assay with [1-14C]arachidonic acid as substrate | Juránek et al., 1999 | |
| ADO | >500 μM | In vitro UPLC-MS-TOF assay with RGS4(2–15) peptide as substrate | Masson et al., 2019 | |
| Monooxygenase | 350 μM (rat) | In vitro heme-NO complex formation assay with L-arginine as substrate | Abu-Soud et al., 1996 | |
| 130 μM (mouse) | In vitro heme-NO complex formation assay with L-arginine as substrate | Abu-Soud et al., 2001 | ||
| NOS3 (eNOS) | 4 μM (bovine) | In vitro heme-NO complex formation assay with L-arginine as substrate | Abu-Soud et al., 2000 | |
| 25 μM (bovine) | In vitro heme-NO complex formation assay with N-hydroxy-L-arginine as substrate | Abu-Soud et al., 2000 | ||
| TH | 16.2 μM (low-activity state); 46.1 μM (high- activity state); | In vitro radioactivity label assay with 3H-tyrosine as substrate | Rostrup et al., 2008 | |
| 12.6–26.7 μM (low-activity state); 28.8–42.9 μM (high-activity state)‡; | In vitro oxygraphic assay with tyrosine as substrate | Rostrup et al., 2008 | ||
| 2.6–3.9 μM (2–3 mmHg, rat) * | In vitro radioactivity label assay with 3H-tyrosine as substrate | Katz, 1980 | ||
| TPH1 | 3.9~12.9 μM (3–10 mmHg, rat) † | In vitro radioactivity label assay with 3H-tryptophan as substrate | Katz, 1980 | |
| PAH | 17 μM | In vitro oxygraphic assay with phenylalanine as substrate | Rostrup et al., 2008 | |
| PAM | 70 μM (rat) | In vitro radioactivity label assay with [α-2H2]-N-acylglycine of different chain length as substrates | McIntyre et al., 2010 | |
| Oxidase | Cytochrome c oxidase | <0.1 μM (rat) | In vitro O2 consumption assay measuring O2 consumption of purified rat mitochondria at low phosphate potential ([ATP]/[ADP]*[Pi]) | Bienfait et al., 1975 |
| 1–3 μM (rat) | In vitro O2 consumption assay measuring O2 consumption of purified rat mitochondria at high phosphate potential | Bienfait et al., 1975 | ||
| 0.5 μM (mouse) | Cellular assay measuring the ‘apparent K (m)’ for O2 or p 50 of respiration in 32D cells using high-resolution respirometry | Scandurra and Gnaiger, 2010 | ||
| AOC3 | 38 μM | In vitro enzymatic assay using purified human AOC3 | Shen et al., 2012 |
-
*
O2-dependent enzymes that are known sensors are highlighted in bold; that are reported to be inhibited under hypoxia are highlighted in a light orange background; that are reported to be associated with positive selections in high-altitude populations are highlighted in red (also see Table 6).
-
†
Km of these enzyme were reported with units as % O2 or mmHg, and calculated according to Mas-Bargues et al., 2019; Place et al., 2017.
-
‡
Combined data for TH1/3/4 splicing isoforms.
Third, the decreased activity of PHDs during hypoxia triggers specific downstream responses (Figure 2A; Majmundar et al., 2010; Kaelin and Ratcliffe, 2008; Ivan and Kaelin, 2017; Schofield and Ratcliffe, 2004). Under normoxia, hydroxylated HIFα is recognized and polyubiquitinylated by the E3 ubiquitin ligase von Hippel-Lindau protein (pVHL), which then leads to proteasome-mediated degradation of HIFα. Under hypoxia, decreased O2 concentration suppresses the activity of PHDs. This allows HIFα to accumulate and translocate to nucleus, where it associates with the constitutively expressed HIF1β and forms the heterodimer transcriptional factor HIF. HIF then recruits transcriptional co-activators p300 and CREP-binding protein (CBP), binds with hypoxia-responsive elements (HREs) on DNA, and subsequently activates its target genes. Products of HIF-regulated genes are involved in multiple cellular and systematic adaptations to hypoxia, including metabolic shift from OXPHOS to glycolysis, redox homeostasis, angiogenesis, and erythropoiesis.
Although all PHD isoforms have similar O2 affinities, their different expression patterns and substrate preferences among the HIFα isoforms lead to differential regulation of hypoxia sensing and response by the PHD-HIF-pVHL pathway. Of the three PHD isoforms, PHD2 exerts the greatest control over the PHD-HIF-VHL pathway response to hypoxia (Berra et al., 2003) and it is ubiquitously expressed across mouse tissues and is the most abundant isoform (Appelhoff et al., 2004; Willam et al., 2006). PHD1 and PHD3 are more tissue-specific, with PHD1 most expressed in testis and PHD3 in heart (Willam et al., 2006). PHD2 favors HIF1α as substrate, while PHD1 and PHD3 favor HIF2α (Table 3; Appelhoff et al., 2004). Interestingly, PHD2 and PHD3 are themselves HIF target genes and can be induced during hypoxia to provide feedback regulation for the PHD-HIF-pVHL pathway (Epstein et al., 2001). The nonoverlapping and complex roles of different PHD isoforms are evidenced by knockout mice: Phd2-/- mice are embryonic lethal due to placental and heart defects, while postnatal whole-body knockout of Phd2 leads to polycythemia, increased angiogenesis, and heart defects (Takeda et al., 2006; Minamishima et al., 2008). Tissue-specific knockout of Phd2 in mouse heart or brain is protective against ischemic cardiac or neural injury, respectively (Kunze et al., 2012; Hölscher et al., 2011). Phd1 knockout mice have altered metabolism in skeletal muscles and overall enhanced hypoxia tolerance (Aragonés et al., 2008); and germline Phd3 knockout mice are hypotensive due to a hypofunctional sympathoadrenal system (Bishop et al., 2008).
There has been interest to identify non-HIF substrates for PHDs and other pathways that are regulated through PHD-catalyzed hydroxylation. In the last two decades, more than 20 non-HIF substrates for PHDs have been reported, and hypoxia-mediated hydroxylation of these proteins alters response of downstream pathways (Cockman et al., 2019; Strowitzki et al., 2019). Most of these substrates were identified through cellular studies, suggesting that PHDs may act upon non-HIF substrates under physiological conditions (Strowitzki et al., 2019; Zheng et al., 2014; Moser et al., 2013; Segura et al., 2016; Guo et al., 2016; Köditz et al., 2007; Xie et al., 2009; Ullah et al., 2017; Deschoemaeker et al., 2015; Rodriguez et al., 2018). One group found in in vitro enzymatic assays that PHDs lacked detectable activities on these non-HIF substrates. This difference in findings between cellular and in vitro enzymatic assays suggests that the action of PHDs on non-HIF substrates requires additional cellular machinery, such as adaptors or post-translational modifications (Cockman et al., 2019).
FIH1 is another 2-OG dioxygenase that is known to sense hypoxia and regulate the HIF pathway (Figure 2A; Lando et al., 2002a; Mahon et al., 2001). Under normoxia, FIH1 catalyzes the asparaginyl hydroxylation of the C-terminal transactivation domain (CTAD) of HIFα (Table 3; Koivunen et al., 2004; Lando et al., 2002b), which is responsible for its binding with the transcriptional coactivator p300/CBP (Lando et al., 2002b; Freedman et al., 2002). FIH1-catalyzed asparaginyl hydroxylation of the CTAD impairs the recruitment of p300/CBP and reduces transcriptional activity of HIF (Lando et al., 2002a; Lando et al., 2002b). In hypoxia, FIH1 activity is also reduced by hypoxia, enabling HIFα to recruit p300/CBP for transcriptional activation of its target genes (Lando et al., 2002a; Lando et al., 2002b). The reported O2 Km value for FIHs is 90 μM, using a HIF1α peptide containing site Asn803 (Table 4; Koivunen et al., 2004). Compared with the Km values of PHDs in similar assays, FIH1 appears to be less sensitive to hypoxia, that is, as O2 levels decrease, PHDs are inhibited before FIH1 (Tian et al., 2011). Thus, FIH1 is considered a fine modulator of the HIF pathway in sensing severe hypoxia. Consistent with the notion that its role is more limited, FIH1 knockout mice have abnormal metabolism but not other HIF-regulated processes (Zhang et al., 2010; Sim et al., 2018). There are also non-HIF substrates identified for FIH1 that may also be regulated in an O2-dependent manner (Table 3; Cockman et al., 2009b; Scholz et al., 2016; Cockman et al., 2009a).
Epigenetic modulators
These include the lysine demethylase (KDM) Jumonji C (JmjC) domain-containing proteins, and the DNA demethylases ten-eleven translocation enzymes (TETs).
JmjC domain-containing proteins contain domains for Fe(II) and 2-OG binding and catalytic activities (Shmakova et al., 2014; Kooistra and Helin, 2012). They also bind with O2 and utilize it as a substrate, therefore having the potential to function as hypoxia sensors if their O2-binding affinities allow (Shmakova et al., 2014). Of the 32 identified JmjC proteins in humans, at least 23 conduct lysine demethylation reactions (Shmakova et al., 2014). Their substrates include both histone lysines (K4, K9, K27, and K36 on histone 3) and some non-histone lysines. (Table 5). Histone methylations affect chromatin structure and compactness and consequently regulate gene expression in either activating or silencing mode (Table 5; Shmakova et al., 2014; Kooistra and Helin, 2012; Walport et al., 2016; Li et al., 2022). H3K4me2/3, H3K9me2, H3K27me3, and H3K36me3 levels increase after hypoxia, possibly due to KDMs acting as O2 sensors and effecting chromatin changes.
Table 5
JmjC domain-containing histone demethylases and their substrates*.
(A = activating transcription, S = silencing transcription).
| KDM class | Members (gene symbol) | Histone lysyl residue substrates | Other substrates |
|---|---|---|---|
| KDM2 | KDM2A | H3K36me1/me2 (A) | p65, NF-κB |
| KDM2B | H3K36me1/me2 (A), H3K4me3 (A) | ||
| KDM3 | KDM3A | H3K9me1/me2 (S) | PGC-1α K224me |
| KDM3B | H3K9me1/me2 (S) | ||
| JJMJD1C | H3K9me1/me2 (S) | ||
| KDM4 | KDM4A | H3K9me2/me3 (S), H3K36me2 (A), H1.4K26me2/me3 | WIZ, CDYL1, CSB, and G9a |
| KDM4B | H3K9me2/me3 (S), H3K36me2 (A), H1.4K26me2/me3 | WIZ, CDYL1, CSB, and G9a | |
| KDM4C | H3K9me2/me3 (S), H3K36me2 (A), H1.4K26me2/me3 | WIZ, CDYL1, CSB, and G9a | |
| KDM4D | H3K9me2/me3 (S) | ||
| KDM4E | H3K9me3 (S) | H3R2me2/me1, H3R8me2/me1, H3R26me2/me1, H4R3me2 | |
| KDM5 | KDM5A | H3K4me2/me3 (A) | |
| KDM5B | H3K4me2/me3 (A) | ||
| KDM5C | H3K4me2/me3 (A) | H3R2me2/me1, H3R8me2, H4R3me2a, ULK1R170me2a | |
| KDM5D | H3K4me2/me3 (A) | ||
| KDM6 | KDM6A | H3K27me2/me3 (S) | |
| KDM6B | H3K27me2/me3 (S) | ||
| KDM6C | |||
| KDM7 | KDM7A | H3K9me1/me2 (S), H3K27me1/me2 (S) | |
| PHF8 | H3K27me1/me2 (S), H4K20me1 | ||
| PHF2 | H3K9me2/me3 (S) | ||
| Jmjc domain only | NO66 | H3K4me2/me3 (A), H3K36me2/me3 (A) | Rpl8 |
| MINA53 | H3K9me3 (S) | Rpl27a | |
| KDM8 | H3K36me2 (A) | NFATc1 | |
| JMJD6 | H3R2me2,H4R3me2/me1, U2AF2/U2AF65, LUC7L2 |
-
*
Known hydroxylation/demethylation sites are indicated.
KDM6A, also known as UTX, catalyzes demethylation at H3K27me2/me3 (Table 5; Hong et al., 2007). In 2019, Chakraborty et al. reported that increase in H3K27me3 levels during hypoxia is HIF-independent and is caused by the direct inhibition of KDM6A due to decreased pO2 under hypoxia (Figure 2A; Chakraborty et al., 2019). The O2 sensitivity of KDM6A was further confirmed by its O2 Km value of 180 μM (Table 4), in a similar range as the PHDs and FIH and the highest among KDM6 members (Chakraborty et al., 2019). Sensing of hypoxia by KDM6A can control cell fate by chromatin reprogramming (Chakraborty et al., 2019). For example, it is reported that in mouse myoblast C2C12 cells, increase in H3K27me3 levels due to inactivation of KDM6A represses the expression of myogenic genes and blocks myogenic differentiation (Chakraborty et al., 2019).
KDM5A catalyzes demethylation at H3K4me2/me3 (Table 5) and was recently reported by Batie et al. to be another hypoxia sensor that could directly regulate cell fate through chromatin reprogramming (Figure 2A; Christensen et al., 2007; Batie et al., 2019). KDM5A also has a relatively low O2 affinity, with a Km ~90 μM (Table 4; Batie et al., 2019). Inactivation of KDM5A by hypoxia results in rapidly increasing H3K4me3 levels, and downstream effects include active transcriptions of genes associated with antiproliferation, antiapoptosis, etc. (Batie et al., 2019). Excitingly, a new study found that KDM5A binding to H3K4me3 is enhanced by PHD1-mediated hydroxylation of H3 at proline residue 16 (H3P16OH) (Liu et al., 2022). This KDM5A-PHD1 axis raises the possibility of other cross-talk between O2 sensors.
KDM4 family enzymes mainly catalyze the demethylation at H3K9me2/me3 and H3K36me2 (Table 5; Hillringhaus et al., 2011). Their O2-binding affinities have also been investigated in vitro: the O2 Km values of KDM4A, KDM4B, KDM4C, and KDM4E are all within the range of 57–197 μM (Table 4; Cascella and Mirica, 2012; Hancock et al., 2017). These Km values suggest the potential of KDM4 members to be hypoxia sensors, but this depends on their cellular roles and the downstream responses of their speculated inhibition during hypoxia (Figure 2B). Of these KDM4 members, cellular activity of KDM4A is reported to show a graded response to O2 concentration in U2OS cells and so does the demethylation levels on H3K9me3 (Hancock et al., 2017). It is also reported that KDM4A regulates the transcription of HIF1α through the H3K9 methylation status at HIF1α locus during hypoxia in tumors (Dobrynin et al., 2017). More studies for the function of other KDM4 demethylases during hypoxia are still needed.
There are also cases where the JmjC-containing KDMs function as hypoxia sensors through non-histone substrates. One such example is KDM3A, a histone demethylase for H3K9me2/1 sites (Table 5), whose activity on H3K9me2 is maintained even under severe hypoxia (0.2% O2), suggesting a high binding affinity with O2 with this substrate (Yamane et al., 2006; Brauchle et al., 2013; Beyer et al., 2008). However, recently Qian et al. discovered that the demethylation activity of KDM3A on a non-histone substrate, peroxisome proliferator-activated receptor gamma coactivator (PGC-1α) K224me, is inhibited by hypoxia (Figure 2A), with Km ~7.6% O2 (~75 μM), high enough to function as a hypoxia sensor under physiological conditions (Table 4; Qian et al., 2019). PGC-1α is a transcriptional coactivator that binds with transcriptional factor nuclear respiratory factor (NRF1/2) for activating transcription of nucleus-encoded mitochondrial genes (Qian et al., 2019; Scarpulla et al., 2012). The inhibited activity of KDM3A causes the accumulation of K224 mono-methylation on PCG-1α, which reduces the interaction between PCG-1α and NRF1/2, decreasing mitochondrial biogenesis (Qian et al., 2019). Another example is KDM5C, a histone demethylase for H3K4me2/me3 sites, which can also function as an arginine demethylase (Figure 2A; Li et al., 2022; Iwase et al., 2007). Its demethylation of ULK1 R170me2s site is inhibited by 1% O2 level in LN229 and several other cell lines (Li et al., 2022). This inhibited demethylation stabilizes ULK1 R170me2s, which further activates ULK1 and induces autophagy as a downstream response (Li et al., 2022). The cases of KDM3A and KDM5C suggest the possibility of other JmjC-containing KDMs to sense and respond to hypoxia through undiscovered non-histone substrates.
Besides KDMs, another set of 2-OG-dependent dioxygenases that act as epigenetic regulators are the TET enzymes (TET1, TET2, and TET3 in humans). These enzymes catalyze the hydroxylation of DNA 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (Ponnaluri et al., 2013; Wu and Zhang, 2017). This facilitates the subsequent demethylation of 5hmC into an unmodified cytosine (Ponnaluri et al., 2013; Wu and Zhang, 2017). Since CpG methylation is typically silencing, TETs tend to promote gene activation. A large variance exists in different reports about the O2 Km values of TET1 and TET2, ranging from 0.31% and 0.53% (3.0 μM and 5.2 μM) using genomic DNA as substrates to 30 μM using oligonucleotides as substrates (Table 4); however, these measurements show much tighter O2 binding of TET1 and TET2 compared with the aforementioned reported hypoxia sensors (KDM3A, KDM5A, and KDM6A) (Laukka et al., 2016; Thienpont et al., 2016). Severe hypoxia, such as 0.5% O2 treatment, is reported to directly impair the cellular activities of TETs, increase DNA hypermethylation, and decrease the expression levels of associated genes (Figure 2A; Thienpont et al., 2016). DNA hypermethylation caused by TETs inhibition also happens during pathophysiological hypoxia found in tumors (Thienpont et al., 2016). Considering their O2 sensitivity, TETs are more likely to function as hypoxia sensors under extreme hypoxic conditions.
Translational modulators
Translational modulators in the 2-OG-dependent dioxygenases include the mRNA hydroxylases and ribosome hydroxylases.
The most abundant RNA modification is N6-methylation of adenosine (m6A), which affects the processing, splicing, translation, and degradation of modified mRNAs (Zaccara et al., 2019; Chen et al., 2019). The dynamics of m6A modification is coordinated by methyltransferases (so-called ‘writers’), demethylases (‘erasers’), and identifiers (‘readers’) (Zaccara et al., 2019; Chen et al., 2019). Two m6A RNA demethylases have been identified thus far: FTO and ALKBH5, with demonstrated demethylase activity in vitro and in vivo, respectively (Zheng et al., 2013; Jia et al., 2011.Zaccara et al., 2019; Zheng et al., 2013; Mauer et al., 2017). Indirect evidence for a sensor role of these enzymes in hypoxia response is that despite their protein levels staying relatively constant, hypoxia leads to m6A accumulation in cancer cells and breast cancer. Thus, it is possible that hypoxia plays a role in direct inhibition of ALKBH5 and/or FTO (Figure 2B). More studies about other possible demethylases for m6A in mRNA and the O2-binding affinities of these enzymes are needed to determine which of them directly sense and respond to hypoxia.
The translational apparatus may itself be targeted by ribosome hydroxylases, which modify the histidyl or prolyl residues of ribosomal subunit proteins (Bundred et al., 2018; Zhuang et al., 2015). These ribosome hydroxylases regulate translation and participate in physiological or disease processes, including cellular growth, skeletal bone formation, tumorigenesis, and immune regulation (Bundred et al., 2018; Zhuang et al., 2015; Ge et al., 2012; Singleton et al., 2014). Currently, three ribosome hydroxylases have been identified: histidyl hydroxylases MINA53 (RIOX2) and NO66 (RIOX1) targeting the 60S large subunits Rpl27a and Rpl8, respectively; and prolyl hydroxylase OGFOD1 targeting the 40S small subunit Rpl23 (Ge et al., 2012; Singleton et al., 2014). Among these, NO66 has its activity inhibited by 0.1–1% O2 in cellular studies (Figure 2B). By contrast, OGFOD1 still retains 80% of its cellular activity even under severe hypoxia (0.2% O2) (Ge et al., 2012; Singleton et al., 2014). While this suggests the potential for NO66 to be a sensor in severe hypoxia, more studies of the O2 affinity of NO66 and the downstream response of its inhibition by hypoxia are needed.
Heme-dependent dioxygenases
Heme prosthetic groups are used by these enzymes for O2 binding and activation (Figure 3—figure supplement 2; Huang and Groves, 2018; Efimov et al., 2011; Raven, 2017). Five known heme-dependent dioxygenases in humans are indoleamine 2,3-dioxygenase (IDO) 1 and 2, tryptophan 2,3-dioxygenase (TDO), and prostaglandin G/H synthase (PGHS) 1 and 2 (Paton and Ntambi, 2009; Efimov et al., 2011; Raven, 2017). Except for IDO2 that has not been measured, these dioxygenases have reported Km values of 10–30 μM (Table 4; Juránek et al., 1999; Kolawole et al., 2015). As reflected by their lower O2 Km values, heme-dependent dioxygenases tend to have stronger O2 binding than 2-OG-dependent dioxygenases.
Both TDO and IDOs catalyze the conversion of L-tryptophan to N-formyl-L-kynurenine (Figure 3C; Thackray et al., 2011). They have similar heme- and substrate-binding pockets, although they share low sequence identity overall, and are believed to be an example of convergent evolution (Ball et al., 2014; Thackray et al., 2011). Both TDO and IDO regulate immune responses, possibly by modifying tryptophan homeostasis. TDO and IDO have distinct tissue expression patterns, with TDO mostly restricted to liver and epidermis, while IDO is found throughout the body and can be induced by certain immune or inflammation signals (Ball et al., 2014). Their expression patterns may further regulate their relative importance in different tissues or toward different stimuli (Ball et al., 2014). The cellular activity of TDO is reported to be inhibited by hypoxia (1–10% O2) in HeLa cells transfected with TDO, while TDO protein level remains unaltered (Elbers et al., 2016). Cellular activity of IDO1 is also decreased by hypoxia (1% O2) in 86HG39 and HeLa cells with unaltered IDO1 protein level (Schmidt et al., 2013). Impaired immune responses are observed in both cases as downstream effects (Figure 2B; Elbers et al., 2016; Schmidt et al., 2013). Further studies are needed to clarify whether hypoxia directly inhibits the enzymatic activities of TDO and IDO1 and how this might trigger downstream responses in a more physiological system.
Lipoxygenases
Lipoxygenases (LOXs) are iron-containing dioxygenases that catalyze the insertion of O2 into polyunsaturated fatty acids (PUFA) and their derivatives, forming hydroperoxyl eicosatetraenoic acid (HPETE) products (Figure 3D). HPETE products are chemically unstable and reduced by peroxidases to hydroxyl eicosatetraenoic acid (HETE) (Biringer, 2020; Ivanov et al., 2010; Kuhn et al., 2015). In humans, there are six known LOXs with arachidonic acid as the most common substrate (Biringer, 2020; Kuhn et al., 2015). These arachidonate lipoxygenases (ALOXs) are named according to the positional specificity in their catalyzed hydroperoxyl reactions as the ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3 (Biringer, 2020; Kuhn et al., 2015). Functions of ALOXs include biosynthesis of inflammatory mediators as well as regulation of cellular redox state (Kuhn et al., 2015).
ALOXs demonstrate that O2-binding affinities can be affected by the specific substrate, as illustrated by ALOX15. The reported O2 Km values of human ALOX12, rat ALOX5, and rabbit ALOX15 are all within the range of 8–26 μM, as measured by biochemical studies with arachidonic acid as the substrate (Table 4; Juránek et al., 1999; Wecksler et al., 2009). While the rabbit ALOX15 reaches its Vmax under normoxia with arachidonic acid as the substrate, its reaction rate with hydroxyl arachidonic acids as substrates still increases with increasing O2 concentration under hyperoxic conditions (Ivanov et al., 2005). This suggests a higher O2 Km value, a lower O2-binding affinity, and the ability to sense the change of O2 concentration from normoxic to hypoxic conditions when hydroxyl arachidonic acids are the substrates for rabbit ALOX15. Similarly, human ALOX15 has O2 Km values for different substrates, namely 24 μM for arachidonic acid and 9.6 μM for linoleic acid. Furthermore, allosteric binding of 12-HEHE to ALOX15 affects its O2 affinity (Wecksler et al., 2009).
The fact that substrates affect O2 affinity is not a mere laboratory curiosity. Multiple ALOX substrates may be involved in hypoxia-related diseases, including pulmonary hypertension and cardiovascular diseases (Mashima and Okuyama, 2015; Zhu and Ran, 2012; Ivanov et al., 2015). Studying the substrate-dependent O2 sensitivity of human ALOXs in various biological contexts will be necessary to ascertain whether and how these enzymes sense and respond to hypoxia in vivo.
Other dioxygenases
Ten other human dioxygenases have been identified (Supplementary file 1). They have the shared property of using an octahedral Fe(II) as the catalytic center.
Among these 10 enzymes, cysteamine (2-aminoethanethiol) dioxygenase (ADO) has been identified as a hypoxia sensor (Masson et al., 2019). ADO catalyzes the oxidation of protein N-terminal cysteines to cysteine sulfinic acid (Figure 3E) and promotes the degradation of the oxidized substrate protein through the N-degron pathway (Masson et al., 2019). Human ADO has a relatively low O2-binding affinity (Km > 500 μM, Table 4, Masson et al., 2019). As a result, even mild hypoxia inhibits ADO activity, allowing stabilization of its substrates, including the regulator of G protein signaling (RGS4/5) and cytokine interleukin (IL)-32 (Masson et al., 2019). During hypoxia, inhibited ADO results in the stabilization of RGS4/5 and subsequently modulates G protein-coupled calcium ion signals and mitogen-activated protein kinase (MAPK) signaling (Figure 2A; Masson et al., 2019). Hypoxia sensing by ADO provides a faster response compared with HIF-mediated transcriptional regulation (Masson et al., 2019).
Monooxygenases
The monooxygenase members can be further classified into iron-dependent, copper-dependent, and flavin-dependent monooxygenases based on their catalytic centers (Table 2).
Iron-dependent monooxygenases
Most monooxygenases utilize iron as the catalytic center for oxygen insertion and can be further divided into heme-dependent and non-heme-dependent ones.
Heme-dependent monooxygenases
In humans, these include cytochrome P450 enzymes, heme oxygenases, and nitric oxide synthases.
Cytochrome P450 enzymes (CYPs), which comprise ~60% of all human monooxygenases (Supplementary file 1), are responsible for the oxidative metabolism of both endogenous and exogenous chemicals (Guengerich, 2007; Danielson, 2002; Poulos and Johnson, 2005). They play an important role in the synthesis and metabolism of hormones, cholesterols, and vitamins, and the clearance and detoxification of xenobiotics (Danielson, 2002; Poulos and Johnson, 2005). Not only do they have diverse biological functions, but their biochemical properties vary widely as well. For instance, the substrate specificity of CYPs ranges from a single substrate (such as CYP19A1) to a diverse repertoire of substrates (Poulos and Johnson, 2005). The O2 sensitivities of CYPs have been assessed for drug clearance in cellular systems or subcellular systems such as liver microsomes (Fradette and Du Souich, 2004). With different drug substrates used in the assays, a wide range of O2 Km values of mixed CYPs have been reported, ranging from 0.5 to 200 μM in mammalian species (Jones, 1981). This is consistent with reports that hypoxia could increase the half-life and/or toxicity of certain drugs. However, it is currently unclear whether CYPs do, in fact, function as hypoxia sensors. Direct evidence is lacking that cellular CYP activity is affected by hypoxia, and their O2 Kms with endogenous substrates are largely unknown. It is known, however, that hypoxia affects the expression levels of some CYPs, suggesting that these CYPs may have a role in hypoxia response (Fradette and Du Souich, 2004). In principle, a given CYP could act as both a hypoxia sensor and downstream effector.
Heme oxygenases (HO) catalyze the degradation of cellular heme to biliverdin, also producing ferrous iron and carbon monoxide (Figure 3F; Yoshida and Migita, 2000). There are two catalytically active human heme oxygenases, HO-1 and HO-2. They do not contain prosthetic heme groups for their catalytic reactions; instead, they bind heme substrates that are used for O2 binding, activation, and reduction (Yoshida and Migita, 2000). Although their O2 Kms are unknown, their estimated dissociation constant (Kd) is 0.012–0.034 μM (Migita et al., 1998). These very high O2-binding affinities suggest that HOs are unlikely to be direct O2 sensors. Instead, the activity of HO-2 may indirectly be regulated by O2 through redox potential, which has implications for whole-body sensing in the carotid body (López-Barneo et al., 2008; Ragsdale and Yi, 2011), thereby affecting whole-body physiological response to hypoxia.
Nitric oxide synthases (NOSs) convert L-arginine into nitric oxide (NO) in two steps, each using one O2 molecule activated by the heme iron (Figure 3G; Daff, 2010). NO is a gas signaling molecule with an array of functions, including regulation of vascular tone, immune defense, neural development, and hypoxia signaling (Moncada and Higgs, 1991; Ho et al., 2012). There are three human NOS enzymes: neuronal NOS (NOS1 or nNOS) is constitutively expressed in nerve, skeletal muscle, and heart muscle cells; inducible NOS (NOS2 or iNOS) is induced in multiple immune cells after stimuli; and endothelial NOS (NOS3 or eNOS) is constitutively expressed in vascular endothelial cells (Daff, 2010).
The O2 Km values for all three NOSs have been reported and are quite different from each other: 350 μM for rat nNOS, 130 μM for mouse iNOS, and 4 μM for bovine eNOS when using L-Arg as substrate (Table 4; Stuehr et al., 2004; Santolini et al., 2001a; Abu-Soud et al., 2000; Abu-Soud et al., 2001; Santolini et al., 2001b; Abu-Soud et al., 1996). (Although measured from different species, these values have been compared with each other to illustrate the different O2 affinities of these three NOSs; Abu-Soud et al., 2001; Semenza, 2005.) These differences in O2 Km values, together with tissue-specific expression patterns of different NOS isoforms, account for their distinct roles in response to hypoxia. The low O2 Km value of eNOS may help eNOS enzymatic activity remain constant across O2 concentration ranges in vascular endothelial cells (Ho et al., 2012; Semenza, 2005). The high Km value of nNOS indicates its enzymatic activity is more dependent on O2 concentrations, and it is reported to have a linear relationship between O2 concentration and its NO-producing activity over the entire physiological O2 range (Santolini et al., 2001a; Semenza, 2005; Elayan et al., 2000). This suggests the activity of nNOS decreases during acute hypoxia, which should be neuroprotective as excessive NO is reported to increase neurotoxicity during acute ischemic stroke (Ho et al., 2012; Dawson and Dawson, 1996). In fact, hyperbaric O2 treatment can increase NO production in rat nNOS and lead to neurotoxicity, a hint that nNOS could also function to sense hyperoxia (Elayan et al., 2000). During chronic hypoxia, nNOS functions more as an effector: upregulation of nNOS expression level leads to the increase of NO production to increase blood flow by vasodilation (Ward et al., 2005). The Km value of iNOS is also high enough for a hypoxia sensor, but iNOS is not typically expressed and needs to be induced by different stimulus in most human tissues, making the condition for it to function as a sensor more complicated (Abu-Soud et al., 2001; Robinson et al., 2011).
In addition, the NOSs can also cross-talk with the PHD-HIF-pVHL pathway by NO-derived cysteine S-nitrosylation of HIF1α and pVHL protein, which can inhibit the binding between hydroxylated HIF1α and pVHL and stabilize HIF1α even when O2 is not limiting (Li et al., 2007; Palmer et al., 2007). This may play a role in immune cells where iNOS can be induced to activate HIF-mediated immune response (Li et al., 2007).
Non-heme Fe-dependent monooxygenases
There are eight identified non-heme Fe-dependent monooxygenases in humans (Supplementary file 1) that utilize several different cofactors for iron coordination. Five use (6R)-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4) as an electron donor and co-substrate, namely tyrosine 3-hydroxylase (TH), tryptophan 5-hydroxylase 1 and 2 (TPH1 and TPH2), phenylalanine-4-hydroxylase (PAH), and alkylglycerol monooxygenase (Bassan et al., 2003; Watschinger et al., 2010). Similar to 2-OG-dependent dioxygenases, the catalytic iron is coordinated in an octahedral mode for binding and activation of O2 (Bassan et al., 2003).
TH catalyzes the hydroxylation of L-tyrosine into L-3,4-dihydroxyphenylalanine (L-DOPA) (Figure 3H), the rate-limiting step in biosynthesis of catecholamines (dopamine, noradrenaline, and adrenaline) (Rostrup et al., 2008). The O2 Km values of TH vary by splice isoform, ranging from 12 to 47 μM across the four splice isoforms as measured in in vitro enzymatic assays (Table 4; Rostrup et al., 2008; Katz, 1980). In cellular assays, the activity of TH in PC12 cells was inhibited when O2 concentration decreased from 139 to 33 μM (Rostrup et al., 2008). These measurements suggest that acute hypoxia inhibiting TH could lead to decreased synthesis of catecholamines (Figure 2B; Raghuraman et al., 2012; Souvannakitti et al., 2009). Since dopamine inhibits the chemotransduction of the carotid body in most mammals, suppression of TH activity during acute hypoxia may sensitize the carotid body to hypoxia (Iturriaga and Alcayaga, 2004; Iturriaga et al., 2009). In contrast to acute hypoxia, chronic or intermittent hypoxia leads to upregulation of TH activity via increased mRNA and/or phosphorylation, countering its decreased enzymatic activity (Kumar et al., 2003; Hui et al., 2003; Schnell et al., 2003).
PAH and TPH, like TH, are also aromatic amino acid hydroxylases, with similar structures and catalytic mechanisms (Bassan et al., 2003). The O2 Km values of human PAH and rat TPH are reported to be 17 uM and 3.9–12.9 uM in enzymatic assay, respectively (Table 4; Katz, 1980). Their potential for hypoxia sensing and responding needs further exploration.
Copper-dependent monooxygenases
Besides iron, copper is also frequently used for O2 binding and activation by oxidizing enzymes. In humans, there are five identified or speculated monooxygenases that use copper as the catalytic center (Supplementary file 1). These enzymes all have two copper ions at their active sites but employ different strategies for O2 binding and activation, depending on whether the two copper irons are in sufficient proximity to be magnetically coupled (Decker and Solomon, 2005; Lewis and Tolman, 2004). The coupled binuclear Cu enzymes such as tyrosinase (TYR) use both copper ions for O2 binding and activation, while the non-coupled binuclear Cu enzymes such as peptidylglycine α-amidating monooxygenase (PAM) and dopamine b-monooxygenase (DβM) use only one copper iron (CuB) for this process (Decker and Solomon, 2005; Lewis and Tolman, 2004). The crystal structure of PAM shows that CuB has a tetrahedral structure, coordinated by two His residues and one Met residues, with the other position for O2 binding (Prigge et al., 2004).
PAM catalyzes the amidation of C-terminal glycines in peptides (Figure 3I), a post-translational modification that may affect substrate stability (Simpson et al., 2015). PAM activity is progressively inhibited from mild (7% O2) to severe (1% O2) hypoxia in mammalian cells (Figure 2B; Simpson et al., 2015). Rat PAM has been shown to have high O2 Km values (100–550 μM), with this wide range attributable to different degrees of substrate hydrophobicity (Table 4; McIntyre et al., 2010). The best-characterized substrates of PAM are endocrine peptides, for example, chromogranin A (CgA), whose amidation by PAM is profoundly suppressed by hypoxia (Simpson et al., 2015; Merkler, 1994). However, the functional consequence of this change in amidation remains unclear (Simpson et al., 2015).
Flavin-dependent monooxygenases
Flavin-dependent monooxygenases utilize a non-covalently bound FAD prosthetic group to activate O2 (Palfey and McDonald, 2010; Romero et al., 2018). Unlike above discussed O2-dependent enzymes whose reaction rates are saturated above an O2 threshold, reaction rates for these enzymes are thought to be directly proportional to O2 concentration. This suggests that decreased O2 concentration from normoxia to hypoxia could decrease reaction rates of these enzymes (Massey, 2002), although it is not clear how their cellular activities are affected by hypoxia.
Oxidases
The oxidase members can be further classified into heme-copper, iron-dependent, copper-dependent, flavin-dependent, and other oxidases based on their catalytic centers (Table 2).
Heme-copper oxidases
Heme-copper oxidases (HCO) are the terminal oxidases in the aerobic respiratory chain that catalyze the 4-electron reduction of O2 to water (Ferguson-Miller and Babcock, 1996; Nolfi-Donegan et al., 2020). In mammals, this is the cytochrome c oxidase (CcO), also known as the Complex IV of the electron transport chain (ETC) in mitochondria (Nolfi-Donegan et al., 2020; Figure 3—figure supplement 3). Mammalian CcOs utilize a hetero-binuclear heme-copper center to activate O2 (Wikström et al., 2018; Namslauer and Brzezinski, 2004). O2 binds with both the heme iron and the copper as a ligand bridge and is then reduced with electrons passed from the reduced form of cytochrome c through other metal prosthetic sites (Wikström et al., 2018; Namslauer and Brzezinski, 2004; Aoyama et al., 2009). Compared with other O2-dependent enzymes, CcO has a high O2 affinity, with O2 Km values measured to be <1 μM in assays using intact cells or purified mitochondria with sufficient substrates (Table 4; Petersen et al., 1974; Bienfait et al., 1975; Gnaiger et al., 1995; Scandurra and Gnaiger, 2010). Based solely on its low Km values, CcO would not be expected to act as hypoxia sensors.
However, CcO appears to be inhibited during hypoxia (1–3% O2) (Chandel et al., 1997; Duranteau et al., 1998). Inhibition of CcO disrupts the ETC, which is related to electron leakage from Complex III and Complex I (Duranteau et al., 1998; Fuhrmann and Brüne, 2017; Hernansanz-Agustín et al., 2017; Guzy et al., 2007), increased mitochondrial produced ROS (Duranteau et al., 1998; Fuhrmann and Brüne, 2017; Hernansanz-Agustín et al., 2017; Guzy et al., 2007; Chandel et al., 2000), and altered downstream HIF, PI3K/Akt, AMPK, and MAPK signaling (Fuhrmann and Brüne, 2017; Guzy et al., 2007; Chandel et al., 2000; Brand, 2016; Kim et al., 2018; Emerling et al., 2005; Kulisz et al., 2002; Emerling et al., 2009). Thus, despite CcO’s low Km values it has hypoxia sensor-like properties. How CcO’s activities are regulated during hypoxia remains to be elucidated.
Iron-dependent oxidases
Iron-dependent oxidases include the desaturases and ferroxidases (Supplementary file 1; Jasniewski and Que, 2018). The structures and catalytic mechanisms are relatively poorly characterized, but the studied ones all have two Fe(II) ions, coordinated by five His/Glu residues from the enzymes, that bind and active O2 (Jasniewski and Que, 2018; Hess et al., 2010; Bertini et al., 2012).
Stearoyl-CoA desaturase 1 (SCD1) catalyzes the formation of the monounsaturated fatty acid oleic acid from the saturated fatty acid stearic acid (Figure 3J; Paton and Ntambi, 2009), thereby playing an important role in lipid metabolism, membrane fluidity, and cell integrity (Paton and Ntambi, 2009), as increased fatty acid saturation could result in lipotoxicity and cell death (Hess et al., 2010; Wang et al., 2006; Green and Olson, 2011). Inhibition of SCD1 activates the unfolded protein response (UPR) through ER stress (Green and Olson, 2011; Volmer et al., 2013). Although the O2 affinity of SCD1 is unknown, hypoxia (1% O2) impairs the cellular activity of SCD1 in A549 and HeLa cells, increasing the saturated fatty acid ratio and thereby altering the cellular lipid composition (Figure 2B; Kamphorst et al., 2013). SCD1 may well be a hypoxia sensor if the decrease in its activity is directly due to reduced O2 concentration.
Copper-dependent oxidases
Copper-dependent oxidases include the copper amine oxidases (CAOs) and lysyl oxidases (LOXs), both of which catalyze oxidative deamination of amines to the corresponding aldehydes, also producing hydrogen peroxide and ammonia (Figure 3K; Finney et al., 2014). Human CAO member AOC3 is reported to have an O2 Km ~38 μM in enzymatic assays (Table 4), and its cellular activity in adipocyte lysate is inhibited by hypoxia in a HIF-independent manner (Figure 2B; Shen et al., 2012; Repessé et al., 2015; Andrés et al., 2001; Morris et al., 1997). The cellular function of AOC3 is not clear since its endogenous substrates are unknown, although in vitro kinetics studies suggest dopamine and cysteamine as potential substrates (Shen et al., 2012).
Flavin-dependent oxidases
Similar to flavin-dependent monooxygenases, this group of oxidases utilize either FAD or FMN for the activation of O2 and have reaction rates proportional to the O2 concentration (Massey, 2002). The >20 members of this group (Table 2, Supplementary file 1) in humans have substrates ranging from small molecules (e.g., fatty acids and amino acids) to protein residues (Romero et al., 2018). Their role in hypoxia sensing and responding is unknown.
Other oxidases
Several other oxidases remain poorly characterized (Supplementary file 1). The dual oxidases DUOX1 and DUOX2 catalyze the formation of H2O2 from O2 molecules with electrons provided by NADPH (Donkó et al., 2005). In HIF1-deficient Caenorhabditis elegans, hypoxia-induced extracellular matrix (ECM) remodeling could be phenocopied by inactivation of BLI-3, the ortholog of human DUOXs. This suggests a potential role of BLI-3 as a hypoxia sensor independent from the HIF pathway (Figure 2B; Vozdek et al., 2018) and raises the possibility that human DUOXs also sense and respond to hypoxia in an HIF-independent manner.
ODE as hypoxia sensors in other organisms
ODEs are evolutionary ancient. The major emergence of ODEs occurred at the separation of terrestrial and marine bacteria, coinciding with the emergence of oxygenic photosynthesis ~3.1 billion years ago (Jabłońska and Tawfik, 2021). Given the importance of hypoxia sensing, the evolutionary conservation of the HIF pathway across metazoans comes as no surprise. HIF1α and PHD2 (EGLN1 in C. elegans) emerged early in evolution, whereas additional HIFα and PHD isoforms emerged later in more complex organisms as context-dependent and fine-tuned hypoxia sensing became necessary (Taylor and McElwain, 2010).
What about plants? Plants have a hypoxia sensor, ADO, shared with metazoans. ADO is a thiol dioxygenase that modulates Arg/N-degron pathways and was found to be a sensor in both humans and Arabidopsis thaliana (Masson et al., 2019). Besides ADO, plant cysteine oxidases (PCOs), homologs of ADO, also function as hypoxia sensors, regulating Arg/N-degron pathways in plants (White et al., 2017; Weits et al., 2014; White et al., 2018).
What about single-celled organisms? In fission yeast, two hypoxia-sensing mechanisms exist that converge on activation of Sre1 (the yeast SREBP homolog), a transcription factor that triggers a downstream hypoxia response. One mechanism is that hypoxia inhibits multiple ODEs that are required for sterol synthesis, and this suppression of sterol synthesis stimulates the cleavage (and hence activation) of Sre1 (Hughes et al., 2005). The other mechanism is that hypoxia inhibits Ofd1, a yeast prolyl 4-hydroxylase-like 2-OG-dependent dioxygenase. Similar to how PHD inhibition allows HIF1α stabilization, Ofd1 inhibition allows Sre1 stabilization (Hughes and Espenshade, 2008). Protozoa also have prolyl hydroxylases that regulate the stability of S-phase kinase-associated protein 1 (Skp1) and alter the cell cycle (Xu et al., 2012).
What about prokaryotes? Prokaryotes also sense O2, although they do not appear to use ODEs as sensors. In nitrogen-fixing bacteria (Rhizobium meliloti), changes in O2 levels impact the kinase activity of FixL, which phosphorylates the transcription factor FixJ to regulate the expression of nitrogen-fixing genes (Monson et al., 1995; Agron et al., 1994). In this case, O2 actually acts as an allosteric binding cofactor that leads to a conformation change of FixL (Monson et al., 1995). Hence, FixL directly interacts with O2 molecules, but O2 is not used as a substrate. In most of all of the above, although the details may differ, the key criteria for a hypoxia sensor are met: (1) direct interaction with O2, (2) utilization of O2 as a substrate except in the case of FixL, and (3) causing a downstream response.
Connection of O2-dependent enzymes to hypoxia adaptations and diseases
Hypoxia is related to many diseases. Decreased O2 at high altitudes can lead to systemic, organismal-level hypoxia and induce acute and chronic mountain sickness (AMC, CMC) (Roach and Hackett, 2001; Villafuerte and Corante, 2016). Systemic hypoxia is also seen in some respiratory diseases and anemic conditions that have disruption in O2 uptake or transport (Lee et al., 2019). Ischemia resulting from the blockage of blood flow leads to cell death and failure of affected tissues, most notably heart (in myocardial infarction) and brain (in ischemic stroke). (Lee et al., 2019) However, other tissues can also be affected, including the intestine, kidney, and skeletal muscle.
As previously mentioned, many O2-dependent enzymes are regulated at a transcriptional, translational, and/or post-translational level in response to hypoxia. Depending on the specific downstream responses and cellular context, a change in enzymatic activity may confer protection or further injury in hypoxia. One of the greatest challenges in understanding the effects of hypoxia is discerning the effects of adaptation and hypoxia tolerance versus maladaptation and hypoxia-mediated tissue injury. Injury and adaptation almost always overlap, either in time, development, or tissue domains.
We conclude below with two scenarios illustrating the role of O2-dependent enzymes in hypoxia adaptation or diseases: (1) positively selected genetic adaptations associated with O2-dependent enzymes in high-altitude populations; and (2) mutations in genes targeted by drugs associated with O2-dependent enzymes for hypoxia-related diseases.
O2-dependent enzymes in hypoxia adaptations of high-altitude populations
Tibetan, Andean, and Ethiopian populations reside at altitudes above 3500 m with a decreased O2 pressure (<60% of sea level) due to hypobaric hypoxia. Distinct genetic adaptations and physiological characteristics have developed within each population to promote survival at altitude (Beall, 2006). These three groups of humans have resided at high altitude for different lengths of time: Andeans for 10,000–15,000 years (Aldenderfer, 2003), Tibetans for over 30,000 years (Qi et al., 2013), and Ethiopians for even longer (Alkorta-Aranburu et al., 2012).
In lowlanders, one of the major adaptations upon exposure to hypoxia is increased red blood cell production (erythropoiesis) (Windsor and Rodway, 2007). Acutely, the increased hemoglobin helps compensate for decreased blood pO2, but in the long run, this increases blood viscosity and thereby increases risk of blood clots and ischemia (Braekkan et al., 2010; Parati et al., 2018). Erythropoiesis during exposure to hypoxia is mainly regulated by EPO, a downstream target of HIF that regulates erythropoiesis by activating EPO receptors on erythroid progenitors in the bone marrow (Haase, 2013).
Do Tibetan highlanders maintain higher levels of hemoglobin compared to populations living at sea level? Somewhat surprisingly, their hemoglobin levels are actually similar to those of lowlanders (Beall et al., 1998; Beall et al., 1997), but they have increased vasodilation and blood flow to compensate for O2 delivery to tissues. Genetic studies have identified variants under positive selection at the EGLN1 (PHD2) and EPAS1 (HIF2A) loci (Haase, 2013; Lorenzo et al., 2014; Bigham et al., 2010; Yi et al., 2010; Simonson et al., 2010; Xu et al., 2011; Yang et al., 2017; Peng et al., 2011; Wuren et al., 2014). One variant of EGLN1 exhibits a lower O2 Km value, which enhances HIF degradation under hypoxia and contributes to blunting of EPO-mediated erythropoiesis (Lorenzo et al., 2014).
Interestingly, positive selection is also observed at the EGLN1 and EGLN2 loci in Andean populations, but unlike Tibetans, Andeans have higher hemoglobin levels than lowlanders (Beall et al., 1998; Bigham et al., 2010; Bigham et al., 2009). Genetic studies suggest that Andeans cope with the risks of augmented erythropoiesis by enhancing cardiovascular function associated with positive selection in BRINP3, NOS2, and TBX5 (Crawford et al., 2017). Among these, NOS2, also known as iNOS (discussed above in the sensor section), synthesizes NO as a gas signaling molecule to modulate vascular tone upon induction of this gene (Moncada and Higgs, 1991; Robinson et al., 2011). How these gene variants mechanistically lead to adaptation awaits further study.
Ethiopians have a distinct adaptation pattern compared with Tibetans and Andeans. Some studies suggest that they maintain both normal blood saturation and hemoglobin concentration (Beall et al., 2002). A genome-wide scan identified HIF pathway-related genes, ARNT2 and THRB, as candidate genes for positive selection with potential roles in the physiological response to hypoxia (Scheinfeldt et al., 2012).
Apart from these relatively well-studied HIF pathway-related genes, there are multiple other genes harboring variants associated with positive selection in these three highlander populations (Table 6). Notably, these include other known hypoxia sensors, such as HIF1AN and KDM5A, as well as potential hypoxia sensors, including KDM4A, HMOX4, SCD, and DUOX2. It will be interesting to further explore how these positively selected variants enhance hypoxia adaptation of highlanders.
Table 6
O2-dependent enzymes encoded by genes associated with positive selection in different high-altitude populations.
-
*
Known hypoxia sensors are highlighted in red.
Pathogenic mutations and drug targets within O2-dependent enzymes for hypoxia-related diseases
Erythrocytosis commonly results from exposure to hypoxia. Genetic mutations in the pathway regulating erythropoiesis can also cause pathogenic erythrocytosis with excessive blood viscosity. Such pathogenic mutations have been found in genes, including (1) VHL, EGLN1 (PHD2), and EPAS1 (HIF2A) that affect EPO production, (2) EPOR and its regulator JAK2 that affect erythroid progenitor maturation, and (3) hemoglobin subunits HBA and HBB that affect O2 delivery and tissue pO2 (Bento, 2018). Specifically, for EGLN1, more than 10 variants have been associated with erythrocytosis onset (Gardie et al., 2014). For example, one such mutation (P317R) has significantly decreased enzymatic activity (Percy et al., 2006). No mutations associated with pathogenic erythrocytosis have been identified in EGLN2 and EGLN3, consistent with the notion that EGLN1/PHD2 is the major isoform involved in HIF-mediated EPO upregulation.
Conversely, chronic kidney diseases (CKDs) lead to diminished EPO production and anemia. In adults, EPO is mainly produced by erythropoietin-producing cells (EPCs) in the kidney (Haase, 2013). The dysfunction of EPCs during CKDs results in EPO deficiency and is a key factor leading to associated anemia (Koury and Haase, 2015). Injectable erythropoiesis-stimulating agents (ESAs), such as recombinant human erythropoietin (rhEPO), are a cornerstone of CKD treatment (Singh et al., 2006; Pfeffer et al., 2009; Portolés et al., 2021). In recent years, PHD inhibitors have been developed as an alternative route to HIF stabilization and subsequent EPO production (Portolés et al., 2021; Gupta and Wish, 2017). Unlike rhEPO, PHD inhibitors ameliorate not only EPO deficiency but also inflammation and altered iron metabolism in CKD, both of which are regulated by HIF (Koury and Haase, 2015; Portolés et al., 2021). Currently, four PHD inhibitors (Roxadustat, Vadadustat, Daprodustat, and Molidustat) have entered or completed phase III clinical trials for treatment of the anemia of CKD (Portolés et al., 2021). Of them, Roxadustat and Daprodustat have been approved for use in Japan and/or China (Dhillon, 2019; Dhillon, 2020).
Open questions for discovering hypoxia sensors within the ODE members
Although we have an in-depth understanding of a small handful of O2 sensors, the potential landscape of hypoxia sensing in humans remains largely uncharted. Even once an ODE is confirmed to be a hypoxia sensor, much remains to be investigated.
At the enzymatic level
Most reported O2 Km values for ODEs are based on in vitro testing of the enzyme. However, in vivo, O2 Km depends on (1) the substrate (e.g., as discussed for ALOX12), and (2) the regulation of the ODE by other proteins and cofactors (e.g., as discussed for HO-2). Regarding (1), for a given ODE, what are its O2 affinities when catalyzing reactions using its various endogenous substrates in vivo? Answering this question requires identifying the in vivo substrates and then measuring O2 Km for each substrate. Regarding (2), how is the O2 Km of the ODE affected by modifying factors (e.g., PTMs and cofactor binding)? Answering this question requires identifying the modifying factors and then measuring O2 Km in the appropriate cellular contexts.
At the cellular level
Most studies of ODEs have focused on individual pathways directly responsible for downstream effects. However, these pathways do not act in isolation but rather as part of a network. Each ODE could have roles in multiple downstream response pathways, feedback loops, and cross-talk with other pathways. Ultimately, a systems-level understanding is needed to capture the complexity of O2 sensing within the cell.
At the tissue level
Currently, most studies of ODEs use cell models in which the O2 level is set to a single level controlled experimentally. However, this ignores the fact that from tissue-to-tissue, O2 levels vary substantially even at baseline. Furthermore, when an organism is exposed to hypoxic stress, the O2 levels from tissue-to-tissue and within a tissue can vary even further due to tissue-level changes such as vasodilation. This raises the question: what is the tissue specificity (and/or cell type specificity) of hypoxia sensors under basal and stressed conditions? An intriguing possibility is that each tissue might have a unique set of ODEs in order to sense and respond to the ongoing fluctuations in O2 concentration during maintenance of homeostasis, in accordance with tissue-specific O2 levels.
At the organismal level
Although an impressive diversity of molecular O2 sensors has been identified, their role in organismal-level adaptations to hypoxia remains unexplored territory. By far the best studied system for hypoxia sensing and response at the organismal level is PHD-HIF-pVHL pathway. Its role in improving O2 transport by increasing EPO synthesis by the kidney is well understood and has been the subject of many reviews (Haase, 2013; Nangaku and Eckardt, 2007).
There are numerous other adaptations to hypoxia that are much less well understood than HIF adaptations. An important one is the regulation of breathing. The mystery in this fundamental adaptation is the basis for gradually increasing breathing volume with time at altitude, such that blood O2 level is restored toward normal. This respiratory adaptation has several different time domains, and each likely has a unique set of sensors and effectors. O2 sensing at the carotid body is the first part of this response, and carotid body chemoreceptors have been the target of numerous attempts to identify molecular O2 sensors and the transduction pathways involved (López-Barneo et al., 2008). O2-sensitive potassium channels, redox sensors, and others have been proposed. Final agreement on the nature of the O2 sensor remains surprisingly elusive, perhaps reflecting that a diversity of O2 sensors take part in shaping the breathing response, not just one.
One of the challenges in linking molecular O2 sensors to responses at the organismal level is that organismal-level responses and adaptations are diverse. Vertebrate animals vary enormously on their tolerance of O2 deprivation. Some vertebrates, such as the crucian carp and the Western painted turtle, can survive for months without O2 (Bickler and Buck, 2007). These animals exceed the hypoxia tolerance of humans by a factor of at least 10,000 (Bickler and Buck, 2007). The molecular switches that orchestrate this impressive capability remain poorly defined. Certainly, if one is searching for molecular O2sensors, animals such as the carp and turtle would be fertile ground.
At the developmental level
Development as a model for changing O2 sensing and response has been little explored. Changes in O2 during development can be dramatic: the intrauterine environment of placental gas exchange has been likened to that of ascent of Mt. Everest, with a rapid increase in O2 upon aerial respiration at birth (Barcroft, 1946; Martin et al., 2010). The changes in O2 availability may signal crucial changes in synaptic physiology in the brain. How O2 sensing is regulated throughout development in accordance with changes in O2 levels is an important question to be answered.
Summary
Aerobic organisms have evolved mechanisms to sense and respond to changes in O2 levels. O2 participates in hundreds of biochemical reactions regulating diverse, essential cellular processes. The enzymes responsible for these reactions directly interact with O2 and may function as hypoxia sensors by transducing the signal of low O2 via a decrease in enzymatic activity (rate or product yield). Here, we summarized and discussed the known and potential hypoxia sensors within each subcategory of O2-dependent enzymes in human, expanding from the well-known PHD enzymes, to the more recently identified sensors within the KDM family, to other enzymes with emerging roles in hypoxia sensing. We also discussed O2-dependent enzymes involved in hypoxia-related evolutionary adaptations and diseases, highlighting their relevance beyond chemical reactions. Much remains to be explored for most O2-dependent enzymes and roles in hypoxia. Are there new hypoxia sensors still to be discovered within O2-dependent enzymes? How do various hypoxia sensors coordinate with each other to regulate downstream cellular responses? What is the mechanism for each tissue to set its own hypoxia sensing threshold based on the specific physiological pO2? Furthermore, how can these discoveries help with hypoxia adaptation and disease treatment? All these questions await future research.
References
-
Reaction mechanisms of mononuclear non-heme iron oxygenasesChemical Reviews 105:2227–2252.https://doi.org/10.1021/cr040653o
-
Nitric oxide binding to the heme of neuronal nitric-oxide synthase links its activity to changes in oxygen tensionJournal of Biological Chemistry 271:32515–32518.https://doi.org/10.1074/jbc.271.51.32515
-
Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthaseThe Journal of Biological Chemistry 275:17349–17357.https://doi.org/10.1074/jbc.M000050200
-
Regulation of inducible nitric oxide synthase by self-generated NOBiochemistry 40:6876–6881.https://doi.org/10.1021/bi010066m
-
Oxygen regulation of expression of nitrogen fixation genes in Rhizobium melilotiResearch in Microbiology 145:454–459.https://doi.org/10.1016/0923-2508(94)90094-9
-
Tissue activity and cellular localization of human semicarbazide-sensitive amine oxidaseThe Journal of Histochemistry and Cytochemistry 49:209–217.https://doi.org/10.1177/002215540104900208
-
Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factorThe Journal of Biological Chemistry 279:38458–38465.https://doi.org/10.1074/jbc.M406026200
-
Turning the oxygen dial: Balancing the highs and lowsTrends in Cell Biology 30:516–536.https://doi.org/10.1016/j.tcb.2020.04.005
-
Tryptophan-catabolizing enzymes - party of threeFrontiers in Immunology 5:485.https://doi.org/10.3389/fimmu.2014.00485
-
Ventilation and hypoxic ventilatory response of Tibetan and Aymara high altitude nativesAmerican Journal of Physical Anthropology 104:427–447.https://doi.org/10.1002/(SICI)1096-8644(199712)104:4<427::AID-AJPA1>3.0.CO;2-P
-
Hemoglobin concentration of high-altitude Tibetans and Bolivian AymaraAmerican Journal of Physical Anthropology 106:385–400.https://doi.org/10.1002/(SICI)1096-8644(199807)106:3<385::AID-AJPA10>3.0.CO;2-X
-
Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxiaIntegrative and Comparative Biology 46:18–24.https://doi.org/10.1093/icb/icj004
-
Genetic basis of congenital erythrocytosisInternational Journal of Laboratory Hematology 40 Suppl 1:62–67.https://doi.org/10.1111/ijlh.12828
-
Structural insights into the ferroxidase site of ferritins from higher eukaryotesJournal of the American Chemical Society 134:6169–6176.https://doi.org/10.1021/ja210084n
-
The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIFThe Journal of Biological Chemistry 283:36542–36552.https://doi.org/10.1074/jbc.M804578200
-
Adaptive responses of vertebrate neurons to hypoxiaThe Journal of Experimental Biology 205:3579–3586.https://doi.org/10.1242/jeb.205.23.3579
-
Hypoxia tolerance in reptiles, amphibians, and fishes: life with variable oxygen availabilityAnnual Review of Physiology 69:145–170.https://doi.org/10.1146/annurev.physiol.69.031905.162529
-
Mitochondrial oxygen affinity as a function of redox and phosphate potentialsBiochimica et Biophysica Acta 376:446–457.https://doi.org/10.1016/0005-2728(75)90166-8
-
The enzymology of human eicosanoid pathways: the lipoxygenase branchesMolecular Biology Reports 47:7189–7207.https://doi.org/10.1007/s11033-020-05698-8
-
Abnormal sympathoadrenal development and systemic hypotension in PHD3-/- miceMolecular and Cellular Biology 28:3386–3400.https://doi.org/10.1128/MCB.02041-07
-
Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signalingFree Radical Biology & Medicine 100:14–31.https://doi.org/10.1016/j.freeradbiomed.2016.04.001
-
Oxygenases: mechanisms and structural motifs for O(2) activationCurrent Opinion in Chemical Biology 5:550–555.https://doi.org/10.1016/s1367-5931(00)00236-2
-
The emerging roles of ribosomal histidyl hydroxylases in cell biology, physiology and diseaseCellular and Molecular Life Sciences 75:4093–4105.https://doi.org/10.1007/s00018-018-2903-z
-
Function and evolution of vertebrate globinsActa Physiologica 211:501–514.https://doi.org/10.1111/apha.12312
-
Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxiaJournal of Cellular and Molecular Medicine 15:1239–1253.https://doi.org/10.1111/j.1582-4934.2011.01258.x
-
Cellular respiration during hypoxia: role of cytochrome oxidase as the oxygen sensor in hepatocytesThe Journal of Biological Chemistry 272:18808–18816.https://doi.org/10.1074/jbc.272.30.18808
-
Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensingThe Journal of Biological Chemistry 275:25130–25138.https://doi.org/10.1074/jbc.M001914200
-
The role of m6A RNA methylation in human cancerMolecular Cancer 18:1–9.https://doi.org/10.1186/s12943-019-1033-z
-
FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteinsAnnals of the New York Academy of Sciences 1177:9–18.https://doi.org/10.1111/j.1749-6632.2009.05042.x
-
Natural selection on genes related to cardiovascular health in high-altitude adapted andeansAmerican Journal of Human Genetics 101:752–767.https://doi.org/10.1016/j.ajhg.2017.09.023
-
NO synthase: structures and mechanismsNitric Oxide 23:1–11.https://doi.org/10.1016/j.niox.2010.03.001
-
The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humansCurrent Drug Metabolism 3:561–597.https://doi.org/10.2174/1389200023337054
-
Tyrosine hydroxylase and regulation of dopamine synthesisArchives of Biochemistry and Biophysics 508:1–12.https://doi.org/10.1016/j.abb.2010.12.017
-
Nitric oxide neurotoxicityJournal of Chemical Neuroanatomy 10:179–190.https://doi.org/10.1016/0891-0618(96)00148-2
-
Dioxygen activation by copper, heme and non-heme iron enzymes: comparison of electronic structures and reactivitiesCurrent Opinion in Chemical Biology 9:152–163.https://doi.org/10.1016/j.cbpa.2005.02.012
-
PHD1 regulates p53-mediated colorectal cancer chemoresistanceEMBO Molecular Medicine 7:1350–1365.https://doi.org/10.15252/emmm.201505492
-
KDM4A regulates HIF-1 levels through H3K9me3Scientific Reports 7:11094.https://doi.org/10.1038/s41598-017-11658-3
-
Dual oxidasesPhilosophical Transactions of the Royal Society B 360:2301–2308.https://doi.org/10.1098/rstb.2005.1767
-
Hypoxia--implications for pharmaceutical developmentsSleep & Breathing = Schlaf & Atmung 14:291–298.https://doi.org/10.1007/s11325-010-0368-x
-
Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytesJournal of Biological Chemistry 273:11619–11624.https://doi.org/10.1074/jbc.273.19.11619
-
Structure and reaction mechanism in the heme dioxygenasesBiochemistry 50:2717–2724.https://doi.org/10.1021/bi101732n
-
Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assayThe Biochemical Journal 401:227–234.https://doi.org/10.1042/BJ20061151
-
Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brainJournal of Neurophysiology 83:2022–2029.https://doi.org/10.1152/jn.2000.83.4.2022
-
Negative impact of hypoxia on tryptophan 2,3-Dioxygenase FunctionMediators of Inflammation 2016:1638916.https://doi.org/10.1155/2016/1638916
-
Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signalingMolecular and Cellular Biology 25:4853–4862.https://doi.org/10.1128/MCB.25.12.4853-4862.2005
-
Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratioFree Radical Biology & Medicine 46:1386–1391.https://doi.org/10.1016/j.freeradbiomed.2009.02.019
-
Human copper-dependent amine oxidasesArchives of Biochemistry and Biophysics 546:19–32.https://doi.org/10.1016/j.abb.2013.12.022
-
Human 2-oxoglutarate-dependent oxygenases: nutrient sensors, stress responders, and disease mediatorsBiochemical Society Transactions 48:1843–1858.https://doi.org/10.1042/BST20190333
-
Role and regulation of prolyl hydroxylase domain proteinsCell Death & Differentiation 15:635–641.https://doi.org/10.1038/cdd.2008.10
-
Effect of hypoxia on cytochrome P450 activity and expressionCurrent Drug Metabolism 5:257–271.https://doi.org/10.2174/1389200043335577
-
Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humansNature Chemical Biology 8:960–962.https://doi.org/10.1038/nchembio.1093
-
Control of mitochondrial and cellular respiration by oxygenJournal of Bioenergetics and Biomembranes 27:583–596.https://doi.org/10.1007/BF02111656
-
Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic β-cells by stearoyl-CoA desaturase and Elovl6American Journal of Physiology. Endocrinology and Metabolism 300:E640–E649.https://doi.org/10.1152/ajpendo.00544.2010
-
Mechanisms of cytochrome P450 substrate oxidation: MiniReviewJournal of Biochemical and Molecular Toxicology 21:163–168.https://doi.org/10.1002/jbt.20174
-
Hypoxia-Inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKDAmerican Journal of Kidney Diseases 69:815–826.https://doi.org/10.1053/j.ajkd.2016.12.011
-
Mitochondrial complex III is required for hypoxia-induced ROS production and gene transcription in yeastAntioxidants & Redox Signaling 9:1317–1328.https://doi.org/10.1089/ars.2007.1708
-
The mitochondrial electron transport and oxidative phosphorylation systemAnnual Review of Biochemistry 54:1015–1069.https://doi.org/10.1146/annurev.bi.54.070185.005055
-
Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase familyThe Journal of Biological Chemistry 286:41616–41625.https://doi.org/10.1074/jbc.M111.283689
-
Characterization of the human prolyl 4-Hydroxylases that modify the hypoxia-inducible factorJournal of Biological Chemistry 278:30772–30780.https://doi.org/10.1074/jbc.M304982200
-
Nitric oxide signaling in hypoxiaJournal of Molecular Medicine 90:217–231.https://doi.org/10.1007/s00109-012-0880-5
-
Comparative biology of oxygen sensing in plants and animalsCurrent Biology 30:1979–1980.https://doi.org/10.1016/j.cub.2020.04.054
-
Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injuryThe Journal of Biological Chemistry 286:11185–11194.https://doi.org/10.1074/jbc.M110.186809
-
2-Oxoglutarate-Dependent OxygenasesAnnual Review of Biochemistry 87:585–620.https://doi.org/10.1146/annurev-biochem-061516-044724
-
Mononuclear copper active-oxygen complexesCurrent Opinion in Chemical Biology 10:115–122.https://doi.org/10.1016/j.cbpa.2006.02.012
-
Neurotransmission in the carotid body: transmitters and modulators between glomus cells and petrosal ganglion nerve terminalsBrain Research. Brain Research Reviews 47:46–53.https://doi.org/10.1016/j.brainresrev.2004.05.007
-
BookNeurotransmitters in carotid body function: the case of dopamine – invited articleIn: Gonzalez C, Nurse CA, Peers C, editors. Arterial Chemoreceptors. Springer. pp. 137–143.https://doi.org/10.1007/978-90-481-2259-2
-
Dual role of oxygen during lipoxygenase reactionsThe FEBS Journal 272:2523–2535.https://doi.org/10.1111/j.1742-4658.2005.04673.x
-
Molecular enzymology of lipoxygenasesArchives of Biochemistry and Biophysics 503:161–174.https://doi.org/10.1016/j.abb.2010.08.016
-
The evolution of oxygen-utilizing enzymes suggests early biosphere oxygenationNature Ecology & Evolution 5:442–448.https://doi.org/10.1038/s41559-020-01386-9
-
Oxidative stress under ambient and physiological oxygen tension in tissue cultureCurrent Pharmacology Reports 2:64–72.https://doi.org/10.1007/s40495-016-0050-5
-
N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTONature Chemical Biology 7:885–887.https://doi.org/10.1038/nchembio.687
-
Hypoxia and drug metabolismBiochemical Pharmacology 30:1019–1023.https://doi.org/10.1016/0006-2952(81)90436-6
-
Affinities of various mammalian arachidonate lipoxygenases and cyclooxygenases for molecular oxygen as substrateBiochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1436:509–518.https://doi.org/10.1016/S0005-2760(98)00159-3
-
Oxygen affinity of tyrosine and tryptophan hydroxylases in synaptosomesJournal of Neurochemistry 35:760–763.https://doi.org/10.1111/j.1471-4159.1980.tb03721.x
-
Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagyCell Death and Differentiation 25:1921–1937.https://doi.org/10.1038/s41418-018-0165-9
-
Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylasesThe Journal of Biological Chemistry 279:9899–9904.https://doi.org/10.1074/jbc.M312254200
-
Catalytic activity of human indoleamine 2,3-dioxygenase (hIDO1) at low oxygenArchives of Biochemistry and Biophysics 570:47–57.https://doi.org/10.1016/j.abb.2015.02.014
-
Molecular mechanisms and potential functions of histone demethylasesNature Reviews. Molecular Cell Biology 13:297–311.https://doi.org/10.1038/nrm3327
-
Anaemia in kidney disease: harnessing hypoxia responses for therapyNature Reviews. Nephrology 11:394–410.https://doi.org/10.1038/nrneph.2015.82
-
Mammalian lipoxygenases and their biological relevanceBiochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1851:308–330.https://doi.org/10.1016/j.bbalip.2014.10.002
-
Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytesAmerican Journal of Physiology. Lung Cellular and Molecular Physiology 282:L1324–L1329.https://doi.org/10.1152/ajplung.00326.2001
-
Activation of tyrosine hydroxylase by intermittent hypoxia: involvement of serine phosphorylationJournal of Applied Physiology 95:536–544.https://doi.org/10.1152/japplphysiol.00186.2003
-
Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET EnzymesThe Journal of Biological Chemistry 291:4256–4265.https://doi.org/10.1074/jbc.M115.688762
-
Structural divergence and distant relationships in proteins: evolution of the globinsCurrent Opinion in Structural Biology 15:290–301.https://doi.org/10.1016/j.sbi.2005.05.008
-
Hypoxia signaling in human diseases and therapeutic targetsExperimental & Molecular Medicine 51:1–13.https://doi.org/10.1038/s12276-019-0235-1
-
Reactivity of dioxygen-copper systemsChemical Reviews 104:1047–1076.https://doi.org/10.1021/cr020633r
-
Carotid body oxygen sensingThe European Respiratory Journal 32:1386–1398.https://doi.org/10.1183/09031936.00056408
-
A genetic mechanism for tibetan high-altitude adaptationNature Genetics 46:951–956.https://doi.org/10.1038/ng.3067
-
Protein hydroxylation catalyzed by 2-oxoglutarate-dependent oxygenasesThe Journal of Biological Chemistry 290:20712–20722.https://doi.org/10.1074/jbc.R115.662627
-
Catalytic mechanisms of fe(ii)- and 2-oxoglutarate-dependent oxygenasesThe Journal of Biological Chemistry 290:20702–20711.https://doi.org/10.1074/jbc.R115.648691
-
Relevance of oxygen concentration in stem cell culture for regenerative medicineInternational Journal of Molecular Sciences 20:1195.https://doi.org/10.3390/ijms20051195
-
The reactivity of oxygen with flavoproteinsInternational Congress Series 1233:3–11.https://doi.org/10.1016/S0531-5131(02)00519-8
-
Evidence for substrate preorganization in the peptidylglycine α-amidating monooxygenase reaction describing the contribution of ground state structure to hydrogen tunnelingJournal of the American Chemical Society 132:16393–16402.https://doi.org/10.1021/ja1019194
-
Defining normoxia, physoxia and hypoxia in tumours-implications for treatment responseThe British Journal of Radiology 87:20130676.https://doi.org/10.1259/bjr.20130676
-
The oxygen and carbon monoxide reactions of heme oxygenaseThe Journal of Biological Chemistry 273:945–949.https://doi.org/10.1074/jbc.273.2.945
-
Endogenous nitric oxide: physiology, pathology and clinical relevanceEuropean Journal of Clinical Investigation 21:361–374.https://doi.org/10.1111/j.1365-2362.1991.tb01383.x
-
THe oxygen sensor protein, fixl, of rhizobium melilotiJournal OF Biological Chemistry 270:5243–5250.https://doi.org/10.1074/jbc.270.10.5243
-
Membrane amine oxidase cloning and identification as a major protein in the adipocyte plasma membraneThe Journal of Biological Chemistry 272:9388–9392.https://doi.org/10.1074/jbc.272.14.9388
-
Hypoxia and the HIF system in kidney diseaseJournal of Molecular Medicine 85:1325–1330.https://doi.org/10.1007/s00109-007-0278-y
-
Partial pressure of oxygen in the human body: a general reviewAmerican Journal of Blood Research 9:1–14.
-
Control of catalysis in flavin-dependent monooxygenasesArchives of Biochemistry and Biophysics 493:26–36.https://doi.org/10.1016/j.abb.2009.11.028
-
S-nitrosothiols signal hypoxia-mimetic vascular pathologyThe Journal of Clinical Investigation 117:2592–2601.https://doi.org/10.1172/JCI29444
-
Clinical recommendations for high altitude exposure of individuals with pre-existing cardiovascular conditions: A joint statement by the European Society of Cardiology, the Council on Hypertension of the European Society of Cardiology, the European Society of Hypertension, the International Society of Mountain Medicine, the Italian Society of Hypertension and the Italian Society of Mountain MedicineEuropean Heart Journal 39:1546–1554.https://doi.org/10.1093/eurheartj/ehx720
-
Biochemical and physiological function of stearoyl-CoA desaturaseAmerican Journal of Physiology. Endocrinology and Metabolism 297:E28–E37.https://doi.org/10.1152/ajpendo.90897.2008
-
Genetic variations in Tibetan populations and high-altitude adaptation at the HimalayasMolecular Biology and Evolution 28:1075–1081.https://doi.org/10.1093/molbev/msq290
-
A trial of darbepoetin alfa in type 2 diabetes and chronic kidney diseaseNew England Journal of Medicine 361:2019–2032.https://doi.org/10.1056/NEJMoa0907845
-
Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational researchFree Radical Biology & Medicine 113:311–322.https://doi.org/10.1016/j.freeradbiomed.2017.10.003
-
A mechanistic overview of TET-mediated 5-methylcytosine oxidationBiochemical and Biophysical Research Communications 436:115–120.https://doi.org/10.1016/j.bbrc.2013.05.077
-
BookStructures of cytochrome P450 enzymesIn: Ortiz de Montellano PR, editors. Cytochrome P450: Structure, Mechanism, and Biochemistry. Springer. pp. 87–114.https://doi.org/10.1007/b139087
-
Genetic evidence of paleolithic colonization and neolithic expansion of modern humans on the tibetan plateauMolecular Biology and Evolution 30:1761–1778.https://doi.org/10.1093/molbev/mst093
-
Differential regulation of tyrosine hydroxylase by continuous and intermittent hypoxiaArterial Chemoreception 758:381–385.https://doi.org/10.1007/978-94-007-4584-1
-
Thiol/Disulfide Redox Switches in the Regulation of Heme Binding to ProteinsAntioxidants & Redox Signaling 14:1039–1047.https://doi.org/10.1089/ars.2010.3436
-
A short history of heme dioxygenases: rise, fall and rise againJournal of Biological Inorganic Chemistry 22:175–183.https://doi.org/10.1007/s00775-016-1412-5
-
Hypoxia inhibits semicarbazide-sensitive amine oxidase activity in adipocytesMolecular and Cellular Endocrinology 411:58–66.https://doi.org/10.1016/j.mce.2015.04.011
-
Frontiers of hypoxia research: acute mountain sicknessJournal of Experimental Biology 204:3161–3170.https://doi.org/10.1242/jeb.204.18.3161
-
Mechanisms of tryptophan and tyrosine hydroxylaseIUBMB Life 65:350–357.https://doi.org/10.1002/iub.1144
-
Oxygen-dependent regulation of nitric oxide production by inducible nitric oxide synthaseFree Radical Biology & Medicine 51:1952–1965.https://doi.org/10.1016/j.freeradbiomed.2011.08.034
-
Same Substrate, Many Reactions: Oxygen Activation in FlavoenzymesChemical Reviews 118:1742–1769.https://doi.org/10.1021/acs.chemrev.7b00650
-
Inhibition of 2-oxoglutarate dependent oxygenasesChemical Society Reviews 40:4364–4397.https://doi.org/10.1039/c0cs00203h
-
Oxygen dependence of tyrosine hydroxylaseAmino Acids 34:455–464.https://doi.org/10.1007/s00726-007-0547-7
-
A kinetic simulation model that describes catalysis and regulation in nitric-oxide synthaseThe Journal of Biological Chemistry 276:1233–1243.https://doi.org/10.1074/jbc.M006858200
-
Differences in three kinetic parameters underpin the unique catalytic profiles of nitric-oxide synthases I, II, and IIIThe Journal of Biological Chemistry 276:48887–48898.https://doi.org/10.1074/jbc.M108666200
-
Cell respiration under hypoxia: facts and artefacts in mitochondrial oxygen kineticsOxygen Transport to Tissue XXXI:7–25.https://doi.org/10.1007/978-1-4419-1241-1
-
Transcriptional integration of mitochondrial biogenesisTrends in Endocrinology & Metabolism 23:459–466.https://doi.org/10.1016/j.tem.2012.06.006
-
Oxygen sensing by HIF hydroxylasesNature Reviews. Molecular Cell Biology 5:343–354.https://doi.org/10.1038/nrm1366
-
New insights into nNOS regulation of vascular homeostasisJournal of Clinical Investigation 115:2976–2978.https://doi.org/10.1172/JCI26792
-
Chromatin and oxygen sensing in the context of JmjC histone demethylasesThe Biochemical Journal 462:385–395.https://doi.org/10.1042/BJ20140754
-
Striking oxygen sensitivity of the peptidylglycine α-amidating monooxygenase (pam) in neuroendocrine cellsThe Journal of Biological Chemistry 290:24891–24901.https://doi.org/10.1074/jbc.M115.667246
-
Correction of anemia with epoetin alfa in chronic kidney diseaseNew England Journal of Medicine 355:2085–2098.https://doi.org/10.1056/NEJMoa065485
-
Update on mechanism and catalytic regulation in the NO synthasesThe Journal of Biological Chemistry 279:36167–36170.https://doi.org/10.1074/jbc.R400017200
-
Encyclopedia of Inorganic and Bioinorganic ChemistryIndoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase, Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, 10.1002/9781119951438.eibc0650.
-
Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitorsThe Journal of Biological Chemistry 286:13041–13051.https://doi.org/10.1074/jbc.M110.211110
-
Chronic mountain sickness: Clinical aspects, etiology, management, and treatmentHigh Altitude Medicine & Biology 17:61–69.https://doi.org/10.1089/ham.2016.0031
-
Hypoxia induces a functionally significant and translationally efficient neuronal NO synthase mRNA variantJournal of Clinical Investigation 115:3128–3139.https://doi.org/10.1172/JCI20806
-
The plant cysteine oxidases from Arabidopsis thaliana are kinetically tailored to act as oxygen sensorsJournal of Biological Chemistry 293:11786–11795.https://doi.org/10.1074/jbc.RA118.003496
-
Oxygen activation and energy conservation by cytochrome c oxidaseChemical Reviews 118:2469–2490.https://doi.org/10.1021/acs.chemrev.7b00664
-
Heme oxygenase: evolution, structure, and mechanismAntioxidants & Redox Signaling 4:603–614.https://doi.org/10.1089/15230860260220102
-
HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligationJournal of Molecular and Cellular Cardiology 41:68–77.https://doi.org/10.1016/j.yjmcc.2006.04.009
-
Oxygen-sensing mechanisms in cellsThe FEBS Journal 287:3888–3906.https://doi.org/10.1111/febs.15374
-
Heights and haematology: the story of haemoglobin at altitudePostgraduate Medical Journal 83:148–151.https://doi.org/10.1136/pgmj.2006.049734
-
TET-mediated active DNA demethylation: mechanism, function and beyondNature Reviews. Genetics 18:517–534.https://doi.org/10.1038/nrg.2017.33
-
A genome-wide search for signals of high-altitude adaptation in tibetansMolecular Biology and Evolution 28:1003–1011.https://doi.org/10.1093/molbev/msq277
-
Mechanism of heme degradation by heme oxygenaseJournal of Inorganic Biochemistry 82:33–41.https://doi.org/10.1016/s0162-0134(00)00156-2
-
Reading, writing and erasing mRNA methylationNature Reviews. Molecular Cell Biology 20:608–624.https://doi.org/10.1038/s41580-019-0168-5
-
Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertensionThe Journal of Physiological Sciences 62:163–172.https://doi.org/10.1007/s12576-012-0196-9
Article and author information
Author details
Steven J Altschuler

Funding
Human Frontier Science Program (LT000908/2020-C)
- Li Li
National Institutes of Health (R38AG070171)
- Susan Shen
National Institutes of Health (R25MH0602)
- Susan Shen
Defense Advanced Research Projects Agency (HR0011-19-2-0018)
- Matthew P Jacobson
- Steven J Altschuler
- Lani F Wu
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We gratefully acknowledge support from the DARPA Panacea program (HR0011-19-2-0018) to LFW; the Human Frontiers Science Program (LT000908/2020-C) to LL; and the National Institute on Aging (R38AG070171) and the National Institute of Mental Health (R25MH0602) to SQS. We thank Heinz Hammerlindl, Sabrina Hammerlindl, Feng Bao, and all members of the Altschuler & Wu labs for their input.
Copyright
© 2023, Li et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,444
- views
-
- 397
- downloads
-
- 43
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 43
- citations for umbrella DOI https://doi.org/10.7554/eLife.87705
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Searching for molecular hypoxia sensors among oxygen-dependent enzymes
eLife 12:e87705.
https://doi.org/10.7554/eLife.87705




{kind=link}
{kind=link}
{kind=link}