PfMORC protein regulates chromatin accessibility and transcriptional repression in the human malaria parasite, Plasmodium falciparum
- Department of Molecular, Cell and Systems Biology, University of California, Riverside, United States
- Stowers Institute for Medical Research, United States
eLife Assessment
This important study examines the role of Microrchidia (MORC) proteins in the human malaria parasite Plasmodium falciparum. Solid experimental results, including genome editing and chromatin profiling methods (ChIP-seq and Hi-C), provide a comprehensive picture of the critical role MORC plays in shaping parasite chromatin. Depletion of MORC results in a lethal collapse of heterochromatin and parasite death, nominating the factor as a new target of antimalarial therapies.
https://doi.org/10.7554/eLife.92499.3.sa0Significance of the findings:
Important: Findings that have theoretical or practical implications beyond a single subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Solid: Methods, data and analyses broadly support the claims with only minor weaknesses
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
The environmental challenges the human malaria parasite, Plasmodium falciparum, faces during its progression into its various lifecycle stages warrant the use of effective and highly regulated access to chromatin for transcriptional regulation. Microrchidia (MORC) proteins have been implicated in DNA compaction and gene silencing across plant and animal kingdoms. Accumulating evidence has shed light on the role MORC protein plays as a transcriptional switch in apicomplexan parasites. In this study, using the CRISPR/Cas9 genome editing tool along with complementary molecular and genomics approaches, we demonstrate that PfMORC not only modulates chromatin structure and heterochromatin formation throughout the parasite erythrocytic cycle, but is also essential to the parasite survival. Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) experiments suggests that PfMORC binds to not only sub-telomeric regions and genes involved in antigenic variation but may also play a role in modulating stage transition. Protein knockdown experiments followed by chromatin conformation capture (Hi-C) studies indicate that downregulation of PfMORC impairs key histone marks and induces the collapse of the parasite heterochromatin structure leading to its death. All together these findings confirm that PfMORC plays a crucial role in chromatin structure and gene regulation, validating this factor as a strong candidate for novel antimalarial strategies.
eLife digest
Malaria is an infectious disease caused by parasites that spread to humans through mosquitoes. The parasite species Plasmodium falciparum accounts for the most fatal forms of the disease and can undergo substantial genetic changes that allow it evade detection by the human immune system. A better understanding of how P. falciparum controls its gene expression could help find new ways for treating this aggressive form of malaria.
One of the key mechanisms by which cells – including parasites – control gene expression is by remodeling the structure of their DNA, which is tightly packed in to a construct known as chromatin. By loosening or condensing regions of chromatin, cells can make certain genes more or less accessible to the machinery responsible for activating them.
Recently, a protein called MORC has been found to play a key role in chromatin remodeling and gene expression in different parasites, plants and animals. However, it remains unclear if MORC also influences chromatin structure of P. falciparum and contributes to the parasite’s ability to evade immune detection.
To investigate this question, Chahine, Gupta, Lenz, Hollin et al. employed a variety of gene editing tools, including CRISPR/Cas9. This revealed that reducing MORC levels caused the structure of chromatin in P. falciparum to collapse, ultimately resulting in the death of the parasite. The team also found that MORC not only directly interacts with the parasite’s chromatin, but also other factors involved in chromatin remodeling and genes that help the parasite evade the immune system.
These findings highlight the essential role of MORC protein in maintaining chromatin structure and the survival of P. falciparum, and offer a new therapeutic target for controlling the spread of this aggressive form of malaria.
Introduction
Malaria is a mosquito-borne infectious disease that is caused by protozoan parasites of the genus Plasmodium. Among the five human-infecting species, Plasmodium falciparum is the deadliest, with over 619,000 deaths in 2023 (WHO, 2023). To adapt to extreme environmental challenges, P. falciparum possesses unique strategies that direct the tightly coordinated changes in gene expression and control transcriptional switching in genes encoded by multigene families to ensure antigenic variation and immune evasion. Changes in gene expression throughout the parasite life cycle are controlled by a surprisingly low repertoire of transcription factors (TFs) encoded in the Plasmodium genome (Coulson et al., 2004; Balaji et al., 2005; De Silva et al., 2008; Yuda et al., 2009; Iwanaga et al., 2012; Sinha et al., 2014; Kafsack et al., 2014; Lesage et al., 2018). The 27 apicomplexan APETALA2 (ApiAP2) DNA-binding proteins are the only well-documented parasite TFs known to contribute to the modulation of gene expression throughout various stages of the parasite’s development (Balaji et al., 2005; Yuda et al., 2009; Iwanaga et al., 2012; Sinha et al., 2014; Kafsack et al., 2014; Yuda et al., 2010; Modrzynska et al., 2017; Gu et al., 2017; Yuda et al., 2020). Since the discovery of the AP2 gene family (Balaji et al., 2005), evidence has alluded to their role as master regulators of gene expression throughout transitory phases of the parasite life cycle. The most well-documented are the gametocyte-specific TFs (AP2-G, AP2-G2) (Sinha et al., 2014; Yuda et al., 2015) with the subsequent discovery of those associated with a transition to sexual differentiation (Yuda et al., 2020) as well as sporozoite (AP2-SP) and liver stages (AP2-L) (Iwanaga et al., 2012; Yuda et al., 2010). The Plasmodium genome, however, encompasses well over 5,000 protein-coding genes suggesting there are most likely other molecular components responsible for gene expression. It is believed that, to offset the relatively low TF range, Plasmodium has evolved additional mechanisms regulating gene expression including mechanisms that use epigenetics factors, RNA binding proteins, or regulate chromatin structure (Bozdech et al., 2003b; Le Roch et al., 2003; Bozdech et al., 2003a; Srinivasan et al., 2004; Silvestrini et al., 2005; Yang et al., 2021). All together these components work in combination to regulate the dynamic organization of DNA, and the cascade of gene expression required for the parasite life cycle progression (Dekker et al., 2002; Dostie et al., 2006; Cui et al., 2008; Dixon et al., 2012; Dembélé et al., 2014; Deng et al., 2015; Bunnik et al., 2019; Batugedara et al., 2020). Despite their importance, the identification and functional characterization of regulatory players controlling chromatin remains a challenge in this intractable organism. However, with the advent of sensitive technologies capable of capturing important chromatin associated regulatory complexes such as ChIP, chromatin conformation capture (Hi-C) technologies, and chromatin enrichment for proteomics (ChEP), we can now solve important facets of molecular components controlling epigenetics and chromatin structure.
MORC belongs to a highly conserved nuclear protein superfamily with widespread domain architectures that link MORCs with signaling-dependent chromatin remodeling and epigenetic regulation across plant and animal kingdoms, including the apicomplexan parasites (Lorković, 2012; Li et al., 2013; Weiser et al., 2017; Singh et al., 2021a). In all organisms, MORC contains several domains that form a catalytically active ATPase. Apicomplexan MORC ATPases are encircled by Kelch-repeat β-propellers as well as a CW-type zinc finger domain functioning as a histone reader (Farhat et al., 2020). In higher eukaryotes, MORCs were first identified as epigenetic regulators and chromatin remodelers in germ cell development. Currently, these proteins are shown to be involved in various human diseases including cancers and are expected to serve as important biomarkers for diagnosis and treatment (Wang et al., 2021). In the apicomplexan parasite, Toxoplasma gondii, TgMORC was shown to recruit the histone deacetylase HDAC3 to particular genome loci to regulate chromatin accessibility, restricting sexual commitment (Farhat et al., 2020; Zhong et al., 2023). TgMORC-depleted cells also resulted in change in a gene expression with up regulation of secondary AP2 factors and a significant shift from asexual to sexual differentiation (Singh et al., 2021a; Farhat et al., 2020; Saksouk et al., 2005; Bougdour et al., 2009; Hillier et al., 2019). Having multiple homologs with T. gondii, Plasmodium AP2 protein conservation is primarily restricted to their AP2 DNA-binding domains (Jeninga et al., 2019; Hiyoshi and Wada, 1990). Recently, ChEP experiments done in P. falciparum identified PfMORC as one of the highest enriched chromatin-bound proteins at different stages of the parasite intraerythrocytic development cycle (IDC) (Batugedara et al., 2020). PfMORC was also detected at a relatively high level throughout the parasite life cycle, including sporozoites and liver stages (Farhat et al., 2020; Singh et al., 2021b), and has been identified in several protein pull-down experiments as targeting both AP2 TFs and epigenetic factors (Singh et al., 2021a; Farhat et al., 2020; Hillier et al., 2019; Subudhi et al., 2023; Bryant et al., 2020; Singh et al., 2024). Immunoprecipitation experiments demonstrated that PfMORC seems to interact with AP2-G2 (Singh et al., 2021a), a TF that plays a critical role in the maturation of gametocytes. However, the genome-wide distribution and exact function of this protein throughout the parasite life cycle remained elusive.
In this study, we apply CRISPR/Cas9 genomic editing technologies to determine the function, genome distribution, and indispensability of PfMORC throughout the parasite’s IDC. Immunoprecipitation of an HA-tagged parasite line validates the role of PfMORC in heterochromatin structure maintenance. Using the downregulation of PfMORC induced through the TetR-DOZI system, we demonstrate the functional significance of PfMORC in heterochromatin stability and gene repression. Immunofluorescence-based assays and ChIP-seq experiments show that PfMORC localizes to heterochromatin clusters at or near var genes with significant overlap with well-known signatures of the parasite heterochromatin post-translational modification (PTM) H3K9-trimethylation (H3K9me3) marks. When PfMORC was downregulated, a level of H3K9me3 was detected at a lower level, demonstrating a possible role of this protein in epigenetic regulation and gene repression. Finally, Hi-C analyses demonstrate that downregulation of PfMORC results in significant dysregulation of chromatin architectural stability resulting in parasite death. All together our work provides significant insight into the role of PfMORC in the maintenance of the pathogens' epigenetics and chromosomal architectural integrity and validates this protein as a promising target for novel therapeutic interventions.
Results
Generation of PfMORC-HA transgenic line shows nuclear localization in heterochromatin clusters
To characterize the role of PfMORC in P. falciparum, we applied the CRISPR/Cas9 genome editing tool to add a 3 X-HA tag at the C-terminal coding region of the Pfmorc locus (PF3D7_1468100) in an NF54 line (Ganesan et al., 2016, Figure 1a, Supplementary file 1). Recovered transgenic parasites were cloned using limiting dilution and the correct incorporation of the tag into the parasite genome was validated via PCR and whole genome sequencing (WGS) (Figure 1b, Supplementary file 2). WGS analysis confirmed the presence of the HA tag at the expected locus with no obvious off-target effect but uncovered an additional nonsense mutation in the gametocyte development protein 1 (gdv1) gene (PF3D7_0935400) at amino acid position 561 out of 599 total. Mutations in gdv1 have been detected in the past and seem to emerge relatively often, indicating a fitness benefit in the in vitro culture system used in the laboratory (Kafsack et al., 2014). The involvement of GDV1 as essential in sexual commitment is well substantiated and further study involving the role of PfMORC in sexual commitment could not be fully addressed (Sinha et al., 2014; Kafsack et al., 2014; Yuda et al., 2015; Filarsky et al., 2018; Yuda et al., 2021; Kent et al., 2018, Supplementary file 2). Despite this finding, we were able to design experiments to determine the role of PfMORC throughout the IDC as no other major detrimental mutations were detected in the genome. We first validated PfMORC protein expression through Western blot analysis. Regardless of potential protein degradation, results showed an expected band size within the ~290 kDa range that was absent in NF54 control compared to our tagged line (Figure 1—figure supplement 1). Immunofluorescence (IFA) analysis of intracellular parasites revealed that PfMORC is localized in the nucleus throughout the parasite IDC in punctate patterns (Figure 1c). A single foci per parasite was detected in the nuclear periphery at the ring stage. An increased number of foci with a more diffuse signal could be detected as the number of DNA copies increased at the schizont stage. Moreover, punctuates were shown to have strong colocalization signals with histone H3K9me3 marks present at the different parasite developmental stages analyzed. This was most evident during the early ring stage of the parasite IDC where the chromatin organization is more compact with a single DNA copy. Altogether, our findings indicated that PfMORC is a true nuclear protein that is most likely associated with P. falciparum heterochromatin cluster(s).
Figure 1 with 1 supplement see all
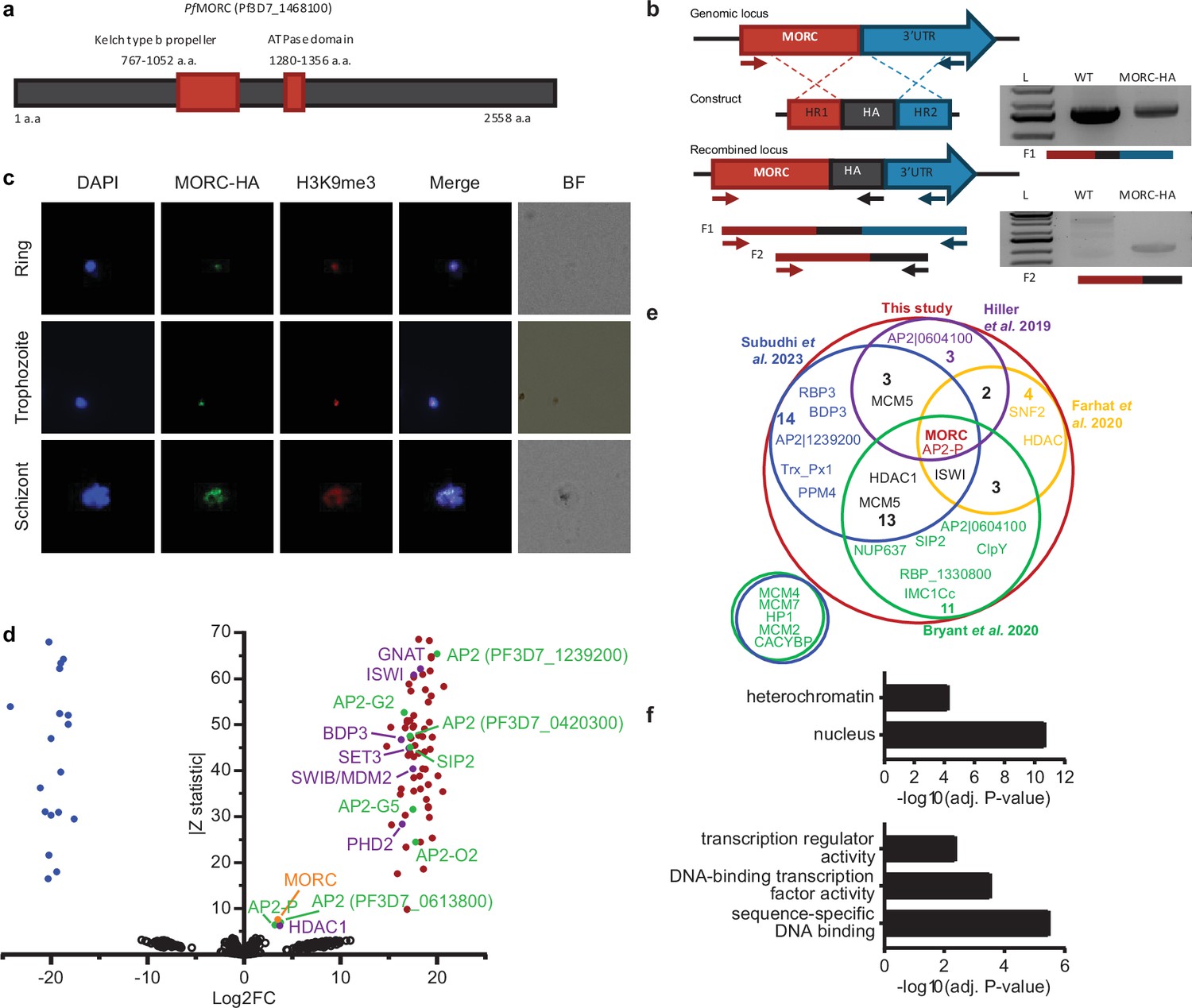
PfMORC-HA is associated with heterochromatin.
(a) Illustration of the PfMORC containing domains including Kelch type b propeller and ATPase domains using InterProScan. (b) Design strategy applied for PfMORC C-terminal HA tagging. PCR amplification of the genomic C-terminus end of Pfmorc region extending towards the 3’UTR (F1) as well as extension from the C-terminus towards the HA flanking sequence (F2) verifies the correct insertion site. NF54 genomic DNA was used as a negative control. (c) Immunofluorescence analysis (IFA) experiment: PfMORC foci (green) expressing co-localization with H3K9me3 marks (red). Cell nuclei are stained with DAPI (blue). BF: brightfield (d) Protein immunoprecipitation: Significance plot representing PfMORC interactome recovered through immunoprecipitation followed by mass spectrometry (IP-MS). Graph lists Microrchidia (MORC) (orange) bindings partners of the highest affinity associated with TF regulation (green) and chromatin remodelers, erasers, and writers (purple). Proteins enriched in the PfMORC-HA samples compared with controls were filtered with log2 FC ≥2 and Z statistic >5. (e) Venn diagram representing overlapping proteins identified among five publications. Values represent the total number of significant proteins identified as overlapping between two subsets. (f) Gene ontology enrichment analysis of the significantly enriched proteins. The top two terms of Cellular Component (top) and top three terms of molecular function (bottom) are represented as -Log10 (adjusted P-value) (Fisher’s exact test with Bonferroni adjustment). Figure 1b was created with BioRender.com.
-
Figure 1—source data 1
Original, unedited gel image for displayed PfMORC-HA results displayed in Figure 1.
- https://cdn.elifesciences.org/articles/92499/elife-92499-fig1-data1-v1.zip
-
Figure 1—source data 2
Original gel image for displayed PfMORC-HA results displayed in Figure 1 with marked ladder and described lanes.
- https://cdn.elifesciences.org/articles/92499/elife-92499-fig1-data2-v1.zip
PfMORC interacting partners include proteins involved in heterochromatin maintenance, transcription regulation, and chromatin remodeling
To confirm the association of PfMORC with proteins involved in heterochromatin cluster(s), we performed immunoprecipitation (IP) followed by mass-spectrometry (MS) analysis using mature stages of PfMORC-HA and parental line as control. MORC was detected with the highest peptides (97 and 113) and spectra (1041 and 1177) counts compared to control conditions (5 and 7 peptides; 16 and 43 spectra) confirming the efficiency of our pull-down. However, considering the relatively large size of the MORC protein (295 kDa) and its weak detection in the control, the log2 FC and Z-statistic after normalization are minimal when compared to smaller proteins that were not identified in the control samples. To define a set of PfMORC-associated proteins, we used the QPROT statistical framework (Choi et al., 2015) to compare proteins detected in PfMORC samples and controls. We identified 73 PfMORC-associated proteins (log2 FC ≥2 and Z statistic >5) (Figure 1d, Supplementary file 3). Additional chromatin remodeling proteins were detected such as ISWI chromatin-remodeling complex ATPase (PF3D7_0624600) and SWIB/MDM2 domain-containing protein (PF3D7_0611400), as well as the epigenetic readers BDP3 (PF3D7_0110500) and PHD2 (PF3D7_1433400) (Hoeijmakers et al., 2019). We also detected a significant enrichment of histone erasers and writers such as a N-acetyltransferase (PF3D7_1020700), HDAC1 (PF3D7_0925700), and SET3 (PF3D7_0827800). Several of which have been detected to interact with MORC in previous studies including the AP2-G5, HDAC1, ELM2, and ApiAp2 proteins (Farhat et al., 2020; Subudhi et al., 2023; Bryant et al., 2020; Singh et al., 2024, Figure 1e). Notably, we observed that many of the detected proteins were enriched at levels comparable to those of the bait proteins such as AP2-P and HDAC1. This may be a result of proteins forming complexes in a one-to-one ratio, leading to similar enrichment levels. Notably, two of these three proteins have been reported to interact with MORC in several studies, further supporting a strong interaction between them. Gene Ontology (GO) enrichment analysis revealed that these proteins are nuclear and are associated with heterochromatin, DNA binding, and transcription factor activity (Figure 1f). Detailed analysis showed the detection of SIP2, involved in heterochromatin formation and chromosome end regulation (Flueck et al., 2010), and eight AP2 transcription factors, including AP2-G2, AP2-O2, AP2-G5. Among the other AP2s detected, five were previously identified by ChIP-seq as Plasmodium falciparum AP2 Heterochromatin-associated Factors (PfAP2-HFs) (Shang et al., 2022). Our findings, corroborated with several published works, indicate that PfMORC interacts with multiple ApiAP2 TFs, chromatin remodelers, and epigenetic players associated with heterochromatin regions.
Genome-wide distribution of PfMORC displays functional roles in stage-specific gene silencing and antigenic variation
Given the multifunctional role of MORC proteins throughout apicomplexan parasites, we further explored the association of PfMORC with chromatin accessibility and stage-specific gene regulation by assessing the genome-wide distribution of PfMORC using chromatin immunoprecipitation followed by deep sequencing (ChIP–seq) in duplicate at the ring, trophozoite, and schizont stages. Inspection of PfMORC binding sites across each chromosome revealed a strong signal for telomeric and subtelomeric regions of the parasite genome, as well as the internal clusters of genes responsible for antigenic variation such as var and rifin (Figure 2a–c, Supplementary file 4). We found that while only 47% of reads mapped to antigenic genes in the ring stage, 95% of reads mapped to the same genes during the trophozoite stage and 82% during the schizont stage. Although a vast majority of the genome is actively transcribed during the trophozoite stage, these antigenic gene families are under tight regulatory control to ensure mutually exclusive expression and participate in immune evasion (Chen et al., 1998; Scherf et al., 1998). This could explain the disparity in the level of PfMORC binding of the coding regions between the ring and trophozoite stages (Figure 2c, Figure 2—figure supplement 1b).
Figure 2 with 1 supplement see all
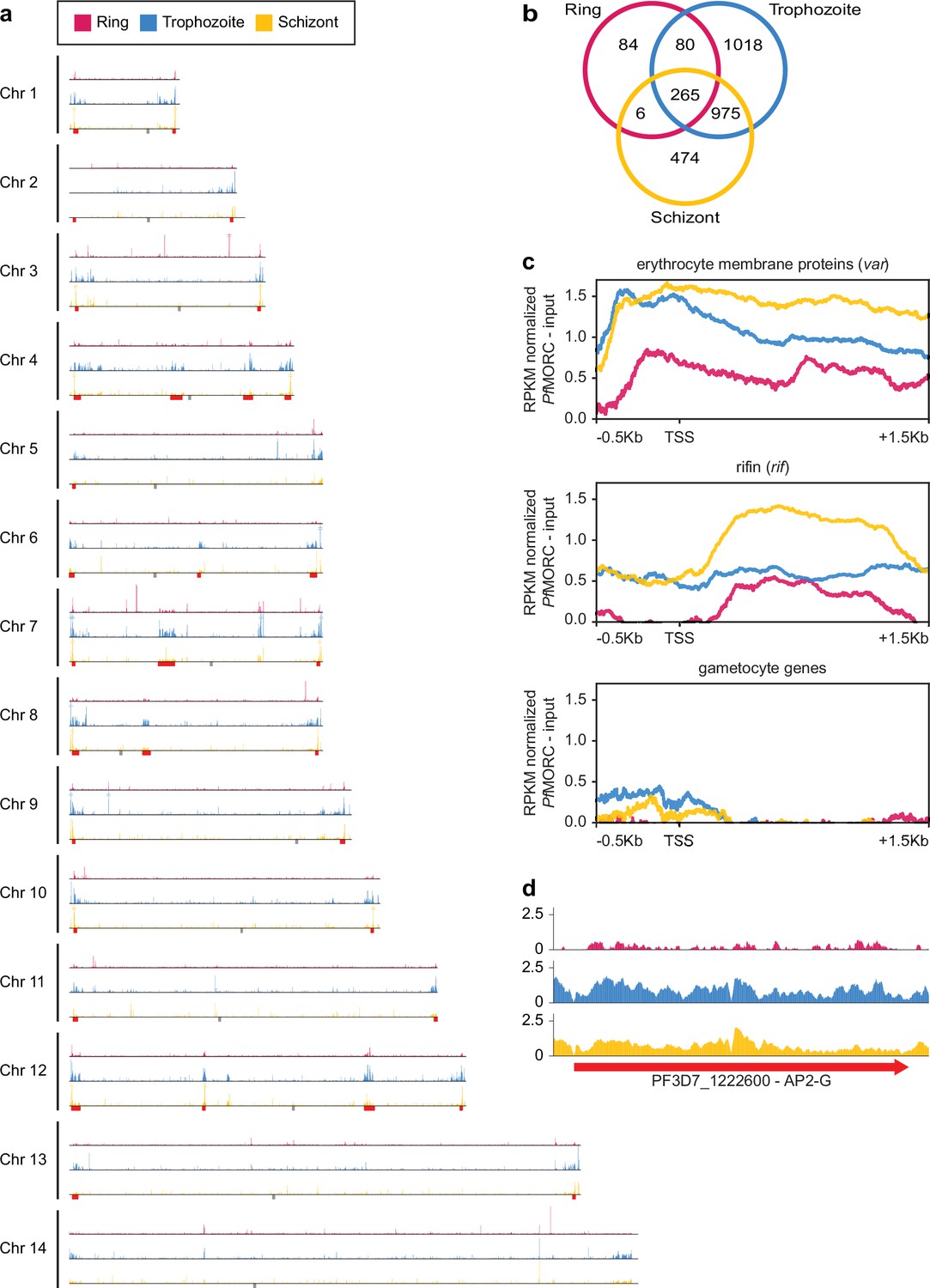
Genome-wide distribution of PfMORC proteins.
(a) Chromosome distribution plots of PfMORC binding show a predisposition for subtelomeric and internal var gene regions (red). Each track is input subtracted, and the per-million read count is normalized before normalizing the track height to allow for direct comparison between stages. Gray boxes indicate the position of centromeres. (b) Overlap of called peaks between time points. (c) Profile plots showing PfMORC coverage from 0.5 kb 5’ of the transcription start site (TSS) to 1.5 kb 3’ of the TSS in the ring, trophozoite, and schizont stages. Each plot includes per-million read count normalized coverage at 1 bp resolution for all genes within the var and rifin gene families as well as gametocyte-specific genes. (d) PfMORC coverage of the gametocyte-specific transcription factor ap2-g.
We also evaluated PfMORC binding of stage-specific gene families including gametocyte-related genes, and merozoite surface proteins (msp) (Meerstein-Kessel et al., 2018). In trophozoite and schizont stages, gametocyte-associated genes contain a mean of <0.5 RPKM normalized reads per nucleotide of PfMORC binding within their promoter region, whereas antigenic gene families such as var and rifin contain ~1.5 and 0.5 normalized reads, respectively (Figure 2b). The difference is even greater within the gene body with gametocyte genes displaying almost no reads mapped more than 200 bp downstream of the TSS and antigenic gene families containing 0.5–1.5 RPKM normalized reads. However, the gametocyte-specific transcription factor AP2-G, known to be repressed during the IDC and required for sexual commitment, deviates from this trend and contains similar levels of PfMORC binding to var genes (Figure 2c and Figure 2—figure supplement 1b, c). These results indicate a major role of PfMORC in controlling AP2-G and sexual differentiation. For some of the msp genes that are usually expressed later during the IDC to prepare the parasite for egress and invasion of new erythrocytes, PfMORC binding was detected in the gene bodies at the trophozoite stage. We also detected a small switch in PfMORC binding sites, moving from their gene bodies to their intergenic regions at the schizont stage (Figure 2—figure supplement 1c). PfMORC most likely moved away from the gene body to the regulatory regions surrounding the transcription start site (TSS) to guide RNA Polymerase and transcription factors and aid in activating expression of genes at the schizont stage that are crucial for egress and invasion. These results provide strong evidence for the direct effects that PfMORC binding has on tightly controlled antigenic and stage-specific genes including crucial invasion and gametocyte genes.
PfMORC is essential for P. falciparum survival
We next sought to confirm the functional relevance of PfMORC protein in parasite survival. While partial PfMORC knockdown using the glmS-ribozyme system had previously been shown to have no significant effect on the parasite survival (Singh et al., 2021b), another study using transposon mutagenesis (piggyBac) identified PfMORC as likely essential (Zhang et al., 2018). To resolve these conflicting results, we applied a complementary approach using the CRISPR/Cas9 gene editing strategy to incorporate an inducible TetR-DOZI system to knockdown (KD) PfMORC (Goldfless et al., 2014) through the administration of anhydrotetracycline (aTC) (Figure 3a, Ganesan et al., 2016; Nasamu et al., 2021). The protein was also modified to include a C-terminal 3 x-HA tag. Parental and transgenic clones were validated via PCR (Figure 3b) and WGS to confirm the correct insertion of the inducible system (Supplementary file 1 and 2) as well as the absence of major mutation that could explain some of the phenotypes observed. While results from our WGS validated our editing strategy without the detection of any obvious deleterious off-target effect, we identified a nonsense mutation in the AP2-G gene (PF3D7_1222600) explaining our inability to obtain mature gametocytes. Protein expression and successful KD was confirmed via western blot with a decrease of ~58% and ~88.2% in PfMORC expression at 24 hpi and 36 hpi, respectively, compared to their aTC-supplemented conditions (Figure 3—figure supplement 1). Downregulation of PfMORC was detected significantly above the level observed by Singh and colleagues (Singh et al., 2021b).
Figure 3 with 1 supplement see all
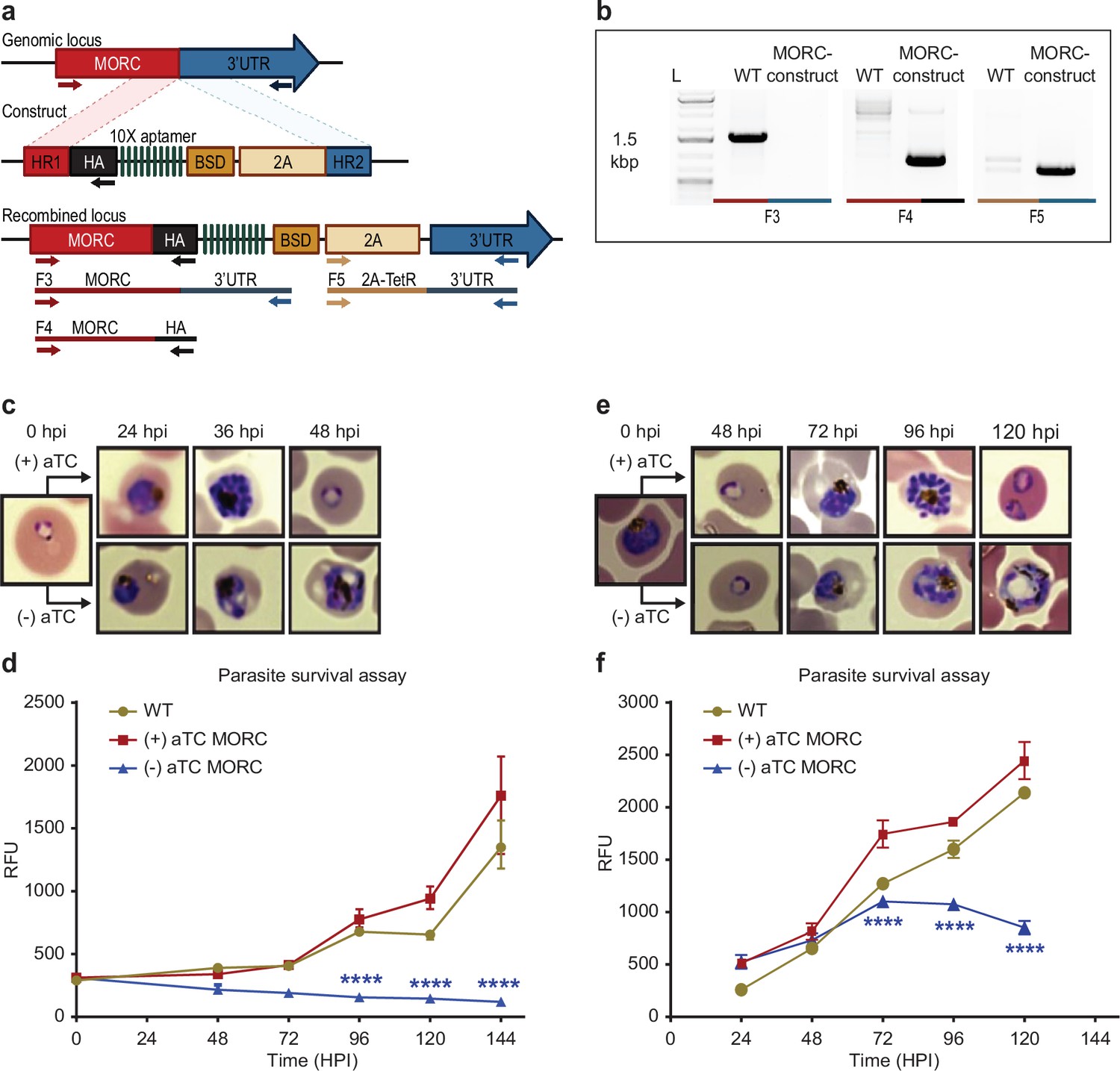
PfMORC is essential for cell survival.
(a) Diagram representation of PfMORC-HA-TetR-DOZI plasmid. (b). PCR amplification is used to verify genomic insertion using primers sets targeting 1.5 kbp of WT Pfmorc genome locus absent in transgenic line (Microrchidia, MORC construct) (F3) as well as verification of HA insertion (F4) and TetR-DOZI system extending along 3’ UTR of the construct (F5). (c) Phenotypic and (d) quantitative analysis of parasite cell progression after aTC withdrawal at the ring stage (0–6 hpi) (2-way ANOVA, n=3, p≤0.0001). (e) Phenotypic and quantitative (f) analysis of parasite cell progression after aTC removal at the trophozoite stage of cell cycle progression (24 hpi) (two-way ANOVA, n=3, p≤0.0001). Figure 3a was created with BioRender.com.
We then performed a phenotypic assay on (+/-) aTC PfMORC-HA-TetR-DOZI parasite cultures. The assay was conducted in replicates of synchronized cultures that were then split into (+) aTC and (-) aTC conditions either at the ring or trophozoite stages on PfMORC-HA-TetR-DOZI clones and WT control lines (Figure 3c–f, Supplementary file 5). Sequential morphological screens were performed through Giemsa-stained smears and monitored by microscope imaging. As opposed to what was observed previously with the glmS-ribozyme system (Singh et al., 2021b), aTC removal at the ring stage induced clear signs of stress and cell cycle arrest in mid- trophozoite and schizont stages of the first intraerythrocytic cycle (Figure 3c–d) compared to (+) aTC PfMORC and WT parasites. Our data showed a ~53%, 77%, and 84% (n=3, p≤0.0001) drop in parasitemia at 48-, 72-, and 120- hr post-invasion (hpi), respectively, when compared to control conditions. aTC supplemented and WT cultures showed unperturbed cell cycle progression, reinvasion, and morphological development. Interestingly, when aTC was withheld at the late trophozoite stages (24–30 hpi), parasites could complete the first cycle and successfully reinvade into new red blood cells (RBCs) (Figure 3e–f). The evident hindered phenotypic response may either be associated with the delay time for the protein to be completely depleted or may suggest a more dispensable role of PfMORC in the schizont stage. However, clear indications of stress and cell cycle arrest were ultimately detected at trophozoite and early schizont stages of the second cell cycle. Quantitative analysis of (-) aTC PfMORC cultures revealed significantly decreased parasitemia of ~37% and 66% (n=3, p≤0.0001) at 96 hpi and 120 hpi, respectively, compared to (+) aTC PfMORC and WT control lines (Figure 3e–f, Supplementary file 5), confirming the importance of the protein at the trophozoite and early schizont stages.
Effect of PfMORC knockdown on parasite transcriptome
To define the effects of PfMORC KD on transcription, we conducted detailed time-course measurements of mRNA levels using RNA-sequencing (RNA-seq) throughout the parasite asexual cycle on (+/-) aTC PfMORC lines. Parasites were first synchronized and aTC was removed from one set of samples. Total RNA was then extracted at the trophozoite (24 hpi) and the schizont (36 hpi) stages to allow for the detection of early and late changes in gene expression after aTC removal. Pairwise correlations analysis (Figure 4—figure supplement 1) between (+/-) aTC PfMORC treated lines at the two different time points confirmed the appropriate reproducibility of our RNA-seq experiments.
Our results identified a relatively low number of differentially expressed genes at 24 hpi with 96 and 93 upregulated and downregulated genes, respectively (FDR <0.05 and log2 FC >0.5 or <–0.5) (Figure 4a, Supplementary file 6). This data is in agreement with the absence of major phenotypic changes observed in (-) aTC PfMORC parasite cultures after 24 hr. GO enrichment analysis indicated upregulation of genes in response to xenobiotic stimulus including several phosphatases, hydrolases, and heat shock proteins suggesting stress sensing of the parasites at this stage (Figure 4c). GO analyses of downregulated genes, on the other hand, were found to be closely associated with regular metabolic processes and intracellular transport mechanisms suggesting dysregulation of regular cellular activity. However, at this stage, it is probable that the limited depletion of PfMORC (58%) has restricted effects at the transcriptional level (See Figure 3—figure supplement 1) and that the observed down-regulated genes are likely due to an indirect effect of cell cycle arrest. At 36 hpi the number of genes that exhibit changes in gene expression were significantly higher with 1319 upregulated and 1150 downregulated (FDR <0.05 and log2 FC >0.5 or <–0.5) in (-) aTC PfMORC conditions, reflecting of a significant decrease of the protein (~88.2%) and confirming the significant changes observed at the phenotypic level (Figure 4b). These results emphasize the cascading effects of PfMORC KD as the parasite progressed throughout its cell cycle. GO analysis indicated an enrichment of upregulated genes involved in multiple pathways including ATP metabolic process, mitochondria, translation, food vacuole, and protein folding (Figure 4d); characteristic of not only stress, but also a clear signal of cell cycle arrest in (-) aTC PfMORC samples. Among upregulated genes, we also observed several var genes and genes exported at the surface of the red blood cell that could be linked to a significant decreased in PfMORC binding of these gene families as well as a disorganization of the heterochromatin cluster(s) at the trophozoite and schizont stages. GO enrichment analysis for genes that were downregulated included many genes involved in DNA replication, chromosome organization, and mitotic spindle further emphasizing a strong cell cycle arrest and absence of potential major compensatory mechanisms for cell division and chromatin organization (Figure 4d). Additional downregulated genes were required for invasion such as several merozoite surface proteins. Our results clearly indicate stress sensing and an arrest in cell cycle progression at the trophozoite and schizont stages (Supplementary file 6, Kiss, 2002).
Figure 4 with 1 supplement see all
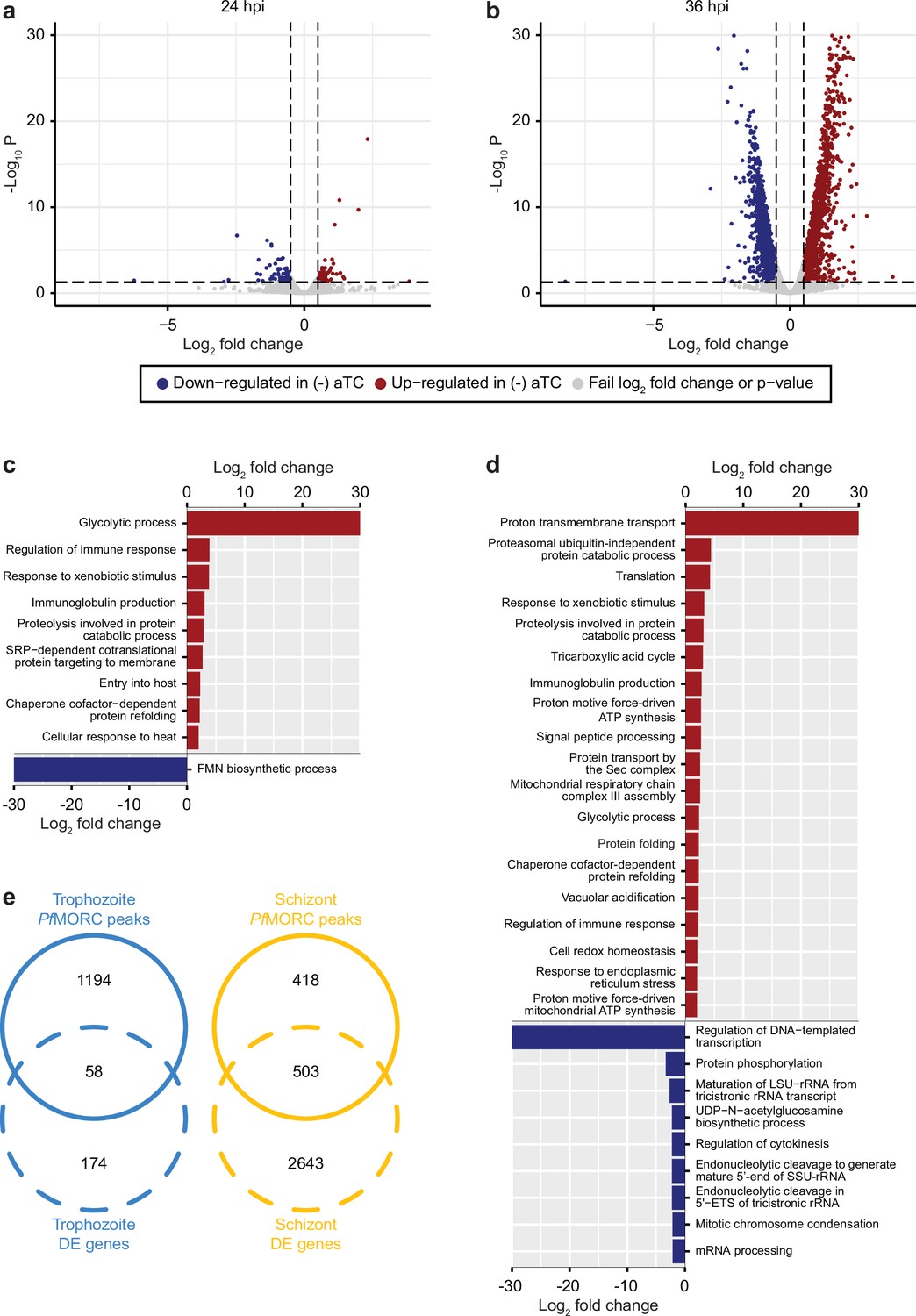
PfMORC knockdown (KD) on parasite transcriptome.
Volcano plots denoting upregulated (red), and downregulated (blue) genes were discovered through differential expression analysis following PfMORC knockdown at (a) 24 hpi (b) 36 hpi. Gene ontology enrichment analysis for upregulated (red) and downregulated (blue) genes at (c) 24 hpi and (d) 36 hpi. (e) Overlap of differentially expressed genes and genes containing significant peaks called by PfMORC Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) analysis at the trophozoite and schizont stages.
Although these results are compelling, the small overlap observed between the ChIP-seq signals and RNA-seq results indicates that major changes observed in gene expression at the schizont stages are not simply the result of reduced PfMORC binding in targeted gene bodies but a combination of direct and indirect effects of the degradation of PfMORC that leads to cell cycle arrest and potential collapse of the heterochromatin (Figure 4e).
PfMORC knockdown erodes antigenic-gene silencing framework
We next sought to analyze the effect of PfMORC down-regulation on the global chromatin landscape. We, therefore, performed a ChIP-seq experiment against histone H3K9me3 and H3K9ac marks in response to PfMORC depletion. Synchronized parasites were equally split between permissive and repressive conditions. At trophozoite (24 hpi) (Figure 5—figure supplement 1) and schizont (36 hpi) stages of development, both control (+aTC) and experiment (-aTC) samples were fixed, and parasites collected for chromatin immunoprecipitation followed by sequencing. The experimental procedure was performed in duplicate between (+/-) aTC PfMORC treated lines. Correlation analysis confirms the reproducibility of our ChIP-seq experiments.
Results revealed no significant changes in histone H3K9ac marks across the genome (data not shown) but a reduction in the heterochromatin landscape in the PfMORC-depleted conditions, specifically in the telomere regions of the chromosomes (Figure 5a). PfMORC KD resulted in significantly reduced (Mann-Whitney U test, p<0.05) H3K9me3 marks within var gene promoters from 200 to 800 bp upstream of the TSS, as well as 400–800 bp downstream of the TSS (Figure 5b–c). This result coincides with our transcriptomic profile and the upregulation of var genes at 36 hpi of (-) aTC samples (Figure 4d, Supplementary file 6). Further analyses revealed similar trends in other gene families associated with parasite reinvasion and immune response typically silenced under WT conditions. Most noticeable was the reduced H3K9me3 coverage across the rifin gene family, which displayed a significant reduction (p<0.05) in all bins from 1000 bp upstream of the TSS to 1000 bp downstream from the end of the gene (Figure 5b–c) in response to PfMORC depletion. There is a clear reduction of H3K9me3 coverage of virulence genes shown to be upregulated following PfMORC knockdown (Figure 5c). Overall, these findings confirm that PfMORC protein depletion not only affects the chromatin landscape but has a significant impact on heterochromatin. PfMORC downregulation leads to the erosion of heterochromatin integrity most likely required for mutually exclusive gene expression and immune evasion within heterochromatin clusters.
Figure 5 with 1 supplement see all
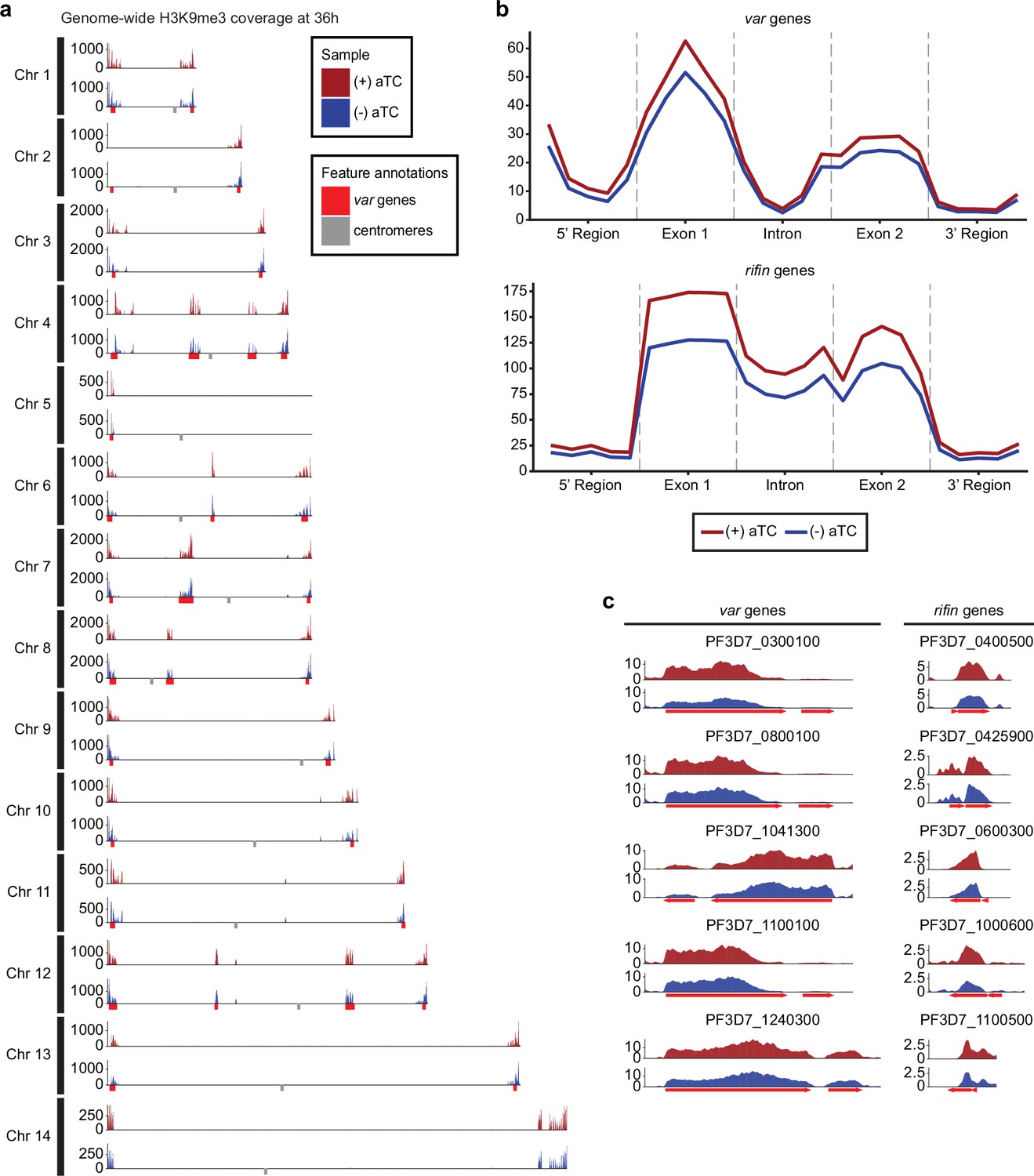
Impact of PfMORC knockdown (KD) on heterochromatin markers.
(a) Genome-wide H3K9me3 coverage of IGG subtracted and per-million normalized (+/-) aTC show similar distribution and concentration within telomeres and antigenic gene clusters highlighted in red. Replicates (n=2) are merged using the mean of the normalized read coverage per base pair. (b) Binned coverage of var and rifin genes from 1 kb upstream of the transcription start site (TSS) to 1 kb downstream of the end. The exons and intron of all genes within these families are split into five equal-sized bins and the 5’ and 3’ regions are binned into five 200 bp bins. Read counts within each bin are per million and bin length is normalized prior to plotting. (c) Coverage of the five most upregulated var and rifin genes as determined by the transcriptomic analysis which shows elevated coverage in (+) aTC cells.
Knockdown of PfMORC expression results in the loss of tightly regulated heterochromatin structures
To better understand the effect that downregulation of PfMORC has on the chromatin structure and investigate whether changes in chromatin accessibility may explain large changes in gene expression observed using RNA-seq, we performed Hi-C on (+/-) aTC PfMORC cultures at 24 hpi (trophozoite) and 36 hpi (schizont). Biological replicates for each sample were collected and used to generate Hi-C libraries with >37 million reads per replicate/sample, ensuring comprehensive coverage of both intrachromosomal and interchromosomal interactions. After processing (pairing, mapping, and quality filtering) the raw sequences via HiC-Pro (Servant et al., 2015), there were approximately 15 million (σ=~10 million) high-quality interaction pairs per sample. Due to the high number of reads and relatively small size of the P. falciparum genome (23.3 Mb) compared to higher eukaryotes, we elected to bin our Hi-C data at 10 kb resolution, which allowed the identification of genome-wide patterns while not introducing much noise by binning at too high of a resolution. A high stratum-adjusted correlation (Figure 6—figure supplement 1), especially at the 24 hpi time point, suggested that the chromatin structure was consistent between biological replicates, therefore, we chose to combine replicates for downstream analyses and visualization.
Because of variation in the number of valid interaction pairs between (+/-) aTC PfMORC samples, 100 iterations of random sampling were performed on samples with higher read count to obtain ~35 million and ~9 million interaction pairs at 24 hpi and 36 hpi, respectively. ICED normalized intrachromosomal heatmaps displayed a high proportion of interactions at distances less than 10% the length of each chromosome as well as strong subtelomeric interactions and internal regions containing genes involved in antigenic variation (Figure 6a–b, Figure 6—figure supplements 2–5) This pattern was similar to those observed previously at various stages of the life cycle (Bunnik et al., 2019; Servant et al., 2015; Bunnik et al., 2018) and confirm that genes involved in antigenic variation are usually confined within dense heterochromatin rich regions to aid the tight control of var genes necessary for their mutually exclusive expression and immune evasion (Chen et al., 1998; Scherf et al., 1998). Data generated at 36 hpi in (-) aTC PfMORC parasite cultures were, however, tumultuous, with a decrease in defined heterochromatin borders and fewer long-range intrachromosomal interactions across the genome. This may indicate a significant loss of chromatin maintenance in (-) aTC PfMORC parasites (Figure 6a–b).
Figure 6 with 7 supplements see all
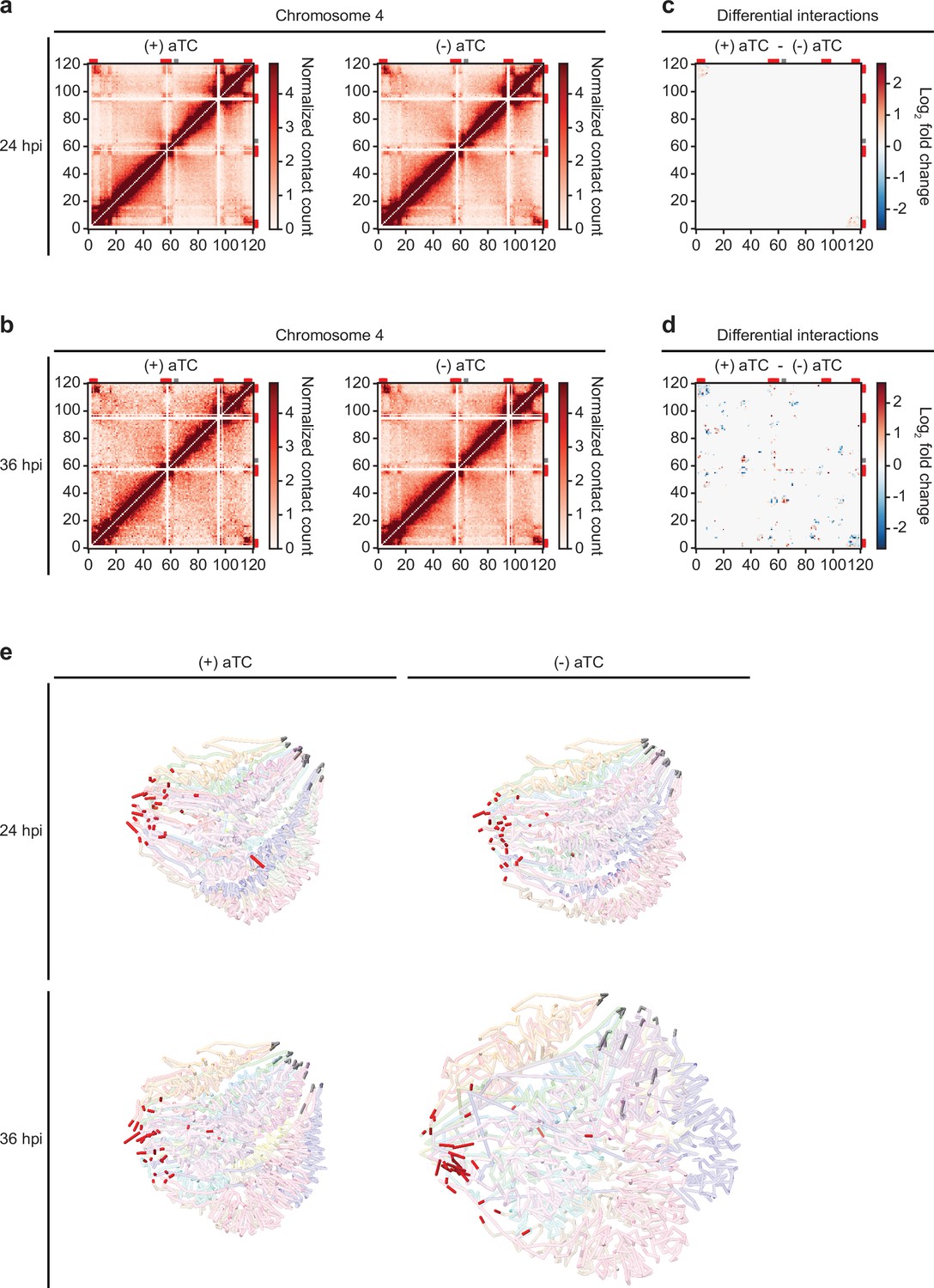
Loss of PfMORC expression correlates with heterochromatin expansion.
Intrachromosomal interaction heatmaps of (+/-) aTC PfMORC for chromosome 4 at (a) 24 hpi and (b) 36 hpi displaying heterochromatin clustering within antigenic (var, rifin, and stevor) gene-dense regions (red). Differential interaction heatmaps highlight changes in chromatin structure following removal of aTC and subsequent PfMORC knockdown at (c) 24 hpi and (d) 36 hpi. (e) Whole-genome 3D models of the chromatin structure at both time points (24 hpi and 36 hpi) and (+/-) aTC.
To pinpoint which regions were most strongly affected by the knockdown of PfMORC, we used Selfish (Ardakany et al., 2019) to identify differential intrachromosomal and interchromosomal interactions in the (+/-) aTC PfMORC samples. Although the 24 hpi and 36 hpi time points shared similarities in loci most highly affected by PfMORC KD on many chromosomes, the changes at 36 hpi were more significant with a higher log2 FC (FDR <0.05) at most loci (Figure 6c–d, Figure 6—figure supplements 6–7). We also observed consistent loss of interactions between most regions containing var genes. These results indicate a failed attempt of the (-) aTC PfMORC parasites to maintain their overall chromatin structure with a significant weakening of the tightly controlled heterochromatin cluster.
We further validated our results using PASTIS to generate coordinate matrices and subsequently visualize a consensus three-dimensional model of the chromosome folding and their overall organization within the nucleus (Varoquaux et al., 2014). This can indicate changes in spatial distance that best describe the interaction data. The overall change in the chromatin 3D structure was clearly shown by our models of (+/-) aTC PfMORC samples (Figure 6e). While the co-localization of centromeres and telomeres in different regions of the nucleus were conserved, the 3D chromatin structure of (-) aTC PfMORC at 36 hpi displayed a clear opening of the chromatin and loss of interactions in most regions. This could explain the large changes in gene expression, including increased expression of all var and rifin genes in the (-) aTC PfMORC line (Supplementary file 6). While heterochromatin maintenance may be essential for tight control of var gene expression, preservation of the overall structure of the chromatin may be necessary to regulate the accurate expression of most P. falciparum transcripts.
Discussion
Despite intensive investigations on the dynamic nature of the Plasmodium chromatin, progress has been slow in capturing the regulatory factors that maintain and control chromatin structure and gene expression throughout parasite development. Preliminary studies performed by our lab and others have identified PfMORC as one of the most abundant chromatin-bound proteins (Singh et al., 2021a; Zhong et al., 2023). MORC proteins from plant to animal cells have a broad range of binding sites within the genome and participate in either DNA methylation establishment or heterochromatin formation (Li et al., 2013; Tencer et al., 2020; Shao et al., 2010; He et al., 2010; Zhang et al., 2019; Luo et al., 2020; Mimura et al., 2010). They have also been shown to co-localize with TFs in unmethylated promoter regions to alter chromatin accessibility and regulate TF binding and gene expression (He et al., 2010; Zhang et al., 2019; Luo et al., 2020; Mimura et al., 2010). For instance, AP2-P (PF3D7_1107800), AP2-G5, ISW1, NUP2, MCM5, and HDAC1 have been shown to interact with MORC in several published works (Farhat et al., 2020; Subudhi et al., 2023; Bryant et al., 2020), including this work. Protein pull-down experiments in Plasmodium spp. and T. gondii studies have detected MORC in complexes with several AP2 TFs. In T. gondii, TgMORC was characterized as an upstream transcriptional repressor of sexual commitment (Farhat et al., 2020; Zhong et al., 2023). In Plasmodium, the role of PfMORC was still poorly understood. Conflicting results on the essentiality of PfMORC using either KD or KO strategies remained unresolved (Singh et al., 2021a; Zhang et al., 2018). Using the CRISPR/Cas9 genome editing tool, we have determined the nuclear localization, genome-wide distribution, and regulatory impacts of PfMORC on the parasite chromatin, transcriptome, and cell cycle development. We now have powerful evidence demonstrating that PfMORC is not only critical for parasite cell cycle progression and its survival but also has a direct role in heterochromatin formation and gene silencing, including regulating immune evasion through antigenic variation. Disparities observed in the essentiality of PfMORC for the parasite survival in previous studies (Singh et al., 2021a) are most likely the results of weak protein disruption highlighting the need for a significant downregulation of PfMORC for true functional analysis. Using IFA and protein pull-downs, we have confirmed that PfMORC localizes to the nucleus and interacts with multiple ApiAP2 TFs, chromatin remodelers, and epigenetic players associated with heterochromatin regions including H3K9me3, SIP2, HDAC1, and the ISWI chromatin-remodeling complex (SWIB/MDM2) (Shang et al., 2022, Figure 1). Although some discrepancies have been observed with other studies, mainly due to the fact that MORC protein was not used as the bait protein, our study validated the interaction of MORC with these partners, supporting that PfMORC is in complex with key heterochromatin regulators. Using a combination of ChIP-seq, protein knock down, RNA-seq, and Hi-C experiments, we also demonstrated that the MORC protein is essential for the tight regulation of gene expression. We can speculate that lack of MORC impacts heterochromatin and chromatin compaction, preventing access to gene promoters from TFs and the general transcriptional machinery in a stage-specific manner. Although additional experiments will be required, our hypothesis is reinforced by the fact that downregulation of the PfMORC significantly reduced H3K9me3 coverage in the heterochromatin cluster(s) and 5’ flanking regions of var and rifin genes. While we were unable to confirm a direct role of PfMORC in the sexual conversion due to a nonsense mutation in AP2-G gene in our transfected lines, its strong interaction with AP2-G during the asexual cell cycle indicates that PfMORC in combination with other epigenetic factors may most likely control AP2-G expression and sexual differentiation. It is also important to recognize that the many pathways affected at the transcriptional level throughout the asexual stages in (-) aTC PfMORC lines are not only the direct results of PfMORC downregulation and decrease of its targeted DNA binding site. They are most likely a combination of direct and indirect effects of PfMORC KD warranted by the cell cycle arrest observed at the phenotypic level and the collapse of the chromatin organization confirmed using the chromatin conformation capture experiment. These direct and indirect effects should be carefully considered, and a combination of functional genomic studies should be completed when interpreting changes in gene expression in mutant-generated lines.
All together our work-demonstrated that in addition to its direct role in heterochromatin formation and antigenic variation, PfMORC may act as a potential repressor to control a set of parasite-specific genes including genes involved in parasite egress and invasion, and antigenic variation between the trophozoite to schizont stage transition (Shang et al., 2022). As our data confirm the importance of PfMORC and the parasite specificity of several interacting partners, it is tempting to speculate that drugs targeting these protein complexes could lead to novel antiparasitic strategies.
Materials and methods
Asexual parasite culture and maintenance
Request a detailed protocolAsexual P. falciparum strain NF54 or 3D7 parasites (MRA-1000, MRA-102, respectively) were propagated in 5% of human O+ erythrocytes and 10 mL of RPMI-1640 medium containing 0.5% Albumax II (Invitrogen), 2 mM L-glutamine, 50 mg/L hypoxanthine, 25 mM HEPES, 0.225% NaHCO3 and 10 mg/mL gentamicin. They were maintained at 37 °C and gassed with a sterile mixture of 5% O2, 5% CO2, and 90% N2(70, 71). Parasite synchronization was achieved using two 5% D-sorbitol treatments 8 hr apart (Trager and Jensen, 1976; Fidock et al., 1998).
Plasmid construction
Request a detailed protocolFlagging of the P. falciparum MORC (Pf3D7_1468100) gene spanning position (Ch14: 2785386–2797934 (-)) was performed using a two-plasmid design to insert a 3 x HA tag. The pCasG-Cas9-sgRNA plasmid vector (gifted by Dr. Sean Prigge) contains the site to express the sgRNA, along with the yDHODH gene as the positive selection marker. The gRNA oligos (Supplementary file 1) were ligated after digestion of the plasmid with BsaI. The pDC2-cam-Cas9-U6 plasmid (gifted by Dr. Marcus Lee) was digested with BamHI and Apal to remove the eGFP tag from the backbone. 453 bp of the C-terminal region of PfMORC and 469 bp of 3’UTR were amplified from P. falciparum genomic DNA with their respective primers (Supplementary file 1). A 3 x HA-tag was fused to the C-terminal region of the amplified product along with the 3’UTR formed through the Gibson assembly master mix (NEB, E2611S). To generate the PfMORC-HA knockdown constructs, a pKDPfAUBL plasmid (gifted by Dr. Sean Prigge) (Rajaram et al., 2020) was digested with AscI and AatII. Homology arms HA1 and HA2 of PfMORC were amplified with respective primers (Supplementary file 1) having 20 bp overhang and inserted into the digested plasmid using a Gibson assembly mix. The resulting pKD-MORC-HA-TetR-DOZI construct was linearized with the EcoRV enzyme prior to transfection. All constructs were confirmed through restriction enzyme digestion and Sanger sequencing.
Plasmids were isolated from 250 mL cultures of Escherichia coli (XL10-Gold Ultracompetent Cells, Agilent Cat. 200314) and 60 µg of pDC2-MORC-HA or linearized pKD-MORC-HA-TetR-DOZI, were used with 60 µg of pCasG-plasmid containing gRNA to transfect 200 µl of fresh red blood cells (RBCs) infected with 3–5% early ring stage parasites. After one erythrocytic cycle, transfected cultures were supplemented with 1.5 µM WR99210 (2.6 nM) (provided by the Jacobus Pharmaceuticals, Princeton, NJ) and 2.5 μg/mL Blasticidin (RPI Corp B12150-0.1). PfMORC-HA-TetR-DOZI transfected parasites were maintained with 500 nM anhydrotetracycline (aTC) (Ganesan et al., 2016; Nasamu et al., 2021). Media and drug selection was replenished every 24 hr for seven consecutive days after which DSM-1 drug selection was halted Once parasites were detected by microscopy, integration of the insert was confirmed by PCR amplification. To generate genetically homogenous parasite lines, the transfected parasites were serially diluted to approximately 0.5% parasite/well, into 96 well plates.
Molecular analysis of the transgenic lines
Request a detailed protocolGenomic DNA (gDNA) was extracted and purified using the DNeasy Blood & Tissue kit (Qiagen) following instructions from the manufacturer. The diagnostic PCR analysis was used to genotype the transfected lines using the primers listed in Supplementary data 1. The PCR amplification was conducted using KAPA HiFi HotStart ReadyMix (Roche) and amplicons were analyzed by gel electrophoresis followed by sequencing. For whole genome sequencing, genomic DNA was fragmented using a Covaris S220 ultrasonicator and libraries were generated using the KAPA LTP Library Preparation Kit (Roche, KK8230). To verify that the insertion was present in the genome at the correct location in both transfected lines, reads were mapped using Bowtie2 (v2.4.4) to the P. falciparum 3D7 reference genome (PlasmoDB, v48), edited to include the insertion sequence in the intended location. Integrative Genomic Viewer (IGV, Broad Institute) was used to verify that reads aligned to the modified sequence.
Variant analysis by genome-wide sequencing
Request a detailed protocolTo call variants (SNPs/indels) in the transfected lines compared to a previously sequenced control 3D7 line, genomic DNA reads were first trimmed to adapters and aligned to the Homo sapiens genome (assembly GRCh38) to remove human-mapped reads. Remaining reads were aligned to the P. falciparum 3D7 genome using bwa (version 0.7.17) and PCR duplicates were removed using PicardTools (Broad Institute). GATK HaplotypeCaller (https://gatk.broadinstitute.org/hc/en-us) was used to call variants between the sample and the 3D7 reference genome for both the transfected lines and the NF54 control. Only variants that were present in both transfected lines but not the NF54 control line were kept. We examined only coding-region variants and removed those that were synonymous variants or were located in var, rifin, or stevor genes. Quality control of variants was done by hard filtering using GATK guidelines.
Immunofluorescence assays
Request a detailed protocolA double-staining immunofluorescence assay was used on NF54 control and transgenic parasite lines of mixed parasite population growth stages. Parasites were washed in incomplete medium prior to fixing onto coverslips with 4% paraformaldehyde for 20 min at RT under darkness. After fixation, samples were washed three to five times with 1 x PBS followed by permeabilization with 0.5% Triton X-100 in PBS for 25 min at RT. Subsequently, samples were subjected to PBS washes and then incubated overnight at 4 °C in blocking buffer (2 mg/ml BSA solution in PBS containing 0.05% Tween-20 solution). Following overnight blocking, samples were washed and incubated for 1 hr at RT with Anti-HA Rb Ab (1:500, Abcam, ab9110) in a blocking buffer. After primary Ab incubation, samples were subject to 3 x washes with wash buffer (1 x PBS containing 0.05% Tween-20) followed by incubation with anti-rabbit DyLight 550 (Abcam ab98489; 1: 500) secondary antibody for 1 hr at room temperature. After incubation and a series of washes with wash buffer, slides were incubated with anti-H3K9me3 antibody, Alexa Fluor 488 conjugate (Millipore 07–442-AF488; 1:100) for 1 hr at RT. Slides are then washed and mounted in Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories, H-1200). Images were acquired using a Keyence BZ-X810 Fluorescence Microscope and were processed through ImageJ.
Western blotting
Request a detailed protocolParasites were synchronized twice at 8 hr intervals between synchronizations. After one cycle, culture was washed 3 x with complete media to remove residual aTC followed by dividing the cultures into two equal conditions. Parasites were grown with or without 500 nM aTC for 24 and 36 hr. after which RBCs were lysed using 0.15% saponin and parasites were collected after being washed with ice-cold 1 X PBS. Proteins were recovered from the lysed parasites after 30 min of incubation in lysis buffer (150 mM NaCl, 0.5 % NP40, 50 mM Tris pH 8, 1 mM EDTA, and protease inhibitors) and 10 s of sonication. Proteins were quantified with Pierce BCA Protein Assay Kit (Thermo Fisher 23227). 20 µg of proteins were loaded onto 3–8% Criterion XT Tris-Acetate Midi Protein Gels (Bio-Rad, 3450129). After migration, proteins were transferred onto a PVDF membrane and the membrane was blocked and then probed overnight with an anti-HA tag antibody (1:2500, Abcam, ab9110) as well as anti-aldolase antibody as a loading control (1:10,000, abcam, ab252953). After primary Ab incubation, blots were subsequently washed 3 x with a washing buffer followed by HRP-labeled Goat anti-Rabbit IgG (H + L) (1:10,000, Novex, A16104). Clarity Western ECL Substrate (Bio-Rad, 1705060) was applied to reveal the blots. Relative abundance of (+/-) aTC PfMORC was calculated by Bio-Rad ChemiDoc image lab software.
Immunoprecipitation followed by MudPIT mass spectrometry
Request a detailed protocolMid- to late-stage asexual parasites were collected following saponin treatment and purified samples were then resuspended into fresh IP buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.5 mM EDTA, 0.5 mM EGTA, 2 mM AEBSF 0.5% Triton X-100, and EDTA-free protease inhibitor cocktail (Roche)). Post-cell lysis solution was homogenized via sonication for 6–9 rounds. The soluble extracts were centrifuged at 13,000 x g for 15 min at 4 °C. The lysates were precleared with Dynabeads Protein A (Invitrogen) for 1 hr at 4 °C. Anti-HA tag antibodies (1:2500, Abcam, ab9110) are added to control and HA-tagged PfMORC precleared protein extract samples for 1 hr at 4 °C under constant rotation followed by the addition of fresh Dynabeads Protein A beads to each sample and incubated overnight at 4 °C. Dynabeads were washed three times with 500 µL of buffer (PBS, 0.05% Tween-20). Proteins were eluted into an elution buffer (50 mM Tris-HCl pH 6.7, 100 mM DTT, and 2% SDS). The eluent was subsequently precipitated overnight in 20% TCA followed by cold acetone washes. The urea-denatured, reduced, alkylated, and digested proteins were analyzed by Multidimensional Protein Identification Technology (MudPIT) on an Orbitrap Elite mass spectrometer coupled to an Agilent 1260 series HPLC, as described previously (Florens and Washburn, 2006).
Proteomics data processing and analysis
Request a detailed protocolTandem mass (MS/MS) spectra were interpreted using ProluCID v.1.3.3 (Xu et al., 2015) against a database consisting of 5527 non-redundant (NR) Plasmodium falciparum 3D7 proteins (PlasmoDB, v42), 36661 NR human proteins (NCBI, 2018-03-30 release), 419 common contaminants (human keratins, IgGs, and proteolytic enzymes), together with shuffled versions of all of these sequences. DTASelect v.1.9 (Tabb et al., 2002) and swallow v.0.0.1, an in-house developed software (https://github.com/tzw-wen/kite copy archived at Wen, 2024) were used to control FDRs resulting in protein FDRs less than 1.86%. All datasets were contrasted against their merged data set, respectively, using Contrast v1.9 (Tabb et al., 2002) and in-house developed sandmartin v.0.0.1 (https://github.com/tzw-wen/kite/tree/master/kitelinux). Our in-house developed software, NSAF7 v.0.0.1 (https://github.com/tzw-wen/kite/tree/master/windowsapp/NSAF7x64), was used to generate spectral count-based label-free quantitation results (Zhang et al., 2010). QPROT (Choi et al., 2015; Choi et al., 2008) was used to calculate values of log2 fold change and Z-statistic to compare two replicate PfMORC affinity purifications to two negative controls. Proteins enriched in the PfMORC-HA samples with values of log2 fold change ≥2 and Z-statistic ≥5 were considered significantly enriched.
ChIP assay
Request a detailed protocolPfMORC-HA and PfMORC KD asexual stage parasites were supplemented with and without aTC along with NF54 parasites (as a control) and were harvested at 24 and 36 HPS and cross-linked with 1% formaldehyde for 10 min at 37 °C followed by quenching with 150 mM glycine and 3 x washing with 1 x PBS. The pellets were resuspended in 1 mL of nuclear extraction buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM AEBSF, and 1 x Protease inhibitor cocktail), and incubated on ice for 30 min before addition of NP-40/Igepal to a final of 0.25%. After lysis, the parasites are subject to homogenization by passing through a 26 G 3/8 needle/syringe to burst the nucleus. After centrifugation for 20 min at 2,500 x g at 4 °C, samples were resuspended in 130 µL of shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris-HCl pH 7.5 and 1 X Protease inhibitor cocktail) and transferred to a 130 µL Covaris sonication microtube. The samples were then sonicated using a Covaris S220 Ultrasonicator for 8 min (Duty cycle: 5%, intensity peak power: 140, cycles per burst: 200, bath temperature: 6 °C). The samples were transferred to ChIP dilution buffer (30 mM Tris-HCl pH 8.0, 3 mM EDTA, 0.1% SDS, 30 mM NaCl, 1.8% Triton X-100, 1 X protease inhibitor tablet, 1 X phosphatase inhibitor tablet) and centrifuged for 10 min at 13,000 rpm at 4 °C, retaining the supernatant. For each sample, 13 μL of protein A agarose/salmon sperm DNA beads were washed three times with 500 µL ChIP dilution buffer by centrifuging for 1 min at 1000 rpm at room temperature. For pre-clearing, the diluted chromatin samples were added to the beads and incubated for 1 hr at 4 °C with rotation, then pelleted by centrifugation for 1 min at 1000 rpm. Before adding antibody,~10% of each sample was taken as input. 2 µg of anti-HA tag (1:2500, Abcam, ab9110) or rabbit polyclonal anti-H3K9me3 or anti-H3K9ac (Millipore no. 07–442 and H0913) antibodies were added to the sample and incubated overnight at 4 °C with rotation. For each sample, rabbit IgG antibody (Cat.689 No. 10500 c, Invitrogen) was used as a negative-control library. Per sample, 25 µL of protein A agarose/salmon sperm DNA beads were washed with ChIP dilution buffer (no inhibitors), blocked with 1 mg/mL BSA for 1 hr at 4 °C, and then washed three more times with buffer. 25 µL of washed and blocked beads were added to the sample and incubated for 1 hr at 4 °C with continuous mixing to collect the antibody/protein complex. Beads were pelleted by centrifugation for 1 min at 1000 rpm at 4 °C. The bead/antibody/protein complex was then washed with rotation using 1 mL of each buffer twice; low salt immune complex wash buffer (1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl), high salt immune complex wash buffer (1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl), and TE wash buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Complexes were eluted from antibodies by adding 250 μL of freshly prepared elution buffer (1% SDS, 0.1 M sodium bicarbonate). 5 M NaCl were added to the eluate, and cross-linking was reversed by heating at 45 °C overnight followed by the addition of 15 μL of 20 mg/mL RNase A with 30 min incubation at 37 °C. After this, 10 μL of 0.5 M EDTA, 20 μL of 1 M Tris-HCl pH 7.5, and 2 μL of 20 mg/mL proteinase K were added to the eluate and incubated for 2 hr at 45 °C. DNA was recovered by phenol/chloroform extraction and ethanol precipitation. DNA was purified using Agencourt AMPure XP beads. Libraries were then prepared from this DNA using the KAPA LTP Library Preparation Kit (Roche, KK8230) and sequenced on a NovaSeq 6000 machine.
ChIP-seq analysis
Request a detailed protocolFastQC (version 0.11.8, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to analyze raw read quality. Any adapter sequences were removed using Trimmomatic (version 0.39, http://www.usadellab.org/cms/?page=trimmomatic). Bases with Phred quality scores below 20 were trimmed using Sickle (version 1.33, https://github.com/najoshi/sickle; najoshi, 2015). The resulting reads were mapped against the P. falciparum genome (v48) using Bowtie2 (version 2.4.4). Using Samtools (version 1.11), only properly paired reads with mapping quality 40 or higher were retained, and reads marked as PCR duplicates were removed by PicardTools MarkDuplicates (version 2.18.0, Broad Institute). Genome-wide read counts per nucleotide were normalized by dividing millions of mapped reads for each sample (for all samples including input) and subtracting IgG read counts from the anti-HA IP counts. Peak calling was performed using MACS3 (version 3.0.0a7) (Zhang et al., 2008) with the upper and lower limits for fold enrichment set to 2 and 50, respectively.
Phenotypic analyses
Request a detailed protocolP. falciparum NF54 line along with transgenic PfMORC-HA-TetR-DOZI line were synchronized, grown for one cycle post-synchronization, and subject to three consecutive washes with 1 X PBS before being supplemented either with or without (aTC) for 48 hr or 72 hr (depending on which stage the parasites were washed). Culture media were replaced daily with or without aTC supplementation.
Quantitative growth assay
Request a detailed protocolSynchronous cultures either of ring or trophozoite stage at 0.5% parasitemia were grown with or without aTC (500 nM) into a 96-well plate in triplicates for each time point. Parasites were collected at every 24 hr time for three cycles and subjected to SYBR green assay (Thermo Fisher, S7523).
RNA-seq library preparation
Request a detailed protocolParasites at the ring, trophozoite, or schizont stage were extracted following saponin treatment before flash freezing. Two independent biological replicates were generated for each time point, culture condition, and line. Total RNA was extracted with TRIzol LS Reagent (Invitrogen) followed by incubation for 1 hr with 4 units of DNase I (NEB) at 37 °C. RNA samples were visualized by RNA electrophoresis and quantified on Synergy HT (BioTek). mRNA was then purified using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490) according to the manufacturer’s instructions. Libraries were prepared using NEBNext Ultra Directional RNA Library Prep Kit (NEB, E7420) and amplified by PCR with KAPA HiFi HotStart Ready Mix (Roche). PCR conditions consisted of 15 min at 37 °C followed by 12 cycles of 98 °C for 30 s, 55 °C for 10 s, and 62 °C for 1 min 15 s, then finally one cycle for 5 min at 62 °C. The quantity and quality of the final libraries were assessed using a Bioanalyzer (Agilent Technology Inc). Libraries were sequenced using a NovaSeq 6000 DNA sequencer (Illumina), producing paired-end 100 bp reads.
RNA-seq data processing and differential expression analysis
Request a detailed protocolFastQC [https://www.bioinformatics.babraham.ac.uk/projects/fastqc/] was used to analyze raw read quality and thus 11 bp of each read and any adapter sequences were removed using Trimmomatic (v0.39) [http://www.usadellab.org/cms/?page=trimmomatic]. Bases were trimmed from reads using Sickle with a Phred quality threshold of 25 (v1.33) [https://github.com/najoshi/sickle] and reads shorter than 18 bp were removed. The resulting reads were mapped against the P. falciparum 3D7 genome (PlasmoDB, v53) using HISAT2 (v2-2.2.1) with default parameters. Uniquely mapped, properly paired reads with a mapping quality of 40 or higher were retained using SAMtools (v1.11) [http://samtools.sourceforge.net/]. Raw read counts were determined for each gene in the P. falciparum genome using BedTools [https://bedtools.readthedocs.io/en/latest/#] to intersect the aligned reads with the genome annotation. Differential expression analysis was performed using DESeq2 to call up- and down-regulated genes (FDR<0.05 and log2 FC>0.5). Volcano plots were made using the R package EnhancedVolcano.
Hi-C library preparation
Request a detailed protocol(+/-) aTC parasites at 24 hpi and 36 hpi were cross-linked using 1.25% formaldehyde for 15 min at 37 °C in 10 mL total volume followed by quenching with 150 mM glycine. Parasites were then washed three times with chilled 1 x PBS on a rocking platform. Parasite nuclei were released using lysis buffer (10 mM Tris-HCl, pH 8.0, 10 mM NaCl, 2 mM AEBSF, 0.25% Igepal CA-630, and 1 X EDTA-free protease inhibitor cocktail (Roche)) and 15 syringe passages through a 26.5-gauge needle. After releasing the crosslinked chromatin with 0.1% SDS, chromatin was digested using MboI restriction enzyme overnight at 37 °C. Thereafter Hi-C libraries were prepared as previously described (Bunnik et al., 2019; Gupta et al., 2021).
Hi-C data processing, differential interaction analysis, and generation of 3D models
Request a detailed protocolPaired-end Hi-C libraries were processed (pairing, mapping, quality filtering, binning, and normalization) using HiC-Pro (Servant et al., 2015). A mapping quality cutoff of 30 was set while aligning to the P. falciparum genome (PlasmoDB, v58), and the resulting reads were binned at 10 kb resolution and ICED-normalized. Stratum-adjusted correlation coefficients were calculated using HiCRep (Yang et al., 2017), then replicates were merged to generate a single consensus sample for each time point and condition. The normalized matrices were per-million read count normalized and maximum values for the heatmap color scale on each chromosome was set to the maximum value for all samples to allow for direct comparisons between each condition and time point. Furthermore, due to the overrepresentation of short-range interactions, the maximum values for each heatmap were also set to the 90th percentile of each chromosome to slightly compress highly interacting regions and enhance the visualization of interactions. Significant intrachromosomal interactions were identified with FitHiC (Ay et al., 2014) to identify the log-linear relationship between contact probability and genomic distance. Differential interchromosomal and intrachromosomal interactions between (+) aTC and (-) aTC were identified using Selfish (Ardakany et al., 2019) with default parameters (FDR <0.05). The maximum and minimum values for the color scales in the differential heatmaps were set to the absolute value of the largest log2 fold change within each chromosome. PASTIS (Varoquaux et al., 2014) was used to generate coordinate matrices from the raw read count matrices output by HiC-Pro, and then visualized as 3D chromatin models in ChimeraX (Goddard et al., 2018), highlighting regions containing var genes, telomeres, and centromeres.
Statistical analyses
Request a detailed protocolParasitemia and the proportion of asexual stages were analyzed using a two-way ANOVA with Tukey’s test for multiple comparisons. Significant differences were indicated as follows: *p<0.05; **p<0.01, ***p<0.001, and ****p<0.0001. Statistical tests were performed with GraphPad Prism. Figures were generated with GraphPad Prism and BioRender.
Data availability
Sequence reads for all sequencing experiments have been deposited in the NCBI Sequence Read Archive with accession PRJNA994684. Original data underlying this manuscript generated at the Stowers Institute can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/LIBPB-2404. The mass spectrometry dataset generated for this study is available from the MassIVE data repository using the identifier MSV000092353. All other data are available in the main text, methods, and supplementary data.
-
NCBI BioProjectID PRJNA994684. PfMORC protein regulates chromatin accessibility and transcriptional repression in the human malaria parasite, P. falciparum.
-
MassiveID MSV000092353. PfMORC protein regulates chromatin accessibility and transcriptional repression in the human malaria parasite, P. falciparum.
-
Stowers InstituteID LIBPB-2404. Index of /publications/LIBPB-2404.
References
-
The chromatin bound proteome of the human malaria parasiteMicrobial Genomics 6:e000327.https://doi.org/10.1099/mgen.0.000327
-
Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasitesThe Journal of Experimental Medicine 206:953–966.https://doi.org/10.1084/jem.20082826
-
Exploring the virulence gene interactome with CRISPR/dCas9 in the human malaria parasiteMolecular Systems Biology 16:e9569.https://doi.org/10.15252/msb.20209569
-
Significance analysis of spectral count data in label-free shotgun proteomicsMolecular & Cellular Proteomics 7:2373–2385.https://doi.org/10.1074/mcp.M800203-MCP200
-
Histone lysine methyltransferases and demethylases in Plasmodium falciparumInternational Journal for Parasitology 38:1083–1097.https://doi.org/10.1016/j.ijpara.2008.01.002
-
Proteomic analysis by multidimensional protein identification technologyMethods in Molecular Biology 328:159–175.https://doi.org/10.1385/1-59745-026-X:159
-
Chromosomes conformation capture coupled with next-generation sequencing (Hi-C) in Plasmodium falciparumMethods in Molecular Biology 2369:15–25.https://doi.org/10.1007/978-1-0716-1681-9_2
-
Epigenetic reader complexes of the human malaria parasite, Plasmodium falciparumNucleic Acids Research 47:11574–11588.https://doi.org/10.1093/nar/gkz1044
-
MORC proteins and epigenetic regulationPlant Signaling & Behavior 7:1561–1565.https://doi.org/10.4161/psb.22460
-
MORC4 promotes chemoresistance of luminal a/b breast cancer via stat3-mediated MID2 UpregulationOncoTargets and Therapy 13:6795–6803.https://doi.org/10.2147/OTT.S260509
-
Two-step colocalization of MORC3 with PML nuclear bodiesJournal of Cell Science 123:2014–2024.https://doi.org/10.1242/jcs.063586
-
Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondiiMolecular and Cellular Biology 25:10301–10314.https://doi.org/10.1128/MCB.25.23.10301-10314.2005
-
Involvement of histone deacetylation in MORC2-mediated down-regulation of carbonic anhydrase IXNucleic Acids Research 38:2813–2824.https://doi.org/10.1093/nar/gkq006
-
Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparumMolecular and Biochemical Parasitology 143:100–110.https://doi.org/10.1016/j.molbiopara.2005.04.015
-
The PfAP2-G2 transcription factor is a critical regulator of gametocyte maturationMolecular Microbiology 115:1005–1024.https://doi.org/10.1111/mmi.14676
-
Analysis of the plasmodium and anopheles transcriptomes during oocyst differentiationThe Journal of Biological Chemistry 279:5581–5587.https://doi.org/10.1074/jbc.M307587200
-
DTASelect and contrast: tools for assembling and comparing protein identifications from shotgun proteomicsJournal of Proteome Research 1:21–26.https://doi.org/10.1021/pr015504q
-
Molecular mechanism of the MORC4 ATPase activationNature Communications 11:5466.https://doi.org/10.1038/s41467-020-19278-8
-
MORC protein family-related signature within human disease and cancerCell Death & Disease 12:1112.https://doi.org/10.1038/s41419-021-04393-1
-
Full-length transcriptome analysis of Plasmodium falciparum by single-molecule long-read sequencingFrontiers in Cellular and Infection Microbiology 11:631545.https://doi.org/10.3389/fcimb.2021.631545
-
Identification of a transcription factor in the mosquito-invasive stage of malaria parasitesMolecular Microbiology 71:1402–1414.https://doi.org/10.1111/j.1365-2958.2009.06609.x
-
Transcription factor AP2-Sp and its target genes in malarial sporozoitesMolecular Microbiology 75:854–863.https://doi.org/10.1111/j.1365-2958.2009.07005.x
-
Female-specific gene regulation in malaria parasites by an AP2-family transcription factorMolecular Microbiology 113:40–51.https://doi.org/10.1111/mmi.14334
-
Mechanisms of triggering malaria gametocytogenesis by AP2-GParasitology International 84:102403.https://doi.org/10.1016/j.parint.2021.102403
-
Model-based analysis of ChIP-Seq (MACS)Genome Biology 9:R137.https://doi.org/10.1186/gb-2008-9-9-r137
Article and author information
Author details
Laurence A Florens

Funding
National Institute of Allergy and Infectious Diseases (R01 AI136511)
- Karine G Le Roch
National Institute of Allergy and Infectious Diseases (R21 AI142506)
- Karine G Le Roch
University of California, Riverside (NIFA-Hatch-225935)
- Karine G Le Roch
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by NIH grants to KGLR (nos. 1R01 AI136511 and R21 AI142506-01) and by the University of California, Riverside to KGLR (no. NIFA-Hatch-225935). This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929).
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.92499. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Chahine, Gupta, Lenz et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,969
- views
-
- 136
- downloads
-
- 15
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 5
- citations for umbrella DOI https://doi.org/10.7554/eLife.92499
-
- 2
- citations for Reviewed Preprint v1 https://doi.org/10.7554/eLife.92499.1
-
- 1
- citation for Reviewed Preprint v2 https://doi.org/10.7554/eLife.92499.2
-
- 7
- citations for Version of Record https://doi.org/10.7554/eLife.92499.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
PfMORC protein regulates chromatin accessibility and transcriptional repression in the human malaria parasite, Plasmodium falciparum
eLife 12:RP92499.
https://doi.org/10.7554/eLife.92499.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}