Genetics: A colourful duplication
A genetic duplication event during evolution allowed male wood tiger moths to have either yellow or white patterns on their wings.
- Centre national de la recherche scientifique (CNRS), France
- Muséum national d'Histoire naturelle, France
The beautiful patterns found on the wings of moths and butterflies can provide important insights into adaptive evolution (Orteu and Jiggins, 2020). Natural selection exerted by predators, and sexual selection exerted by females, have led to the emergence of an incredible diversity of new colours and patterns (Briolat et al., 2019; Robertson and Monteiro, 2005). While some of the genes involved in this diversification have recently been identified (Van’t Hof et al., 2016; Livraghi et al., 2021), it is still unclear whether the emergence of new colours and patterns is generally driven by similar sets of genes or by the evolution of new ones.
Within some species, individuals can have strikingly different colour patterns on their wings. For instance, male wood tiger moths can have either yellow or white hindwings, with both types of males usually occurring within the same geographical location (Figure 1A). Understanding the genetic mechanisms that allow different coloured individuals to co-exist within a population – a phenomenon called colour polymorphism – can help identify how new traits emerged over the course of evolution (Llaurens et al., 2017). Now, in eLife, Chris Jiggins, Johanna Mappes and co-workers – including Melanie Brien (University of Helsinki) and Anna Orteu (University of Cambridge) as joint first authors – report which genes determine whether a male wood tiger moth will develop white or yellow hindwings (Brien et al., 2023).
Figure 1
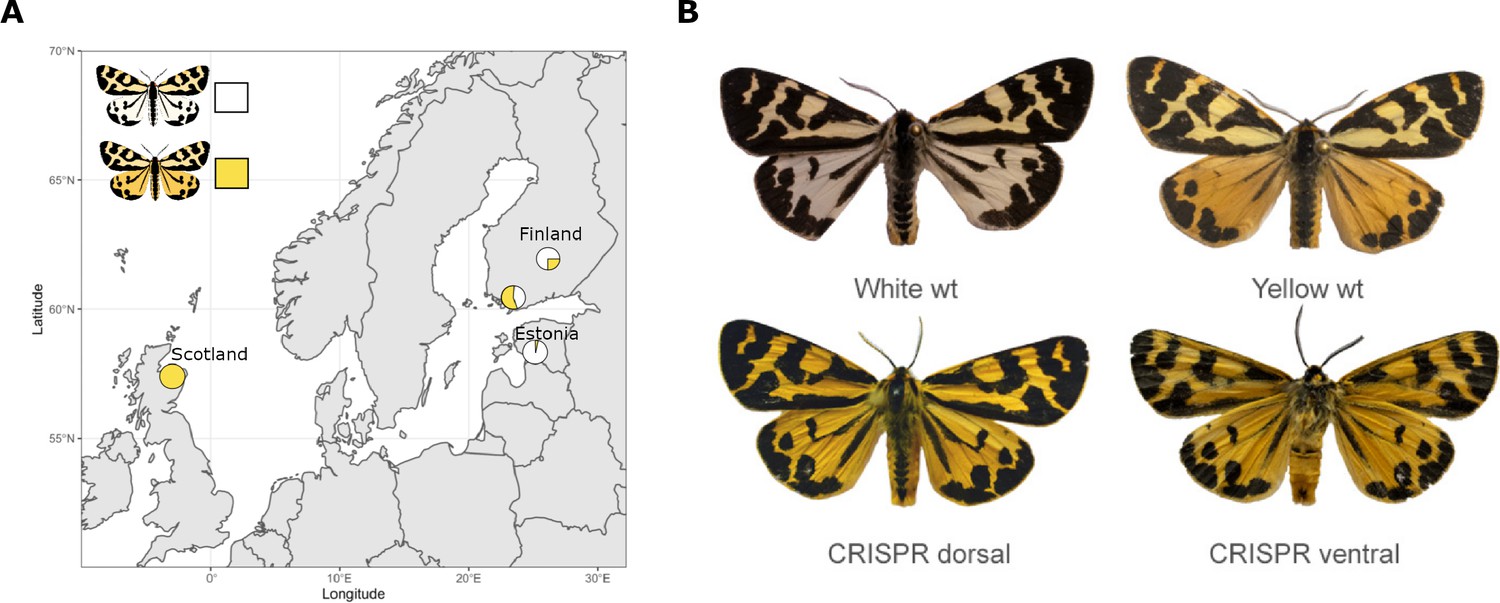
Finding the gene responsible for colour polymorphism in wood tiger moths.
(A) The proportion of male wood tiger moths that have yellow or white hindwings varies between geographical locations. For instance, 40–70% of males living in Finland and 97% of males living in Estonia have white hindwings, whereas all male moths in Scotland have yellow hindwings. (B) To investigate if the gene valkea was responsible for this colour polymorphism, Brien et al. used the genetic tool CRISPR-Cas9 to modify its sequence in wild-type (wt) moths with white hindwings (top left). This caused the mutant males to display yellow instead of white on the back (bottom left) and front (bottom right) of their hindwings, similarly to wild-type yellow moths (top right). The forewings of the genetically modified moths were also more yellow than wild-type white males, which is likely due to the CRISPR-Cas9 modification also introducing a mutation in to the full-length copy of the yellow-e gene.
Image credit: Adapted from Figure 1 and Figure 4 of Brien et al., 2023.
Like detectives working on a complex case, the team patiently gathered several lines of evidence to find the genetic variations responsible for this colour polymorphism. By crossing yellow females with white males, they were able to identify a region of the genome that is associated with colour differences in male offspring. This region contains 21 genes, including four from the yellow gene family, and is a different size in yellow and white males. In white males, this part of the genome consists of a large duplicated area which contains both a full-length copy and truncated copy of the yellow-e gene. Brien et al. hypothesized that the truncated gene is responsible for the white phenotype in the wood tiger moth, and named the suspected gene valkea, the Finnish word for white.
Next, Brien et al. studied the genes expressed in the wings of caterpillars and pupae before they grow into adult wood tiger moths. Several genes were found to be differentially expressed in the white and yellow moths: as expected, valkea was only turned on in white males. Interestingly, the full-length copy of the yellow-e gene was also overexpressed in the wings of the white males during this phase of development.
To confirm that valkea controls wing colour, Brien et al. introduced a ‘guide’ that allowed the gene editing tool CRISPR-Cas9 to specifically modify the DNA sequence of the valkea gene. More than 1000 eggs from white moths were injected with the valkea-specific guide, but only six individuals reached adulthood, with four out of five male adults developing partially yellow hindwings (Figure 1B). Unfortunately, the guide also targeted the full-length copy of the yellow-e gene in addition to valkea, making it difficult to determine the respective role each of these genes play in colour polymorphism.
The findings of Brien et al. suggest that the hindwing colour of male wood tiger moths is determined by genes from the yellow family, which are known to regulate wing colour in other insects (Wittkopp et al., 2002). This highlights how the diverse range of colour patterns seen in winged insects are determined by only a small number of genes (Zhang et al., 2017). The results also confirm the important role of gene duplication in driving the evolution of new traits (Martin and Reed, 2010).
The re-use and duplication of the yellow-e gene can be interpreted as evolutionary tinkering (Jacob, 1977). The tinkering of such a small set of genes demonstrates how a few genetic variations can generate such a striking diversity of phenotypes. Nevertheless, the small set of genetic variations that can be targeted by selection also exert strong constraints on adaptive evolution, potentially limiting the range of colours and patterns that can emerge over the course of evolution.
Distinguishing the respective roles of similar genes is challenging. The work of Brien et al. is a promising step in investigating the exact changes within and among gene copies that led to individual members of a species developing one trait or another. This type of work on polymorphism could be combined with research investigating how gene duplications impact the evolution of phenotypes between different species. The results of these studies could highlight the different evolutionary paths responsible for the diverse colours and patterns seen on the wings of moths and butterflies.
References
-
Diversity in warning coloration: selective paradox or the norm?Biological Reviews of the Cambridge Philosophical Society 94:388–414.https://doi.org/10.1111/brv.12460
-
wingless and aristaless2 define a developmental ground plan for moth and butterfly wing pattern evolutionMolecular Biology and Evolution 27:2864–2878.https://doi.org/10.1093/molbev/msq173
-
Publisher Correction: The genomics of coloration provides insights into adaptive evolutionNature Reviews Genetics 21:461–475.https://doi.org/10.1038/s41576-020-0249-5
-
Female Bicyclus anynana butterflies choose males on the basis of their dorsal UV-reflective eyespot pupilsProceedings of the Royal Society B 272:1541–1546.https://doi.org/10.1098/rspb.2005.3142
-
Evolution of yellow gene regulation and pigmentation in DrosophilaCurrent Biology 12:1547–1556.https://doi.org/10.1016/s0960-9822(02)01113-2
Article and author information
Author details
Publication history
Copyright
© 2023, Llaurens
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 448
- views
-
- 25
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Genetics: A colourful duplication
eLife 12:e92763.
https://doi.org/10.7554/eLife.92763




{kind=link}