Intestinal microbiome dysbiosis increases Mycobacteria pulmonary colonization in mice by regulating the Nos2-associated pathways
- Department of Tuberculosis, The First Affiliated Hospital of Xinxiang Medical University, China
- Department of Pathogenic Biology, School of Basic Medical Science, Xinxiang Medical University, China
eLife Assessment
This important study shows the effect of gut dysbiosis on the colonization of mycobacteria in the lung. The data with comprehensive analysis of gene expression profiles in the lung with dysbiotic mice is compelling and goes beyond the current state of the art. However, the mechanistic insight, where the lung epithelial cell line was used, and the experiments with Mtb infection are currently incomplete.
https://doi.org/10.7554/eLife.99282.3.sa0Significance of the findings:
Important: Findings that have theoretical or practical implications beyond a single subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Compelling: Evidence that features methods, data and analyses more rigorous than the current state-of-the-art
Incomplete: Main claims are only partially supported
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
Increasing researches reveal gut microbiota was associated with the development of tuberculosis (TB). How to prevent or reduce Mycobacterium tuberculosis colonization in the lungs is a key measure to prevent TB. However, the data on gut microbiota preventing Mycobacterium colonization in the lungs were scarce. Here, we established the clindamycin-inducing intestinal microbiome dysbiosis and fecal microbial transplantation models in mice to identify gut microbiota’s effect on Mycobacterium’s colonization in the mouse lungs and explore its potential mechanisms. The results showed that clindamycin treatment altered the diversity and composition of the intestinal bacterial and fungal microbiome, weakened the trans-kingdom network interactions between bacteria and fungi, and induced gut microbiome dysbiosis in the mice. Gut microbiota dysbiosis increases intestinal permeability and enhances the susceptibility of Mycobacterium colonization in the lungs of mice. The potential mechanisms were gut microbiota dysbiosis altered the lung transcriptome and increased Nos2 expression through the ‘gut–lung axis’. Nos2 high expression disrupts the intracellular antimicrobial and anti-inflammatory environment by increasing the concentration of nitric oxide, decreasing the levels of reactive oxygen species and Defb1 in the cells, and promoting Mycobacteria colonization in the lungs of mice. The present study raises a potential strategy for reducing the risks of Mycobacteria infections and transmission by regulating the gut microbiome balance.
Introduction
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (Mtb), with approximately 10 million people being infected each year and 1.6 million deaths in 2021 (Furin et al., 2019; Bagcchi, 2023). The epidemic of TB has brought heavy economic and social burdens to countries around the world. According to the World Health Organization (WHO) Global TB Report 2022, the global spending on essential TB services for TB diagnosis, treatment, and prevention has exceeded $6.4 billion (Global tuberculosis report 2023, 2023). Besides, drug-resistant forms of Mtb are currently on course to be the world’s deadliest pathogens, responsible for a quarter of deaths due to antimicrobial resistance (Furin et al., 2019). The prevalence of drug-resistant Mtb is also a major threat to the control of TB. Hence, it has been an urgent task for researchers to develop more effective measures for the prevention and treatment of TB.
Recent advances in gut microbiota explorations have led to improved knowledge of the ‘gut–lung axis’. Some studies have shown that intestinal microbiota dysbiosis may affect the occurrence and development of respiratory diseases including asthma, chronic obstructive pulmonary disease, respiratory pathogens infection, etc. by the ‘gut–lung axis’ (Keely et al., 2012; Barcik et al., 2020; Wang et al., 2023). The documents about the relationship between gut microbiota and TB have increased in recent years, and increasing evidence supports the presence of intestinal microbiota dysbiosis in TB patients. A case–control study of pulmonary TB in pediatric patients showed that the abundance of Prevotellaceae and Enterococcaceae was increased, and the abundance of Oscillospiraceae and Bifidobacteriaceae was decreased in TB patients compared with healthy control subjects (Li et al., 2019). Maji et al. have found that the levels of Provetalla decreased and Bacteroides increased in TB patients (Maji et al., 2018). Recently, our teams’ study in active TB patients showed the abundance of Bacteroidetes significantly altered compared with health subjects (Han et al., 2024). Another study also showed the phylum of Bacteroidetes altered in the feces samples of patients with recurrent TB (Luo et al., 2017). All these researches indicated that Mtb infection can lead to the alteration of intestinal microbiota composition.
However, the causal relations between Mtb infection and intestinal microbiota dysbiosis have not been fully elucidated. Some factors including alcohol, smoking, HIV infection, and diabetes have been proven to cause gut microbiota dysbiosis, and these also are important risk factors for TB (Naidoo et al., 2019). A recent study has shown that the mice fed with a high-fat diet-induced gut microbiota dysbiosis increased the risk of developing active TB in mice (Arias et al., 2019). Another study found Firmicutes/Bacteroidetes ratio increased in a murine model of type 2 diabetes induced by an energy-dense diet, and increased the susceptibility to Mtb infection (Sathkumara et al., 2021). These findings support a viewpoint that the changes in the gut microbiome are a contributing factor in Mtb infection pathogenesis and its clinical presentation. Many documents supposed that gut microbiota dysbiosis could modulate immunity and inflammation reaction at distal sites such as the lungs, reduce colonization resistance by external pathogens, and promote TB susceptibility (Naidoo et al., 2019; Osei Sekyere et al., 2020). Hence, modulation of the gut microbiota and balance of the gut–lung axis was a potential avenue for TB prevention and management.
Based on our previous studies and other reports, we found the effect of Mtb infection on the gut Bacteroidetes was most significant. Hence, in the present study, clindamycin (CL), an antibiotic that selectively disorders anaerobic Bacteroidetes (You et al., 2019), was used to establish a mouse model of intestinal microbiota dysbiosis. We uncovered the important role of gut microbiome dysbiosis affecting Mycobacteria colonization in the mouse lungs using the technology of fecal microbiota transplantation (FMT). Furthermore, based on the ‘gut–lung axis’ theory, we performed a transcriptomic analysis of the mice lungs after Mycobacteria infection. We aim to explore the potential role and mechanism of gut microbiome dysbiosis enhancing Mycobacteria colonization in the lungs of mice.
Results
Clindamycin increases Mycobacterium smegmatis colonization in the lungs of mice
According to Figure 1a procedure, we established a clindamycin-treated mice model to assess the effects of clindamycin-associated gut dysbiosis on Mycobacterium smegmatis (MS) colonization in the lungs of mice. The results showed that the mice with clindamycin treated presented a significantly higher colonization of MS in the lungs than that of control mice (Figure 1b, Figure 1—source data 1). The size of the cecum of CL-treated mice was markedly more dilated compared with the control groups (Figure 1c, Figure 1—source data 1). To assess the effect of CL on gut mucosal damage and permeability, the intestinal fatty acid binding protein (i-FABP, a marker of enterocyte death) and lipopolysaccharides (LPS, an endotoxin, as a marker of gut permeability) in serum were investigated with enzyme-linked immunosorbent assay (ELISA) technology. The results showed that the levels of i-FABP and LPS were significantly higher in the CL group than in the control group (CON) (Figure 1d, e, Figure 1—source data 1). Pathological sections of intestinal tissue showed the intestinal epithelial tissues of mice with no obvious alteration after clindamycin treatment vs the CON group (Figure 1—figure supplement 1). H&E staining of paraffin-embedded lung sections revealed that there was more diffused inflammation and inflammatory cell infiltration in the CL-treated mice compared with that of the CON group animals after infecting with MS (Figure 1—figure supplement 1). All these results indicated that CL treatment damages the enterocyte, increases gut permeability, promotes the fermentation of cecum contents, and increases MS colonization in the lungs of mice.
Figure 1 with 1 supplement see all
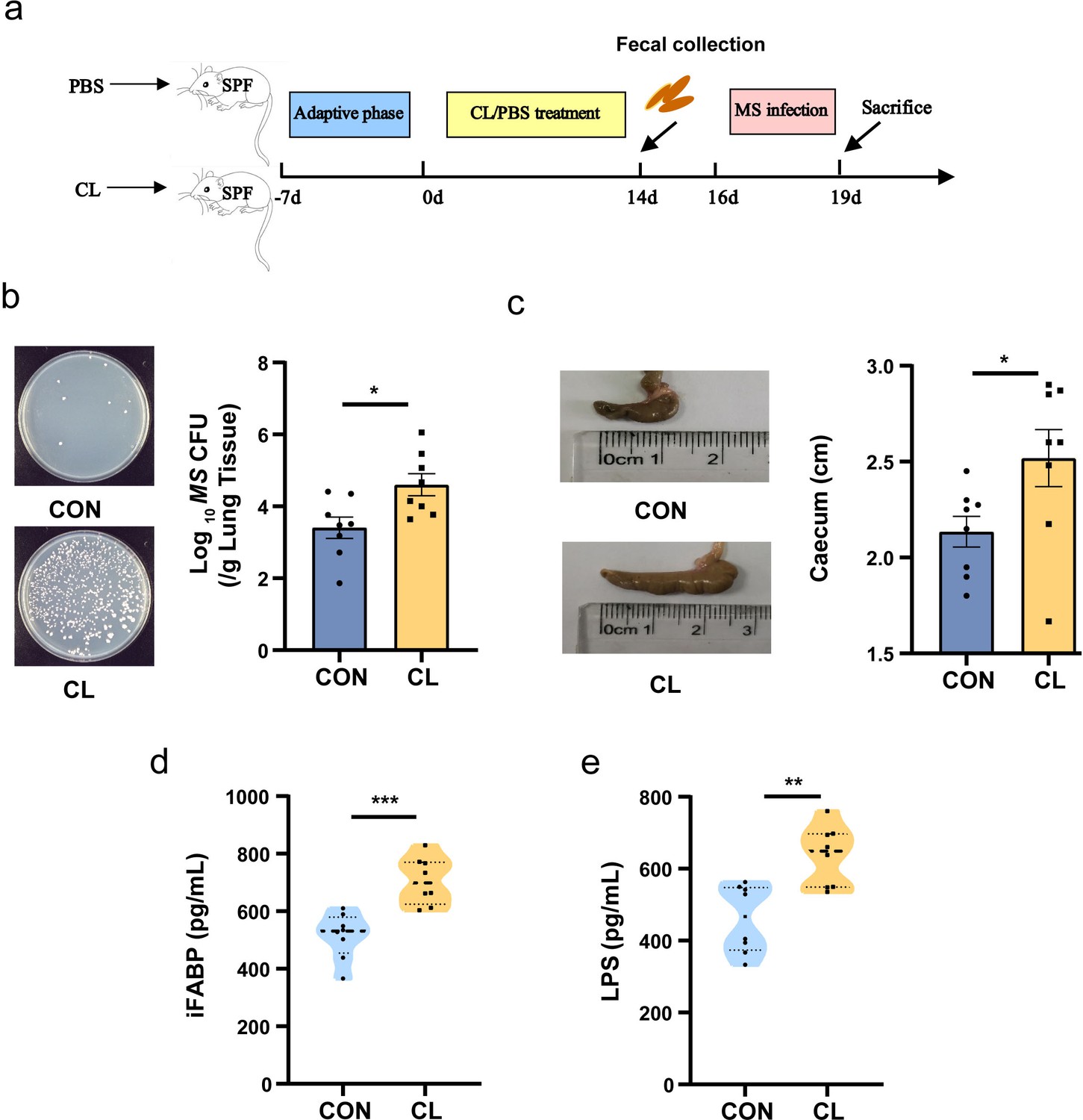
Clindamycin increases MS pulmonary colonization in mice.
(a) The experimental procedures of clindamycin-inducing gut microbiota dysbiosis model in mice. (b) The colonization of MS in the lungs of mice after infection at 72 hr. (c) The length of cecum after clindamycin treatment. (d) The level of iFABP in serum. (e) LPS concentration in serum. CL: clindamycin treatment group (n = 8), CON: control group (n = 8). MS: Mycobacterium smegmatis, iFABP: intestinal fatty acid binding protein, LPS: lipopolysaccharides. *p < 0.05, **p < 0.01, ***p < 0.001.
-
Figure 1—source data 1
Effect of clindamycin on MS pulmonary colonization and intestinal permeability of mice.
- https://cdn.elifesciences.org/articles/99282/elife-99282-fig1-data1-v1.xlsx
Altered the diversity and composition of the gut microbiome in mice
To assess the effect of CL treatment on the gut microbiome of mice, we investigated the alteration of gut microbiota (bacterial) and mycobiota (fungi) by 16S rRNA and ITS2 amplicon sequencing, respectively. For the gut bacterial analysis, the α-diversity was significantly decreased in the CL group compared with the CON group (Figure 2a, Figure 2—source data 1). The β-diversity based on the weighted UniFrac distance showed that the CL group samples were separated from the CON group (R = 0.8488, p = 0.001) (Figure 2b). It indicated that the intestinal bacterial microbial community structure was a significant difference between the CL-treated mice and the control mice. At the phylum level, we observed a remarkable decrease of Bacteroidota and a significant increase of Firmicutes and Proteobacteria in the CL group compared to the control group (Figure 2c, Supplementary file 1). At the genus level, the CL group had a significantly higher relative abundance of Bacteroides, Lactobacillus, Escherichia–Shigella, and Faecalibaculum, and a lower abundance of Dubosiella, Alloprevotella, Akkermansia, and Bifidobacterium compared with that in the CON group (Figure 2d, Supplementary file 1).
Figure 2 with 1 supplement see all
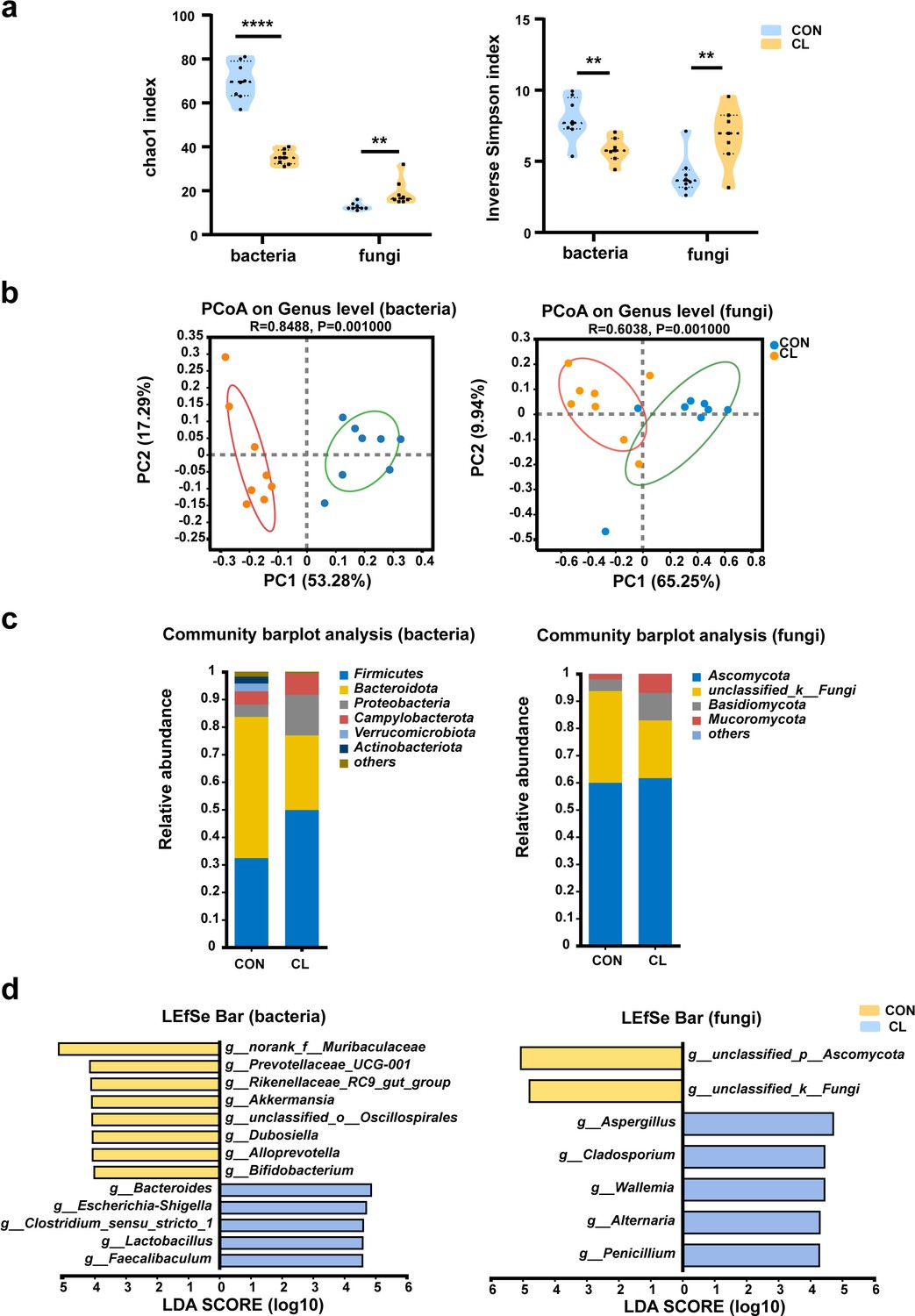
Clindamycin altered the diversity and composition of the gut microbiome in mice.
(a) Chao1 and inverse Simpson indices of gut microbiota. (b) Principal coordinate analysis (PCoA) of gut microbiota based on weighted Unifrac distance. (c) The composition distribution of gut microbiome (bacteria and fungi) at the phylum level. (d) The linear discriminant analysis effect size (LEfSe) analysis of the differentially abundant gut microbiota between the CL and CON groups at the genera level (linear discriminant analysis [LDA] > 4, p < 0.05). CL: clindamycin treatment group (n = 8), CON: control group (n = 8). **p < 0.01, ****p < 0.0001.
-
Figure 2—source data 1
Alpha diversity index of gut microbiome.
- https://cdn.elifesciences.org/articles/99282/elife-99282-fig2-data1-v1.xlsx
For the fungal mycobiota analysis, the Chao1 and inverse Simpson indices of the CL group was significantly increased compared to that of the CON group (Figure 2a). The principal coordinate analysis (PCoA) showed that the samples were significantly distinguished between two groups (R = 0.6038, p = 0.001) (Figure 2b), which suggested that the community of intestinal mycobiota in the two groups is different. At the phylum level, the fungal microbiota was dominated by the Ascomycota and Basidiomycota in two groups, and Ascomycota showed no obvious differences between the two groups. However, the relative abundance of Basidiomycota significantly increases in the CL group compared with the CON group (Figure 2c, Supplementary file 1). The LEfSe analysis at the genus level showed that the relative abundance of Aspergillus, Penicillium, Cladosporium, and Alternaria was enriched in the CL group vs the CON group (Figure 2d, Supplementary file 1). These results are consistent with the Wilcoxon rank-sum test analysis (Figure 2—figure supplement 1). Taken together, all these data indicated that CL treatment altered the diversity and composition of the intestinal microbiome (including bacterial and fungi) in the mice and induced gut microbiome dysbiosis.
Clindamycin weakens the trans-kingdom networks construction of gut bacterial and fungi
To evaluate the effect of clindamycin treatment on gut microbiome balance, we analyzed the fungi-to-bacteria species ratio with ITS/16S. The results show that the ITS/16S ratio of the CL group was significantly increased compared with the CON group (Figure 3a, Figure 3—source data 1). We also assessed the alterations in the ratio of the dominant phyla in the gut microbiome. The results showed that the ratio of Firmicutes/Bacteroidota in the CL group was increased, and Ascomycota/Basidiomycota was significantly decreased compared with that of the CON group (Figure 3b, c, Figure 3—source data 1). These results revealed that clindamycin treatment not only disturbed the intestinal equilibrium between bacteria and fungi but also destroyed the balance among the dominant bacteria (or fungi). Then, to assess the interplay between bacteria and fungi, we performed the trans-kingdom networks analysis at the genus level. The results showed that the trans-kingdom networks were altered in the CL group vs that in the CON group. In the control group, the bacteria and fungi were closely related to each other, gathering in a cluster and forming a more complex network (Figure 3d). There were 154 nodes including 45 fungi and 109 bacteria, and 810 edges; the relative connectedness was 5.26 (Table 1). However, the complexity of the trans-kingdom network in the CL group was dramatically decreased, and the interplay between bacteria and fungi was also weakened (Figure 3e). A total of 110 nodes including 62 fungi and 48 bacteria gathered into clusters in this network. The edges decreased to 357 and the relative connectedness was 3.25 in the CL group network (Table 1). We also found that the interplay ratio between fungi in the CL group was 52.10%, which was much higher than that of the CON group (7.53%) (Table 1). All these results indicated that clindamycin treatment not only altered the proportion of bacteria and fungi, but also weakened the trans-kingdom networks, changed the interactions between bacteria and fungi, and resulted in gut microbiome dysbiosis.
Figure 3
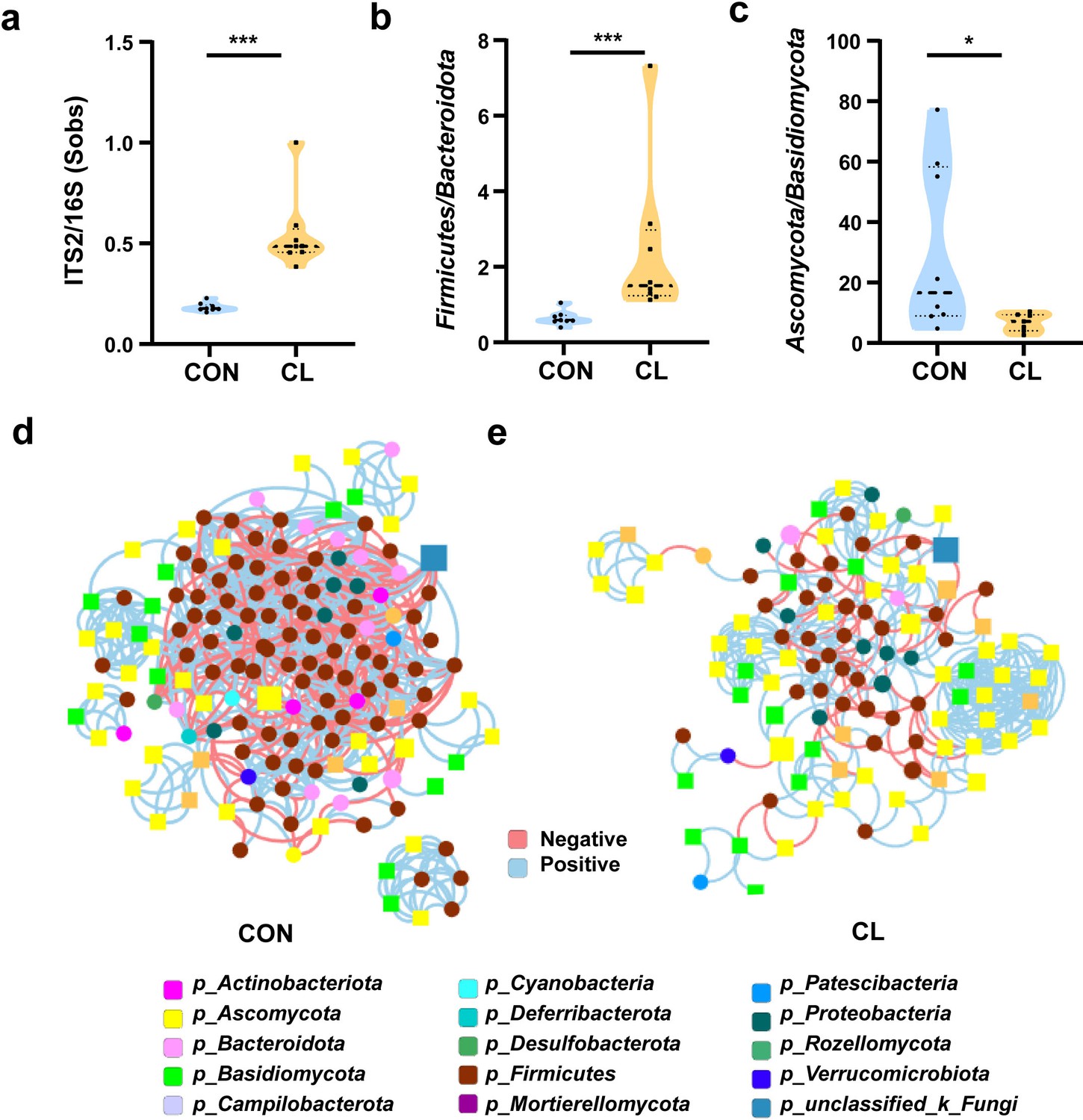
Clindamycin weakens the trans-kingdom network construction of gut bacteria and fungi.
(a) The ratio of ITS2/16S at the genus level. (b) The relative abundant ratio of Firmicutes/Bacteroidota. (c) The relative abundant ratio of Ascomycota/Basidiomycota. (d) The trans-kingdom correlation networks of the CON at the genus level. (e) The trans-kingdom correlation networks of the CL at the genus level. CL: clindamycin treatment group (n = 8), CON: control group (n = 8). *p < 0.05; ***p < 0.001.
-
Figure 3—source data 1
Effects of clindamycin on the gut bacteria and fungi microbiota in mice.
- https://cdn.elifesciences.org/articles/99282/elife-99282-fig3-data1-v1.xlsx
Table 1
Parameters of the trans-kingdom abundance correlation networks.
| CON group | CL group | |
|---|---|---|
| Nodes (n) (fungi/bacteria) | 154 (45/109) | 110 (62/48) |
| Edges (n) | 810 | 357 |
| Relative connectedness* | 5.26 | 3.25 |
| Bacteria–bacteria (%) | 78.77 | 22.69 |
| Bacteria–fungi (%) | 13.70 | 25.21 |
| Fungi–fungi (%) | 7.53 | 52.10 |
-
*
The relative connectedness of the network was calculated as the ratio of edges (the number of significant interactions) and nodes (the number of genera).
Fecal microbial transfer from clindamycin-treated mice donors to antibiotics-treated conventional mice enhances MS colonization
We performed an FMT model to further verify the relationship of gut microbiota dysbiosis with the susceptibility to MS infection in the mice’s lungs. The experimental design for the FMT is shown in Figure 4a. First, we analyzed the α-diversity of gut bacterial and fungal microbiota after FMT with the Chao1 and inverse Simpson indices. The results show that the α-diversity of gut bacterial microbiota was significantly decreased, and the α-diversity of gut fungal microbiota was significantly increased in the CL-FMT group compared with that in the CON-FMT group (Figure 4—figure supplement 1, Figure 4—figure supplement 1—source data 1). Then we performed PCoA analysis to distinguish the gut microbiome alteration between CL- and CON-recipient mice. The results show a significant separation of gut bacterial and fungal microbiota between CT- and AD-recipient samples (p = 0.007 and p = 0.042, Figure 4b). The fungal-to-bacterial species ratio was significantly increased in the CL- vs the CON-recipient groups (p < 0.05, Figure 4c, Figure 4—source data 1). To calculate the changes of the dominant phyla in the gut microbiome, the results show that Firmicutes/Bacteroidota in the CL-FMT group was decreased, and Ascomycota/Basidiomycota was no obvious different compared with that of the CON-FMT group (Figure 4d, e, Figure 4—source data 1). The trans-kingdom network analyses between bacteria and fungi showed the complexity of the microbiome network was significantly reduced and the interactions between bacteria and fungi were also weakened in the CL-FMT vs the CON-FMT groups (Figure 4f). The above results showed that the trends of the gut microbiome in recipient mice were consistent with those in the donor mice. Then, the Venn diagram was used to assess the gut microbiome profile of FMT mice at the genus level. The results showed that 85.11% (40/47) of bacterial genera and 52.38% (33/63) of fungi genera present in the CL inocula were successfully transferred to the CL-recipient mice, and 91.45% (107/117) of bacteria genera and 56.36% (31/55) of fungi genera in the CON inocula were also successfully transferred to the CON-recipient mice, respectively (Figure 4g). These results indicated that the FMT model was successfully established, and the CL-recipient mice showed similar characteristics of gut microbiota dysbiosis with the CL group.
Figure 4 with 2 supplements see all
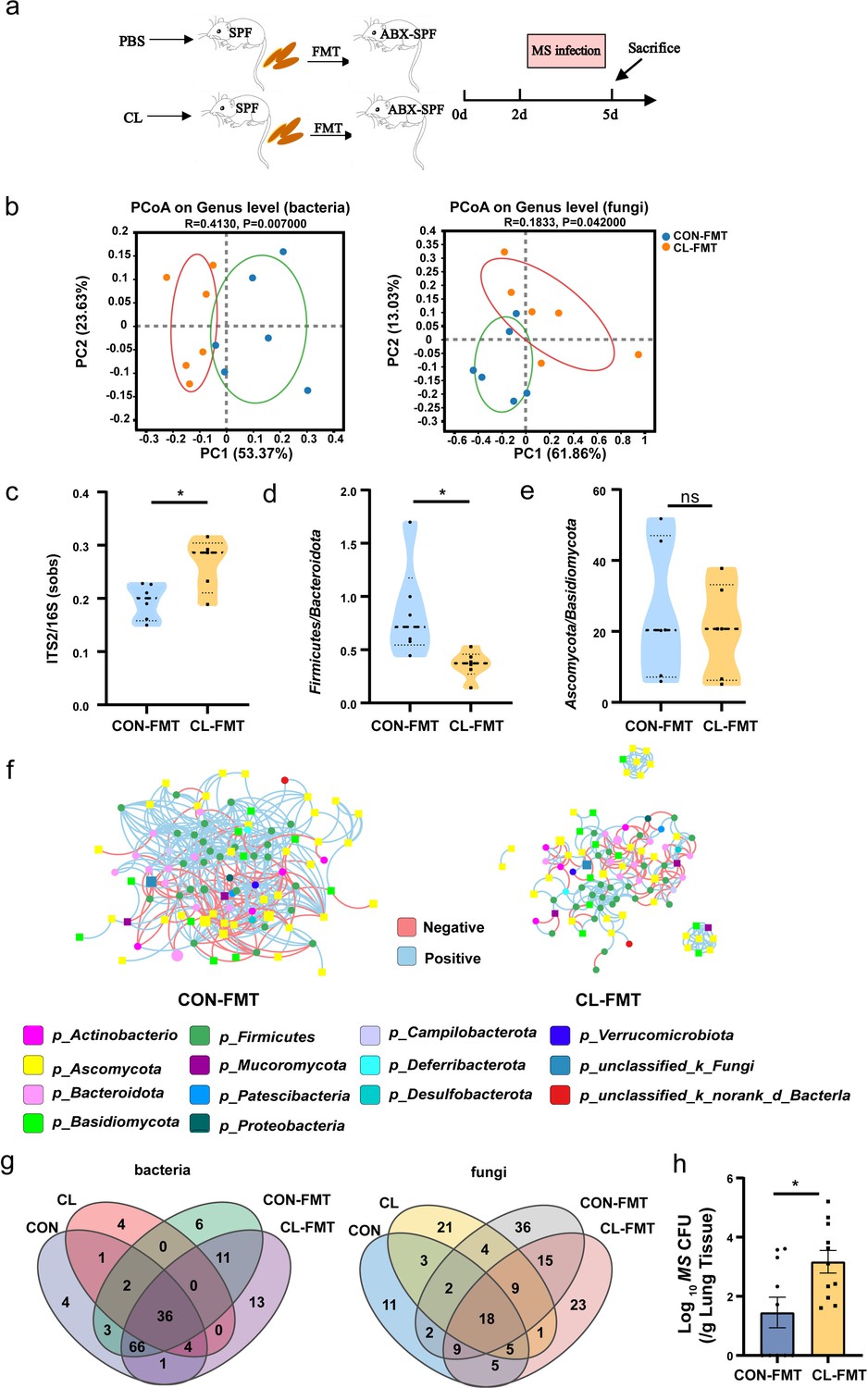
Gut microbiota dysbiosis enhances the susceptibility of MS pulmonary colonization in mice.
(a) The experimental procedures of the feces microbiota transplantation. (b) The principal coordinate analysis (PCoA) of gut microbiota based on weighted Unifrac distance. (c) The ITS2/16S diversity ratio at the genus level. (d) The relative abundant ratio of Firmicutes/Bacteroidota. (e) The relative abundant ratio of Ascomycota/Basidiomycota. (f) The trans-kingdom correlation networks of CON-FMT and CL-FMT groups at the genus level. (g) The Venn diagram of gut bacteria and gut fungi in different groups at the genus level. (h) The load of MS in the lungs of mice after infection at 72 hr. FMT: fecal microbiota transplantation, CON-FMT: the fecal microbiota of the control group was transplanted (n = 6), CL-FMT: the fecal microbiota of the clindamycin treatment group was transplanted (n = 6). MS, Mycobacterium smegmatis. *p < 0.05.
-
Figure 4—source data 1
Effects of FMT on the gut bacteria and fungi microbiota in mice.
- https://cdn.elifesciences.org/articles/99282/elife-99282-fig4-data1-v1.xlsx
Then, we assessed the susceptibility of MS in the lungs of FMT mice, the results showed that the burden of MS in the lungs of CL-recipient mice was significantly rise than that in the CON-recipient mice after 72 hr of MS infection (Figure 4h, Figure 4—source data 1). However, the size of the cecum showed no significant differences between the two groups (Figure 4—figure supplement 2, Figure 4—figure supplement 1—source data 1). The level of iFABP and LPS also significantly increased in the CL-recipient mice compared with the CON-recipient mice after FMT (Figure 4—figure supplement 2, Figure 4—figure supplement 1—source data 1). The pathological sections of intestinal tissue showed the intestinal epithelial tissues of mice have no significant damage after FMT (Figure 4—figure supplement 2, Figure 4—figure supplement 1—source data 1). Altogether, our data suggested that gut microbiota dysbiosis increases intestinal permeability and enhances the susceptibility of MS colonization in the lungs of mice.
The gut microbiota dysbiosis altered the lung transcriptome and increased Nos2 expression
To further explore the potential mechanisms by which intestinal microbiota dysbiosis affects MS colonization in the lungs of mice, we performed a transcriptome analysis of the mice’s lung tissue. The results showed that there were 1191 upregulated differentially expressed genes (DEGs) and 1013 downregulated DEGs in the CL vs the CON groups (false discovery rate [FDR] <0.05, and fold change [FC] >1, Figure 5—figure supplement 1). Compared with the CON-FMT group, 274 DEGs were upregulated and 32 DEGs were downregulated in the CL-FMT group (Figure 5—figure supplement 1). Then, we screened the overlapping DEGs between the two comparison sets including CL vs CON and CL-MFT vs CL-FMT. The Venn diagram showed that 93 upregulating DEGs and 5 downregulating DEGs were commonly expressed among these groups (Figure 5a). Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to clarify the function of these 98 DEGs. The top 30 biological processes enriched by GO showed that CL and CL-FMT groups mainly affected the immune response and inflammatory response, including ‘defense response’, ‘response to bacterium’, ‘cellular response to interleukin-1’, and ‘cellular response to lipopolysaccharide’ (Figure 5b). The enriched molecular functions of these DEGs were some cytokine and protease involving the immune defense response, including ‘cytokine activity’, ‘chemokine activity’, ‘nitric-oxide synthase binding’, and ‘CXCR chemokine receptor binding’ (Figure 5b).
Figure 5 with 2 supplements see all
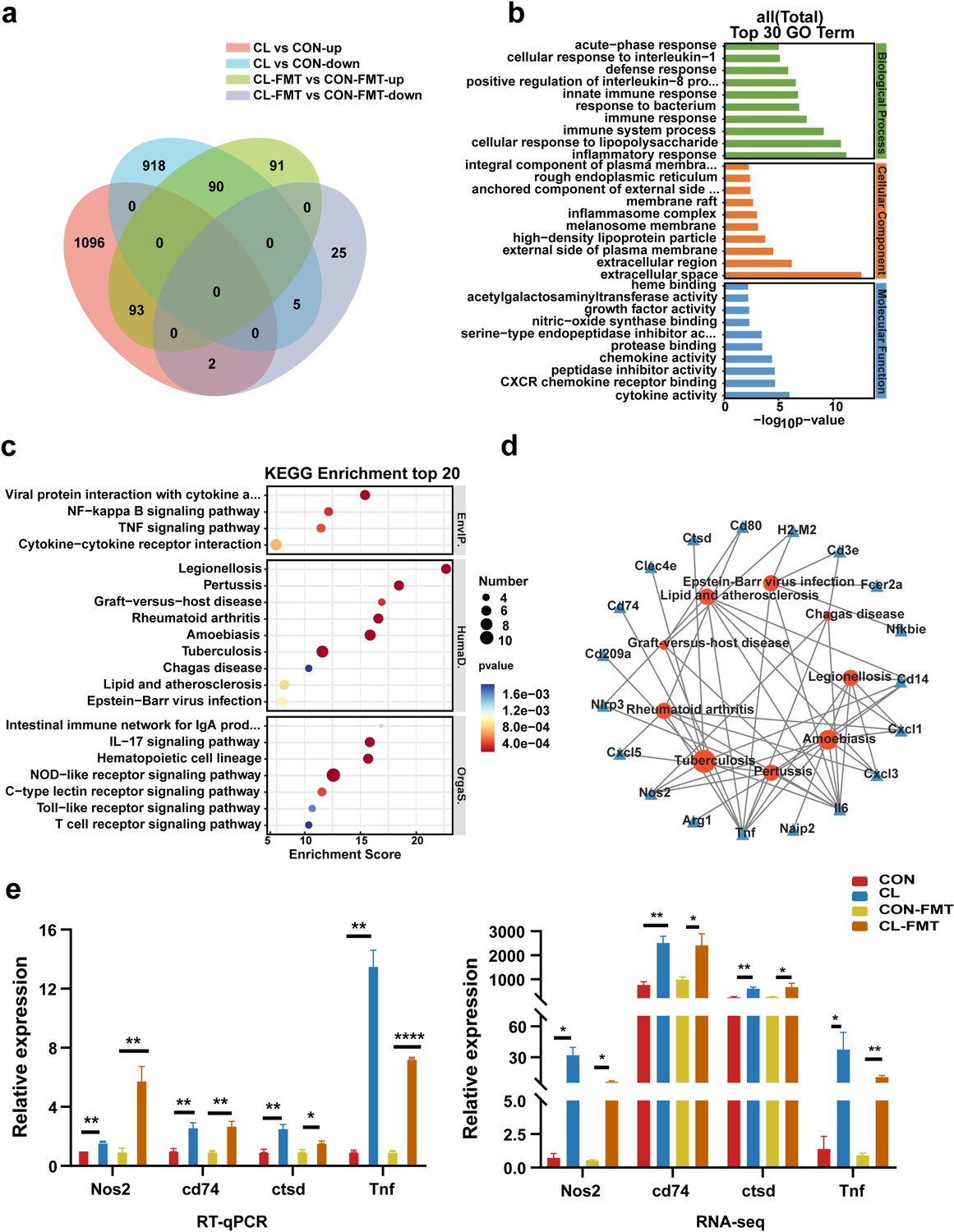
Gut microbiota dysbiosis altered the lung transcriptome of mice.
(a) The Venn diagram of DEGs in the different groups. (b) The top 30 Gene Ontology (GO) enrichment analyses of DEGs. (c) The top 20 Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEGs. (d) Interaction network analysis of selected DEGs and significant KEGG pathways related to human diseases. (e) The expression levels of Nos2, Ctsd, Cd74, and Tnf with RT-qPCR and RNA-seq. DEGs: differentially expressed genes, CL: clindamycin treatment group, CON: control group; CON-FMT: the fecal microbiota of the control group was transplanted, CL-FMT: the fecal microbiota of the clindamycin treatment group was transplanted. *p < 0.05, **p < 0.01, **** p<0.0001.
-
Figure 5—source data 1
The results of RT-qPCR and RNA-seq of Nos2, Ctsd, Cd74, and Tnf.
- https://cdn.elifesciences.org/articles/99282/elife-99282-fig5-data1-v1.xlsx
The top 20 KEGG pathways enrichment analyses at classification level 1 indicated that these DEGs are mainly involved in environmental information processing, human diseases, and organismal systems (Figure 5c and Supplementary file 2). At classification level 2 of the KEGG analysis, we found six pathways relating to infectious disease, and seven pathways involving the immune system among the enrichment top 20 pathways (Figure 5c and Supplementary file 2). Interestingly, among the six pathways of infectious disease, there were three pathways involving bacterial infectious disease, including TB, legionellosis, and pertussis (Figure 5c). In addition, the pathway of ‘graft-versus-host disease’ is also enriched in these DEGs (Figure 5c).
For the future screening of the potential regulatory genes correlated with TB, nine KEGG pathways related to human disease were selected for interaction network analysis with DEGs. The results showed that eight genes were strongly associated with TB (Figure 5d). Among those genes, seven genes including Nos2, Cd14, Tnf, Cd74, Clec4e, Ctsd, and Il6 are upregulating expression and the Cd209a gene is the downregulating expression in the CL and CL-FMT groups (Figure 5—figure supplement 2, Figure 5—figure supplement 1—source data 1). It is worth noting that the Nos2 gene was an enriched expression in both GO and KEGG analyses. Then, we performed an RT-qPCR to assess the validity of the transcriptome. The results revealed that the genes of Nos2, Ctsd, Cd74, and Tnf were increased in CL-FMT and CL groups compared with that in control groups (Figure 5e, Figure 5—source data 1), which indicated that the RNA-seq data is reliable. In summary, our data revealed that gut microbiota dysbiosis significantly altered the transcriptomic profiling in the lung tissue of mice, and increased the expression of Nos2 genes.
Nos2 regulates the expression of nitric oxide, reactive oxygen species, and Defb1
Based on the above transcriptomic results, we speculate that the upregulating expression of Nos2 induced by gut microbiota dysbiosis may play an important role in MS pulmonary colonization in mice. Hence, an overexpression Nos2 vector, Nos2-pcDNA3.1, was constructed and transfected into A549 cells. Then the cells were infected with MS to verify the effect of Nos2 overexpression on MS colonization ability. The results showed that the expression level of Nos2 was significantly raised in A549 cells after transfection with the Nos2-pcDNA3.1 plasmid (Figure 6a, Figure 6—source data 1). MS colonization density was significantly increased in A549 cells with Nos2-pcDNA3.1 plasmid after infection MS 6, 12, and 24 hr (Figure 6b, Figure 6—source data 1), which suggested that Nos2 overexpression increases the infection susceptibility of MS to A549 cells.
Figure 6
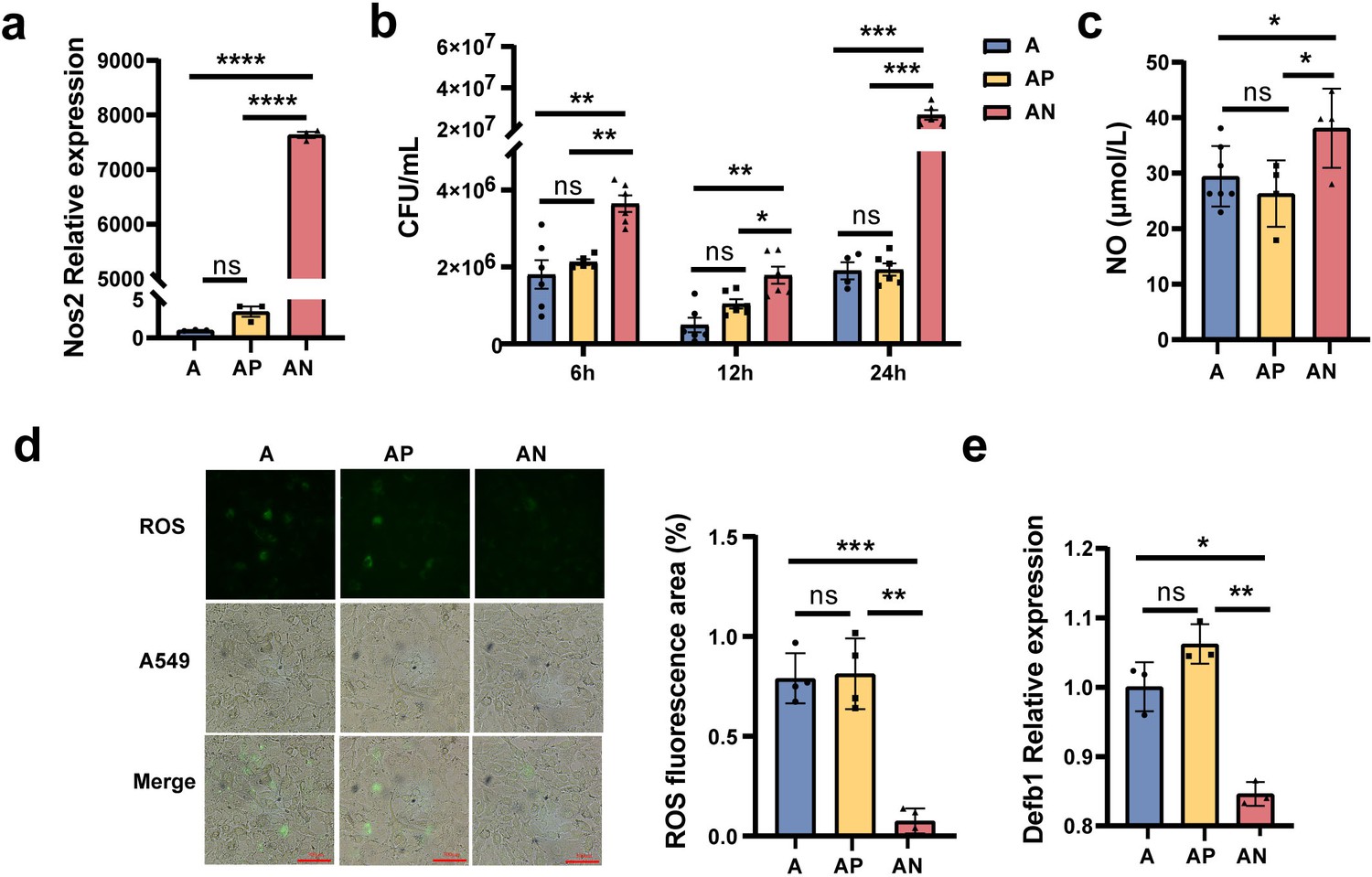
Nos2 regulates the expression of nitric oxide (NO), reactive oxygen species (ROS), and Defb1.
(a) The expression levels of the Nos2 in the A549 cells. (b) The load of Mycobacterium smegmatis (MS) in the A549 cells at different times. (c) The NO concentration in the A549 cells. (d) The concentration of ROS in the A549 cells. (e) The expression levels of Defb1 in A549. A: A549 cells control, AP: transfected A549 cells with blank pcDNA3.1plasmid, AN: transfected A549 cells with Nos2-pcDNA3.1 plasmid. *p < 0.05, **p < 0.01, ***p < 0.001, "****" p<0.0001.
-
Figure 6—source data 1
Effect of Nos2 on the expression of nitric oxide (NO), reactive oxygen species (ROS), Defb1 and the colonization of MS in cells.
- https://cdn.elifesciences.org/articles/99282/elife-99282-fig6-data1-v1.xlsx
Subsequently, we explored the potential molecular mechanism by which Nos2 overexpression increased MS colonization ability. Nos2 is a key enzyme required for nitric oxide (NO) synthesis, so we detected the level of NO in A549 cells. The results showed that the NO level significantly increased in the A549 cells with transfection of the Nos2-pcDNA3.1 plasmid (Figure 6c, Figure 6—source data 1). Reactive oxygen species (ROS) play an important role in pathogens invade and colonize host organs (André et al., 2022). To explore whether Nos2 regulates ROS production in A549 cells, ROS concentration in cells was detected by the fluorescent probe 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA). As fluorescence microscopy shown in Figure 6d, ROS concentration in A549 cells transfected with Nos2-pcDNA3.1 was significantly lower than that in the control A549 and A549 cells transfected with pcDNA3.1 empty vector (Figure 6d, Figure 6—source data 1). In addition, we also detected the expression of human β-defensin-1 (Defb1), an important antimicrobial peptide (Zhu et al., 2003), by RT-qPCR. The results showed that Nos2 overexpression significantly reduced the level of Defb1 in A549 cells (Figure 6e, Figure 6—source data 1). Taken together, our results revealed that Nos2 overexpression disrupts the intracellular antimicrobial and anti-inflammatory environment by increasing the concentration of NO, decreasing the levels of ROS and Defb1, and resulting in the enhancement of MS pulmonary colonization capacity in mice.
Discussion
TB remains a major health challenge globally. How to effectively limit or reduce Mtb colonization in the host is a potential strategy to prevent this disease transmission and development. Recent studies have shown that the gut microbiome can affect distant organs via the ‘gut–lung axis’, and gut microbiota dysbiosis is a potential factor inducing respiratory diseases including TB (Waldman and Balskus, 2018; Ma et al., 2022). Previous studies have shown that the gut microbiota was significantly different in TB patients compared with healthy humans and revealed gut microbiota dysbiosis, especially Bacteroidota, and Firmicutes, is strongly associated with the development of TB (Maji et al., 2018; Luo et al., 2017). However, how the gut microbiota affects TB development by the ‘gut–lung axis’ remains unclear. In the present study, we established a mouse model of gut microbiota dysbiosis and an FMT model using clindamycin treatment and revealed the potential mechanisms of intestinal microbiota dysbiosis promoting MS pulmonary colonization in the mouse by upregulating Nos2 gene-associated pathways.
Antibiotics can induce intestinal microbiota dysbiosis, which in turn affects host immunity and leads to increased susceptibility and deterioration of a variety of diseases (Fishbein et al., 2023). Previous research has verified that gut Bacteroidota dysbiosis is strongly associated with the development of TB (Du et al., 2022; Huang et al., 2019; Yunusbaeva et al., 2024). Hence, we select clindamycin, an antibiotic that selectively disrupts anaerobic Bacteroidota (You et al., 2019), to treat the mice in this study. Our results show that the abundance of Bacteroidota was significantly reduced and the abundance of Firmicutes was increased in the CL group compared with the CON group. This result is consistent with the previous study which reported that CL is effective in clearing Bacteroidota (You et al., 2019). Bacteroidota is a consortium of many commensal bacterial species responsible for major fermentation processes, glycolipid production, and promoting systemic Th1 immune responses (Mazmanian et al., 2005).
The fungi are also important components of the gut microbiome. The gut fungi mycobiota dysbiosis is related to many diseases (Azzam et al., 2020). In the present study, we found that the gut fungi mycobiota balance of mice was disrupted by CL, especially, Aspergillus and Cladosporium significant increase in the CL vs the CON groups. Aspergillus and Cladosporium are opportunistic pathogens that usually cause lung infections in immunocompromised patients (Wheeler et al., 2017; Sandoval-Denis et al., 2015). Bacteria and fungi harbor together in the gastrointestinal tract and occupy the same ecological niche. They interact with each other and develop complex ecological networks through these interactions. Homeostasis of microbial ecological networks limits pathogen invasion and infection (Lozupone et al., 2012). The ratio of ITS2/16S rRNA can reflect the diversity and composition structural alterations of the fungi–bacterial microbiota (Lemoinne et al., 2020). In general, antibiotics are directed against bacteria and do not affect fungi. However, our data revealed that CL treatment significantly altered the diversity and composition structure of the fungi–bacterial microbiota, and reduced the trans-kingdom network complexity and interaction between bacteria and fungi. These suggested that the alterations of gut bacteria can affect the composition and diversity of fungi. Previous documents had also reported that any changes in gut bacteria would inadvertently cause alterations in the gut fungal community, and targeted fungal interventions would also cause changes in the gut bacterial community (Haak et al., 2021; Sovran et al., 2018; Charlet et al., 2018; van Tilburg Bernardes et al., 2020). Hence, gut microbiota should be taken as a whole rather than a single genus to explain the relationship between gut microbiota and associated diseases.
Since most of the microbes in the gut are non-culturable, the FMT is considered to be an ideal model to study the function of gut microbiota. The success or not of FMT is influenced by many factors, such as host intestinal microbiota, immunity, and genetic factors (Porcari et al., 2023). During the FMT, not all microbiota in the donor feces have the same colonization ability in the receptors. Some research has revealed that the colonization success rate of Bacteroidetes is higher than that of Firmicutes (Hintze et al., 2014). In the present study, we found that the ratio of Firmicutes/Bacteroidetes in the CL treatment group increased compared to the control group (Figure 3b). However, after FMT, the ratio of Firmicutes/Bacteroidetes decreased in the CL-FMT group vs the CON-FMT group (Figure 4d). We speculated that the possible reason for this difference was that the colonization of Firmicutes decreased in the CL-FMT receptor group after transplantation. In contrast, the colonization of Bacteroides increased, resulting in a decrease in the proportion of Firmicutes/Bacteroides in the CL-FMT group. However, we considered the gut microbiota as a whole in the analysis of the experimental results. After FMT, we found that 85.11% of bacterial genera and 52.38% of fungi genera present in the CL inocula were successfully transferred to the CL-recipient mice, and 91.45% of bacteria genera and 56.36% of fungi genera in the CON inocula were also successfully transferred to the CON-recipient mice, respectively (Figure 4g). The trans-kingdom network analyses between bacteria and fungi showed that the trends of the gut microbiome in recipient mice were consistent with those in the donor mice. Therefore, the inconsistent results between Figures 3b and 4d will not affect this study’s findings, and the FMT model established in this study is successful.
The FMT results revealed that gut commensal bacteria dysbiosis can promote MS colonization in the lungs of mice, but the underlying mechanisms remain not fully understood. Recent studies have suggested that changes in the ‘gut–lung axis’ are a contributing factor in the pathogenesis and clinical manifestations of Mtb infection (Yang et al., 2022; Alvarado-Peña et al., 2023.) What is the substance mediating the interaction between the lung and the gut? There have been a variety of results in the different studies. Yang et al. have found that the gut microbiota via modulation of lncRNA-mediated protective immunity against Mtb, and they found Bacteroides fragilis directly upregulated expression of lncRNA and promoted anti-TB immunity (Yang et al., 2022). However, another clinical study on TB found that the expression level of iNOS in the plasma of new-onset pulmonary TB patients was significantly higher than that of healthy humans (Chinta et al., 2016). In the present study, we found that Nos2 expression significantly rises in the CL and CL-FMT groups. Hence, we speculated that the gut microbiota may increase MS colonization in mouse lungs by upregulation of Nos2 gene expression and associated pathways. Lung epithelial cells, as first responders during Mtb infection, have been shown to play an important role in TB pathogenesis (Scordo et al., 2016; Ryndak and Laal, 2019). Our results also confirmed that overexpression of Nos2 in A549 cells (a human alveolar epithelial cell) can enhance MS colonization in these cells.
Nos2, also termed iNOS, is a homodimeric enzyme that is induced by immune stimulation in an independent of intracellular Ca2+ manner and plays an important role in infection, inflammation, immune regulation, and the control of intracellular bacterial pathogen infection (Chakravortty and Hensel, 2003). Compared with Nos1 and Nos3, Nos2 has the highest efficiency for production NO (Fischer et al., 2002). NO has direct antibacterial effects and immune-modulating function to intracellular pathogens and was considered a key molecule in controlling pathogens infections (Chakravortty and Hensel, 2003). It has been verified that reactive nitrogen intermediates produced by murine cells could inhibit Mtb infection (Scanga et al., 2001). The use of Nos2 knock-out mice and NO synthase inhibitors is impaired in the control of Mtb growth (MacMicking et al., 1997; Chan et al., 1995). However, our data found that the high concentration of NO increased the colonization of MS in A549 cells. This result seems to contradict that NO has antibacterial activity. Some studies have also shown that NO can promote bacterial growth. Gusarov et al. reported that NO increases the resistance of bacteria to a broad spectrum of antibiotics, enabling the bacteria to survive and share habitats with antibiotic-producing microorganisms (Gusarov et al., 2009). Cole et al. showed that host cell-derived NO promoted the escape of listerolysin-dependent bacteria from phagocytic vacuoles into the cytoplasm by inhibiting the proton-pumping activity of V-ATPase and delaying phagolysosome fusion (Cole et al., 2012; Bogdan, 2012). Another study has shown that the maximal level of NO produced by human macrophages was not bactericidal or bacteriostatic to Mtb, and the number of viable mycobacteria was increased in macrophages that produced NO, and this is correlated with the expression of nitrate reductase (Jung et al., 2013). Therefore, we speculate that intracellular NO synthesized by Nos2 has dual roles, including bactericidal at high concentrations and promoting bacterial growth at low concentrations.
ROS are commonly present in various habitats occupied by living organisms and the production of ROS appears as a very ancient host strategy for coping with pathogens (Rhen, 2019; Roy et al., 2017). ROS has shown potent antimicrobial activity against many pathogens including bacteria, fungi, and viruses. The accumulation of ROS is required for killing or inhibiting bacteria (Hong et al., 2019). NO interferes with antimicrobial killing by suppressing ROS accumulation (Gusarov et al., 2009). Hence, we assess the level of ROS in the overexpression of Nos2 plasmids A549 cells. The results show that the ROS concentration in A549 cells transfected with Nos2-pcDNA3.1 was significantly lower than that in the control A549 and A549 cells transfected with pcDNA3.1 empty vector. This suggested that the promotion of bacterial proliferation by Nos2 is dependent on lower ROS levels. The crosstalk between Nos2 and ROS has been investigated in other research. Zheng et al. reported that the lower ROS level induced by Nos2a significantly inhibited E. piscicida proliferation and infection in zebrafish (Zheng et al., 2023). Another study has also shown that Nos2-produced ROS have an important role in maintaining homeostasis of the gut microbiota and defense against bacterial translocation (Matziouridou et al., 2018). Therefore, regulating the Nos2–ROS pathway may enhance the host’s defense ability against pathogens.
β-defensin-1 is an antimicrobial peptide, which is mainly produced by various epithelial cells and is an important part of the innate immune response (Flores Saiffe Farías et al., 2015). Defb1 is also expressed in the respiratory epithelium cells and protects the airways from respiratory pathogens (Diamond et al., 2008). Previous research has found that Defb1 has 98% killing activity against active Mtb H37Rv (Álvarez et al., 2018), and also has an effective antibacterial effect against dormant mycobacteria (Sharma et al., 2017). Consistent with the above results, our data revealed that there was a strong negative correlation between Defb1 gene expression and MS colonization in A549 cells. The present study was mainly performed at the gene level and lacked verification at the protein level. We will verify the role of the Nos2–Defb1 pathway on MS colonization at the protein levels in future studies.
Altogether, the crosstalk between the gut microbiome and the lungs through the ‘gut–lung axis’ is complex and may involve multiple mechanisms, including immune response, metabolic disorders, cytokines, inflammation, etc. However, in the present study, we found that gut microbiome dysbiosis induced by clindamycin disturbs the gut equilibrium between bacteria and fungi, alters the lung transcriptome, and increases Nos2 expression. Then, the microbiota dysbiosis could enhance the pulmonary colonization of MS in mice by regulating the Nos2–NO, Nos2–ROS, and Nos2–Defb1 pathways (Figure 7). In the future, we need more experiments to explore the relationship between the intestinal microbiome and the Nos2-associated pathways in the lung and to further explore new targets for the prevention and treatment of TB.
Figure 7

Mechanisms of the intestinal microbiome dysbiosis effect on the colonization of MS in the mouse lungs.
Gut microbiome dysbiosis and gut permeability-increasing disrupt the lung transcriptome, and increase Nos2 expression through the ‘gut–lung axis’. Nos2 high expression weakens the intracellular antimicrobial and anti-inflammatory environment by increasing the concentration of NO, decreasing the levels of ROS and Defb1 in the cells, and promoting MS colonization in the lungs of mice. MS: Mycobacterium smegmatis.
Study limitations
This study has some limitations that need to be mentioned. First, due to the lack of experimental conditions in our laboratory that meet biosafety standards, we did not choose a wild-type Mtb strain to develop a mouse infection model, which may lead to the failure of the establishment of TB granuloma and other TB pathology icons and these also weaken the clinical significance of this study. Second, alveolar epithelial cells are one of the early contacting cells in Mtb infection, we only select alveolar epithelial cells (A549) to explore the colonization mechanism of intestinal microbiota affecting MS in vivo. This study lacks the choice of Mtb infection target cells–alveolar macrophages as the research object. Hence, future studies need to choose wild-type Mtb to establish animal infection models and alveolar macrophages for in vitro experiments to explore the regulation function of Nos2 expression on NO, ROS, and Defb1, which will resolve these limitations of the study.
Conclusions
In conclusion, the present study reveals that (1) clindamycin treatment induces gut microbiome dysbiosis, disturbs the gut equilibrium between bacteria and fungi, reduces the interactions among bacterial–fungal trans-kingdom, and increases intestinal permeability. (2) The intestinal microbiome dysbiosis alters the lung transcriptome, increases Nos2 expression, and enhances MS colonization in the lungs of mice. (3) Intestinal microbiota could promote the pulmonary colonization of MS in mice by regulating the expression of NO, ROS, and Defb1 through Nos2-associated pathways, and changing the intracellular antimicrobial and anti-inflammatory environment. Hence, regulating the gut microbiome balance may be a potential strategy for reducing the risks of Mtb infections and transmission.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Mycobacterium smegmatis) | Mycobacterium smegmatis; MS | ATCC | ATCC 19420 Cat#700084 | |
| Cell line (Homo sapiens) | Human alveolar basal epithelial cell (A549) | Servieebio Biochemical Technology Co., Ltd | RRID:CVCL_0023 | |
| Transfected construct (mouse) | pcDNA3.1-Nos2 | Youbio Biochemical Technology Co., Ltd | NCBI GenBank accession No: NM_010927 | |
| Commercial assay or kit | All-in-One Script RT premix | Kryptoner Mei Biotechnology Co., Ltd | #MR0502-2 | |
| Commercial assay or kit | TB Green Premix Ex Taq II | TaKaRa Biotechnology Co., Ltd | #RR820A | |
| Commercial assay or kit | Nitric Oxide (NO) assay kit | Nanjing Jiancheng Biotechnology Research Institute Co., Ltd | #A012-1 | |
| Commercial assay or kit | Reactive oxygen species (ROS) detection kit | Beyotime Biotechnology Co., Ltd | #S0033S | |
| Commercial assay or kit | The commercial ELISA kits of iFABP | Shanghai Enzyme-linked Biotechnology Co., Ltd | #TW9968 | |
| Commercial assay or kit | The commercial ELISA kits of LPS | Shanghai Enzyme-linked Biotechnology Co., Ltd | #TW12543 | |
| Chemical compound, drug | Clindamycin | Dalian Meilun Biotechnology Co., Ltd | CAS: 18323-44-9 | 1 mg/ml |
| Chemical compound, drug | Ampicillin | Shanghai Maclin Biochemical Technology Co., Ltd | CAS: 69-52-3 | 1 mg/ml |
| Chemical compound, drug | Metronidazole | Shanghai Maclin Biochemical Technology Co., Ltd | CAS: 443-48-1 | 1 mg/ml |
| Chemical compound, drug | Neomycin | Shanghai Maclin Biochemical Technology Co., Ltd | CAS: 1405-10-3 | 1 mg/ml |
| Chemical compound, drug | Vancomycin | Shanghai Maclin Biochemical Technology Co., Ltd | CAS: 1404-90-6 | 0.05 mg/ml |
| Other | Middlebrook 7H9/7H10 | Qingdao Haibo Biotechnology Co., Ltd | #HB6270/HB9128 | Mycobacterium culture media |
Mouse husbandry and antibiotic treatment
Request a detailed protocolSpecific pathogen-free (SPF) C57BL/6 mice (6–8 weeks) were purchased from SiBeiFu (Beijing) Biotechnology Co., Ltd (No. SCXK (Beijing) 2019-0010). The mice were reared in an animal laboratory with a temperature of 22 ± 2°C and a relative humidity of 50 ± 5%, for 1 week before the model started. After adaptive feeding, mice were randomly divided into two groups, including a control group (CON) and a clindamycin group (CL), each group had 10 mice. During the experimental procedure, the CON group was fed with phosphate buffer saline (PBS). Whereas the CL group received 10 mg/kg clindamycin by oral gavage once a day for 14 days. Each oral gavage treatment did not exceed 200 μl, and treatment was stopped 2 days before the mycobacteria infection. All animal experiments were conducted according to the Declaration of Helsinki and were approved by the Animal Care and Use Ethics Committee of Xinxiang Medical University (No. EC-023-098).
Bacterial strains and infection
Request a detailed protocolDue to the strong infectivity of Mtb, MS, a model strain for the study of TB, was used in the present experiments. MS was grown in Middlebrook 7H9 liquid medium supplemented with glycerin 0.5% and Tween 80 0.05%. After 18–24 hr of cultivation in a shaking incubator at 37°C, MS were centrifuged at 5000 rpm for 5 min, and the resulting pellet was suspended in sterile PBS to the concentration of 2 × 107 CFU per ml. Figure 1a shows the experiment design procedure. For infection, mice were anesthetized by injection of 4% chloral hydrate (7 μl/g) and infected intranasally with 107 CFU of MS. The mice were euthanized after infection 72 hr, and the left lung of each mouse was extracted and homogenized in PBS with 0.1% Tween 20. Tenfold serial dilutions were made in PBS with 0.1% Tween 20 and plated on Middlebrook 7H10 Agar plates, and colonies of MS were counted after 5 days of incubation at 37°C.
Fecal sample DNA extraction, 16S rRNA, and ITS sequencing
Request a detailed protocolFresh stool samples from each mouse were collected aseptically and the total genomic DNA from fecal samples was extracted using the Quick-DNA Kit for feces (QIAGEN, Germany) according to the manufacturer’s instructions. The quality and concentration of DNA were determined using NanoDrop ND-2000 spectrophotometer (Thermo Scientific Inc, USA) and 1.0% agarose gel electrophoresis. Each DNA sample amplifies the hypervariable V3–V4 regions of the bacterial 16S rRNA genes using the primers 338F (5′-ACTCCTACGGAGGCAGCAGCAGCA-3′) and 806R (5′-GGACTACHVGGTWTAAT-3′). The ITS2 regions of the fungi were amplified using primers ITS3F (5′-GCATCGATGAAGAACGCAGC-3′) and ITS4R (5′-TCCTCCGCTTATTGATATGC-3′). The PCR products were purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using Quantus Fluorometer (Promega, USA). Purified amplicons were pooled in equimolar amounts, and paired-end sequenced on an Illumina MiSeq PE300 platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd (Shanghai, China).
Serum iFABP and LPS measurement
Request a detailed protocolThe blood samples of all mice were collected via Eyeball, and the serum was separated by centrifugation at 1000 × g for 20 min. Then, ELISA technology was used to measure the iFABP and LPS, according to the manufacturer’s instructions. The commercial ELISA kits were obtained from Shanghai Enzyme-linked Biotechnology Co., Ltd (Shanghai, China) including iFABP (catalog number TW9968) and LPS (catalog number TW12543).
Fecal microbial transplantation
Request a detailed protocolTo establish the gut microbiota depletion mouse model, SPF mice were treated with a cocktail of antibiotics (1 mg/ml ampicillin, 1 mg/ml metronidazole, 1 mg/ml neomycin, and 0.5 mg/ml vancomycin) by oral gavage daily for 14 days. All mice were randomly separated into two groups, the CON-recipient group (n = 10) and the CL-recipient group (n = 10). Each group of mice was randomly housed in two cages with five mice in each cage. The fecal microbial transplantation procedure is shown in Figure 4a. Fecal samples from mice of the CON and CL groups were collected in sterile containers. Then 1 g fecal sample was suspended in 5 ml sterile PBS, followed by the vortexes, sedimentation, and filtrate with a 100-μm cell strainer. The suspension of the same group was mixed as microbiota donors and immediately administered to the mice by oral gavage. 200 μl of the supernatant containing fecal microbiota from either CON or CL donors was transferred to microbiota-depleted mice by oral gavage every day, for 14 days. All recipient mice were infected intranasally with 107 CFU MS after stopping FMT 2 days, and 3 days later of the infection, all mice were sacrificed, and the colonic contents and lung tissue samples were collected for further analysis.
Lung histological assessment
Request a detailed protocolAfter the MS-infected mice were sacrificed, the lung tissues were perfused with sterile PBS and fixed in 4% paraformaldehyde for 3 days, followed by paraffin embedding. For histopathological analysis, 5 μm sections were cut and stained using a standard H&E protocol. Motic EasyScan whole-slide scanner was used for scanning histological sections and images were analyzed using Matic DSAaaistant Lite.
Lung tissue RNA extraction and RNA sequencing
Request a detailed protocolThe lung tissue from each mouse was separated aseptically and quickly stored in liquid nitrogen for subsequent RNA extraction. Total RNA was extracted from lung tissue using TRIzol reagent (Dingguo Changsheng Biotechnology Co., Ltd, Beijing, China) according to the manufacturer’s protocol. RNA quantity and quality were determined using the NanoDrop 2000 Spectrophotometer (Thermo Scientific, USA). The Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) was used to assess RNA integrity. RNA-seq libraries were prepared using VAHTS Universal V6 RNA-seq Library Prep Kit following the manufacturer’s recommendations. Then the paired-end RNA-seq libraries were sequenced with the Illumina HiSeq X Ten platform (2 × 150 bp read length) by OE Biotech Co., Ltd (Shanghai, China). After the quality control, clean reads performed bioinformatics analysis.
Quantification of gene expression using RT-qPCR
Request a detailed protocolTotal RNA was extracted from mouse lungs or A549 cells using the Trigol (Dingguo Changsheng Biotechnology Co., Ltd, Beijing, China) according to the manufacturer’s protocol, and was reversed transcribed using the kit from All-in-One Script RTpremix (Kryptoner Mei Co., Ltd, Zhengzhou, China). Then RT-qPCR was performed with TB Green Premix Ex Taq II (TaKaRa Biotechnology, China) to evaluate the amount of mRNA expression according to the manufacturer’s recommendations. Subsequently, PCR products were detected on a sequence detection system. The primer sequences of RT-qPCR are listed in Supplementary file 2 in this study. The relative gene expression levels were calculated using the 2−ΔΔCt method.
Construction of Nos2 overexpression plasmid and transfection into A549 cells
Request a detailed protocolHuman alveolar basal epithelial cell lines (A549) were purchased from Servieebio Biochemical Technology Co., Ltd (Wuhan), and cell lines have been authenticated with STR Short Tandem Repeat (STR) profiling. All the cell culture supernatants were found to be negative for mycoplasma contamination. To enhance Nos2 expression levels in A549 cells, the recombinant plasmid Nos2-pcDNA3.1 was constructed. Based on the nucleotide sequences of Nos2 (GenBank accession No: NM_010927), the forward and reverse primers Nos2F/Nos2R (Supplementary file 2) were used for cloning the open reading frame and inserted into pcDNA3.1 expression vector. The insert orientation was confirmed by separate XhoI and BamHI digests followed by agarose gel electrophoresis. The Nos2-pcDNA3.1 plasmid was mixed with lipo 8000 (Beyotime, Shanghai, China) and then transfected into A549 cells via electroporation. Twenty-four hours post-transfection, transfection efficiency was determined using RT-qPCR to ensure Nos2 overexpression in A549 cells.
MS infects in A549 cells
Request a detailed protocolA549, a human alveolar basal epithelial cell, was used to assess the infection capacity of MS. A549 cells were inoculated into 24-well plates with a density of 2 × 105 cells per well, and were cultured in RPMI1640 medium (Gibco Laboratories, USA) supplemented with 10% fetal bovine serum (Sangon Biotech Co., Ltd, Shanghai, China) and 100 U/ml penicillin/streptomycin (Beyotime, Shanghai, China) at 37°C. Nos2-pcDNA3.1 was transfected into A549 cells when the cell density reached 70–80%. After 24 hr, the supernatant was replaced with fresh medium containing MS and cultured for 6, 12, and 24 hr. Then the wells were washed three times with PBS to remove unattached bacteria. Subsequently, A549 cells were lysed with 1 ml 0.1% Triton X-100 (Shanghai Beyotime Biotechnology Co., Ltd, Shanghai, China) per well to prepare appropriate dilutions, which were plated on MiddleBrook7H10 Agar plates cultured for standard colony counts.
Detection of NO, ROS, and Defb1
Request a detailed protocolGeneration of NO in the A549 cells was detected by Nitric Oxide (NO) assay kit (Nanjing Jiancheng Biotechnology Research Institute Co., Ltd, Nanjing, China). Briefly, Cells were seeded into 24-well plates at a density of 2 × 105 cells per well, and Nos2-pcDNA3.1 was transfected into A549 cells when the cell density reached 70–80%. After 24 hr, the cells were harvested by trypsinization and centrifuged at 1000 rpm for 10 min. Then NO concentration was detected with the spectrophotometric method according to the manufacturer’s protocols. ROS levels were measured using the DCFH-DA. Briefly, after 24 hr post-transfection of Nos2-pcDNA3.1, DCFH-DA was added to the culture medium at a final concentration of 10 μmol/ml, and incubated for 1 hr at 37°C. The cells were washed twice with PBS and then stained with 10 μmol/ml DCFH-DA at 25°C for 30 min in the dark room. Images were acquired by a confocal microscope (Nikon, Japan). For ROS quantification, A549 cells were collected, rinsed twice with PBS, and suspended in 10 μmol/ml DCFH-DA for 30 min. After incubation, fluorescence was detected at 485 nm (excitation) and 530 nm (emission) using a microplate reader. All these analyses were conducted in three replicates. The expression of Defb1 was detected using RT-qPCR, and the primer sequences of RT-qPCR are listed in Supplementary file 2.
Bioinformatics analysis
Request a detailed protocolFor microbiome analysis, the bioinformatics data were analyzed using the Majorbio Cloud platform (https://cloud.majorbio.com). The α-diversity at the genus level was assessed according to the Chao1 and inverse Simpson indices. The β-diversity was calculated by PCoA. A permutational analysis of variance was performed to assess the variation in the taxonomic structure of microbiota communities between groups. The Wilcoxon rank-sum test was used to assess the different structures of microbiota communities between groups. LEfSe analyses were performed to compare different biomarkers between groups. The trans-kingdom network figures were built using the package igraph (version 1.2.6). For transcriptomic analysis, the bioinformatics data were analyzed using the OE Cloud platform (https://www.oebiotech.com). Differential expression analysis was performed using the DESeq2 q value <0.05 and foldchange >2 was set as the threshold for significantly DEGs. Venn diagram and volcano plot of DEGs were performed to explore gene expression patterns. The functional enrichment of the above DEGs was conducted using the GO database (http://www.geneontology.org/) and the KEGG (http://www.genome.jp/kegg/).
Statistical analysis
Request a detailed protocolAll statistics were performed using GraphPad Prism 8.0. If the data followed a normal distribution, unpaired Student’s t-tests were used to compare various parameters between the two groups. If the data did not follow a normal distribution, a non-parametric Mann–Whitney U test, and Wilcoxon rank-sum test were used to compare the results. One-way analysis of variance was used for three or more groups of data. The graphs were made with GraphPad Prism 8.0 or R package (version 3.6.2). p-values of <0.05 were set as a threshold for statistical significance.
Data availability
16s RNA / ITS and transcriptome sequencing data have been deposited in NCBI under the accession codes PRJNA1091926 and PRJNA1099882. All data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for the figures and figures supplements.
-
NCBI BioProjectID PRJNA1091926. SrRNA gene sequencing and ITS gene sequencing of mice.
-
NCBI BioProjectID PRJNA1099882. RNA-Seq of mice.
References
-
The microbiome and the gut-lung axis in tuberculosis: interplay in the course of disease and treatmentFrontiers in Microbiology 14:1237998.https://doi.org/10.3389/fmicb.2023.1237998
-
Human β-defensin 1 update: potential clinical applications of the restless warriorThe International Journal of Biochemistry & Cell Biology 104:133–137.https://doi.org/10.1016/j.biocel.2018.09.007
-
The battle for oxygen during bacterial and fungal infectionsTrends in Microbiology 30:643–653.https://doi.org/10.1016/j.tim.2022.01.002
-
WHO’s global tuberculosis report 2022The Lancet Microbe 4:e20.https://doi.org/10.1016/S2666-5247(22)00359-7
-
Inducible nitric oxide synthase and control of intracellular bacterial pathogensMicrobes and Infection 5:621–627.https://doi.org/10.1016/s1286-4579(03)00096-0
-
Host defense peptides in the oral cavity and the lung: similarities and differencesJournal of Dental Research 87:915–927.https://doi.org/10.1177/154405910808701011
-
Mediators of asthma: nitric oxidePulmonary Pharmacology & Therapeutics 15:73–81.https://doi.org/10.1006/pupt.2001.0332
-
Antibiotic perturbations to the gut microbiomeNature Reviews. Microbiology 21:772–788.https://doi.org/10.1038/s41579-023-00933-y
-
Predicting functional regulatory SNPs in the human antimicrobial peptide genes DEFB1 and CAMP in tuberculosis and HIV/AIDSComputational Biology and Chemistry 59 Pt A:117–125.https://doi.org/10.1016/j.compbiolchem.2015.09.002
-
Systemic proinflammation after Mycobacterium tuberculosis infection was correlated to the gut microbiome in HIV-uninfected humansEuropean Journal of Clinical Investigation 49:e13068.https://doi.org/10.1111/eci.13068
-
Pulmonary-intestinal cross-talk in mucosal inflammatory diseaseMucosal Immunology 5:7–18.https://doi.org/10.1038/mi.2011.55
-
Alternation of gut microbiota in patients with pulmonary tuberculosisFrontiers in Physiology 8:822.https://doi.org/10.3389/fphys.2017.00822
-
Gut microbiota: a new insight into lung diseasesBiomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie 155:113810.https://doi.org/10.1016/j.biopha.2022.113810
-
The microbiome and tuberculosis: state of the art, potential applications, and defining the clinical research agendaThe Lancet. Respiratory Medicine 7:892–906.https://doi.org/10.1016/S2213-2600(18)30501-0
-
Mycobacterium tuberculosis, antimicrobials, immunity, and lung-gut microbiota crosstalk: current updates and emerging advancesAnnals of the New York Academy of Sciences 1467:21–47.https://doi.org/10.1111/nyas.14300
-
Salmonella and reactive oxygen species: a love-hate relationshipJournal of Innate Immunity 11:216–226.https://doi.org/10.1159/000496370
-
Physiological role of reactive oxygen species as promoters of natural defensesFASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology 31:3729–3745.https://doi.org/10.1096/fj.201700170R
-
Mycobacterium tuberculosis primary infection and dissemination: a critical role for alveolar epithelial cellsFrontiers in Cellular and Infection Microbiology 9:299.https://doi.org/10.3389/fcimb.2019.00299
-
Cladosporium species recovered from clinical samples in the United StatesJournal of Clinical Microbiology 53:2990–3000.https://doi.org/10.1128/JCM.01482-15
-
A murine model of tuberculosis/type 2 diabetes comorbidity for investigating the microbiome, metabolome and associated immune parametersAnimal Models and Experimental Medicine 4:181–188.https://doi.org/10.1002/ame2.12159
-
Activity of human beta defensin-1 and its motif against active and dormant Mycobacterium tuberculosisApplied Microbiology and Biotechnology 101:7239–7248.https://doi.org/10.1007/s00253-017-8466-3
-
The bidirectional gut-lung axis in chronic obstructive pulmonary diseaseAmerican Journal of Respiratory and Critical Care Medicine 207:1145–1160.https://doi.org/10.1164/rccm.202206-1066TR
-
Immunity to commensal fungi: detente and diseaseAnnual Review of Pathology 12:359–385.https://doi.org/10.1146/annurev-pathol-052016-100342
-
Excess fermentation and lactic acidosis as detrimental functions of the gut microbes in treatment-naive TB patientsFrontiers in Cellular and Infection Microbiology 14:1331521.https://doi.org/10.3389/fcimb.2024.1331521
-
Mycobacterium bovis bacille Calmette-Guérin (BCG) enhances human beta-defensin-1 gene transcription in human pulmonary gland epithelial cellsActa Pharmacologica Sinica 24:907–912.
Article and author information
Author details
Funding
Science and Technology Research Project of Henan Province (242102521045)
- Fan Yang
Science and Technology Research Project of Henan Province (242102310202)
- JunWei Cui
Project of Health Commission of Henan Province (LHGJ20230525)
- JunWei Cui
Xinxiang Medical University (Open Project of the Institute of Tuberculosis XYJHB20210)
- Fan Yang
Henan Provincial Health Commission (Tuberculosis Capacity Improvement Project 2023-68)
- JunWei Cui
The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors wish to acknowledge funding from the Science and Technology Research Project of Henan Province (grant 242102521045, 242102310202), the Project of Health Commission of Henan Province (LHGJ20230525), and the Open Program of the Institute of Tuberculosis, Xinxiang Medical University (grant XYJHB202104).
Ethics
All animal experiments were conducted according to the Declaration of Helsinki and were approved by the Animal Care and Use Ethics Committee of Xinxiang Medical University (No. EC-023-098).
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.99282. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2024, Han et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,058
- views
-
- 95
- downloads
-
- 7
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 1
- citation for umbrella DOI https://doi.org/10.7554/eLife.99282
-
- 1
- citation for Reviewed Preprint v1 https://doi.org/10.7554/eLife.99282.1
-
- 5
- citations for Version of Record https://doi.org/10.7554/eLife.99282.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Intestinal microbiome dysbiosis increases Mycobacteria pulmonary colonization in mice by regulating the Nos2-associated pathways
eLife 13:RP99282.
https://doi.org/10.7554/eLife.99282.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}