Plant Reproduction: Long story short
- University of Lausanne, Switzerland
The vast majority of flowering plants are hermaphrodites, with both male and female parts in each of their flowers (Renner, 2014). This strategy is clearly a successful one, coming with several likely advantages including that, by producing both pollen and ovules, each flower has more opportunities to pass genes on to the next generation (Charnov et al., 1976). Although hermaphrodites might also benefit from the possibility of self-fertilization (and many do), most hermaphrodites do all they can to avoid ‘selfing’ because it is often detrimental to a plant’s fitness (Charlesworth and Willis, 2009; Barrett, 2002). Hermaphroditic plants have thus evolved a wonderful array of mechanisms that help them to promote outcrossing.
One of these mechanisms, known as heterostyly, involves male and female parts of the flower (the stamen and the stigma) being in different places in different flowers. The stigma sits on the end of a stalk called the style. Some flowers have long styles and hold their stamens, which produce pollen, deep in the floral tube, whereas others have short styles and hold their stamens much higher in the floral tube (Figure 1A). This means that pollen from a long-styled plant is more likely to pollinate a short-styled flower, and vice versa. Long- and short-styled plants may also differ in the size and colour of their pollen grains and the texture of their stigmas, and outcrossing is only possible between different morphs (Barrett, 2002). Remarkably, despite its complexity, heterostyly has evolved repeatedly in flowering plants and is found in at least 30 families, including the primroses (genus Primula).
Figure 1
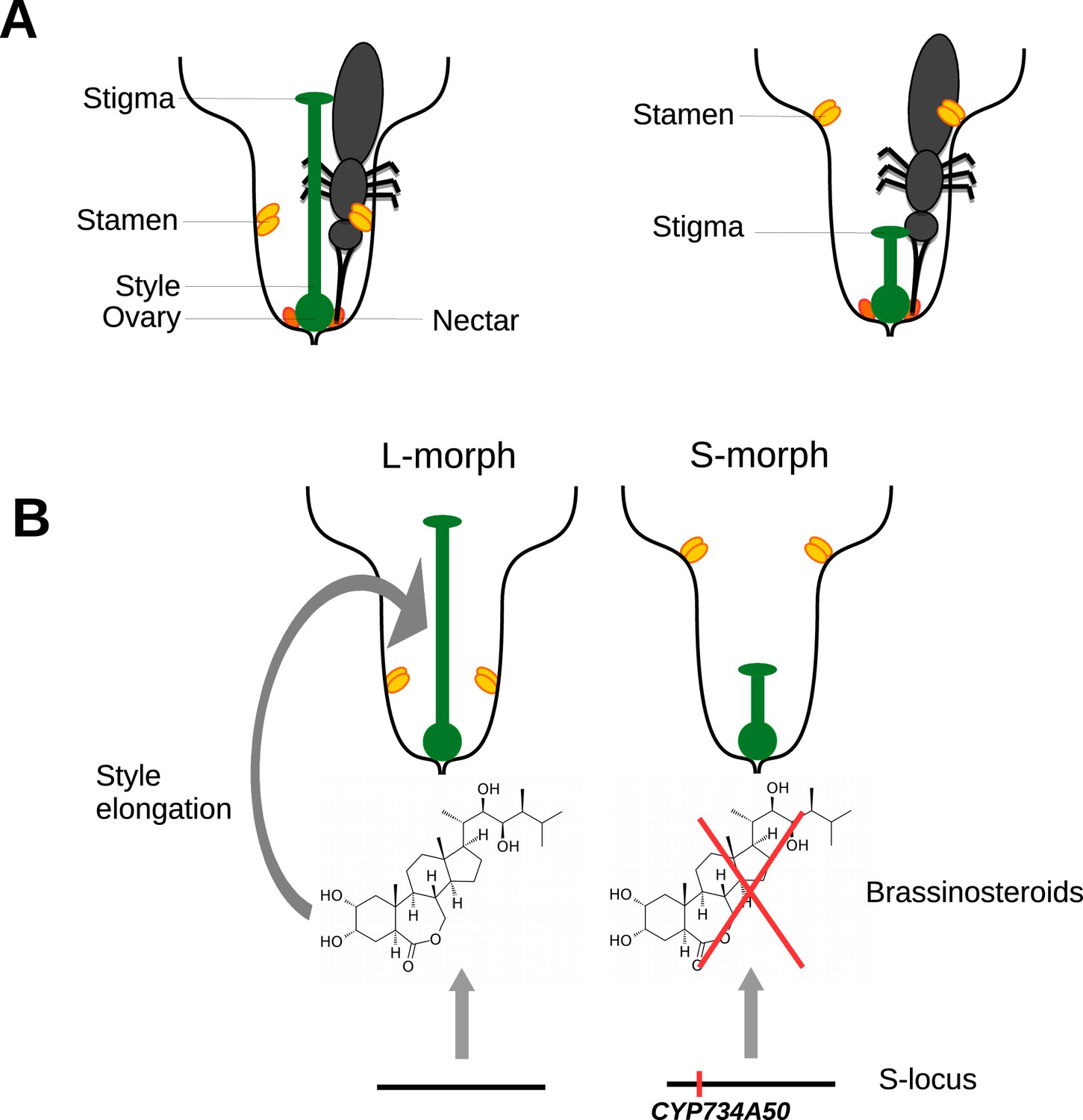
Heterostyly in primroses.
(A) Schematic diagrams of a long-styled (left) and short-styled flower (right) from a distylous species such as Primula vulgaris. When an insect enters a long-styled flower it would pick up pollen mainly on its head. If this insect then visits a short-styled flower, the stigma would pick up the pollen from the insect’s head. Meanwhile, the high stamens would deposit fresh pollen on to the insect’s abdomen for effective transfer to the stigmas of long-styled flowers. (B) Huu et al. identified CYP730A50 as a duplicated gene that alters style length in Primula flowers. Flowers without CYP730A50 (left) produce brassinosteroids, which cause the cells in the style to elongate and produce a long style. On the other hand, flowers with this gene (right) in the S-locus suppress the production of brassinosteroids, which leads to a short style.
Charles Darwin was fascinated by heterostyly (Darwin, 1877) and we now know that it is caused by the expression of several genes that are inherited together as a single genetic unit (locus). However, working out how these genes influence the development of flowers has been something of a holy grail in the field. Now, in eLife, Michael Lenhard at the University of Potsdam and colleagues – including Cuong Nguyen Huu as first author – have identified the gene that is responsible for the different lengths of styles in primroses, and shown how it works (Huu et al., 2016).
Huu et al. found that a gene called CYP734A50 is only present in short-styled flowers in several species of primrose, and estimates of transcript read coverage suggest that only one copy of the gene is present in these flowers. CYP734A50 encodes an enzyme that degrades plant hormones called brassinosteroids, which are known to promote cell elongation (Ohnishi et al., 2006). Huu et al. found that a particular brassinosteroid, castasterone, is present at higher levels in long styles than in short styles, and that treating short styles with another brassinosteroid increased style length (Figure 1B). Moreover, virus-induced silencing of CYP734A50 in young plants with short styles resulted in the production of flowers with longer styles.
It has been thought for a long time, based on theoretical reasoning and comparative analysis, that the first mutation in the evolution of heterostyly was a mutation that reduced style length, and that the expression of this mutated gene should be dominant over the expression of its homologous gene at the style-length locus (Lloyd and Webb, 1992a; 1992b). The results of Huu et al. are consistent with the idea that the expression of the CYP734A50 gene in short-styled individuals is genetically dominant. But how did this dominance come about?
Huu et al. show that CYP734A50 is the result of the duplication of a gene that degrades brassinosteroids more generally. The duplication event occurred early in the evolution of the Primula genus, and the duplicated copy then evolved to be expressed only in styles. The presence or absence of the duplication has been maintained in the genus by a process called negative frequency-dependent selection, whereby the fitness of a trait (e.g. short styles) increases if it becomes rare, thus preventing its loss.
In his short autobiography, Darwin reflected that "I do not think anything in my scientific life has given me so much satisfaction as making out the meaning of the structure of heterostylous flowers". Like so much of his work, the hypothesis that he put forward has stood the test of time and remains the favoured explanation for how and why heterostyly evolved (Barrett, 2002). By identifying the gene that controls style length, the work of Huu et al. is an important step towards understanding the underlying mechanisms involved in heterostyly. A future challenge is to understand the evolutionary history of the other genes involved in heterostyly and to work out what roles they play in flower development.
References
-
The genetics of inbreeding depressionNature Reviews Genetics 10:783–796.https://doi.org/10.1038/nrg2664
-
BookThe evolution of heterostylyIn: Barrett SCH, editors. Evolution and Function of Heterostyly. Berlin: Springer Verlag. pp. 151–178.https://doi.org/10.1007/978-3-642-86656-2_6
-
BookThe selection of heterostylyIn: Barrett SCH, editors. Evolution and Function of Heterostyly. Berlin: Springer Verlag. pp. 179–207.https://doi.org/10.1007/978-3-642-86656-2_7
-
The relative and absolute frequencies of angiosperm sexual systems: dioecy, monoecy, gynodioecy, and an updated online databaseAmerican Journal of Botany 101:1588–1596.https://doi.org/10.3732/ajb.1400196
Article and author information
Author details
Publication history
Copyright
© 2016, Pannell et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,686
- views
-
- 228
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Plant Reproduction: Long story short
eLife 5:e20314.
https://doi.org/10.7554/eLife.20314
Further reading
-
- Chromosomes and Gene Expression
- Immunology and Inflammation
Genome-wide association studies (GWAS) have identified hundreds of genetic signals associated with autoimmune disease. The majority of these signals are located in non-coding regions and likely impact cis-regulatory elements (cRE). Because cRE function is dynamic across cell types and states, profiling the epigenetic status of cRE across physiological processes is necessary to characterize the molecular mechanisms by which autoimmune variants contribute to disease risk. We localized risk variants from 15 autoimmune GWAS to cRE active during TCR-CD28 co-stimulation of naïve human CD4+ T cells. To characterize how dynamic changes in gene expression correlate with cRE activity, we measured transcript levels, chromatin accessibility, and promoter–cRE contacts across three phases of naive CD4+ T cell activation using RNA-seq, ATAC-seq, and HiC. We identified ~1200 protein-coding genes physically connected to accessible disease-associated variants at 423 GWAS signals, at least one-third of which are dynamically regulated by activation. From these maps, we functionally validated a novel stretch of evolutionarily conserved intergenic enhancers whose activity is required for activation-induced IL2 gene expression in human and mouse, and is influenced by autoimmune-associated genetic variation. The set of genes implicated by this approach are enriched for genes controlling CD4+ T cell function and genes involved in human inborn errors of immunity, and we pharmacologically validated eight implicated genes as novel regulators of T cell activation. These studies directly show how autoimmune variants and the genes they regulate influence processes involved in CD4+ T cell proliferation and activation.
-
- Chromosomes and Gene Expression
- Developmental Biology
Differentiation of female germline stem cells into a mature oocyte includes the expression of RNAs and proteins that drive early embryonic development in Drosophila. We have little insight into what activates the expression of these maternal factors. One candidate is the zinc-finger protein OVO. OVO is required for female germline viability and has been shown to positively regulate its own expression, as well as a downstream target, ovarian tumor, by binding to the transcriptional start site (TSS). To find additional OVO targets in the female germline and further elucidate OVO’s role in oocyte development, we performed ChIP-seq to determine genome-wide OVO occupancy, as well as RNA-seq comparing hypomorphic and wild type rescue ovo alleles. OVO preferentially binds in close proximity to target TSSs genome-wide, is associated with open chromatin, transcriptionally active histone marks, and OVO-dependent expression. Motif enrichment analysis on OVO ChIP peaks identified a 5’-TAACNGT-3’ OVO DNA binding motif spatially enriched near TSSs. However, the OVO DNA binding motif does not exhibit precise motif spacing relative to the TSS characteristic of RNA polymerase II complex binding core promoter elements. Integrated genomics analysis showed that 525 genes that are bound and increase in expression downstream of OVO are known to be essential maternally expressed genes. These include genes involved in anterior/posterior/germ plasm specification (bcd, exu, swa, osk, nos, aub, pgc, gcl), egg activation (png, plu, gnu, wisp, C(3)g, mtrm), translational regulation (cup, orb, bru1, me31B), and vitelline membrane formation (fs(1)N, fs(1)M3, clos). This suggests that OVO is a master transcriptional regulator of oocyte development and is responsible for the expression of structural components of the egg as well as maternally provided RNAs that are required for early embryonic development.




{kind=link}