Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMurim ChoiSeoul National University, Seoul, Republic of Korea
- Senior EditorMurim ChoiSeoul National University, Seoul, Republic of Korea
Reviewer #1 (Public review):
The authors describe a massively parallel reporter assays (MPRA) screen focused at identifying polymorphisms in 5' and 3' UTRs that affect translation efficiency and thus might have a functional impact on cells. The topic is of timely interest, and indeed, several related efforts have recently been published and preprinted (e.g., https://pubmed.ncbi.nlm.nih.gov/37516102/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10635273/). This study has several major issues with the results and their presentation.
Major comments:
• The main issue remains that it appears that the screen has largely failed, and the reasons for that remain unclear, which make it difficult to interpret how useful is the resulting data. The authors mention batch effects as a potential contributor. The authors start with a library that includes ~6,000 variants, which makes it a medium-size MPRA. But then, only 483 pairs of WT/mutated UTRs yield high confidence information, which is already a small number for any downstream statistical analysis, particularly since most don't actually affect translation in the reporter screen setting (which is not unexpected). It is unclear why >90% of the library did not give high-confidence information. The profiles presented as base-case examples in Fig. 2B don't look very informative or convincing. All the subsequent analysis is done on a very small set of UTRs that have an effect, and it is unclear to this reviewer how these can yield statistically significant and/or biologically-relevant associations.
• From the variants that had an effect, the authors go on to carry out some protein-level validations, and see some changes, but it is not clear if those changes are in the same direction was observed in the screen. In their rebuttal the authors explain that they largely can not infer directionality of changes form the screen, which further limits its utility.
• It is particularly puzzling how the authors can build a machine learning predictor with >3,000 features when the dataset they use for training the model has just a few dozens of translation-shifting variants.
Comments on revisions:
It appears that the authors have extracted the information they could from the problematic dataset they obtained. Repeating the experiments in a cleaner setting, obtaining data for the >6000 UTRs they intended will allow the authors to achieve the goals they set out to achieve in establishing the screen.
Author response:
The following is the authors’ response to the current reviews.
Public Reviews:
Reviewer #1 (Public review):
The authors describe a massively parallel reporter assays (MPRA) screen focused at identifying polymorphisms in 5' and 3' UTRs that affect translation efficiency and thus might have a functional impact on cells. The topic is of timely interest, and indeed, several related efforts have recently been published and preprinted (e.g., https://pubmed.ncbi.nlm.nih.gov/37516102/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10635273/). This study has several major issues with the results and their presentation.
Major comments:
• The main issue remains that it appears that the screen has largely failed, and the reasons for that remain unclear, which make it difficult to interpret how useful is the resulting data. The authors mention batch effects as a potential contributor. The authors start with a library that includes ~6,000 variants, which makes it a medium-size MPRA. But then, only 483 pairs of WT/mutated UTRs yield high confidence information, which is already a small number for any downstream statistical analysis, particularly since most don't actually affect translation in the reporter screen setting (which is not unexpected). It is unclear why >90% of the library did not give high-confidence information. The profiles presented as base-case examples in Fig. 2B don't look very informative or convincing. All the subsequent analysis is done on a very small set of UTRs that have an effect, and it is unclear to this reviewer how these can yield statistically significant and/or biologically-relevant associations.
• From the variants that had an effect, the authors go on to carry out some protein-level validations, and see some changes, but it is not clear if those changes are in the same direction was observed in the screen. In their rebuttal the authors explain that they largely can not infer directionality of changes form the screen, which further limits its utility.
• It is particularly puzzling how the authors can build a machine learning predictor with >3,000 features when the dataset they use for training the model has just a few dozens of translation-shifting variants.
We recognize that RNA distribution within polysomes is inherently less stable than the associated protein components. This instability has been noted in previous studies, including those cited by the reviewer, which used RNA from bulk polysomes to infer the translatome without fractionation. Acknowledging this limitation, we purposely adopted a conservative strategy: (i) performing gross fractionation of polysomes, and (ii) collaborating with biostatisticians at the Institute of Statistical Science, Academia Sinica, to design a conservative yet optimized analysis pipeline that minimized batch effects.
This approach proved robust: representative cases in Fig. 2B clearly demonstrate distinct distributions of reference and alternative alleles. From our high-confidence dataset, we applied a well-established statistical framework specifically designed to accommodate multiple influencing factors in relatively small datasets (Elements of Statistical Learning by Hastie, Tibshirani, and Friedman). We further conducted sensitivity analyses to select an optimal QC cutoff across a range of stringencies, ensuring maximal reliability of our results. We have therefore successfully shortlisted UTR variants which have strong effect on translation.
Building upon these conservative measures, we developed a predictive model for translation effects of UTR variants. Importantly, this model was validated not only with our internal test dataset but also with independent external datasets. In addition, the sequence features identified by the model were validated through reporter assays and in vivo CRISPR editing. These external and functional validations establish the generalizability and robustness of our approach.
A more detailed analysis of the directionality of changes in translation efficiency is under active investigation. These results will be reported in a separate manuscript currently in preparation.
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
The authors describe a massively parallel reporter assays (MPRA) screen focused on identifying polymorphisms in 5' and 3' UTRs that affect translation efficiency and thus might have a functional impact on cells. The topic is of timely interest, and indeed, several related efforts have recently been published and preprinted (e.g., https://pubmed.ncbi.nlm.nih.gov/37516102/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10635273/). This study has several major issues with the results and their presentation.
Major comments:
(1) The main issue is that it appears that the screen has largely failed, yet the reasons for that are unclear, which makes it difficult to interpret. The authors start with a library that includes approximately 6,000 variants, which makes it a medium-sized MPRA. But then, only 483 pairs of WT/mutated UTRs yield highconfidence information, which is already a small number for any downstream statistical analysis, particularly since most don't actually affect translation in the reporter screen setting (which is not unexpected). It is unclear why >90% of the library did not give high-confidence information. The profiles presented as basecase examples in Figure 2B don't look very informative or convincing. All the subsequent analysis is done on a very small set of UTRs that have an effect, and it is unclear to this reviewer how these can yield statistically significant and/or biologically relevant associations.
To make sure our final results are technically and statistically sound, we applied stringent selection criteria and cutoffs in our analytics workflow. First, from our RNA-seq dataset, we filtered the UTRs with at least 20 reads in a polysome profile across all three repeated experiments. Secondly, in the following main analysis using a negative binomial generalized linear model (GLM), we further excluded the UTRs that displayed batch effect, i.e. their batch-related main effect and interaction are significant. We believe our measure has safeguarded the filtered observations (UTRs) from the (potential) high variation of our massively parallel translation assays and thus gives high confidence to our results.
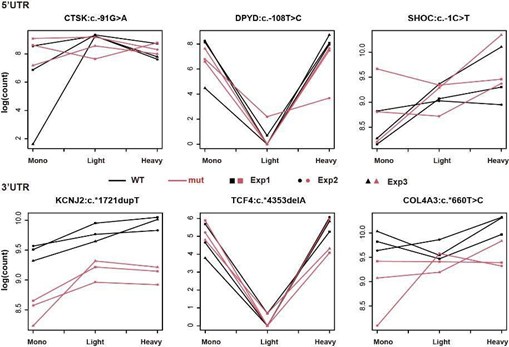
Regarding the interpretation of Figure 2B, since we aimed to identify the UTRs whose interaction term of genotype and fractions is significant in our generalized linear model, it is statistically conventional to doublecheck the interaction of the two variables using such a graph. For instance, in the top left panel of Figure 2B (5'UTR of ANK2:c.-39G>T), we can see that read counts of WT samples congruously decreased from Mono to Light, whereas the read counts of mutant samples were roughly the same in the two fractions – the trend is different between WT and mutant. Ergo, the distinct distribution patterns of two genotypes across three fractions in Figure 2B offer the readers a convincing visual supplement to our statistics from GLM.
In contrast to Figure 2B, the graphs of nonsignificant UTRs (shown below) reveal that the trends between the two genotypes are similar across the 'Mono and Light' and 'Light and Heavy' polysome fractions. Importantly, our analysis remains unaffected by differential expression levels between WT and mutant, as it specifically distinguishes polysome profiles with different distributions. This consistent trend further supports the lack of interaction between genotype and polysome fractions for these UTRs.
Author response image 1.
Examples of non-significant UTR pairs in massively parallel polysome profiling assays.
(2) From the variants that had an effect, the authors go on to carry out some protein-level validations and see some changes, but it is not clear if those changes are in the same direction as observed in the screen.
To infer the directionality of translation efficiency from polysome profiles, a common approach involves pooling polysome fractions and comparing them with free or monosome fractions to identify 'translating' fractions. However, this method has two major potential pitfalls: (i) it sacrifices resolution and does not account for potential bias toward light or heavy polysomes, and (ii) it fails to account for discrepancies between polysome load and actual protein output (as discussed in https://doi.org/10.1016/j.celrep.2024.114098 and https://doi.org/10.1038/s41598-019-47424-w). Therefore, our analysis focused on the changes within polysome profiles themselves. 'Significant' candidates were identified based on a significant interaction between genotype and polysome distribution using a negative binomial generalized linear model, without presupposing the direction of change on protein output.
(3) The authors follow up on specific motifs and specific RBPs predicted to bind them, but it is unclear how many of the hits in the screen actually have these motifs, or how significant motifs can arise from such a small sample size.
We calculated the Δmotif enrichment in significant UTRs versus nonsignificant UTRs using Fisher’s exact test. For example, the enrichment of the Δ‘AGGG’ motif in 3’ UTRs is shown below:
Author response table 1.

This test yields a P-value of 0.004167 by Fisher’s exact test. The P-values and Odds ratios of Δmotifs in relation to polysome shifting are included in Supplementary Table S4, and we will update the detailed motif information in the revised Supplementary Table S4.
(4) It is particularly puzzling how the authors can build a machine learning predictor with >3,000 features when the dataset they use for training the model has just a few dozens of translation-shifting variants.
We understand the concern regarding the relatively small number of translation-shifting variants compared to the large number of features. To address this, we employed LASSO regression, which, according to The Elements of Statistical Learning by Hastie, Tibshirani, and Friedman, is particularly suitable for datasets where the number of features 𝑝𝑝 is much larger than the number of samples 𝑁𝑁. LASSO effectively performs feature selection by shrinking less important coefficients to zero, allowing us to build a robust and generalizable model despite the limited number of variants.
(5) The lack of meaningful validation experiments altering the SNPs in the endogenous loci by genome editing limits the impact of the results.
Following the reviewer’s suggestion, we assessed the endogenous mutant effect by generating CRISPR knock-in clones carrying the IRF6:c.-4609G>A variant. We showed that this G>A variant generate a deleterious upstream open reading frame, which dramatically reduced protein expression of the main open reading frame (Fig. 7B-D). The genome editing further demonstrated the G>A variant reduced endogenous IRF6 protein expression to 23% or 44% in two independent clones. We have incorporated the genome editing results in the revised main text and the new Figure 7E&F:
“To further validate the endogenous effect of the novel upstream ATG (uATG), we generated CRISPR knockin clones carrying the IRF6:c.-4609G>A variant and examined its impact on gene expression. The introduction of the uATG reduced RNA levels to 88% and 37% of the wild-type in two independent clones (Fig. 7E), and protein levels to 44% and 23%, respectively (Fig. 7F), resulting in an overall reduction of translation efficiency to 50–62%.“ (p.18)
Reviewer #2 (Public Review):
Summary:
In their paper "Massively Parallel Polyribosome Profiling Reveals Translation Defects of Human DiseaseRelevant UTR Mutations" the authors use massively parallel polysome profiling to determine the effects of 5' and 3' UTR SNPs (from dbSNP/ClinVar) on translational output. They show that some UTR SNPs cause a change in the polysome profile with respect to the wild-type and that pathogenic SNPs are enriched in the polysome-shifting group. They validate that some changes in polysome profiles are predictive of differences in translational output using transiently expressed luciferase reporters. Additionally, they identify sequence motifs enriched in the polysome-shifting group. They show that 2 enriched 5' UTR motifs increase the translation of a luciferase reporter in a protein-dependent manner, highlighting the use of their method to identify translational control elements.
Strengths:
This is a useful method and approach, as UTR variants have been more difficult to study than coding variants. Additionally, their evidence that pathogenic mutations are more likely to cause changes in polysome association is well supported.
Weaknesses:
The authors acknowledge that they "did not intend to immediately translate the altered polysome profile into an increase or decrease in translation efficiency, as the direction of the shift was not readily evident. Additionally, sedimentation in the sucrose gradient may have been partially affected by heavy particles other than ribosomes." However, shifted polysome distribution is used as a category for many downstream analyses. Without further clarity or subdivision, it is very difficult to interpret the results (for example in Figure 5A, is it surprising that the polysome shifting mutants decrease structure? Are the polysome "shifts" towards the untranslated or heavy fractions?)
Our approach, combining polysome fractionation of the UTR library with negative binomial generalized linear model (GLM) analysis of RNA-seq data, systematically identifies variants that affect translational efficiency. The GLM model is specifically designed to detect UTR pairs with significant interactions between genotype and polysome fractions, relying solely on changes in polysome profiles to identify variants that disrupt translation. Consequently, our analytical method does not determine the direction of translation alteration.
Following the massively parallel polysome profiling, we sought to understand how these polysome-shifting variants influence the translation process. To do this, we examined their effects on RNA characteristics related to translation, such as RBP binding and RNA structure. In Figure 5A, we observed a notable trend in significant hits within 5’ UTRs—they tend to increase ΔG (weaker folding energy) in response to changes in polysome profiles, regardless of whether protein production increases or decreases (Fig. 3).
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
Minor comments:
(1) Figure 3A - the claim that 5'UTR variants had a stronger effect than 3'UTR is based on the two UTRs with the strongest effect. It is unclear how these differences between 5' and 3'UTRs are significant.
We carried out a Wilcoxon rank-sum test to examine the mut/WT fold change of translation efficiency between the 3’ and 5’ UTR variants. The results showed that the 5’ UTR variants exhibited a greater change of translation efficiency. We have inserted this result in the revised Figure 3C and refers to this figure in the main text: “Furthermore, we observed that 5’ UTR variants had a greater impact on translation activity relative to 3’ UTR variants (Fig. 3C).” (p. 12)
(2) Figures 2B and S1, S2 - what is the meaning of less signal for a light chain and a similar signal for a heavy chain? How can this situation, while being a significant difference between the profiles, lead to a biologically relevant difference in eventual protein output?
Taking 3’UTR ACADSB:c.*4177G>A (bottom-left panel in Figure 2B) as an example: WT transcripts have less read count (in the unit of log(CPM)) compared with the transcripts carrying the mutant UTR in the light polysome-containing fraction, whereas the read counts of the two genotypes are approximately the same in the heavy polysome-containing fraction.
In line with our reply to Reviewer 1’s major comment 1, we aimed to identify the UTRs whose interaction term of genotype and fractions is significant in our generalized linear model (GLM). That is, the UTR pairs whose WT and mutant have different trends across the fractions (Mono to Light & Light to Heavy) are our targets. In Figure 2B, 3’UTR ACADSB:c.*4177G>A is a perfect example of our significant hits, as it displays the clear distinction of the trends of the two genotypes across three fractions.
It is widely known that the alteration of polysome profiling distribution indicates the change of translational efficiency. Our GLM model helped us identify the UTR pairs whose WT and mutant have different polysome profiling patterns and thus likely have distinct translational efficiency. Nevertheless, since we only had limited polysome fractions in our experiments, we further validated our significant hits and confirmed the direction of regulation using luciferase reporter assay.
(3) The paragraph starting with "Even with the high confidence dataset, we did not intend to immediately translate the altered polysome profile into an increase or decrease in translation efficiency" is confusing. The whole premise of the screen used by the authors is that polysome profiling is a useful proxy for estimating levels of translation, so claiming that it doesn't necessarily measure translation is counterintuitive.
In line with our reply to the last question, our goal is to use the alteration of polysome profiling patterns as a proxy for the change of translational efficiency. However, due to the limited number of fractions in our experiment, we could not directly infer the direction of regulation, i.e. increase or decrease of translational efficiency, of the statistically significant variants. That is why we refrained from making any conclusion about the direction of the regulation for the significant hits and proceed to validate them using luciferase reporter assay.
(4) Figure S5A - this is normalized to the nucleotide distribution in 5' or 3'UTRs? Is this statistic being applied to 27 SNPs in 3'UTRs?
To identify sequence features associated with altered polysome association, we systematically analyzed both significant and nonsignificant UTRs for nucleotide and motif-level changes. Fisher’s exact test was employed to evaluate whether specific nucleotide or motif alterations were enriched or depleted in polysome-shifting UTRs, compared to nonsignificant UTR pairs. For example, in the case of nucleotide C (see table below; also Table S4 and new Fig. S6A), only four significant 3’ UTRs involved a change in C, resulting in a significant depletion of this nucleotide change among polysome-shifting 3’ UTRs (odds ratio = 0.22, p = 0.0069). Expanding this approach to all 1-7 nt motifs, we identified multiple motif and nucleotide changes that were significantly associated with altered polysome association.
Author response table 2.

(5) "uATG in the 5' UTR was not identified by the model as a widespread feature explaining polysome shifting". Is this because of the method of ribosome profiling or because of the sequences in the library? Can having more sequences in the library specifically looking at 5'UTR give more power for such an effect to emerge?
Our assay design accounted for the presence of upstream ATG codons and the strength of adjacent Kozak sequences. However, additional factors known to influence the function of upstream open reading frames (uORFs)—such as the reading frame of the uORF relative to the main coding sequence, and the use of nonATG initiation codons—were not systematically included. As a result, the current assay may have limited sensitivity in detecting uORF-related regulatory effects. A dedicated design specifically tailored to uORF variants is likely to enhance the detection power and better capture their contribution to translational control.
(6) Figure 7B- it is not clear whether the luciferase reporter and the GFP reporter in the library function in a similar manner; is it creating out-of-frame or in of in frame uORF? Also, it is not clear if the differences are statistically significant.
In the MPRA library, the IRF6 uORF is out of frame relative to the GFP coding sequence. To directly assess its translational impact, we employed a luciferase reporter assay by fusing luciferase downstream of the IRF6 uORF. These constructs revealed a significant reduction in protein production, as shown in Figures 3 and 7B–F. Although the clinically relevant IRF6 uORF is out-of-frame with the main ORF, we engineered an inframe uORF variant to validate translation initiation at the upstream ATG (uATG) (Fig. 7B-D). The in-frame construct confirmed uATG usage and led to a significant reduction in luciferase protein expression. Together, these results support the conclusion that the IRF6:c.-4609G>A variant gives rise to an active uORF that suppresses translation of the main ORF.
Reviewer #2 (Recommendations For The Authors):
(1) It would be helpful for the authors to subcategorize their data in ways that they consider meaningful and interpretable (e.g. shifts from all monosome to heavy, all heavy to monosome/free, etc.) Relatedly, what do the authors think the functional meaning is when a given transcript has high mono/heavy occupancy but low light occupancy (like what is shown in Figure 2B for ANK2) in the polysome profiling experiment? It is not apparent why a transcript with a high ribosome occupancy (heavy) would also have light occupancy (light).
From the amplicon sequencing data, we obtained read counts for each UTR variant across the monosome, light, and heavy polysome fractions. Notably, this approach does not preserve the original relative abundance of transcripts among the three fractions. That is, despite a greater abundance of mRNAs in the heavy polysome fraction, comparable numbers of sequencing reads were recovered from the monosome and light fractions. As a result, this method is not suitable for interpreting the global directionality of translational shifts but is well-suited for detecting relative differences in polysome association. Therefore, our experimental and analytical design—combining targeted amplicon sequencing with generalized linear modeling (GLM)—was optimized to identify UTR variants that alter polysome association, independently of absolute transcript abundance in each fraction.
(2) The method put forward in Figure 2 would be more convincing if there was data showing reproducibility in the massively parallel reporter assay. Perhaps the mut/WT ratio for all transcripts can be plotted against each other and a statistical test of correlation can be performed.
Thank you for pointing this out. To demonstrate the reproducibility of our massively parallel reporter assay, we have plotted scatter plots of the  ratios of all transcripts (summing the monosome, light, and heavy fractions) across different batches using our high-confidence dataset. We calculated the Pearson correlation coefficients and corresponding p-values for these comparisons. The results show strong correlation between each batch, supporting the reproducibility of our assay. We have incorporated this analysis in the main text as well as Supplemental Figure 3: “Pearson correlation analysis revealed R coefficients ranging from 0.59 to 0.71 for the mut-to-WT transcript ratios across three independent experiments (Supplemental Fig. 3).”
ratios of all transcripts (summing the monosome, light, and heavy fractions) across different batches using our high-confidence dataset. We calculated the Pearson correlation coefficients and corresponding p-values for these comparisons. The results show strong correlation between each batch, supporting the reproducibility of our assay. We have incorporated this analysis in the main text as well as Supplemental Figure 3: “Pearson correlation analysis revealed R coefficients ranging from 0.59 to 0.71 for the mut-to-WT transcript ratios across three independent experiments (Supplemental Fig. 3).”
(3) The dots in Figure 2B indicate separate experiments, but the y-axis is log(counts). Values could be normalized (perhaps a ratio of mut/WT) for comparison between experiments.
We aimed to compare UTR distribution across polysome fractions and recognized the importance of presenting the distribution patterns for both genotypes. This approach allows us to more clearly illustrate the differences or similarities in polysome association between the two genotypes.
(4) When describing the 5' UTRs used for the validation experiments in Figure 3, more information about the 5' UTR sequence used is necessary. It is not clear how much or what part of the 5' UTRs were removed, or why this was necessary considering the same experiment was conducted using full-length UTRs.
In the initial library design, technical limitations of bulk oligonucleotide synthesis constrained the UTRs to 155 nucleotides, comprising 115-nt of endogenous human UTR sequence flanked by 20-nt priming sites on both ends. Variants were centered at the 58th nucleotide within the 115-nt UTR sequence. When one flanking region of the native UTR was shorter than 57 nt, the variant was shifted accordingly toward the shorter arm to maintain the 115-nt UTR length (Fig. 2A).
Given that endogenous UTRs in the human genome are often longer than 155 nt, we further evaluated the functional consequences of variants within full-length UTR sequences (Fig. 3B). While the mutant effects observed in the library setting were largely recapitulated, their magnitude was diminished in the full-length context, likely due to the increased sequence and structural complexity.
To clarify the experimental design related to Figure 3, we modified the text as the following: “The variants significantly altering the polysome profile were then individually validated by means of high-sensitivity luciferase reporter assays (Fig. 3A). To that end, we resynthesized both the variant and corresponding wildtype alleles in the same library format - 115-nt native UTR segments centered on the variant and flanked by 20-nt priming sites. These UTRs were then cloned upstream (5’) or downstream (3’) of the firefly luciferase coding sequence, depending on their genomic location.” (p. 11)
(5) The conclusions from inserting RBP-binding motifs into 5' UTRs and assaying translational output (Figure 4) would be strengthened by including luciferase reporters containing endogenous 5' UTRs containing these motifs, and versions where the motifs are disrupted.
Several variants that altered translation efficiency were validated in their native sequence contexts, including 5’ UTR variants in DMD and NF1 that affect SRSF1/2 binding sites, as well as a 3’ UTR variant in AL049650.1 that impacts a KHSRP binding site (Fig. 3 and Supplemental Figs. S1 & S2). To address the functional relevance of these variants within their native regulatory landscapes, we have incorporated the following clarification into the text (p. 13): “This observation is consistent with additional findings where variants that create or disrupt specific RBP binding sites—such as SRSF1/2 (e.g., in DMD and NF1; Fig. 2 and Supplementary Fig. S4) and KHSRP (e.g., in AL049650.1; Fig. 2 and Supplementary Figs. S4 & S5)—led to significant changes in translation efficiency within their native UTR contexts.”
(6) Figure 5C shows that 5' UTR SNPs that form an uAUG are associated with greater structural changes, but this does not "indicate" that "structure‐modifying UTR variants may control primary ORF translation partly by interfering with translation initiation from a uORF." The data presented in Figure 5 and luciferase/polysome data presented previously do not distinguish whether translation is occurring at an uAUG or canonical AUG. The statement quoted above is speculative and it should be clear that it is a hypothesis generated by the data and is not conclusive.
We appreciate the reviewer’s suggestion. We have therefore modified our text to: ”Therefore, while changes in uATG may not be common explanatory factors for polysome-shifting mutations, our results suggest that structure-modifying UTR variants may control primary ORF translation partly by interfering with translation initiation from a uORF.” (p. 14)
Minor points/questions
(1) The authors should clarify whether during library construction for massively parallel polysome profiling the 3' UTR constructs contain a common 5' UTR? Likewise, do the 5' UTR constructs contain a common 3' UTR? Perhaps the lack of a 5' UTR in the 3' UTR constructs, which is implied by Figure 2A, would influence differences seen between 3' UTR pairs (and likewise for 5' UTR pairs).
There are short common 5’ UTRs appended to the 3’ UTR library, and likewise, a common short 3’ UTR is included in the 5’ UTR library. The common 5’ UTR comprises partial sequences from the CMV promoter and the plasmid backbone of pEGFP-N1 vector. The common 3’ UTR includes sequences from the pEGFP-N1 backbone and a short polyadenylation signal from HBA1 (hemoglobin subunit alpha 1). While we cannot entirely rule out potential crosstalk between 5’ and 3’ UTRs, the design ensures that all constructs are compared in a controlled and consistent context, enabling valid pairwise comparisons between variant and wildtype alleles.
To clarify the library design, we have revised the main text to include this explanation:
“The entire library of UTR oligonucleotides (UTR library) was subsequently ligated upstream or downstream of an enhanced GFP (EGFP) coding region, along with a CMV promoter and a common UTR sequence on the opposite end. Cells transfected with the UTR library were treated with cycloheximide 14 hours post transfection and then subjected to polysome fractionation (see Methods).” (p.11)
“The variants significantly altering the polysome profile were then individually validated through highsensitivity luciferase reporter assays (Fig. 3A). To this end, we resynthesized both the variant and corresponding wildtype alleles in the same library format - 115-nt native UTR segments centered on the variant and flanked by 20-nt priming sites. These UTRs were then cloned upstream (5’) or downstream (3’) of the firefly luciferase coding sequence, depending on their genomic location. As the initial library design, the test UTR segment differs only by one nucleotide, while a shared short UTR fragment is present on the opposite end of the coding sequence to ensure consistency across constructs (Fig. 2A).” (p. 12)
(2) The lines connecting the polysome distribution points make the plots appear busy and difficult to read, the data would be easier to interpret if they were removed.
We employed a generalized linear model (GLM) to identify the variants that altered the polysome association of the corresponding transcripts. Statistically speaking, we were looking for the variants which led to significant interaction between genotype and polysome fractions. Ergo, displaying the lines as it is in our plots offers readers a convincing visualization of the interaction: lines from WT and Mut groups were not parallel, which indicates the interaction between genotype and polysome fractions. Moreover, showing the lines from three batches of experiments also helps us ascertain the reproducibility of our experiments. Taken all together, the presence of the lines makes our plots even more informative.



