B-cell Lymphoma: When 19 is greater than 92
- Johns Hopkins University School of Medicine, United States
- UT Southwestern, United States
MicroRNAs are short, single-stranded RNA molecules that silence genes by targeting messenger RNA molecules. Examples of abnormal microRNA expression and function have been observed across the full spectrum of human cancers. These findings, coupled with data showing that microRNAs can promote or inhibit the formation of tumours in cell lines and animal models, have led to a widespread acceptance that some microRNAs function as bona fide oncogenes and tumour suppressors. In particular, the miR-17-92 cluster—a group of six microRNAs all encoded by the same gene—has been studied extensively since researchers first discovered that it is overexpressed in Burkitt’s lymphoma, diffuse B-cell lymphoma and multiple types of solid tumours (Ota et al., 2004).
The miR-17-92 cluster is transcribed as a single primary transcript that is subsequently processed into six mature microRNAs: miR-17, miR-18, miR-19a, miR-20, miR-19b, and miR-92a (Figure 1). The assembly of multiple microRNAs into polycistronic transcripts has occurred repeatedly during evolution, but the functional significance of this type of organization has remained largely unexplored. Now, in eLife, Lin He of the University of California, Berkeley and colleagues—including Virginie Olive and Erich Sabio as joint first authors—provide new insights into the functional relationships between the individual components of the miR-17-92 cluster (Olive et al., 2013).
Figure 1
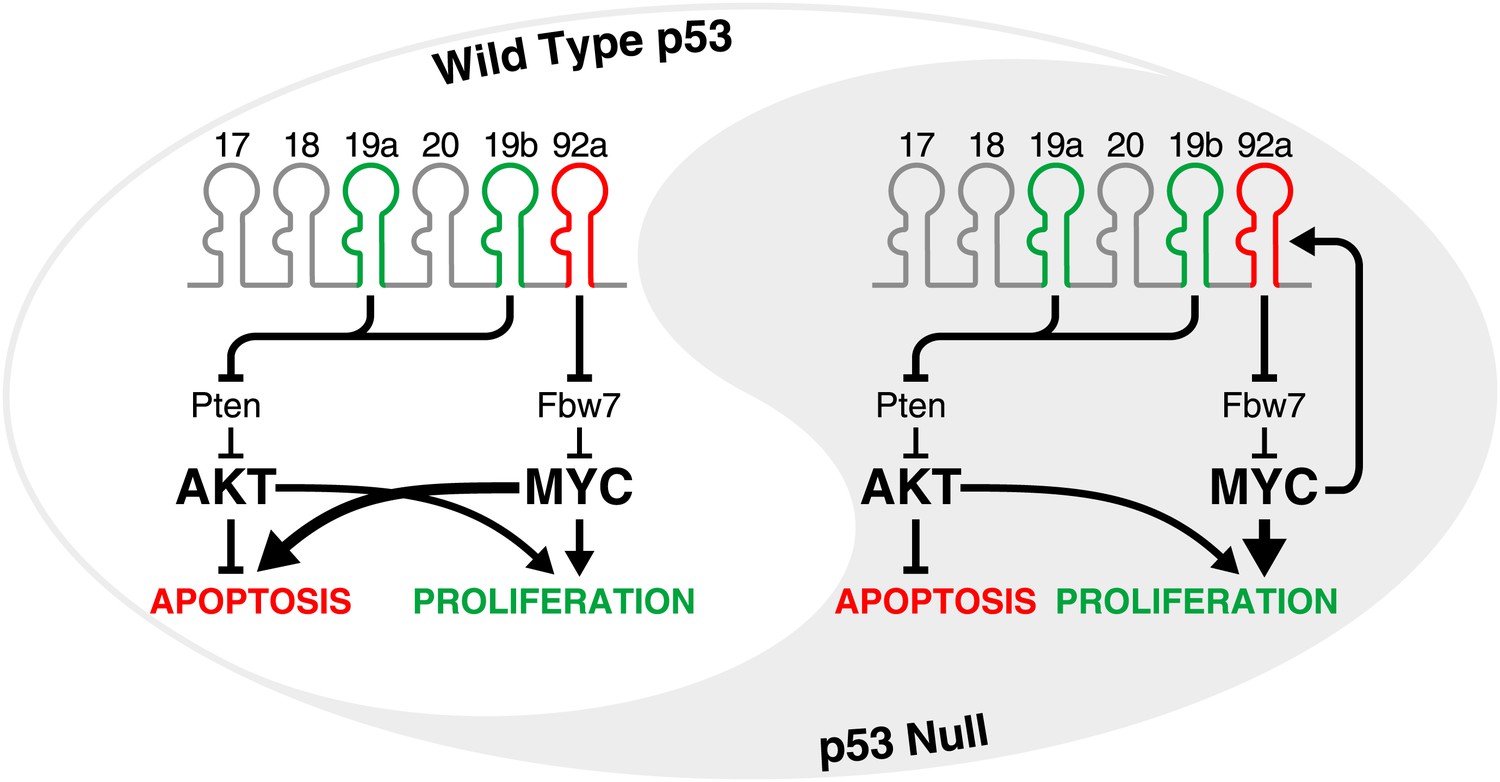
The yin and yang of miR-92a in tumours with and without an intact p53 pathway.
The oncogenic activity of miR-17-92 in the development of B-cell lymphomas is largely attributable to miR-19-mediated suppression of the tumour suppressor gene Pten, and the consequent activation of the PI3K/Akt pathway which inhibits apoptosis. At the same time, miR-92a causes hyperactivation of MYC by downregulating Fbw7 (a gene that encodes an enzyme that facilitates MYC degradation). In cells with an intact p53 pathway (left), high levels of MYC promote proliferation but also engage the p53 pathway to trigger apoptosis, allowing miR-92a to counteract the pro-proliferative and pro-survival effects of miR-19. In the p53-deficient state (right), the hyperactivation of MYC does not lead to apoptosis. Instead, miR-19 and miR-92a both promote the growth of tumours. Moreover, since MYC directly activates the transcription of miR-17-92, a feed-forward loop is established which may amplify these effects.
The oncogenic activity of miR-17-92 was first directly demonstrated by He and co-workers using Eμ-myc mice (He et al., 2005). MYC is a transcription factor that is involved in the regulation of a large number of genes and hyperactivation or amplification of the Myc gene occurs in many cancers. Eμ-myc transgenic mice overexpress the Myc gene in B lymphocytes, causing them to develop B-cell lymphomas by 4–6 months of age (Adams et al., 1985). He et al. showed that miR-17-19b—a truncated form of the cluster that does not contain miR-92a—accelerated the development of B-cell lymphoma when expressed in hematopoietic progenitor cells from Eμ-myc mice. Further experiments pinpointed miR-19 (represented by miR-19a and miR-19b in the cluster) as the key oncogenic component of the cluster in this model (Olive et al., 2009; Mu et al., 2009) (Figure 1).
Olive, Sabio, He and co-workers have now further delineated the contributions of individual microRNAs within miR-17-92 to the oncogenic activity of the cluster. Surprisingly, they found that miR-92a counteracts the effects of miR-19 during the growth and development of B-cell lymphomas. This antagonistic function for miR-92a was first suggested by experiments which showed that the truncated cluster (which lacks miR-92a) exhibited significantly stronger oncogenic activity than the full cluster in Eμ-myc B cells. Similarly, mutations that inactivated miR-92a strongly enhanced the oncogenic effects of the full cluster. In pre-malignant B cells from Eμ-myc mice, and also in mouse embryonic fibroblasts, the expression of miR-92a triggered programmed cell death (apoptosis), thereby counterbalancing the effects of miR-19 (which suppresses apoptosis). Intriguingly, however, the overexpression of miR-92a also increased the proliferation of fibroblasts and B cells. This dichotomy can be at least partially explained by the ability of miR-92a to reduce expression of the gene Fbw7, which encodes an enzyme that facilitates the degradation of the MYC protein. Thus, the increase in MYC levels resulting from miR-92a expression simultaneously stimulates proliferation and engages the p53 tumour suppressor pathway, resulting in apoptosis (Figure 1, left).
While the miR-92a-Fbw7-MYC axis clearly contributes to the apoptosis induced by miR-92a, it is unlikely that this mechanism can fully explain the ability of this microRNA to induce cell death. For example, elevation of MYC protein in fibroblasts does not cause apoptosis as efficiently as expressing miR-92a. Additionally, Olive et al. did not demonstrate that Eμ-myc lymphoma cells with enforced miR-92a expression have higher MYC levels than lymphoma cells without enforced miR-92a. Further studies will be necessary to establish precisely how this pathway influences the growth and development of B-cell lymphomas, and whether miR-92a leads to apoptosis via other pathways.
Olive et al. suggest that the ratio of miR-19 to miR-92a determines the overall oncogenic potency of the miR-17-92 cluster. Interestingly, they found that miR-19 is more highly induced than miR-92a in pre-malignant and malignant Eμ-myc B cells compared to normal B cells. A similar phenomenon is evident in human Burkitt’s lymphoma cell lines. These observations suggest that the processing of miR-19 and miR-92a from the miR-17-92 primary transcript or the stability of the individual mature miRNAs is differentially regulated in normal B cells compared to lymphoma cells. Elucidation of this mysterious post-transcriptional regulatory pathway, which itself may be oncogenic, will undoubtedly be a priority for future work.
Given that the pro-apoptotic activity of miR-92a depends on the presence of an intact p53 pathway, it is important to consider the effects of expressing this microRNA when p53 is absent, as is the case in a large fraction of human tumours. In the absence of p53, the ability of miR-92a to enhance levels of the protein MYC will undoubtedly serve a pro-tumorigenic agenda. Indeed, miR-92a has been shown to promote the proliferation and survival of liver and colon cancer cell lines (Shigoka et al., 2010; Tsuchia et al., 2011). Moreover, given that MYC leads to increased transcription of miR-17-92 (O’Donnell et al., 2005), it is conceivable that the stabilization of MYC by miR-92a may result in a feed-forward loop that leads to even greater activation of MYC. In such a setting, miR-92a may work with, rather than against, miR-19, with both of these microRNAs promoting the formation and growth of tumours through independent pathways (Figure 1, right).
This work also begins to address a broader question: why have microRNA clusters evolved? One might expect that co-transcribed microRNAs would generally work in concert and, indeed, there are many such examples. For instance, both microRNAs within the miR-199-214 cluster work together to promote cardiac remodelling (El Azzouzi et al., 2013). Nevertheless, the work of Olive, Sabio and colleagues—who include co-workers from the UK, China and universities across the US—demonstrates that co-transcription does not necessitate cooperation. The extension of their work to other clusters and other in vivo models will elucidate whether this intricate balance is unique to the miR-17-92 cluster or if it can be generalized to other microRNAs and other species.
References
-
miR-19 is a key oncogenic component of mir-17-92Genes Dev 23:2839–2849.https://doi.org/10.1101/gad.1861409
Article and author information
Author details
Publication history
Copyright
© 2013, Zeitels and Mendell
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 468
- views
-
- 45
- downloads
-
- 1
- citation
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 1
- citation for umbrella DOI https://doi.org/10.7554/eLife.01514
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
B-cell Lymphoma: When 19 is greater than 92
eLife 2:e01514.
https://doi.org/10.7554/eLife.01514




{kind=link}