A component of the mir-17-92 polycistronic oncomir promotes oncogene-dependent apoptosis
- University of California, Berkeley, United States
- University of Cambridge, United Kingdom
- University of California, San Francisco, United States
- Duke University, United States
- Center for Systems Biology, University of Texas at Dallas, United States
- The Third Military Medical University, China
- Northwestern University Feinberg School of Medicine, United States
Abstract
mir-17-92, a potent polycistronic oncomir, encodes six mature miRNAs with complex modes of interactions. In the Eμ-myc Burkitt’s lymphoma model, mir-17-92 exhibits potent oncogenic activity by repressing c-Myc-induced apoptosis, primarily through its miR-19 components. Surprisingly, mir-17-92 also encodes the miR-92 component that negatively regulates its oncogenic cooperation with c-Myc. This miR-92 effect is, at least in part, mediated by its direct repression of Fbw7, which promotes the proteosomal degradation of c-Myc. Thus, overexpressing miR-92 leads to aberrant c-Myc increase, imposing a strong coupling between excessive proliferation and p53-dependent apoptosis. Interestingly, miR-92 antagonizes the oncogenic miR-19 miRNAs; and such functional interaction coordinates proliferation and apoptosis during c-Myc-induced oncogenesis. This miR-19:miR-92 antagonism is disrupted in B-lymphoma cells that favor a greater increase of miR-19 over miR-92. Altogether, we suggest a new paradigm whereby the unique gene structure of a polycistronic oncomir confers an intricate balance between oncogene and tumor suppressor crosstalk.
https://doi.org/10.7554/eLife.00822.001eLife digest
The role of genes, in very simple terms, is to be transcribed into messenger RNA molecules, which are then translated into strings of amino acids that fold into proteins. Each of these steps is extremely complex, and a wide range of other molecules can speed up, slow down, stop or otherwise disrupt the expression of genes as protein products. Genes can also code for nucleic acids that are not translated into proteins, such as microRNAs. These are small RNA molecules that can reduce the production of proteins by repressing the translation step and/or by partially degrading the messenger RNA molecules.
mir-17-92 is a gene that exemplifies much of this complexity. It codes for six different microRNAs in a single primary transcript, and has been implicated in a number of cancers, including lung cancer, Burkitt’s lymphoma and other forms of lymphomas and leukemia. One of six microRNAs has a longer evolutionary history than the remaining five: mir-92 is found in vertebrates, chordates and invertebrates, whereas the other five are only found in vertebrates. However, it is not known how or why the mir-17-92 gene evolved to code for multiple different microRNAs.
Olive et al. have studied how these mir-17-92 microRNAs functionally interact in mice with Burkitt’s lymphoma, a form of cancer that is associated with a gene called c-Myc being over-activated. Mutations in this gene promote the proliferation of cells, and in cooperation with other genetic lesions, this ultimately leads to cancer. mir-17-92 is implicated in this cancer because it represses the process of programmed cell death (which is induced by the protein c-Myc) that the body employs to stop tumors growing.
Olive et al. found that deleting one of the six microRNAs, miR-92, increased the tendency of the mir-17-92 gene to promote Burkitt’s lymphoma. By repressing an enzyme called Fbw7, miR-92 causes high levels of c-Myc to be produced. While this leads to the uncontrolled proliferation of cells that promotes cancer, it also increases programmed cell death, at least in part, by activating the p53 pathway, a well-known tumor suppression pathway. The experiments also revealed that the action of miR-92 and that of one of the other microRNAs, miR-19, were often opposed to each other. These findings have revealed an unexpected interaction among different components within a single microRNA gene, which acts to maintain an intricate balance between pathways that promote and suppress cancer.
https://doi.org/10.7554/eLife.00822.002Introduction
MicroRNAs (miRNAs) are a class of small, non-coding RNAs that regulate post-transcriptional gene repression in a variety of developmental and pathological processes (Ambros, 2004; Zamore and Haley, 2005; Bartel, 2009; Kim et al., 2009). Due to their small size and the imperfect nature of target recognition, miRNAs have the capacity to regulate many target mRNAs through translational repression and mRNA degradation, thereby acting as global regulators of gene expression (Lewis et al., 2005; Filipowicz et al., 2008). Unlike mammalian protein-coding genes that follow the one-transcript, one-protein paradigm, many miRNA genes are expressed as polycistronic primary transcripts, generating multiple mature miRNAs under the same transcriptional regulation (Megraw et al., 2007). miRNA polycistrons further expand the gene regulatory capacity, since different miRNA components can confer specific yet overlapping biological effects, and their functional interactions can yield unusual complexity.
Polycistronic miRNAs often exhibit pleiotropic biological functions with unique gene regulatory mechanisms (Megraw et al., 2007). One of the best example is mir-17-92, a potent oncomir (i.e., miRNA oncogene), whose genomic amplification and aberrant overexpression have been observed in many human tumors including Burkitt’s lymphoma, diffuse large B-cell lymphoma (DLBCL), and lung cancer (Lu et al., 2005; Mendell, 2008). mir-17-92 regulates multiple cellular processes during tumor development, including proliferation, survival, angiogenesis, differentiation, and metastasis (He et al., 2007; Uziel et al., 2009; Conkrite et al., 2011; Nittner et al., 2012). As a polycistronic oncomir, mir-17-92 produces a single precursor that yields six individual mature miRNAs (Figure 1A, Figure1—figure supplement 1A) (Tanzer and Stadler, 2004). Based on the seed sequence homology, the six mir-17-92 components are categorized into four miRNA families (Figure 1A, Figure 1—figure supplement 1A): miR-17 (miR-17 and 20), miR-18, miR-19 (miR-19a and 19b), and miR-92a (we will designate miR-92a as miR-92 in the remainder of our paper). Interestingly, miR-92 has a more ancient evolutionary history compared to the other mir-17-92 components (Tanzer and Stadler, 2004). miR-92 is evolutionarily conserved in vertebrates, chordates, and invertebrates, while the remaining mir-17-92 components are only found in vertebrates (Figure 1—figure supplement 1B,C). Conceivably, the distinct mature miRNA sequence of each mir-17-92 component determines the specificity of the target regulation. However, the functional significance of the mir-17-92 polycistronic gene structure remains largely unknown.
Figure 1 with 1 supplement see all
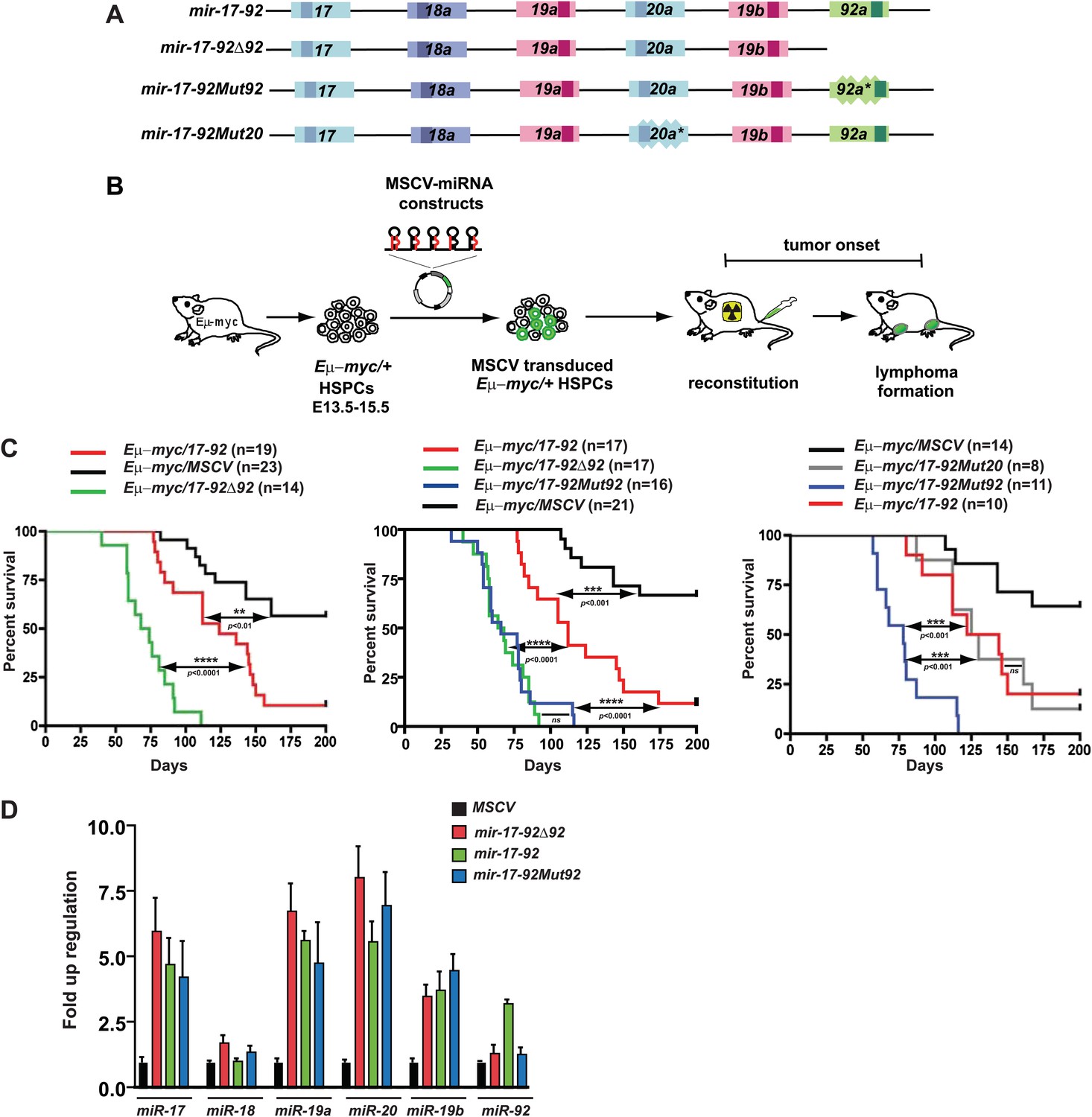
miR-92 negatively regulates the mir-17-92 oncogenic activity in the Eμ-myc B-lymphoma model.
(A) The gene structure of the mir-17-92 polycistron and its mutated derivatives. Light colored boxes, pre-miRNAs; dark colored boxes, mature miRNAs. Homologous miRNA components are indicated by the same color. (B) Schematic representation of the adoptive transfer protocol using Eμ-myc hematopoietic stem and progenitor cells (HSPCs). Eμ-myc/+ HSPCs were extracted from E13.5–E15.5 mouse embryos, infected with MSCV retroviral vectors overexpressing mir-17-92 and its derivatives, and finally transplanted into lethally irradiated recipient mice. Lymphoma onset of the adoptive transferred mice was monitored to evaluate the oncogenic collaboration between c-Myc and a specific miRNA. (C) miR-92 deficiency specifically accelerates the oncogenic activity of mir-17-92 in the Eμ-myc model. Using the Eμ-myc adoptive transfer model, we compared the oncogenic effects between mir-17-92 and mir-17-92Δ92 and observed a significant acceleration of tumor onset in Eμ-myc/mir-17-92Δ92 mice (p<0.0001, left). When the oncogenic effects of mir-17-92, mir-17-92Δ92 and mir-17-92Mut92 were compared in the same adoptive transfer model, mir-17-92Δ92 and mir-17-92Mut92 similarly accelerated Eμ-myc-induced lymphomagenesis compared to mir-17-92 (p<0.0001 for both comparisons, middle). Deficiency of miR-20 failed to affect the oncogenic cooperation between mir-17-92 and Eμ-myc, having minimal effects on tumor onset (right). (D) The mutation of miR-92 has minimal effects on the levels of the remaining mir-17-92 components. Eμ-myc B-lymphoma cells were infected with MSCV retrovirus overexpressing mir-17-92, mir-17-92Δ92 and mir-17-92Mut92 at an MOI (multiplicity of infection) of 1. Expression levels of miR-17, 18a, 19a, 20a, 19b and 92 were subsequently determined using Taqman miRNA assays. Error bars indicate standard deviation (n = 3). **p<0.01, ***p<0.001, ****p<0.0001.
The structural analogy to prokaryotic operons has led to the speculation that the co-transcribed mir-17-92 components can collectively contribute to oncogenesis. However, our studies reveal an unexpected functional interaction among mir-17-92 components. In the Eμ-myc mouse B-cell lymphoma model, while the intact mir-17-92 acts as an oncogene, its miR-92 component negatively regulates the oncogenic cooperation with c-Myc. This effect, at least in part, results from the ability of miR-92 to yield aberrant c-Myc dosage, which promotes a strong coupling between oncogene stress and p53-dependent apoptosis. Surprisingly, miR-92 functionally antagonizes miR-19, a key oncogenic mir-17-92 component, in the context of c-Myc-induced oncogenesis. During B-cell transformation, this miR-19:miR-92 antagonism is disrupted to favor a greater increase of miR-19 than miR-92. Thus, the polycistronic mir-17-92 employs an antagonistic interaction among its encoded miRNA components to confer an intricate crosstalk between the oncogene and tumor suppressor networks.
Results
Since mir-17-92 is overexpressed in human Burkitt’s lymphomas (Tagawa et al., 2007), we set out to functionally dissect mir-17-92 components in the Eμ-myc model of Burkitt’s lymphoma (Figure 1B). The Eμ-myc mice carry a c-myc transgene downstream of the immunoglobulin (Ig) heavy chain enhancer Eμ (Langdon, 1986; Adams et al., 1985), which functionally resembles the Ig-MYC translocations that occur frequently in Burkitt’s lymphomas (Tagawa et al., 2007). The resulting B-cell specific, aberrant c-Myc activation promotes excessive proliferation, yet also evokes potent, p53-dependent apoptosis (Schmitt et al., 2002; Hemann et al., 2003). Thus, c-Myc-induced apoptosis enables a self-defense mechanism against malignant transformation, producing B-lymphomas with a late onset (Lowe et al., 2004). In our adoptive transfer model (Olive et al., 2009), Eμ-myc/+ hematopoietic stem and progenitor cells (HSPCs) were transplanted into lethally irradiated recipient mice, generating chimeric mice that faithfully recapitulated the late tumor onset of the Eμ-myc transgenic mice (Figure 1B).
When Eμ-myc/+ HSPCs were infected with MSCV (murine stem cell virus) retrovirus to overexpress the intact mir-17-92 oncomir, we observed a considerable acceleration in tumor onset compared to the Eμ-myc/MSCV control mice (p<0.01, Figure 1C). Unexpectedly, the oncogenic cooperation between c-Myc and mir-17-92 was significantly stronger when miR-92 was deleted within this oncomir (Figure 1C). The average survival of Eμ-myc/17-92Δ92 mice was 66 days, significantly shorter than that of Eμ-myc/17-92 mice (112 days, p<0.0001). mir-17-92Δ92 carried a deletion of miR-92 pre-miRNA and its flanking sequences, which might alter the expression of the remaining mir-17-92 components (Figure 1D, Figure 1—figure supplement 1D). We then engineered a 12-nucleotide miR-92 seed mutation within mir-17-92 to abolish the functional miR-92 with minimal disruption to the overall gene structure. The resulting mir-17-92Mut92 phenocopied mir-17-92Δ92 in vivo (Figure 1C), significantly enhancing the oncogenic cooperation with c-Myc without altering the level of any remaining mir-17-92 components (Figure 1D, Figure 1—figure supplement 1D). This unexpected effect was specifically attributable to miR-92. Mutations of miR-20 or miR-17 failed to affect oncogenesis in the Eμ-myc model (Figure 1C, Figure 1—figure supplement 1D and data not shown), and mutations of both miR-19 miRNAs nearly abolished this oncogenic cooperation (Olive et al., 2009). This finding suggests that, although mir-17-92 acted as a potent oncogene as a whole, its miR-92 component confers an internal negative regulation on its oncogenic cooperation with c-Myc. This effect of miR-92 clearly contrasts with that of miR-19, a key oncogenic mir-17-92 component that promotes c-Myc-induced lymphomagenesis by repressing apoptosis (Mu et al., 2009; Olive et al., 2009; Mavrakis et al., 2010).
In the Eμ-myc model, a strong oncogenic lesion often leads to the B-cell transformation at an earlier developmental stage (Hemann et al., 2003). The greater oncogenic activity of mir-17-92Mut92 in comparison with mir-17-92 was consistent with mir-17-92Mut92 preferentially transforming IgM negative progenitor B-cells, and mir-17-92 frequently transforming IgM positive B-cells (Figure 2A; Table 1). In comparison to Eμ-myc/17-92 mice, both Eμ-myc/17-92Δ92 and Eμ-myc/17-92Mut92 mice developed more aggressive B-lymphomas, characterized by massive lymph node enlargement, splenic hyperplasia, leukemia, and widespread dissemination into visceral organs outside of the lymphoid compartment (Figure 2B, data not shown).
Figure 2
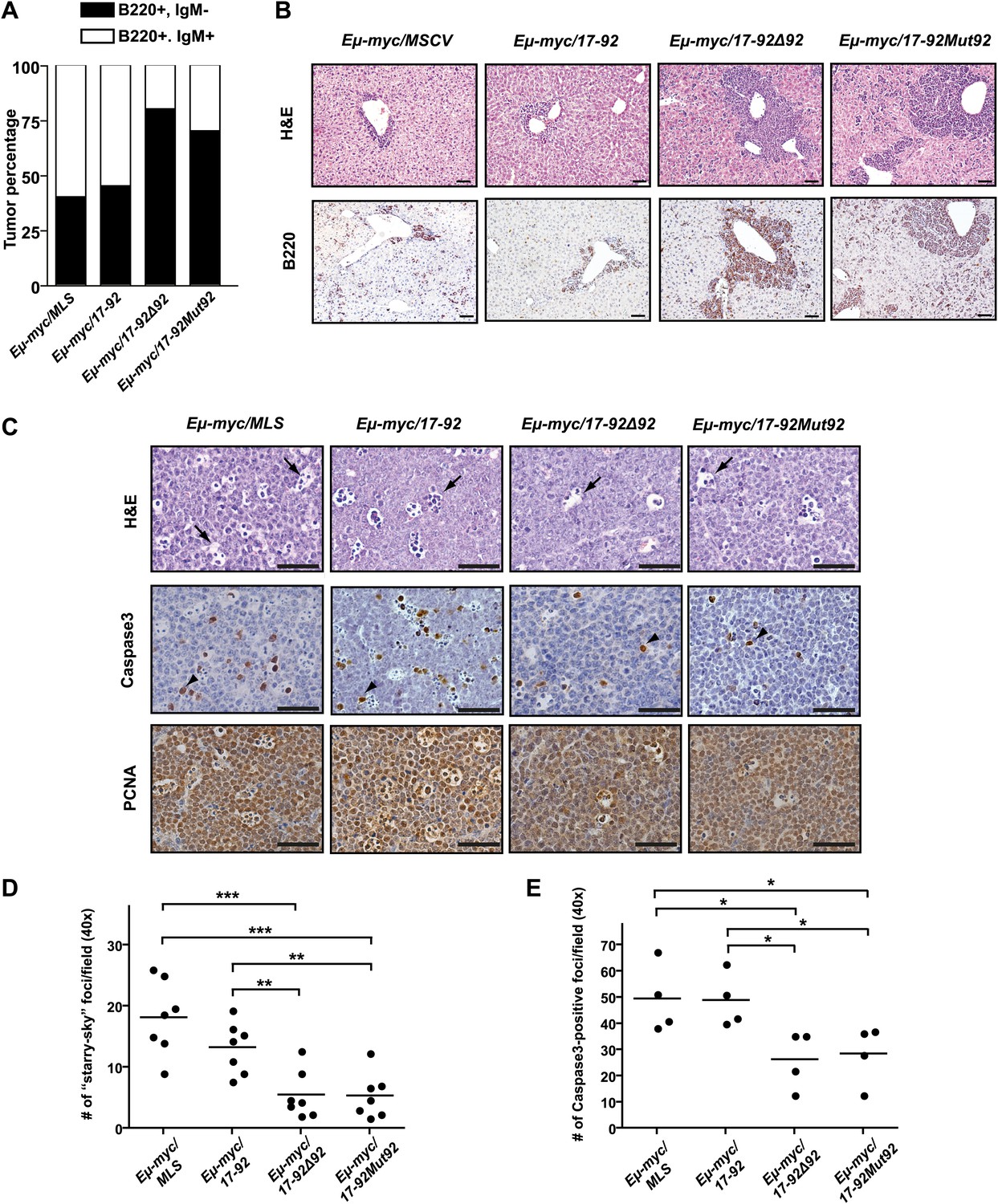
The miR-92 deficient mir-17-92 cooperates with c-Myc to promote highly aggressive B-lymphomas.
(A) The percentage of IgM positive and IgM negative B-lymphomas was calculated for each genotype (Eμ-myc/MSCV, n = 10; Eμ-myc/17-92, n = 9; Eμ-myc/17-92Δ92, n = 10; Eμ-myc/17-92Mut92, n = 10). (B) The Eμ-myc/17-92Mut92 and Eμ-myc/17-92Δ92 mice developed high grade B-lymphomas that were frequently disseminated into the liver. When compared to Eμ-myc/MSCV and Eμ-myc/17-92 mice, Eμ-myc/17-92Mut92 and Eμ-myc/17-92Δ92 lymphomas gave rise to more liver dissemination, as indicated by H&E and B220 staining. (C) Eμ-myc/17-92Mut92 and Eμ-myc/17-92Δ92 lymphomas exhibited a decreased apoptosis compared to Eμ-myc/MSCV or Eμ-myc/17-92 lymphomas. Representative lymphomas were stained for H&E, cleaved caspase-3 and PCNA. Arrow, ‘starry sky’ feature of apoptotic lymphoma cells; arrowhead, apoptotic cells with positive staining for cleaved caspase-3; scale bar, 50 μm. (D and E) Apoptosis was quantitatively measured in representative lymphomas of each genotype using the ‘starry sky’ features (D) and cleaved caspase-3 staining (E). *p<0.05, **p<0.01, ***p<0.001.
Table 1
Flow cytometric immunophenotyping of Eμ-myc lymphomas with enforced expression of different mir-17-92 derivatives
https://doi.org/10.7554/eLife.00822.006| Genotype | n | Percentage (%) | Immunotype |
|---|---|---|---|
| Eμ-myc/MSCV | 4 | 40 | B220+, IgM−, CD19+, CD4−, CD8− |
| 6 | 60 | B220+, IgM+, CD19+, CD4−, CD8− * | |
| Eμ-myc/17–92 | 4 | 40 | B220+, IgM−, CD19+, CD4−, CD8− |
| 5 | 50 | B220+, IgM+, CD19+, CD4−, CD8− † | |
| 1 | 10 | B220−, IgM−, CD19−, CD4+, CD8+ | |
| Eμ-myc/17–92Mut92 | 7 | 70 | B220+, IgM−, CD19+, CD4−, CD8− |
| 3 | 30 | B220+, IgM+, CD19+, CD4−, CD8− ‡ | |
| Eμ-myc/1792Δ92 | 8 | 80 | B220+, IgM−, CD19+, CD4−, CD8− |
| 2 | 20 | B220+, IgM+, CD19+, CD4−, CD8− § |
-
*
1 out of 6 samples predominantly contains IgM+ cells, with a small percentage of IgM− cells.
-
†
3 out of 5 samples predominantly contain IgM+ cells, with a small percentage of IgM− cells.
-
‡
1 out of 3 samples predominantly contains IgM+ cells, with a small percentage of IgM− cells.
-
§
1 out of 2 samples predominantly contains IgM+ cells, with a small percentage of IgM− cells.
During Myc-induced tumorigenesis, aberrant c-Myc dosage yields simultaneous induction of proliferation and apoptosis, imposing a unique selective pressure for pro-survival lesions (Evan and Vousden, 2001). Thus, we compared the extent of Myc-induced apoptosis in the Eμ-myc/17-92, Eμ-myc/17-92Δ92, Eμ-myc/17-92Mut92, and control Eμ-myc/MSCV lymphomas. The control Eμ-myc/MSCV lymphomas invariably exhibited a high proliferation index accompanied by extensive cell death, as evidenced by the widespread ‘starry sky’ pathology (Figure 2C,D) and cleaved caspase 3 staining (Figure 2C,E). The potent oncogenic activity of mir-17-92Δ92 and mir-17-92Mut92 was consistent with the strong reduction of apoptosis in the lymph node tumors. In comparison, the intact miR-92 significantly attenuated the repression of c-Myc-induced apoptosis by mir-17-92 in vivo (Figure 2C–E).
We next investigated the effect of miR-92 alone in regulating c-Myc-induced apoptosis. In the Eμ-myc model, miR-92 overexpression significantly enhanced c-Myc-induced apoptosis in vivo (Figure 3A,B, Figure 3—figure supplement 1A), consistent with a rapid depletion of miR-92-infected cells in premalignant Eμ-myc B-cells (Figure 3—figure supplement 1B). Similar miR-92 effects on c-Myc-induced apoptosis were observed in vitro. The R26MER/MER mouse embryonic fibroblasts (MEFs) carry a switchable variant of Myc, MycERT2, downstream of the constitutive Rosa26 promoter, which allows acute activation of the MycER transgene by 4-OHT (4-Hydroxytamoxifen) induced nuclear translocation (Murphy et al., 2008). The R26MER/MER MEFs recapitulate c-Myc-induced apoptosis in vitro, as activated MycERT2 induces p53-dependent apoptosis in response to serum starvation (Murphy et al., 2008). Enforced miR-92 expression in R26MER/MER MEFs invariably enhanced Myc-induced apoptosis (Figure 3C, Figure 3—figure supplement 1C).
Figure 3 with 1 supplement see all
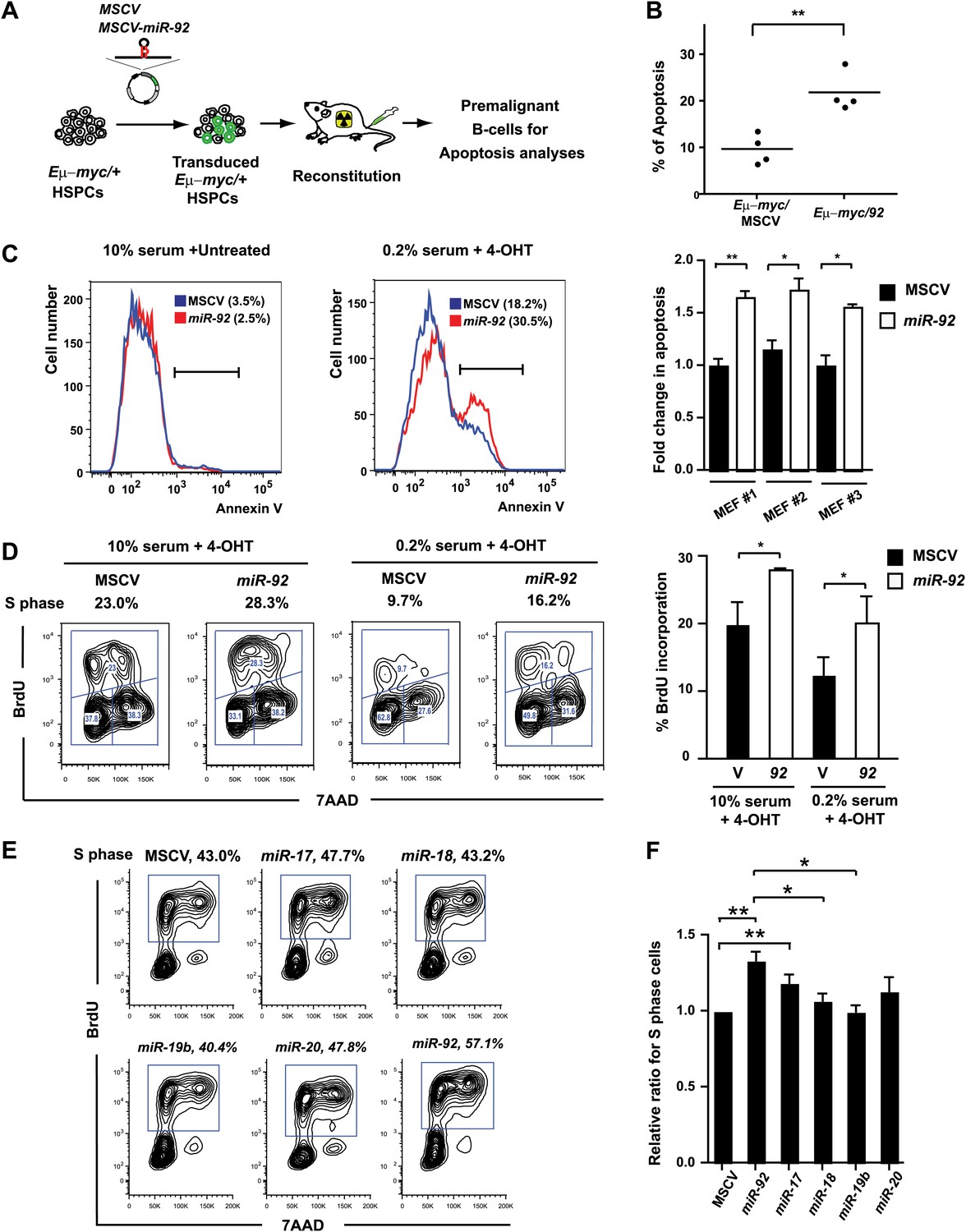
miR-92 enhances both c-Myc-induced apoptosis and c-Myc-induced proliferation.
(A) The schematic representation of the adoptive transfer model to evaluate the miR-92 effects on the Eμ-myc premalignant B-cells in vivo. (B) miR-92 overexpression enhances the apoptotic response in the premalignant Eμ-myc B-cells in vivo. Using the Eμ-myc adoptive transfer model, we generated well-controlled Eμ-myc/MSCV and Eμ-myc/92 mice reconstituted from donor matched Eμ-myc HSPCs. Premalignant Eμ-myc splenic B-cells were isolated from the Eμ-myc/MSCV and Eμ-myc/92 mice 6 weeks after reconstitution. The in vivo apoptosis was measured by the level of caspase activation using Red-VAD-FMK, a fluorescently labeled caspase inhibitor that specifically bound to cleaved caspases. The percentage of Eμ-myc B-cells positive for cleaved caspases was shown for four independent experiments. (C) Enforced miR-92 expression in R26MER/MER MEFs significantly enhanced c-Myc-induced apoptosis. miR-92 overexpressing and the control R26MER/MER MEFs were serum starved, and the MycERT2 transgene was activated by 4-OHT treatment. The level of apoptosis of each MEF was measured using Annexin V staining before (left) and after (middle) 4-OHT treatment and serum starvation. Quantification of c-Myc-induced apoptosis was performed in three independent MEF lines that overexpressed MSCV or miR-92 (right panel, error bars represent SEM). (D) Enforced miR-92 expression in R26MER/MER MEFs significantly enhanced c-Myc-induced proliferation. Proliferative effects of miR-92 was measured by BrdU incorporation in MycERT2 activated R26MER/MER MEFs. miR-92 cooperated with c-Myc to promote BrdU incorporation in both 10% (left) and 0.2% (middle) serum culture conditions. Quantification of BrdU incorporation was performed in two independent experiments (right). (E) miR-92 is a potent mir-17-92 component to promote primary B-cell proliferation. The proliferative effects of all mir-17-92 miRNAs were measured individually in primary B-cells using BrdU incorporation. (F) The quantification of BrdU incorporation in experiments described in (E) was performed in four independent experiments. Error bars represent standard deviation, *p<0.05, **p<0.01.
In addition to promoting c-Myc-induced apoptosis, miR-92 unexpectedly enhanced c-Myc-induced cell proliferation. A significant increase of BrdU incorporation was observed in R26MER/MER MEFs overexpressing miR-92, both under normal culture conditions and, more evidently, under serum starvation (Figure 3D). The same proliferative effect of miR-92 was also observed in primary B-cells. Comparison of the proliferative effect of each mir-17-92 component in bone marrow derived primary B-cells revealed that the miR-92 component yielded one of the strongest effects (Figure 3E,F). In addition, miR-92 deficiency significantly compromised the ability of mir-17-92 to promote cell cycle progression in B-cells (Figure 3—figure supplement 1D). Interestingly, strong proliferative effects have been reported for nearly all mir-17-92 components, yet the exact cell type and biological context can select specific components as the predominant drivers for cell proliferation. Taken together, our data suggest that miR-92 is a unique mir-17-92 component that functionally couples c-Myc-induced cell proliferation and c-Myc-induced apoptosis in the B-cell compartment.
To investigate the molecular mechanism underlying miR-92 functions, we performed microarray analyses comparing gene expression profiles of R26MER/MER MEFs overexpressing miR-92 or the control MSCV vector. These MEFs were serum starved and 4-OHT treated to trigger strong Myc-induced apoptosis. miR-92-upregulated genes were significantly enriched for the cell cycle pathway, including ccnd1, ccnb1, ccnb2, cdc25b, cdc25c, and cdk4 (Figure 4A,B), consistent with the ability of miR-92 to promote Myc-induced cell proliferation. Genes upregulated by miR-92 were also enriched for the p53 pathway, including the classic p53 target mdm2, as well as the pro-apoptotic p53 targets—noxa, bax, puma, perp, and bid (Figure 4A,B, Figure 4—figure supplement 1A). Since aberrant c-Myc activation triggered a p53-dependent apoptotic response (Lowe et al., 2004), our observation is consistent with miR-92 further enhancing p53 activation downstream of c-Myc. Interestingly, p21, a canonical p53 target, was not induced by miR-92 in the MycERT2 activated R26MER/MER MEFs (Figure 4—figure supplement 1A). It is likely that the transcriptional repression of p21 by c-Myc renders p21 irresponsive to p53 activation under this biological context (Heasley et al., 2002). Using real-time PCR, we validated the ability of miR-92 to induce cell cycle genes and activate p53 targets in both R26MER/MER MEFs, as well as primary B-cells (Figure 4C, Figure 4—figure supplement 1A,B). Hence, the molecular signature imposed by miR-92 overexpression is consistent with its functional readout.
Figure 4 with 1 supplement see all

miR-92 induces apoptosis through the activation of the p53 pathway.
(A) The genes upregulated by miR-92 were enriched for the cell cycle pathway and the p53 pathway. Microarray analyses compared gene expression profiles of serum starved and 4-OHT treated R26MER/MER MEFs overexpressing either miR-92 or a control MSCV vector (n = 3). The differentially expressed genes were defined as those with at least 1.5-fold expression level change using SAM (Significance analysis of microarrays, false discovery rate <1%). Pathway analyses were performed on upregulated and downregulated genes using the KEGG database. (B) The heatmaps of the miR-92 upregulated genes enriched for the cell cycle and p53 pathways. (C) Components of the cell cycle and p53 pathways were upregulated upon miR-92 overexpression in both MEFs (left) and primary B-cells (right). The quantitation of gene expression was performed using real time PCR. (D) miR-92 overexpression induces the accumulation of Arf and p53 proteins in MEFs and primary B-cells from bone marrow. Western analyses were performed on the R26MER/MER MEFs (left) and primary B-cells (right) that overexpressed miR-92 or a control MSCV vector in two independent experiments. The infected R26MER/MER MEFs were assayed at 6 hr after serum starvation and 4-OHT treatment; the infected primary B-cells were collected 72 hr post infection. (E) The apoptotic effect of miR-92 requires an intact p53 pathway. We infected R26MER/MER MEFs with two MSCV retrovirus, MSCV-p53shRNA and MSCV-92, to obtain doubly infected cells. Knocking down p53 in R26MER/MER MEFs abolished the ability of miR-92 to enhance c-Myc-induced apoptosis, as measured by Annexin V staining (two left panels). The percentage of apoptotic MEFs of each experimental condition was quantitatively measured (right). (F) The induction of the p53 pathway components by miR-92 is dependent on an intact p53. Knocking down p53 in R26MER/MER MEFs abolished the ability of miR-92 to induce pro-apoptotic p53 targets and other canonical p53 targets, including noxa, perp and mdm2. Error bars represent standard deviation, *p<0.05, **p<0.01, ***p<0.001.
The activation of the p53 pathway by c-Myc is essential for the induction of the apoptotic response in the Eμ-myc model (Schmitt et al., 2002). A major mechanism that governs Myc-induced p53 activation is the transcriptional induction of the gene encoding Arf, which inhibits Mdm2-mediated p53 ubiquitination and degradation (Lowe et al., 2004; Campaner and Amati, 2012). The ability of miR-92 to enhance c-Myc-induced apoptosis and to increase the expression of p53 targets raised the possibility that miR-92 overexpression activates p53 possibly through elevated Arf. In both R26MER/MER MEFs and wild-type primary B-cells, miR-92 overexpression alone caused significant accumulation of Arf mRNA and protein (Figure 4C,D, Figure 4—figure supplement 1C), consistent with the rapid stabilization of the p53 protein (Figure 4D, Figure 4—figure supplement 1C) without alteration of p53 mRNA (Figure 4—figure supplement 1D). Notably, the ability of miR-92 to induce p53 activation occurred not only in 4-OHT treated R26MER/MER MEFs with MycERT2 activation, but also in untreated R26MER/MER MEFs with normal c-Myc level. This was clearly demonstrated by the elevation of p53 protein level, as well as the increased p53 target expression (Figure 4—figure supplement 1B,C).
The induction of p53 by miR-92 prompted us to investigate the functional importance of p53 in miR-92-induced apoptotic response. Knockdown of p53 in R26MER/MER MEFs not only led to a suppression of c-Myc-induced apoptosis, but also completely abolished the effect of miR-92 to enhance c-Myc-induced apoptosis (Figure 4E). These findings suggested that an intact p53 pathway is required for the apoptotic effect of miR-92. Consistently, the miR-92 induction of the pro-apoptotic genes, including noxa, perp, and mdm2, also was mediated by the intact p53 (Figure 4F). Thus, aberrant c-Myc activation triggers an apoptotic response through p53 activation; and co-expression of miR-92 with c-Myc leads to an even stronger p53 activation, and subsequently apoptotic response.
Our findings suggest parallels between c-myc and miR-92: both are potent oncogenes that promote excessive cell proliferation coupled with p53-dependent apoptosis, and both are capable to induce expression of cell cycle genes (ccnb1, ccnd1, cdk4, and cdc25) (Lowe et al., 2004; Campaner and Amati, 2012) and p53 pathway components (Arf, puma, noxa, perp, and mdm2) (Lowe et al., 2004; Campaner and Amati, 2012). The functional analogy between c-Myc and miR-92, as well as the molecular overlap between their downstream pathways, led us to investigate the effect of miR-92 on c-Myc. Intriguingly, miR-92 expression significantly enhanced c-Myc protein level both in MEFs and in primary B-cells (Figure 5A), without affecting the c-myc mRNA level (Figure 5—figure supplement 1A, data not shown). Consistent with the stabilization of endogenous c-Myc, miR-92 overexpression in R26MER/MER MEFs stabilized the MycERT2 protein (Figure 5B). The dosage of c-Myc protein is crucial for its biological readout (Murphy et al., 2008). While c-Myc dosage determines the extent of cell cycle gene induction and cell proliferation, it also regulates the degree of p53 activation and subsequent apoptosis (Murphy et al., 2008) (Figure 5—figure supplement 1B). Thus, the ability of miR-92 to induce aberrant c-Myc accumulation likely constitutes the molecular basis for its ability to promote both cell proliferation and p53-dependent apoptosis.
Figure 5 with 1 supplement see all
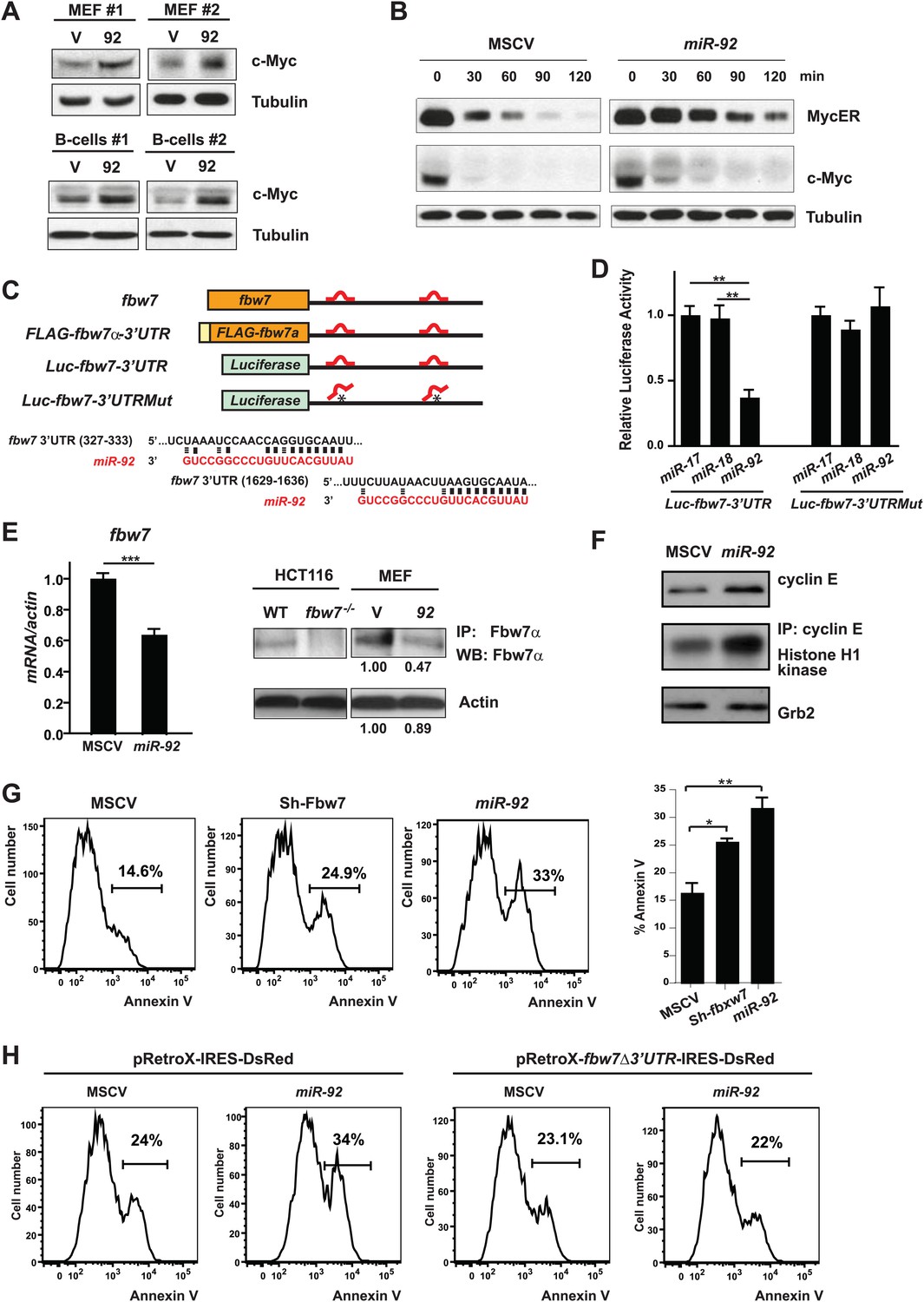
miR-92 promotes the accumulation of c-Myc protein through repressing Fbw7.
(A) miR-92 enhances the accumulation of c-Myc protein in synchronized R26MER/MER MEFs (upper), as well as primary B-cells (lower). The miR-92 overexpression and the control R26MER/MER MEFs were synchronized by serum starvation and were collected 12 hr after being released into serum culture conditions to determine the c-Myc protein level. This synchronization approach in R26MER/MER MEFs has provided us with the most consistent measurement for c-Myc protein level, because it is regulated in a cell-cycle-dependent manner. (B) miR-92 overexpression decreases the turnover of c-Myc protein. Serum-synchronized R26MER/MER MEFs that overexpress either miR-92 or the control MSCV vector were released into the serum for 6 hr, treated with cycloheximide, collected at the indicated time points, then analyzed by western blot to determine the levels of MycER and the endogenous c-Myc protein. (C) Schematic representation of the two miR-92 binding sites in the murine fbw7 3′UTR. Additionally, a luciferase reporter and a FLAG tagged fbw7 ORF were each placed upstream of a wild-type fbw7 3′UTR, or a mutated fbw7 3′UTR that abolished the predicted miR-92 binding. (D) The expression of Luc-fbw7-3′UTR was specifically repressed by miR-92 in Dicer−/− HCT116, while mutations of the two putative miR-92 binding sites within the fbw7-3′UTR (Luc-fbw7-3′UTRMut) abolished this repression. (E) The endogenous fbw7 gene was downregulated by miR-92 post-transcriptionally. Both the endogenous fbw7 mRNA (left) and the endogenous Fbw7 protein (right) were repressed upon miR-92 overexpression in R26MER/MER MEFs. Due to the lack of a proper antibody to detect endogenous Fbw7 in regular western analysis, we demonstrated the downregulation of endogenous Fbw7 by miR-92 using immunoprecipitation followed by immunoblotting with a polyclonal anti-Fbw7 antibody. (F) miR-92 enhances the accumulation of Cyclin E protein. Overexpression of miR-92 increased the accumulation of Cyclin E protein, which was further confirmed by the increased Cyclin E-dependent kinase activity. (G) The knockdown of fbw7 resembles the effect of miR-92 to enhance c-Myc-induced apoptosis. Knocking down fbw7 in R26MER/MER MEFs enhanced c-Myc-induced apoptosis, partially recapitulating the phenotype caused by miR-92 overexpression. Apoptosis was quantitatively measured by Annexin V staining in two independent lines of R26MER/MER MEFs upon serum starvation and 4-OHT treatment. (H) Overexpression of fbw7 abolished the apoptotic effects of miR-92 in R26MER/MER MEFs. R26MER/MER MEFs were doubly infected by pRetro-fbw7αΔ3′UTR-IRES-dsRed and MSCV-miR-92. The c-Myc-induced apoptosis was quantitatively measured by Annexin V staining in doubly infected R26MER/MER MEFs upon serum starvation and 4-OHT treatment. Error bars represent standard deviation). *p<0.05; **p<0.01.
Based on our findings, we speculated that miR-92 targets could include negative regulators of c-Myc protein accumulation. Therefore, we searched genes known to negatively regulate c-myc for the presence of putative miR-92 binding sites. Using the Targetscan and RNA22 algorithms (Lewis et al., 2005; Miranda et al., 2006; Bartel, 2009), we identified eight candidate miR-92 targets, each of which contained one or more predicted miR-92 binding sites in the 3′ untranslated region (3′UTR). Real-time PCR analysis of these candidate genes confirmed fbw7 (F-box and WD repeat domain-containing 7) as a likely target of miR-92 (Figure 5C, Figure 5—figure supplement 1C). fbw7, which contains two miR-92 target sites within its 3′UTR (Figure 5C), is the substrate recognition component of an SCF-type E3 ubiquitin ligase that mediates the degradation of several proto-oncoproteins, including Myc, Cyclin E, c-Jun, and Notch (Welcker and Clurman, 2008; Crusio et al., 2010; Wang et al., 2012). A luciferase reporter or a FLAG-tagged fbw7-encoding ORF (open reading frame), when fused to the wild-type fbw7 3′ UTR, were both significantly repressed in a miR-92 dependent manner (Figure 5D, Figure 5—figure supplement 1D). Yet enforced miR-92 expression failed to repress the luciferase reporter that contained an fbw7 3′UTR with two mutated miR-92 binding sites (Figure 5D), suggesting that miR-92 binding to fbw7 3′UTR is required for this repression. Furthermore, miR-92 effectively repressed endogenous Fbw7 protein level, as demonstrated by the decreased fbw7 mRNA level and Fbw7 immunoprecipitation (Figure 5E). Consistent with fbw7 as an important target for miR-92, enforced miR-92 expression upregulated multiple Fbw7 substrates at their protein levels, including c-Myc and cyclinE (Figure 5F). Observations from our in vivo experiments also supported this post-transcriptional regulation of Fbw7 by miR-92, as we observed an inverse correlation between the level of miR-92 and fbw7 when comparing Eμ-myc/17-92Δ92 and Eμ-myc/17-92 lymphoma cells (Figure 5—figure supplement 1E).
fbw7 has previously been postulated as a potential miR-92 target based on the presence of miR-92 target sites (Mavrakis et al., 2011), yet it remains unclear how fbw7 mediated the pro-apoptotic effects of miR-92, given its well-characterized functions as a tumor suppressor. Recent findings indicate that the acute inactivation of tumor suppressor Fbw7 imposes a strong oncogenic stress to induce p53-dependent apoptosis, conferring a selective advantage to cells with deficient p53 function (Minella et al., 2007; Onoyama et al., 2007; Matsuoka et al., 2008; Grim et al., 2012). This p53-dependent apoptosis is, at least in part, due to an aberrant increase of c-Myc dosage (Onoyama et al., 2007; Matsuoka et al., 2008). These findings suggested that a major mechanism through which miR-92 enhanced the c-Myc protein level, and subsequently, c-Myc-induced apoptosis, could be through its direct repression of Fbw7. In support of this hypothesis, miR-92 overexpression significantly increased the c-Myc protein level in wild-type Hct116 cells, but not in FBW7−/− Hct116 cells (Figure 5—figure supplement 1F), suggesting FBW7 was essential for miR-92 to induce c-MYC increase. Functionally, acute fbw7 knockdown in R26MER/MER MEFs partially phenocopied the effect of miR-92 to enhance c-Myc-induced apoptosis (Figure 5G, Figure 5—figure supplement 1G); while overexpression of an fbw7α open reading frame (ORF), albeit above its physiological level, completely abolished this apoptotic effect of miR-92 (Figure 5H, Figure 5—figure supplement 1H). Nevertheless, it is still likely that additional mechanisms downstream of miR-92 also promote its apoptotic effects, because fbw7 knockdown largely recapitulated the extent of c-Myc upregulation by miR-92 (Figure 5—figure supplement 1G), yet only partially phenocopied its pro-apoptotic effects (Figure 5G). In addition, overexpression of fbw7 above its physiological level might amplify the extent of functional interactions between fbw7 and miR-92 in regulating apoptosis. Despite these caveats, our results strongly argue that the miR-92-Fbw7 axis constitutes a major mechanism underlying the pro-apoptotic effects of miR-92.
Downregulation of Fbw7 by miR-92 significantly enhanced the protein level of c-Myc in R26MER/MER MEFs and in primary B-cells (Figure 5A). It is conceivable that the ability of miR-92 to repress Fbw7 in vivo could similarly enhance the c-Myc accumulation in the Eμ-myc/92 premalignant B-cells, promoting rapid cell proliferation and a p53-dependent apoptotic response. Unfortunately, due to technical limitations, we were not able to demonstrate an increased c-Myc protein level as a result of miR-92 overexpression in the Eμ-myc premalignant B-cells. There was a significant depletion of the Eμ-myc/92 premalignant B-cells due to excessive apoptosis (Figure 3—figure supplement 1B), making it difficult to collect enough cells to analyze the c-Myc protein level by western analyses. Similarly, we could not obtain enough cells to compare the protein level of c-Myc in the premalignant B-cells from the Eμ-myc/17-92, Eμ-myc/17-92Δ92, and Eu-myc/MSCV animals. Nevertheless, our functional studies in cell culture, combined with the inverse expression correlation between fbw7 and miR-92 in vivo, strongly argue the importance of the miR-92-Fbw7-Myc axis to promote the pro-apoptotic effects of miR-92.
In the context of the c-Myc cooperation, mir-17-92 encodes miRNA components with opposing biological functions. While miR-19 miRNAs repress c-Myc-induced apoptosis to promote Eμ-myc lymphomagenesis (Mu et al., 2009; Olive et al., 2009), miR-92 enhances c-Myc-induced apoptosis to attenuate the tumorigenic effects. Consistent with the opposing effects of miR-19 and miR-92, co-expression of these two miRNAs as a dicistron attenuated the apoptotic effect of miR-92 in premalignant Eμ-myc B-cells in vivo (Figure 6A,B, Figure 6—figure supplement 1A). A similar antagonistic interaction was also observed in R26MER/MER MEFs; and introducing a miR-19b mutation within mir-19b-92 dicistron abolished this interaction (Figure 6C). Since miR-19 represses pten to promote the PI3K/AKT pathway, the activation of AKT signaling would lead to increased phosphorylation of Mdm2, thus destabilizing p53 to dampen the apoptotic response induced by miR-92 (Gottlieb et al., 2002; Ogawara et al., 2002). Consistent with this hypothesis, we observed a decreased p53 induction and an unaltered c-Myc level when miR-92 was co-expressed with miR-19 (Figure 6D, Figure 6—figure supplement 1B).
Figure 6 with 1 supplement see all
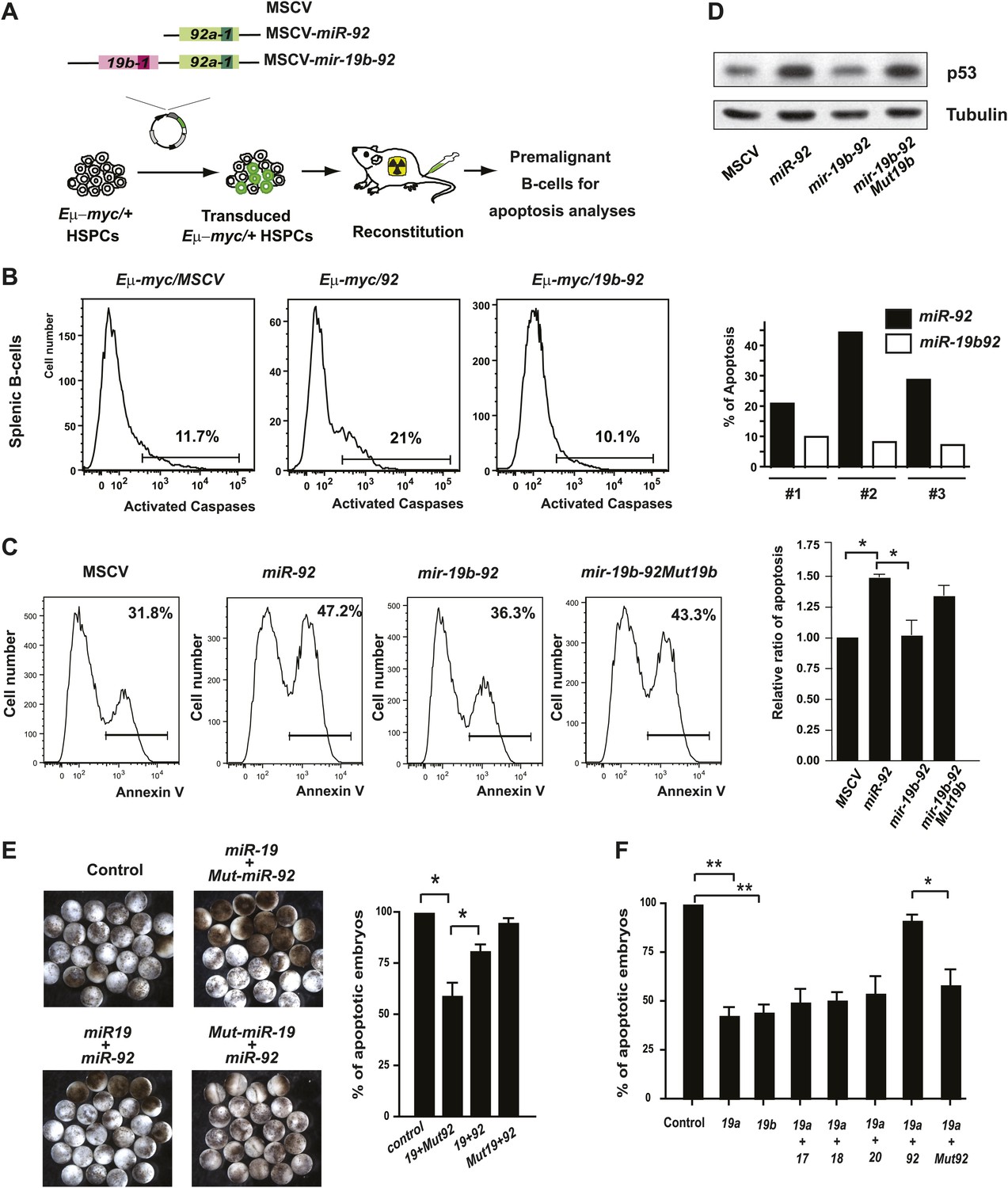
The antagonistic interaction between miR-19 and miR-92 regulates the balance between proliferation and apoptosis.
(A) The schematic representation of the Eμ-myc adoptive transfer model to evaluate the functional interaction between miR-92 and miR-19 in vivo. Light colored boxes, pre-miRNAs; dark colored boxes, mature miRNAs. (B) miR-19 antagonizes the apoptotic effects of miR-92 in vivo. miR-92 overexpression in the Eμ-myc adoptive transfer model enhanced apoptosis in premalignant Eμ-myc splenic B-cells, while the mir-19b-92 dicistron expression abolished this apoptotic effect (left three panels). A quantitative analysis of apoptosis by FACS was shown for three independent, well-controlled experiments (right). (C) miR-19b dampens the miR-92-induced apoptosis in MycERT2 activated R26MER/MER MEFs. R26MER/MER MEFs were infected by miR-92, mir-19b-92, mir-19b-92Mut19b and the MSCV control vector, and were subsequently serum starved and treated with 4-OHT to activate MycERT2. Apoptosis in these samples was measured quantitatively using Annexin V staining (left four panels). The extent of apoptosis induced by MSCV, miR-92, mir-19b-92, mir-19b-92Mut19b was normalized to that of MSCV infected R26MER/MER MEFs and then averaged from four independent experiments (right). (D) miR-19b dampens the miR-92-induced p53 activation. R26MER/MER MEFs that overexpress the indicated constructs (miR-92, mir-19b-92Mut19b and mir-19b-92) were collected 48 hr after infection and then analyzed by western blot to determine the level of p53 protein. (E) miR-92 and miR-19 exhibit antagonistic effects to regulate hydroxyurea (HU)-induced cell death in Xenopus embryos. Representative images of HU-treated Xenopus embryos that were co-injected with human Ago2 and the indicated miRNA mimics (left). Co-injection of miR-92 dampened the cell survival effects of miR-19 on HU-induced apoptosis (right, n = 3, with >20 embryos in each group). (F) miR-92 exhibits a specific antagonistic interaction with miR-19. Injection of miR-19a or miR-19b rescued HU-induced apoptosis in Xenopus embryos. Co-injection of miR-92, but not a mutated miR-92, or other mir-17-92 components, dampened the cell survival effect of miR-19 (n = 3, with >20 embryos in each group). Error bars represent standard deviation, *p<0.05; **p<0.01.
This miR-19:miR-92 antagonism appears to be conserved evolutionarily. In Xenopus laevis, miR-19 and miR-92 have identical sequence to their mammalian orthologs (Figure 1—figure supplement 1B). Based on Targetscan and RNA22 miRNA target prediction algorisms (Lewis et al., 2003, 2005; Miranda et al., 2006; Grimson et al., 2007), their target specificity is also conserved for key miRNA targets, although the exact binding sites may or may not be conserved (Olive et al., 2009) (Figure 6—figure supplement 1C). In addition, the biological functions of miR-19 and miR-92 exhibit evolutionary conservation between Xenopus laevis and mammals. Individual injection of miR-19 promoted cell survival of hydroxyurea-treated Xenopus embryos, while co-injection of miR-19a and miR-92 significantly attenuated this pro-survival effect (Figure 6E). This functional antagonism was specific for miR-92 and miR-19, since co-injection of other mir-17-92 components or a mutated miR-92 did not yield any functional interactions in combination with miR-19 (Figure 6F).
Given the opposing biological effects of miR-19 and miR-92 during c-Myc-induced lymphoma development, differential regulation of these two miRNA families could determine the oncogenic activity of mir-17-92. Under normal physiological conditions, this miR-19:miR-92 antagonism could attenuate the detrimental oncogenic signaling by inducing apoptosis in cells with inappropriate mir-17-92 induction. During malignant transformation, and particularly during c-Myc-induced oncogenesis, this miR-19:miR-92 antagonism could be disrupted to favor cell survival. Using real time PCR analyses, we compared the relative abundance of miR-19a, miR-19b, and miR-92 in normal splenic B-cells, premalignant Eμ-myc B-cells, and Eμ-myc lymphomas (Figure 7A). Comparing to normal splenic B-cells, the levels of all three mature miRNA species were elevated in both premalignant and malignant Eμ-myc B-cells, possibly due to transcriptional activation of mir-17-92 by c-Myc (Donnell et al., 2005). However, the miR-19 to miR-92 ratios significantly increased during c-Myc-induced lymphomagenesis (Figure 7B). In other words, when normalized to the respective miRNA levels in normal splenic B-cells, mature miR-19 (including miR-19a and miR-19b) exhibited a greater increase in premalignant and malignant Eμ-myc B-cells than mature miR-92 (Figure 7A–C). This differential increase was most evident in premalignant Eμ-myc B-cells; the fully transformed Eμ-myc B-lymphoma cells exhibited a lesser difference (Figure 7A,B). This observation is consistent with premalignant Eμ-myc B-cells having an intact p53-dependent apoptotic response, thus a stronger selective pressure for a greater miR-19:miR-92 ratio. In comparison, most Eμ-myc B-lymphomas have a defective p53 response, hence a less strong selective pressure to maintain a high miR-19:miR-92 ratio. We also validated this observation using northern analysis. Comparing normal splenic B-cells and multiple Eμ-myc lymphoma cells, the levels of the mature miR-19a, miR-19b and miR-92 were all elevated in transformed B-cells; however, the degree of increase for miR-19a and miR-19b was significantly higher than that of miR-92 (Figure 7C). This differential increase of miR-19 and miR-92 was also observed in human Burkitt’s lymphoma cell lines when compared to normal B-cells isolated from the periphery blood (Figure 7D). More importantly, this phenomenon was not limited to c-myc driven B-lymphomas. In the LT2-MYC murine model of hepatocellular carcinoma (HCC), where tumor development was initiated by tetracycline-inducible c-Myc expression, miR-19a and miR-19b also exhibited a stronger increase than miR-92 when comparing tumor cells and the normal counterpart (Figure 7E).
Figure 7
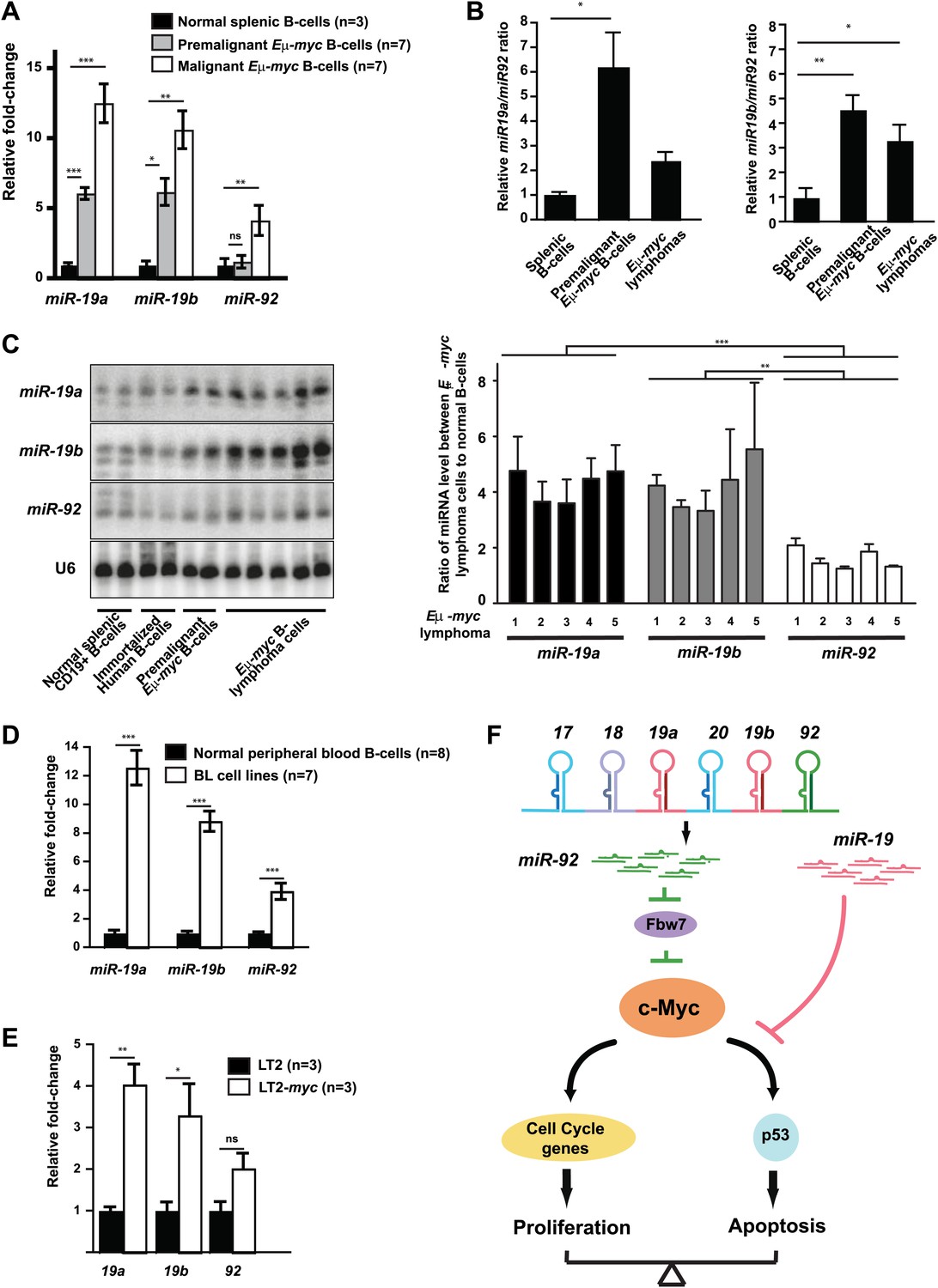
The miR-19:miR-92 antagonism is disrupted during malignant transformation.
(A and B) Compared to normal splenic B-cells, premalignant and malignant Eμ-myc B-cells favored a greater increase in mature miR-19 (miR-19a and miR-19b) than miR-92. The purified normal splenic B-cells, premalignant Eμ-myc bone marrow B-cells and malignant Eμ-myc B-lymphoma cells were subjected to Taqman miRNA assays to determine the expression level of miR-19a, miR-19b and miR-92. Comparing premalignant/malignant Eμ-myc B-cells vs normal splenic B-cells, all three miRNAs exhibited an increased level, although the increase in miR-19a or miR-19b was significantly higher than that of miR-92 (A). In the same experiment, the relative ratios for miR-19a:miR-92 and miR-19b:miR-92 were measured for all normal splenic B-cells and Eμ-myc B-cells (B). (C) Mature miR-19 and miR-92 are differentially expressed in normal splenic B-cells and Eμ-myc B-lymphoma cells. The normal splenic B-cells, immortalized human B-cells, premalignant Eμ-myc/+ B-cells, and Eμ-myc/+ B-lymphoma cells were subjected to Northern analysis. Compared to normal splenic B-cells, both malignant and premalignant Eμ-myc/+ B-cells favored a greater increase of miR-19 than miR-92. (D) Compared to normal B-cells isolated from peripheral blood, human Burkitt’s lymphoma cell lines favor a greater increase in mature miR-19 than miR-92. (E) Compared to normal livers (LT2), mouse hepatocellular carcinomas caused by the inducible c-Myc over-expression (LT2-myc) favor a greater increase in mature miR-19 than miR-92. (F) A diagram describes our proposed model to explain the functional interactions between miR-92 and miR-19 in c-Myc-induced B-lymphomagenesis. Aberrant c-Myc expression couples rapid proliferation and p53-dependent apoptosis. miR-92 overexpression further increases c-Myc dosage to strengthen this coupling, at least in part by repressing Fbw7. This miR-92 effect ensures a potent mechanism to eliminate premalignant c-Myc overexpressing cells. Interestingly, miR-92 and can be antagonized by the survival effects of the miR-19 miRNAs encoded by the same mir-17-92 miRNA polycistron. Taken together, while miR-19 miRNAs repressed c-Myc-induced apoptosis to promote the oncogenic cooperation between mir-17-92 and c-Myc, miR-92 exhibits a negative regulation. Thus, the antagonistic interactions between miR-92 and miR-19 confer an intricate crosstalk between proliferation and apoptosis. Error bars represent standard deviation, *p<0.05; **p<0.01, ***p<0.001.
These observations were consistent with a previous finding, where the inducible c-myc activation in a human Burkitt’s lymphoma cell line induced both miR-19a and miR-19b to a greater extent than miR-92 (Donnell et al., 2005). Although miR-19 and miR-92 are co-transcribed from the mir-17-92 precursor, the differential increase of miR-19 vs miR-92 occurs in multiple c-Myc-driven tumor types. Thus, the relative abundance of miR-19 and miR-92 could constitute an important molecular basis to regulate the initiation and progression of c-Myc-induced tumor development.
Discussion
The unique polycistronic structure of mir-17-92 constitutes the basis for its pleiotropic functions and the complex mode of interactions among its miRNA components. A high level of mir-17-92 in normal or premalignant cells could lead to suboptimal consequences that are counter-balanced through an intrinsic negative regulation by miR-92 (Figure 7F). As we demonstrated in vitro where miR-92, by directly downregulating Fbw7, enhances c-Myc protein level to promote apoptosis, the ability of miR-92 to repress Fbw7 in vivo could similarly constitute a major mechanism to enhance c-Myc-induced apoptosis. This effect of miR-92 is a double edged sword in c-Myc driven tumors, as its overexpression gives rise to a strong and obligated coupling between excessive proliferation and a potent, p53-dependent apoptosis (Figure 7F). This coupling is consistent with the previous observation that a lower level of constitutive c-Myc acts more effectively to promote tumor initiation, while a higher level of c-Myc is selected by the terminal tumors with defective apoptosis machinery (Murphy et al., 2008). Therefore, mir-17-92 encodes an internal component to confer a negative regulatory feedback on its oncogenic activity, imposing a strong selection for anti-apoptotic lesions to shape the path of malignant transformation. More interestingly, c-Myc transcriptionally activates mir-17-92 that encodes miR-92 (Hemann et al., 2005), which in turn enhances c-Myc dosage, at least in part, by repression Fbw7. It is possible that aberrant c-Myc activation triggers a positive feedback loop to further increase c-Myc dosage to strengthen the apoptotic response and to eliminate cells with oncogenic potential. It is worth noting that the miR-92 apoptotic effect described in this study depends on an intact p53 response. Consequently, in terminal Eμ-myc B-lymphoma cells that often carry a defective p53 response, miR-92 failed to enhance c-Myc-induced apoptosis (Mu et al., 2009).
The functional readout of miR-92 heavily depends on cell types and biological contexts. It is important to recognize that miR-92 is not a tumor suppressor miRNA. Like c-Myc, miR-92 elicits potent oncogene stress to engage tumor suppressor response, at least in part, by activating p53. In the premalignant Eμ-myc/92 B-cells, the effect of miR-92 to repress Fbw7 most likely results in an increase of c-Myc level, which coupled with the intact p53 response to strongly sensitize the cells to miR-92-induced apoptosis. Under other contexts when proliferation becomes a rate-limiting event for oncogenesis, or when p53-dependent apoptosis is compromised, miR-92 could render a pro-proliferative effect that is strictly oncogenic (Tsuchida et al., 2011). Likewise, the functional readout of other mir-17-92 components also heavily depends on cell types and biological contexts. miR-19 promotes c-Myc-induced B-lymphomas by repressing apoptosis (Mu et al., 2009; Olive et al., 2009), yet has little effects in promoting Rb-deficient retinoblastomas (Conkrite et al., 2011); miR-17 allows the bypass of Ras-induced senescence by promoting proliferation (Hong et al., 2010), yet fails to affect c-Myc-induced lymphomas, possibly due to its functional redundancy with c-Myc.
Both cooperative and antagonistic interactions operate among subsets of mir-17-92 components. The miR-19:miR-92 antagonism constitutes a novel mechanism to confer an intricate balance between oncogene signaling and innate tumor suppressor responses (Figure 7F). This balance can be disrupted in premalignant and malignant cells that exhibit c-Myc overexpression, as an increase in the miR-19:miR-92 ratio is likely to favor the suppression of c-Myc-induced apoptosis and to promote oncogenesis. Although all mir-17-92 components are co-transcriptionally regulated, different changes of miR-19 vs miR-92 during oncogenesis could be a result of differential miRNA biogenesis and/or turn-over. It has been shown that specific RNA-binding proteins, such as hnRNP A1, promote the processing of a specific mir-17-92 component, miR-18 (Guil and Cáceres, 2007). Future studies are likely to reveal important mechanisms underlying cell type- and context-dependent differential regulation of mir-17-92 components, which will generate important insights on the biology of polycistronic miRNAs.
Our current study mostly focuses on the antagonistic interaction between miR-19 and miR-92 in c-Myc driven oncogenesis, yet it reveals a more general mechanism underlying the structural function relationship of polycistronic miRNAs. It is likely that the complex interactions among polycistronic miRNA components can coordinate and balance a multitude of cellular and molecular processes during normal development and disease. Interestingly, in the case of mir-17-92, miR-92 has a different evolutionary history compared to the other mir-17-92 components. miR-92 is evolutionary conserved in Deuterostome (including vertebrates and chordates), Ecdysozoa (including flies and worms), and Lophotrochozoa, yet the remaining mir-17-92 components are only found in vertebrates (Figure 1—figure supplement 1C). The functional antagonism between the more ancient miR-92 and the newly evolved mir-19 might result from the convergence of these two separate evolutionary paths at the origin of vertebrates. This antagonism could evolve to regulate cell proliferation and cell death downstream or independent of c-Myc in both normal development and disease. Thus, our studies suggest a novel mechanism by which a crosstalk between oncogene and tumor suppressor pathways has been hardwired through evolution into the unique gene structure of a polycistronic oncomir.
Materials and methods
Molecular cloning
Request a detailed protocolmir-17-92 Δ92 and mir-17-92 were amplified by PCR and subsequently cloned into the XhoI and EcoRI sites of the MSCV retrovirus vectors. In these vectors, miRNAs were placed downstream of the LTR promoter, which is followed either by a SV40-GFP cassette (for all in vivo experiments), a PGK-Puro-IRES-GFP cassette, or a SV40-CD4 cassette (for in vitro experiments) (Hemann et al., 2005). To construct MSCV-17-92Mut92, MSCV-17-92Mut20, and MSCV-17-92Mut19b vectors, a 12-nucleotide mutation was introduced into the seed region of the mature miR-92, miR-20, or miR-19b using the Quikchange XL mutagenesis kit (200521; Stratagene) and the following primers:
Mut20 primers: GACAGCTTCTGTAGCACTAAtaaacaataatcGCAGGTAGTGTTTAGTTATC and GATAACTAAACACTACCTGCGATTATTGTTTATTAGTGCTACAGAAGCTGTC.
Mut92 primers: CAATGCTGTGTTTCTGTATGGTtaacattaacatCCGGCCTGTTGAGTTTG and CAAACTCAACAGGCCGGATGTTAATGTTAACCATACAGAAACACAGCATTG.
Mut19b primers: CTGTGTGATATTCTGCTGacatttaagtacCAAAACTGACTGTGGTAGTG and CACTACCACAGTCAGTTTTGGTACTTAAATGTCAGCAGAATATCACACAG.
The loss of miR-92, miR-20 or miR-19b expression and the intact expression level of the remaining mir-17-92 components were validated using the TaqMan MicroRNA Assays (4427975; Applied Biosystems, Foster City, CA). mir-19b-92, mir-19bMut92, and mir-19b-92Mut19b were similarly amplified by PCR (ACTGCTCGAGAGCTTCGGCCTGTCGCCC and GTAGAATTCATGTATCTTGTAC) from the mir-17-92, mir-17-92Mut92, and mir-17-92Mut19b construct described above and subsequently cloned into the XhoI and EcoRI sites of the MSCV retrovirus vectors.
To construct the MSCV-Shp53 vector, shRNA against p53 was placed downstream of the LTR promoter of the MSCV-SV40-HuCD4 retroviral vector (Xue et al., 2007). MSCV-Shfbw7 construct was kindly provided by Dr Hans Guido Wendel (Mavrakis et al., 2011). To construct the pRetroX-fbw7-IRES-DsRedExpress (Xu et al., 2010), fbw7α ORF was placed downstream of the LTR promoter followed by an IRES-DsRed cassette.
Adoptive transfer of Eμ-myc HSPCs for lymphomagenesis
Request a detailed protocolThe hematopoietic stem and progenitor cells (HSPCs) were isolated from E13.5-E15.5 Eμ-myc/+ mouse embryos and were transduced with MSCV alone or MSCV vectors expressing various mir-17-92 derivatives. The MSCV retroviral vector used in our adoptive transfer model contains a SV40-GFP cassette that allows us to monitor transduced HSPCs both in vitro and in vivo. Infected HSPCs were subsequently transplanted into an 8- to 10-week-old, lethally irradiated C57BL/6 recipient mice. Tumor onset was subsequently monitored by weekly palpation, and tumor samples were either collected into formalin for histopathological studies, or prepared as single cell suspension for FACS analysis and for cell culture studies. Both the Eμ-myc/+ mice and the recipient mice are on C57BL/6 background.
LT2-MYC mouse liver tumor model
Request a detailed protocolThe LT2-MYC mouse model for human hepatocellular carcinoma (HCC) is a double transgenic mouse model, in which the tetracycline transactivator protein (tTA) is driven by the hepatocyte-specific promoter, the liver activator protein (LAP) promoter, while the human c-MYC gene is driven by the tetracycline response element (TRE). The LT2-MYC model exhibits ‘dox-off’ regulation, where c-Myc expression is turned on in hepatocytes in the absence of doxycycline.
LT2-MYC mice taken off doxycycline-containing food, between 3–5 weeks of age, develop distinct tumor nodules around 8–12 weeks on an average (Kistner et al., 1996; Shachaf et al., 2004). Total RNA was extracted from liver tumor samples from three independent mice, as well as normal livers from the doxycycline treated LT2 mice. Total RNAs were prepared using Trizol (15596018; Invitrogen) and subjected to real time PCR analyses as described below.
Cell culture and retroviral infection
Request a detailed protocolPrimary murine B-cells were prepared from bone marrows of 4- to 6-week-old mice and were cultured in RPMI with 10% fetal bovine serum (FBS), 50 μM beta-mercaptoethanol (M3148; Sigma) and 2 ng/ml Il-7 (407-ML-005; R&D). R26MER/MER and R26MER/+ MEFs were kindly provided by Gerald Evan’s laboratory. MEFs were cultured in DMEM with 10% fetal bovine serum. Eμ-myc tumor cells were derived from lymphomas from the terminal-stage Eμ-myc animals. Eμ-myc lymphoma cells overexpressing various mir-17-92 derivatives were cultured in 45% DMEM, 45% IMDM with 10% fetal bovine serum, and 50 μM β-mercaptoethanol (M3148; Sigma) on irradiated NIH-3T3 feeder cells. Immortalized human B-cell lines were cultured in RPMI with 10% FBS and 90 μM beta-mercaptoethanol. Dicer-deficient Hct116 cells, kindly provided by Dr Bert Vogelstein (Cummins et al., 2006), and Fbxw7-deficient Hct116 cells (Grim et al., 2012) were cultured in McCoy’s 5A media with 10% fetal bovine serum. Human Burkitt’s lymphoma cell lines, including BL41, BL2, MutuI, Daudi, Raji (provided by Dr Terry Rabbitts), Manca, and Jiyoje were cultured in RPMI with 10% FBS.
Mouse primary B-cell cultures or MEFs were infected by MSCV retroviruses expressing various mir-17-92 derived miRNA clusters, shRNA against p53 (Xue et al., 2007), shRNA against fbw7 (Mavrakis et al., 2011), or fbw7 cDNA (pRetroX-fbw7-IRES-DsRedExpress). In Figure 4E,F, double infection was performed to obtain R26MER/MER MEFs that co-expressed shRNA p53 and miR-92. In this experiment, MEFs were initially infected with an ecotropic MSCV-p53shRNA-SV40huCD4 retrovirus to a nearly 100% infection efficiency, as validated by FACS analysis using huCD4 antibody. The second infection was achieved using an amphotropic MSCV-miR-92-PGK-Puro-IRES-GFP retrovirus. Doubly infected cells were then selected using puromycin. In Figure 5H, double infection of R26MER/MER MEFs with pRetroX-fbw7-IRES-DsRedExpress and MSCV-miR-92-PGK-Puro-IRES-GFP were similarly performed. For all experiments with primary murine B-cells, bone marrow cells were cultured for 48 hr before retroviral infection and collected or analyzed 72 hr after infection. After 5 days in culture, the percentage of B220-positive cell is 100%. In Figure 3E, Figure 3—figure supplement 1, B-cells were infected with MSCV retrovirus containing a PGK-Puro-IRES-GFP cassette. FACS analysis was performed after gating on the GFP-positive population. In Figure 5A, the collected B-cells were infected with retrovirus containing SV40-CD4 cassette. Infected cells were purified with Human CD4 Micro-beads (130-045-101; Miltenyi Biotec) using MACS Purification Columns MS (130-042-201; Miltenyi Biotec).
The collection of normal and malignant B-cells in vivo
Request a detailed protocolNormal mouse B-cells were isolated from the spleen or the bone marrow of 4- to 6-week-old C57B/6J mice, using CD19 Micro-Beads (Miltenyi Biotec) or by negative selection (Easysep 19754; STEMCELL). Similarly, premalignant Eμ-myc B-cells were extracted from the bone marrow of 5- to 6-week-old Eμ-myc transgenic mice. Malignant Eμ-myc B-cells were extracted from the lymph node tumors of terminal-stage Eμ-myc mice. In addition, the normal human B-cells from peripheral blood were FACS sorted from the peripheral blood of healthy donors.
Histopathology and immunotyping
Request a detailed protocolMouse tissue samples were fixed in formalin (SF100-4; Fisher), embedded in paraffin (AC41677-0020; Fisher), sectioned into 5 µm tissue samples, and stained with hematoxylin and eosin (7211 & 7111, Fisher). For caspase-3 (AF835, 1:200; R&D Systems), PCNA (MS-106P, 1:200; Lab Vision Corp.), and B220 (14-0452-85, 1:100; eBioscience) detection, representative sections were deparaffinized and rehydrated in graded alcohols before subjected to antigen retrieval treatment with 10 mM sodium citrate buffer 10 min in a pressure cooker. Detection of antibody staining was carried out following standard procedures from the avidin-biotin immunoperoxidase methods. Diaminobenzidine (002014, Invitrogen) was used as the chromogen and hematoxylin as the nuclear counter stain. Quantitation of apoptosis was evaluated by counting the number of starry sky foci in three fields (40X) from seven representative animals of each genotype, as well as by counting the number of caspase-3 positive cells in three fields (40X) from five representative animals of each genotype.
To determine the cell surface markers of the lymphoma cells harvested from the animals, cells were resuspended in 10% FBS/PBS to reach a concentration of 107 cells/ml. 20 μl of this cell suspension was stained with antibodies diluted in 10% FBS/PBS for 1 hr. Subsequently, cells were washed with 2% FBS/PBS and resuspended in 10% FBS/PBS for flow cytometry analysis. Antibodies used for FACS analyses include PE anti-mouse IgM (12-5790, 1:200; eBioscience), APC-Cy7 anti-mouse B220 (552094, 1:200; BD Pharmingen), APC-Cy7 anti-mouse CD4 (552051, BD Pharmingen, 1:200), PE anti-mouse CD8 (553032, 1:200; BD Pharmingen), PE anti-mouse CD25 (553866, 1:200; BD Pharmingen), and APC anti-mouse CD19 (115511, 1:100; Biolegend).
Apoptosis assays and proliferation assays
Request a detailed protocolSubconfluent MSCV- or miR-92-infected R26MER/MER MEFs were induced and serum starved by incubating the cells with 100 nM of 4-hydroxytamoxifen (H6278; Sigma) in DMEM with 0.2% fetal bovine serum for 12–24 hr before harvesting the cells for apoptosis analyses using APC-Annexin V antibody (550475, 1:50; BD Pharmingen) and 7AAD staining solution (559925; BD Pharmingen). To evaluate the apoptotic effects of miR-92 in our adoptive transfer model in vivo, we collected premalignant Eμ-myc B-cells from spleen or bone marrow of well-controlled Eμ-myc/92 and Eμ-myc/MSCV mice at 5 weeks after adoptive transfer and measured the extent of apoptosis by FACS. Apoptosis in GFP-positive B220-positive premalignant B-cells was measured using the Caspase Detection Kit (Calbiochem, Red-VAD-FMK) following the manufacturer’s instructions. To quantitate cell proliferation, 10 μM of BrdU was used to label primary B-cells for 4 hr and MEFs for 30 min. The percentage of BrdU-positive cells was determined using the Flow BrdU kit (552598; BD Pharmingen).
Real time PCR and western analyses
Request a detailed protocolTaqMan MicroRNA Assays (Applied Biosystems) were used to measure the level of mature miRNAs, including miR-17, 18, 19a, 20, 19b, and 92 (4427975; ABI). mRNA level for perp (GACCCCAGATGCTTGTTTTC, GGGTTATCGTGAAGCCTGAA), noxa (GGAGTGCACCGGACATAACT, TGAGCACACTCGTCCTTCAA), puma (GCGGCGGAGACAAGAAGA, AGTCCCATGAAGAGATTGTAC), p21 (ACGGTGGAACTTTGACTTCG, CAGGGCAGAGGAAGTACTGG), bax (GTTTCATCCAGGATCGAGCAG, CCCCAGTTGAAGTTGCCATC), mdm2 (CTCTGGACTCGGAAGATTACAGCC, CCTGTCTGATAGACTGTCACCCG), p53 (AACCGCCGACCTATCCTTAC, TCTTCTGTACGGCGGTCTCT), ccnb1 (AAGGTGCCTGTGTGTGAACC, GTCAGCCCCATCATCTGCG), ccnb2 (GCCAAGAGCCATGTGACTATC, CAGAGCTGGTACTTTGGTGTTC), cdc20 (AGACCACCCCTAGCAAACCT, GACCAGGCTTTCTGATGCTC), cdc25b (ATTCTCGTCTGAGCGTGGAC, GCTGTGGGAAGAACTCCTTG), fbw7 (CGGCTCAGACTTGTCGATACT, CTTGATGTGCAACGGTTCAT), gtse1 (GCTTTGCCTGTGAGAGGAAG, CACTCTGGGATCCCTTTTCA), bid (CTGCCTGTGCAAGCTTACTG, GTCTGGCAATGTTGTGGATG), pten (CACAATTCCCAGTCAGAGGCG, GCTGGCAGACCACAAACTGAG), bim (ACCACTATCTCAGTGCAATGGCTTCC, CGGTAATCATTTGCAAACACCCTCCTTG), cdk4 (TGGTACCGAGCTCCTGAAGT, GTCGGCTTCAGAGTTTCCAC), c-myc (GTGCTGCATGAGGAGACACCGCC, GCCCGACTCCGACCTCTTGGC), Pirh2 (TGCAGTGCATCAACTGTGAA, CAAACAGGTGGCAAATACTGC), Ppp2r5d (CCGTGATGTTGTCACTGAGG, ACTCTGCTCCTGTGGGATTC), Dyrk2 (CCAGCAACGCTACCACTACA, AACAGCTGCTGAACCTGGAT), Romo1 (ATTCGGAGTGAGACGTCGAG, TGACGAAGCCCATCTTCAC), Pak2 (TTGGCTTTGATGCTGTTACG, CACTGCCTGAGGGTTCTTCT), Trpc4ap (CGCAAATGTCCTTCCTCTTC, GCCAGCATCAGGATTACCAG), and Axin1 (AGGACGCTGAGAAGAACCAG, CTGCTTCCTCAACCCAGAAG) were determined using real time PCR analyses with SYBR (KK4605; Kapa Biosystems). Actin (GATCTGGCACCACACCTTCT, GGGGTGTTGAAGGTCTCAAA) was used as a normalization control in all our real time PCR analysis with SYBR. U6 snRNA assay (4427975; ABI) was used as a normalization control in all our TaqMan MicroRNA Assays (Applied Biosystems).
For western analyses, all samples were directly collected into Laemmli buffer. p53 (1C12; Cell Signaling), Arf (5-C3-1; Novus), and c-Myc (1472-1; Epitomics) antibodies were used at 1:1000 dilution. FLAG (M2; Sigma) and Tubulin (12G10) were used at 1:2500 dilution. HRP conjugated secondary antibodies (Santa Cruz Biotechnology, sc-2004 sc-2005 and sc-2006) were used at 1:5000.
Microarray analyses
Request a detailed protocolThree independent R26MER/MER MEF lines were infected by MSCV vector alone or by MSCV vector encoding miR-92. These MEFs were induced and serum starved by incubating the cells with 100 nM of 4-hydroxytamoxifen (H6278; Sigma) in DMEM with 0.2% fetal bovine serum for 12 hr before harvesting the cells for RNA preparation. Total RNAs were prepared using Trizol (15596018; Invitrogen), and subjected to microarray analysis using Affymetrix chip Mouse 430_2. To identify differentially expressed genes that could be regulated by miR-92, we used gcRMA in the bioconductor package (Wu et al., 2004) and SAM (Significance Analysis of Microarrays) (Tusher et al., 2001) for statistical analysis of our microarray data. Gene expression signals were estimated from the probe signal values in the CEL files using statistical algorithm gcRMA. This data processing at the probe level includes background signal subtraction and quantile normalization to facilitate the comparison among microarrays. SAM was then used to identify the genes with significant expression level alterations between miR-92 overexpressing MEFs and the control MEFs. The genes with at least 1.5-fold expression level change and FDR <1% were regarded as differentially expressed genes. Pathway analyses were performed on upregulated and downregulated genes using the KEGG database (Dennis et al., 2003).
Xenopus embryo apoptosis assays
Request a detailed protocolXenopus laevis eggs were collected, fertilized, and embryos cultured by standard procedures. The miR-19b mimics were produced from the annealing products of 5′UGUGCAAAUCCAUGCAAAACUGA3′ and 5′AGUUUUGCAGGUUUGCAUCCAUU3′ (IDT).
The miR-17 mimics were produced from the annealing products of 5′CAAAGUGCUUACAGUGCAGGUAGU3′ and 5′UACUGCAGUGAAGGCACUUGUAG3′(IDT).
The miR-18 mimics were produced from the annealing products of 5′UAAGGUGCAUCUAGUGCAGAUAG3′ and 5′ACUGCCCUAAGUGCUCCUUCUG3′(IDT).
The miR-19a mimics were produced from the annealing products of 5′AGUUUUGCAUAGUUGCACUA3′ and 5′UGUGCAAAUCUAUGCAAAACUGA3′(IDT).
The miR-20 mimics were produced from the annealing products of 5′UAAAGUGCUUAUAGUGCAGGUAG3′ and 5′ACUGCAUAAUGAGCACUUAAAGU3′(IDT).
The miR-92 mimics were produced from the annealing products of 5′UAUUGCACUUGUCCCGGCCUG3′ and 5′AGGUUGGGAUUUGUCGCAAUGCU3′(IDT).
The annealing of miRNA mimics were performed by combining two complimentary RNA oligos at a stock concentration of 1 μg/μl, heating the oligos to 80°C for 1 min, and then cooling down to room temperature to allow duplexes to form. The same was done for generating the mutated miR-19 mimics (Mut-miR-19), by annealing 5′UCAGGUAAUCCAUGCAAAACUGA3′ and 5′AGUUUUGCAGGUUACCUUCGAUU3′, and mutated miR-92 mimics (Mut-miR-92) by annealing 5′UUAUCGACUUGUCCCGG3′ and 5′GGUUGGGAUUGGUUCGA 3′.
Xenopus embryos were injected into both cells at the two-cell stage with 2 ng of each RNA (Walker and Harland, 2009). The pcDNA3-myc-AGO2 vector, kindly provided by Dr Greg Hannon, was cut using ScaI; and the synthetic hAGO2 mRNAs were transcribed using mMessage mMachine T7 kit (Ambion). When indicated, a total of 0.5 ng hAGO2 mRNA (Liu et al., 2004) was injected into two-cell stage embryos either alone or with 2 ng of each miRNA (Lund et al., 2011). The embryos were then treated with hydroxyurea (H8627; Sigma) at a final concentration of 5 mM from stage 3 until stage 10. Apoptotic embryos were scored as those containing any apoptotic cells based on morphological changes.
Luciferase assays
Request a detailed protocolA luciferase reporter fused with the fbw7 3′UTR was kindly provided by Dr Hans-Guido Wendel (Mavrakis et al., 2011). In this psiCHECK-2 based reporter, the fbw7 3′UTR was cloned downstream of the Renilla luciferase reporter, and a separate firefly luciferase cassette was used as a transfection control. Because the two predicted miR-92 binding sites are close to each end of the 3′UTR, we mutated the miR-92 binding sites by PCR using the following primers:
3′UTR-Fbw7-Mut-Xho1-F (GATCTCGAGCAAGACGACTCTCTAAATCCAACTATTCTTT) and 3′UTR-Fbw7-mut-Not1-R (ATGCGGCCGCAACACATTTAGTTATAAGAAAATAAAATTT). The PCR fragment was subsequently cloned into the XhoI and Not1 sites of the psiCHECK-2 vector. The reporter construct, together with 50 nM miR-92 mimics, was transfected into Dicer-deficient Hct116 cells (Cummins et al., 2006), with transfection of miR-17 or miR-18 as negative controls. Luciferase activity of each construct was determined by dual luciferase assay (E19100; Promega) 48 hr post-transfection following the manufacturer’s instructions. The miR-17 mimics were produced by annealing 5′CAAAGUGCUUACAGUGCAGGUAGU3′ and 5′UACUGCAGUGAAGGCACUUGUAG3′. The miR-18 mimics were produced by annealing 5′UAAGGUGCAUCUAGUGCAGAUAG3′ and 5′ACUGCCCUAAGUGCUCCUUCUG3′.
The miR-92 mimics were produced by annealing 5′UAUUGCACUUGUCCCGGCCUG3′ and 5′AGGUUGGGAUUUGUCGCAAUGCU3′.
Fbw7α immunoprecipitation and western analyses
Request a detailed protocolBecause Fbxw7-substrate degradation was regulated in a cell-cycle-dependent manner, we used serum starvation synchronized MEFs to study Fbw7 regulation by miR-92 during cell cycle progression. MEFs were made quiescent by serum starvation; then Fbw7 expression was examined following release into serum. Cells were lysed in NP-40 buffer supplemented with protease inhibitors. Lysates were normalized and immunoprecipitated with polyclonal anti-Fbw7 antibody kindly provided by Dr Bruce Clurman (Grim et al., 2008), followed by immunoblotting with polyclonal anti-Fbw7 antibody (A301-720A; Bethyl Laboratories). Wild-type and FBW7−/− Hct116 cells were used, respectively, as positive and negative controls.
The construction of the pFLAG-Fbw7α-3′UTR plasmid was previously described (Xu et al., 2010). The construct was transfected into the Dicer-deficient Hct116 cells together with 50 nM of miR-92 mimics or siRNA against GFP as indicated. Anti-FLAG (M2; Sigma) antibody was used to detect the FLAG-Fbw7α by western blot 48 hr after transfection.
Cyclin E-dependent kinase assays
Request a detailed protocolCyclin E-CDK complexes were immunoprecipitated from MSCV or miR-92 infected Rosa26MER/MER MEFs extracts using affinity-purified polyclonal antibody, provided by Dr Bruce Clurman (Minella et al., 2008). Cyclin E immunoprecipitates were then incubated with purified histone subunit H1 (Sigma) and (gamma-32P)ATP to measure cyclin E-dependent kinase activity. The anti-Grb2 monoclonal (BD Biosciences) antibody was used a normalization control.
References
-
The colorectal microRNAomeProc Natl Acad Sci USA 103:3687–3692.https://doi.org/10.1073/pnas.0511155103
-
The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18aNat Struct Mol Biol 14:591–596.https://doi.org/10.1038/nsmb1250
-
Biogenesis of small RNAs in animalsNat Rev Mol Cell Biol 10:126–139.https://doi.org/10.1038/nrm2632
-
Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic miceProc Natl Acad Sci USA 93:10933–10938.https://doi.org/10.1073/pnas.93.20.10933
-
miRGen: a database for the study of animal microRNA genomic organization and functionNucleic Acids Res 35:D149–D155.https://doi.org/10.1093/nar/gkl904
-
Akt enhances Mdm2-mediated ubiquitination and degradation of p53J Biol Chem 277:21843–21850.https://doi.org/10.1074/jbc.M109745200
-
miR-19 is a key oncogenic component of mir-17-92Genes Dev 23:2839–2849.https://doi.org/10.1101/gad.1861409
-
Molecular evolution of a microRNA clusterJ Mol Biol 339:327–335.https://doi.org/10.1016/j.jmb.2004.03.065
-
Significance analysis of microarrays applied to the ionizing radiation responseProc Natl Acad Sci USA 98:5116–5121.https://doi.org/10.1073/pnas.091062498
-
The miR-17∼92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastomaProc Natl Acad Sci USA 106:2812–2817.https://doi.org/10.1073/pnas.0809579106
-
A model based background adjustment for oligonucleotide expression ArraysJohns Hopkins University, Dept of Biostatistics Working Papers at, http://biostats.bepress.com/jhubiostat/paper1.
-
Ribo-gnome: the big world of small RNAsScience 309:1519–1524.https://doi.org/10.1126/science.1111444
Article and author information
Author details
Funding
American Cancer Society (123339-RSG-12-265-01-RMC)
- Lin He
National Cancer Institute (R00 CA126186)
- Lin He
Tobacco-Related Disease Research Program (21RT-0133)
- Lin He
The Leukemia and Lymphoma Society (LLS, 3423-13)
- Virginie Olive
National Instititute of Health (F31 CA165825-02)
- Erich Sabio
National Cancer Institute (R01 CA139067)
- Lin He
National Cancer Institute (1R21CA175560-01)
- Lin He
National Institute of Health (R01HL098608)
- Alex C Minella
National Heart, Lung and Blood Institute (R01HL098608)
- Alex C Minella
US Department of Defense (W81XWH-12-1-0272)
- Andrei Goga
National Institutes of Health (5R01CA170447)
- Andrei Goga
The Leukemia and Lymphoma Society (LLS, 1531)
- Andrei Goga
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank members of the He Lab for their help and input. Particularly, we thank C Fulco, I Jiang, Y Chen, R Song, E Ho, J Cisson, YJ Choi and C Lin for technical assistance and stimulating discussions. We also thank H Nolla and A Valeros for advice on our FACS analysis, thank J Choi for microarray analyses, and P Margolis for proofreading our manuscript. We thank SW Lowe, J Mendell, B Clurman, M Schlissel, M Junttila, L Soucek, HG Wendel, A Ventura, GJ Hannon, DS Sandeep, T Rabbitts, B Vogelstein, J Mao and M Burger for sharing reagents and helpful discussions. We are particularly grateful for B Olive and B Colpo for their support during this study. Finally, we would like to dedicate this work to the memory of Gisele Cocher, whom we lost during the preparation of this manuscript. Her unconditional love and kindness shape who we are; her courage and support will always be with us. LH is a Searle Scholar supported by the Kinship Foundation.
Ethics
Animal experimentation: Our experimentation is conducted to the highest ethical standards, and we follow the guidelines established by the University of California, Berkeley’s Animal Care and Use Committee (ACUC). The animal protocol detailing the experimental procedures with laboratory mice was carefully reviewed and approved by Animal Care and Use Committee (ACUC) at the University of California at Berkeley. Our Animal Use protocol number is R316-0613BR.
Copyright
© 2013, Olive et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,485
- views
-
- 264
- downloads
-
- 70
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 70
- citations for umbrella DOI https://doi.org/10.7554/eLife.00822
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A component of the mir-17-92 polycistronic oncomir promotes oncogene-dependent apoptosis
eLife 2:e00822.
https://doi.org/10.7554/eLife.00822




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}