Retinoblastoma Protein: Disruptive influence
- University of Manchester, United Kingdom
With more than 11,000 papers published on it, one would assume that we know all there is to know about retinoblastoma protein (Rb). This protein has been the subject of intense study ever since it was realized that the inactivation of Rb is a fundamental event in the development of cancer. However, it has been difficult to determine what Rb does in normal cells because it appears to be involved in a wide range of processes, including transcriptional regulation, chromatin remodelling, the cell cycle and apoptosis. Now, in eLife, Keigo Araki, Yoichi Taya and colleagues at the National University of Singapore report an intriguing new aspect of Rb. They show that, under certain conditions, Rb can move from the cell nucleus—where it is usually found—to the cytoplasm, where it disrupts the basic units of muscle (Araki et al., 2013).
This finding is important because it could help explain why late-stage cancer patients often encounter progressive muscle weakness and atrophy that cannot be reversed by increased calorie intake (reviewed in Fearon et al., 2012). It has been estimated that this condition—which is called cachexia, and which also involves changes to the neuroendocrine system, the immune system and adipose tissue–is the direct cause of death for approximately 20% cancer patients, so obtaining a better understanding of the molecular mechanisms responsible for it is of paramount importance. According to the current understanding of cachexia, various inflammatory cytokines lead to the up-regulation of enzymes called E3 ubiquitin ligases, which then mediate the breakdown of the proteins that make up sarcomeres, which are the basic building blocks of muscle. At the same time, the expression of the genes that code for skeletal muscle are down-regulated.
The new results demonstrate that Rb can destabilize the sarcomeric proteins in the muscle cells of cancer patients: by interacting with mDia1 (a protein that nucleates the formation of actin filaments in the cytoplasm), Rb may influence both the assembly of actin filaments and the generation of force by these filaments (Figure 1). It remains to be seen whether the interaction between mDia1 and Rb is the consequence of the activity of E3 ligases or of altered cellular energy balance, or if it precedes these stages in the weakening of skeletal muscle. Indeed, it has been suggested that the filaments are more susceptible to degradation after they have been dislodged from the sarcomere (Solomon and Goldberg, 1996). The equivalent of mDia1 in C. elegans is known to be important in the attachment of actin filaments to a structure called the Z-line, which marks the edge of the sarcomere (Figure 1; Mi-Mi et al., 2012). It is conceivable, therefore, that the co-localization of mDia1 and Rb near the Z-line in human cells may lead to the degradation of actin filaments and enhance the breakdown of proteins within the cells.
Figure 1
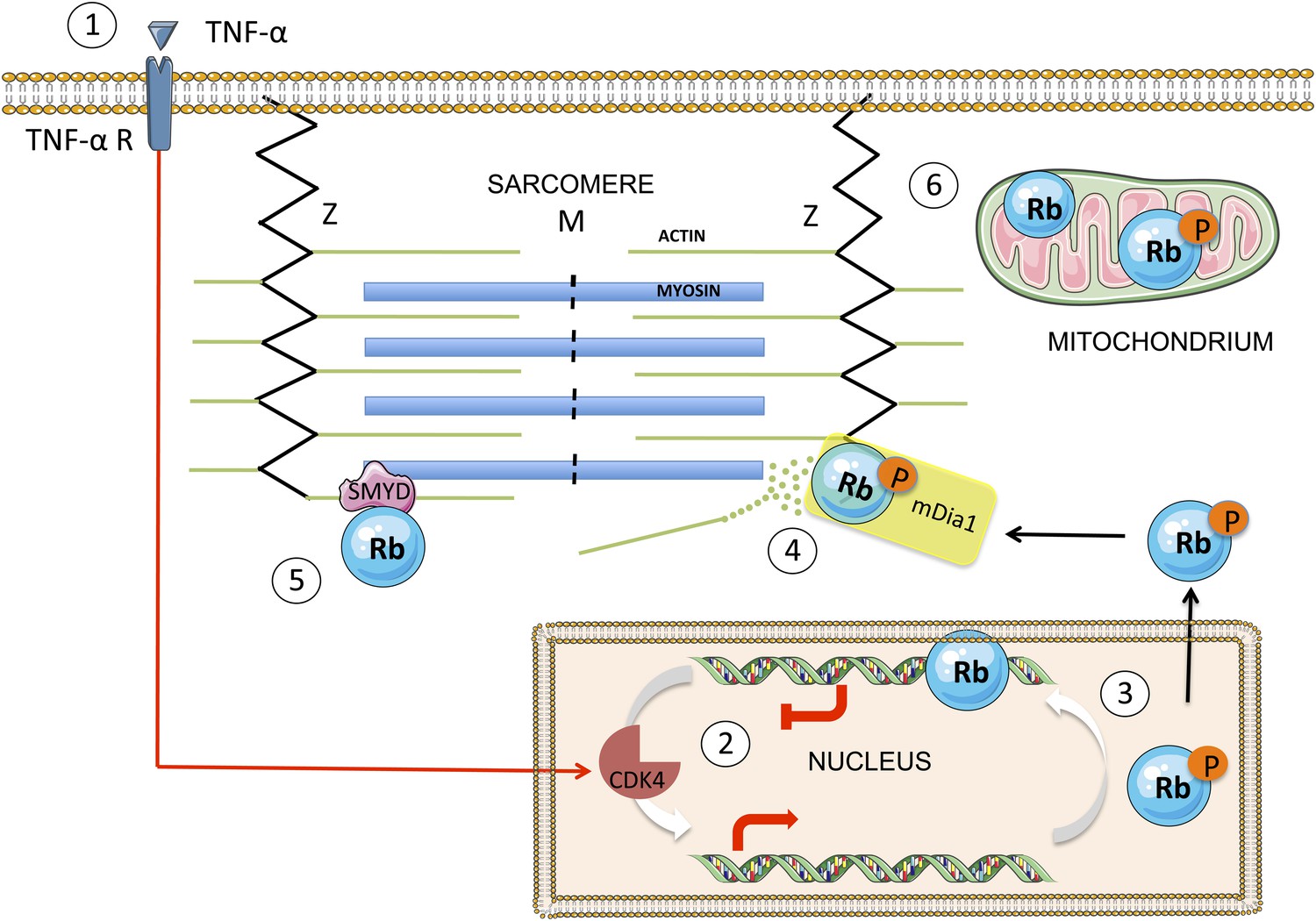
A simplified scheme showing the translocation of Rb from the nucleus to the cytoplasm, and the effect it has on sarcomeres.
TNF-α (tumour necrosis factor-alpha) is a cytokine that may, in certain cases, kill cancer cells. When TNF-α binds to its receptor on the surface of a normal cell (1), it triggers a signal transduction cascade that leads to the phosphorylation of Rb in the nucleus (2) by the enzyme CDK4. The phosphorylated Rb (3) separates from the DNA, allowing transcription of E2F target genes, and moves to the cytoplasm, where it binds to mDia1 at the Z line (4). This leads to the destabilization of the actin filaments in the sarcomere and to impaired muscle function. The enzyme SMYD methylates the Rb, which contributes to sarcomere stability (5). There is also evidence that Rb accumulates in the mitochondria, where it seems to play a role in apoptosis (6). The figure was produced using Servier Medical Art (http://www.servier.com).
A considerable amount of evidence about the diverse range of roles performed by Rb in the cytoplasm has emerged in recent years. For example, it has been demonstrated that Rb can be localized in mitochondria and also shown that it is involved in inducing apoptosis (Hilgendorf et al., 2013). Moreover, in various cells Rb is methylated by an enzyme called SMYD2, which enhances the phosphorylation of Rb and the recruitment of additional co-factors (Saddic et al., 2010; Cho et al., 2012). And in separate studies of skeletal muscle cells, SMYD1 and SMYD2 have been found to be vital for forming complexes with myosin, myosin chaperones and titin. Loss of SMYD function is also known to lead to the disruption of sarcomere (Just et al., 2011; Donlin et al., 2012; Li et al., 2013).
These data place Rb in a protein interaction network, which now includes both myosin and actin filaments of skeletal muscle, along with their regulatory factors. We can expect exciting times in unravelling how this stress responsive pathway is fine-tuned to sense the balance between healthy cells, sarcomere breakdown and apoptosis. It will be intriguing to study how these cytoplasmic functions of Rb relate to its role as a transcriptional regulator in the nucleus and how Rb is distributed between different cellular compartments.
With these newly identified functions, Rb has managed to surprise us once again. The loss of Rb has long been associated with cancer. Now it appears that native Rb can also have detrimental effects for patients away from cancer tissue. And while the presence of Rb in the cytoplasm of cells in skeletal muscles can have damaging consequences, we still do not know if there is an underlying physiological role for Rb in the healthy turnover sarcomeres. Ultimately, of course, we hope it will be possible to minimize cancer-related muscle wasting by modifying the behaviour of Rb in skeletal muscle cells.
References
-
Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organizationGenes & Development 26:114–119.https://doi.org/10.1101/gad.177758.111
-
The myosin-interacting protein SMYD1 is essential for sarcomere organizationJournal of Cell Science 124:3127–3136.https://doi.org/10.1242/jcs.084772
-
Smyd1b is required for skeletal and cardiac muscle function in zebrafishMolecular Biology of the Cell 24:3511–3521.https://doi.org/10.1091/mbc.E13-06-0352
-
Z-line formins promote contractile lattice growth and maintenance in striated muscles of C. elegansThe Journal of Cell Biology 198:87–102.https://doi.org/10.1083/jcb.201202053
-
Methylation of the retinoblastoma tumor suppressor by SMYD2The Journal of Biological Chemistry 285:37733–37740.https://doi.org/10.1074/jbc.M110.137612
-
Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extractsThe Journal of Biological Chemistry 271:26690–26697.https://doi.org/10.1074/jbc.271.43.26690
Article and author information
Author details
Publication history
Copyright
© 2013, Cossu et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 374
- views
-
- 21
- downloads
-
- 1
- citation
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 1
- citation for umbrella DOI https://doi.org/10.7554/eLife.01779
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Retinoblastoma Protein: Disruptive influence
eLife 2:e01779.
https://doi.org/10.7554/eLife.01779




{kind=link}