MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin
- University of California, Los Angeles, United States
- National Institutes of Health, United States
- Louisiana State University Health Sciences Center, United States
- Brain Research Institute, The David Geffen School of Medicine, University of California, Los Angeles, United States
Abstract
Parkinson's disease (PD) genes PINK1 and parkin act in a common pathway that regulates mitochondrial integrity and quality. Identifying new suppressors of the pathway is important for finding new therapeutic strategies. In this study, we show that MUL1 suppresses PINK1 or parkin mutant phenotypes in Drosophila. The suppression is achieved through the ubiquitin-dependent degradation of Mitofusin, which itself causes PINK1/parkin mutant-like toxicity when overexpressed. We further show that removing MUL1 in PINK1 or parkin loss-of-function mutant aggravates phenotypes caused by loss of either gene alone, leading to lethality in flies and degeneration in mouse cortical neurons. Together, these observations show that MUL1 acts in parallel to the PINK1/parkin pathway on a shared target mitofusin to maintain mitochondrial integrity. The MUL1 pathway compensates for loss of PINK1/parkin in both Drosophila and mammals and is a promising therapeutic target for PD.
https://doi.org/10.7554/eLife.01958.001eLife digest
Parkinson's disease is the second most common neurodegenerative disorder. Symptoms include tremors, rigidity, and slowness, as well as dementia and depression. While most cases of Parkinson's disease have no known genetic cause, mutations in either of two genes—PINK1 or parkin—are known to lead to the disease.
PINK1 and parkin belong to a single pathway that regulates the structure and function of mitochondria, the organelles that generate energy inside cells. Identifying inhibitors of this pathway is critically important for development of future therapies. In addition, previous studies showed that mice with mutations in PINK1 or parkin, as opposed to those in humans and flies, display subtle signs of Parkinson's disease: the fact that these are weak suggests that other unknown proteins or cellular pathways might compensate for loss of the genes.
Yun et al. have now identified one such protein by showing that an increase in the level of a Protein called MUL1 counteracts the deleterious effects due to the loss of PINK1 or parkin in fruit flies. MUL1 is a mitochondrial protein that regulates another protein called mitofusin; the role of mitofusin is to promote the fusion of mitochondria. Conversely, removing MUL1 from PINK1 or parkin mutant worsens the symptoms because MUL1 is no longer present to compensate for the defects.
Yun et al. also show that MUL1 operates through a pathway that is independent of PINK1/parkin. Moreover, this pathway is found in both flies and mouse neurons, which suggest that it has been conserved during evolution. The work of Yun et al. also suggests that MUL1 as a potential therapeutic target for Parkinson's disease.
https://doi.org/10.7554/eLife.01958.002Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder and there is no cure for this progressive illness (Guo, 2012). Mutations in PINK1, a mitochondria-localized serine–threonine kinase, and Parkin, an E3 ubiquitin ligase, lead to autosomal recessive forms of the disease (Kitada et al., 1998; Valente et al., 2004). Genetic studies in Drosophila first demonstrated that PINK1 and parkin act in the same genetic pathway, with PINK1 positively regulating parkin, to regulate mitochondrial integrity and function (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). Mitochondrial morphology is maintained by a balance between two opposing actions, mitochondrial fusion that is promoted by mitofusin (mfn) and mitochondrial fission that is controlled by Dynamin-related protein 1 (Drp1) (Chan, 2012; Nunnari and Suomalainen, 2012). Genetic studies in Drosophila have shown that downregulation of mfn or overexpression of drp1 suppresses multiple phenotypes associated with lack of PINK1 or parkin, including defects in mitochondrial integrity, cell death, tissue health, and flight ability (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008). Parkin ubiquitinates Mfn and promotes Mfn degradation (Poole et al., 2010; Ziviani et al., 2010). However, it is not clear if increased mfn or decreased drp1 levels are sufficient to cause the phenotypes observed in PINK1 or parkin mutants.
In addition to mitochondrial dynamics, the PINK1/Parkin pathway promotes mitophagy, selective autophagic degradation of defective mitochondria in mammalian cells. Accumulation of mitochondrial damage can result in loss of mitochondrial membrane potential. This leads to recruitment of Parkin to the depolarized mitochondria, ultimately resulting in autophagic degradation of these mitochondria (Narendra et al., 2008; Ding et al., 2010; Gegg et al., 2010; Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010; Okatsu et al., 2010; Tanaka et al., 2010; Vives-Bauza et al., 2010; Chan et al., 2011). Parkin-mediated mitophagy also occurs in mouse cortical neurons and heart muscle (Cai et al., 2012; Chen and Dorn, 2013). An important step during this process is Parkin-dependent ubiquitination of Mfn and other substrates, followed by their proteasome-dependent degradation (Tanaka et al., 2010; Chan et al., 2011). Relevant to PD, PINK1 and parkin mutant fibroblasts from PD patients also show deregulation of mitochondrial dynamics and modest defects in the clearance of mitochondria (Rakovic et al., 2011, 2013).
An important puzzle in the field of PD research is why mice lacking PINK1 or parkin bear only subtle phenotypes related to dopaminergic neuronal degeneration or mitochondrial morphology change (Palacino et al., 2004; Perez and Palmiter, 2005; Perez et al., 2005; Kitada et al., 2007; Frank-Cannon et al., 2008; Gautier et al., 2008; Gispert et al., 2009; Kitada et al., 2009; Akundi et al., 2011). This raises the possibility that other mechanisms may compensate for loss of PINK1 or parkin. Indeed, when parkin is knocked down in adult dopaminergic neurons rather than during development, more striking neuronal degeneration is observed (Dawson et al., 2010; Shin et al., 2011; Lee et al., 2012). However, the molecular mechanisms by which loss of PINK1/parkin function can be compensated are not known.
Mitochondrial ubiquitin ligase 1 (MUL1), also known as mitochondrial-anchored protein ligase (MAPL) (Neuspiel, 2008), mitochondrial ubiquitin ligase activator of NF-kB (MULAN) (Li et al., 2008), or growth inhibition and death E3 ligase (GIDE) (Zhang et al., 2008), was identified as an E3 protein ligase by three independent groups. Work in mammalian systems shows that MUL1 has small ubiquitin-like modifier (SUMO) ligase activity, stabilizing Drp1 (Harder et al., 2004; Braschi et al., 2009), or ubiquitin ligase activity, degrading Mfn (Lokireddy et al., 2012). As expected from a protein with these proposed biochemical activities, MUL1 expression in mammalian cells results in smaller and more fragmented mitochondria (Li et al., 2008; Neuspiel, 2008). However, the consequences of loss of MUL1 in vivo have not been reported in any organism.
In this study, we show that overexpression of mfn is sufficient to recapitulate many PINK1/parkin mutant phenotypes, underlining the central importance deregulation of this protein has for PD pathogenesis. Expression of wild-type MUL1, but not a ligase-dead version, suppresses PINK1 or parkin mutant phenotypes, and those due to mfn overexpression in Drosophila. Conversely, removing MUL1 in PINK1 or parkin null mutants results in enhanced phenotypes as compared with the single mutants, suggesting that MUL1 acts in parallel to the PINK1/parkin pathway. MUL1 physically binds to Mfn and promotes its ubiquitin-dependent degradation. MUL1, but not a ligase-dead version, also regulates Mfn levels and mitochondrial morphology in human cells. Experiments in Drosophila and mammalian systems suggest that MUL1 regulates mfn through a pathway parallel to that of PINK1/parkin pathway. Finally, knockdown of MUL1 from parkin knockout mouse cortical neurons augments mitochondrial damage and induces neurodegeneration-like phenotypes than does removing either gene alone. Together, these results suggest that MUL1 plays an important compensatory function in organisms or cells lacking PINK1/parkin.
Results
Overexpression of MUL1, but not a ligase-dead form, suppresses PINK1 and parkin mutant phenotypes in dopaminergic neurons and muscle
We identified MUL1 as a novel suppressor of PINK1/parkin mutant phenotypes. Human MUL1 contains two transmembrane (TM) domains and a highly conserved C-terminal ring finger (RNF) domain. Topological studies suggest that the two TM domains anchor the protein to the mitochondrial outer membrane, with the RNF domain facing the cytosol (Li et al., 2008). Drosophila MUL1 (CG1134) encodes a protein with a similar domain structure, and 52% amino acid similarity to human MUL1 (Figure 1A,B).
Figure 1 with 1 supplement see all

Overexpression of MUL1, but not MUL1 LD, suppresses PINK1/parkin mutant phenotypes.
(A) Protein domain organization of Drosophila MUL1. TM1, TM2, and RNF represent transmembrane domains 1 and 2, and the RING Finger domain, respectively. The position of the mutation in the ligase dead (LD) version of MUL1 is marked with a red asterisk. (B) Sequence alignment of MUL1 in various species in the highly conserved RNF domain. A highly conserved histidine residue (marked as red) was mutated to alanine in MUL1 LD, ablating ligase activity. (C–C″) Dopaminergic neurons stained with an anti-TH antibody in red and mitochondria labeled with mitoGFP in green. Neurons in the PPL1 cluster are shown. While mitochondria in wild-type dopaminergic neurons are dispersed (C), mitochondria in PINK1 mutant dopaminergic neurons are clumped (C′, white arrow heads). This phenotype is suppressed by MUL1 overexpression driven by TH-Gal4 (C″). Scale bars: 10 µm. (D–E″’’ and J–M″’’) Confocal images of the IFM from thoraces double labeled with mitoGFP and phalloidin (red) (D–D″, J–J″, L–L″), or double labeled with mitoGFP and TUNEL (red) with lower magnification (E–E″, K–K″, M–M″). Scale bars: 5 µm. MUL1 overexpression is driven by Mef2-Gal4. In wild-type (D), mitochondria have a regular size and shape, and are localized in between myofibrils. In PINK1 mutants (D′), mitochondrial size becomes irregular, and the GFP signal is reduced. Large mitochondrial clumps also appear. PINK1 mutant muscle is TUNEL-positive (E′). (F–F″) Touidine blue staining of muscle. Compared with the wild-type (F), PINK1 mutant muscle shows vacuolation indicating muscle degeneration (F′). These PINK1 mutant phenotypes (D′, E′, F′) are almost completely suppressed by MUL1 overexpression (D″, E″, F″). (Ga–Ga″, Gb–Gb″) EM images of mitochondria in muscle. (Gb–Gb″) Single mitochondrion (outlined with dashed lines) from white boxes in Ga–Ga″. Scale bars: 1 µm (Ga–G″a) 0.5 µm (Gb–G″b). In wild-type (Ga and Gb), mitochondria have compact and organized cristae whereas mitochondria from PINK1 mutants (Ga′, Gb′) are swollen with fragmented cristae, and this is rescued by MUL1 overexpression (Ga″, Gb″). (H) Images of thoraces. Arrows point to thoracic indentations due to muscle degeneration. Compared with WT, PINK1 mutants have thoracic indentation due to muscle degeneration. MUL1 overexpression, but not MUL1 LD overexpression, suppresses PINK1 mutant thoracic indentation. (I) qPCR analysis shows that MUL1 and MUL1 LD mRNA are expressed at similar levels in muscles. The data are shown as the mean ± SEM from three experiments (RNA from ten 5-day-old fly thoraces for each genotype). The statistical analysis was done using One-way ANOVA with Tukey' multiple comparisons test. ns: not statistically significant. MUL1 LD overexpression in the PINK1 mutant background does not suppress the formation of mitochondrial clumps (J″) or TUNEL-positivity (K″). (L–M″) Overexpression of MUL1, but not MUL1 LD, suppresses parkin mutant phenotypes.
We overexpressed MUL1 in various tissues using the UAS/GAL4 system (Brand and Perrimon, 1993). Drosophila contains clusters of dopaminergic (DA) neurons in the adult brain. In wild-type DA neurons, mitochondria are dispersed in the cytosol (Figure 1C). In contrast, PINK1 mutant DA neurons show abnormally clumped mitochondria (Figure 1C′, arrowheads) (Park et al., 2006), which can be suppressed by overexpression of MUL1 (Figure 1C″).
We further characterized MUL1's effects on PINK1/parkin mutants in thoracic indirect flight muscle (IFM), which consists of well-organized muscle fibers, in which mitochondria fill spaces between myofibrils. PINK1 null mutant flies have severe defects in mitochondrial morphology, including an overall reduction in mitochondria-targeted GFP (mitoGFP) signal and the presence of large mitoGFP clumps (Figure 1D–D′,E–E′). PINK1 mutant muscle also shows extensive TUNEL-positive cell death (Figure1E–E′), muscle vacuolation, and degeneration (Figure 1F–F′). In addition, when examined under the electron microscopy (EM) level, many mitochondria are swollen with broken cristae (Figure 1Ga–Ga′,Gb–Gb′). At the level of the whole organism, PINK1 mutants show a thoracic indentation due to IFM degeneration (Figure 1H). Strikingly, MUL1 overexpression almost completely rescues all of the above PINK1 mutant phenotypes (Figure 1D″–F″,Ga″–Gb″,H).
To determine if the E3 ligase activity of MUL1 is required for suppression of PINK1 mutant phenotypes, we generated a ligase-dead form of Drosophila MUL1 (MUL1 LD) in which histidine 307, a highly conserved residue within the RNF domain, was mutated to alanine (Figure 1A,B). This mutation has been shown to abolish ligase activity of mammalian MUL1 (Zhang et al., 2008); in vitro ubiquitination assays confirm that Drosophila MUL1 LD lacks ligase activity (Figure 1—figure supplement 1). The expression levels of MUL1 and MUL1 LD in muscles are comparable (Figure 1I), and no mitochondrial clumps or muscle cell death are observed when MUL1 or MUL1 LD is overexpressed in wild-type animals (Figure 1J,J′,K,K′). Expression of MUL1 LD does not suppress PINK1 mutant phenotypes (Figure 1H,J″,K″). Overexpression of MUL1 (Figure 1L′,M′), but not MUL1 LD (Figure 1L″,M″), also suppressed parkin null mutant phenotypes. Thus, MUL1 is a robust suppressor of PINK1/parkin mutants and this requires MUL1's ligase activity.
MUL1 regulates mitochondrial morphology in Drosophila
As an E3 ligase anchored onto the mitochondrial outer membrane, MUL1 has been shown to have multiple substrates including Drp1 and Mfn (Braschi et al., 2009; Lokireddy et al., 2012). However, the consequences of loss of MUL1 have not been reported in any organism. The P element, MUL1EY12156 (MUL1EY), inserted at 20 bp upstream of the MUL1 start codon (Figure 2A), is a partial loss-of-function allele with reduced mRNA expression (Figure 2B–C). We performed imprecise excision of this P element and generated a large deletion allele, MUL1A6. MUL1A6, hereafter called the MUL1 mutant, produces no detectable transcript (Figure 2B–C), and therefore is a null allele. Flies homozygous for MUL1A6 are viable. We also generated two independent RNAi constructs that target two different locations in the MUL1 coding region. Flies expressing these constructs (MUL1 RNAi lines) show the same phenotypes (see below) and reverse the suppression of PINK1 mutant phenotypes observed upon MUL1 overexpression (Figure 2D).
Figure 2
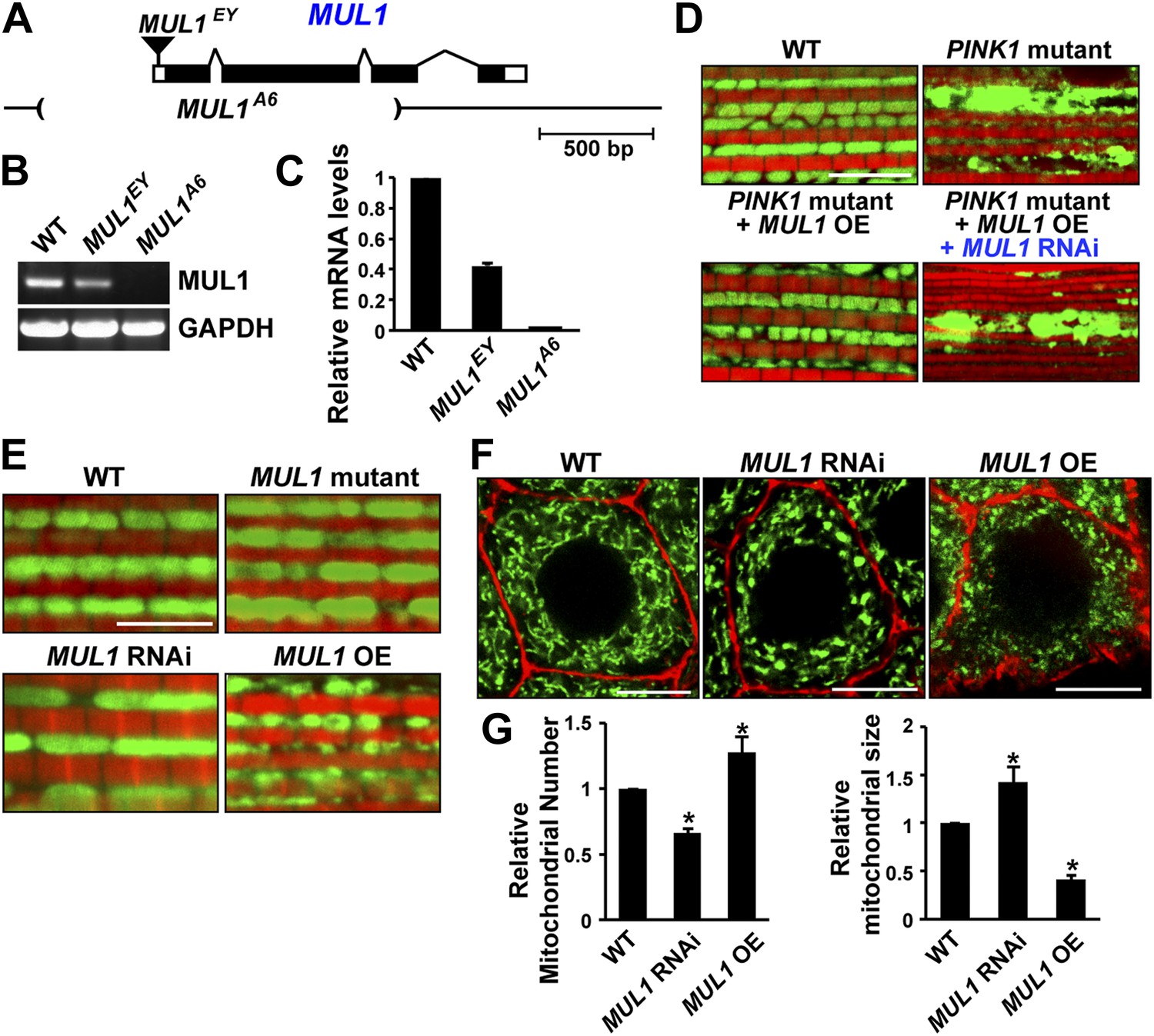
MUL1 regulates mitochondrial morphology.
(A) A schematic depicting the Drosophila MUL1 genomic region (cytological location 64A4). MUL1 coding and untranslated regions (dark and open rectangles, respectively) are depicted. The P element, MUL1EY, inserted in the 5′ UTR, is shown as an inverted triangle. The deleted region in the MUL1A6 allele is indicated by parentheses. (B) RT PCR shows that flies carrying the MUL1EY allele have detectable but reduced levels of MUL1 transcripts. However, no MUL1 transcript is detected in flies homozygous for the MUL1 deletion, MUL1A6. (C) qPCR shows that MUL1EY allele has approximately a 60% reduction of MUL1 transcript compared to the wild-type (WT). No MUL1 transcript is detected in flies homozygous for MUL1A6. (D) MUL1 RNAi line reverses the suppression of PINK1 mutant mitochondrial phenotypes due to MUL1 overexpression. (E) Muscle fibers stained with mitoGFP in green and actin in red. Compared with the WT, flies homozygous for the MUL1 deletion or expressing MUL1 RNAi show slightly elongated mitochondria. In contrast, when MUL1 is overexpressed using the Mef2-Gal4 driver, mitochondria are significantly smaller. (F) Salivary glands, with cell boundaries labeled with rhodamine phalloidin in red, and mitoGFP in green. In WT, mitochondria are tubular and evenly distributed. In contrast, in cells expressing MUL1 RNAi (driven by OK6-Gal4) mitochondria are fewer in number and found in clumps. In contrast, MUL1 overexpression (also driven by OK6-Gal4) results in fragmented mitochondria and irregular cell boundaries. (G) Quantification of mitochondrial number and size in salivary glands (mean ± SEM, n > 6 larvae for each genotype). * Significantly different from wild-type, p<0.05 (One-way ANOVA with Tukey's multiple comparisons test).
We examined phenotypes due to loss-of-function and overexpression of MUL1 in the IFM, which is a cellular syncytium, and in salivary glands, in which a number of individual cells contain an extensive tubular mitochondrial reticulum. Cells from MUL1 null mutant flies, or flies in which MUL1 RNAi is expressed, have mildly elongated mitochondria, while those from flies overexpressing MUL1 have small and fragmented mitochondria (Figure 2E–G). Thus, Drosophila MUL1 has a mild pro-fission function, as with mammalian MUL1 (Braschi et al., 2009).
MUL1 binds to Mfn and negatively regulates its levels through ubiquitination
Next, we asked whether Drp1, Mfn or both serve as MUL1 targets. Previous work suggested that MUL1 positively regulates Drp1's pro-fission activity through sumoylation-dependent protein stabilization (Harder et al., 2004; Braschi et al., 2009). Surprisingly, overexpression of MUL1 did not change Drp1 levels (Figure 3A). In contrast, overexpression of MUL1 led to a reduction in Mfn levels (Figure 3B). Analysis of larval lysates from mutants showed that loss of MUL1 results in an increase in Mfn levels, as does loss of PINK1 or parkin, which serve as positive controls (Figure 5—figure supplement 1). Knockdown of MUL1 in Drosophila S2 cells also resulted in an increase in Mfn levels (Figure 3C).
Figure 3 with 1 supplement see all

MUL1 physically binds to Mfn, and promotes ubiquitination-mediated Mfn degradation.
(A and B) Western blots and quantifications of Drp1 and Mfn levels in vivo. Analysis of lysates from thoraces show that MUL1 overexpression reduces Mfn levels (B) but not Drp1 levels (A). The data are shown as the mean ± SEM from three experiments (each experiment was done with lysate from 8 thoraces for each genotype). The statistical analysis was done using One-way ANOVA with Tukey's multiple comparisons test. ns: not statistically significant. ** Significantly different, p<0.01. (C) Western blots of Mfn levels in S2 cells either not treated or treated with control, PINK1, parkin or MUL1 RNAi. Quantification of relative Mfn levels shows that there is an increase in Mfn levels in cells treated with RNAi to PINK1, parkin, or MUL1 (mean ± SEM, ** Significantly different from cells not treated with RNAi, p<0.01, One-way ANOVA with Tukey's multiple comparisons test). (D) Co-immunoprecipitation using lysates from S2 cells transfected with the indicated constructs. The INPUT represents 2% of total lysate to monitor protein expression (top panel). MUL1-GFP is co-immunoprecipitated with Mfn-myc using both anti-GFP and anti-Myc antibodies. Mfn-myc also co-immunoprecipitates with HA-Parkin, which serves as a positive control. The interaction between Mfn-Myc and MUL1-GFP was specific, as confirmed by separate immunoprecipitation control experiments (Figure 3—figure supplement 1). (E) Mfn ubiquitination levels in S2 cells. S2 cells are treated with dsRNA designed to silence various genes and transfected with Mfn-Flag. Immunoprecipitation was performed with anti-Flag antibody, and Western blots were probed with anti-Ubiquitin antibody and an anti-Flag antibody. Relative ubiquitination levels compared to control are shown below (mean ± SEM). ** Significantly different from control, p<0.01 (One-way ANOVA with Tukey's multiple comparisons test). In S2 cells, Mfn is highly ubiquitinated. RNAi of MUL1 or parkin results in reduced levels of ubiquitnated Mfn. Two independent MUL1 RNAs are utilized to knockdown MUL1, which yield the same results. (F) In PINK1 mutant thoraces, where Mfn levels are increased, MUL1 overexpression (driven by Mef2-Gal4) reduces the increased Mfn levels. Relative Mfn levels compared to control are shown below (mean ± SEM). ** Significantly different, p<0.01 (One-way ANOVA with Tukey's multiple comparisons test).
Next, we determined if MUL1 and Mfn interact physically, and if MUL1 regulates Mfn levels through ubiquitination. Overexpressed MUL1 co-immunoprecipitated with overexpressed Mfn from Drosophila S2 cell lysates. Parkin was used as a positive control and co-immunoprecipitated with Mfn as previously reported (Poole et al., 2010; Ziviani et al., 2010). These physical interactions are specific to MUL1 and Mfn rather than the tags utilized (Figure 3—figure supplement 1). To assay ubiquitination, Flag-tagged Mfn was expressed in S2 cells exposed to dsRNA targeting MUL1 or parkin in the presence of the proteasome inhibitor MG132 (Figure 3E). In control cells, highly ubiquitinated Mfn was observed. When cells were treated with two different dsRNAs against MUL1, ubiquitinated Mfn levels were dramatically reduced, similar to those observed in parkin RNAi-treated cells, which serve as a positive control. Finally, we observed that the increased Mfn levels seen in the PINK1 mutant flies were reduced when MUL1 was overexpressed (Figure 3F), strengthening our argument that MUL1 suppresses PINK1 mutant phenotypes through reduction of Mfn levels. Together, these results suggest that MUL1 suppresses PINK1/parkin phenotypes by reducing Mfn levels through its ubiquitination-dependent degradation.
mfn overexpression, but not loss of drp1, results in phenotypes similar to those of PINK1 or parkin mutants; and these phenotypes are suppressed by MUL1 overexpression
Previous studies showed that downregulation of mfn or overexpression of drp1 could suppress PINK1 and parkin mutant phenotypes in Drosophila (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008). Parkin has also been shown to bind and ubiquitinate Mfn, promoting Mfn degradation (Gegg et al., 2010; Poole et al., 2010; Tanaka et al., 2010; Ziviani et al., 2010; Chan et al., 2011; Glauser et al., 2011). While increased Mfn levels are observed in PINK1 or parkin mutants (Poole et al., 2010; Ziviani et al., 2010), it is unclear if these increased mfn levels are sufficient to cause the phenotypes observed in PINK1 or parkin mutants. It is also unclear if a decrease in the levels of drp1, which can result in increased mitochondrial size through loss of fission, results in a phenotypically equivalent effect.
To address these questions, we generated transgenic flies carrying UAS-mfn (also called Marf, CG3869) and obtained two drp1 null alleles, drp11 and drp12 (Verstreken et al., 2005). Overexpression of mfn under the control of the muscle-specific (mef2) GAL4 driver resulted in organismal lethality. To circumvent this lethality, we also generated a new Gal4 driver, IFM-GAL4, in which GAL4 expression is driven specifically in the IFM (Figure 4F–J for IFM-GAL1, vs Figure 4A–E for mef2-GAL4), using regulatory sequences from the flightin gene. Since the IFMs are not required for viability, knockdown of essential genes using IFM-GAL4 does not cause lethality in flies (data not shown).
Figure 4
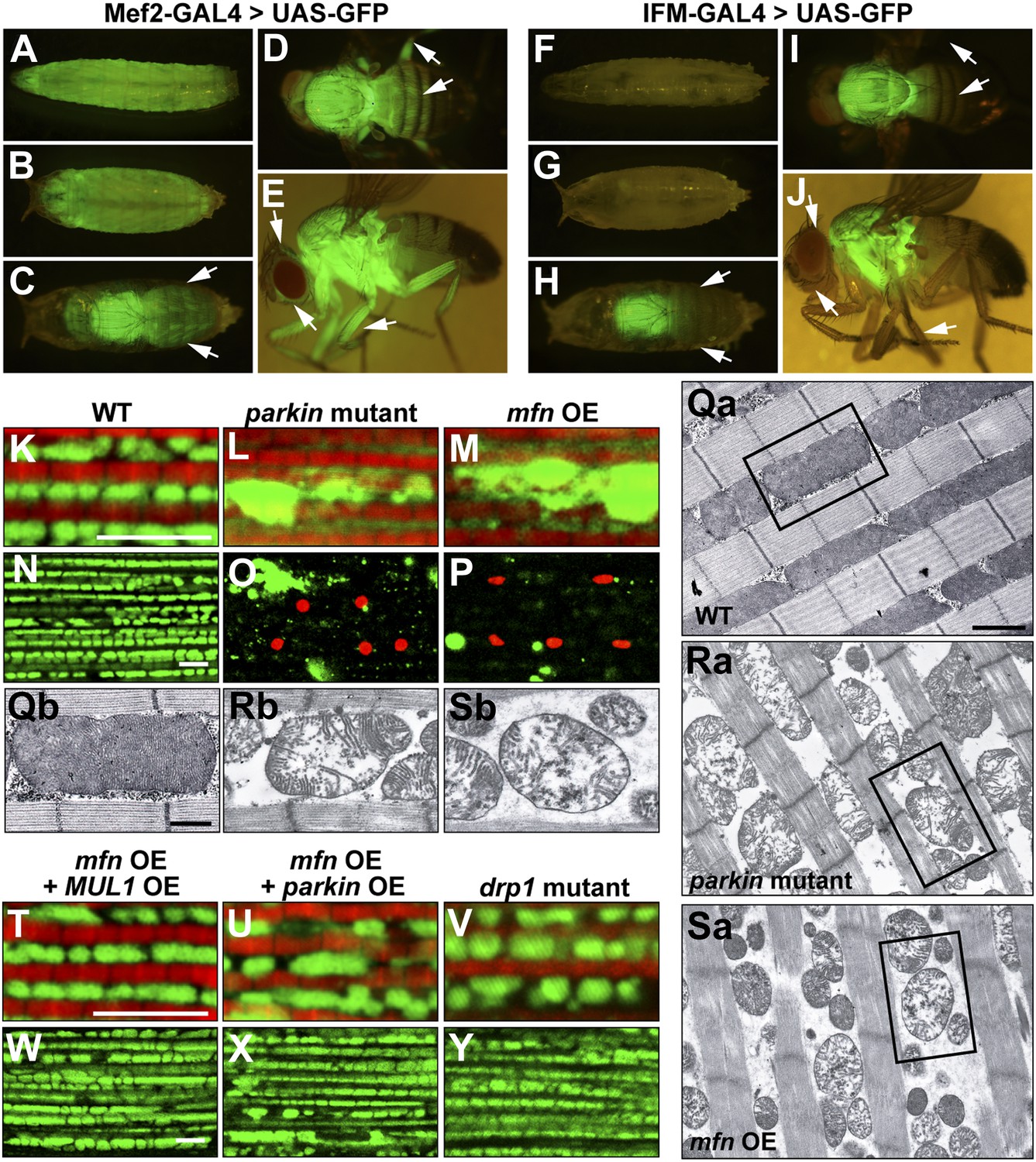
Generation and expression of the IFM-GAL driver; mfn overexpression, but not loss of drp1, induces PINK1/parkin-mutant like pathology.
(A–J) Different developmental stages of flies expressing GFP under Mef2-Gal4 (A–E) or IFM-Gal4 (F–J). (A) Third instar larvae show GFP expression in whole body muscles. (B) At the early pupal stage, GFP is expressed in a similar pattern as in larvae. However, the GFP expression pattern become more specific at the late pupal stage (C), in which the strongest GFP signal is seen in the thorax, and a weaker signal is observed in the head and abdomen (arrows). (D) In an adult fly, dorsal view shows GFP signal in the thorax, upper abdomen and legs. (E) GFP is also expressed in adult head and legs, marked with arrows. (F) Flies expressing GFP under IFM-Gal4 show no GFP expression in third instar larvae, or in early pupae (G). (H) GFP is strongly expressed only in the thorax at the late pupal stage, but not in other areas (arrows). (I) In the adult fly, GFP signal is highly concentrated in the thorax. No GFP expression in abdomen and legs is observed, arrows. (J) In contrast to GFP expression under Mef2-Gal4, IFM-Gal4 does not express in adult head or legs, as indicated with arrows. (K–P, T–Y) Confocal images of muscle double labeled with mitoGFP (green) and phalloidine (red) (K–M, T–V), or those labeled with mitoGFP and TUNEL (red) at lower magnification (N–P, W–Y), respectively. (Qa–Sb) EM images of mitochondria in muscle. Single mitochondrion from the black-boxed area in Qa, Ra, Sa is shown in Qb, Rb, Sb. Scale bars: 1 µm (Qa, Ra, Sa) and 0.5 µm (Qb, Rb, Sb). Compared with wild-type (K and N), parkin null mutant (L and O) shows overall reduced levels of mitoGFP signal, large mitochondrial clumps, and muscle cell death. Similar phenotypes are observed with mfn overexpression (M and P), and these phenotypes are suppressed by MUL1 overexpression (T and W). As a control, parkin overexpression also suppresses phenotypes due to mfn overexpression (U and X). Importantly, drp1 null (drp11/drp12) mutant muscle does not have any mitochondrial clumping or TUNEL-positivity seen in loss of parkin function or mfn overexpression (V and Y). mfn overexpression is driven by IFM-Gal4. Scale bars: 5 µm.
Interestingly, overexpression of mfn in the IFM results in phenotypes (Figure 4M,P,Sa,Sb) similar to those of PINK1 or parkin mutants; mitoGFP clumps, TUNEL-positive muscle cell death, and broken mitochondrial cristae when examined at the EM level (Figure 1D′,E′,G′a,G′b). In contrast, while loss of drp1 results in an increase in mitochondrial size, no muscle cell death or degeneration is observed (Figure 4V,Y). Importantly, MUL1 overexpression (Figure 4T,W), as with parkin overexpression (Figure 4U,X), suppressed the phenotypes associated with mfn overexpression. Together, these results show that overexpression of mfn, but not loss of drp1, leads to phenotypes similar to those due to lack of PINK1 or parkin, suggesting a direct link between increased Mfn levels and pathology.
MUL1 acts in parallel to the PINK1/Parkin pathway
Our observations that MUL1 overexpression suppresses PINK1 or parkin mutant phenotypes, and that both Parkin and MUL1 promote Mfn degradation, suggest two possible scenarios of how MUL1 and PINK1/parkin interact. MUL1 may be a downstream target of the PINK1/parkin pathway and upstream of mfn. Alternatively, MUL1 could function in a parallel pathway to PINK1/Parkin, but with action on a common target such as Mfn. Characterization of double null mutants provides an effective way of distinguishing these possibilities. If MUL1 functions in the same pathway as PINK1, double null mutants of PINK1 and MUL1 would be expected to show the same phenotype as the single mutant alone, as is observed in the case of PINK1 parkin double mutants (Clark et al., 2006; Park et al., 2006). Conversely, if MUL1 and PINK1/parkin act in parallel pathways, the phenotypes of double null mutants may be stronger than those of single mutants.
We generated PINK1 MUL1 and parkin MUL1 double mutants. Several lines of evidence show that double mutants have significantly enhanced phenotypes as compared to those of single mutants alone. First, PINK1 MUL1 and parkin MUL1 double null mutants show a high frequency of pupal lethality as compared with single mutants (data not shown), while double null mutants of PINK1 parkin have the same level of viability as single mutants (Clark et al., 2006; Park et al., 2006). Second, a thoracic indentation observed in PINK1 or parkin null mutants is much more severe in PINK1 MUL1 and parkin MUL1 double null mutants. In contrast, PINK1 parkin double null mutants show the same degree of thoracic indentation as PINK1 or parkin single mutants alone (Figure 5A–G). Third, at the cellular level, PINK1 MUL1 and parkin MUL1 double null mutants have highly elongated and interconnected mitochondria, as determined using anti-mitochondrial ATPase antibodies. These mitochondrial phenotypes are very different from those of PINK1, parkin, or MUL1 mutants (Figure 5I–O). PINK1 parkin double null mutants show similar mitochondrial morphology phenotypes as PINK1 or parkin single mutants alone (Figure 5O vs Figure 5J,M). Fourth, ATP levels in parkin MUL1 double null mutants were further reduced compared to those of parkin or MUL1 single null mutants (Figure 5Q). Fifth, the ability of parkin overexpression to rescue PINK1 mutants is not dependent on MUL1, and MUL1 overexpression can still suppress PINK1 mutants in the absence of parkin (Figure 5—figure supplement 1). Sixth, knockdown of both MUL1 and parkin in S2 cells further reduces Mfn ubiquitination below levels seen with knockdown of MUL1 or parkin alone (Figure 5R). Seventh, PINK1 MUL1 and parkin MUL1 double null mutants have higher Mfn levels as compared to single null mutants of MUL1, PINK1, or parkin (Figure 5—figure supplement 1). Finally, knockdown of mfn in the background of parkin MUL1 double mutants almost completely rescues the thoracic indentation and mitochondrial phenotypes of parkin MUL1 double mutants (Figure 5H,P). These genetic observations, in combination with biochemical findings that MUL1 physically interacts with Mfn, and that loss of MUL1 results in decreased ubiquitination of endogenous Mfn and increased Mfn levels, indicate that MUL1 acts in parallel to the PINK1/parkin pathway to regulate a common target Mfn.
Figure 5 with 1 supplement see all
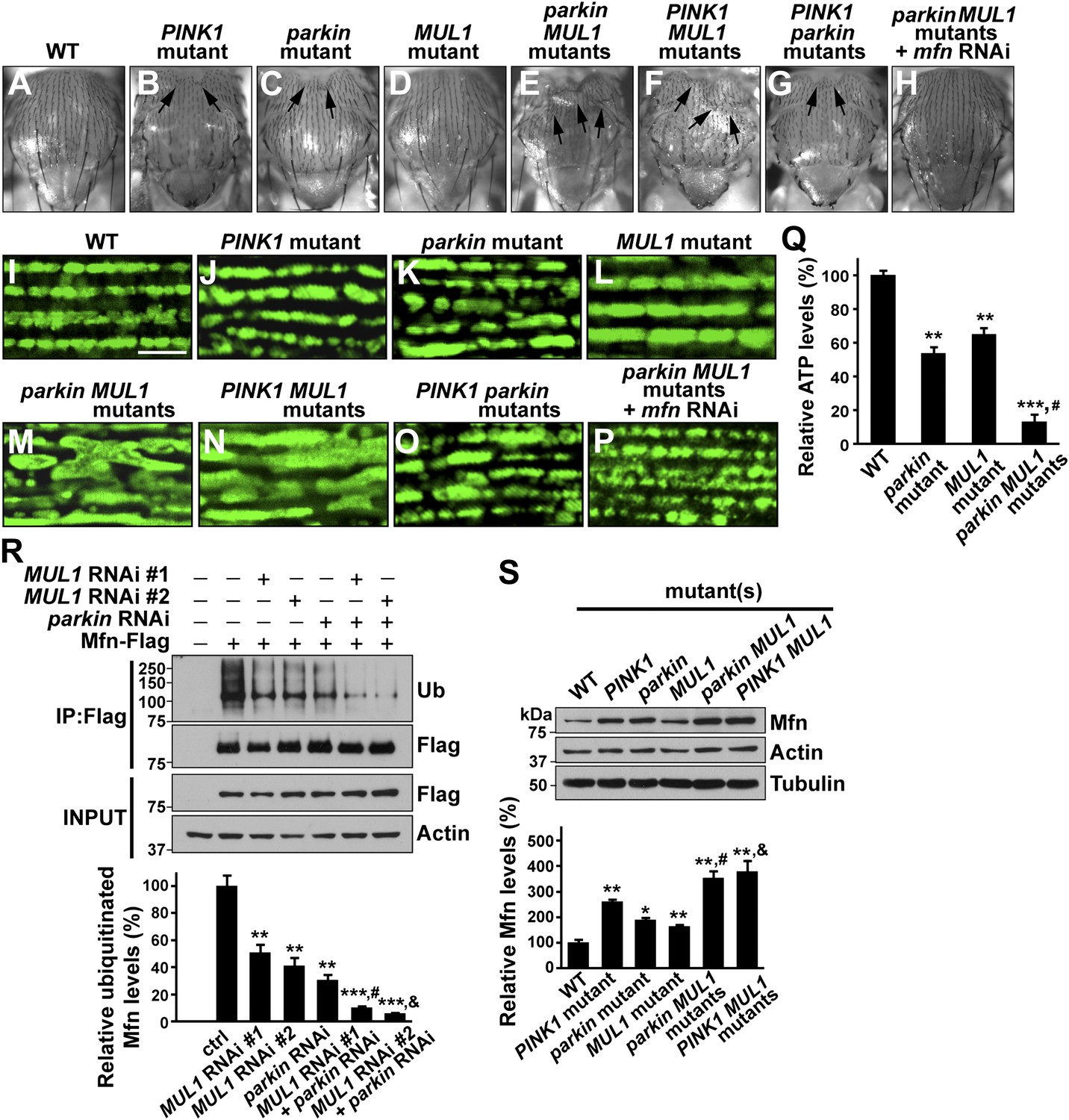
MUL1 acts in parallel to the PINK1/parkin pathway.
(A–H) Images of thoraces of various mutants. Arrows point to thoracic indentations due to muscle degeneration. PINK1 MUL1 and parkin MUL1 double mutants have more severe thoracic indentation compared to either mutant alone. Remarkably, the severe thoracic indentation phenotype in parkin MUL1 double mutants is almost completely suppressed when mfn is also knocked down. (I–P) Mitochondria are labeled using an anti-ATP synthase antibody in the IFM. While PINK1, parkin, and MUL1 mutant show slightly elongated mitochondrial morphology, PINK1 MUL1 and parkin MUL1 double mutants exhibit highly elongated and interconnected mitochondria. These phenotypes can be suppressed by mfn knockdown. Instead of using mitoGFP, we utilized anti-ATPase antibodies that allow better visualization of the enhancement phenotypes seen with double mutants. (Q) Relative ATP levels in whole flies of various mutants (mean ±SEM from three experiments, five 5-day-old flies for each genotype). ** and *** significantly different from wild-type, p<0.01 and p<0.001, respectably (One-way ANOVA with Tukey's multiple comparisons test). # Significantly different from parkin mutant and MUL1 mutant, both p<0.01 (Two-way ANOVA with Tukey's multiple comparisons test). (R) In vivo ubiquitination assay of Mfn. S2 cells were treated with the indicated RNAi, transfected with Flag-Mfn, and treated with proteasome inhibitor MG132. Immunoprecipitations were performed using anti-Flag antibody, and western blots were probed with antibodies against anti-Ubiquitin antibody (P4D1) or anti-Flag antibody. Relative ubiquitination levels compared to control are shown in the lower panel (mean ± SEM). ** and *** Significantly different from control, p<0.01 and p<0.001, respectably (One-way ANOVA with Tukey's multiple comparisons test). # Significantly different from MUL1 RNAi #1 and parkin RNAi, both p<0.01. & Significantly different from MUL1 RNAi #2 and parkin RNAi, p<0.001 and p<0.01, respectably (Two-way ANOVA with Tukey's multiple comparisons test). (S) Western blot analysis of Mfn levels in vivo and quantification (mean ± SEM from three experiments, eight third instar larvae for each genotype). * and ** significantly different from wild-type, p<0.05 and p<0.01, respectably (One-way ANOVA with Tukey's multiple comparisons test). # Significantly different from parkin mutant and MUL1 mutant, both p<0.01. & Significantly different from PINK1 mutant and MUL1 mutant, both p<0.01 (Two-way ANOVA with Tukey's multiple comparisons test).
The roles of MUL1 in regulating mitochondrial morphology and mfn levels are conserved in human cells
Next, we asked if MUL1-mediated mitochondrial morphology and Mfn regulation is conserved in human cells. We expressed human MUL1 and MUL1 LD in HeLa cells (Figure 6F). Cells expressing MUL1 or MUL1 LD are GFP-positive and marked with asterisks (Figure 6A–D″). Cells expressing GFP-MUL1 showed peri-nuclear mitochondrial clustering (Figure 6A–B, asterisks), and mitochondria appeared small and globular in shape as compared to those in untransfected, GFP-negative cells (Figure 6B–B″). MUL1 LD neither causes mitochondrial clustering nor alters mitochondrial morphology (Figure 6C–D″, asterisks).
Figure 6
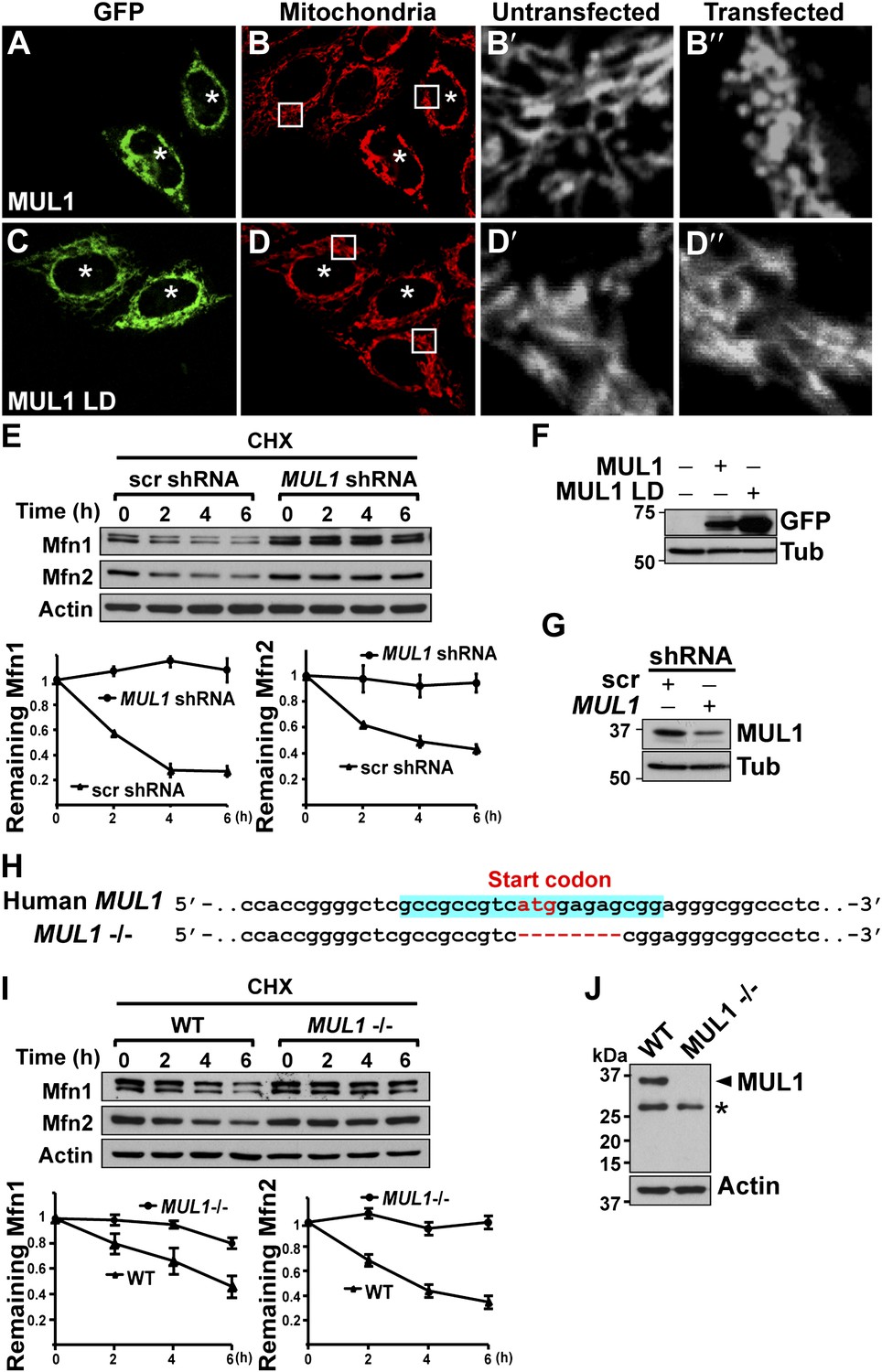
MUL1’s function in mitochondrial morphology and Mfn levels is conserved in human cells.
(A–D‴) HeLa cells transfected with GFP-MUL1 (A–B″) or GFP-MUL1 LD (C–D″) are marked with asterisks, while cells not transfected serve as internal controls. Mitochondria are labeled with mitotracker in red (B and D). (B′ and B″, D′ and D″) Higher magnification images of mitochondria within white boxes in B and D. Cells expressing GFP-MUL1 have clustered mitochondria in the perinuclear region (B). Mitochondria are also small and fragmented (B″), as compared to cells not expressing GFP-MUL1 (B′). Importantly, GFP-MUL1 LD does not result in localization of mitochondria to the perinuclear region (D) or in mitochondrial fragmentation (D′). (E) Western blot analysis of Mfn1 and Mfn2 levels after CHX treatment. HeLa cells expressing scrambled shRNA or MUL1 shMUL1 are treated with CHX. Mfn1 and 2 levels at each time point are normalized with Actin. The relative portion of remaining Mfn1 and 2 as compared to time point 0 was calculated and plotted (E). In cells expressing MUL1 shRNA, Mfn1 and 2 levels after CHX treatment are more stable than those in cells expressing scrambled shRNA. (F) Expression of transfected GFP-MUL1 and GFP-MUL1 LD in HeLa cells, as detected using anti-GFP antibody. (G) Western blot analysis of endogenous MUL1 levels in HeLa cells stably expressing scrambled shRNA and MUL1 shRNA. MUL1 shRNA expressing cells have reduced levels of endogenous MUL1. (H) Human MUL1 sequence and deletion in MUL1 knockout (MUL1−/−) HeLa cells, generated using the CRISPR/Cas 9 system. Sequences targeting MUL1 are highlighted in blue. Red letters indicate start codon. Red dashes represent deleted bases. Deleted eight base pairs include the start codon of MUL1. (I) Western blot analysis of Mfn1 and Mfn2 levels in wild-type and MUL1−/− HeLa cells treated with CHX for the indicated time. Remaining Mfn1 and Mfn2 levels at each time point were plotted below. (J) Western blot showing no MUL1 expression in MUL1−/− HeLa. Arrowhead points to MUL1 protein. Asterisk indicates a non-specific band.
Mammals have two Mfn proteins, Mfn1 and Mfn2, both able to promote mitochondrial fusion (Chen et al., 2003; Eura et al., 2003). We monitored the fate of Mfn1 and Mfn2 in control HeLa cells and HeLa cells stably expressing small hairpin RNA against MUL1 (MUL1 shRNA) (Figure 6E,G). When cells were exposed to the protein synthesis inhibitor cycloheximide (CHX), Mfn1 and Mfn2 levels gradually decreased in control cells, but were dramatically stabilized in cells with decreased levels of MUL1 (Figure 6E). To confirm the above result, we generated MUL1 knockout HeLa cells (Figure 6H), which contain a deletion including the start codon in the MUL1 genomic region, using the CRISPR/Cas 9 system (Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013). Two independent anti-MUL1 antibodies confirmed no MUL1 expression in MUL1 knockout cells (Figure 6J and data not shown). Similar to what was observed for the MUL1 shRNA, Mfn1 and Mfn2 levels were also dramatically stabilized in MUL1 knockout cells (Figure 6I). Together, these results suggest that the role of MUL1 in regulating Mfn stability and mitochondrial morphology is conserved in human cells.
MUL1 does not affect Parkin-mediated mitophagy
The PINK1/Parkin pathway mediates mitophagy in HeLa cells (Narendra et al., 2008, 2010), mouse cortical neurons, and heart muscle (Cai et al., 2012; Chen and Dorn, 2013). When cells are treated with a mitochondrial uncoupler, mitochondria lose their membrane potential. This leads to recruitment of Parkin to the depolarized mitochondria, ultimately resulting in autophagic degradation of these mitochondria. Because of the genetic interactions observed between MUL1 and PINK1/parkin in Drosophila, we asked if MUL1 was able to modulate Parkin-mediated mitophagy.
We induced mitophagy by exposing HeLa cells to antimycin A, which inhibits electron transport and depolarizes the mitochondrial membrane. Wild-type, MUL1 knockout, and PINK1 knockout (as a control) HeLa cells were transfected with YFP-Parkin and treated with DMSO or antimycin A for indicated time. In wild-type, without antimycin A treatment, Parkin mainly localizes in the cytosol (Figure 7A). However, following antimycin A treatment for 3 hrs, most Parkin was translocated to the mitochondria (Figure 7B,D). After 24 hrs of antimycin A treatment, Parkin was found dispersed in the cytosol and mitochondria were no longer observed, indicating that mitophagy had occurred (Figure 7C,E). To ensure there was no delay in mitophagy, we also assayed cells that were treated with antimycin A for 6, 12, 16 and 20 hrs (Figure 7F, data not shown). In all cells, mitophagy was observed at least 16 hrs after antimycin A treatment. No significant differences were observed in the fraction of Parkin recruited to mitochondria, or the fraction of mitochondria that underwent mitophagy, at any of these time points, for wild-type and MUL1 knockout cells (Figure 7B–F). Knockdown of MUL1 using shRNA also had no effect on Parkin translocation or mitophagy (Figure 7—figure supplement 1). As a control, PINK1 knockout HeLa cells showed almost no Parkin localization to mitochondria and lack of mitophagy (Figure 7B–F). Similar results were obtained when cells were treated with carbonyl cyanide m-chlorophenylhydrazone (CCCP), which uncouples mitochondrial membrane potential (data not shown). Finally, MUL1 overexpression also had no effect on Parkin translocation (Figure 7G–H) and did not block mitophagy (data not shown). Thus, neither loss of MUL1 nor its overexpression altered Parkin translocation or mitophagy. These results are consistent with MUL1 acting in parallel to PINK1/parkin in Drosophila, and suggest that MUL1 regulates mitochondrial health through a distinct pathway.
Figure 7 with 1 supplement see all

Neither MUL1 knockout nor overexpression affects Parkin-mediated mitophagy.
(A–C) HeLa cells (control, MUL1 knockout or PINK1 knockout) were transfected with YFP-Parkin, treated with either DMSO or antimycin A, and immunostained with an anti-Tom20 antibody which labels mitochondria. (A) HeLa cells treated with DMSO as a control. (B) Following treatment with antimycin A for 3 hrs, Parkin is recruited to mitochondria, as shown by co-localization of Parkin and the mitochondrial marker. In MUL1 null cells, Parkin recruitment to mitochondria is not affected, whereas in PINK1 null cells (positive control), Parkin recruitment to mitochondria is abolished. (C) After 24 hrs of antimycin A treatment, Parkin returns to the cytosol and the mitochondrial signal disappears. In MUL1 null cells, mitochondrial disappearance occurs similarly as with WT, whereas in PINK1 null cells (positive control), mitochondria are not eliminated. (D–E) Quantification of cells with Parkin recruited to mitochondria after 3 hrs of antimycin A treatment (D) and with few or no mitochondria after 24 hr of antimycin A treatment (E) and after 16 and 20 hrs of antimycin A treatment (F). The data are shown as the mean ± SEM from three experiments (n ≥ 100 for each genotype). *** Significantly different from wild-type, p<0.001. ns: not statistically significant (One-way ANOVA with Tukey's multiple comparisons test). While Parkin translocation and mitochondrial disappearance are significantly blocked in PINK1 knockout cells, there is no significant difference between HeLa cells and MUL1 knockout cells in these processes. (G) HeLa cells stably expressing YFP-Parkin and mito-RFP are transfected with Flag-MUL1, treated with DMSO or antimycin A, and immunostained with anti-Flag antibody. 3-hour antimycin A treatment causes Parkin localization to mitochondria in cells with or without MUL1 expression. (H) Quantification of cells with Parkin recruited to mitochondria after 1.5 or 3 hrs Antimycin A treatment. Both 1.5 and 3 hrs of antimycin A treatments results in similar levels of Parkin recruitment to mitochondria. The data are shown as the mean ± SEM from three experiments (n ≥ 100 for each genotype). ns: not statistically significant (One-way ANOVA with Tukey's multiple comparisons test).
Loss of both MUL1 and parkin aggravates mitochondrial damage and induces degeneration-like phenotypes in mouse cortical neurons
To further test the hypothesis that MUL1 functions in parallel to the PINK1/parkin pathway in mammalian cells, we investigated the effects of depleting both MUL1 and parkin. As HeLa cells do not express Parkin, we turned to cultured mature cortical neurons. GFP-MUL1 localizes to the mitochondria in cell bodies and axons of the primary cortical neurons (Figure 8A, Figure 8—figure supplement 1).
Figure 8 with 2 supplements see all
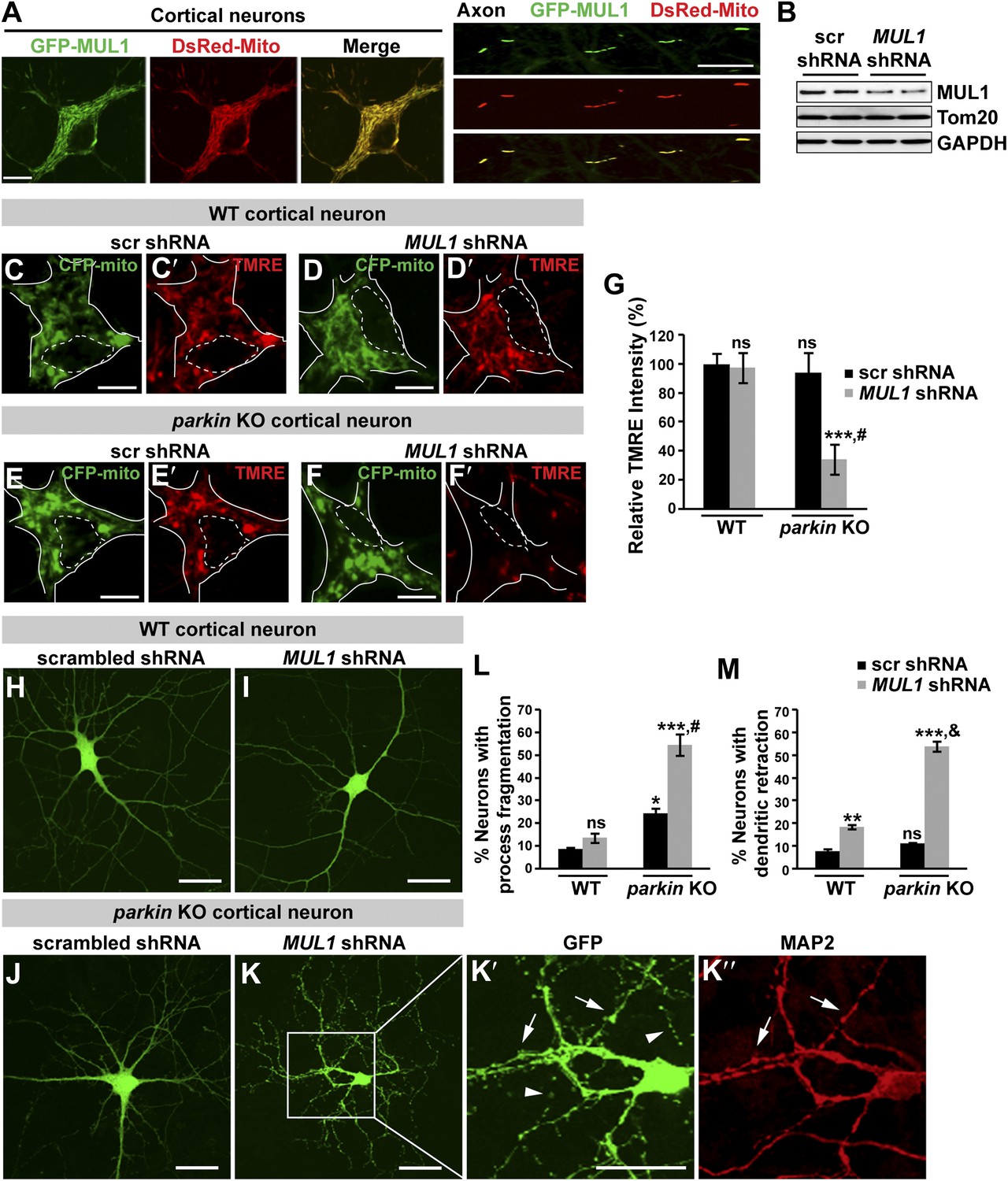
Loss of both MUL1 and parkin aggravates mitochondrial damage and induces degeneration-like phenotypes in mouse cortical neurons.
(A) MUL1 targets mitochondria in the cell bodies and axons of mouse primary cortical neurons. Neuronal mitochondria were labeled by DsRed-Mito or stained with an antibody against mitochondrial marker, TOM20 or Cytochrome C (Figure 8—figure supplement 1). (B) Levels of endogenous MUL1 in neurons transfected with scrambled or MUL1 shRNA. Note that partial suppression of endogenous MUL1 may reflect relative low transfection rate (20%) in the neuronal culture. (C–F′) Mitochondria in live cortical neurons were co-labeled by expressing CFP-mito, which targets all mitochondria, and by loading fluorescent dye TMRE, which stains healthy mitochondria dependent upon membrane potential (Δψm). Loading TMRE also labels mitochondria in glia in the culture. The edges of neuron cell bodies are marked with white solid lines, and the nuclei are outlined with white dashed lines. In contrast to other neurons, parkin knockout neurons with MUL1 knockdown show reduced TMRE intensity (F and F′), indicating decreased Δψm. Scale bars: 10 µm. (G) Quantification of relative TMRE intensity. TMRE intensity measured from each group of neurons was normalized to WT neurons transfected with scrambled shRNA. The data are shown as the means ± SEM from three experiments. (n ≥ 12 for each group). *** Significantly different from wild-type neurons transfected with scrambled shRNA, p<0.001. ns: not statistically significant (One-way ANOVA with Tukey's multiple comparisons test). # Significantly different from wild-type neurons transfected with MUL1 shRNA and parkin KO neurons transfected with scrambled shRNA, p<0.001 and p<0.01, respectively (Two-way ANOVA with Tukey's multiple comparisons test). (H–M) MUL1 knockdown in parkin KO neurons results in enhanced fragmentation of neurites. Representative wild-type (H and I) and parkin KO (J–K″) cortical neurons transfected with scrambled or MUL1 shRNA and labeled with GFP (confirming transfection of shRNA and labeling axons and dendrites). (K′–K″) Higher magnification of a white box in K showing the soma and proximal dendrites labeled with an anti-MAP2 antibody (red). Arrows point to the GFP- and MAP2-labeled dendrites, and arrowheads indicate GFP-labeled but MAP2-negative fragmented axons. Scale bars: 20 µm. (L and M) Quantitative analysis showing enhanced process fragmentation (L) and dendritic retraction (M). The data are shown as the means ± SEM from three experiments (process fragmentation phenotype: n ≥ 115 for each genotype, dendritic retraction: n ≥ 127 for each phenotype). *, **, and *** Significantly different from wild-type neurons transfected with scrambled shRNA, p<0.05, p<0.01, and p<0.001, respectively. ns: not statistically significant (One-way ANOVA with Tukey's multiple comparisons test). # Significantly different from wild-type neurons transfected with MUL1 shRNA and parkin KO neurons transfected with scrambled shRNA, both p<0.001. & Significantly different from wild-type neurons transfected with MUL1 shRNA and parkin KO neurons transfected with scrambled shRNA, both p<0.001 (Two-way ANOVA with Tukey's multiple comparisons test).
The proper maintenance of the mitochondrial inner membrane potential (Δψm) depends on the physiological function of the mitochondrial respiratory chain, and is crucial for generating ATP. Dissipation of the membrane potential is a strong indication of unhealthy mitochondria, which can lead to severe mitochondrial dysfunction and subsequent cell death. The Δψm can be measured using a fluorescent dye tetramethyl rhodamine ethyl ester (TMRE). We used two independent MUL1 shRNAs to suppress endogenous MUL1 expression in cortical neurons (Figure 8B). Cortical neurons expressing CFP-mito, from either wild-type mice co-expressing two independent MUL1 shRNA (Figure 8D–D′), or parkin gene KO mice co-expressing a scrambled shRNA (Figure 8E–E′), showed no significant decrease in the intensity of TMRE fluorescence (Figure 8C,C′,G). However, parkin KO neurons co-expressing MUL1 shRNA showed a significant reduction of Δψm (Figure 8F–F′,G), suggesting that proper MUL1 expression in primary cortical neurons is required to compensate for loss of parkin in maintaining mitochondrial integrity.
Next, we asked if MUL1 knockdown alters Mfn2 levels in mouse cortical neurons. Neurons from wild-type or parkin KO mice were transfected with either scrambled shRNA or MUL1 shRNA, followed by co-staining with anti-Cytochrome C and anti-Mfn2 antibodies. Relative Mfn2 intensity in individual neurons was analyzed by calculating the ratio of Mfn2 to Cytochrome C (Figure 8—figure supplement 2). Compared to wild-type neurons transfected with scrambled shRNA, wild-type neurons with MUL1 shRNA, or parkin KO neurons with either scrambled shRNA or MUL1 shRNA had an increased intensity ratio of Mfn2 to Cytochrome C. This suggests that MUL1's role in regulating Mfn2 levels is also conserved in neurons.
Finally, we investigated if loss of either MUL1 or parkin, or loss of both, has any impact on cultured primary cortical neuron morphology. MUL1 knockdown in cells from wild-type mice resulted in a minor increase in dendritic retraction but no significant process fragmentation as compared with wild-type cells (Figure 8H,L,M). Cortical neurons from parkin KO mice showed slightly increased process fragmentation but no dendritic retraction (Figure 8J,L,M), as compared with wild-type cells. In contrast, MUL1 knockdown in parkin KO neurons resulted in a dramatic increase in the number of neurons with dendritic and axonal fragmentation and retraction (Figure 8K–K″,L–M; process fragmentation: total number of neurons examined: n > 115 and n > 127 each genotype for fragmentation and dendritic retraction analysis, respectively), indicative of early neurodegeneration. The observed phenotypes were confirmed using a second MUL1 shRNA in parkin KO neurons (data not shown). These observations suggest that MUL1 acts in parallel to the PINK1/parkin pathway to ensure mitochondrial integrity and function, thus maintaining neuronal health in primary cortical neurons.
Discussion
In summary, we identified MUL1 as a robust suppressor of PINK1/parkin mutant phenotypes in Drosophila. MUL1 overexpression, but not expression of a ligase-dead version, strongly suppresses PINK1 and parkin mutant phenotypes. The mechanism of this suppression is unique in that MUL1 does not act on PINK1 or parkin, nor does it function as a downstream target. Rather, MUL1 acts by suppressing mfn in parallel to the PINK1/parkin pathway (Figure 9B). mfn is crucial for actions downstream of PINK1/parkin to maintain mitochondrial function and tissue health (Figure 9B), as overexpression of mfn leads to pathology similar to lack of PINK1/parkin function. We hypothesize that the increase in the Mfn level needs to reach a threshold, such as that observed in the PINK1/parkin mutant backgrounds, but not in the MUL1 null background, in order for overt muscle cell degeneration to occur (Figure 9B). Biochemically, MUL1 binds to Mfn, and loss of MUL1 results in decreased ubiquitination of Mfn and increased Mfn levels. These observations suggest that MUL1 may directly ubiquitinate Mfn, leading to its degradation (Figure 9A). Alternatively, MUL1 may act via an intermediary that promotes Mfn ubiquitination and degradation. In Drosophila, overexpression of MUL1 almost completely suppresses all aspects of the PINK1/parkin null phenotypes. Thus, treatments that manipulate MUL1 expression or activity may have potential as therapeutics strategies.
Figure 9

Models for how MUL1 interacts with PINK1/parkin.
(A) Schematic depictions of how MUL1, PINK1, Parkin, and Mfn interact in the mitochondria. In mammalian cells, upon mitochondrial damage (CCCP or antimycin A treatment), PINK1 is stabilized onto the mitochondrial OM of damaged mitochondria, with its kinase domain facing the cytosol (Zhou et al., 2008). PINK1 recruits Parkin onto the OM, either through direct phosphorylation or indirect interaction with other proteins (not depicted here) (Jin and Youle, 2012). Parkin then ubiquitinates multiple substrates on the OM, including Mfn. MUL1, a mitochondrial OM-anchored ligase with its RNF domain facing the cytosol, also mediates ubiquitination of Mitofusin. (B) The PINK1/parkin pathway and MUL1 act in parallel to regulate mfn, and maintain mitochondrial function and tissue health. Reducing either PINK1/parkin or MUL1 leads to increased levels of Mfn. Significant elevation of Mfn leads to mitochondrial dysfunction and tissue damage, similar to what is observed in PINK1/parkin mutants. Loss of both PINK1/parkin and MUL1 leads to significantly higher Mfn levels, associated with severe mitochondrial dysfunction and tissue damage. OM: mitochondrial outer membrane; IM: mitochondrial inner membrane.
In addition to showing that overexpression of MUL1 compensates for lack of PINK1/parkin by downregulating Mfn levels, we have identified an evolutionarily conserved pathway and provide compelling evidence showing that endogenous levels of MUL1 normally compensates for lack of PINK1 or parkin in Drosophila and in mammals. Removal of MUL1 in the PINK1 or parkin null background significantly aggravates the phenotypes due to lack of PINK1 or parkin alone. Flies lacking MUL1, PINK1 or parkin are viable, but PINK1/MUL1 or parkin/MUL1 double mutants manifest increased lethality with much more severe muscle degeneration, reduced ATP levels, defective mitochondrial morphology and increased Mfn levels. In addition, while parkin KO mature mouse cortical neurons or MUL1 knockdown neurons show only mild neuronal phenotypes, neurons with parkin KO and MUL1 KD show significantly diminished mitochondrial membrane potential, indicating mitochondrial dysfunction. They also show neurodegeneration-like phenotypes including axonal and dendritic fragmentation, and reduced mitochondrial distribution along processes. Finally, human HeLa cells, which have little or no endogenous Parkin, show a dramatic stabilization of Mfn when MUL1 is eliminated.
Our findings may help to address an important puzzle in the field of PD research: why do PINK1 or parkin knockout mice, or even parkin/DJ-1/PINK1 triple knockout mice, bear only subtle phenotypes related to dopaminergic neuronal degeneration or mitochondrial morphology changes (Palacino et al., 2004; Perez and Palmiter, 2005; Perez et al., 2005; Kitada et al., 2007; Frank-Cannon et al., 2008; Gautier et al., 2008; Gispert et al., 2009; Kitada et al., 2009; Akundi et al., 2011). Our studies provide genetic and cellular clues that suggest compensation by MUL1 may contribute to the subtle phenotypes in PINK1 or parkin mutant mice. It will be interesting to determine whether PINK1/MUL1 or parkin/MUL1 double knockout mice show more severe PD-related pathology. Regarding PD therapies, optimizing the function of MUL1 is likely to be beneficial for PINK1/PARKIN patients; upregulating MUL1 may rescue the pathology due to lack of PINK1 or PARKIN. In contrast, downregulating MUL1 and/or mutations in MUL1 may lead to disruption of this compensatory pathway in maintaining mitochondrial integrity and function, and result in accelerated disease progression.
Why do cells have multiple E3 ubiquitin ligases acting on a common target? Mfn is localized to the mitochondrial outer membrane (OM) (Figure 9A) and is a key molecule that regulates mitochondrial fusion in response to various cellular processes. Due to its importance, the level of Mfn is expected to be tightly regulated, and this may require several E3 ubiquitin ligases and deubiquitinases that respond to different stimuli (Gegg et al., 2010; Park et al., 2010; Leboucher et al., 2012; Lokireddy et al., 2012; Anton et al., 2013; Fu et al., 2013). In the case of mitochondrial damage, Parkin translocates to depolarized mitochondria before it degrades Mfn (Figure 9A), thus preventing damaged mitochondria from fusing with healthy ones. As an E3 ligase anchored on the OM (Li et al., 2008; Figure 9A), MUL1 is constantly present in the vicinity of Mfn, thus mediating Mfn clearance either constitutively or in a regulated manner in response to different stress signals. It is also possible that multiple E3 ligases work in a concerted way to ensure constant Mfn levels. In our study, CHX treatment leads to the stabilization of Mfn levels in HeLa cells lack of MUL1. However, steady-state Mfn levels in these cells are not strongly affected. This may result from the existence of other pathways for Mfn regulation, such as direct transcriptional feedback regulation on Mfn expression, activities of deubquitinases, and additional E3 ligases. Similar considerations may explain the viability and apparently mild phenotypes of MUL1 mutant flies. More severe phenotypes may be uncovered in flies lacking MUL1 in response to specific stresses that cannot be buffered by other components.
A recent study reports that MUL1 promotes mitophagy, when muscle wasting is stimulated in mice (Lokireddy et al., 2012). To monitor mitophagy, this study measured mitochondrial DNA content and emission of a mitochondrial fluorescent protein that changes color in an acidic environment such as the lysosome (Lokireddy et al., 2012). However, since these methods do not directly visualize mitochondrial fate, it is possible that the observations may reflect early signs of mitochondrial dysfunction or turnover of the indicator protein, rather than clearance of the mitochondria. Also, it is unknown whether MUL1 interacts with the PINK1/parkin pathway to regulate the mitochondrial clearance. Our results show that overexpression or lack of MUL1 does not affect Parkin-mediated mitophagy induced by mitochondrial damage in HeLa cells. This further strengthens our hypothesis that MUL1 acts in PINK1/parkin-independent pathway for regulating mitochondrial quality control.
Given our observations, it will be interesting to ask if human mutations in mfn1 or mfn2 that decrease their abilities to be targeted for ubiquitin-dependent degradation, or mutations in MUL1, result in susceptibility for PD. It will also be interesting to see if polymorphisms in MUL1 that affect MUL1 expression levels or activity occur in PD patients. In this regard, it is worth noting that MUL1 forms a complex with VPS35 and VPS26 (Braschi et al., 2010). Since mutations in VPS35 have been identified in multiple PD families (Vilarino-Guell et al., 2011; Zimprich et al., 2011; Kumar et al., 2012; Lesage et al., 2012), it will be particularly interesting to determine if PD-associated mutations in VPS35 have effects on MUL1-dependent degradation of Mfn. Finally, our observation that overexpression of mfn alone is sufficient to recapitulate key phenotypes associated with loss of PINK1 or parkin suggests that inhibition of mfn may have important therapeutic potential for PD.
Materials and methods
Molecular biology and constructs
Request a detailed protocolTo generate UAS-MUL1, an EST clone from the Drosophila Genome Research Center (DGRC), AT15655, was subcloned into the UASt vector using EcoR1 and Xho1. The Drosophila MUL1 ligase-dead mutant (MUL1 LD) was generated by mutating H307 to A via site-specific mutagenesis (Stratagene QuikChange II XL Kit; Stratagene, La Jolla, CA). To generate UAS-mfn, the EST clone from DGRC, RE04414, was subcloned into the UASt vector. For UAS-MUL1-GFP, UAS-mfn-myc, and UAS-HA-parkin, each gene's coding region was fused to a different tag using the gateway cloning system (Invitrogen, Carlsbad, CA). To silence MUL1 and drp1, the coding regions of MUL1 and drp1 transcripts were targeted using a synthetic microRNA-based technology (Chen et al., 2006; Ganguly et al., 2008). PCR products of these miRNA precursors were cloned into pUASt. To generate IFM-GAL4, the regulatory region of the flightin gene was used. All constructs made were confirmed by DNA sequencing. To map MUL1 imprecise excision lines, breakpoints were determined by genomic PCR followed by DNA sequencing. pAC-mfn-Flag was a gift from Dr Alexander J Whitworth (Ziviani et al., 2010). Human MUL1 cDNA (BC010101) was purchased from ATCC and cloned into a pEGFP vector (Clontech, San Jose, CA) to generate GFP-MUL1. Flag-MUL1 was generated by replacing the GFP tag with a Flag tag. Human MUL1 LD was generated by mutating H319 to A, which corresponds to Drosophila MUL1 H307A. Human MUL1 shRNA constructs were purchased from OriGene. The MUL1 shRNA sequences are 5′-CTTCAAGTCCTGCGTCTTTCTGGAGTGTG-3′ and 5′-GAAGGAGCTGTGCGGTCTGTTAAAG AAAC-3′.
Drosophila genetics and strains
Request a detailed protocolCaSpeR-HA-drp1 flies were a gift from Dr Hugo J Bellen (Verstreken et al., 2005). MUL1EY12156, TRiP parkin RNAi, UAS-mitoGFP, Mef2-GAL4, OK6-GAL4 and TH-GAL4 flies were obtained from the Bloomington Drosophila Stock Center. PINK15, parkin25, dpk21, UAS-drp1 and UAS-mfn RNAi flies have been previously described (Clark et al., 2006; Deng et al., 2008). For experiments involving transgenic flies, constructs were injected into w1118 and multiple independent fly lines were generated and analyzed (Rainbow Transgenic Flies, Inc.). The deletion mutant MUL1A6 was generated by imprecise excision of MUL1EY12156 using previously described methods (Gross et al., 2013). Drosophila strains were largely maintained in a 25°C humidified incubator.
RNA isolation, cDNA synthesis, and quantitative PCR (qPCR)
Request a detailed protocolRNA was isolated from whole flies using the Macherry-Nagel Nucleospin RNA II kit. cDNA synthesis was performed using the Clontech RNA to cDNA EcoDry Premix Kit, using a combination of Oligo-dT and random hexamer priming. Quantitative PCR was performed using the BioRadiTaq Fast Sybr Green enzyme mix, 10 µl reactions in triplicate, on a Roche Light Cycler 480. Standard curves were generated for MUL1 and two control genes, rpl32 and eIF1α. Table 1.
Table 1
Primer sequences for qPCR
| Primers | Sequence |
|---|---|
| MUL1-F | GCTATTGGTGAACTGGAGTTGGA |
| MUL1-R | AGCTTGAGTATCGTCGTTGTCTT |
| rpl32-F | TATGCTAAGCTGTCGCACAAATG |
| rpl32-R | GAACTTCTTGAATCCGGTGGGC |
| eIF1α-F | ACTTCGCAAGAAGGTGTGGATTA |
| eIF1α-R | GTACGTCTTCAGGTTCCTGGC |
Reverse transcription PCR (RT-PCR)
Request a detailed protocolTotal RNA was prepared as described above. RT-PCR was performed using Titanium One-Step RT-PCR Kit according to the manufacturer's instructions (Promega, Madison, WI). Primers used for RT-PCR are as follows in Table 2.
Table 2
Primer sequences for RT-PCR
| Primers | Sequence |
|---|---|
| MUL1 RT-F | ACACGAATCCGT TGCACTG |
| MUL1 RT-R | GCTCGTAGTTGTCGTAGACC |
Immunofluorescence and confocal microscopy
Request a detailed protocolFor analysis of muscle, thoraces of 1- to 2-day-old-adult flies were dissected and fixed in 4% paraformaldehyde in phosphate buffered saline (PBS). After thoraces were washed three times in PBS, muscle fibers were isolated and stained with rhodamine phalloidin (Invitrogen, 1:1000) in PBS+1% Triton X-100. For antibody staining, muscle fibers were permeabilized in PBS+0.1% Triton X-100, blocked in 5% normal goat serum in PBS, and incubated in primary and secondary antibodies diluted in 5% normal goat serum in PBS. For analysis of dopaminergic neurons, brains of 3-day-old male flies were dissected and fixed in 4% paraformaldehyde in PBS. Blocking, primary and secondary antibody staining were performed as described previously (Yun et al., 2008). To analyze mitochondria in salivary glands, salivary glands of third instar larvae were dissected, fixed in 4% paraformaldehyde in PBS, and stained with rhodamine phalloidin. The following primary antibodies were used: mouse anti-ATP Synthase (Mitosciences, Eugene, OR), chicken anti-HA (Millipore, Billerica, CA), mouse anti-Tyrosine Hydroxylase (Immunostar Hudson, WI). All images were taken on a Zeiss LSM5 confocal microscope.
TUNEL assay
Request a detailed protocolAdult male flies were aged for 5 days at 25°C. Thoraces of the flies were dissected and fixed in 4% paraformaldehyde in PBS. Muscle fibers were dissected and subsequently permeabilized and blocked in T-TBS-3% BSA (T-TBS: 0.1% Triton X-100, 50 mM Tris-Cl [pH 7.4], 188 mM NaCl). After blocking, TUNEL staining was carried out using an In Situ Cell Death Detection Kit according to the manufacturer's instructions (Roche, Switzerland).
Embedding, sections, Toluidine blue staining, and transmission electron microscopy
Request a detailed protocolThoraces from 3-day-old male flies were dissected, fixed in paraformaldehyde/glutaraldehyde, postfixed in osmium tetraoxide, dehydrated in ethanol, and embedded in Epon. After polymerization of Epon, blocks were cut to generate 1.5-µmthick sections using a glass knife, or 80-nm thick sections using a diamond knife on a microtome (Leica, Germany). Toluidine blue was used to stain 1.5-µm -thick tissue sections. Thin sections (80-nm thick) were stained with uranyl acetate and lead citrate, and examined using a JEOL 100C transmission electron microscope (UCLA Brain Research Institute Electron Microscopy Facility). At least six thoraces were examined in each sample.
Quantification of mitochondrial number and size in salivary glands
Request a detailed protocolImages were taken on a Zeiss LSM5 confocal microscope. Each cell in the image was outlined, and the outlined area was analyzed for mitochondrial number, average size and total area using the Analyze Particles function in ImageJ software (NIH). N = 8
Drosophila lysate preparation and western blotting
Request a detailed protocolThoraces from adult flies or whole animals were homogenized in RIPA buffer containing protease inhibitors (Roche). Total protein concentration was measured using a Bradford assay kit (Bio-Rad, Hercules, CA), and the same amount of protein was loaded onto SDS-polyacryamide gels. The following primary antibodies were used for Western blots: mouse anti-myc (Millipore), mouse anti-HA (Millipore), mouse anti-Tubulin (Sigma, St. Louis, MO), rabbit anti-Actin (Sigma), mouse anti-Porin (mitosciences), and rabbit anti-Mfn (a generous gift from Dr Alexander J Whitworth).
S2 cell culture, transfection, and RNAi treatment
Request a detailed protocolS2 cells were cultured in Schneider's Drosophila Medium (Gibco, Grand Island, NY) with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). Cells were seeded a day before transfection, and transfections were performed using the Effectene kit according to the manufacturer's recommendations (Qiagen, Valencia, CA). pAC-GAL4 was transfected along with UAS-Mfn-myc, UAS-HA-parkin, and UAS-MUL1-GFP for protein expression. UAS vector was used as empty vector. Cells were harvested 2 days after transfection. Double-stranded RNA (dsRNA) against coding regions of GFP, PINK1, parkin, MUL1, and mfn were generated using the T7 RiboMax express RNAi system (Promega). Primers that were used to generate dsRNAs are described below. S2 cells were seeded and treated with dsRNAs in serum-free medium for 40 min. After dsRNA treatment, complete medium was added to the culture, and the culture was incubated for 2–3 days. Table 3.
Table 3
Primer sequences for the generation of dsRNA templates
| Primer | Sequence |
|---|---|
| GFP-F (control) | TAATACGACTCACTATAGGGTGAACCGCATCGAGCTGAA |
| GFP-R (control) | TAATACGACTCACTATAGGGACTTGTACAGCTCGTCCATG |
| PINK1-F | TAATACGACTCACTATAGGGAATGTGACTTCTCCAGCGA |
| PINK1-R | TAATACGACTCACTATAGGGTCGTAGCGTTTCATCAGCAG |
| parkin-F | TAATACGACTCACTATAGGGGTACGCAAAATGCTGGAGCT |
| parkin-R | TAATACGACTCACTATAGGGTAGAGGCTTGGAGGCTTCAT |
| MUL1 #1-F | TAATACGACTCACTATAGGGCCACCAAGTCCACGCTTATT |
| MUL1 #1-R | TAATACGACTCACTATAGGGTGATCCTGGGACAGAGTGTG |
| MUL1 #2-F | TAATACGACTCACTATAGGGGATTGTGAAGCTGCATGAGC |
| MUL1 #2-R | TAATACGACTCACTATAGGGAACACATGGTCGAAGAGGGA |
Co-immunoprecipitation
Request a detailed protocolS2 cells were lysed in RIPA buffer containing protease inhibitors (Roche), and Western blots were performed with 2% of lysates to check protein expression. Immunoprecipitations were performed with the rest of lysate using Dynabeads (Invitrogen) according to the manufacturer's instructions. Proteins bound to beads were eluted in SDS sample buffer, and Western blots were performed. Primary antibodies used include mouse anti-Myc (Millipore), rabbit anti-GFP (Invitrogen), rabbit anti-HA (Sigma), and rabbit anti-Actin (Sigma).
In vivo ubiquitination assay in S2 cells
Request a detailed protocolAfter treatment with dsRNA for 2 days, S2 cells were transfected with Mfn-Flag and incubated for 24 hr. Before harvest, cells were treated with the proteasome inhibitor MG132 (Millipore) for 4 hr. Cells were lysed and boiled in SDS lysis buffer (1% SDS, 150 mM NaCl, 10 mM Tris–HCl, pH 8.0) with protease inhibitors (Roche) for 10 min. Dilution buffer (10 mM Tris–HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton) was added, and immunoprecipitations were performed using mouse anti-Flag antibody (Sigma). After immunoprecipitations, Western blots were probed with mouse anti-ubiquitin (Covance). Mouse anti-FK1 (Enzo Life Sciences, Farmingdale, NY) and anti-FLAG (Sigma) antibodies were used.
Protein purification and in vitro ubiquitination assay
Request a detailed protocolFor in vitro ubiquitination assay, the glutathione S-transferase (GST)-tagged expression vectors pGex-MUL1 and pGEX-MUL1 LD were generated. GST fusion proteins (GST-MUL1 and GST-MUL1 LD) were expressed in E. coli and purified from inclusion body. The in vitro ubiquitination assay was performed using the following buffer: 25 mM Tris (pH 7.5), 5 mM MgCl2, 100 mM NaCl, 1 mM DTT, 0.05 mM MG132, 2 mM ATP, and 0.125 µg/µl Ubiquitin, with E1 (Rabbit, 0.5 µg/ml), E2 (extract from E. coli expressing UbcH5C), and presence or absence of GST-MUL1, or GST-MUL1 LD (as indicated). Reaction mixtures were incubated at 30°C for 2 hr, and reactions were terminated by boiling in SDS loading buffer.
Mammalian cell culture, transfection, and western blotting
Request a detailed protocolHeLa cells that did or did not overexpress parkin were generous gifts from Dr David C Chan (Chan et al., 2011). Cells were cultured in Dulbeco's modified Eagle's medium (DMEM, Gibco) containing 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). Cells were plated a day before transfections, and transfections were performed using the Effectene kit (Qiagen) or X-tremeGENE 9 DNA Transfection Reagent (Roche) according to the manufacturer's recommendations. After transfections, Z-VAD-FMK (Santa Cruz Biotechnology, Santa Cruz, CA) was added to cultures every 24 hr to inhibit apoptosis. Cells were harvested 48 hr later and lysed in RIPA buffer containing protease inhibitors (Roche). Western blots were performed with the following primary antibodies: rabbit anti-human MUL1 (Sigma), mouse anti-Mfn1 (Abcam), mouse anti-Mfn2 (Abcam), rabbit anti-Actin (Sigma), and mouse anti-Porin (Mitosciences).
Parkin-mediated mitophagy assays
Request a detailed protocolHeLa cells that did or did not stably express MUL1 shRNA were seeded in chamber slides, respectively, and transfected with YFP-Parkin one day later. 24 hrs after transfection, cells were treated with DMSO or 40 µg/ml Antimycin A (Sigma) for 1.5, 3, 24, or 48 hrs as indicated to dissipate mitochondrial membrane potential. For MUL1 overexpression, HeLa cells stably expressing YFP-Parkin and mitoRFP (a kind gift from Dr. Mark R Cookson) were seeded and transfected with Myc-MUL1 1 day later. Cells were treated with DMSO or 80 µg/ml Antimycin A for 1.5 or 3 hrs. After treatment of Antimycin A, cells were fixed in 10% Formalin solution (Sigma), permeabilized with 0.1% Triton X-100 in PBS, and blocked in PBS containing 5% fetal bovine serum. Primary and secondary antibody staining were performed in 5% fetal bovine serum + PBS. The following primary antibodies were used: mouse anti-Tom20 (BD), mouse anti-Flag (Sigma), rabbit anti-GFP (Invitrogen), and rabbit anti-Parkin (Abcam, Cambridge, MA). More than 100 cells for each experiment were counted for quantification, and the experiments were repeated twice. PINK1 knockout cells were a generous gift from Dr. Richard Youle. Western blot analysis confirmed that there is no PINK1 expression in PINK1 knockout cells (Narendra et al., 2013; personal communication).
Generation of MUL1 knockout (MUL1−/−) HeLa cells using CRISPR/Cas 9 system
Request a detailed protocolMUL1 knockout HeLa cells were generated using the CRISPR/Cas system as previously described (Cong et al., 2013). Briefly, MUL1 targetting sequence 5′-GCCGCCGTCA TGGAGAGCGG-3′ was inserted into pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene). HeLa cells were seeded a day before transfection. Cells were transfected with the construct using X-tremeGENE 9 DNA transfection reagent (Roche) following manufacturer's instructions. 2 days after the transfection, cells were diluted and split into 48 well plates. Each colony was screened for deletions in MUL1 by PCR and sequencing using a set of primers 5′-CGCCTCGAACCTGACACATAATAGG-3′ and 5′-GTCTGTAAAGCAAGGAGTG GAGTGG-3′. Two MUL1 knockout cells were isolated. Both MUL1 knockout cells have deletions including the start codon of MUL1, one with 228 base pair deletion and another with 8 base pair deletion. Both deletions result in frame shift and early termination of protein translation. Further western blot analysis using two different anti-MUL1 antibodies (Sigma) confirmed that there is no MUL1 expression in MUL1−/− cells.
Protein turnover
Request a detailed protocolHeLa cells that express scrambled shRNA or MUL1 shRNA were treated with cycloheximide (Sigma) for 0, 2, 4, 6 hrs. After cycloheximide treatment, cells were harvested and lysed. Protein concentration of each lysate was determined by Bradford assay (Bio-Rad), and an equal amount of total protein was subjected to Western blot. Blots were probed with anti-Mfn1 (Abcam), anti-Mfn2 (Abcam) and Actin (Sigma) antibodies. Levels of Mfn2 and Actin were quantified using ImageJ.
Mouse cortical neuronal culturing, transfection, and immunocytochemistry
Request a detailed protocolAnimal care and use were carried out in accordance with NIH guidelines, NIH Manual 3040-2, Guide for the Care and Use of Laboratory Animals (National Research Council), Institutional Animal Care and Use Committee Guidebook (ARENA and OLAW) and approved by the NIH, NINDS/NIDCD Animal Care and Use Committee on 3/5/2012 (ASP# 1303-9).
The work and submission for publication was approved by the Intramural Program of NINDS, NIH. Dissection of embryonic mouse brains and isolation of cortical neurons and plating were designed to be very quick with minimal enzymatic, mechanical, chemical, and oxidative damage, as described by Cai et al. (2012). Cortices were dissected from E18-19 mouse embryos. Cortical neurons were dissociated by papain (Worthington) and plated on glial beds at a density of 50,000 cells per cm2 on polyornithine (Sigma) and Matrigel (BD Biosciences)-coated coverslips. Neurons were grown overnight in plating medium (5% FBS, insulin, glutamate, G5, 1 x B27 and beta-mercaptoethanol) supplemented with 100 x L-glutamine in Neurobasal (Invitrogen). Starting at DIV2, cultures were maintained in conditioned medium with half-feed changes of neuronal feed (1 × B27, 100 × L-glutamine and beta-mercaptoethanol in Neurobasal) every 3 days. Neurons were transfected with various constructs at DIV7-8 using calcium phosphate and processed for immunocytochemistry 72 hr (DIV10-11) post transfection.
For immunostaining, cultured cells were fixed with 4% formaldehyde (Electron Microscopy Sciences) and 4% sucrose (Sigma) in 1X phosphate-buffered saline (PBS) at 4°C for 30 min, washed three times with PBS for 5 min each, and then incubated in 0.2% saponin, 5% normal goat serum (NGS), and 2% bovine serum albumin (BSA) in PBS for 1 hr. Fixed cultures were incubated with primary antibodies in PBS with 1% BSA and 0.05% saponin at 4°C overnight. Cells were washed four times with PBS at RT for 5 min each, incubated with secondary fluorescent antibodies at 1:400 dilution in PBS with 1% BSA and 0.05% saponin for 60 min, re-washed with PBS, and then mounted with Fluoro-Gel anti-fade mounting medium (EMS) for imaging. Sources of antibodies are as follow: polyclonal antibodies against TOM20 (Santa Cruz), MUL1 (Sigma), Mfn2 (Cell Signaling, Danvers, MA); monoclonal antibodies against MAP2 (Millipore), GAPDH (Research Diagnostic, Hackensack, NJ), CytC (BD Biosciences, San Jose, CA); Alexafluor 546 and 633-conjugated secondary antibodies (Invitrogen). Confocal images were obtained using an Olympus Fluoview FV1000 microscope, oil immersion 63X objective (NA-1.45) with sequential-acquisition settings. Images were acquired using the same settings below saturation at a resolution of 1024X1024 pixels (12 bit). Z stacks were acquired using a step size of 0.37 µm from top to bottom, and brightest point projections were made. For quantification, data were obtained from at least three independent experiments and the number of cells used for quantification is indicated in the figures. All statistical analyses were performed using One-way or Two-way ANOVA with Tukey's multiple comparison test and are presented as mean ± SEM.
TMRE (tetramethyl rhodamine ethyl ester) staining
Request a detailed protocolTo access mitochondrial potential on a single cell basis, mature cortical neurons DIV (10–11), both from wild-type (WT) and parkin knockout (KO), were incubated with the cationic lipophilic compound TMRE (50 nM) for 20 min in a 37°C CO2 incubator. Post treatment, cells were washed three times with imaging media and mounted for imaging. Confocal images were obtained using an Olympus confocal oil immersion 63x objective with the sequential-acquisition setting. The images were acquired within 30 min. For fluorescent quantification, image acquisition settings were below saturation at a resolution of 1024 × 1024 pixels (12 bit). Five to six sections were taken from the top-to-bottom of the specimen and brightest point projections were made. Morphometric measurements were performed using NIH ImageJ. Measured data were imported into Excel software for analysis. The thresholds in all images were set to similar levels. Fluorescence intensity of TMRE was expressed in corrected total cell fluorescence (CTCF) values. The mean intensity of TMRE in the soma of each group was normalized as a percentile ratio relative to that in WT cells expressing scrambled shRNA. Data were obtained from at least three independent experiments and the number of cells used for quantification is indicated in the figures. All statistical analyses were performed using one-way or two-way ANOVA with Tukey's multiple comparisons test and are presented as mean ± SEM.
References
-
Targeted gene expression as a means of altering cell fates and generating dominant phenotypesDevelopment 118:401–415.
-
Vps35 mediates Vesicle transport between the mitochondria and PeroxisomesCurrent Biology 20:1310–1315.https://doi.org/10.1016/j.cub.2010.05.066
-
Fusion and fission: interlinked processes critical for mitochondrial healthAnnual Review of Genetics 46:265–287.https://doi.org/10.1146/annurev-genet-110410-132529
-
Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagyHuman Molecular Genetics 20:1726–1737.https://doi.org/10.1093/hmg/ddr048
-
Identifying microRNA regulators of cell death in DrosophilaMethods in Molecular Biology 342:229–240.https://doi.org/10.1385/1-59745-123-1:229
-
Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic developmentThe Journal of Cell Biology 160:189–200.https://doi.org/10.1083/jcb.200211046
-
The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in DrosophilaProceedings of the National Academy of Sciences of the United States of America 105:14503–14508.https://doi.org/10.1073/pnas.0803998105
-
Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial primingThe Journal of Biological Chemistry 285:27879–27890.https://doi.org/10.1074/jbc.M110.119537
-
Parkin deficiency increases vulnerability to inflammation-related nigral degenerationThe Journal of Neuroscience 28:10825–10834.https://doi.org/10.1523/JNEUROSCI.3001-08.2008
-
Regulation of mitophagy by the Gp78 E3 ubiquitin ligaseMolecular Biology of the Cell 24:1153–1162.https://doi.org/10.1091/mbc.E12-08-0607
-
ubiquilin antagonizes presenilin and promotes neurodegeneration in DrosophilaHuman Molecular Genetics 17:293–302.https://doi.org/10.1093/hmg/ddm305
-
Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stressProceedings of the National Academy of Sciences of the United States of America 105:11364–11369.https://doi.org/10.1073/pnas.0802076105
-
Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagyHuman Molecular Genetics 19:4861–4870.https://doi.org/10.1093/hmg/ddq419
-
PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1Nature Cell Biology 12:119–131.https://doi.org/10.1038/ncb2012
-
Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1Journal of Neurochemistry 118:636–645.https://doi.org/10.1111/j.1471-4159.2011.07318.x
-
X11/Mint genes control Polarized localization of axonal membrane proteins in vivoThe Journal of Neuroscience 33:8575–8586.https://doi.org/10.1523/JNEUROSCI.5749-12.2013
-
Drosophila as a model to study mitochondrial dysfunction in Parkinson's diseaseCold Spring Harbor Perspectives in Medicine 2:a009944.https://doi.org/10.1101/cshperspect.a009944
-
PINK1- and Parkin-mediated mitophagy at a glanceJournal of Cell Science 125:795–799.https://doi.org/10.1242/jcs.093849
-
Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient miceProceedings of the National Academy of Sciences of the United States of America 104:11441–11446.https://doi.org/10.1073/pnas.0702717104
-
Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout miceJournal of Neurochemistry 111:696–702.https://doi.org/10.1111/j.1471-4159.2009.06350.x
-
Frequency of the D620N Mutation in VPS35 in Parkinson DiseaseArchives of Neurology pp. 1–5.https://doi.org/10.1001/archneurol.2011.3367
-
Animal models of Parkinson's disease: vertebrate geneticsCold Spring Harbor Perspectives in Medicine 2:a009324.https://doi.org/10.1101/cshperspect.a009324
-
Parkin is recruited selectively to impaired mitochondria and promotes their autophagyThe Journal of Cell Biology 183:795–803.https://doi.org/10.1083/jcb.200809125
-
PINK1 rendered temperature sensitive by disease-associated and engineered mutationsHuman Molecular Genetics 22:2572–2589.https://doi.org/10.1093/hmg/ddt106
-
Mitochondrial dysfunction and oxidative damage in parkin-deficient miceThe Journal of Biological Chemistry 279:18614–18622.https://doi.org/10.1074/jbc.M401135200
-
Parkin-deficient mice are not a robust model of parkinsonismProceedings of the National Academy of Sciences of the United States of America 102:2174–2179.https://doi.org/10.1073/pnas.0409598102
-
The PINK1/Parkin pathway regulates mitochondrial morphologyProceedings of the National Academy of Sciences of the United States of America 105:1638–1643.https://doi.org/10.1073/pnas.0709336105
-
Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neuronsThe Journal of Biological Chemistry 288:2223–2237.https://doi.org/10.1074/jbc.M112.391680
-
Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by ParkinThe Journal of Cell Biology 191:1367–1380.https://doi.org/10.1083/jcb.201007013
-
VPS35 mutations in Parkinson diseaseAmerican Journal of Human Genetics 89:162–167.https://doi.org/10.1016/j.ajhg.2011.06.001
-
PINK1-dependent recruitment of Parkin to mitochondria in mitophagyProceedings of the National Academy of Sciences of the United States of America 107:378–383.https://doi.org/10.1073/pnas.0911187107
-
Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by ParkinProceedings of the National Academy of Sciences of the United States of America 103:10793–10798.https://doi.org/10.1073/pnas.0602493103
-
Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machineryProceedings of the National Academy of Sciences of the United States of America 105:7070–7075.https://doi.org/10.1073/pnas.0711845105
-
The kinase domain of mitochondrial PINK1 faces the cytoplasmProceedings of the National Academy of Sciences of the United States of America 105:12022–12027.https://doi.org/10.1073/pnas.0802814105
-
A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson diseaseAmerican Journal of Human Genetics 89:168–175.https://doi.org/10.1016/j.ajhg.2011.06.008
-
Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusinProceedings of the National Academy of Sciences of the United States of America 107:5018–5023.https://doi.org/10.1073/pnas.0913485107
Article and author information
Author details
Funding
McKnight Endowment Fund for Neuroscience
- Ming Guo
National Institutes of Health (R01, K02, P01)
- Ming Guo
Ellison Medical Foundation (Senior Scholar Award)
- Ming Guo
Esther A. and Joseph Klingenstein Fund (Klingenstein-Simons Fellowship Award in Neurosciences)
- Ming Guo
The American Parkinson Disease Association
- Ming Guo
Glenn Family Foundation
- Ming Guo
National Institute of Neurological Disorders and Stroke (Intramural research program)
- Zu-Hang Sheng
The Chinese Scholarship Council Fellowship
- Huan Yang
NIH-DBT Khorana Nirenberg Scholarship
- Rajat Puri
UCLA Dissertation Fellowship
- Jina Yun
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank H Bellen, D Chan, M Cookson, A Whitworth, and R Youle for reagents, H Mcbride for communicating unpublished results, H Huang and BA Hay for generating IFM-Gal4 lines, H Deng, H Huang, Y Sun, and B Al-Anzi for technical assistance, BA Hay, L Leung, P Patel, and CY Lee for comments on the manuscript, and F Laski, L Dreier and N Freimer for use of their equipment. This work was supported by a UCLA Dissertation Fellowship to JY, the NIH-DBT Khorana Nirenberg Scholarship to RP, the Chinese Scholarship Council Fellowship to HY, the intramural research program of NIH/NINDS to Z-HS., and NIH (R01, K02, P01), the McKnight Neuroscience Foundation, the American Parkinson's Disease Association, the Klingenstein Fellowship Award in the Neurosciences, the Kenneth Glenn Family Foundation, and the Ellison Medical Foundation Senior Scholar Award to MG.
Ethics
Animal experimentation: Animal care and use were carried out in accordance with NIH guidelines, NIH Manual 3040-2, Guide for the Care and Use of Laboratory Animals (National Research Council), Institutional Animal Care and Use Committee Guidebook (ARENA and OLAW) and approved by the NIH, NINDS/NIDCD Animal Care and Use Committee on 3/5/2012 (ASP# 1303-9).The work and submission for publication was approved by the Intramural Program of NINDS, NIH.
Copyright
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Metrics
-
- 7,885
- views
-
- 863
- downloads
-
- 297
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 297
- citations for umbrella DOI https://doi.org/10.7554/eLife.01958
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin
eLife 3:e01958.
https://doi.org/10.7554/eLife.01958




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}