Apoptosis: Keeping inflammation at bay
- The Weizmann Institute of Science, Israel
The cells in our bodies are genetically programmed to undergo a natural process of self-destruction called apoptosis, after which the dying cell is removed by cells that have the ability to engulf them (‘phagocytes’). The membrane of the dying cell is still intact as it is engulfed by the phagocyte, so its contents do not come into contact with other nearby cells. Apoptosis does not trigger inflammation, whereas another form of cell death called necrosis—in which the cell membrane is ruptured—is often associated with inflammation (Kerr et al., 1972).
Necrosis causes inflammation because some components of the dying cell that are capable of triggering inflammation come into contact with healthy cells nearby (Rock and Kono, 2008). At first it was assumed that the only reason why apoptosis did not cause inflammation was that all the contents of the dying cell remained inside the membrane and the phagocyte. However, it was later discovered that apoptosis can actually block inflammation (Voll et al., 1997; Fadok et al., 1998). Initial observations suggested that this anti-inflammatory effect is triggered when the phagocytes are exposed to phosphatidylserine—a molecule on the surface of apoptotic cells that has a central role in phagocytosis (Huynh et al., 2002). It seemed, therefore, that these anti-inflammatory changes could be induced only in cells intimately associated with the dying cell (Figure 1A).
Figure 1
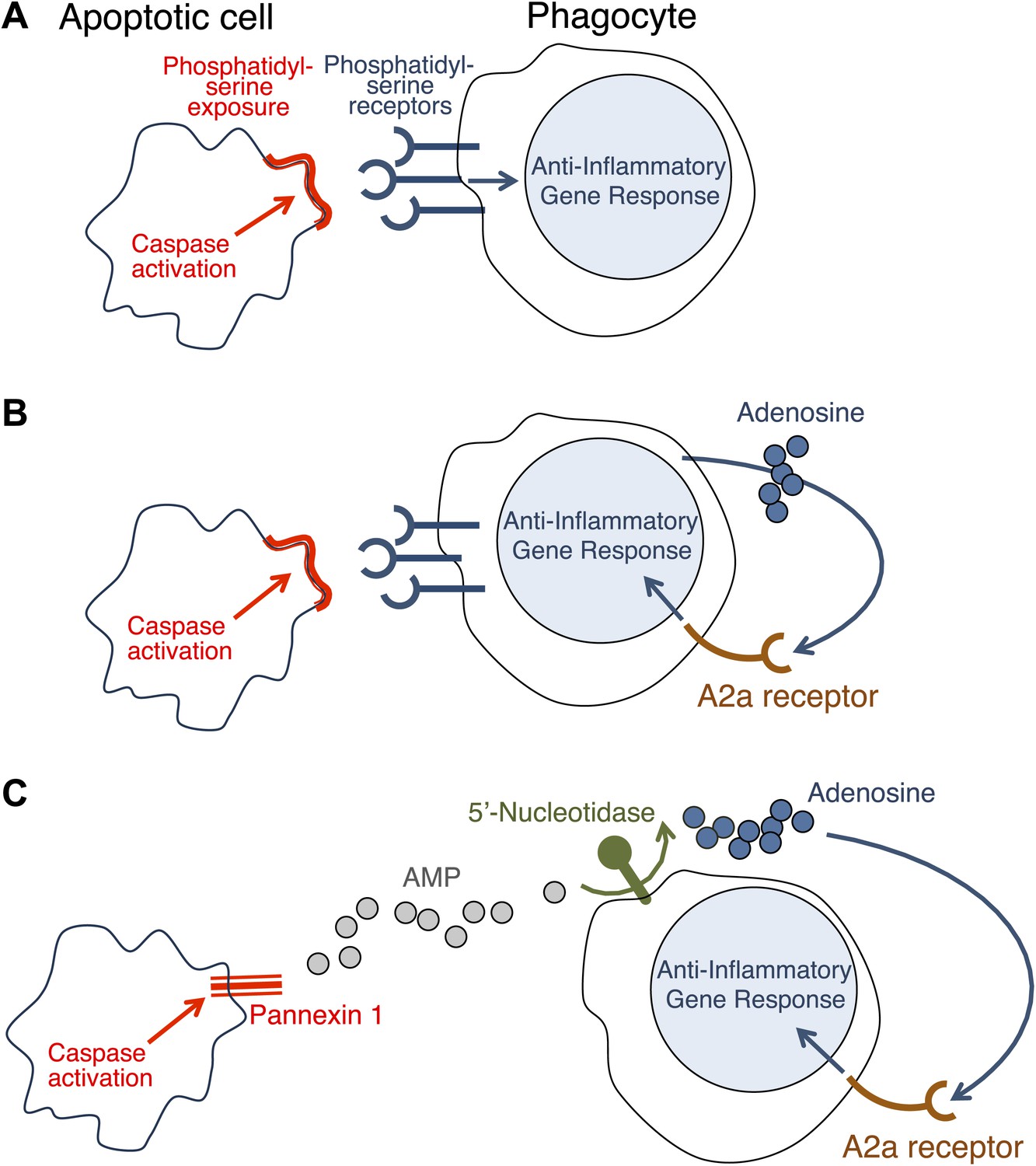
How do apoptotic cells trigger an anti-inflammatory response in phagocytes?
(A) Phosphatidylserine molecules on the surface of an apoptotic cell can bind to phosphatidylserine receptors on the surface of a phagocyte and previously it was suggested that this triggered an anti-inflammatory gene response. (B) It was also suggested that the direct apoptotic cell–phagocyte interaction shown in A also results in the release of adenosine by the phagocyte: this adenosine can bind to A2a receptors on the surface of the phagocyte and trigger an anti-inflammatory gene response. (C) Yamaguchi et al. found that the apoptotic cell releases a molecule called adenosine monophosphate (AMP) that is converted to adenosine by a 5′-nucleotidase on the surface of the phagocyte. The adenosine can then trigger an anti-inflammatory gene response by binding to A2a receptors. Enzymes called caspases play a central role in apoptosis in a variety of ways. The action of these caspases is required for the exposure of phosphatidylserine on the surface of the apoptotic cells (A and B); they also activate a channel protein called pannexin-1 to allow the release of AMP (C).
Now, in eLife, Shigekazu Nagata and co-workers at Kyoto University and the Osaka Bioscience Institute—including Hiroshi Yamaguchi as first author—report that apoptotic cells release a molecule called adenosine that can activate an anti-inflammatory gene response in phagocytes (Yamaguchi et al., 2014). They have also shown that adenosine activates this response by stimulating the A2a adenosine receptor in phagocytes.
Similar results have been reported before (Sitkovsky and Ohta, 2005; Köröskényi et al., 2011), but it had been thought that the adenosine was generated by the phagocytes as a consequence of their uptake of the apoptotic cells (Figure 1B). Yamaguchi et al. now show that the adenosine comes from the apoptotic cells themselves, with the phagocytes having only a secondary role in its production. The first step involves enzymes called caspases—which have a central role in apoptosis—cleaving a membrane channel protein called pannexin-1 in the dying cells, and thereby activating it. This results in the release of adenosine monophosphate (AMP) from the dying cells. A 5′-nucleotidase expressed by the phagocytes then removes a phosphate group from the AMP to yield adenosine. The adenosine then binds to the A2a receptor on the phagocytes to trigger an anti-inflammatory gene response (Figure 1C).
Adenosine is not the only soluble molecule released by apoptotic cells to perform a specific role. For example, various other molecules—including lysophosphatidylcholine and the nucleotides ATP and UTP—act as ‘find me’ signals that attract phagocytes towards apoptotic cells (Hochreiter-Hufford and Ravichandran, 2013). Another example is an iron-binding glycoprotein called lactoferrin that inhibits the translocation of certain white blood cells, thereby apparently contributing to the anti-inflammatory effect of apoptosis (Bournazou et al., 2009).
To what extent do the soluble molecules released by apoptotic cells have an effect on cells remote from the site of death? And how does the contribution of these molecules to the anti-inflammatory consequences of apoptosis compare with the contribution that results from direct contact between the dying cell and the cell engulfing it? Nagata and co-workers report that in a mouse model of inflammation (zymosan-induced peritonitis), deletion of either the Pannexin-1 gene or the A2a gene prolongs the inflammation. These findings support the notion that (in this experimental model) adenosine derived from apoptotic cells contributes significantly to the restriction of inflammation. More precise cell-type-specific targeting of these molecules (and other molecules that have anti-inflammatory effects) should lead to an improved understanding of their relative contributions to immune regulation in specific pathological situations.
References
-
Apoptotic human cells inhibit migration of granulocytes via release of lactoferrinJournal of Clinical Investigation 119:20–32.https://doi.org/10.1172/JCI36226
-
Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestionCold Spring Harbor Perspectives in Biology 5:a008748.https://doi.org/10.1101/cshperspect.a008748
-
Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammationJournal of Clinical Investigation 109:41–50.https://doi.org/10.1172/JCI11638
-
Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kineticsBritish Journal of Cancer 26:239–257.https://doi.org/10.1038/bjc.1972.33
-
The inflammatory response to cell deathAnnual Reviews of Pathology 3:99–126.https://doi.org/10.1146/annurev.pathmechdis.3.121806.151456
-
The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors?Trends in Immunology 26:299–304.https://doi.org/10.1016/j.it.2005.04.004
Article and author information
Author details
Publication history
- Version of Record published: March 25, 2014 (version 1)
Copyright
© 2014, Wallach and Kovalenko
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,053
- views
-
- 211
- downloads
-
- 20
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Apoptosis: Keeping inflammation at bay
eLife 3:e02583.
https://doi.org/10.7554/eLife.02583
Further reading
-
- Cell Biology
Erythropoiesis and megakaryopoiesis are stringently regulated by signaling pathways. However, the precise molecular mechanisms through which signaling pathways regulate key transcription factors controlling erythropoiesis and megakaryopoiesis remain partially understood. Herein, we identified heat shock cognate B (HSCB), which is well known for its iron–sulfur cluster delivery function, as an indispensable protein for friend of GATA 1 (FOG1) nuclear translocation during erythropoiesis of K562 human erythroleukemia cells and cord-blood-derived human CD34+CD90+hematopoietic stem cells (HSCs), as well as during megakaryopoiesis of the CD34+CD90+HSCs. Mechanistically, HSCB could be phosphorylated by phosphoinositol-3-kinase (PI3K) to bind with and mediate the proteasomal degradation of transforming acidic coiled-coil containing protein 3 (TACC3), which otherwise detained FOG1 in the cytoplasm, thereby facilitating FOG1 nuclear translocation. Given that PI3K is activated during both erythropoiesis and megakaryopoiesis, and that FOG1 is a key transcription factor for these processes, our findings elucidate an important, previously unrecognized iron–sulfur cluster delivery independent function of HSCB in erythropoiesis and megakaryopoiesis.
-
- Biochemistry and Chemical Biology
- Cell Biology
Hibernation is a period of metabolic suppression utilized by many small and large mammal species to survive during winter periods. As the underlying cellular and molecular mechanisms remain incompletely understood, our study aimed to determine whether skeletal muscle myosin and its metabolic efficiency undergo alterations during hibernation to optimize energy utilization. We isolated muscle fibers from small hibernators, Ictidomys tridecemlineatus and Eliomys quercinus and larger hibernators, Ursus arctos and Ursus americanus. We then conducted loaded Mant-ATP chase experiments alongside X-ray diffraction to measure resting myosin dynamics and its ATP demand. In parallel, we performed multiple proteomics analyses. Our results showed a preservation of myosin structure in U. arctos and U. americanus during hibernation, whilst in I. tridecemlineatus and E. quercinus, changes in myosin metabolic states during torpor unexpectedly led to higher levels in energy expenditure of type II, fast-twitch muscle fibers at ambient lab temperatures (20 °C). Upon repeating loaded Mant-ATP chase experiments at 8 °C (near the body temperature of torpid animals), we found that myosin ATP consumption in type II muscle fibers was reduced by 77–107% during torpor compared to active periods. Additionally, we observed Myh2 hyper-phosphorylation during torpor in I. tridecemilineatus, which was predicted to stabilize the myosin molecule. This may act as a potential molecular mechanism mitigating myosin-associated increases in skeletal muscle energy expenditure during periods of torpor in response to cold exposure. Altogether, we demonstrate that resting myosin is altered in hibernating mammals, contributing to significant changes to the ATP consumption of skeletal muscle. Additionally, we observe that it is further altered in response to cold exposure and highlight myosin as a potentially contributor to skeletal muscle non-shivering thermogenesis.




{kind=link}