Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine
- Walter and Eliza Hall Institute of Medical Research, Australia
- University of Melbourne, Australia
- Medical Research Council Laboratory of Molecular Biology, United Kingdom
- Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Australia
Abstract
Malaria inflicts an enormous burden on global human health. The emergence of parasite resistance to front-line drugs has prompted a renewed focus on the repositioning of clinically approved drugs as potential anti-malarial therapies. Antibiotics that inhibit protein translation are promising candidates for repositioning. We have solved the cryo-EM structure of the cytoplasmic ribosome from the human malaria parasite, Plasmodium falciparum, in complex with emetine at 3.2 Å resolution. Emetine is an anti-protozoan drug used in the treatment of ameobiasis that also displays potent anti-malarial activity. Emetine interacts with the E-site of the ribosomal small subunit and shares a similar binding site with the antibiotic pactamycin, thereby delivering its therapeutic effect by blocking mRNA/tRNA translocation. As the first cryo-EM structure that visualizes an antibiotic bound to any ribosome at atomic resolution, this establishes cryo-EM as a powerful tool for screening and guiding the design of drugs that target parasite translation machinery.
https://doi.org/10.7554/eLife.03080.001eLife digest
Each year, malaria kills more than 600,000 people, mostly children younger than 5 years old. Humans who have been bitten by mosquitoes infected with malaria-causing parasites become ill as the parasites rapidly multiply in blood cells. Although there are several drugs that are currently used to treat malaria, the parasites are rapidly developing resistance to them, setting off an urgent hunt for new malaria drugs.
Developing new antimalarial medications from scratch is likely to take decades—too long to combat the current public health threat posed by emerging strains of drug-resistant parasites. To speed up the process, scientists are investigating whether drugs developed for other illnesses may also act as therapies for malaria, either when used alone or in combination with existing malaria drugs.
Certain antibiotics—including one called emetine—have already shown promise as antimalarial drugs. These antibiotics prevent the parasites from multiplying by interfering with the ribosome—the part of a cell that builds new proteins. However, humans become ill after taking emetine for long periods because it also blocks the production of human proteins.
Tweaking emetine so that it acts only against the production of parasite proteins would make it a safer malaria treatment. To do this, scientists must first map the precise interactions between the drug and the ribosomes in parasites. Wong et al. have now used a technique called cryo-electron microscopy to examine the ribosome of the most virulent form of malaria parasite. This technique uses very cold temperatures to rapidly freeze molecules, allowing scientists to look at molecular-level details without distorting the structure of the molecule—a problem sometimes encountered in other techniques.
The images of the parasitic ribosome taken by Wong, Bai, Brown et al. show that emetine binds to the end of the ribosome where the instructions for how to assemble amino acids into a protein are copied from strands of RNA. In addition, the images revealed features of the parasitic ribosome that are not found in the human form. Drug makers could exploit these features to improve emetine so that it more specifically targets the production of proteins by the parasite and is less toxic to humans.
https://doi.org/10.7554/eLife.03080.002Introduction
Malaria is responsible for an estimated 627,000 annual deaths worldwide, with the majority of victims being children under 5 years of age (WHO, 2012). At present there is no licensed malaria vaccine and parasites have developed resistance to all front-line anti-malarial drugs. As such, there is an urgent need for novel therapeutics that can be used as monotherapies or as partner drugs for combinatorial regimes (Kremsner and Krishna, 2004). An alternative to novel candidates is the repurposing or repositioning of clinically approved drugs that can be used in combination with known anti-malarials, such as chloroquine, antifolates, and artemisinin, to increase their useable lifespan by reducing resistance (Grimberg and Mehlotra, 2011).
The etiological agents for malaria are a family of unicellular protozoan pathogens of the genus Plasmodium. The parasite has a complex two-host lifecycle with a sexual stage occurring in the mosquito vector and an asexual stage in the human host. It is during the asexual blood stage that disease symptoms in humans first appear, including those associated with severe malaria, and it is often at this stage that the need for clinical intervention becomes apparent (Miller et al., 2002). Much of malaria pathology is the result of exponential growth of the parasite within erythrocytes, and given the critical role that protein synthesis plays in this, the translational machinery is an attractive drug target.
Protein translation in the parasite is focused on three centers (Jackson et al., 2011): the cytoplasmic ribosome, responsible for the vast majority of protein synthesis, and organellar ribosomes of the mitochondrion and non-photosynthetic plastid, termed the apicoplast (McFadden et al., 1996). In addition, and unusually for a eukaryotic cell, Plasmodium species have two distinct types of cytoplasmic ribosome that differ in their ribosomal RNA (rRNA) composition. These are expressed at different stages of the lifecycle, one predominantly in the mosquito vector and the other in the mammalian host, with evidence that both can occur simultaneously for limited periods (Waters et al., 1989).
Antibiotics known to target the apicoplast ribosome, such as the macrolide azithromycin, demonstrate a delayed-death effect, whereby treated parasites die in the second generation of drug exposure, and therefore have slow clinical onset (Dahl and Rosenthal, 2007 ; Goodman et al., 2007 ). However, because anti-malarial treatment at the blood-stage requires rapid intervention, we focused on the dominant, blood stage-specific cytoplasmic ribosome from the most virulent form of Plasmodium, P. falciparum (Pf80S) (Waters et al., 1989), as inhibition of cytosolic translation would be expected to be direct and fast-acting. Pf80S is both a candidate for development of novel therapeutics that target specific differences between itself and its counterpart in the human cytosol, and also for repurposing of anti-protozoan inhibitors, such as emetine (Matthews et al., 2013).
In this present study, we solved the structure of Pf80S–emetine complex at 3.2 Å resolution and built a fully-refined all-atom model. This represents, to our knowledge, the first structure of an entire eukaryotic ribosome at atomic resolution solved by electron cryo-microscopy (cryo-EM). Pf80S has a broad distribution of Pf-specific elements across its surface, with particularly long rRNA expansion segments (ESs) in the small subunit. The atomic structure of Pf80S in complex with emetine reveals the molecular basis of this clinically relevant anti-protozoan translation inhibitor. In doing so, we establish cryo-EM as a powerful tool for structure-based drug design.
Results
Cytoplasmic ribosomes were isolated from the 3D7 strain of P. falciparum parasites maintained in human erythrocytes (Figure 1A,B). Limitations in parasite culture volume, yielding ∼1010 parasitized red blood cells and low yield of sample material (1 g of parasites yielded 0.35 mg Pf80S), precluded an ability to crystallize Pf80S to solve the structure by conventional X-ray crystallography. We therefore exploited recent advances in direct electron detection and statistical image processing (Bai et al., 2013; Allegretti et al., 2014) to determine the structure by cryo-EM at an overall resolution of 3.2 Å (Figure 1C–E, Figure 1—figure supplement 1).
Figure 1 with 1 supplement see all
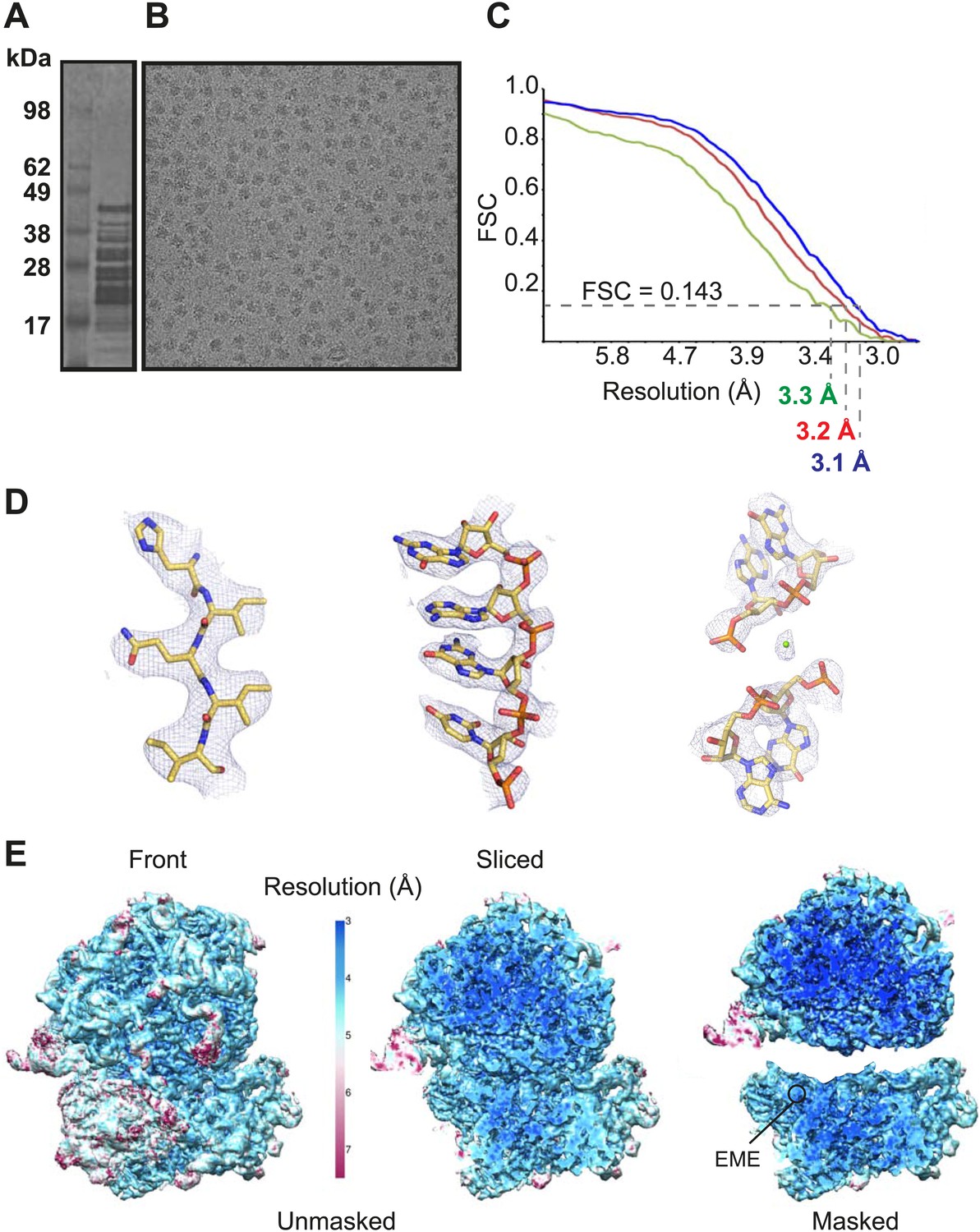
Cryo-EM data and processing.
(A) Sucrose gradient purification of Pf80S ribosomes. (B) Representative electron micrograph showing Pf80S particles. (C) Fourier Shell Correlation (FSC) curves indicating the overall resolutions of unmasked (red), Pf40S masked (green) and Pf60S masked (blue) reconstructions of the Pf80S–emetine complex. (D) Representative density with built models of a β-strand with well-resolved side chains (left), an RNA segment with separated bases (middle), and a magnesium ion (green sphere) that is coordinated by RNA backbone phosphates. (E) Density maps colored according to local resolution for the unmasked Pf80S (left) and masked Pf40S and Pf60S subunits (right).
Protein side chains and RNA bases were clearly resolved in our maps (Figure 1D). The use of model building and refinement tools that were adapted from X-ray crystallography (Amunts et al., 2014) led to a near-complete atomic model with excellent geometrical properties (Figure 2A,B; Table 1). The ribosome model comprises the large (Pf60S) and small subunit (Pf40S) with a total of 74 proteins (Tables 2 and 3) as well as the 5S, 5.8S, 18S, and 28S rRNAs and a tRNA bound at the E-site. The head region of Pf40S has weaker density than the rest of the ribosome due to the inherent flexibility at the neck (centered around h28). This meant that eS31, located in the beak of the 40S head (Rabl et al., 2011), could not be positioned accurately, and has therefore been omitted from the final model. Using base-pair information extracted directly from the atomic model it was possible to revise secondary structure diagrams for P. falciparum rRNA (Figure 2—figure supplements 1–3), facilitating comparison with rRNA of other species.
Figure 2 with 3 supplements see all

Structure of the Pf80S ribosome.
Overview of Pf80S atomic model showing views facing (A) tRNA entry side and (B) tRNA exit side. rRNAs are shown in gray, proteins numbered according to Ban et al. (2014). (C and D) Pf40S and Pf60S subunits are colored in yellow and blue respectively. Flexible regions are shown in red and at a resolution of 6 Å. Pf-specific expansion segments (ESs) relative to human ribosomes are labeled. Their numbering is as described for the human cytoplasmic ribosome (Anger et al., 2013).
Table 1
Refinement and model statistics
| Pf80S–emetine | |
|---|---|
| Data collection | |
| Particles | 105,247 |
| Pixel size (Å) | 1.34 |
| Defocus range (μm) | 0.8–3.8 |
| Voltage (kV) | 300 |
| Electron dose (e− Å−2) | 20 |
| Pf60S | Pf40S | |
|---|---|---|
| Model composition | ||
| Non-hydrogen atoms | 124,509 | 68,858 |
| Protein residues | 6,244 | 4,106 |
| RNA bases | 3,460 | 1,682 |
| Ligands (Zn2+/Mg2+/emetine) | 5/163/0 | 1/67/1 |
| Refinement | ||
| Resolution used for refinement (Å) | 3.1 | 3.3 |
| Map sharpening B-factor (Å2) | −60.3 | −79.9 |
| Average B factor (Å2) | 113.1 | 143.2 |
| Rfactor* | 0.2294 | 0.257 |
| Fourier Shell Correlation† | 0.86 | 0.854 |
| Rms deviations | ||
| Bonds (Å) | 0.006 | 0.007 |
| Angles (°) | 1.20 | 1.29 |
| Validation (proteins) | ||
| Molprobity score | 2.45 (96th percentile) | 2.73 (95th percentile) |
| Clashscore, all atoms | 3.65 (100th percentile) | 4.23 (100th percentile) |
| Good rotamers (%) | 90.0 | 86.0 |
| Ramachandran plot | ||
| Favored (%) | 90.4 | 85.4 |
| Outliers (%) | 2.4 | 4.2 |
| Validation (RNA) | ||
| Correct sugar puckers (%) | 97.3 | 97.5 |
| Good backbone conformations (%) | 71.1 | 70.0 |
-
*
Rfactor = Σ||Fobs| − ||Fcalc|/Σ|Fobs|.
-
†
FSCoverall = Σ(Nshell FSCshell)/Σ(Nshell), where FSCshell is the FSC in a given shell, Nshell is the number of ‘structure factors’ in the shell. FSCshell = Σ(Fmodel FEM)/(√(Σ(|F|2model)) √(Σ(F2EM)).
Table 2
Ribosomal proteins of the Pf40S subunit
| Protein names | Uniprot ID | PlasmoDB ID | Chain ID | Built residues | Extensions compared to human | Total number of residues |
|---|---|---|---|---|---|---|
| eS1 | RS3A_PLAF7 | PF3D7_0322900 | B | 24–233 | 245–262 | 262 |
| uS2 | RSSA_PLAF7 | PF3D7_1026800 | C | 10–204 | – | 263 |
| uS3 | Q8IKH8_PLAF7 | PF3D7_1465900 | D | 4–39; 65–78; 97–193; 207–216 | – | 221 |
| uS4 | Q8I3R0_PLAF7 | PF3D7_0520000 | E | 2–186 | – | 189 |
| eS4 | Q8IIU8_PLAF7 | PF3D7_1105400 | F | 2–258 | – | 261 |
| uS5 | Q8IL02_PLAF7 | PF3D7_1447000 | G | 39–262 | – | 272 |
| eS6 | Q8IDR9_PLAF7 | PF3D7_1342000 | H | 1–160; 170–213 | 249–306 | 306 |
| uS7 | Q8IBN5_PLAF7 | PF3D7_0721600 | I | 7–118; 128–195 | – | 195 |
| eS7 | Q8IET7_PLAF7 | PF3D7_1302800 | J | 3–190 | – | 194 |
| uS8 | O77395_PLAF7 | PF3D7_0316800 | K | 2–130 | – | 130 |
| eS8 | Q8IM10_PLAF7 | PF3D7_1408600 | L | 5–120; 161–213; 216–218 | 154–163 | 218 |
| uS9 | Q8IAX5_PLAF7 | PF3D7_0813900 | M | 6–143 | – | 144 |
| uS10 | Q8IK02_PLAF7 | PF3D7_1003500 | N | 21–118 | – | 118 |
| eS10 | Q8IBQ5_PLAF7 | PF3D7_0719700 | O | 11–89 | – | 137 |
| uS11 | Q8I3U6_PLAF7 | PF3D7_0516200 | P | 25–151 | – | 151 |
| uS12 | O97248_PLAF7 | PF3D7_0306900 | Q | 2–145 | – | 145 |
| eS12 | RS12_PLAF7 | PF3D7_0307100 | R | 22–78; 85–100; 111–135 | 10–16 | 141 |
| uS13 | Q8IIA2_PLAF7 | PF3D7_1126200 | S | 12–139 | – | 156 |
| uS14 | C0H4K8_PLAF7 | PF3D7_0705700 | T | 7–54 | – | 54 |
| uS15 | Q8IDB0_PLAF7 | PF3D7_1358800 | U | 3–151 | – | 151 |
| uS17 | O77381_PLAF7 | PF3D7_0317600 | V | 6–25; 36–161 | – | 161 |
| eS17 | Q8I502_PLAF7 | PF3D7_1242700 | W | 3–83; 97–110 | – | 137 |
| uS19 | C0H5C2_PLAF7 | PF3D7_1317800 | X | 21–95; 103–123 | – | 145 |
| eS19 | Q8IFP2_PLAF7 | PF3D7_0422400 | Y | 15–168 | 1–19 | 170 |
| eS21 | Q8IHS5_PLAF7 | PF3D7_1144000 | Z | 11–82 | – | 82 |
| eS24 | Q8I3R6_PLAF7 | PF3D7_0519400 | 1 | 3–122 | – | 133 |
| eS25 | Q8ILN8_PLAF7 | PF3D7_1421200 | 2 | 35–42; 58–84; 97–102 | – | 105 |
| eS26 | O96258_PLAF7 | PF3D7_0217800 | 3 | 2–96 | – | 107 |
| eS27 | Q8IEN2_PLAF7 | PF3D7_1308300 | 4 | 7–82 | – | 82 |
| eS28 | Q8IKL9_PLAF7 | PF3D7_1461300 | 5 | 2–29; 37–66 | – | 67 |
| eS30 | RS30_PLAF7 | PF3D7_0219200 | 6 | 6–48 | – | 58 |
| eS31 | Q8IM64_PLAF7 | PF3D7_1402500 | – | Not built | – | 149 |
Table 3
Ribosomal proteins of the Pf60S subunit
| Protein names | Uniprot ID | PlasmoDB ID | Chain ID | Built residues | Extensions compared to human | Total number of residues |
|---|---|---|---|---|---|---|
| uL2 | Q8I3T9_PLAF7 | PF3D7_0516900 | D | 2–248 | – | 260 |
| uL3 | Q8IJC6_PLAF7 | PF3D7_1027800 | E | 2–381 | – | 386 |
| uL4 | Q8I431_PLAF7 | PF3D7_0507100 | F | 6–395 | 373–411 | 411 |
| uL5 | Q8IBQ6_PLAF7 | PF3D7_0719600 | G | 8–51; 64–85; 92–106; 124–166 | – | 173 |
| uL6 | Q8IE85_PLAF7 | PF3D7_1323100 | H | 2–186 | – | 190 |
| eL6 | Q8IDV1_PLAF7 | PF3D7_1338200 | I | 9–151; 158–221 | 110–118; 139–143; 174–182 | 221 |
| eL8 | Q8ILL2_PLAF7 | PF3D7_1424400 | J | 40–46; 54–131; 147–283 | 11–24;279–283 | 283 |
| uL13 | Q8IJZ7_PLAF7 | PF3D7_1004000 | K | 1–201 | – | 202 |
| eL13 | Q8IAX6_PLAF7 | PF3D7_0814000 | L | 2–212 | 134–141; 168–174 | 215 |
| uL14 | Q8IE09_PLAF7 | PF3D7_1331800 | M | 8–139 | – | 139 |
| eL14 | Q8ILE8_PLAF7 | PF3D7_1431700 | N | 5–150 | 1–18 | 165 |
| uL15 | C6KT23_PLAF7 | PF3D7_0618300 | O | 2–148 | – | 148 |
| eL15 | C0H4A6_PLAF7 | PF3D7_0415900 | P | 2–205 | – | 205 |
| uL16 | Q8ILV2_PLAF7 | PF3D7_1414300 | Q | 2–101; 118–206 | – | 219 |
| uL18 | Q8ILL3_PLAF7 | PF3D7_1424100 | R | 5–126; 141–185; 189–250; 271–293 | – | 294 |
| eL18 | C0H5G3_PLAF7 | PF3D7_1341200 | U | 5–184 | – | 184 |
| eL19 | C6KSY6_PLAF7 | PF3D7_0614500 | T | 2–182 | – | 182 |
| eL20 | Q8IDS6_PLAF7 | PF3D7_1341200 | S | 2–187 | – | 184 |
| eL21 | Q8ILK3_PLAF7 | PF3D7_1426000 | V | 4–158 | – | 161 |
| uL22 | Q8IDI5_PLAF7 | PF3D7_1351400 | W | 4–154; 197–215 | – | 203 |
| eL22 | Q8IB51_PLAF7 | PF3D7_0821700 | X | 40–136 | 4–18; 34–38 | 139 |
| uL23 | Q8IE82_PLAF7 | PF3D7_1323400 | Y | 88–188 | 13–34; 57–67 | 190 |
| uL24 | O77364_PLAF7 | PF3D7_0312800 | Z | 2–122 | – | 126 |
| eL24 | Q8IEM3_PLAF7 | PF3D7_1309100 | 0 | 8–69 | – | 162 |
| eL27 | Q8IKM5_PLAF7 | PF3D7_1460700 | 1 | 2–126;132–146 | – | 146 |
| eL28 | Q8IHU0_PLAF7 | PF3D7_1142500 | 2 | 2–69; 77–82; 86–98; 103–119 | – | 127 |
| uL29 | Q8IIB4_PLAF7 | PF3D7_1124900 | 3 | 3–121 | – | 124 |
| eL29 | C6S3J6_PLAF7 | PF3D7_1460300 | 4 | 2–67 | – | 67 |
| uL30 | O97250_PLAF7 | PF3D7_0307200 | 5 | 35–257 | – | 257 |
| eL30 | Q8IJK8_PLAF7 | PF3D7_1019400 | 6 | 8–105 | – | 108 |
| eL31 | Q8I463_PLAF7 | PF3D7_0503800 | 7 | 15–88; 95–116 | – | 120 |
| eL32 | Q8I3B0_PLAF7 | PF3D7_0903900 | 8 | 2–126 | – | 131 |
| eL33 | Q8IHT9_PLAF7 | PF3D7_1142600 | 9 | 35–137 | 1–35 | 140 |
| eL34 | Q8IBY4_PLAF7 | PF3D7_0710600 | a | 2–107 | – | 150 |
| eL36 | Q8I713_PLAF7 | PF3D7_1109900 | b | 2–27; 38–106 | 5–10 | 112 |
| eL37 | C0H4L5_PLAF7 | PF3D7_0706400 | c | 2–90 | – | 92 |
| eL38 | Q8II62_PLAF7 | PF3D7_1130100 | d | 2–31; 36–77 | – | 87 |
| eL39 | C0H4H3_PLAF7 | PF3D7_0611700 | e | 2–30; 38–51 | – | 51 |
| eL40 | Q8ID50_PLAF7 | PF3D7_1365900 | f | 1–51 | – | 52 |
| eL41 | C6S3G4_PLAF7 | PF3D7_1144300 | g | 3–39 | 1–14 | 39 |
| eL43 | RL37A_PLAF7 | PF3D7_0210100.1 | h | 2–86 | – | 96 |
| eL44 | RL44_PLAF7 | PF3D7_0304400 | i | 2–96 | – | 104 |
Currently, high resolution structures of eukaryotic ribosomes have been solved using X-ray crystallography and are limited to just three structures; the individual subunits from a ciliated protozoan, Tetrahymena thermophila (Klinge et al., 2011; Rabl et al., 2011), and the complete 80S ribosome from the yeast Saccharomyces cerevisiae (Ben-Shem et al., 2011). These models have been used to interpret lower resolution structures solved by cryo-EM of other species including the yeast Kluyveromyces lactis (Fernandez et al., 2014), Drosophila melanogaster (Anger et al., 2013), Trypanosoma brucei (Hashem et al., 2013), as well as human ribosomes (Anger et al., 2013) and provide the basis of the nomenclature used for describing the structures.
To examine overall architectural differences, we compared the model of Pf80S to yeast 80S (Ben-Shem et al., 2011). Perhaps the largest difference is the absence of RACK1 (Figure 1A,B), which associates with the head of the 40S in the vicinity of the mRNA exit channel (Sengupta et al., 2004; Rabl et al., 2011) and has been identified in all eukaryotic ribosome structures solved to-date. RACK1 serves as a signaling scaffold that can recruit other proteins to the ribosome and may link the ribosome with signal transduction pathways, thus allowing translation regulation in response to stimuli. It has also been proposed that RACK1 promotes the docking of ribosomes at sites where local translation is required (Nilsson et al., 2004).
PfRACK1 is conserved with its human homolog with an identity of 60%. The binding site on the ribosome, which comprises eS17, uS3, and 18S rRNA helices h39 and h40 (Figure 1A,B), also appears highly conserved (Rabl et al., 2011). However, the C-terminus of uS3 is not resolved in our structure and probably only becomes ordered upon binding RACK1. The absence of PfRACK1 as an integral member of the small subunit indicates either a different mode of interaction between the ribosome and PfRACK1 in Plasmodium compared to humans, or that under the culturing conditions used PfRACK1 is not expressed, or expressed in a form that does not interact with the ribosome. In yeast, RACK1 has been shown to be present in both a ribosome- and a non-ribosome-bound form dependent on growth conditions (Baum et al., 2004). If the interaction between PfRACK1 and the Pf40S is weaker than in other organisms, the possibility that PfRACK1 dissociated during purification and grid preparation cannot be discounted.
The yeast 80S structure was also solved in the presence of STM1, a translation repressor protein, that binds to the head region of the 40S and blocks mRNA entry and binding of tRNA to the A- and P-sites (Rabl et al., 2011). The human and D. melanogaster structures also co-purified with an STM1-like protein (SERBP1 and VIG2 respectively) (Anger et al., 2013). Pf80S is not bound by a suppressor molecule, as also observed for the T. brucei structure (Hashem et al., 2013), and hence reflects a ribosome capable of active translation.
Pf80S co-purifies with a tRNA bound to the E-site. Although the density is not well resolved, presumably as a result of low and mixed occupancy, it could be interpreted by positioning a model of tRNAMet. The presence of tRNA helps to partially stabilise the L1 stalk near the elbow of the tRNA, however the stalk remains considerably flexible and is averaged out of the high-resolution reconstruction.
Perhaps due to the absence of RACK1 and/or STM1 or the presence of an E-site tRNA, the head of Pf40S adopts an orientation with respect to the body that is different to the yeast structure, with uS11 at the beak of the small subunit displaced by more than 10 Å. The root mean square deviation (RMSD) of the two small subunits is 2.9 Å2, however if the head and body are superimposed independently this improves to 1.0 Å2 and 1.5 Å2 respectively. The structure of Pf60S superimposes with the yeast 60S with a RMSD of 1.6 Å2. The largest morphological differences in this subunit result from a cluster of rRNA helices (ES7AL, ES15L, and ES7CL) protruding at the solvent side.
Given the potential of Pf80S as a drug target, we sought to describe its detailed structure in comparison to its direct counterpart in the human cytoplasm, where a 4.8 Å cryo-EM 80S structure represents the highest resolution solved to-date (Anger et al., 2013). Therefore, all protein extensions and rRNA expansion segments (ESs) are annotated on the basis of comparison with human ribosomes. While the core of the Pf80S and human ribosome are conserved, the periphery of the ribosomes differs extensively in the nature and length of rRNA ESs and protein extensions. The constraints on rRNA expansion appear to be fewer than on protein extension, as rRNA contributes greater to the mass difference between species.
Compared to human ribosomes, P. falciparum typically has shorter ESs, some of which are entirely absent in the large subunit (ES7D-HL, ES9AL, ES10L, ES20L, ES30L) (Table 4). The functions, if any, of many of these ESs are not well known. ES7E, which is highly conserved in vertebrates, is implicated in selenoprotein synthesis by binding the SBP2 protein that specifically recruits the selenocysteine-specific tRNA and elongation factor (Kossinova et al., 2014). While P. falciparum does utilize selenocysteine, it is incorporated into very few proteins (Lobanov et al., 2006) and there is no homolog of SBP2, providing a possible explanation for why ES7E is not present in Plasmodium.
Table 4
Comparison of ESs in Pf80S and human cytoplasmic ribosomes
| rRNA | ES | Helix | Comparison between Pf80S and human ribosomes |
|---|---|---|---|
| 18S | ES2S | Shorter loop in Pf80S | |
| ES3S | A | Conserved | |
| B | Truncated in Pf80S | ||
| ES13S | Conserved | ||
| ES6S | A | Expanded in Pf80S | |
| B | Truncated in Pf80S | ||
| C | Conserved | ||
| D | Expanded in Pf80S | ||
| E | Conserved | ||
| ES7S | Expanded in Pf80S | ||
| ES14S | Conserved | ||
| ES9S | Expanded in Pf80S | ||
| ES10S | Expanded in Pf80S | ||
| ES12S | Helix truncated in Pf80S | ||
| 28S | ES3L | Conserved | |
| ES4L | Conserved | ||
| ES5L | Conserved | ||
| ES7L | A | Truncated in Pf80S | |
| B | Truncated. Loop in Pf80S forms a novel interaction with eL14 | ||
| B1 | Pf-specific ES | ||
| C | Present | ||
| D–H | Absent from Pf80S | ||
| ES8L | H28 | Expanded in Pf80S | |
| ES9L | A | Absent in Pf80S | |
| H30 | Conserved | ||
| H31 | Conserved | ||
| ES10L | Absent in Pf80S | ||
| ES12L | Expanded in Pf80S | ||
| ES15L | A | Truncated in Pf80S | |
| ES19L | Truncated in Pf80S | ||
| ES20L | A | Absent in Pf80S | |
| B | Conserved in Pf80S | ||
| ES26L | Expanded in Pf80S | ||
| ES27L | A–C | Not present in Pf80S model, predicted divergence between Pf and human cytoplasmic ribosomes | |
| ES30L | Absent in Pf80S | ||
| ES31L | A | Conserved | |
| B | Expanded in Pf80S | ||
| C | Conserved | ||
| ES34L | Pf-specific ES | ||
| ES36L | Pf-specific ES | ||
| ES39L | A | Conserved; preceding loop in Pf80S forms a short helix (3 base pairs) with the 5′ end of the 5.8S rRNA | |
| B | Conserved | ||
| ES41L | Conserved |
The largest Pf-specific ESs are concentrated in the 18S rRNA, with ES6S and ES9S being particularly extended (Figure 2C,D; Figure 2—figure supplement 1). These ESs, like those described in both the human (Anger et al., 2013) and Trypanosoma brucei (Hashem et al., 2013) ribosome structures, are highly flexible and, in our structure, are only partly visible using a map filtered at 6 Å (Figure 2C,D). We have therefore not included these sections in our atomic model. ES10S is located at the top of the 40S head and has been partially built.
P. falciparum ribosomes resemble those of T. brucei in that both have large ES6S and ES7S, although these are slightly larger in T. brucei (Hashem et al., 2013). ES6S is in contact with ribosomal components that form part of the mRNA entry and exit sites and was therefore suggested as being involved in translation initiation (Jenner et al., 2012). Recently, ES6/7S have been implicated in binding of the conserved translation initiation factor eIF3 based on superposition with a mammalian 43S complex (Hashem et al., 2013). Almost 90 nucleotides of ES6AS are averaged out of our high-resolution reconstruction indicating this stalk is highly flexible, perhaps acting in a manner similar to the P stalk (known as the L7/L12 stalk in prokaryotes) by recruiting factors necessary for translation (in this case eIF3). The other large ES of the 18S rRNA, ES9S, is positioned at the head of the 40S. Given both the intrinsic mobility of the head and presumably the ES itself, there is no density for this ∼150 nucleotide Pf-specific element and the role it plays remains unclear.
The sites of Pf-specific elements are broadly distributed across the solvent-accessible surface of the ribosome, although the region surrounding the exit tunnel is conserved (Klinge et al., 2011) and undecorated with ESs and protein extensions (Figure 2C,D). The subunit interface and eukaryotic-specific bridges, which in addition to having structural roles help transmit information to coordinate activity during translation (Ben-Shem et al., 2011), are generally highly conserved in Pf80S. There are a couple of examples of stabilizing interactions that are not observed in human ribosomes. Firstly, eL41, the smallest ribosomal protein, bridges the two subunits (Ben-Shem et al., 2011) and has a 14-residue Pf-specific N-terminal extension that reaches into a pocket formed by 18S rRNA of the small subunit and tightly anchors the protein (Figure 3A). Secondly, an additional small bridge (∼200 Å2) is formed between the platform of Pf40S and the region around the L1 stalk by the C-terminal helix extension of eL8 interacting with the C-terminal helix of eS1 (Figure 3B).
Figure 3
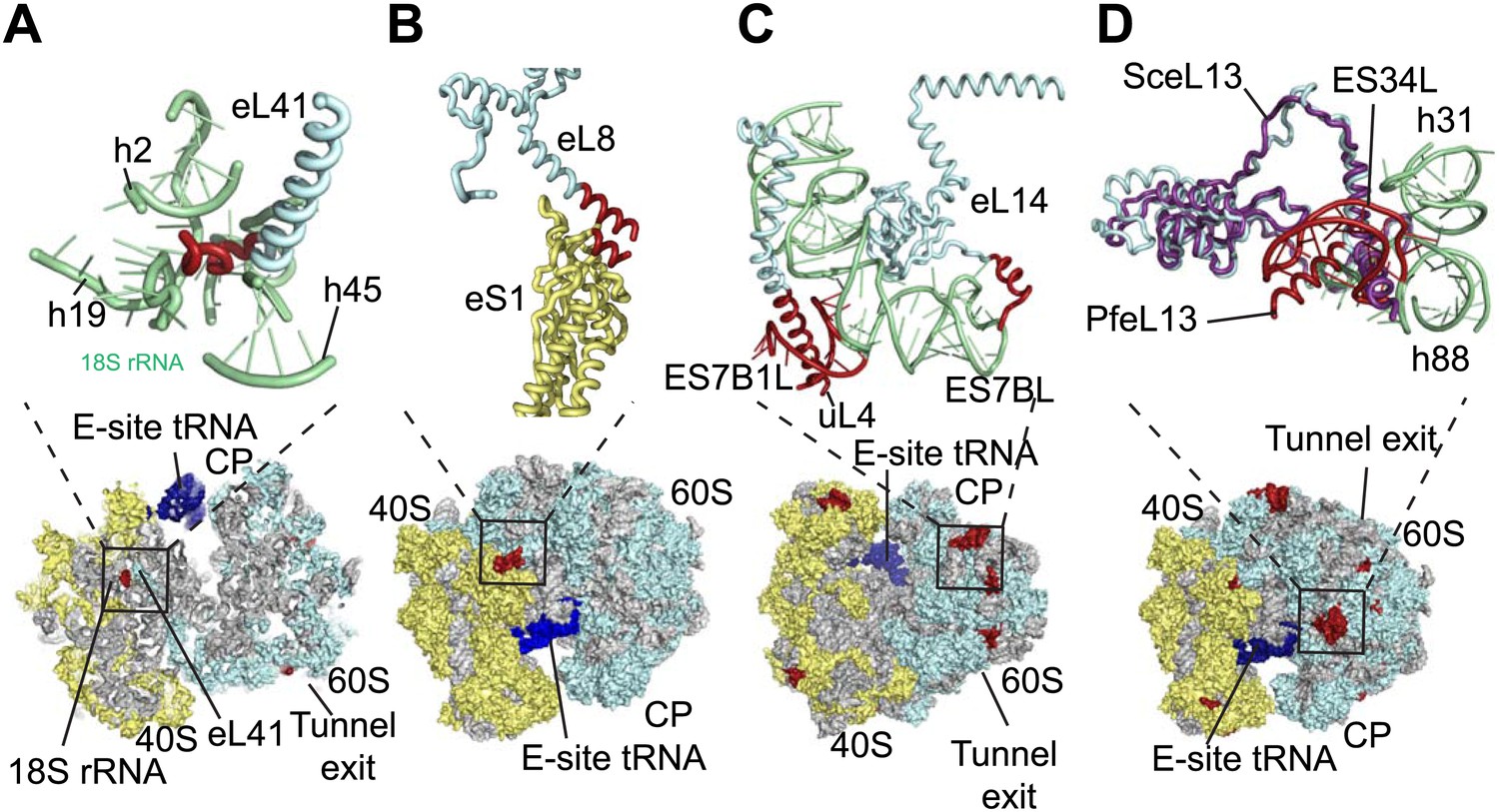
Details of Pf-specific protein extensions and rRNA ESs near the (A and B) subunit interface (C) P stalk and (D) the L1 stalk.
Pf-specific elements are shown in red.
Further ordered Pf-specific elements are concentrated near the L1 and P stalks of Pf60S. Directly above the P stalk, the Pf-specific ES7B1L forms a diverted part of ES7CL that is stabilized by several electrostatic interactions with a C-terminal helix extension of uL4 (Figure 3C). Towards the back of the P-stalk, the C-terminal helix extension of eL14 caps the stem loop of ES7BL (Figure 3C). On the opposite side of the ribosome, near the E-site tRNA, the Pf-specific stem loop ES34L is positioned directly above the L1 stalk (Figure 3D). This ES appears to have caused a 60° rotation of the C-terminal helix of eL13 relative to its position in human ribosomes (Figure 3D). The tip of the helix is displaced by ∼28 Å away from the L1 stalk and now stabilizes the interaction between ES34L and the loop of h22. Since the L1 stalk is required for coordinating the movement of tRNAs and the P stalk is required for coordinating the movement of translation factors during the various steps of protein synthesis (Gonzalo and Reboud, 2003), the expanded mass around the stalks of Pf80S may have functional implications for translation in P. falciparum.
The ability to determine atomic-resolution structures of Pf80S provides a platform for investigating the action of anti-malarial therapeutics that target the ribosome. The clinically used, broad-spectrum eukaryotic translation inhibitor emetine (Figure 4A) (Grollman, 1968), has been reported to act as a translocation inhibitor targeting the ribosome (Jimenez et al., 1977; Dinos et al., 2004), although its precise mode of action is unknown. Emetine is a natural product alkaloid from the plant Carapichea ipecacuanha, and an approved medicine for the treatment of amoebiasis (Goodwin et al., 1948). Although its toxicity associated with chronic usage in humans has limited its clinical use against malaria in its current formulation (Dempsey and Salem, 1966), emetine does demonstrate potent antimalarial activity with a 50% inhibitory concentration (IC50) of 47 nM against the blood stage of multidrug resistant strains of P. falciparum (Matthews et al., 2013). Moreover, the immediate therapeutic effect it offers by rapid killing of blood stage parasites may warrant re-consideration of the use of emetine or its derivatives for short periods during acute malaria infection (James, 1985).
Figure 4
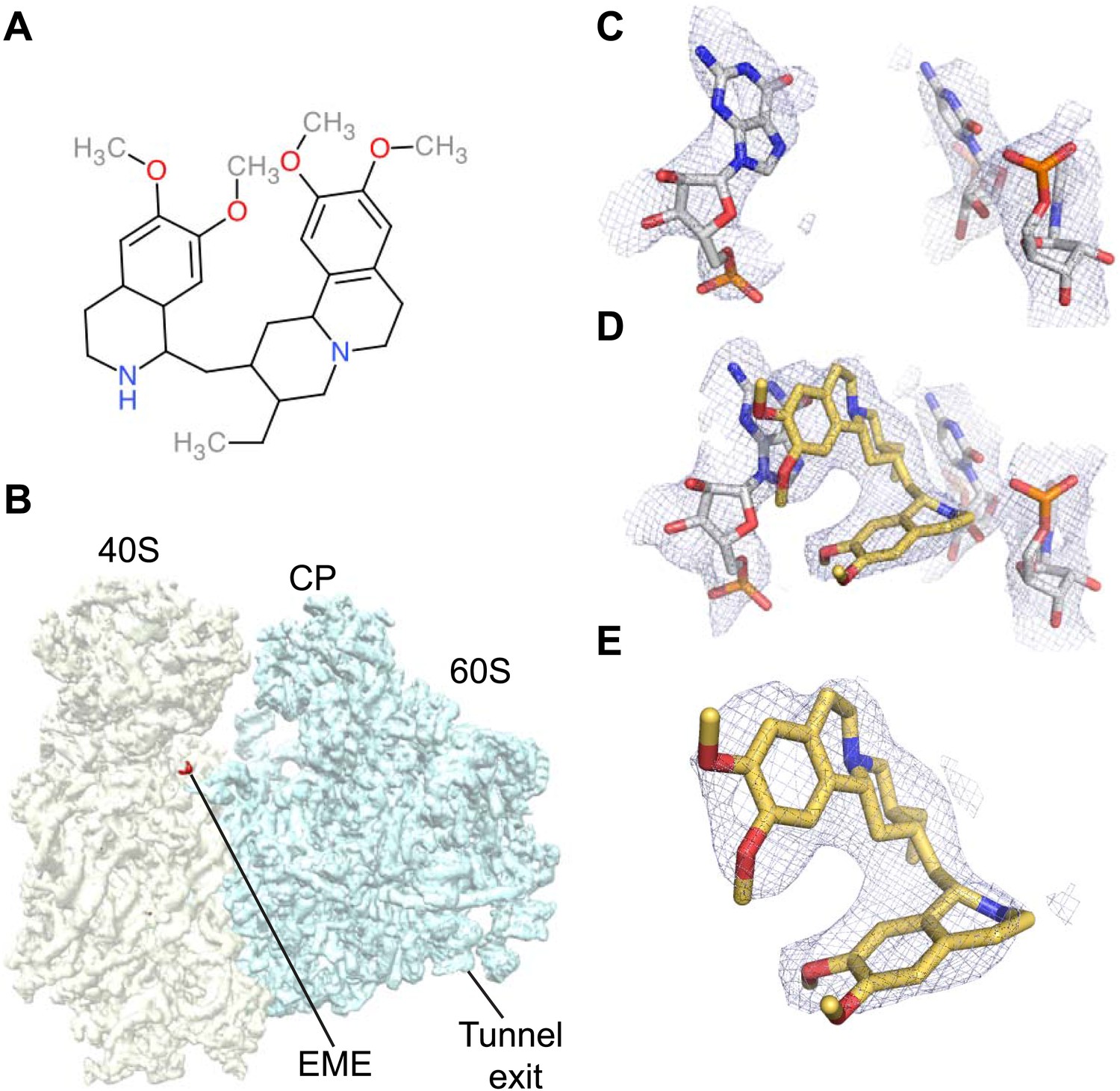
Emetine binds to the E-site of the Pf40S subunit.
(A) 2D chemical structure of emetine. (B) A 4.5 Å filtered difference map (red density) at 5 standard deviation overlaid with the Pf80S map filtered at 6 Å (blue and yellow for Pf60S and Pf40S respectively), showing the emetine density at the E-site of the Pf40S. The emetine binding site in (C) empty and (D) emetine-bound structures, with (E) density for emetine alone at 3.2 Å.
Incubation of purified Pf80S with a 1 mM emetine solution prior to cryo-EM grid preparation, led to a 3.2 Å resolution structure of the complex. Using soft masking, the resolution for the large subunit improved to 3.1 Å, with the small subunit at 3.3 Å (Figure 1C). A difference map was calculated from the reconstructions with and without emetine and showed a single, continuous feature near the E-site of Pf40S with a shape and size congruent with a single emetine molecule when thresholded at 5 standard deviations, and with a maximum value of 11 standard deviations (Figure 4B). At this position in our map, the density provided sufficient detail to confidently model the emetine molecule (Figure 4C–E). The emetine binding pocket is formed at the interface between 18S rRNA helices 23, 24, 45, and the C-terminus of uS11 (Figure 5A). Comparison with the unliganded map showed that binding of emetine does not induce changes to the pocket (Figure 4C,D). The benzo[a]quinolizine ring of emetine mimics a base-stacking interaction with G973 of h23 and its ethyl group forms a hydrophobic interaction with C1075 and C1076 of h24, whereas the isoquinoline ring is stacked against the C-terminal Leu151 of uS11 (Figure 5B,C). The interaction is stabilized by a hydrogen bond formed between the NH group of the isoquinoline ring in emetine and an oxygen atom on the backbone of U2061 of h45 (Figure 5B,C). Although there is no high-resolution structure of the human cytoplasmic ribosome, comparison of the emetine binding site in Pf80S with the equivalent region in the 4.8 Å human structure (Anger et al., 2013) revealed that each of the core binding elements are conserved (Figure 5—figure supplement 1) indicating that emetine likely binds to the cytoplasmic host ribosomes in the same way, potentially accounting for the observed cytotoxicity in humans.
Figure 5 with 1 supplement see all
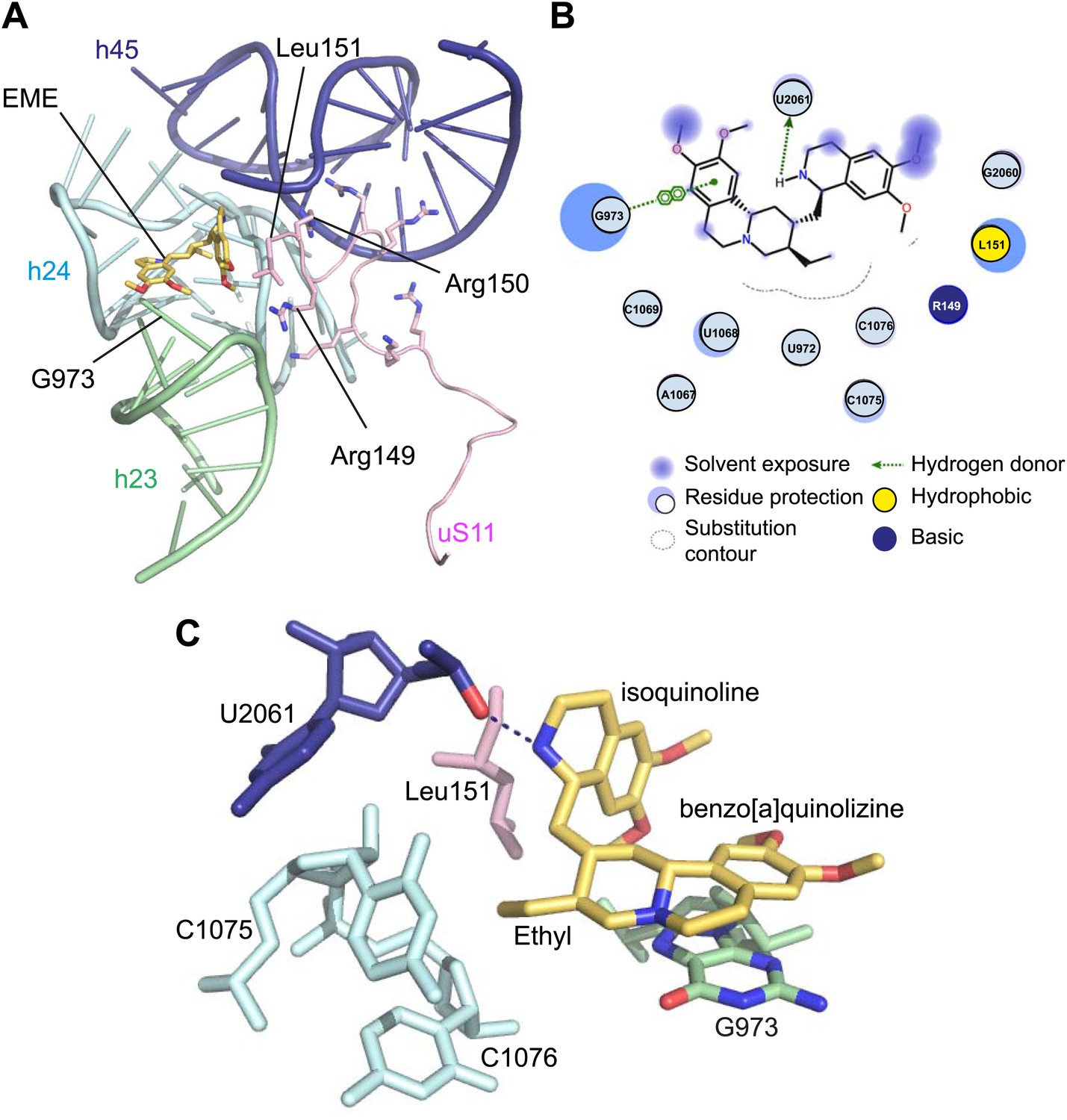
Molecular details of the emetine–ribosome interaction.
(A) Overview of emetine at the binding interface formed by the three conserved rRNA helices and uS11. h23 is in green, h24 in cyan, h45 in blue, uS11 in pink, and emetine in yellow. (B) 2D representation showing the interaction of emetine with binding residues. Substitution contour represents potential space for chemical modification of emetine. (C) Residues in physical contact with emetine. Hydrogen bond is indicated as dashes.
The identified binding site is consistent with mutations of Arg149 and Arg150 of uS11 in Chinese hamster ovary (CHO) cells that have been found to confer resistance to emetine (Madjar et al., 1982). At the emetine-binding pocket, h24 is sandwiched between the apexes of h23 and h45. The C-terminus of uS11 adopts a long coil with seven basic residues (residues 141–151; RKKSGRRGRRL), which form electrostatic interactions with the phosphate backbones of h45, h23 and h24, thereby stabilizing the conformation of this coil together with the 18S rRNA (Figure 5A). This would explain the molecular basis for resistance whereby mutations of the C-terminal arginine residues of uS11 destabilize h23 and h45, disrupting the binding pocket.
The mode of binding of emetine resembles the way in which pactamycin, previously thought to be a unique class of antibiotic, binds to the bacterial 30S (Brodersen et al., 2000). In both structures the guanine base at the tip of h23 (G973 in Pf; G693 in bacteria) forms a stacking interaction with the hydrophobic rings of either compound. Moreover, the two cytosine bases of h24 (C1075 and 1076 in Pf; C795 and 796 in bacteria) are each involved in drug binding (Brodersen et al., 2000; Figure 6). The hydrogen bond to the backbone of h45 and the hydrophobic interaction with Leu151 of uS11 are specific to the Pf80S–emetine interaction. In the 30S-pactamycin complex, the last base of the E-site codon of the mRNA was displaced 12.5 Å compared to the native path of mRNA (Brodersen et al., 2000) thereby blocking mRNA/tRNA entry into the E-site during the translocation step of protein synthesis (Dinos et al., 2004). Based on these structures, emetine appears to elicit its inhibitory effect by the same mechanism as pactamycin.
Figure 6
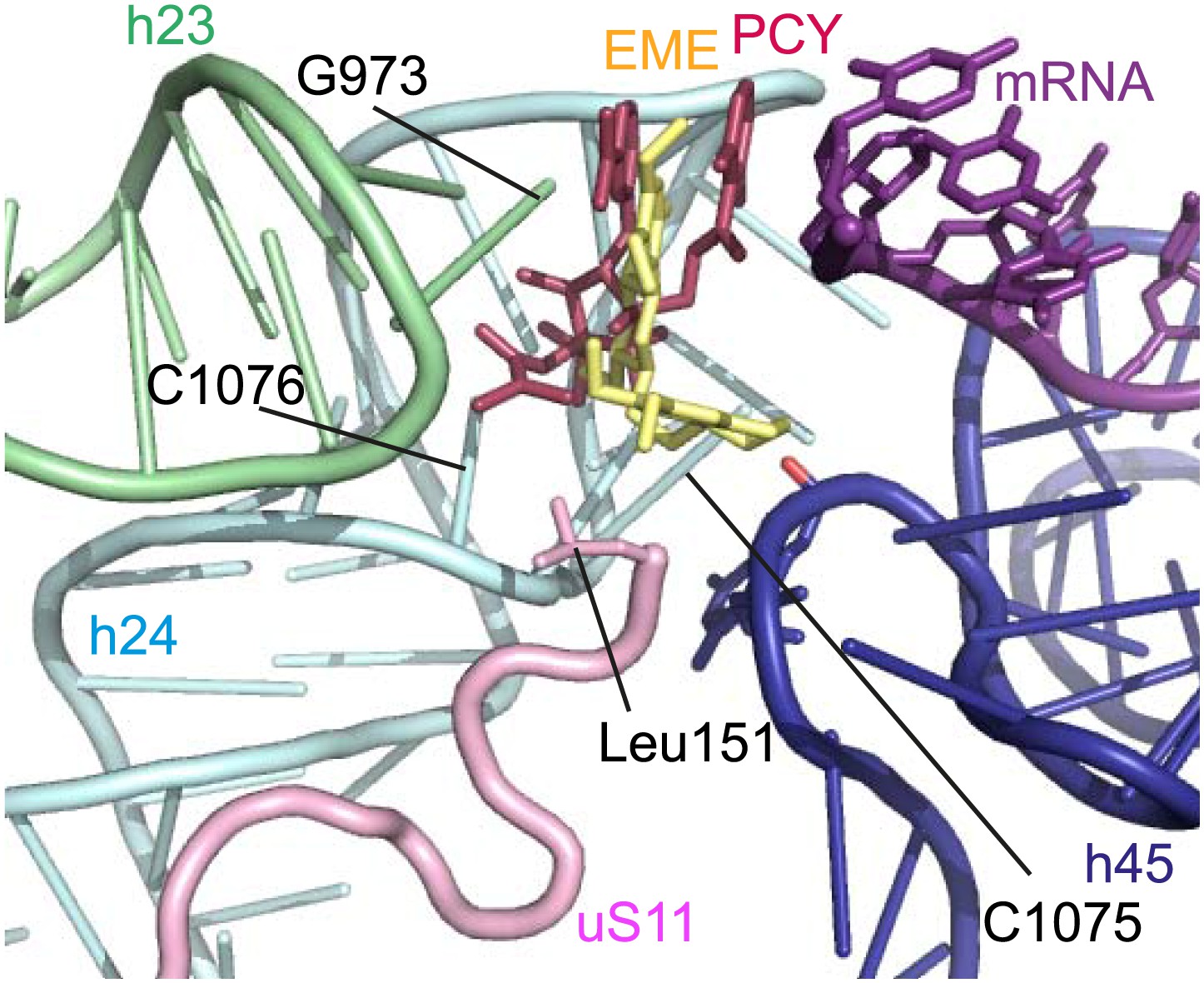
Comparison with pactamycin.
Superposition of emetine and pactamycin at the Pf40S emetine binding pocket. Emetine and pactamycin are shown in yellow and red respectively.
Discussion
The resolution revolution in cryo-EM (Kühlbrandt, 2014) is the product of a new generation of sensors that detect electrons directly (without first converting to light) and have improved quantum efficiencies. These cameras are fast enough to follow beam-induced movement of the particles caused by irradiation with electrons. Statistical movie processing can compensate for this movement allowing for structures to be solved at atomic precision. We have harnessed these technological advances to determine the first structure of a ribosome from a parasite at atomic resolution. Previously, structures of eukaryotic cytosolic 80S ribosomes at a similar resolution had only been possible using X-ray crystallography (Ben-Shem et al., 2011). From the reconstruction of Pf80S–emetine complex at 3.2 Å, we determined a stereochemically accurate all-atom model using recent developments in model building, refinement, and validation (Amunts et al., 2014).
The structure of Pf80S further demonstrates the diversity of ribosome structures among eukaryotes, especially in terms of the location and nature of ESs at the periphery, while maintaining a conserved core. The observation of Pf-specific features could serve as the basis for exploring their functional relevance as an essential, first step towards finding efficacious and clinically safe anti-malarial drugs. An alternative to drug development against Pf-specific ribosomal elements is the repurposing of existing antibiotics as anti-malarials. By determining the structure of Pf80S in both a liganded and unliganded state, we were able to locate the binding site of the anti-protozoan inhibitor, emetine, using an unbiased difference map. That emetine and pactamycin share a binding pocket in eukaryotic ribosomes could not be predicted based on the chemical structures of the drug molecules only. Pactamycin itself has been shown to have potent antiprotozoal activity against both drug-susceptible and drug-resistant strains of P. falciparum (Otoguro et al., 2010). Chemical modifications to pactamycin have yielded analogs that maintain antimalarial activity but with reduced cytotoxicity against mammalian cells (Lu et al., 2011). Similarly, an emetine derivative, dehydroemetine, which differs by the presence of a double bond next to the ethyl group of benzo[a]quinolizine ring, exhibits less toxic effects than the parental compound while maintaining anti-parasitic properties (Dempsey and Salem, 1966; Chintana et al., 1986). This suggests that compounds targeting the emetine/pactamycin binding site are amenable to optimization, potentially leading to drugs more suited to clinical use. The Pf80S–emetine structure reveals an edge centered on the ethyl group of the molecule that could be subjected to modification to increase the affinity of emetine for the binding pocket (Figure 5B, labelled as the ‘contour edge’). Although based on the similarity with the binding site in humans it is unlikely that emetine can be structurally modified to not bind the mammalian system, as demonstrated in the case of dehydroemetine modifications can reduce its cytotoxicity. Although the mechanism for such reduced cytotoxicity mediated by pactamycin and emetine analogs is not known, it may be possible that these derived compounds selectively target tumor/parasite cells that are rapidly dividing, whereby protein synthesis is more sensitive to drug action in these cells. As in the case of antibiotics repurposed as antitumor agents, there is a clinical role for eukaryotic antibiotics that target systems with differential rates of translation provided usage is carefully directed. In malaria, eukaryotic antibiotics, such as emetine, could be used in combination with the slow-acting, but more specific apicoplast-targeting antibiotics (Dahl and Rosenthal, 2007).
This work demonstrates the power of contemporary cryo-EM for drug discovery. A drug, with a previously unknown binding site, can be visualized inside a macromolecular complex that is almost 10,000 times larger in molecular weight and at a level of detail comparable to that obtained by X-ray crystallography. By avoiding the need for crystallization one of the bottlenecks of solving a structure is bypassed. It allows structures to be solved from very small sample quantities, with sample heterogeneity improved through image processing. As such, cryo-EM is of particular use for solving the structures of macromolecules in their native state, isolated from pathogenic organisms where culturing large quantities is not possible.
In summary, our cryo-EM analyses reveal the first structure of a ribosome from a parasite at atomic resolution, along with detailed insights into the molecular basis of a known anti-protozoan translation inhibitor. Finally, it demonstrates that cryo-EM offers an attractive route towards the development of new compounds that target macromolecules by facilitating structure–activity relationships in otherwise intractable biological systems.
Materials and methods
Parasite culture and ribosome purification
Request a detailed protocolWild-type 3D7 strain of P. falciparum parasites were maintained in human erythrocytes (blood group O) at a hematocrit of 4% with 10% Albumax. Saponin lysed parasite pellets were incubated with lysis buffer (20 mM Hepes, pH 7.4, 250 mM KCl, 25 mM Mg(CH3COO)2, 0.15% Triton, 5 mM 2-mecaptoethanol) at 4°C for 1 hr. Ribosomes were purified by ultracentrifugation initially with a sucrose cushion (20 mM Hepes pH 7.4, 1.1 M sucrose, 40 mM KCH3COO, 10 mM NH4CH3COO, 10 mM Mg(CH3COO)2, and 5 mM 2-mecaptoethanol) followed by a 10–40% sucrose gradient separation step using the same buffer.
Electron microscopy
Request a detailed protocolAliquots of 3 μl of purified Pf80S at a concentration of ∼160 nM (∼0.5 mg/ml) were incubated for 30 s on glow-discharged holey carbon grids (Quantifoil R1.2/1.3), on which a home-made continuous carbon film (estimated to be ∼30 Å thick) had previously been deposited. Grids were blotted for 2.5 s and flash frozen in liquid ethane using an FEI Vitrobot. For the empty Pf80S sample, grids were transferred to an FEI Titan Krios electron microscope that was operated at 300 kV. Images were recorded manually during two non-consecutive days on a back-thinned FEI Falcon II detector at a calibrated magnification of 135,922 (yielding a pixel size of 1.03 Å). Defocus values in the final data set ranged from 0.7 to 3.9 µm.
To prepare the Pf80S–emetine sample, purified Pf80S at 160 nM was incubated with a 1 mM solution of emetine in 20 mM Hepes pH7.4, 40 mM KCH3COO, 10 mM NH4CH3COO, 10 mM Mg(CH3COO)2, and 5 mM 2-mecaptoethanol for 15 min at 25°C prior to blotting and freezing as described above. Pf80S–emetine grids were transferred to an FEI Tecnai Polara electron microscope that was operated at 300 kV. Images were recorded manually during two non-consecutive days on a back-thinned FEI Falcon II detector at a calibrated magnification of 104,478 (yielding a pixel size of 1.34 Å). Defocus values in the final data set ranged from 0.8 to 3.8 µm.
During the data collection sessions of both samples, all images that showed signs of significant astigmatism or drift were discarded. An in-house built system was used to intercept the videos frames from the detector at a rate of 17 s−1 for the Krios and 16 s−1 for the Polara microscope.
Image processing
Request a detailed protocolWe used RELION (version 1.3-beta) for automated selection of 126,727 particles from 1310 micrographs for the empty Pf80S sample; and 158,212 particles from 1081 micrographs for the Pf80S–emetine sample. Contrast transfer function parameters were estimated using CTFFIND3 (Mindell and Grigorieff, 2003). All 2D and 3D classifications and refinements were performed using RELION (Scheres, 2012). To discard bad particles, we used a single round of reference-free 2D class averaging with 100 classes for both data sets, and a single round of 3D classification with four classes for the Pf80S–emetine data set. The final refinement for the empty Pf80S and Pf80S–emetine sample contained 72,293 and 105,247 particles, respectively. A 60 Å low-pass filtered cryo-EM reconstruction of the yeast cytoplasmic 80S ribosome (EMDB-2275 [Ben-Shem et al., 2010]) was used as an initial model for the 3D refinement.
For the correction of beam-induced movements, we used statistical movie processing as described previously (Bai et al., 2013), with running averages of five movie frames, and a standard deviation of 1 pixel for the translational alignment. To further increase the accuracy of the movement correction, we used the beta version of RELION-1.3 to fit linear tracks through the optimal translations for all running averages, and included neighboring particles on the micrograph in these fits. In addition, we employed a resolution and dose-dependent model for the radiation damage, where each frame is weighted with a different B-factor as was estimated from single-frame reconstructions. These procedures yielded maps with an overall resolution of 3.4 Å for the empty Pf80S and 3.2 Å for Pf80S–emetine.
Reported resolutions are based on the gold-standard FSC = 0.143 criterion (Chen et al., 2013) and were corrected for the effects of a soft mask on the FSC curve using high-resolution noise substitution (Chen et al., 2013). Soft masks were made by converting atomic models into density maps, binarizing those, and adding cosine-shaped edges. Prior to visualization, all density maps were corrected for the modulation transfer function (MTF) of the detector, and then sharpened by applying a negative B-factor (Table 1) that was estimated using automated procedures (Rosenthal and Henderson, 2003).
In order to locate emetine in the Pf80S–emetine reconstruction, we calculated a difference map between the reconstructions of empty Pf80S and Pf80S–emetine. To this purpose, the two MTF-corrected and B-factor sharpened maps were aligned with respect to each other using the ‘Fit in Map’ functionality in UCSF Chimera (Pettersen et al., 2004 ), and the empty Pf80S map was re-interpolated on the Cartesian grid of the Pf80S–emetine map prior to subtraction of the maps in RELION. For visualization purposes, the resulting difference map was low-pass filtered at 4.5 Å and the threshold was set at 5 standard deviations as calculated within the area of the Pf80S ribosome (Figure 4B). At this threshold, only one continuous U-shaped feature was visible. The highest difference density inside this feature extended to 11 standard deviations in the difference map.
Local resolution variations in all reconstructions were estimated using ResMap (Kucukelbir et al., 2014). Presumably due to unresolved structural heterogeneity the local resolution in the small ribosomal subunit was typically worse than in the large ribosomal subunit. Therefore, for the Pf80S–emetine structure, we performed two additional ‘focussed’ refinements, where we masked out the large or the small subunit at every iteration. This gave rise to two maps (Figure 1E) with improved density for either the small subunit (at an overall resolution of 3.3 Å) or the large ribosomal subunit (at an overall resolution of 3.1 Å), and these maps were used for the refinement of the atomic model as described below.
Model building and refinement
Request a detailed protocolRibosomal protein sequences from the 3D7 strain of P. falciparum were taken from PlasmoDB (The Plasmodium Genome Database Collaborative, 2001) and used as template sequences to obtain homology models generated from I-TASSER (Roy et al., 2010). Homology models were fitted into the reconstructed map of Pf80S using Chimera (Pettersen et al., 2004). Each protein was then subjected to a jiggle-fit and extensively rebuilt with sidechains placed into the map density using Coot v.0.8 (Emsley et al., 2010). The sequences of the Pf80S rRNAs were obtained from PlasmoDB (The Plasmodium Genome Database Collaborative, 2001) and aligned using Clustal Omega (Sievers et al., 2011) with the rRNA sequences extracted from the Saccharomyces cerevisae (Sc) 80S structure (PDB ID: 3U5B and 3U5D) (Ben-Shem et al., 2011). Conserved regions without insertions or deletions were extracted from the yeast structure, mutated and renumbered. These conserved sections were then connected by de novo building of RNA. The complete rRNA was then manually rebuilt in Coot to optimize the fit to density. Building was aided by secondary structure predictions downloaded from the Comparative RNA Website (Cannone et al., 2002).
The model was refined using REFMAC v.5.8, which was modified for structures determined by cryo-EM (Murshudov et al., 2011; Amunts et al., 2014). The Pf80S atomic model was refined as separate 60S and 40S subunits in the two maps that were obtained for either subunit in the focused refinements of the cryo-EM reconstructions. Structure factors for the (Fourier-space) refinement in REFMAC were obtained by cutting out sections of the corresponding maps with a 3 Å radius from the center of each atom in the model, and structure factor phases were not altered during refinement.
Throughout refinement, reference and secondary structure restraints were applied to the ribosomal proteins using the Sc80S structure as a reference model (Nicholls et al., 2012). Base pair and parallelization restraints obtained using LIBG were also applied throughout refinement (Amunts et al., 2014). The stereochemistry of the rRNA model was further improved using the ERRASER-PHENIX pipeline (Chou et al., 2013). Ramachandran restraints were not applied during refinement to preserve backbone dihedral angles for validation.
The R-factor and average overall Fourier shell correlation were monitored during refinement (Table 1) and the final model was validated using MolProbity (Chen et al., 2010). For cross-validation against over-fitting, we randomly displaced the atoms of our final model (with an RMSD of 0.5 Å) and performed a fully restrained refinement against a map that was reconstructed from only one of the two independent halves of the data that were used in our gold-standard FSC procedure. We then calculated FSC curves between the resulting model and the half-map against which it had been refined (FSCwork), as well as the FSC curve between that model and the other half-map (FSCtest). The observation that the FSCwork and FSCtest curves nearly overlap demonstrates the absence of overfitting of the model (Figure 1—figure supplement 1).
Data availability
-
Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetinePublicly available at Electron Microscopy Data Bank.
-
Plasmodium falciparum 80S ribosomePublicly available at Electron Microscopy Data Bank.
-
Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine; large subunitPublicly available at RCSB Protein Data Bank.
-
Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine; small subunitPublicly available at RCSB Protein Data Bank.
-
Ribosome structures at near-atomic resolution from thirty thousand cryo-EM particlesPublicly available at Electron Microscopy Data Bank.
-
The structure of the eukaryotic ribosome at 3.0 Å resolutionPublicly available at RCSB Protein Data Bank.
-
The structure of the eukaryotic ribosome at 3.0 Å resolutionPublicly available at RCSB Protein Data Bank.
-
The structure of the eukaryotic ribosome at 3.0 Å resolutionPublicly available at RCSB Protein Data Bank.
-
The structure of the eukaryotic ribosome at 3.0 Å resolutionPublicly available at RCSB Protein Data Bank.
-
Structure of the human 40S ribosomal proteinsPublicly available at RCSB Protein Data Bank.
-
Structure of the human 60S ribosomal proteinsPublicly available at RCSB Protein Data Bank.
-
Structure of the H. sapiens 40S rRNA and E-tRNAPublicly available at RCSB Protein Data Bank.
-
Structure of the H. sapiens 60S rRNAPublicly available at RCSB Protein Data Bank.
References
-
A new system for naming ribosomal proteinsCurrent Opinion in Structural Biology 24:165–169.https://doi.org/10.1016/j.sbi.2014.01.002
-
Crystal structure of the eukaryotic ribosomeScience 330:1203–1209.https://doi.org/10.1126/science.1194294
-
MolProbity: all-atom structure validation for macromolecular crystallographyActa Crystallographica Section D, Biological Crystallography 66:12–21.https://doi.org/10.1107/S0907444909042073
-
In vitro studies on the sensitivity of local Entamoeba histolytica to anti-amoebic drugsThe Southeast Asian Journal of Tropical Medicine and Public Health 17:591–594.
-
Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplastAntimicrobial Agents and Chemotherapy 51:3485–3490.https://doi.org/10.1128/AAC.00527-07
-
An enzymatic electrocardiographic study on toxicity of dehydroemetineBritish Heart Journal 28:505–511.https://doi.org/10.1136/hrt.28.4.505
-
Features and development of CootActa Crystallographica Section D, Biological Crystallography 66:486–501.https://doi.org/10.1107/S0907444910007493
-
The puzzling lateral flexible stalk of the ribosomeBiology of the Cell 95:179–193.https://doi.org/10.1016/S0248-4900(03)00034-0
-
The effects of anti-bacterials on the malaria parasite Plasmodium falciparumMolecular and Biochemical Parasitology 152:181–191.https://doi.org/10.1016/j.molbiopara.2007.01.005
-
The chemotherapy of amoebiasis; introduction and methods of biological assayBritish Journal of Pharmacology and Chemotherapy 3:44–48.https://doi.org/10.1111/j.1476-5381.1948.tb00351.x
-
Expanding the antimalarial drug Arsenal-now, but how?Pharmaceuticals 4:681–712.https://doi.org/10.3390/ph4050681
-
Inhibitors of protein biosynthesis. V. Effects of emetine on protein and nucleic acid biosynthesis in HeLa cellsThe Journal of Biological Chemistry 243:4089–4094.
-
Protein translation in Plasmodium parasitesTrends in Parasitology 27:467–476.https://doi.org/10.1016/j.pt.2011.05.005
-
Crystal structure of the 80S yeast ribosomeCurrent Opinion in Structural Biology 22:759–767.https://doi.org/10.1016/j.sbi.2012.07.013
-
Quantifying the local resolution of cryo-EM density mapsNature Methods 11:63–65.https://doi.org/10.1038/nmeth.2727
-
Emetine resistance in chinese hamster ovary cells is associated with an altered ribosomal protein S14 mRNAProceedings of the National Academy of Sciences of the United States of America 79:1003–1007.https://doi.org/10.1073/pnas.79.4.1003
-
Accurate determination of local defocus and specimen tilt in electron microscopyJournal of Structural Biology 142:334–347.https://doi.org/10.1016/S1047-8477(03)00069-8
-
REFMAC5 for the refinement of macromolecular crystal structuresActa Crystallographica Section D, Biological Crystallography 67:355–367.https://doi.org/10.1107/S0907444911001314
-
Low-resolution refinement tools in REFMAC5Acta Crystallographica Section D, Biological Crystallography 68:404–417.https://doi.org/10.1107/S090744491105606X
-
Promising lead compounds for novel antiprotozoalsThe Journal of Antibiotics 63:381–384.https://doi.org/10.1038/ja.2010.50
-
UCSF Chimera–a visualization system for exploratory research and analysisJournal of Computational Chemistry 25:1605–1612.https://doi.org/10.1002/jcc.20084
-
RELION: implementation of a Bayesian approach to cryo-EM structure determinationJournal of Structural Biology 180:519–530.https://doi.org/10.1016/j.jsb.2012.09.006
-
Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EMNature Structural & Molecular Biology 11:957–962.https://doi.org/10.1038/nsmb822
Article and author information
Author details
Funding
National Health and Medical Research Council (APP1024678, APP1053801)
- Wilson Wong
- Melanie Condron
- Yan Hong Tan
- Jake Baum
Human Frontier Science Program (RGY007½011)
- Yan Hong Tan
- Jake Baum
Medical Research Council (MC_UP_A025_1013, MC_U105184332)
- Xiao-chen Bai
- Alan Brown
- Israel S Fernandez
- Sjors HW Scheres
OzeMalar
- Wilson Wong
Wellcome Trust (WT096570, 100993/Z/13/Z)
- Alan Brown
- Israel S Fernandez
- Jake Baum
Department of Industry, Innovation, Science, Research and Tertiary Education, Australian Government (FT100100112)
- Wilson Wong
- Jake Baum
Australian Research Council (FT100100112)
- Wilson Wong
- Jake Baum
Victorian State Government Operational Infrastructure Support
- Jake Baum
European Commission (EU FP7 Marie Curie)
- Xiao-chen Bai
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank S Ralph, B Sleebs, D Wilson, E Zuccala, G McFadden, A Cowman, J Rayner, A Ruecker, M Delves, R Sinden, S Chen, C Savva, J Grimmett, T Darling, G Murshudov, and P Emsley for helpful discussions and experimental assistance; and V Ramakrishnan for comments on the manuscript.
Copyright
© 2014, Wong et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 13,618
- views
-
- 1,456
- downloads
-
- 298
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 298
- citations for umbrella DOI https://doi.org/10.7554/eLife.03080
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine
eLife 3:e03080.
https://doi.org/10.7554/eLife.03080




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}