Transcription mediated insulation and interference direct gene cluster expression switches
- University of Oxford, United Kingdom
- European Molecular Biology Laboratory, Germany
Abstract
In yeast, many tandemly arranged genes show peak expression in different phases of the metabolic cycle (YMC) or in different carbon sources, indicative of regulation by a bi-modal switch, but it is not clear how these switches are controlled. Using native elongating transcript analysis (NET-seq), we show that transcription itself is a component of bi-modal switches, facilitating reciprocal expression in gene clusters. HMS2, encoding a growth-regulated transcription factor, switches between sense- or antisense-dominant states that also coordinate up- and down-regulation of transcription at neighbouring genes. Engineering HMS2 reveals alternative mono-, di- or tri-cistronic and antisense transcription units (TUs), using different promoter and terminator combinations, that underlie state-switching. Promoters or terminators are excluded from functional TUs by read-through transcriptional interference, while antisense TUs insulate downstream genes from interference. We propose that the balance of transcriptional insulation and interference at gene clusters facilitates gene expression switches during intracellular and extracellular environmental change.
https://doi.org/10.7554/eLife.03635.001eLife digest
A DNA double helix is made up of two DNA strands, which in turn are made of molecules that are each known by a single letter—A, T, C, or G. The sequence of these ‘letters’ in each DNA strand contains biological information.
Genes are sections of DNA that can be ‘expressed’ to produce proteins and RNA molecules. To express a gene, the DNA strands in the double helix must first be partially separated so that one of them can be used as a template to build an RNA molecule in a process called transcription. Either of the DNA strands in a helix can be used as an RNA template, but contain different genes and are read in opposite directions. One of the two strands is called the ‘sense’ strand, the other the ‘antisense’ strand.
The RNA molecule does not transcribe a whole DNA strand; instead, it transcribes a section of DNA, known as a transcription unit, which contains at least one gene. The end of a transcription unit is marked by certain signals that stop transcription. However, some transcription units in a DNA strand overlap, so there must be some way that the transcription machinery can sometimes ignore these stop signals.
The activity of some genes is linked to the activity of their immediate neighbours. Furthermore, some genes are expressed in different amounts in response to changes in environmental conditions. Researchers have previously suggested that there must be some form of switch that controls when these genes are expressed.
Nguyen et al. now engineer start and stop signals at a neighbouring pair of genes, called HMS2 and BAT2, in yeast. When one gene is switched on, the other is switched off and which gene is active depends on the diet of the yeast cells.
On the antisense DNA strand opposite to HMS2 is another gene, SUT650. Nguyen et al. show that when this gene is transcribed, the transcription of HMS2 on the other DNA strand is blocked. This has the knock-on effect of turning on BAT2. Conversely, transcribing HMS2 switches off SUT650 and BAT2 because the end of HMS2 overlaps with the beginning of both SUT650 and BAT2. Switching between different genes relies on loops that physically link the start and stop signals of the gene to be transcribed while ignoring the start and stop signals for neighbouring genes.
Proteins called transcription factors can bind to DNA and affect whether a gene is transcribed. Nguyen et al. found that a transcription factor that binds near the start of the HMS2 gene helps to control which DNA strand is transcribed. When transcription factors do not bind to the start of HMS2, antisense transcription—and the expression of SUT650—occurs instead.
Overall, Nguyen et al. show that the transcription process itself makes up part of a switch that can control the expression of several genes on both the sense and antisense strands of a DNA double helix. This may also explain how many other, more complex, gene networks are activated in response to changes in the environment.
https://doi.org/10.7554/eLife.03635.002Introduction
Genome-wide mapping of RNA transcripts in the budding yeast Saccharomyces cerevisiae has revealed an extensive array of coding and non-coding transcripts, giving rise to a genome that is heavily interleaved. Individual genes can possess multiple, overlapping transcripts, in the sense and the antisense orientations with respect to the pre-mRNA, as well as poly-cistronic transcripts, which span neighbouring genes (Kapranov et al., 2007; Berretta et al., 2008; Pelechano et al., 2013). Genome-wide mapping of nascent transcription, using techniques such as NET-seq, shows that transcription commonly extends into and over the intergenic regions of both convergent and tandemly arranged genes (Churchman and Weissman, 2011). For tandemly arranged genes, transcription into the promoter of the downstream gene would be expected to interfere with its expression by a variety of mechanisms, including modifying the local chromatin environment and interference by removal of transcription factors (Martens et al., 2004; Martianov et al., 2007; Hainer et al., 2011). Furthermore, extensive transcription antisense to (Venters and Pugh, 2009; Murray et al., 2012), and into the promoter of (Perocchi et al., 2007; Xu et al., 2009), the canonical coding transcript is also implicated in modulating gene expression by similar mechanisms (Hongay et al., 2006; Camblong et al., 2007; Uhler et al., 2007; Houseley et al., 2008; Pinskaya et al., 2009; Xu et al., 2011; van Werven et al., 2012; Castelnuovo et al., 2013). An interleaved genome with overlapping transcription units requires that polyadenylation and transcription termination signals in the sense and antisense orientations are by-passed but it is not clear how this is achieved. In addition, questions are commonly raised about whether transcription of these overlapping transcription units is contemporaneous.
It is now clear that much transcription is organised into biologically relevant temporal windows within phenomena such as the metabolic cycle (Tu and McKnight, 2006). Indeed, periodic or cycling expression of genes can be detected using fluorescent reporters or dual-labelled RNA FISH in cultures of asynchronous cells (Laxman et al., 2010; Silverman et al., 2010), or in the absence of cell division (Slavov et al., 2011). This periodic expression is a result of synchronization of respiratory and glycolytic activities into robust oscillations in oxygen consumption, characterized by phase-specific transcript signatures involving over 3000 genes, known as the Yeast Metabolic Cycle (YMC) (Klevecz et al., 2004; Tu et al., 2005, 2007; Soranzo et al., 2009; Slavov et al., 2011; Cai and Tu, 2012). In the long-period YMC, a single cell alternates between periods of high (oxidative (OX) phase) or low oxygen consumption (reductive building (RB) and charging (RC) phases), the residence time in each phase being nutrient-dependent. For exponentially growing cells in batch culture, the majority of cells in the population will be in the OX phase of the YMC (Slavov et al., 2011). Transcript levels for genes that cycle in the YMC will change as the cell moves through these phases; at any time, some cells in an asynchronous population will contain a transcript and some will not. These shifts through transcriptional states are robust but not invariable, as YMC-regulated genes switch on and off in response to cues prompted by both regulated and erratic changes in the intracellular and extracellular environment.
In addition to the partitioned gene expression patterns during the YMC, genomic spatial arrangements also contribute to regulatory relationships between genes, albeit on a smaller scale (Cohen et al., 2000; Lee and Sonnhammer, 2003; Batada et al., 2007). While genes displaying functional relatedness, such as the GAL genes, are regulated similarly by possessing the same cis-acting sequences (i.e. the UASGAL), co-expression of clustered genes in S. cerevisiae is largely independent of similarly controlled transcription, gene orientation, and/or shared regulatory sequences. However, members of adjacent gene pairs or clusters are more likely to belong to the same functional pathway than expected by chance (Cohen et al., 2000; Lee and Sonnhammer, 2003; Batada et al., 2007). The mechanism by which these clustered genes are regulated remains largely elusive.
Here we show that overlapping transcription, in both the sense and antisense orientations, constitutes an additional layer of regulation at clustered genes by managing state-switching in response to environmental change. We use a simple carbon source shift coupled with NET-seq to define genes whose transcription increases or decreases >threefold, after transfer from glucose (GLU)- to galactose (GAL)-containing media and show a remarkable enrichment for genes whose transcripts cycle during the YMC. The majority of these genes have no functional associations with transcription factors that might mediate repression or induction in GLU or GAL, but are organised in clusters and subject to overlapping sense and antisense transcription. We exemplify this mode of gene regulation, state-switching by transcriptional interference and insulation, at the HMS2:BAT2 tandem gene cluster. By engineering promoters and terminators at HMS2:BAT2, we demonstrate the formation of alternative transcription units underlies state-switching. We suggest that overlapping transcription and the formation of alternative transcription units, associated with temporally segregated gene expression, will be a general feature of gene regulation.
Results
Genome-wide response to a change in carbon source reveals genes whose transcripts cycle in the YMC
The question we address in this work is whether transcriptional interference can explain the switching on and off of genes and thus altered gene expression in response to environmental change. We used a change in carbon source by shifting exponentially growing cells from glucose- (GLU) to galactose-containing media (GAL) for 3 h and analysed the native transcripts associated with elongating RNA polymerase II (NET-seq) (Churchman and Weissman, 2011) to identify genes whose transcription is altered during this environmental change (Supplementary file 1A). 10.45% (551) of ORF-Ts (open reading frame—transcripts) showed a >threefold increase and 9.99% (527) showed a >threefold decrease in transcription in GAL relative to GLU (Supplementary file 1B). By comparing the NET-seq output with a microarray of Poly(A)+ RNA, we show that for the majority (≈88%) of genes, the change in transcript levels on the GLU to GAL shift reflects altered transcription rather than altered transcript stability (Supplementary file 1A). Gene ontology (GO) analysis revealed highly significant associations to growth, quiescence, and transcripts that cycle in the YMC, particularly during the oxidative (OX) and the reductive charging (RC) phases of the YMC (Figure 1A; Supplementary file 1C,D). The genes whose transcription decreases in GAL are enriched (467; 88.6% p < 1x10−5) for OX phase genes that are mainly involved in ribosome biosynthesis and growth (Figure 1B). By contrast, for genes whose transcription increases in GAL, there is significant enrichment (392; 71.1%, p < 1 × 10−5) for genes whose expression peaks in the reductive charging phase (RC) of the YMC (Figure 1B). These genes are associated with stress resistance, metabolism, and quiescence. 33.1% (891) of the 2691 annotated OX- and RC-regulated genes also show >threefold change on the GLU to GAL shift, supporting a shared regulatory mechanism between the YMC and the GLU to GAL shift (Figure 1D; Supplementary file 1C). In addition to ORF-Ts, many of the non-coding transcription events are regulated by the GLU to GAL shift. These include antisense transcription reads (to ORF-Ts), which increase in GAL compared to GLU (Figure 1E) and 25.7% of the 1772 annotated non-coding transcripts, the stable unannotated transcripts (SUTs), or cryptic unstable transcripts (CUTs), which show >threefold change in transcription on the GLU to GAL shift (Supplementary file 1E).
Figure 1 with 4 supplements see all

Reciprocal switching of expression by carbon source reveals links to the YMC.
(A) The distribution of all ORF-Ts into different YMC phases. OX, oxidative phase; RB, reductive building phase; RC, reductive charging phase of the YMC. (B, C, F) YMC profiles (SCEPTRANS; http://moment.utmb.edu/cgi-bin/main_cc.cgi) (Kudlicki et al., 2007) showing cycling expression at 124 genes with the (B) most reduced and (C) most increased transcription (NET-seq) after a switch from glucose (GLU) to galactose (GAL) for 3 h and (F) at the HMS2:BAT2 locus. (D) Overlap between all YMC classes and all ORF-Ts showing >threefold change in transcription on the GLU to GAL shift. (E) NET-seq reads on sense or antisense strands genome-wide in GLU (red) or GAL (blue) for ORF-Ts with peak expression in phases of the YMC indicated. (G) Strand-specific TIF-seq (Pelechano et al., 2013), microarray and NET-seq data at the HMS2:BAT2 locus. Profiles from cells cultured in GLU or GAL on the Watson strand (top) or Crick strand (bottom) are shown. The TIF-seq data indicate each different transcript isoform, microarray data indicate levels of steady-state poly(A)+ RNA (blue—darker colour for more RNA) and the NET-seq data (normalized, unique, and clipped to 3′OH) display transcript reads (scale 0–30) associated with elongating RNAPII. Screen shots are displayed using IGV (Robinson et al., 2011; Thorvaldsdottir et al., 2013).
Global analysis reveals distinct organisation and regulation
We used a computer simulation to obtain the expected number of occurrences of genome-wide features, by randomly shuffling genes, their orientation, YMC status, and expression levels (data from Supplementary file 1A, codes in Source code 1). Selected data from this analysis are shown in Supplementary file 1F. In summary, pairs of ORF-Ts in the convergent orientation occur more often than expected, while tandemly arranged ORF-Ts occur less often than expected. YMC genes are clustered, with 503 (16.9%) YMC genes flanked by non-cycling genes, 1431 (48.4%) with a single adjacent YMC partner either upstream or downstream and 1023 (34.5%) with a neighbouring cycling gene both upstream and downstream. Compared to more isolated genes, ORF-T clusters are more likely to show increased transcription in GLU or GAL than expected, suggesting that clustering is associated with some aspect of their regulation. In addition, ORF-Ts flanked by annotated SUTs or CUTs (in any orientation) occur more often than expected and show a twofold to fourfold higher median expression upon change from GLU to GAL than the average median expression obtained through the simulations. This is consistent with CUTs and SUTs playing a role in modulating ORF-T transcription, particularly on environmental change (Xu et al., 2011). Convergent overlapping arrangements occur three times more frequently than expected, whether the feature is a non-coding transcript (NC) such as a SUT or a CUT, or an ORF-T, with a median overlap of 92 bp for convergent ORF-Ts and 462 bp for ORF-Ts with non-coding transcripts. Thus clustering with overlapping transcription and transcriptional regulation are common features of the yeast genome.
As the genes whose transcription changed during the GLU/GAL shift are enriched for OX and RC YMC genes, we examined whether the nature of the flanking gene influences transcription, focusing on the tandem, and divergent combinations of these genes (Supplementary file 1F). OX genes, regardless of the nature (OX or RC) or orientation (divergent or tandem) of the upstream feature showed higher than expected transcriptional rates in GLU, but not in GAL, suggesting they are activated in GLU but not repressed in GAL. Similarly, RC genes are more likely to show higher than expected transcriptional rates in GAL. The behaviour of RC genes in GLU, however, does appear to be dependent on the orientation of the upstream OX gene. With a divergent upstream OX gene, the transcription of the downstream RC pair is not enriched or depleted in GLU. However, if the OX gene is in tandem, the RC gene is repressed in GLU. This raises the possibility that at the OX.RC tandem pair, transcription of the OX gene is mediating transcriptional interference and repressing the transcription of the RC gene in GLU. We note that the downstream gene in a tandem RC.RC pair also shows significant repression in glucose. To investigate further, we examined three different data sets: (i) three groups of tandemly arranged genes; OX.RC, RC.OX, and non-cycling (Supplementary file 1G); (ii) three sets of 20 consecutive ORF-Ts from Supplementary file 1A showing a sevenfold increase, no change or a sevenfold decrease on the GLU/GAL shift (Supplementary file 1H); and (iii) GLU/GAL-regulated genes with an annotated antisense CUT or SUT that is also GLU/GAL-regulated (Supplementary file 1I). From this analysis, we picked tandem gene clusters where the transcription of the target gene is regulated by GAL/GAL shift or not, and examined how neighbouring genes behave (Figure 1—figure supplements 1–4; Supplementary file 1H; Supplementary file 1J). The tandem partner of a GLU/GAL-regulated gene is often also regulated by the GLU/GAL shift and the YMC, while non-regulated genes are less likely to be in tandem and if they are, are surrounded by genes that are also not regulated by the GLU/GAL shift or the YMC. Common features of the regulated tandem clusters include reciprocal transcription in GLU and GAL, reciprocally cycling transcripts in the YMC, reciprocal AS transcription to OX genes, di-cistronic transcripts, and/or antisense transcripts spanning promoters that could mediate temporal transcriptional interference and thus facilitate cycling transcription. We chose the OX.RC pair HMS2:BAT2 for further study, as all the above characteristics are present and because the relatively abundant HMS2 antisense transcript, SUT650, can be detected experimentally in both GLU and GAL (Figure 1F,G).
Characterisation of transcripts and transcription at the HMS2:BAT2 locus
We examined the relationship between HMS2 and BAT2 (transcripts A and D respectively). BAT2 transcripts peak in the RC phase of the YMC and in GAL, reciprocal to HMS2 sense transcripts, which peak in the OX phase of the YMC and in GLU (Figure 1F,G and Figure 2—figure supplement 1A–C). Interestingly, there is also a reciprocal relationship between HMS2 and its antisense SUT650 (transcripts A and B respectively). Dual-labelled RNA FISH revealed a degree of temporal separation in the production of the HMS2 and SUT650 transcripts. In multiple analyses, no examples of FISH signals for both HMS2 and SUT650 at the site of transcription in the nucleus are observed in either GLU or GAL. Cells with nascent transcripts (arrows, Figure 2A) often have the opposite transcript type or both SUT650 and HMS2 transcripts in the cytoplasm, suggesting they are changing state. 35.5 ± 7.9% of the cells contained both transcripts in GLU, 22.8 ± 10.5% of all cells lacked both the SUT650 and HMS2 sense transcripts. The remaining cells contained either HMS2 or SUT650 transcripts but not both (Figure 2A). For cells containing a single type of transcript, more cells contained antisense transcripts in GAL than GLU while more cells contained sense transcripts in GLU than GAL (Figure 2B). Thus, the increase in levels of SUT650 transcripts in GAL reflects an increase in the number of cells expressing SUT650 and a concomitant decrease in the number of cells expressing HMS2, rather than a change in the levels of transcripts within a fixed number of cells in the population. We conclude that the expression of HMS2 and SUT650 is temporally separated, similar to the PHO84 sense and antisense transcripts (Castelnuovo et al., 2013). This suggests potential co-regulation between BAT2 and SUT650 and reciprocal regulation of HMS2 and BAT2.
Figure 2 with 1 supplement see all
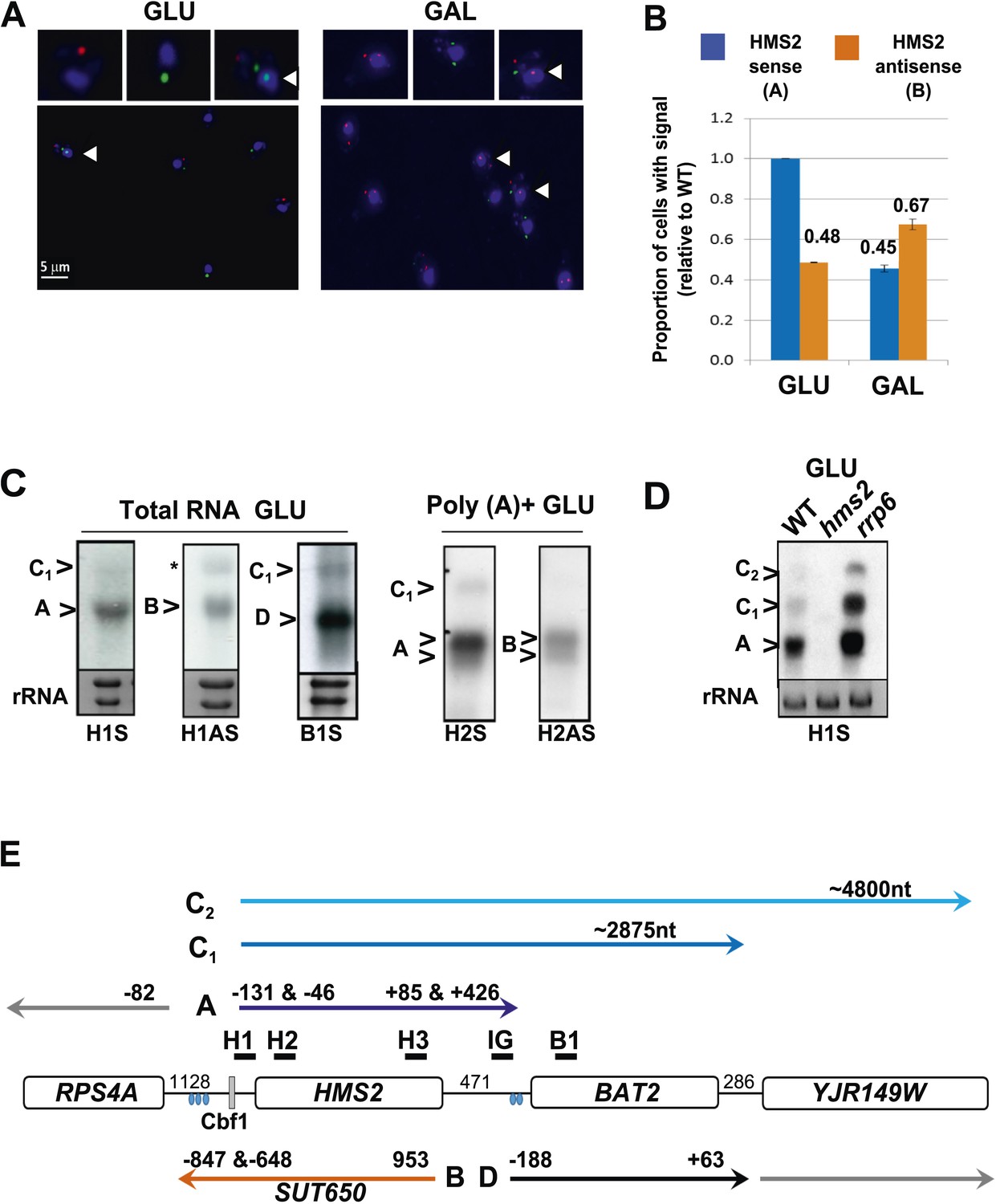
Characterization of transcripts around the HMS2:BAT2 locus (A).
Visualizing HMS2 transcripts using RNA fluorescence in situ hybridization (RNA FISH) in single cells using a combination of four, 50 nt DNA probes labelled with four fluorophores, either Cy5 (sense) or Cy3 (antisense), hybridised to paraformaldehyde-fixed yeast cells. DAPI (blue) marks the nucleus. Smaller boxes are zoomed images of selected cells in the field of view. Cells with nascent nuclear transcription event are marked with arrows. The images presented here are part of larger data sets. The graph (B) represents the proportion of sense- and antisense-expressing cells between growth conditions from different experiments. A total of ≈250 cells were assessed in each growth condition. The mean (MAX) signal from an hms2Δ strain prepared at the same time was used to threshold the signal intensity. Error bars are SEM, n = 2, Figure 2—source data 1A. (C) Northern blots of total and poly(A)+ selected RNA from WT cells probed with HMS2 (H) and BAT2 (B) sense (S)- and HMS2 antisense (AS)-specific probes (positions indicated in Figure 2E). Note the HMS2 antisense probe shows cross-hybridization with the 25S rRNA and is marked * in this and subsequent figures. Ethidium bromide-stained rRNA is used as a loading control. (D) Northern blot of total RNA showing HMS2 sense transcripts in strains indicated. (E) Map of transcripts (A to D) around the HMS2 locus showing approximate length, initiation sites, and termination sites relative to the ATG for each gene (see also Figure 2—figure supplement 1). The Cbf1 transcription factor binding site is shown (grey boxes). Different transcripts are coloured: blue for HMS2, orange for SUT650, black for BAT2, and grey for RPS4A and YJR149W. Transcripts are shown on the top or bottom of the schematic to reflect linked expression. Thick black lines indicate probe position for Northern blots, with the name of the probe above.
-
Figure 2—Source data 1
(A) Source data for Figure 2B, (B) Source data for Figure 2—Supplement 1B.
- https://doi.org/10.7554/eLife.03635.009
A number of different methods revealed heterogeneity at both the 5′ and 3′ ends of the HMS2 and SUT650 transcripts and longer RNA species (C1 and C2) on the HMS2 sense strand extending through the coding region of BAT2 and beyond, consistent with low-level read-through transcripts initiating at the HMS2 promoter (Figure 2C–E and Figure 2—figure supplement 1D,E). These read-through transcripts increase upon loss of nuclear exosome (rrp6Δ) (Figure 2D). Thus HMS2 is at the head of a contiguous gene cluster including the downstream genes BAT2 and YJR149W (Figure 2E). The 3′ UTR of the HMS2 transcript is of variable length. Polyadenylation (pA) occurs either 85 nt (proximal) or 426 nt (distal) downstream from the HMS2 stop codon. The distal pA site is 45 nt upstream of the BAT2 initiation codon, such that this longer transcription event is likely to interfere with initiation of BAT2 transcription. Additional transcriptional interference might be mediated through the production of the longer HMS2-BAT2 di-cistronic RNA species.
In summary, analysis of transcripts around HMS2 and BAT2 supports (i) an overlapping, reciprocally related and temporally segregated, sense–antisense pair (SAP) at HMS2, (ii) reciprocally expressed sense transcripts at HMS2 and BAT2, and (iii) low-level read-through di-cistronic transcripts (HMS2:BAT2 and SUT650:RPS4A). We ask if overlapping transcription in the sense and antisense orientations at HMS2 contributes to the reciprocal regulation of HMS2 and BAT2, by dissociating one transcription unit from the other.
HMS2 transcription interferes with BAT2 transcription
Insertion of the ADH1 transcription terminator (ADH1t) into the middle to HMS2, to separate HMS2 transcription from BAT2, allows us to test directly whether the HMS2 sense transcription over the HMS2:BAT2 intergenic region represses BAT2 transcription (Figure 3A). We confirmed that ADH1t efficiently terminates the HMS2 transcript (Figure 3B,C) and that there are no detectable transcripts over the HMS2 3′ region (Figure 3D). The strain with ADH1t inserted showed a ≈twofold increase in BAT2 transcript levels compared to WT in both GLU and GAL (Figure 3B,C) supporting a role for HMS2 sense transcription in repressing the BAT2 promoter. We conclude that HMS2 sense transcription interferes with BAT2 transcription.
Figure 3
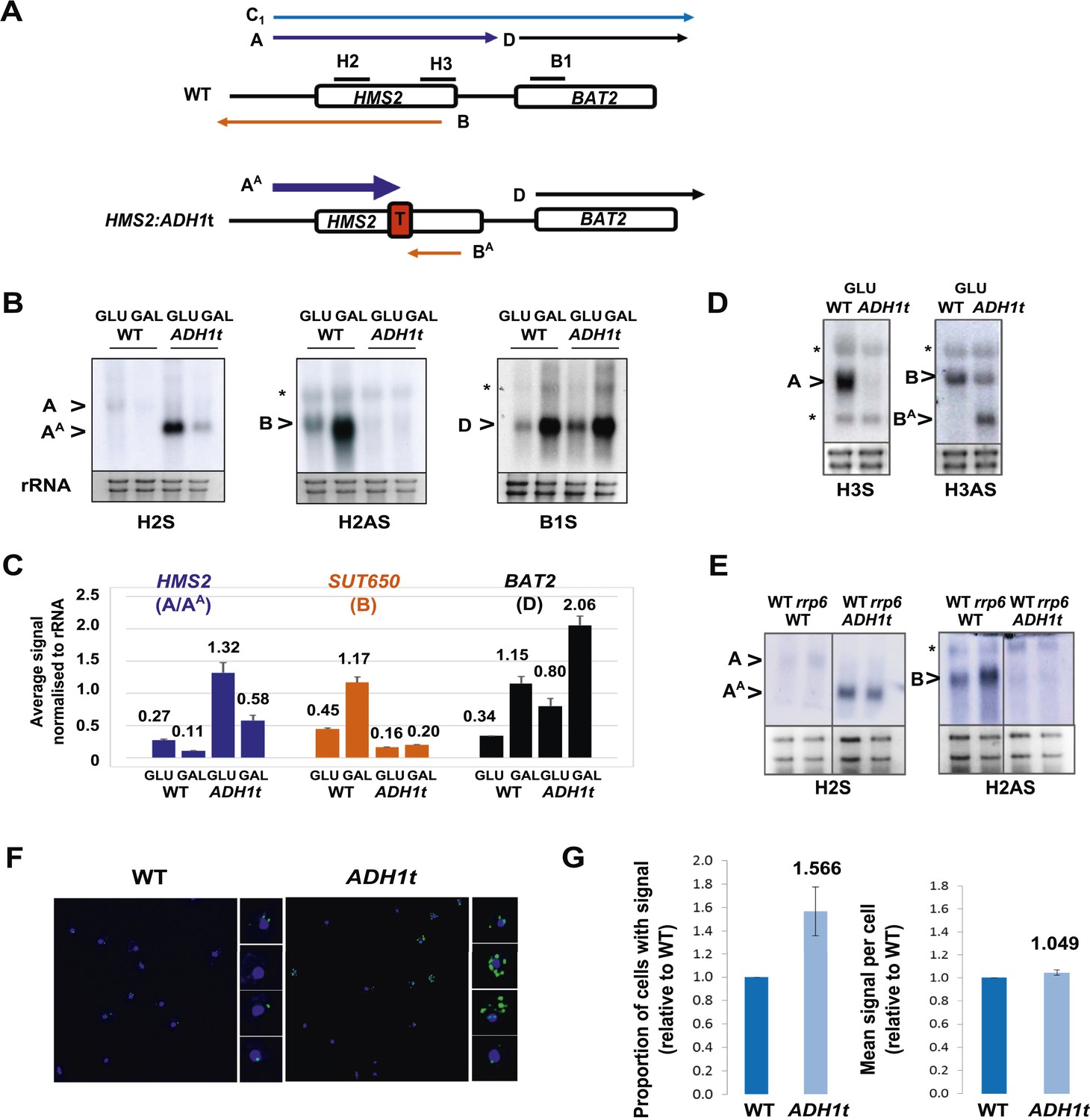
HMS2 mediates transcriptional interference of BAT2.
(A) Schematic showing constructs and transcripts at the WT HMS2:BAT2 locus and after insertion of the ADH1 terminator (T). (B) Exemplarily autoradiographs of Northern blots of total RNA prepared from the constructs in (A) cultured in glucose or after 60 min in galactose probed for the HMS2 sense, the SUT650 antisense, and the BAT2 sense transcripts. (C) Quantitation of autoradiographs showing average signal normalized to rRNA for the transcripts indicated. n = 2, errors are SEM, Figure 3—source data 1A. (D, E) Exemplarily autoradiographs of Northern blots of total RNA prepared from the strains indicated containing the constructs in (A) cultured in glucose probed for the regions indicated. (F) Visualizing HMS2 sense transcripts using fluorescence in situ hybridization (FISH) in single cells using a combination of four, 50 nt DNA probes labelled with four Cy5 fluorophores (green, sense), hybridised to paraformaldehyde-fixed yeast cells. The nucleus is shown in blue (DAPI). Smaller boxes are zoomed images of select cells in the field of view. The images presented here are part of a larger data set. (G) The graphs represent the proportion of sense-expressing cells in the WT compared to HMS2:ADH1t and the mean signal per cell. A total of ≈500 cells were assessed for each strain. Error bars are SEM; n = 4, Figure 3—source data 1A,C).
-
Figure 3—source data 1
- https://doi.org/10.7554/eLife.03635.012
SUT650 transcription limits the number of cells expressing HMS2
Insertion of the ADH1t into the HMS2 coding region resulted in a ≈fourfold increase in levels of a truncated HMS2 sense transcript (AA) (Figure 3A,B,D) and blocks SUT650 antisense transcription, resulting in the truncated antisense transcript (BA) (Figure 3A,D). We confirm there are no antisense transcripts over the 5′ region of HMS2 (Figure 3B,E). In the absence of SUT650, expression of the HMS2:ADH1t transcript remains sensitive to the change in carbon source, showing a similar decrease to full-length HMS2 1 h after transfer from GLU to GAL (Figure 3B,C). This suggests that the carbon source responsive signal is received at the HMS2 promoter and that reciprocal switching of HMS2 and SUT650 is a function of sense transcription. However, as the levels of HMS2 sense transcripts are higher without the antisense transcript, we asked if antisense transcription plays a role in limiting the number of cells containing sense transcript using RNA FISH (Figure 3F). When grown in glucose, optimal conditions for HMS2 sense transcription, we observed an increase in the number of HMS2:ADH1t cells containing sense transcript compared to WT (Figure 3G), consistent with a role for antisense transcription in reducing the number of sense transcription initiation events, leading to a decrease in the number of cells in the population that contain the sense transcript. We conclude that SUT650 antisense transcription does not influence the environmental responsiveness of HMS2 transcription but acts to limit the number of cells in the population that respond.
SUT650 antisense transcription insulates BAT2 from interference by HMS2
Next, we asked whether transcription of SUT650 antisense might prevent HMS2 sense transcription from interfering with BAT2, thus insulating BAT2. To ablate SUT650 whilst ensuring that the regulatory elements in the flanking sequences between HMS2 and BAT2 remained intact, we removed the transcription start sites (TSS) for SUT650 located between +953 and +1038 relative to the HMS2 ATG, near the end of the HMS2 coding region. We chose to do this by substituting the complete coding region of HMS2 with that of URA3, to ensure a natural ORF between the HMS2 promoter and terminator, rather than mutating the regions containing multiple TSSs for SUT650 within the ORF (Figure 4A). We note that insertion of the URA3 ORF could affect transcript stability, although the nature of the promoter may be the primary determinant of transcript stability (Bregman et al., 2011; Trcek et al., 2011), so we focused on transcript size and relative abundance. We observed two clear effects. First, loss of the antisense transcript (B), which could be explained either by introduction of a unidirectional terminator in the URA3 ORF coding region or loss of TSSs for SUT650 (Figure 4B–D and Figure 4—figure supplement 1). Second, an increase in the read-through transcripts (C1U) relative to the HMS2:URA3 sense transcript (AU) (Figure 4B–D). We confirmed these were read-through transcripts by inserting a terminator (T) at the 3′ region of the HMS2:URA3 hybrid gene (Figure 4A) and observing loss of the read-through transcripts and increased levels of a shorter transcript (AUT) (Figure 4E,F). We note that transcripts initiated at the HMS2 promoter remain sensitive to a change in carbon source in the absence of antisense transcription (Figure 4D), as we observed using HMS2:ADH1t (see Figure 3) and consistent with a regulatory input at the promoter. The increase in the read-through transcript (C1U) relative to the HMS2:URA3 sense transcript (AU) in the absence of antisense transcription suggests a role for antisense transcription in attenuating sense transcription and preventing interference at the BAT2 promoter. Consistent with this, a twofold decrease in BAT2 sense transcript is observed in the antisense-less HMS2:URA3 strain relative to WT in GLU, concomitant with a threefold increase in AU and C1U (Figure 4B,C). Taken together, the effect of blocking (Figure 3) or increasing (Figure 4) HMS2 transcription on levels of BAT2 transcript supports transcriptional interference by HMS2 at the BAT2 promoter and a reciprocal relationship between HMS2 and BAT2. We sought evidence for transcriptional interference resulting from increased levels of AU and C1,2U. Chromatin immunoprecipitation (ChIP) analysis at the HMS2:BAT2 intergenic region reveals reduced initiation-related modifications (H3K4me3 and H3K56ac) and increased elongation-related modifications (H3K79me3 and H3K36me3) in the HMS2:URA3 hybrid gene, consistent with transcriptional interference leading to repression of BAT2 transcription initiation (Figure 4G). We conclude that SUT650 antisense transcription (i) changes the proportion of cells expressing HMS2 and (ii) controls the amount of overlapping and read-through transcription from HMS2 and thus levels of BAT2 transcript. We envisage that at any one time in a single cell, formation of the HMS2 and SUT650 transcription units (TUs) is mutually exclusive, but the formation of the SUT650 TU and the BAT2 TU is not. Thus formation of the SUT650 transcription unit insulates BAT2 from interference by HMS2 transcription, as the HMS2 TU cannot form when SUT650 is expressed.
Figure 4 with 1 supplement see all
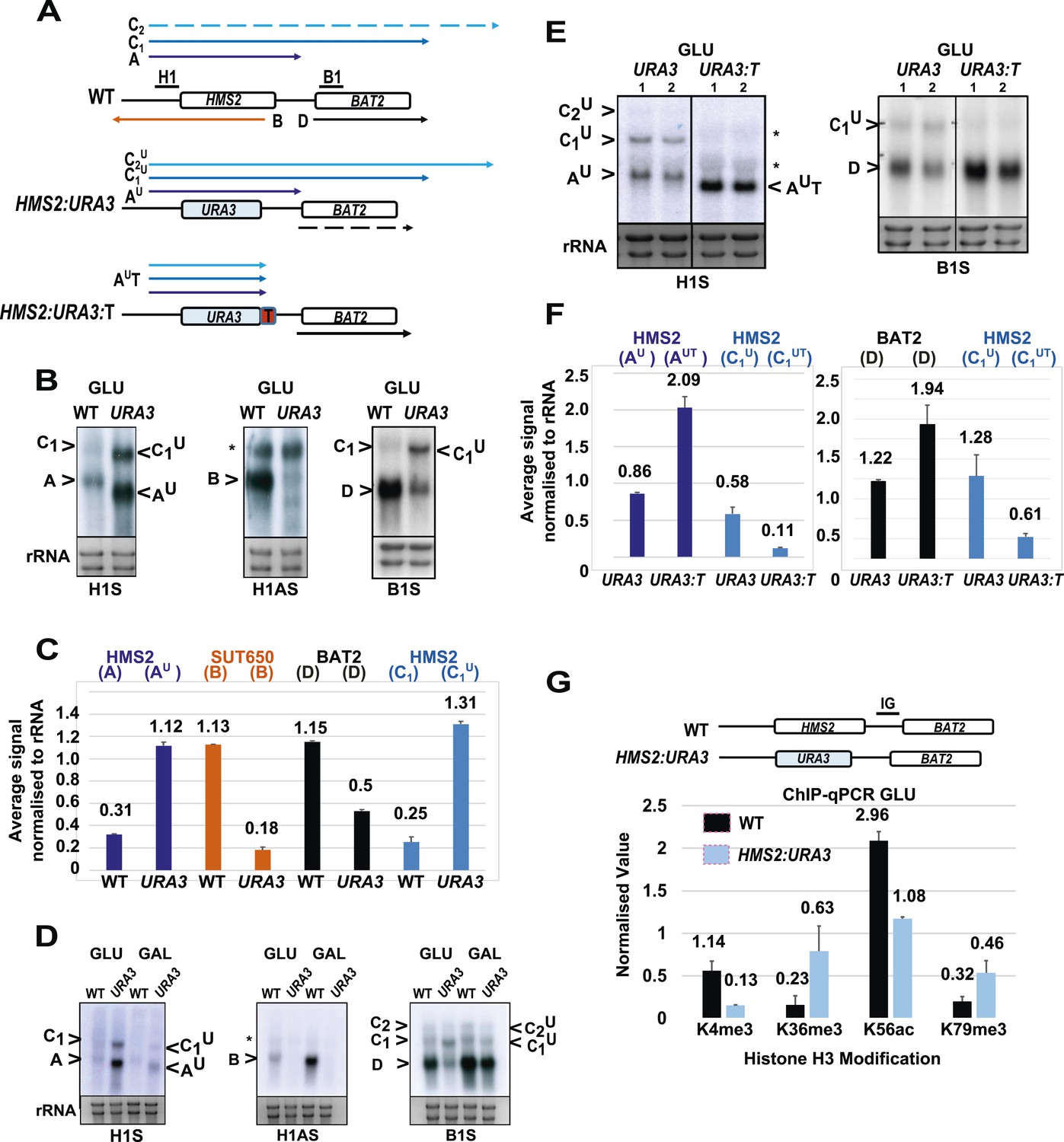
SUT650 antisense transcription insulates BAT2 from interference by HMS2.
(A) Schematic showing transcripts after replacement of the HMS2 coding region (top) with the URA3 coding region (middle) or URA3 plus a transcription terminator (T) (bottom). Transcripts resulting from the URA3 insertions are identified with a superscript U or UT. (B, D, E) Northern blots of total RNA in strains with the HMS2 coding region replaced with the URA3 coding region in glucose (B), after 1 h in galactose (D) or in glucose with a terminator (T) inserted after URA3 (E). In (E), samples were run on the same gel but intervening tracks removed. (C) Quantitation of transcripts for WT and HMS2:URA3 in glucose, n = 2, errors are SEM, Figure 4—source data 1A. (F) Quantitation of transcripts in (E) for HMS2:URA3 and HMS:URA3:T in glucose, n = 2, errors are SEM, Figure 4—source data 1B. (G) Chromatin immunoprecipitation (ChIP-qPCR) at the HMS2:BAT2 intergenic region (IG) in strains indicated using antibodies with the specificities indicated. Signals were normalized to Histone H3 and then the signal in the coding region of TUB2. Error bars are SEM for n = 2; Figure 4—source data 2.
-
Figure 4—source data 1
- https://doi.org/10.7554/eLife.03635.014
-
Figure 4—source data 2
Source data for Figure 4G.
- https://doi.org/10.7554/eLife.03635.015
HMS2 transcription represses the SUT650 antisense transcript and BAT2
To address whether HMS2 transcription influences SUT650 antisense transcription, we replaced the HMS2 promoter with the inducible GAL1 promoter (pGAL1) (Figure 5A). When cultured in glucose, the GAL1 promoter is repressed but becomes induced about 2 h after transfer to galactose (Figure 5B). SUT650 is normally induced during the GLU-GAL shift (see Figure 1). However in the pGAL1:HMS2 strain, an antisense transcript (BG) is evident in GLU and remains until the sense transcript is induced by GAL (Figure 5C,D). This suggests that sense transcription represses the antisense transcript. We used the inducible pGAL1:HMS2 system in conjunction with the Anchor Away technique (Haruki et al., 2008) to deplete the nucleus of the Gal4p activator during growth in GAL. Removal of Gal4p by treatment with rapamycin (Rap) for 1 h, after activation of pGAL1:HMS2 for 3 h with GAL, abolishes the HMS2 transcript (AG) and leads to restoration of the antisense transcript (BG) (Figure 5E,F). This confirms that the reduction in the antisense transcript is a consequence of sense transcription, rather than the change in condition (GLU to GAL). To determine whether antisense transcription had any effect on pGAL1:HMS2 sense transcript induction rates, the antisense--less HMS2:URA3 construct was placed under the control of the GAL1 promoter (Figure 5A,B). The sense transcript profile in poly(A)+-enriched RNA was examined using a probe for the HMS2:BAT2 intergenic region (probe IG), a region transcribed in both constructs. No changes in induction rate were apparent, with the sense transcript only detectable after 2 h in both strains, signifying that antisense transcription does not impair sense activation kinetics (Figure 5B). Rather, this finding is compatible with our observation that without antisense, more cells would express the sense transcript (see Figure 3F,G). Activation of the pGAL1:HMS2 sense transcript results in up to 3.5-fold repression of BAT2 (Figure 5F), consistent with pGAL1:HMS2 sense polyadenylation at 426 nt, just upstream of the BAT2 ATG, interfering with BAT2 transcription initiation in galactose (see Figure 2—figure supplement 1). We conclude that formation of the HMS2 sense transcription unit represses the formation of the SUT650 and BAT2 transcription units, regardless of conditions.
Figure 5 with 1 supplement see all
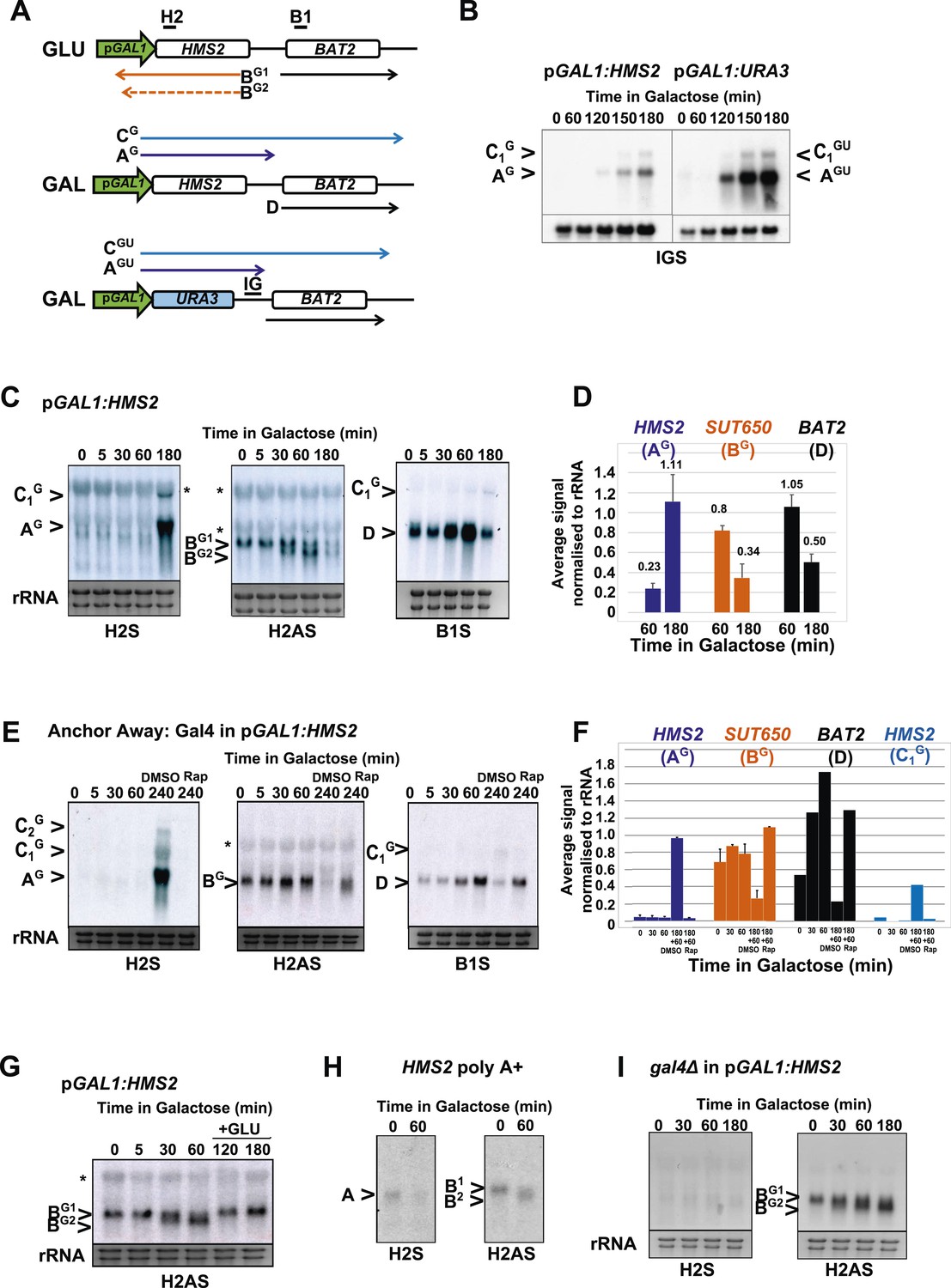
HMS2 sense transcription represses the HMS2 antisense transcript and BAT2.
(A) Schematic showing transcripts when expression of HMS2 is regulated by the GAL1 promoter (pGAL1) in glucose (antisense-dominant state) or galactose (sense-dominant state) or when the HMS2 coding region is substituted with the URA3 coding region. (B, C, E, G–I) Northern blots showing total RNA (C, E, G, I) or poly(A)+ RNA (B, H) from cells expressing pGAL1:HMS2 cultured in glucose (GLU) or induced for the times indicated in galactose (GAL) in the genetic backgrounds indicated. (D, F) Quantitation of the experiments exemplified in (C) and (E) respectively, n = 2, error are SEM, Figure 5—source data 1A,B,C. (E, F) Anchor Away of Gal4 is achieved after incubation with rapamycin in DMSO for 1 h, added 3 h after induction with galactose (240 Rap). The control culture (240 DMSO) was treated with DMSO. See also Figure 5—figure supplement 1.
-
Figure 5—source data 1
- https://doi.org/10.7554/eLife.03635.018
Antisense transcript isoforms are condition-specific and inherent to HMS2
There are two species of antisense transcript during induction of pGAL1:HMS2; a long species (BG1), dominant until 30 min after the shift to GAL, and a shorter species (BG2), also detectable after 30 min of growth in GAL and which becomes the dominant antisense transcript form after 1 h (Figure 5C). 3′end RACE revealed alternative polyadenylation sites as one cause for the observed size difference, with the long form terminating upstream of the Gal4p binding sites at approx. 500 nt upstream of the ATG in the GAL1 promoter and the shorter form terminating midway through the GAL1 promoter, 195 nt upstream of the GAL1 ATG, and downstream of the Gal4p binding sites (Figure 5—figure supplement 1). Upon glucose repression of induced pGAL1:HMS2, levels of the long form of antisense transcript were recovered (Figure 5G). Antisense transcript shortening after transfer to GAL is an inherent property of the natural SUT650 antisense transcript (Figure 5H). To ask if this was a function of transcription factor binding to the promoter, we used the pGAL1:HMS2 construct and showed that shortening of the antisense transcript is independent of Gal4 binding at pGAL1 and remains for 3 h in GAL in the absence of sense transcription (Figure 5I). This rules out a potential roadblock by Gal4 as an explanation for the use of different antisense polyadenylation/termination sites in pGAL1. We conclude that SUT650 antisense transcription is normally repressed by HMS2 transcription but remains stably expressed in GLU or GAL, varying in the use of polyadenylation sites, when sense transcription is inhibited.
Transcription factors contribute to sense/antisense switching at HMS2:SUT650
To see if loss of transcription factors associated with the native HMS2 gene, Cbf1 (MacIsaac et al., 2006), and Fkh1 (Venters et al., 2011), influence the balance of sense and antisense transcription at HMS2, we used cbf1Δ, fkh1Δ, and fkh2Δ strains, regulators of nutrient availability, oxidative growth, and the cell cycle (Mitchell and Magasanik, 1984; Mellor et al., 1990; Kent et al., 1994; Zhu et al., 2000; Morillon et al., 2003; Tsankov et al., 2011). While under standard growth conditions in GLU these mutant strains showed normal expression of HMS2 and SUT650, growth in nutrient-limited GLU-based medium depleted for tryptophan resulted in loss of the HMS2 transcript and accumulation of the short isoform of the SUT650 antisense transcript (B2) in the cbf1Δ and fkh1Δ strains (Figure 6A). In contrast, the fkh2Δ strain showed reduced levels of SUT650 and increased HMS2 under these growth conditions. This supports transcription factors controlling the balance of sense, and thus antisense transcription, at this locus during the cell cycle (Fkh factors) (Granovskaia et al., 2010) and during oxidative growth in metabolic cycles (Cbf1) (Tsankov et al., 2011). We ablated a number of non-essential transcription factors, including Gln3, with putative binding sites at the 3′ region of HMS2 (MacIsaac et al., 2006) but observed no significant change in levels of SUT650, HMS2, or BAT2 transcripts with the exception of a small increase in the HMS2:BAT2 read-through transcript (C) in a gln3Δ strain in minimal medium (Figure 6—figure supplement 1). We propose that transcription factors signal to the HMS2 promoter (Input) to mediate state-changing (antisense-dominant to sense-dominant) in response to environmental conditions (Figure 6B). As we could find no direct evidence for regulation of SUT650 either by TFs (Figure 6 and Figure 6—figure supplement 1) or by the GLU to GAL shift (see Figure 5) but only by transcriptional interference resulting from activation of HMS2 transcription, we examined OX and RC genes genome-wide to look for common themes in their regulation. We asked what factors are significantly enriched (p < 0.01) at promoters of RC and OX genes, using a large data set of 202 factors (Venters et al., 2011). We observed marked differences (Supplementary file 1K). Of particular interest are the predominance of chromatin modulators and components at RC gene promoters, suggesting that RC promoters are particularly sensitive to chromatin-mediated repression (Tirosh and Barkai, 2008). These features, coupled with the distinct behaviour of OX and RC genes in GLU and GAL, particularly OX.RC genes in tandem (Supplementary file 1F), lead us to propose chromatin-mediated repression of RC genes by transcriptional interference. In addition, antisense transcription into OX promoters, for example by SUT650 at HMS2, may also limit OX gene transcription.
Figure 6 with 1 supplement see all
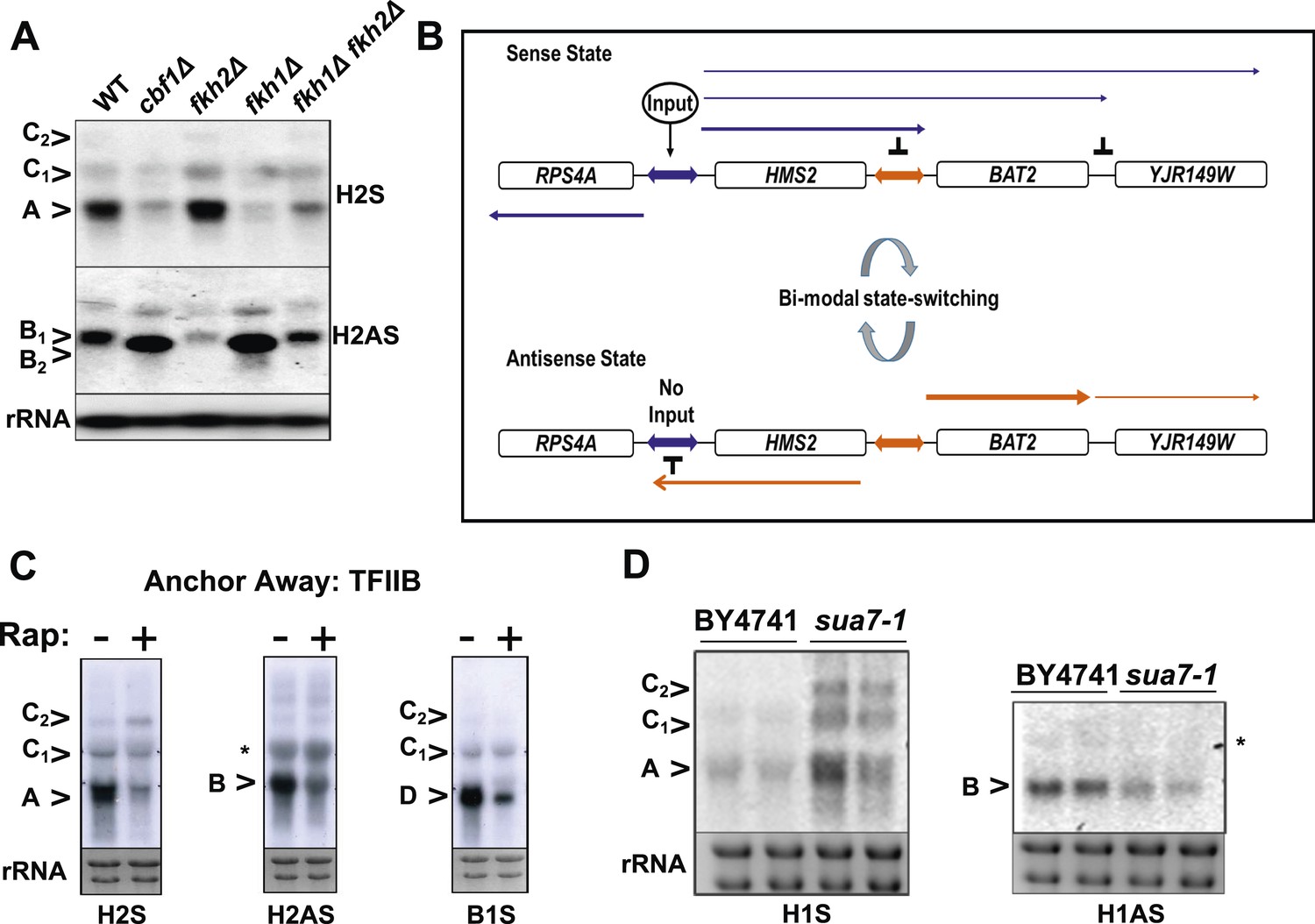
General and specific transcription factors control state switching at HMS2.
(A, C, D) Northern blots showing sense or antisense-dominant state at HMS2 in strains lacking general or specific transcription factors. (A) Strains were cultured in YPD depleted for tryptophan. (B) Model for state-switching between a sense-dominant state and an antisense-dominant state by transcription factors (Input) at the divergent promoters between RPS4A and HMS2 (blue double arrow) and HMS2 and BAT2 (orange double arrow). During growth in glucose (high input), cells cycle between the sense-dominant state and the antisense-dominant state with the majority of cells existing in the sense-dominant state. During growth in galactose (low input), cells cycle between the sense-dominant state and the antisense-dominant state with the majority of cells existing in the antisense-dominant state. (C) Anchor Away of TFIIB (Sua7) is achieved after incubation with rapamycin (Rap) (+) in DMSO for 1 h or DMSO alone (−). (D) Biological replicates for the WT strain BY4741 and the isogenic strain expressing the sua7-1 allele are shown.
TFIIB (Sua7) is required to limit the sense-dominant state at HMS2
We assessed a role for a number of essential transcription factors using the Anchor Away technique (Haruki et al., 2008) but only upon nuclear depletion of TFIIB (Sua7) did we observe an increase in the HMS2:BAT2 read-through transcript (C) (Figure 6C). However, this occurred together with reduced levels of HMS2 (A), BAT2 (D), and SUT650 (B) transcripts, as expected upon depletion of an essential transcription factor. To explore this further, we used a well-characterised allele of SUA7 (sua7-1) (E62K) (Pinto et al., 1994), associated specifically with the 3′ region of genes, polyadenylation/transcription termination and higher order structures (gene loops) in chromatin (Medler et al., 2011). Remarkably, we observed an increase in HMS2 transcript (A) and sense read-through transcripts (C), coupled with a decrease in the SUT650 antisense transcript (B) (Figure 6D). Thus, TFIIB may have a specific role in determining the antisense transcription unit by establishing directionality of transcription over the region (Tan-Wong et al., 2012). For instance, by determining an antisense transcription unit, TFIIB aids in sense transcription termination, preventing ‘read-through’ over the intergenic region between HMS2 and BAT2. The capacity to read through transcription regulatory regions such as promoters and terminators is an essential component of a model for gene regulation by transcriptional interference such as that developed for the HMS2:BAT2 locus. To test this experimentally, we inserted a new transcription unit into HMS2 to disrupt the HMS2 and SUT650 transcription units and asked what mutations would restore transcription between the beginning and end of these TUs.
Engineering HMS2 reveals the balance between sense and antisense states and demonstrates the plasticity of transcription units
The HMS2 locus was engineered by insertion of the pTEF.KanMX.TEFt expression cassette (Longtine et al., 1998) at position +650 bp (Figure 7A). In this construct (HMS2:TEF:Kan), there are two promoters in tandem (HMS2 and TEF) and two terminators in tandem (TEF and HMS2). This is sufficient to disrupt the HMS2 and SUT650 transcription units. The SUT650 antisense promoter is functional and produces a truncated antisense transcript (BK) that ends in the vicinity of TEFt (Figure 7A,B). Thus TEFt functions bi-directionally to terminate SUT650 (BK), the sense transcript from pTEF, expressing the kanamycin resistance (KanMX) transcript (K), and the sense transcript (AK) initiated at the HMS2 promoter and terminating at TEFt (Figure 7A,B). pTEF is a well-characterised (Steiner and Philippsen, 1994) strong bi-directional promoter and produces an antisense transcript (E) extending into pHMS2. To confirm this, we inserted ADH1t directly upstream of pTEF (Figure 7A). This resulted in increased levels of a truncated sense transcript (AA) and considerably reduced levels of antisense transcript (E) (Figure 7B).
Figure 7 with 1 supplement see all
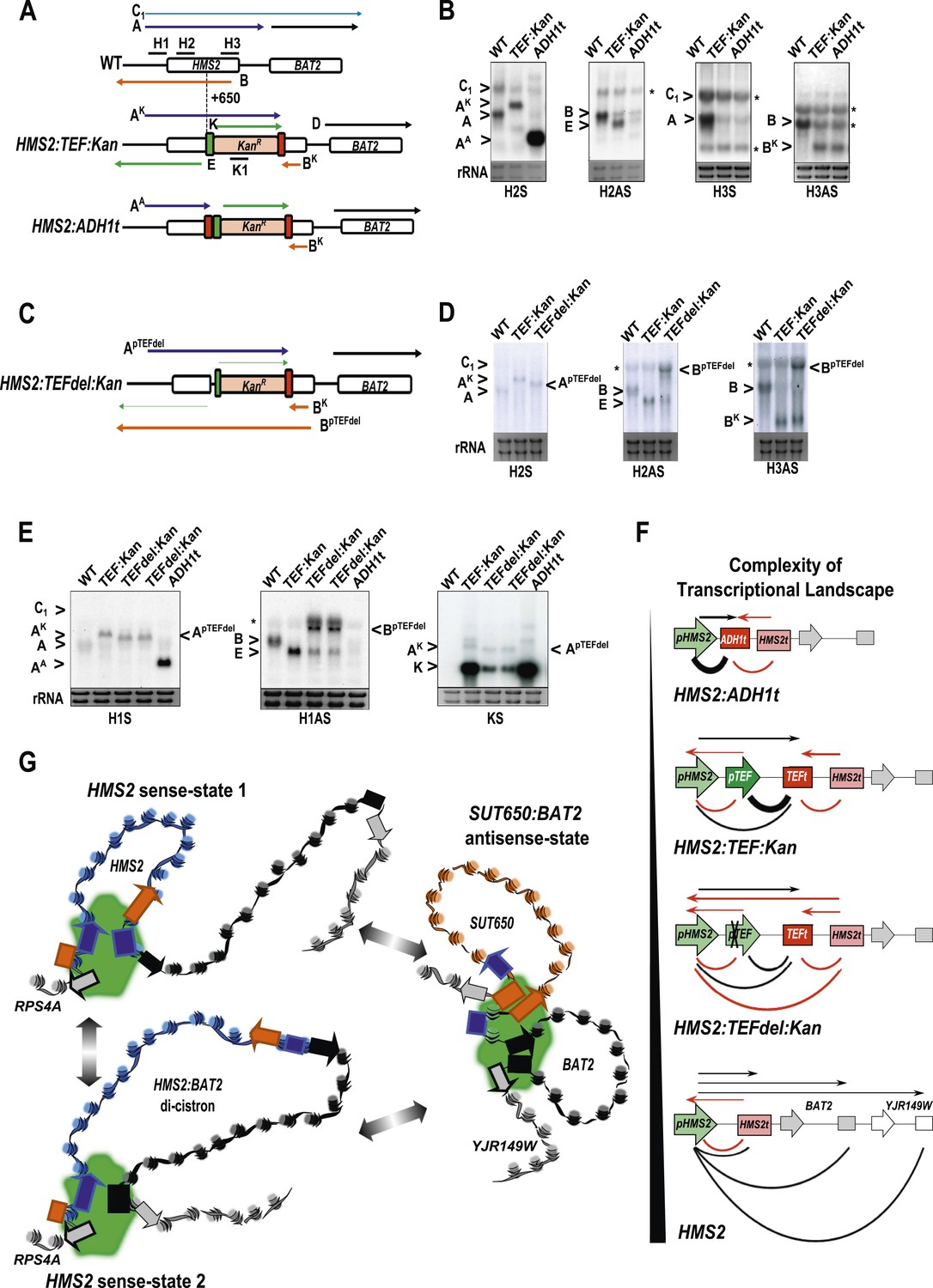
The plasticity of transcription units.
(A, C) Schematic of constructs showing transcripts and position of probes. Transcripts resulting from the pTEF:kanMX:TEFt insertion, the same construct with the ADH1 terminator (pink box), or the TEF:kanMX:TEF insertion with a deleted TEF promoter (p) are identified with a superscript K, A, or pTEFdel, respectively (see Figure 7—figure supplement 1 for more details). (B, D, E) Northern blots in the strains indicated showing the conditional nature of the TEF terminator (t) when the pTEF is compromised. (F) Modelling transcription in an interleaved genome demonstrating an increase in complexity of the transcriptional landscape as the strength of terminators and promoters decreases. Constructs representing each state are indicated; grey and white boxes are downstream TUs. Solid directional boxes are promoters; squares are terminators. Deeper colour indicates increased strength. Arrows on top of diagrams represent the transcripts initiating or terminating at pHMS2 and HMS2t (black sense; red antisense). Curved lines under diagrams show the extent of each individual transcription unit. Transcription units are envisaged as dynamic with any one region only involved in one TU at any time in an individual cell (see Figure 7G). Weakening of pTEF in the HMS2:TEF:Kan construct results in a level of transcription complexity (number of different TUs) similar to the native HMS2 locus. (G) Schematic showing the dynamic alternative transcription units at the HMS2:SUT650:BAT2 locus. Directional boxes are promoters, squares are terminators coloured coded according to the nature of the TU to which they belong (blue, HMS2; black, BAT2; Orange, SUT650; Grey, RPS4A and YJR149W). HMS2 forms alternative TUs with its own terminator (sense-state 1) or with the BAT2 terminator (sense-state 2—a di-cistronic transcription unit) excluding the SUT650, BAT2, and YJR149W promoters from a region where transcription can occur (green shading). RPS4A, an OX gene expressed divergently from HMS2 is active. The sense state predominates in the OX phase of the YMC and during growth on GLU. Any promoter or terminator excluded from the transcription machinery is neutral to the transcription process, explaining how both promoters and terminators are subject to extensive read-through transcription. The sense-state toggles to the alternative antisense-state, predominant in GAL and during the RC phase of the YMC. Here, the formation of the SUT650 transcription unit excludes the HMS2 and RPS4A promoters from the transcription machinery. This relieves interference of BAT2 and YJR149W by HMS2 sense transcription. One transcription unit is envisaged to exist in one cell at any one time. The antisense state is the default, requiring transcription factor-dependent activation at HMS2 to switch to the sense state.
We reasoned that by introducing mutations into the TEF promoter, we would be able to ask how disabling pTEF affects the HMS2 and SUT650 transcription units. Using the HMS2:TEF:Kan construct as a starting point, mutations were introduced into the TEF promoter, including deletions of putative transcription factor binding sites (Figure 7C and Figure 7—figure supplement 1). A long deletion (HMS2.TEFdel1:Kan), removing all the TEF promoter sequences upstream of the TATA box, results in a significant reduction in expression of the KanMX cassette itself (transcript K) and in levels of the antisense transcript (E) (Figure 7D,E and Figure 7—figure supplement 1). Remarkably, in this strain, we observed a new antisense transcript from the 3′ end of HMS2 that reads through TEFt (BpTEFdel) (Figure 7C–E). We confirmed that the BpTEFdel transcript spans the 3′ region of HMS2 using the H3AS probe (Figure 7D). Thus, the disabled weak pTEFdel1 resulted in reduced functionality at TEFt allowing BpTEFdel, initiating at the SUT650 antisense promoter, to read through TEFt, the KanMX cassette and into the HMS2 promoter (Figure 7C–E). This experiment illustrates (i) the conditional nature of the bi-directional TEF terminator and (ii) the remarkable plasticity of interleaved transcription units. We suggest that the pTEF.KanMX.TEFt expression cassette is strongly expressed (compare levels of transcripts AK and K in Figure 7E), effectively preventing the formation of the HMS2:SUT650 transcription units. When the TEF promoter is disabled, reducing expression of the pTEF.KanMX.TEFt expression cassette, the SUT650 antisense transcription unit can form, resulting in the appearance of the long antisense transcript. Alternatively, reduced KanMX sense transcription decreases the ability of a terminator to stop antisense transcription from the SUT650 antisense promoter. This illustrates the complex relationships that control levels of expression in the interleaved yeast genome (Figure 7F).
Discussion
We propose a two-state model for transcription at HMS2 and BAT2 that combines the effects of temporal separation, overlapping interfering transcription able to bypass typical termination and promoter signals, and insulation from interference by the formation of an antisense transcription unit (Figure 7G). There are two states: a sense-dominant state (blue transcripts) and an antisense-dominant state (orange transcripts) with respect to HMS2 (see Figures 6B and 7G). Transcription factors (Input) at the HMS2 promoter regulate sense transcription and loss of input results in the antisense-dominant state. SUT650 transcription insulates BAT2 from interference by HMS2, reinforcing the antisense dominant state-switch. We propose that transcription in the HMS2 sense orientation transmits the state-defining regulatory signals to neighbouring genes through mechanisms of cis-acting transcriptional interference, thus repressing BAT2, YJR149W, and SUT650. By these means, reciprocal regulation in cycles or on environmental change could be achieved.
More generally, how far a transcription event extends into the regulatory sequences of its SAP, or the regulatory sequences of an upstream or downstream gene may determine whether that region is competent to make a state-switch or not and respond to environmental or metabolic cues. Our genome-wide analysis supports many more examples of the type of organisation we observe at HMS2:BAT2. Moreover, the HMS2:BAT2 cluster is conserved in a range of yeast strains expected of a functional linkage. There are many loci at which di-cistronic read-through transcripts are evident (Pelechano et al., 2013), raising the possibility that transcriptional interference leading to reciprocal expression of genes in tandem arrays is a much more widespread phenomenon in yeast.
A di-cistronic transcript means that RNA polymerase has to read through the region determining polyadenylation and termination (the terminator). Moreover, antisense transcription arising from divergent promoters may have to bypass the terminator of the upstream gene. This work indicates the capacity of a terminator to function is conditional on the strength of the proximal promoter. We suggest that this reflects the role for promoters in defining the beginnings and ends of TUs (El Kaderi et al., 2009; O'Sullivan et al., 2004); stronger promoters preferentially selecting the proximal terminator.
The role of Sua7 in preventing read-through transcription is intriguing given its association with the 3′ region of genes, polyadenylation/transcription termination, and higher order structures in chromatin (Medler et al., 2011). This raises the possibility of an antisense transcription unit-specific function for TFIIB at the 3′ ends of genes, characterised here using the sua7-1 allele. A simple scenario in which 3′end TFIIB directs transcription by promoting both antisense transcription and physically terminating sense transcription is compatible with the interference/insulation model proposed here. It is also plausible that alternative higher order structures linking the sense or antisense promoters to one of a possible number of terminators could also explain dynamic state-switching between sense and antisense or multi-cistronic transcripts (Murray et al., 2012) (Figure 7G). Only one structure would exist in each cell at any one time leading to temporal separation of the different TUs. By this model only the juxtaposed regions would function in transcription initiation and termination, allowing other promoters or terminators between these regions to be ignored (Figure 7F,G). In either case, our data support the idea that the functions of these linked regulatory elements are coupled through the act of transcription. Thus, the interleaved architecture of the yeast genome not only lends itself to high degrees of transcriptional overlap, but more importantly, to an equally high degree of transcription unit interdependency by co-opting transcription itself as a regulatory mechanism.
While this study has been predominantly conducted using a single locus approach in yeast, the implications of this work could be far reaching. For instance, establishing wider, biologically relevant transcriptional landscapes through sense/antisense state-switching could be pertinent to aspects of chronobiology such as circadian rhythms (Kramer et al., 2003; Feng and Lazar, 2012), the mitotic/meiotic cell cycles (Morris and Vogt, 2010), and other developmental processes in higher eukaryotes. This work places the sense/antisense relationships in a broader biological context and sheds light on how transcription might work as the nexus in more extensive networks that are organized in both spatial and temporal dimensions.
Materials and methods
All experiments were performed at least in duplicate to ensure that the trends observed were reproducible. Briefly, unless otherwise stated, cells were grown to exponential phase in rich medium (YP) supplemented with 2% glucose (YPD) or 2% galactose (YPGal). The strains of S. cerevisiae used for this study are shown in Supplementary file 1L. The Anchor Away technique exploits the high affinity interaction between the FK506 Binding Protein (FKBP12) and FKBP12 Rapamycin binding (FRB) domain of human mTOR protein in order to deplete target proteins from the nucleus in a time-dependent manner (Haruki et al., 2008). Control and treated cells were harvested after 1 h of DMSO or rapamycin exposure. Total RNA extracts were acquired by hot phenol extraction and enriched for polyadenylated transcripts using Oligotex‐dT as directed by the manufacturer (QIAGEN NV.). The concentration of RNA extract was measured and strandardised to 1 µg/μl using a nanodrop spectrophotometer. The protocol for rapid amplification of cDNA 3′ ends (3′ RACE) used in this study was modified from the methods provided by the 3′ RACE System for Rapid Amplification of cDNA Ends kit (Invitrogen Carlsbad, CA). Primers used are shown in Supplementary file 1M. Northern blotting was done as described in Murray et al. (2012). In vitro transcription with T7 RNA polymerase and radiolabelling was used to create strand-specific probes for Northern blotting. Primers are shown in Supplementary file 1N. Hybridization of RNA to strand-specific, high resolution tiling arrays was performed as described in Perocchi et al. (2007). Data analysis was performed by members of the Lars Steinmetz group at the European Molecular Biology Labs in Heidelberg, Germany (http://steinmetzlab.embl.de/cgi-bin/viewMellorLabArray.pl?showSamples=502_Glu1Vs503_Gal1&type=heatmap&gene=hms2). Native elongating transcript sequencing, (NET-seq) was done and analysed as described in Churchman and Weissman (2011). To assess the genome-wide significance of our observations, all genes in Supplementary file 1A were characterised according to their orientation with respect to their upstream (5P divergent, 5P tandem) or their downstream gene (3P, convergent), their own, and their neighbouring genes′ gene type (ORF, CUT, SUT, other) and YMC status (OX, oxidative; RB, reductive building; RC, reductive charging; NA/NC, non-cycling) and the number of genes in each category and median expression level in glucose and galactose determined. Genes that were located more than 1,000 bp from their neighbouring genes were excluded from the analysis. Computer simulations were performed by shuffling the strand location, gene type, YMC status, and expression levels of individual genes on every chromosome while keeping distances between genes and the total number of genes on each strand and of each type constant (Source code 1–MATLAB codes). Expression levels were simulated by randomly selecting recorded expression levels. After each simulation run, the number of genes and the median expression levels were calculated for each category. Simulations were run 10,000 times. Overlapping genes were defined as being positioned on the opposite strand and having 3′ ends covering the same region of the genome. Enrichment analysis for factors was performed using the published data set by Venters et al. (2011). The dual-labelled single molecule RNA Fluorescence In Situ hybridization (FISH) protocol was adapted from Zenklusen and Singer (2010). Fluorescently labelled DNA probes to detect sense or antisense transcripts are shown in Supplementary file 1O. Image acquisition was performed with a DeltaVision CORE: Wide-field fluorescence deconvolution imaging microscope. Using a 100x objective lens, 31 ‘z-stacks’ were collected. For each stack, an exposure time of 0.5 s for DAPI and 1 s for Cy3 (TRITC) and Cy5 filters was applied. Images were captured using a CoolSNAP HQ camera (Photometrics, Tucson, AZ). To facilitate image analysis, 3D data sets were compressed into 2D images by using a maximum projection, using hms2Δ cells to determine threshold signal for expression. Images displayed as 2D compressions of z-stacks 12–22 (11 stacks), which included most of the nucleus and cytoplasm. Error bars represent standard error of the mean. Chromatin immunoprecipitation (ChIP) was performed as described by Morillon et al. (2003, 2005). Primers used for RT qPCR are displayed in Supplementary file 1P.
References
-
Driving the cell cycle through metabolismAnnual Review of Cell and Developmental Biology 28:59–87.https://doi.org/10.1146/annurev-cellbio-092910-154010
-
Bimodal expression of PHO84 is modulated by early termination of antisense transcriptionNature Structural & Molecular Biology 20:851–858.https://doi.org/10.1038/nsmb.2598
-
Gene looping is conferred by activator-dependent interaction of transcription initiation and termination machineriesThe Journal of Biological Chemistry 284:25015–25025.https://doi.org/10.1074/jbc.M109.007948
-
Clocks, metabolism, and the epigenomeMolecular Cell 47:158–167.https://doi.org/10.1016/j.molcel.2012.06.026
-
Intergenic transcription causes repression by directing nucleosome assemblyGenes & Development 25:29–40.https://doi.org/10.1101/gad.1975011
-
Genome-wide transcription and the implications for genomic organizationNature Reviews. Genetics 8:413–423.https://doi.org/10.1038/nrg2083
-
Chromatin structure modulation in Saccharomyces cerevisiae by centromere and promoter factor 1Molecular and Cellular Biology 14:5229–5241.
-
A genomewide oscillation in transcription gates DNA replication and cell cycleProceedings of the National Academy of Sciences of USA 101:1200–1205.https://doi.org/10.1073/pnas.0306490101
-
Genomic gene clustering analysis of pathways in eukaryotesGenome Research 13:875–882.https://doi.org/10.1101/gr.737703
-
Evidence for a complex of transcription factor IIB with poly(A) polymerase and cleavage factor 1 subunits required for gene loopingThe Journal of Biological Chemistry 286:33709–33718.https://doi.org/10.1074/jbc.M110.193870
-
CPF1, a yeast protein which functions in centromeres and promotersThe EMBO Journal 9:4017–4026.
-
Regulation of glutamine-repressible gene products by the GLN3 function in Saccharomyces cerevisiaeMolecular and Cellular Biology 4:2758–2766.
-
Gene loops juxtapose promoters and terminators in yeastNature Genetics 36:1014–1018.https://doi.org/10.1038/ng1411
-
Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin DNucleic Acids Research 35:e128.
-
Characterization of sua7 mutations defines a domain of TFIIB involved in transcription start site selection in yeastThe Journal of Biological Chemistry 269:30569–30573.
-
Metabolic cycling in single yeast cells from unsynchronized steady-state populations limited on glucose or phosphateProceedings of the National Academy of Sciences of USA 107:6946–6951.https://doi.org/10.1073/pnas.1002422107
-
Metabolic cycling without cell division cycling in respiring yeastProceedings of the National Academy of Sciences of USA 108:19090–19095.https://doi.org/10.1073/pnas.1116998108
-
Sequence and promoter analysis of the highly expressed TEF gene of the filamentous fungus Ashbya gossypiiMolecular & General Genetics 242:263–271.https://doi.org/10.1007/BF00280415
-
Integrative Genomics Viewer (IGV): high-performance genomics data visualization and explorationBriefings in Bioinformatics 14:178–192.https://doi.org/10.1093/bib/bbs017
-
Two strategies for gene regulation by promoter nucleosomesGenome Research 18:1084–1091.https://doi.org/10.1101/gr.076059.108
-
Metabolic cycles as an underlying basis of biological oscillationsNature Reviews. Molecular Cell Biology 7:696–701.https://doi.org/10.1038/nrm1980
-
Cyclic changes in metabolic state during the life of a yeast cellProceedings of the National Academy of Sciences of USA 104:16886–16891.https://doi.org/10.1073/pnas.0708365104
-
A role for noncoding transcription in activation of the yeast PHO5 geneProceedings of the National Academy of Sciences of USA 104:8011–8016.https://doi.org/10.1073/pnas.0702431104
-
Antisense expression increases gene expression variability and locus interdependencyMolecular Systems Biology 7:468.https://doi.org/10.1038/msb.2011.1
Article and author information
Author details
Funding
Wellcome Trust (WT089156MA)
- Jane Mellor
National Institutes of Health
- Lars M Steinmetz
Oxford Biodynamics (ALRNEI1)
- Jane Mellor
European Commission (Epigenesys FP7 Network)
- Jane Mellor
Deutsche Forschungsgemeinschaft
- Lars M Steinmetz
Natural Sciences and Engineering Research Council of Canada
- Tania Nguyen
Fundação para a Ciência e a Tecnologia
- Ana Serra Barros
Wellcome Trust (Graduate Studentships)
- Harry Fischl
- Françoise S Howe
- David Brown
Engineering and Physical Sciences Research Council (Graduate Studentships)
- Ronja Woloszczuk
- Struan C Murray
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors would like to thank Ilan Davis, Micron Oxford, and Dan Larsen for help with RNA FISH, Athar Ansari, and Mike Hampsey for strains containing the sua7-1 allele and helpful advice, Stirling Churchman for help and advice with the NET-seq technique, Anitha Nair for excellent laboratory support, Benjamin Schuster-Böckler for advice regarding the computer simulations, and the following for funding; Keble College and the Clarendon Fund (to T.N.) and a Wellcome Trust Strategic Award (091911) supporting advanced microscopy at Micron Oxford (http://micronoxford.com)
Copyright
© 2014, Nguyen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,161
- views
-
- 423
- downloads
-
- 38
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 38
- citations for umbrella DOI https://doi.org/10.7554/eLife.03635
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Transcription mediated insulation and interference direct gene cluster expression switches
eLife 3:e03635.
https://doi.org/10.7554/eLife.03635




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}