Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae
- University of California, Berkeley, United States
Abstract
Heterochromatin exerts a heritable form of eukaryotic gene repression and contributes to chromosome segregation fidelity and genome stability. However, to date there has been no quantitative evaluation of the stability of heterochromatic gene repression. We designed a genetic strategy to capture transient losses of gene silencing in Saccharomyces as permanent, heritable changes in genotype and phenotype. This approach revealed rare transcription within heterochromatin that occurred in approximately 1/1000 cell divisions. In concordance with multiple lines of evidence suggesting these events were rare and transient, single-molecule RNA FISH showed that transcription was limited. The ability to monitor fluctuations in heterochromatic repression uncovered previously unappreciated roles for Sir1, a silencing establishment factor, in the maintenance and/or inheritance of silencing. In addition, we identified the sirtuin Hst3 and its histone target as contributors to the stability of the silenced state. These approaches revealed dynamics of a heterochromatin function that have been heretofore inaccessible.
https://doi.org/10.7554/eLife.05007.001eLife digest
A single cell from a plant, an animal or another eukaryote can contain several meters of DNA. In order to fit this length inside the nucleus of the cell, the DNA is wrapped around proteins called histones to form a compact structure known as chromatin.
Chromatin exists in two forms: loosely packed chromatin tends to contain the genes that are expressed in cells, whereas highly compacted chromatin (also called heterochromatin) silences the expression of most nearby genes. Adding small chemical markings to histone proteins can alter how much the chromatin is compacted, and newly formed cells can inherit these markings whenever a cell divides. This enables the new cell to essentially ‘remember’ which genes were active and which were repressed in the original cell. However, no one has measured how stable heterochromatin is over multiple cell divisions; as such it is unclear if genes that should remain silent are occasionally expressed after a cell has divided a number of times.
Budding yeast is a well-established model for studying heterochromatin. The genome of this single-celled organism has distinct regions of highly compacted chromatin, which are established and maintained by a number of different proteins.
Dodson and Rine have developed a new technique to detect when gene silencing is lost in individual yeast cells, even if the loss of silencing only occurs for a brief period. This approach revealed that genes in supposedly ‘silent’ heterochromatin were still expressed but only very rarely—in about 1 in every 1000 cell divisions.
Dodson and Rine's findings suggest that when silencing is lost, it is promptly re-established with the aid of a protein called Sir1. In addition, the new technique revealed that Sir1 helps to prevent losses of silencing in the first place. As such, the Sir1 protein appears to have previously unappreciated roles in the maintenance and the inheritance of heterochromatin in dividing yeast cells. Dodson and Rine also discovered that a protein called Hst3—which acts to remove chemical markings from histone proteins—also helps stabilize the silenced state. With this technique in hand, it is now possible to test any molecule, environment, or cellular process for its potential effect on the stability of gene silencing.
https://doi.org/10.7554/eLife.05007.002Introduction
Heterochromatin is a heritable, condensed chromatin structure that silences the expression of most genes within or near it. Phenomena such as clonal inheritance of inactivated X-chromosomes in female mammals, as well as position-effect variegation in Drosophila and yeast, demonstrate the remarkable ability of cells to propagate heterochromatic repression through mitosis. As an epigenetic state, heterochromatic gene repression provides a means for genetically identical cells to differentiate into stable, distinct cell types. However, despite its significance, little is known about the dynamics of heterochromatic repression and which factors contribute to or disrupt its stability.
In Saccharomyces cerevisiae, heterochromatin forms at the silent mating-type loci, HML and HMR, through the recruitment and subsequent spreading of Sir proteins (Grunstein and Gasser, 2013). DNA elements known as the E and I silencers flank each locus and nucleate complexes of Sir2, Sir3 and Sir4. Sir complexes then deacetylate histones and bind nucleosomes throughout the region, thereby rendering HML and HMR transcriptionally silenced and largely inaccessible to DNA-interacting proteins. Since each locus contains either a or α mating-type information, as does the MAT locus, heterochromatic repression of HML and HMR ensures that the MAT genotype is the only determinant of whether haploids mate as a or α cells.
Following its initial establishment, Sir-mediated heterochromatin can be maintained through the G1, G2 and M phases and inherited through S phase. Sir2, Sir3 and Sir4 are essential for all aspects of silencing (Rine and Herskowitz, 1987). Thus, mutants lacking any of these proteins express HML and HMR to the level of the transcriptionally active MAT locus. In contrast, mutants lacking Sir1 exhibit a bistable silencing phenotype (Pillus and Rine, 1989; Xu et al., 2006). Within a population of sir1 cells, HML and HMR exist in one of two phenotypic states: silenced or expressed. Each state is heritable for multiple cell divisions, demonstrating the epigenetic nature of Sir-mediated heterochromatin and inspiring the notion that Sir1 functions in the establishment of silencing, but not the maintenance or inheritance thereafter.
Notably, rare switches occur between the two expression states of HML and HMR in sir1 mutants, during which silencing is either lost or established. If Sir1 functioned exclusively in establishment, then losses of silencing should also occur in wild-type cells, yet no such event has been detected. Wild-type expression levels of genes at the HML and HMR loci are 1000-fold lower than the expression levels of the same genes when at the MAT locus, and efforts to detect expression of HML and HMR by any molecular method have shown the expression signal is indistinguishable from background noise. Moreover, 100% of MATa cells respond to α-factor, and diploids homozygous at the MAT locus are completely unable to sporulate. Thus, by all previous molecular criteria, the silent mating-type loci are transcriptionally inert. However, heterochromatin undergoes regular exchange of at least some of its structural components with newly synthesized molecules of the same proteins (Cheng and Gartenberg, 2000; Festenstein et al., 2003; Cheutin et al., 2003; Ficz et al., 2005) and is subject to perturbations, such as its replication in S phase. These fluctuations in heterochromatin structure imply that either the mechanism of silencing compensates for these changes and flawlessly reassembles each cell cycle, or that there are rare, as yet undetected losses of silencing resulting from heterochromatin dynamics.
To address whether RNA polymerase ever succeeds in transcribing silent chromatin at HML and HMR, we designed an assay capable of detecting short-lived gene expression with single-cell resolution. By capturing the consequences of transcription with a permanent, heritable mark, we detected transient losses of silencing at HML and HMR in wild-type cells, characterized the nature of these losses, and identified genetic determinants of heterochromatin stability.
Results
To determine the stability of gene repression in heterochromatin, we placed the gene encoding the Cre recombinase under control of the α2 promoter at either HMLα or HMRα (Figure 1A). RNA measurements made by quantitative RT-PCR showed that cre was as repressed as the native α2 gene at this location (Figure 1B). On chromosome V of both the HMLα::cre and HMRα::cre strains, we integrated a sequence in which two loxP sites flanked the RFP gene and the selectable drug marker kanMX (Figure 1A). The loxP–RFP-kanMX-loxP sequence resided downstream of the strong GPD promoter and upstream of a promoterless GFP gene. Thus, cells carrying this RFP-GFP cassette were RFP-positive, drug resistant, and GFP-negative. However, in the event that cre repression were lost, the resulting Cre protein could mediate recombination at the loxP sites, thereby excising the RFP and kanMX genes and positioning the GFP gene adjacent to the promoter (Figure 1A). It should be noted that the recombination event would be essentially irreversible in that the excised DNA lacked an origin of replication and would thereby be lost upon cell division. Therefore, the transient expression of cre would trigger a permanent switch to GFP expression and drug sensitivity in that cell and in all of its descendants. By convention, we refer to cells expressing RFP as red, and cells expressing GFP as green.
Figure 1
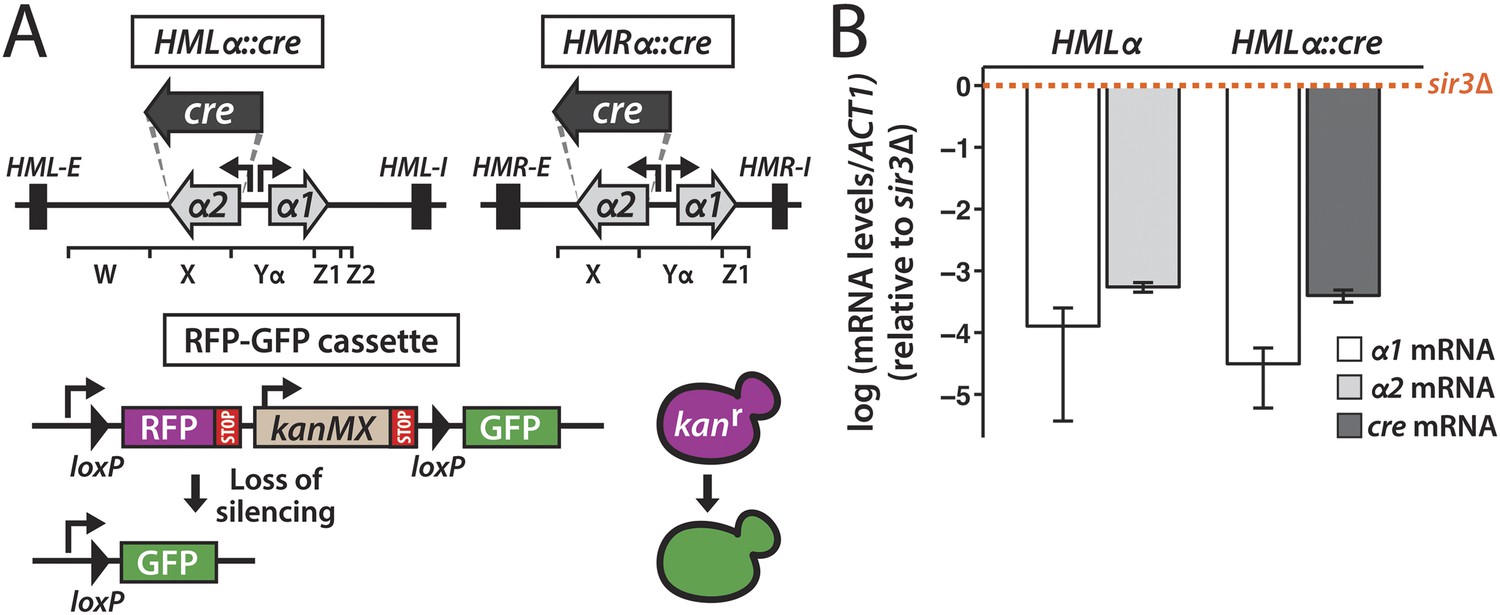
Cre-based assay.
(A) Design of assay to detect short-lived gene expression. The gene coding for the Cre recombinase was integrated at either HMLα or HMRα, replacing the α2 coding region, and was expressed from the native α2 promoter. Black rectangles denote the E and I silencers of HML and HMR. The W, X, Yα, Z1 and Z2 regions of the mating-type loci are indicated by brackets. Loss of cre silencing induces Cre-mediated recombination of the loxP sites (arrowheads) in the RFP-GFP reporter cassette, causing a switch from RFP to GFP expression and a loss of G418 resistance. (B) Quantitative RT-PCR analysis of α1 and α2 mRNA levels in a strain containing wild-type HMLα (JRY9623), and α1 and cre mRNA levels in a strain containing HMLα::cre (JRY9625). To determine the fold repression, mRNA levels were also measured in HMLα and HMLα::cre strains lacking Sir3 (JRY9624 and JRY9626). All strains carried matΔ and hmrΔ mutations to relieve the repression of α1 by the a1-α2 heterodimer in sir3Δ cells (Strathern et al., 1981). mRNA values were normalized to ACT1 mRNA values. Expression levels are shown relative to the corresponding sir3Δ values (set to 1) (orange dotted line). The fold repression of α2 and cre did not significantly differ (p = 0.12; Student's t test). Data are means ± standard deviation (SD) (n = 3).
Transient losses of silencing detected at HML and HMR
After plating cells with the cre gene at either HML or HMR on non-selective medium, we imaged the fluorescence of the resulting colonies. We first tested whether the expression of cre from the α2 promoter would produce enough recombinase to cause an efficient RFP-to-GFP switch by adding the Sir2 inhibitor nicotinamide (NAM) to the medium. The nicotinamide-induced derepression of either HMLα::cre or HMRα::cre resulted in entirely green colonies (Figure 2A and Figure 2—figure supplement 1). In contrast, cells that contained the RFP-GFP cassette but lacked any source of the cre gene grew into colonies that were entirely red (Figure 2A and Figure 2—figure supplement 1). These observations were consistent with the quantification of GFP fluorescence by flow cytometry (Figure 2B). Therefore, expression of the cre gene at either HML or HMR was both necessary and sufficient for the switch from RFP to GFP expression. We then plated HMLα::cre and HMRα::cre cells on medium lacking nicotinamide to determine the stability of gene silencing under normal conditions. The resulting colonies were predominantly red, indicating the majority of cells maintained the repression of cre throughout growth of the colony (Figure 2A). Within these colonies, however, we observed discrete green sectors, each of which represented a loss-of-silencing event that occurred in a cell at the vertex of the sector (Figure 2A and Figure 2—figure supplement 1). Although the switch to GFP expression was irreversible, some of the smaller sectors did not extend to the edge of the colony, likely due to a combination of genetic drift (Hallatschek et al., 2007) and the nature of three-dimensional growth of the colony. GFP expression was also detectable by flow cytometry in a fraction of HMLα::cre and HMRα::cre cells (Figure 2B). Thus, silent chromatin at HML and HMR was transcriptionally dynamic, exhibiting losses of silencing below the level of detection by all previous assays.
Figure 2 with 1 supplement see all
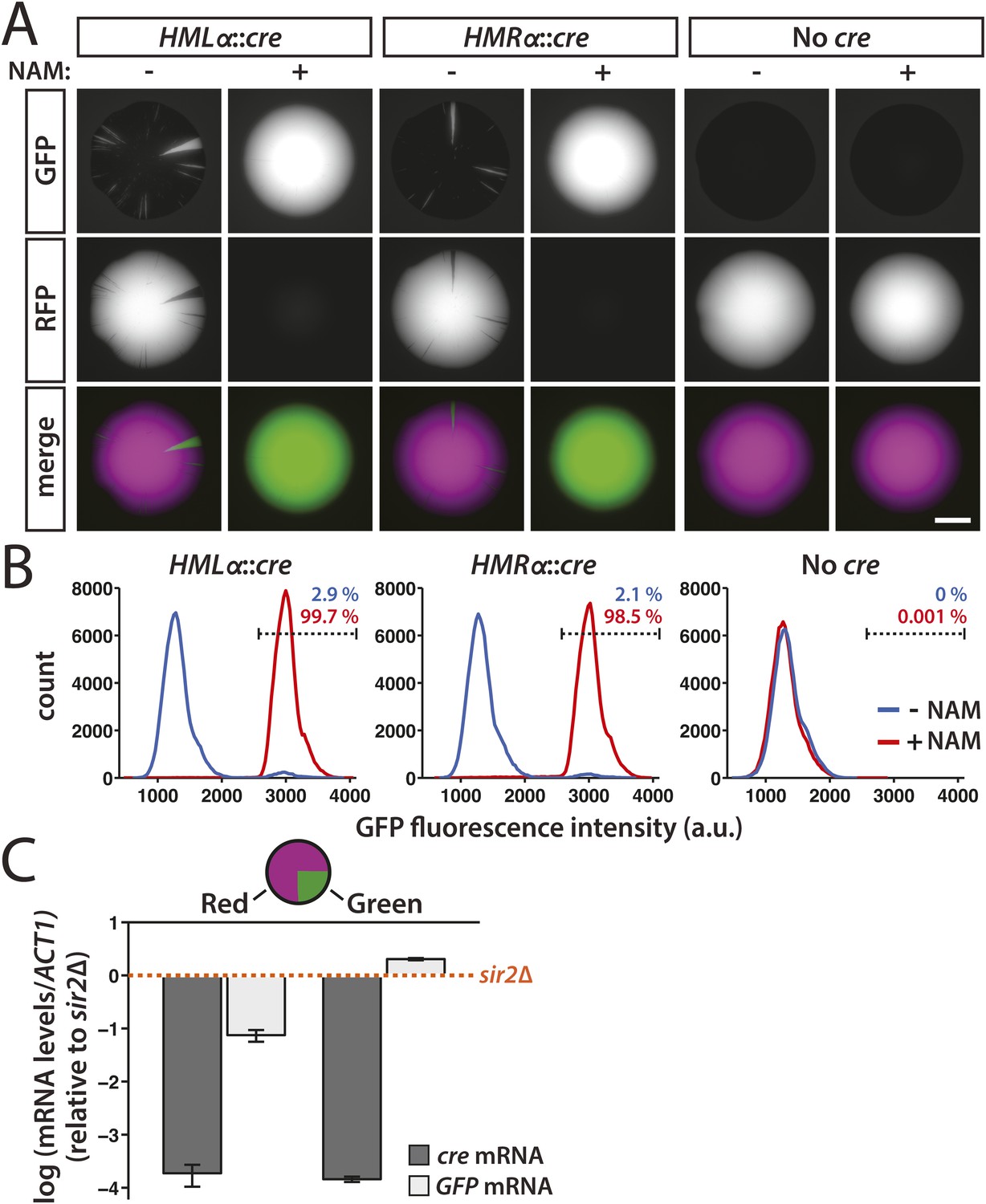
Transient transcription of HML and HMR captured by the Cre-based assay.
(A) Colonies derived from single cells containing the RFP-GFP cassette in addition to either HMLα::cre, HMRα::cre, or no source of the cre gene (No cre). Colonies of each genotype were grown in either the presence or absence of 5 mM nicotinamide (NAM). Additional colonies are shown in Figure 2—figure supplement 1. Scale bar, 2 mm. (B) Flow cytometry measurements of GFP fluorescence intensity in individual cells from within a single colony. Cells containing the RFP-GFP cassette and either HMLα::cre, HMRα::cre or no cre gene were plated on Complete Supplement Mixture (CSM) −Trp in the presence (red) or absence (blue) of 5 mM NAM. On day 6 of growth, representative colonies of each genotype and condition were resuspended in synthetic complete (SC) medium and grown to early log phase for analysis by flow cytometry. Each distribution represents approximately 105 cells. The percentage of GFP-positive cells shown for each sample was determined by a gate (dotted line) that was set based on the intensity profile of the HMLα::cre (+NAM) sample. (C) Quantitative RT-PCR measurements of HMLα::cre transcription in red and green sectors. To determine whether silencing loss was transient, cre and GFP mRNA levels were quantified in the GFP-expressing (green) and RFP-expressing (red) regions of a colony of cells containing HMLα::cre and the RFP-GFP cassette (JRY9628). Three independent experiments were performed, each on a different colony with a large green sector. Cells from the red and green regions of each colony were grown to log phase in YPD and harvested for RNA isolation. As a positive control for HMLα::cre expression, mRNA levels were also measured in a sir2Δ mutant (JRY9633). All mRNA values were normalized to ACT1 mRNA values. Expression levels are shown relative to the corresponding sir2Δ values (set to 1) (orange dotted line). The fold repression of HMLα::cre did not significantly differ between the red and green samples (p = 0.43; Student's t test). Data are means ± SD (n = 3).
At least two lines of evidence indicated that these losses of silencing were transient and did not arise from mutations in SIR genes. First, cre and GFP mRNA levels were measured in both red and green regions from within the same colony. If the loss-of-silencing event that resulted in the green sector were temporary, then the level of HMLα::cre repression would be indistinguishable between red and green sectors. Alternatively, if the loss of silencing were permanent, then cre mRNA would be detectable in cells from the green sector. Consistent with the efficiency of heterochromatin formation, which occurs de novo in approximately 1–2 cell divisions (Osborne et al., 2009), HMLα::cre repression was fully restored in the descendants of a cell that experienced a loss of silencing (Figure 2C). The slight difference in GFP expression between wild-type cells from the green sector and sir2Δ cells may have been due to the constitutive presence of Cre protein in the sir2Δ mutant, which could have potentially disrupted transcription by binding the loxP site between the promoter and the GFP gene. In addition, the GFP mRNA values in the red population likely reflected a low level of GFP expression since colonies were grown in the absence of G418 selection and therefore RFP-to-GFP switches occurred at a low rate. As secondary confirmation that losses of silencing were transient, we have never detected green sectors in colonies of a wild-type strain containing the GFP gene under control of the α2 promoter at HMLα (unpublished observations).
The pattern of colony fluorescence revealed the history of individual lapses in silencing, with the size of each green sector corresponding to how early the silencing loss occurred during colony growth. Colonies that were half red/half green reflected a loss of cre repression in either the mother or daughter cell of the first division that gave rise to the colony (Figure 3A). Therefore, the frequency of half-sectored colonies equaled the rate of RFP-to-GFP switches per cell division. In the HMLα::cre strain, the frequency of half-sectored colonies was 1.6 × 10−3 (Table 1 and Figure 3B). Thus, for every thousand cells, one to two cells temporarily failed to repress HMLα. Silencing of HMRα was slightly more stable than HMLα, with a lower rate of 7 × 10−4 losses per division (p = 0.003; Student's t test) (Table 1 and Figure 3B).
Figure 3
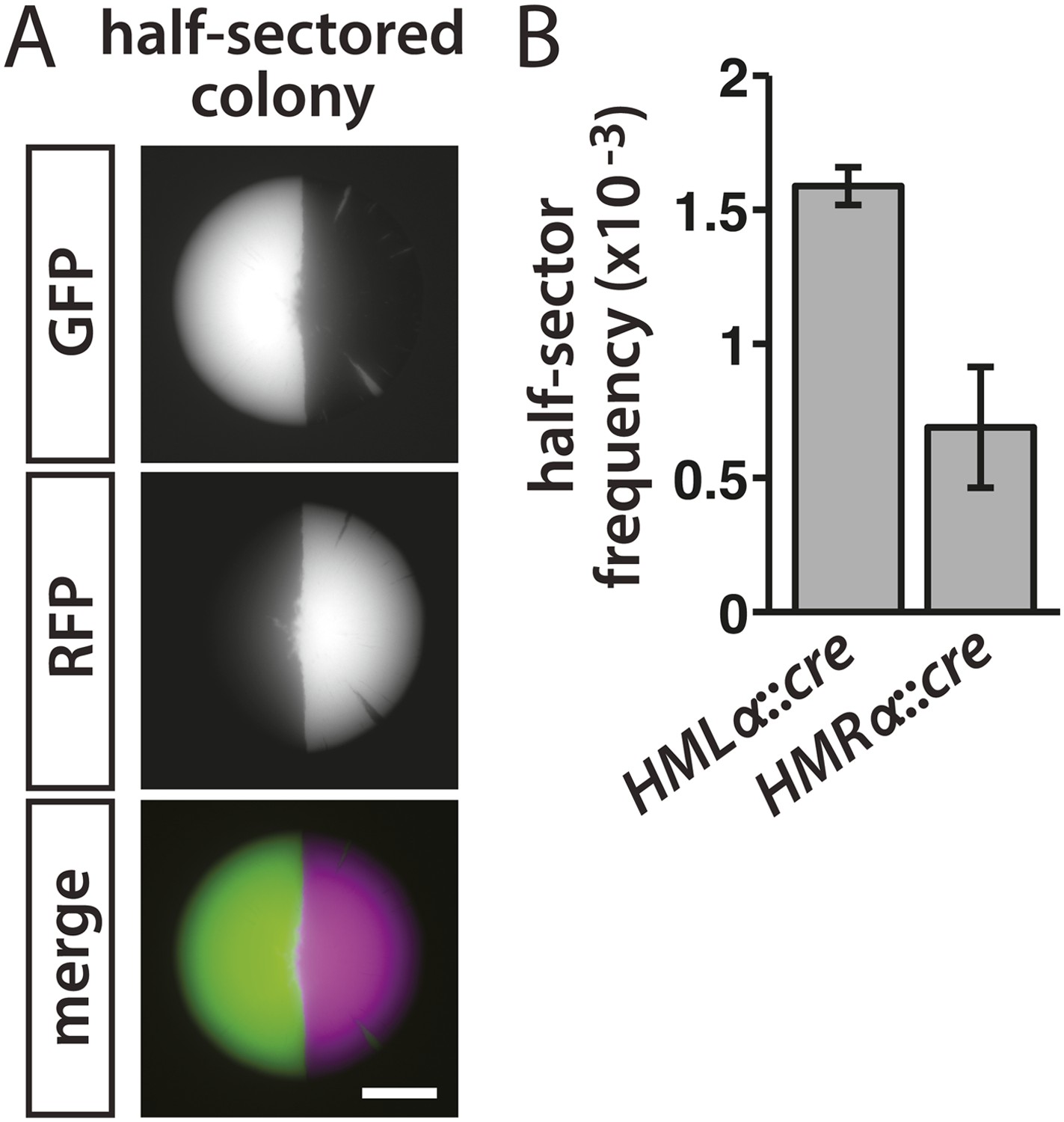
Rates of silencing loss as measured by half-sector frequency.
(A) Example of a half-sectored colony derived from a single cell containing HMLα::cre and the RFP-GFP cassette. Scale bar, 1 mm. (B) Frequency of half-sectored colonies. The half-sector frequency of a strain containing the cre gene at HMLα (JRY9628) was significantly different (p = 0.003; Student's t test) from the half-sector frequency of a strain containing the cre gene at HMRα (JRY9629). Data are means ± SD (n = 3).
Table 1
Frequency of half-sectored colonies for each strain that showed a sectoring phenotype
| Figure | Strain | Relevant genotype | Half-sector frequency |
|---|---|---|---|
| 3 | JRY9628 | matΔ HMLα::cre | 0.00158 ± 0.00007 |
| JRY9629 | matΔ HMRα::cre | 0.0007 ± 0.0002 | |
| 5 | JRY9739 | MATa HMLα::cre sir1Δ | 0.055 ± 0.007 |
| JRY9740 | MATa HMRα::cre sir1Δ | 0.061 ± 0.002 | |
| 6 | JRY9731 | MATα/matΔ HMLα/HMLα::cre | 0.00037 ± 0.00004 |
| JRY9732 | MATα/matΔ HMLα/HMLα::cre sir1Δ/SIR1 | 0.0007 ± 0.0002 | |
| JRY9734 | MATα/matΔ HMLα/HMLα::cre sir3Δ/SIR3 | 0.0016 ± 0.0005 | |
| JRY9735 | MATα/matΔ HMLα/HMLα::cre sir4Δ/SIR4 | 0.0013 ± 0.0003 | |
| 7 | JRY9636 | matΔ HMLα::cre hst3Δ | 0.0111 ± 0.0005 |
| JRY9639 | MATa HMLα::cre hht1-hhf1Δ hht2-hhf2Δ [HHT2-HHF2] | 0.0033 ± 0.0004 | |
| JRY9640 | MATa HMLα::cre hht1-hhf1Δ hht2-hhf2Δ [hht2K56R-HHF2] | 0.0111 ± 0.0008 | |
| JRY9641 | MATa HMLα::cre hht1-hhf1Δ hht2-hhf2Δ [hht2K56Q-HHF2] | 0.009 ± 0.002 | |
| JRY9736 | MATa HMLα::cre hst3Δ hht1-hhf1Δ hht2-hhf2Δ [HHT2-HHF2] | 0.0096 ± 0.0008 | |
| JRY9737 | MATa HMLα::cre hst3Δ hht1-hhf1Δ hht2-hhf2Δ [hht2K56R-HHF2] | 0.009 ± 0.001 | |
| JRY9738 | MATa HMLα::cre hst3Δ hht1-hhf1Δ hht2-hhf2Δ [hht2K56Q-HHF2] | 0.0105 ± 0.0004 |
-
All values represent the mean of three independent experiments ± standard deviation. Full dataset is shown in Table 1—source data 1. All strains contained a copy of the RFP-GFP cassette. See Supplementary file 1 for full genotypes.
-
Table 1—source data 1
Half-sector frequencies from three independent experiments.
- https://doi.org/10.7554/eLife.05007.008
Transient transcription of silent chromatin was restricted to low levels and was captured effectively by the Cre-based assay
In principle, the rates of RFP-to-GFP switches could be compatible with at least three possible distributions of cre expression. In one model, cre is transcribed at a low level in many cells, but the resulting level of Cre protein is sufficient to catalyze recombination of the RFP-GFP cassette in only a subset of those cells. Alternatively, cre transcription could be completely absent in most cells, but in a low fraction of cells, occur at the same level as in a sir- mutant. Finally, cre transcription may be absent in most cells, and limited even in the small fraction of cells that switch from RFP to GFP expression.
Thus far, efforts to detect transcription at HML and HMR have relied on quantitative RT-PCR and other population-based assays that report the average level of RNA for all the cells in a sample. However, advances in RNA imaging now enable the quantification of individual transcripts in single cells (Itzkovitz and van Oudenaarden, 2011). Using a version of fluorescence in situ hybridization (FISH) with single-molecule sensitivity (Raj et al., 2008), we hybridized fixed cells with two sets of probes distinguished by their fluorophore label (Figure 4A). One probe set was specific to the cre RNA expressed from HMLα::cre. As an internal control to account for sample variation, the other probe set was specific to KAP104 RNA, which encodes a protein involved in nuclear transport. We measured cre and KAP104 transcripts by imaging and quantifying fluorescent spots, each representative of a single RNA molecule.
Figure 4 with 2 supplements see all
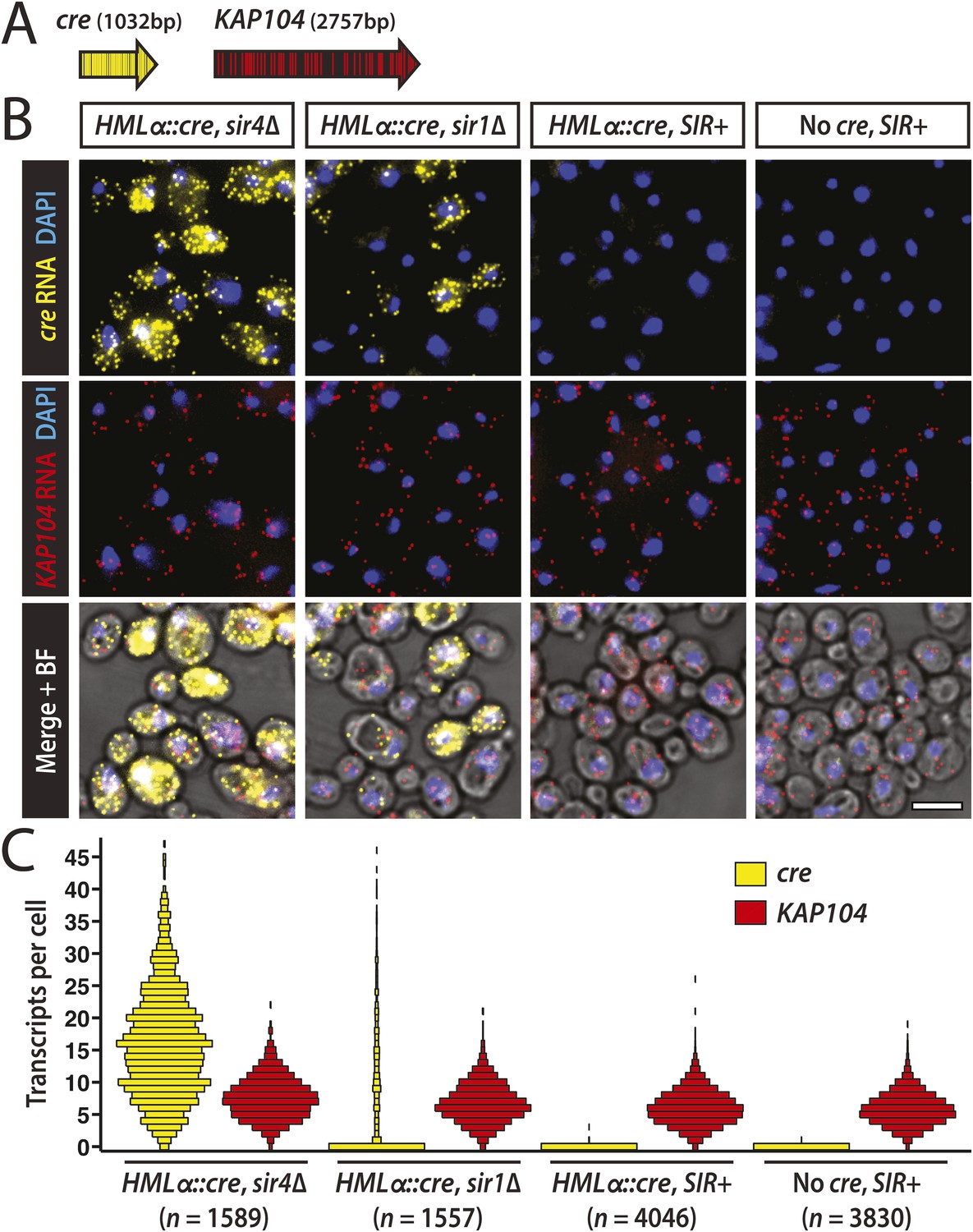
Single-molecule imaging of HMLα::cre transcription.
(A) Schematic of the cre and KAP104 coding sequences (black arrows), with colored lines representing sites targeted by the 20-nucleotide FISH probes (see Supplementary file 4 for probe sequences). To detect both RNA sequences in the same cell, the cre-specific and KAP104-specific probes were differentially labeled. (B) Single-molecule RNA FISH of cre (yellow) and KAP104 (red) transcripts shown as maximum-intensity projections of z-stacks. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Merged images were superimposed on the corresponding brightfield (BF) micrographs. Larger fields are shown in Figure 4—figure supplement 1. Scale bar, 5 μm. (C) Distributions of cre and KAP104 RNA abundance determined by FISH. Each violin plot was normalized so that maximum bin width was the same across all samples. Data shown are from a representative replicate.
As a positive control, we examined cre transcription in a mutant lacking Sir4, a protein integral to heterochromatin structure. As expected, HML was transcriptionally active in the sir4Δ mutant (Figure 4B,C and Figure 4—figure supplement 1), with a mean of 12 cre transcripts per cell (±2 standard error of the mean [SEM] calculated from three independent experiments), and 98% (±1 SEM) of cells containing at least one RNA molecule (Table 2). Transcripts of the constitutively active KAP104 gene were present in 99.0% (±0.3 SEM) of cells (Table 2, Figure 4B,C and Figure 4—figure supplement 1). In concordance with previous studies (Zenklusen et al., 2008; Gandhi et al., 2011), KAP104 expression approximated a Poisson distribution, suggesting that most of the cell-to-cell variation in KAP104 RNA levels could be explained by stochastic, constitutive transcription. In contrast, the variance of cre RNA abundance was large in relation to the mean (Table 2), perhaps reflecting an alternative mode of transcription.
Table 2
Summary of single-molecule RNA FISH statistics
| RNA | Genotype | Mean spots per cell | Variance | % Cells with >0 spots | % Cells with >1 spot |
|---|---|---|---|---|---|
| cre | HMLα::cre, sir4Δ | 12 ± 2 | 53 ± 9 | 98 ± 1 | 95 ± 2 |
| HMLα::cre, sir1Δ | 5 ± 1 | 58 ± 13 | 46 ± 7 | 41 ± 7 | |
| HMLα::cre, SIR+ | <0.005 | – | 0.42 ± 0.07 | 0.04 ± 0.01 | |
| No cre, SIR+ | <0.003 | – | 0.3 ± 0.1 | 0 | |
| KAP104 | HMLα::cre, sir4Δ | 7.3 ± 0.4 | 11 ± 1 | 99.0 ± 0.3 | 97.2 ± 0.8 |
| HMLα::cre, sir1Δ | 7.1 ± 0.2 | 10.8 ± 0.9 | 99.28 ± 0.04 | 97.5 ± 0.1 | |
| HMLα::cre, SIR+ | 6.1 ± 0.2 | 7.8 ± 0.3 | 99.1 ± 0.2 | 96.5 ± 0.8 | |
| No cre, SIR+ | 5.9 ± 0.5 | 7.1 ± 0.5 | 99.0 ± 0.7 | 96 ± 2 |
-
All values represent the mean of three independent experiments ± standard error of the mean. Full dataset is shown in Table 2—source data 1.
-
Table 2—source data 1
Single-molecule RNA FISH values from three independent experiments.
- https://doi.org/10.7554/eLife.05007.013
In a wild-type strain with HMLα::cre, cre transcripts were absent in nearly every cell, whereas KAP104 transcripts were present at a level comparable to that in sir4Δ cells (Figure 4B,C and Figure 4—figure supplement 1). The distribution of KAP104 expression in wild-type cells was slightly lower than the distribution in the sir4Δ mutant, which may be explained by our observation that sir4Δ cells were slightly larger than wild type (Figure 4—figure supplement 2), as noted before for sir2Δ mutants (Moretto et al., 2013). Therefore, HML was completely silent in the vast majority of cells, indicating the Cre-based assay was sensitive to rare events of transcription. Although we detected an apparent cre signal in 4.2 × 10−3 (±0.7 × 10−3 SEM) wild-type cells, we also detected an apparent cre signal at a frequency of 3 × 10−3 (±1 × 10−3 SEM) in cells that lacked the cre gene, which were imaged to control for hybridization specificity (Table 2, Figure 4B,C and Figure 4—figure supplement 1). The wild-type strain and the negative control strain were distinguishable, however, by the frequency of cells containing more than one spot of cre signal (Table 2). We observed five wild-type cells across all three replicates (13,695 wild-type cells total) that contained more than one cre spot, ranging from 2 to 4 spots per cell, whereas we never detected more than one cre spot per cell in the negative control strain (13,722 No-cre cells total). Together, the RNA FISH analysis and the Cre-based assay converged on the same striking conclusion: the low rate of RFP-to-GFP switches reflected the complete absence of HMLα::cre transcription in the vast majority of cells.
There was no evidence of any wild-type cells containing cre RNA levels typical of sir4Δ cells, suggesting that in the rare cells that lost silencing, the duration of transcription was short, presumably due to continuous nucleation of silent chromatin by Sir1 and other factors. Silencing in individual sir1 cells was previously measured by indirect bioassays (Pillus and Rine, 1989; Xu et al., 2006), and with respect to transcription, the expression levels of HML and HMR underlying the two phenotypically distinct cell types in a sir1 population have never been established. RNA FISH allowed us to evaluate sir1Δ cells at the level of transcription on a per cell basis to determine whether these two states were molecularly equivalent to the levels of HML expression seen in sir4Δ and wild-type cells. In the absence of Sir1, 46% (±7 SEM) of cells expressed HML (Table 2), exhibiting a range in cre RNA levels that resembled the sir4Δ mutant (Figure 4B,C and Figure 4—figure supplement 1). The remaining 54% of sir1Δ cells lacked detectable cre RNA molecules, but expressed KAP104 at normal levels (Figure 4B,C and Figure 4—figure supplement 1). Therefore, in approximately half of all cells lacking Sir1, HML was fully repressed, whereas the other half expressed HML at levels indistinguishable from those in a sir4Δ mutant.
Effects of Sir protein availability on the dynamics of silencing
Although Sir1 was defined by its role in the establishment of silencing, and is not required for maintenance or inheritance, the possibility that it contributes to these other processes has never been directly tested. Such a test would require a comparison of the rate of silencing loss in wild-type cells to the rate of silencing loss in the population of sir1 cells in which HML was silenced. Whereas past measurements in wild-type cells were confounded by the role of Sir1 in establishment (Xu et al., 2006), the Cre-based assay is sensitive enough to record losses of silencing before re-establishment occurs.
To determine whether Sir1 contributes to the maintenance or inheritance of silencing, we sporulated a sir1 hemizygote containing HMLα::cre and the RFP-GFP cassette and then imaged the fluorescence of colonies derived from the meiotic products. If the rate of silencing loss were unaffected by the absence of Sir1, then the sectoring patterns would be indistinguishable between colonies arising from SIR1 cells and colonies arising from sir1Δ cells in which HMLα::cre was repressed. However, green sectors were notably more abundant in the colonies of spores lacking Sir1 (Figure 5A, Figure 5—figure supplement 1). Moreover, the frequency of half-sectored colonies in a sir1Δ haploid was 35-fold above the frequency of half-sectored colonies in a wild-type haploid (Table 1 and Figure 5B), indicating that cells lost silencing at HML at a strikingly higher rate when Sir1 was absent. In strains containing HMRα::cre instead of HMLα::cre, sir1Δ colonies showed elevated sectoring and a higher abundance of half sectors in comparison to wild-type colonies, as well (Table 1 and Figure 5). Thus, beyond its role in establishment, Sir1 contributed to either the maintenance or inheritance of silencing at both HMLα and HMRα. Consistent with the RNA FISH measurements, which revealed a large population of sir1Δ cells that lacked detectable cre transcripts (Table 2 and Figure 4C), many of the cells within the sir1Δ colonies remained red (Figure 5A). Therefore, sir1Δ cells that were in the silenced state exhibited the same level of repression as wild-type cells, but transitioned to a transcriptionally active state more often than wild-type cells.
Figure 5 with 1 supplement see all
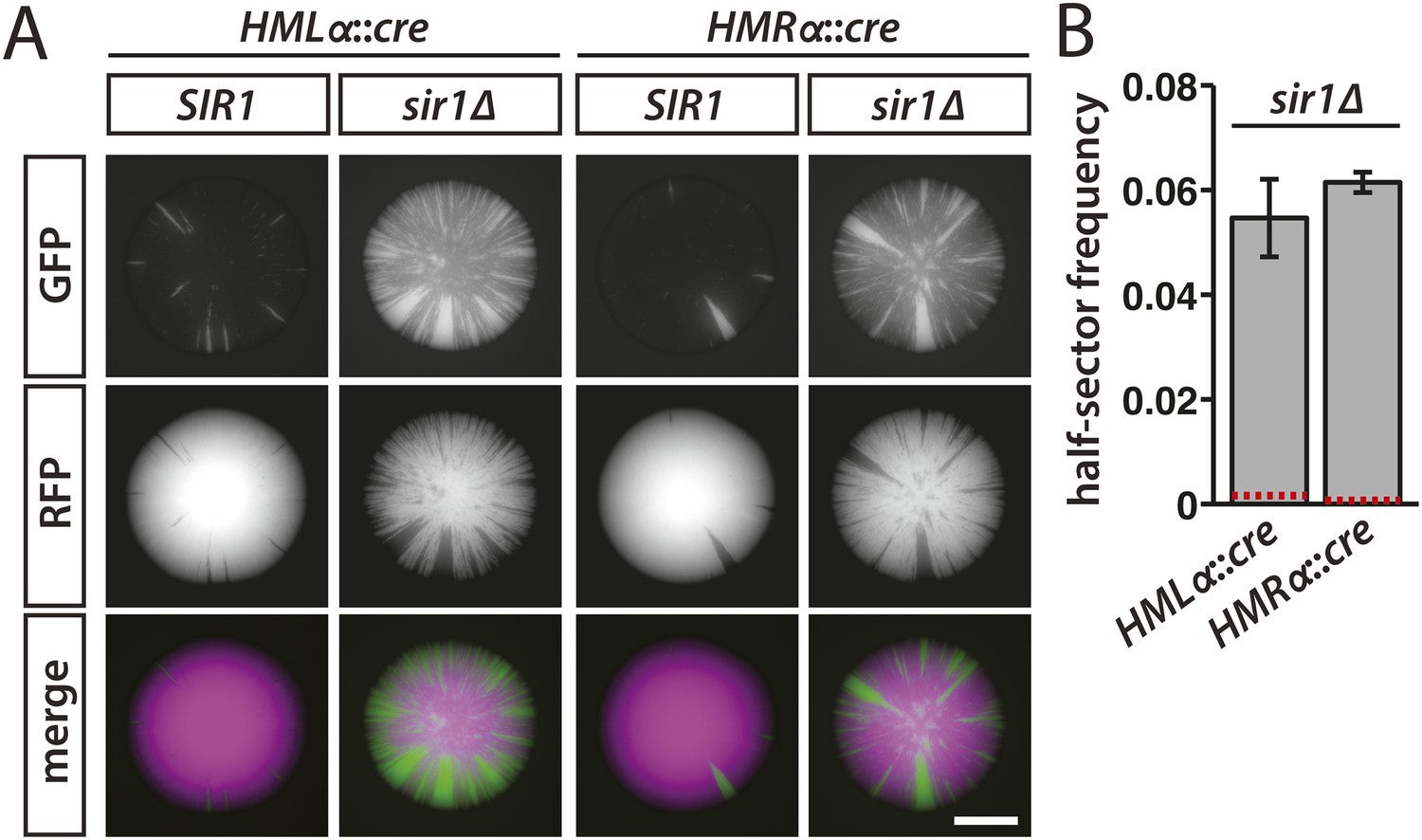
Increased silencing loss in cells lacking Sir1.
(A) Fluorescence of colonies derived from SIR1 and sir1Δ cells containing the RFP-GFP cassette and either HMLα::cre or HMRα::cre. All colonies shown were grown from MATa spores of JRY9729 and JRY9730 tetrad dissections. Additional colonies are shown in Figure 5—figure supplement 1. Scale bar, 1 mm. (B) Frequencies of half-sectored colonies for haploid strains carrying the sir1Δ mutation and either HMLα::cre (JRY9739) or HMRα::cre (JRY9740). For both HMLα::cre- and HMRα::cre-containing strains, the half-sector frequency of the sir1Δ mutant was significantly higher (p = 2 × 10−4 for HMLα::cre, p = 7 × 10−7 for HMRα::cre; Student's t test) than the half-sector frequency of wild type (red dotted lines represent the rates shown in Figure 3B). Data are means ± SD (n = 3).
Sir1 facilitates the recruitment of Sir2, Sir3 and Sir4 and therefore helps maintain a local concentration of heterochromatin components at HML and HMR. To test whether the dynamics of silencing were sensitive to the availability of Sir proteins, HMLα::cre expression was monitored in diploids hemizygous for individual SIR genes. While measuring silencing loss in diploids, we noticed that a wild-type diploid with one copy of HMLα::cre and the RFP-GFP cassette showed considerably reduced colony sectoring and a fourfold decrease (p = 1 × 10−5; Student's t test) in the frequency of half-sectored colonies in comparison to an isogenic haploid (Table 1), indicating a ploidy effect on the rate of RFP-to-GFP switching. Compared to the wild-type diploid, colonies of the sir1, sir3 and sir4 hemizygotes showed a modest yet consistent increase in the number of green sectors (Figure 6A and Figure 6—figure supplement 1) and a higher occurrence of half sectors (Table 1 and Figure 6B). Therefore, reducing the dose of the genes encoding Sir3 or Sir4, two structural components of heterochromatin, or Sir1, a protein that recruits these components, rendered silencing at HML less stable. Altered gene dosage of SIR1 and SIR4 also affects the silencing of a sensitized HMR::ADE2 allele (Sussel et al., 1993). Hemizygosity for SIR2 had no perceivable impact on colony sectoring (Figure 6A and Figure 6—figure supplement 1).
Figure 6 with 1 supplement see all
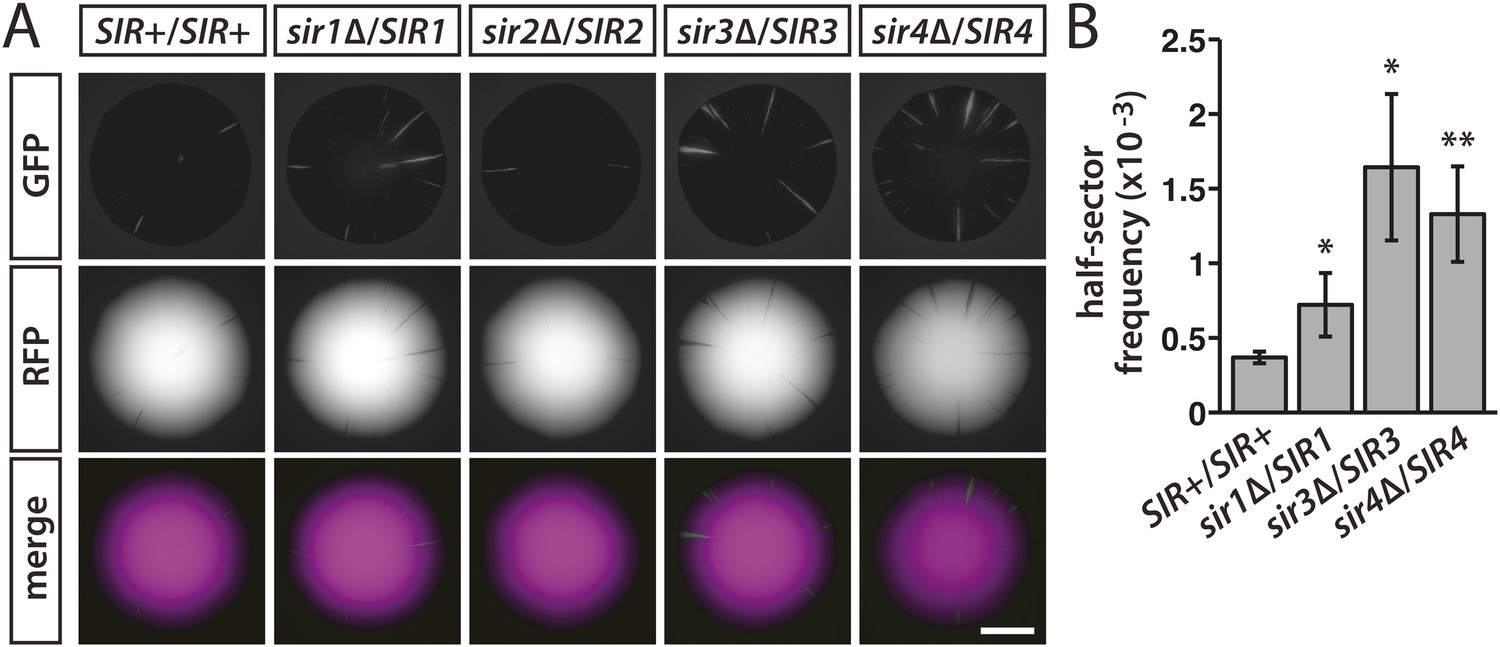
Stability of silencing at HML in diploids hemizygous for individual SIR genes.
(A) Fluorescence of colonies derived from single diploid cells containing one copy of HMLα::cre, one copy of the RFP-GFP cassette, and the indicated SIR genotypes. Additional colonies are shown in Figure 6—figure supplement 1. Scale bar, 2 mm. (B) Frequencies of half-sectored colonies for a wild-type diploid and different sir hemizygotes. The half-sector frequencies of the sir1, sir3 and sir4 hemizygotes were significantly higher (*p < 0.05, **p < 0.01; Student's t test) than the half-sector frequency of the wild-type diploid. Data are means ± SD (n = 3). The frequency of half-sectored colonies was not determined for the sir2 hemizygote.
A novel role for the sirtuin Hst3 in the stabilization of silencing at HML
Sir2 is the only member of the sirtuin family of NAD+-dependent deacetylases previously shown to have a role in silencing at HML or HMR. As measured by mating efficiency, silencing is indistinguishable between wild type and mutants lacking any of the four HST genes (Brachmann et al., 1995; Yang et al., 2008). However, Hst3 and Hst4 have been shown to contribute to the silencing of subtelomeric genes (Brachmann et al., 1995; Yang et al., 2008), as well as silencing of a URA3 reporter in a plasmid-borne HMR cassette (Grünweller and Ehrenhofer-Murray, 2002). Since nicotinamide inhibits all five sirtuins in yeast (Imai et al., 2000; Landry et al., 2000a, 2000b; Smith et al., 2000; Tanner et al., 2000; Tanny and Moazed, 2001), we tested whether other sirtuins, in addition to Sir2, were involved in the nicotinamide-induced loss of silencing observed in Figure 2. As expected, colonies of cells with HMLα::cre and the RFP-GFP cassette turned completely green in the absence of Sir2, a protein essential for the nucleation and maintenance of heterochromatin (Figure 7A and Figure 7—figure supplement 1A). The colonies of hst1Δ, hst2Δ and hst4Δ mutants exhibited sectoring patterns that were indistinguishable from wild-type colonies (Figure 7A and Figure 7—figure supplement 1A). In contrast, the hst3Δ mutant showed a striking increase in colony sectoring and a sevenfold greater frequency of half-sectored colonies compared to wild type (Table 1, Figure 7A,B and Figure 7—figure supplement 1A). Thus, the Hst3 deacetylase contributed to the stability of silenced chromatin at HML. Whereas silencing defects have not been detected at subtelomeric genes in either the hst3Δ or hst4Δ single mutant, the hst3Δ hst4Δ double mutant shows a measurable phenotype (Brachmann et al., 1995; Yang et al., 2008). Therefore, it is likely that the deletion of HST4 would enhance the sectoring phenotype of the hst3Δ mutant shown in Figure 7A.
Figure 7 with 2 supplements see all
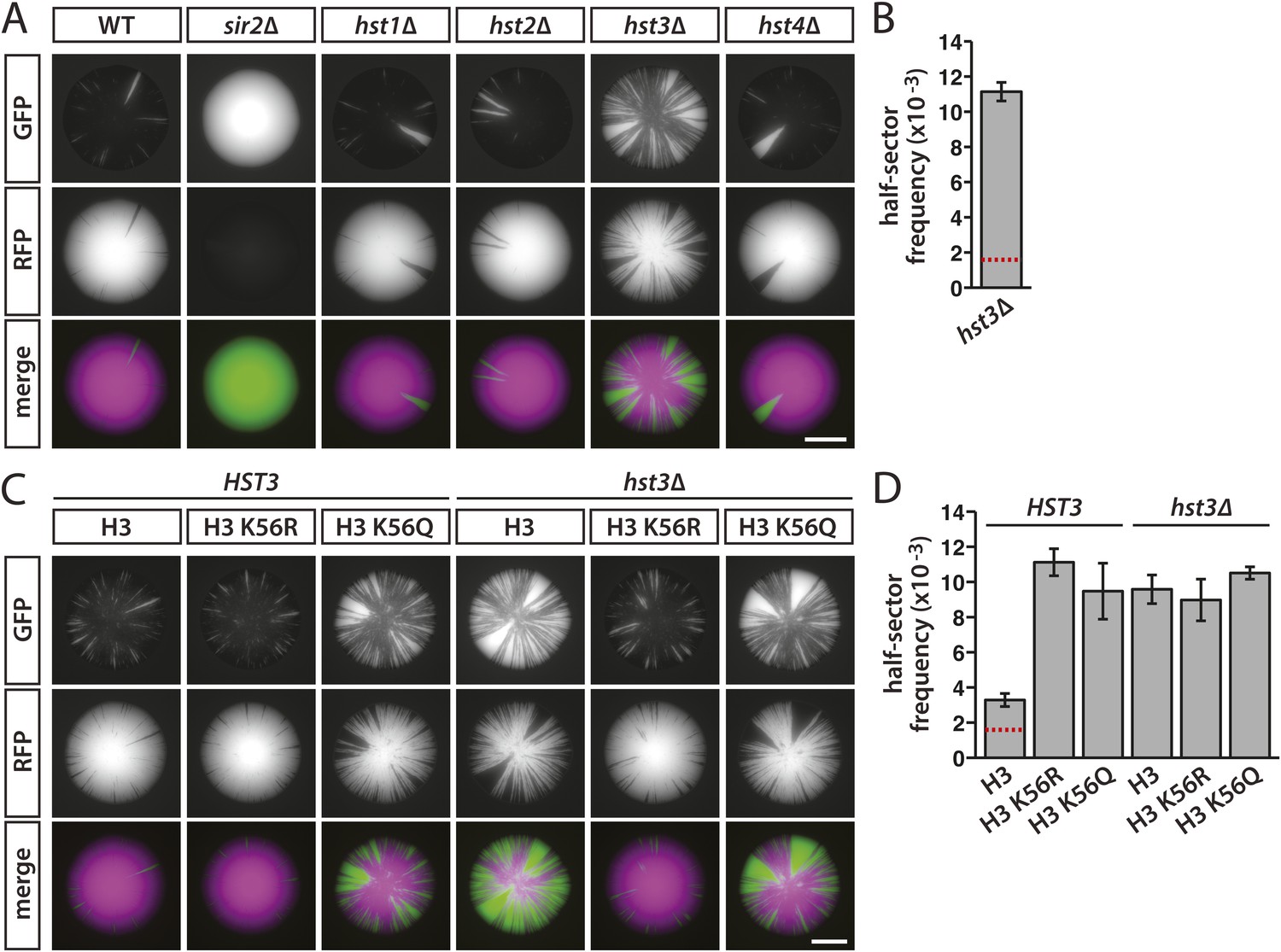
Effects of sirtuin gene deletions and histone H3 K56R or K56Q substitutions on the stability of silencing at HML.
(A) Fluorescence of colonies derived from single cells containing HMLα::cre, the RFP-GFP cassette, and the indicated deletions. Additional colonies are shown in Figure 7—figure supplement 1A. Scale bar, 2 mm. (B) Frequency of half-sectored colonies for the hst3Δ mutant shown in panel A. The half-sector frequency of the hst3Δ mutant was significantly greater (p = 6 × 10−6; Student's t test) than the half-sector frequency of wild type (red dotted line represents the rate shown in Figure 3B). Data are means ± SD (n = 3). The frequency of half-sectored colonies was not determined for the hst1Δ, hst2Δ or hst4Δ mutant. (C) Fluorescence of colonies derived from single cells containing HMLα::cre, the RFP-GFP cassette, hht1-hhf1Δ and hht2-hhf2Δ mutations, and a plasmid-borne copy of either HHT2-HHF2, hht2K56R-HHF2 or hht2K56Q-HHF2. Additional colonies are shown in Figure 7—figure supplement 1B. Scale bar, 2 mm. (D) Frequency of half-sectored colonies for the genotypes shown in panel C. The half-sector frequency of the wild-type strain containing only one copy of the histone H3–H4 gene pair was significantly higher (p = 0.001; Student's t test) than the half-sector frequency of the strain shown in Figure 3B containing both copies of the histone H3–H4 gene pair (red dotted line) (see Figure 7—figure supplement 2). Data are means ± SD (n = 3).
Hst3 regulates the deacetylation of acetylated lysine 56 on histone H3 (H3 K56-ac) (Celic et al., 2006; Maas et al., 2006; Yang et al., 2008). To determine whether Hst3 stabilized silencing through deacetylation of H3 K56-ac or through deacetylation of yet undiscovered substrates, we measured the effects of amino acid substitutions that either mimic (K56Q) or prevent (K56R) the acetylation of H3 K56. Although these histone gene mutations were previously analyzed for their effects on silencing, the outcome varies depending on the locus and the assay (Hyland et al., 2005; Xu et al., 2007; Miller et al., 2008; Yu et al., 2011). In our strains, with cre under the native α2 promoter at HML, colonies of the K56Q mutant showed a dramatic increase in sectoring, whereas the K56R mutant was similar to wild type (Figure 7C and Figure 7—figure supplement 1B). Consistent with the possibility that the hst3Δ silencing defects were due to an increase in acetylated histone H3 K56, the sectoring phenotypes suggested that neutralizing the positive charge at this position on histone H3 impaired the stability of repression. By half-sector analysis, however, both the K56Q and K56R mutants showed a significant increase (p = 3 × 10−3 and p = 9 × 10−5, respectively; Student's t test) in the rate of silencing loss compared to wild type (Table 1 and Figure 7D). The relatively high frequency of half-sectored colonies in the histone H3 K56R mutant was unexpected due to the similarity in sectoring patterns between histone H3 K56R mutant colonies and wild-type colonies (Figure 7C and Figure 7—figure supplement 1B). However, the histone H3 K56R substitution has previously been shown to decrease silencing under various conditions (Xu et al., 2007; Yu et al., 2011). Differences between sectoring patterns and half-sector frequencies are considered further in the ‘Discussion’.
Cells expressing the H3 K56Q mutant produced similar patterns of colony sectoring regardless of whether Hst3 was present (Figure 7C and Figure 7—figure supplement 1B). This phenotype was slightly less severe than that of hst3Δ cells expressing wild-type H3 K56 (Figure 7C and Figure 7—figure supplement 1B), perhaps because glutamine was not quite as disruptive to silencing as acetylated lysine at this residue. In addition, the H3 K56R substitution in hst3Δ cells restored silencing to wild-type levels, as determined by overall colony sectoring (Figure 7C and Figure 7—figure supplement 1B). Therefore, blocking the acetylation of residue 56 suppressed the hst3Δ sectoring phenotype. Collectively, the sectoring patterns suggested that Hst3 promoted the stability of silencing through the deacetylation of histone H3 K56-ac. Whereas the deletion of HST3 caused a threefold increase in the half-sector frequency of wild type (p = 3 × 10−4; Student's t test), it did not significantly affect the half-sector frequency of the H3 K56Q mutant (p = 0.3; Student's t test) or the surprisingly high half-sector frequency of the H3 K56R mutant (p = 0.06; Student's t test) (Table 1 and Figure 7D). Thus, the state of histone H3 K56 affected the stability of silencing at HML, and amino acid substitutions of this residue were epistatic to the hst3Δ mutation.
Colonies derived from cells in which the only source of histone H3–H4 genes was a plasmid-borne copy of HHT2-HHF2 exhibited more sectoring than colonies of cells containing both HHT1-HHF1 and HHT2-HHF2 at their native chromosomal loci (Figure 7—figure supplement 2). Assuming the decrease in gene copy number led to a decrease in protein abundance, silencing was likely affected by levels of histones H3 and H4 that were either limiting or improperly balanced with the levels of histones H2A and H2B. Consistent with these possibilities, histone dosage has previously been shown to affect various functions of heterochromatin (Moore et al., 1979; Moore et al., 1983; Venditti et al., 1999).
In principle, the effect of histone H3–H4 gene dosage should sensitize silencing in all genetic backgrounds. Whereas the reduction from two copies of the histone H3–H4 gene pair to one copy did enhance the sectoring phenotype of hst3Δ colonies (compare panels A and C in Figure 7 and panels A and B in Figure 7—figure supplement 1), it did not increase the half-sector frequency in the hst3Δ background as it did in the wild-type background (Table 1 and Figure 7B,D). Therefore, the degree to which histone H3–H4 gene dosage affected silencing, at least during the first cell division of colony growth, depended on the genetic background.
Live-cell imaging revealed a variety of switching patterns
Whereas colony sectors revealed the history of heterochromatin dynamics, we sought to also capture these events in real time at single-cell resolution to determine whether observable losses of silencing exhibited a fixed pattern or were more stochastic. Therefore, we monitored the fluorescence of cells containing HMLα::cre and the RFP-GFP cassette as they divided in a chamber supplied with fresh medium. Most cells remained red throughout the duration of the time course. Consistent with the sectors observed within colonies, rare RFP-to-GFP switches occurred in a subset of the lineages (see Video 1 for an example). The rate of switching observed with live-cell imaging was within 10% of the rate of switching determined by the half-sector assay.
Video 1
RFP-to-GFP switch captured in real time.
Time-lapse imaging of wild-type cells containing HMLα::cre and the RFP-GFP cassette. Images were taken every 10 min for 13 hr.
The time lag between the expression of cre and the ultimate maturation of GFP limits the temporal resolution from such analyses. Nonetheless, a survey of the pattern of switch events allowed some useful inferences. For each RFP-to-GFP switch, we determined whether cells were unbudded, small-budded, or large-budded at the moment when GFP expression was initially detected. We observed a fluorescent switch in 24 different cells, nine of which were unbudded and thus in the G1 phase during the onset of GFP expression (Figure 8A,D). Multiple occurrences of both patterns 1 and 2 (Figure 8D) indicated that silencing loss followed by recombination was not restricted to either the mother or daughter cell (Given that pattern 3 was supported by only one example, we refrained from offering an interpretation of this event).
Figure 8 with 1 supplement see all
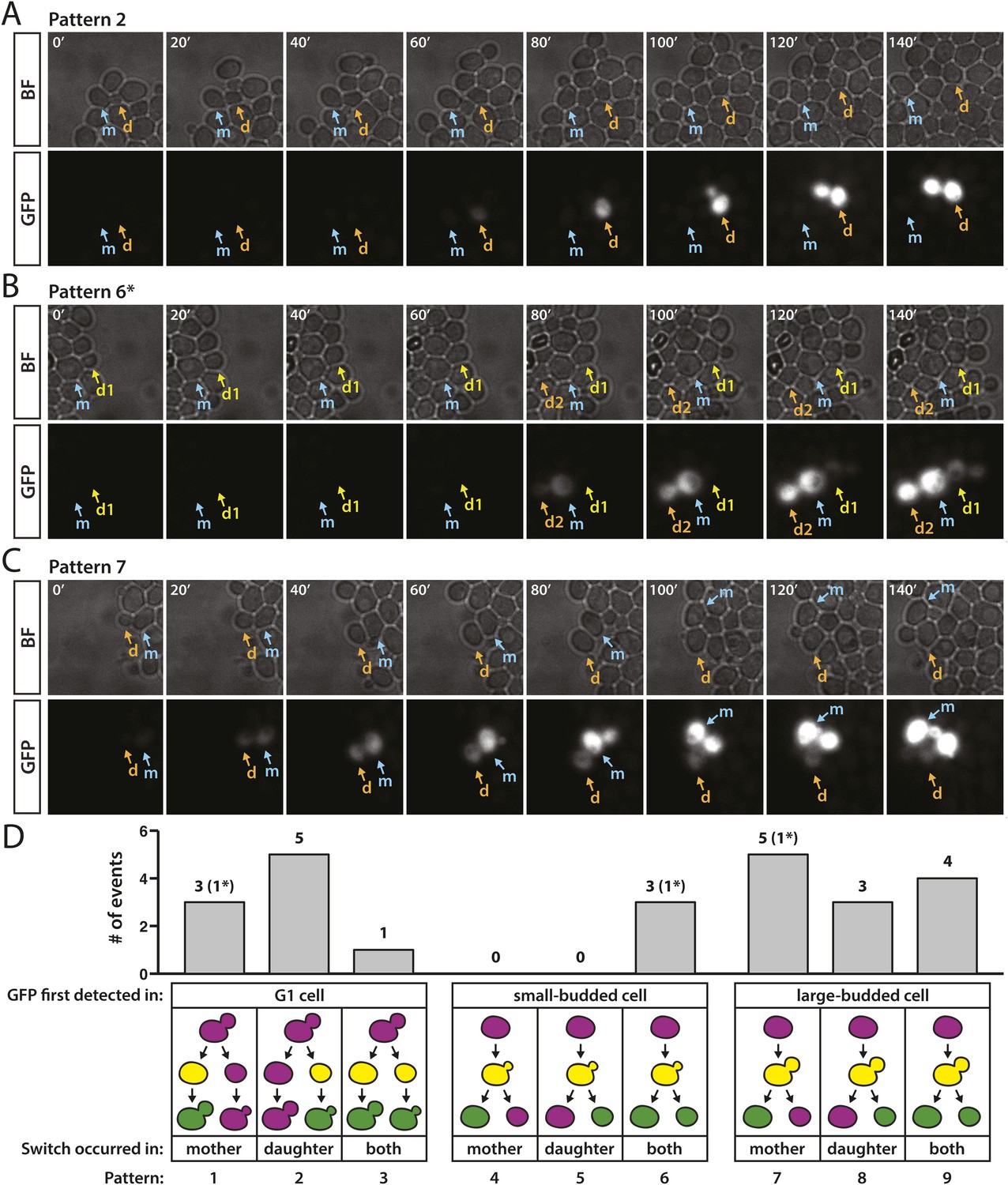
Patterns of RFP-to-GFP switches observed in real time.
(A–C) Brightfield (BF) and GFP fluorescence montages (shown in 20-min intervals, 140 min total) showing different switching patterns of wild-type cells containing HMLα::cre and the RFP-GFP cassette. Labeled arrows track the mother (m) and daughter (d) cells of interest. (A) GFP expression first detected in an unbudded daughter cell. (B) GFP expression first detected in a small-budded cell. Both the mother (m) and daughter (d2) cells remained GFP-positive (pattern 6). *A second RFP-to-GFP switch occurred in the daughter (d1) of the preceding cell division (see Figure 8—figure supplement 1, pedigree B). (C) GFP expression first detected in a large-budded cell. Only the mother (m) cell remained GFP-positive. (D) Distribution of different switch patterns. RFP-to-GFP switches were initially categorized by bud morphology at the time that GFP expression was first detected (depicted as yellow cells in the cartoons since RFP was still present). Events in each bud category were then classified according to whether the mother cell, daughter cell, or both underwent recombination of the RFP–GFP cassette, as determined by whether they continued to express GFP (depicted as green cells in the last phase of the pedigree cartoons). Asterisks denote the number of events within a particular category that were associated with a second switch in a directly related cell (see Figure 8—figure supplement 1).
Of the remaining cells that switched, three cells had small buds when GFP fluorescence was first detected and 12 had large buds (Figure 8D). All three small-budded cells gave rise to mothers and daughters that both remained green (pattern 6) (Figure 8B,D). Since the bud emerges during early S phase, and there were no cases of only the mother or daughter cell remaining green following the onset of GFP expression in a small-budded cell (patterns 4 and 5), it was likely that recombination of the loxP sites occurred prior to DNA replication. Alternatively, recombination may have occurred shortly after DNA replication at both newly synthesized copies of the RFP-GFP cassette. In contrast, 8 of the 12 cells that expressed detectable GFP as large-budded cells gave rise to mother-daughter pairs in which only the mother or daughter remained green (patterns 7 and 8) (Figure 8C,D), indicating that Cre catalyzed recombination after S phase and acted on only one of the two RFP-GFP cassettes. Switches that followed patterns 7 and 8 also demonstrated that the time between recombination, which occurred some point after DNA replication, and GFP detection, which occurred when a large bud was present, spanned only a fraction of a complete cell cycle.
For three of the RFP-to-GFP switches, we observed a second switch in the daughter cell that arose from the immediately preceding cell division (Figure 8B,D and Figure 8—figure supplement 1). These paired switches, which occurred either simultaneously or within 40 min of each other, could have reflected either two loss-of-silencing events in directly related cells, or rather one loss-of-silencing event that happened when the mother cell and its daughter still shared a cytoplasm, assuming there was perdurance of the Cre protein in both cells and a delay in recombination. These patterns are considered further in Figure 8—figure supplement 1 and the ‘Discussion’.
Discussion
Several lines of evidence establish the dynamic nature of heterochromatin structure. The incorporation of newly synthesized Sir3 into silenced chromatin during G1-phase arrest suggests that at least some Sir3 binding to nucleosomes is transient (Cheng and Gartenberg, 2000). Similarly, fluorescence-recovery-after-photobleaching experiments have shown that heterochromatin protein 1 (HP1), a structural component of heterochromatin in other eukaryotes, can rapidly associate and dissociate from heterochromatic loci (Cheutin et al., 2003; Festenstein et al., 2003). However, the consequences of these structural fluctuations on the underlying functions of heterochromatin remain poorly understood. Here, we showed that heterochromatin dynamics had a functional impact on the transcriptional repression imparted by Sir proteins. Moreover, we have established a route to a comprehensive understanding of all factors influencing heterochromatin dynamics.
Inherent dynamics of heterochromatic gene silencing
At a low rate, the heterochromatic loci HML and HMR underwent losses of silencing that had escaped detection by all previous efforts, due to the combination of their rarity and transience. Independent observations indicated that silencing was promptly re-established following its loss. First, HMLα::cre was fully silenced in the descendants of cells that switched to GFP expression, showing that cre expression was only temporary. Secondly, single-molecule RNA FISH did not detect any wild-type cells expressing HMLα::cre to the average level of sir4Δ cells, implying that when silencing was lost in wild-type cells, it was restored before cre transcription could reach that level. These data, taken together with the inability of less sensitive assays to capture expression of HML or HMR, suggested that losses of silencing were rare and short-lived.
The detection of silencing loss in a subset of wild-type cells supported a model where Sir proteins and the transcriptional machinery constantly compete for access to the silent mating-type loci. Such a concept is consistent with the dynamic exchange of structural components in heterochromatin, the continuous recruitment of Sir proteins by the silencers (Cheng and Gartenberg, 2000), and the ability of increased levels of a transcriptional activator to overcome the silenced state (Aparicio and Gottschling, 1994; Ahmad and Henikoff, 2001; Xu et al., 2006). This model could explain why losses of silencing were more frequent when cells contained only half the normal gene dosage of SIR1, SIR3 or SIR4. Perhaps losses of silencing at HML and HMR resulted from stochastic fluctuations in the local concentration of silencing factors. Alternatively, specific perturbations such as DNA replication may have disrupted the binding of Sir proteins to nucleosomes, thereby creating an opportunity for RNA polymerase to access heterochromatin.
Defining the roles of Sir1 in silencing
The contribution of Sir1 to silencing in wild-type cells was previously underappreciated. Heritability of the repressed state in a subset of sir1Δ cells showed that Sir1 was not absolutely required for the maintenance or inheritance of silenced chromatin, and suggested that Sir1 primarily functioned in establishment (Pillus and Rine, 1989). However, the presumed role of Sir1 seemed paradoxical under the assumption that wild-type cells never lose silencing and thus do not undergo events requiring re-establishment. This point of confusion can now be resolved by two key findings. First, the Cre-based assay revealed that transient transcription of HML and HMR occurred at a low rate in wild-type cells. Therefore, Sir1 may serve a purpose in the recovery of silencing on the rare occasion that it is lost. Secondly, losses of silencing occurred more often in cells lacking Sir1, indicating that Sir1 contributed to either the maintenance or inheritance of silencing, or both.
Previous measurements of silencing established that two phenotypic states exist within a population of sir1 cells (Pillus and Rine, 1989; Xu et al., 2006), yet the levels of expression in each of the two subpopulations remained unresolved. For example, in sir1Δ cells containing a fluorescent reporter under control of the URA3 promoter at HML, the fluorescence intensity profile of each subpopulation shifted in relation to the profiles of wild-type cells and sir3Δ cells depending on whether the URA3 trans-activator Ppr1 was present (Xu et al., 2006). By directly measuring the transcription of a gene from the native α2 promoter at HML with single-cell and single-molecule resolution, we showed that the silenced subset of sir1Δ cells was fully repressed, whereas the expressed subset of sir1Δ cells transcribed cre to the level of sir4Δ cells.
Together, the Cre-based assay and the RNA FISH measurements resolved two separable aspects of silencing: the level of repression and the rate at which it is lost. Deletion of SIR1 affected the rate of silencing loss, but not the level of repression since approximately half of all sir1 cells completely lacked cre transcripts. Moreover, the persistence of RFP expression within regions of sir1Δ colonies containing either HMLα::cre or HMRα::cre and the RFP-GFP cassette underscored the remarkable ability of heterochromatin to template its own replication for multiple cell divisions.
Is there a role for a dynamic dimension to heterochromatic gene silencing?
In principle, the dynamic nature of silenced chromatin could result from stochastic processes intrinsic to, for example, the binding constants of Sir proteins for their nucleosomes and serve no useful role. Alternatively, the stability of silencing at HML and HMR could be tuned high enough to achieve cell-type specificity, yet just low enough to allow for events such as DNA replication or mating-type interconversion. Haploid cells of a homothallic strain can switch mating types through repair of a double-stranded DNA break at MAT using HML or HMR as a template. Therefore, successful switching requires that the invading MAT strand, along with the accessory proteins involved in recombinational repair, gain access to the heterochromatic donor sequence. We speculate that the dynamics of silenced chromatin may contribute to the efficiency of mating-type interconversion.
Role of the Hst3 sirtuin and its histone H3 target in heterochromatin stability
The stability of silencing at HML depended in part on Hst3 and the deacetylation of H3 K56-ac. H3 K56 localizes to the DNA entry/exit region of the nucleosome (Luger et al., 1997), and in the acetylated state it is thought to promote transient DNA ‘breathing’, during which the DNA partially unwraps from the nucleosome (Neumann et al., 2009; Simon et al., 2011; North et al., 2012). Furthermore, H3 K56-ac is important for transcriptional activation (Williams et al., 2008; Värv et al., 2010), and telomeric heterochromatin is more accessible to the bacterial dam DNA methylase in various H3 K56 mutants (Xu et al., 2007). Collectively, these observations suggest that H3 K56-ac destabilizes the nucleosome conformation and thereby renders the DNA more accessible to RNA polymerase.
H3 K56 acetylation and Hst3 expression are both regulated by the cell cycle. Whereas H3 K56 acetylation peaks during S phase (Masumoto et al., 2005), Hst3 expression does not peak until G2/M phase (Spellman et al., 1998; Celic et al., 2006; Maas et al., 2006). Therefore, heterochromatin may be especially susceptible to transcription during the time after K56-acetylated histone H3 is deposited into chromatin, but before Hst3 removes the modification.
A tractable tool for measuring the dynamics of heterochromatic repression
As previous measurements of HML silencing were unable to distinguish hst3 mutants from wild type, our detection of a stability phenotype in this mutant underscored the importance of measuring fluctuations in repression for a fuller understanding of heterochromatin. The ability of this assay to capture the existence of transient events through their conversion to permanent, heritable marks can now allow a comprehensive evaluation of the contribution of any and all genes to heterochromatin dynamics. By preserving a historical record of transcription, the Cre-based assay also provides the opportunity to measure the effects of transient environmental or cellular stresses on silencing subsequent to their occurrence. Such analyses will inform our understanding of the mechanisms by which cells maintain integrity of the epigenome.
Measuring the frequency of half-sectored colonies allowed for quantification of the rate at which silencing was lost. In general, half-sector frequencies were consistent with the overall patterns of colony sectoring. However, strains bearing the histone H3 K56R substitution, for example, were indistinguishable from wild type by sectoring pattern, yet showed a significant increase in the frequency of half-sectored colonies. While we do not fully understand why silencing in histone H3 K56R mutants appeared to be less stable at the two-cell stage of colony growth than at later stages, the complexities of colony development may offer a simple explanation. The physiology of cells during the first cell division of colony growth, at which point silencing was measured by half-sector analysis, would be expected to differ from the physiology of cells during later divisions. Therefore, it is possible that the histone H3 K56R mutation rendered heterochromatin more sensitive to the physiological state of cells. Overall, both assays were informative, and sectoring patterns in particular provided the opportunity to survey heterochromatin dynamics throughout the various microenvironments and metabolic states of colonies.
A direct comparison could not be made between the rate of RFP-to-GFP switching, as determined by half-sector analysis, and the frequency of wild-type cells expressing HMLα::cre, as determined by FISH. Whereas half-sector analysis measured events throughout the duration of an entire cell cycle, the FISH method measured expression in a snapshot of time. Furthermore, the background levels of FISH signal in the negative control strain, though very low, limited quantitative interpretation of the low cre RNA levels in wild-type cells. The FISH measurements did show, however, that any detectable transcription of HMLα::cre in wild-type cells was both rare and restricted to relatively low levels. These observations implied that a small number of cre transcripts produced enough recombinase to catalyze the RFP-to-GFP switch. Further experiments would be necessary to determine the number of cre RNAs sufficient for recombination.
Capturing losses of silencing in real time
The live-cell imaging of pedigrees of wild-type cells endowed with the Cre-based assay reflected our first efforts to visualize in real time the silencing loss events whose history is written in the number and size of green colony sectors. In principle, RNA FISH by itself could provide better time resolution, but morphological classification of fixed spheroplasts using brightfield images was unreliable, especially with the high density of cells needed to capture rare events. Despite the time lag between loss-of-silencing events and the production of the GFP chromophore, several points were clear. First, there was no obvious mother-daughter bias as to which cell contained the more labile HML locus. Secondly, the expression of cre and the subsequent switches from red to green could occur within the same cell cycle. Overall, the variety of patterns we observed were consistent with the possibility that losses of silencing occur at multiple points during the cell cycle, but such conclusions rely on assumptions for which we have no independent verification. Knowing the extent of variation in the time lag between HMLα::cre expression and recombination would help solidify further interpretation. Intriguingly, there were three pedigrees in which a red daughter, born from a mother that switched to green, itself switched to green. Given the low probability of a switch, two switches in such closely related cells implied there was either perdurance of the Cre protein following a single loss-of-silencing event, or some ‘heritable’ instability shared between these two cells, as has been previously suggested in a different context (Kaufmann et al., 2007). Future experiments such as live-cell imaging of transcription should reveal the exact timing of silencing loss and address whether the history of a lineage affects stability.
Materials and methods
Yeast strains
Request a detailed protocolThe strains, plasmids, and oligonucleotides used in this study are listed in Supplementary files 1, 2 and 3, respectively. All strains were derived from the W303 background. Deletions were made using one-step integration of gene disruption cassettes (Longtine et al., 1998; Goldstein and McCusker, 1999; Gueldener et al., 2002) and confirmed by PCR of the 5′ and 3′ junctions. For analysis of HHT2 (histone H3) mutations, the URA3-marked plasmid (pJR2657) in JRY9638 was replaced with a TRP1-marked plasmid (either pJR2759, pJR3212 or pJR3213) by plasmid shuffle to produce JRY9639, JRY9640 and JRY9641. Using the hst3Δ::K.l.LEU2 fwd/rev primers and pUG73 (Gueldener et al., 2002), HST3 was deleted in each of these strains to create JRY9736, JRY9737 and JRY9738.
The cre gene from bacteriophage P1 was integrated at HML by transformation of a sir4Δ strain containing HMLα::K.l.URA3 and counter-selection on medium containing 5-fluoroorotic acid (5-FOA). First, the K.l.URA3 gene was amplified by PCR from pUG72 (Gueldener et al., 2002) using the HMLα::K.l.URA3 fwd/rev primers and integrated at HMLα, replacing the coding sequence of the α2 gene. Then, the cre gene was amplified from pSH47 (Güldener et al., 1996) using the HMLα::cre fwd/rev primers, and the resulting PCR product was transformed into the strain containing HMLα::K.l.URA3. 5-FOA was used to select for HMLα::cre cells, which no longer contained K.l.URA3. Integration of cre was confirmed by sequencing. The resulting strain was mated with a SIR4 strain and sporulated to obtain a haploid containing both SIR4 and the HMLα::cre allele.
To construct the HMRα::cre allele, the K.l.URA3 gene was amplified by PCR from pUG72 (Gueldener et al., 2002) using the HMRa::K.l.URA3 fwd/rev primers and integrated at HMRa in a sir4Δ strain, thus replacing the coding sequence of the a1 gene with K.l.URA3. Then, the HMRα::cre fwd/rev primers were used to amplify a fragment of HMLα::cre from JRY9628 genomic DNA that spanned from the X region to the Z1 region (see Figure 1A), and the resulting PCR product was transformed into the strain containing HMRa::K.l.URA3. 5-FOA was used to select for HMRα::cre cells, which no longer contained K.l.URA3. Integration of cre was confirmed by sequencing. The resulting strain was mated with a SIR4 strain and sporulated to obtain a haploid containing both SIR4 and the HMRα::cre allele.
For integration of the RFP-GFP cassette at the URA3 locus, the PGPD-loxP-yEmRFP-TCYC1-kanMX-loxP-yEGFP-TADH1 sequence in pJR3214 was amplified by PCR using the ura3Δ::RFP-GFP fwd/rev primers. pJR3214 contains the following features: the ampR-containing fragment of pUG73 (Gueldener et al., 2002) resulting from digestion with restriction enzymes NdeI and NotI (2301 bp); the GPD promoter from p413-GPD (Mumberg et al., 1995); a loxP site (5′-ATAACTTCGTATAGCATACATTATACGAAGTTAT-3′), separated from PGPD by a SalI restriction site (GTCGAC); immediately downstream of the first loxP site is the yEmRFP gene (Keppler-Ross et al., 2008), followed by a BglII restriction site (AGATCT) and the CYC1 terminator from pSH63 (Gueldener et al., 2002); the kanMX cassette from pUG6 (Güldener et al., 1996), separated from TCYC1 by a StuI restriction site (AGGCCT); adjacent to the kanMX cassette is a second loxP site (5′-ATAACTTCGTATAGCATACATTATACGAAGTTAT-3′); and immediately following the second loxP site is an EcoRV restriction site (GATATC) and the yEGFP-TADH1 sequence from pKT127 (Sheff and Thorn, 2004).
The version of the RFP-GFP cassette marked with hphMX instead of kanMX (in strains JRY9729 and JRY9730) was constructed by transforming strains containing the kanMX-marked RFP-GFP cassette with PCR product amplified from pAG32 (Goldstein and McCusker, 1999) using the PTEF fwd and TTEF rev primers.
Colony growth and imaging
Request a detailed protocolAll colonies were imaged using a Zeiss Axio Zoom.V16 microscope equipped with ZEN software (Zeiss, Jena, Germany), a Zeiss AxioCam MRm camera and a PlanApo Z 0.5× objective. The colonies shown in Figures 2, 6 and 7 (and the associated figure supplements) were imaged on day 6 of growth, whereas the colonies shown in Figures 3 and 5 (and the associated figure supplements) were imaged on day 3 of growth. Images were assembled using Photoshop (Adobe Systems, San Jose, CA).
In preparation for colony analysis, red cells were selected using medium containing G418 (Geneticin) (Life technologies, Grand Island, NY). Due to the complete loss of silencing in sir2Δ cells, JRY9633 was unable to grow in the presence of G418 and was therefore grown on nonselective medium. Cells were then grown to log phase in Complete Supplement Mixture (CSM) −Trp (Sunrise Science Products, San Diego, CA), and serial dilutions were performed to spread approximately 10 cells/plate (CSM −Trp, 1% agar). To inhibit sirtuin activity, 200 μl of 0.5 M nicotinamide was spread onto CSM −Trp, 1% agar plates prior to plating cells.
For Figure 5 and Figure 5—figure supplement 1, red cells of JRY9729 and JRY9730 were selected using medium containing both G418 (Life technologies) and Hygromycin B (Sigma–Aldrich, St. Louis, MO), and then sporulated for 4–5 days at room temperature on 1% potassium acetate. The resulting tetrads were dissected on CSM −Trp plates and grown into colonies for imaging.
For half-sector analysis, red cells were selected using G418 and then grown to log phase in CSM −Trp. Serial dilutions were performed to plate approximately 100 cells/plate (CSM −Trp). On day 3 of growth, plates were scanned, face up, with a Typhoon Trio (GE Healthcare Life Sciences, Buckinghamshire, England) using the 488-nm laser and 520-nm emission filter to detect GFP fluorescence. Since colonies of cells containing the H3 K56R substitution grew notably slower than colonies of other genotypes, plates were scanned on day 4 of growth for JRY9640 and JRY9737. Colonies that were half green or completely green were manually counted. The total number of colonies was determined with Matlab (MathWorks, Natick, MA) by applying a threshold and converting scans to a binary image (code available upon request). Then, the following equation was used to calculate the half-sector frequency:
# of half-sectored colonies/(# of total colonies–# of fully green colonies)
Three independent experiments were performed for each genotype. See Table 1—source data 1 for the total number of colonies, as well as the number of half-sectored colonies and fully green colonies determined for every experiment.
Flow cytometry
Request a detailed protocolColonies were grown as described above, scraped off the agar surface and resuspended in synthetic complete (SC) medium (Sunrise Science Products). After growth overnight, cultures were diluted back to 0.01 OD600 and then grown in SC to approximately 0.2 OD600. Cells were harvested by centrifugation, and then washed and resuspended in PBS, pH 7.4 on ice. A FC-500 flow cytometer (Beckman–Coulter, Brea, CA) was used to measure the GFP fluorescence intensity of 105 cells/sample. Using FlowJo software (Tree Star, Ashland, OR) cells were gated based on forward and side scatter, and the data were exported as FCS files. Data analysis was performed with Matlab (Mathworks).
RNA preparation for quantitative RT-PCR
Request a detailed protocolTotal RNA was isolated from log-phase cells using hot acidic phenol (Collart and Oliviero, 2001). RNA was digested with DNase I (Roche Diagnostics, Indianapolis, IN) and then purified by phenol-chloroform extraction and precipitation with isopropanol. cDNA was synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA) and oligo(dT) primers. Quantitative PCR of cDNA was performed using the Thermo Scientific DyNAmo HS SYBR Green qPCR Kit (Fisher Scientific, Chicago, IL) and a Mx3000P machine (Stratagene, acquired by Agilent Technologies, Santa Clara, CA). Samples were analyzed in technical triplicate for three independent RNA preparations.
Single-molecule RNA FISH
Request a detailed protocolRNA FISH was performed as described (Raj et al., 2008) using JRY9630, JRY9631, JRY9632 and JRY4012. All probes (see Supplementary file 4 for probe sequences) were designed, synthesized and labeled by Stellaris (Biosearch Technologies, Novato, CA). Probes targeting cre were coupled to Quasar 670 dye (Biosearch Technologies) and probes targeting KAP104 were coupled to CAL Fluor Red 590 dye (Biosearch Technologies). Each probe mix (5 nmol) was dissolved in 100 μl of TE buffer, pH 8.0. For hybridization, 1:10 dilutions of the cre and KAP104 probe stocks were made, and 1 μl of each probe dilution was added to 50 μl of hybridization buffer.
Cells were grown in CSM to 0.2–0.3 OD600 and then fixed and hybridized using the protocol as described (Youk et al., 2010) with additional guidance (Raj, 2013) and the following modification to the spheroplasting procedure: cells were digested with 3 μl of zymolyase (2.5 mg/ml) at 30°C and monitored by phase contrast microscopy until approximately 80% of cells appeared to be digested (35–50 min) (L Teytelman, personal communication, January 2013). The cre and KAP104 probes were hybridized to cells overnight at 30°C in 10% formamide buffer. Samples were then washed, stained with DAPI, washed again and resuspended in 10 μl of glucose-oxygen-scavenging (GLOX) solution without enzymes. Prior to imaging, 10 μl of GLOX solution with enzymes (1% [vol/vol] catalase [Sigma-Aldrich C3515], 1% [vol/vol] glucose oxidase [Sigma-Aldrich G2133], 2 mM Trolox [Sigma-Aldrich 238813]) was added to the sample.
Images were acquired with a DeltaVision RT microscope and softWoRx software (Applied Precision, Issaquah, WA) using a 60 × /1.40 oil-immersion objective (Olympus, Tokyo, Japan), a CoolSNAP HQ CCD camera (Photometrics, Tuscon, AZ) and the following filters from Chroma Technology (Bellows Falls, VT): ET402/15x, ET455/50m (DAPI); ET555/25x, ET605/52m (CAL Fluor Red 590); and ET645/30x, ET705/72m (Quasar 670). Series of z-stacks were acquired with a step size of 0.2 μm. Cell boundaries were hand-drawn using ImageJ software (NIH, Bethesda, MD). Spots were detected and analyzed using Matlab (MathWorks). First, a three-dimensional Laplacian of Gaussian filter (Raj et al., 2008) was applied to raw images of cre and KAP104 RNA. Then, a fluorescence intensity threshold was selected to identify spots within an expected range of sizes. Given that some sir4Δ and sir1Δ cells contained a relatively large number of cre transcripts, some of which appeared to overlap each other in a maximum-intensity projection of z-stacks, spot detection was performed in three dimensions to resolve individual spots (code adapted from L Teytelman and available upon request). Within each experiment (three independent experiments were performed), the same threshold values (one for cre RNA images and one for KAP104 RNA images) were applied across all samples.
Live-cell imaging
Request a detailed protocolCells containing HMLα::cre and the RFP-GFP cassette (JRY9628) were grown to mid-log phase in CSM −Trp, sonicated once for 5 s at a power output of 10%, and then imaged over time using the CellASIC ONIX Microfluidic Platform (EMD Millipore, Hayward, CA). Growth was restricted to a single focal plane within chambers of the Y04C Microfluidic Plate for Haploid Yeast (EMD Millipore). CSM −Trp flowed through the chambers continuously at a pressure of 3 psi. Brightfield and fluorescence images of 46 different fields were taken every 10 min for 13 hr using MetaMorph software (Molecular Devices, Sunnyvale, CA) and an Eclipse Ti microscope (Nikon Instruments, Melville, NY) equipped with a Clara Interline CCD camera (Andor Technology, Belfast, Northern Ireland) and a CFI Apo TIRF 60 × /1.49 oil-immersion objective (Nikon Instruments), which was heated to 30°C using an objective heater (Bioptechs, Butler, PA). Exposure times for yEGFP and yEmRFP fluorescence were each 100 ms. Image analysis and video assembly were performed using ImageJ software (NIH).
References
-
Yeast heterochromatin is a dynamic structure that requires silencers continuouslyGenes & Development 14:452–463.https://doi.org/10.1101/gad.14.4.452
-
Preparation of yeast RNACurrent Protocols in Molecular Biology, Chapter 13:Unit13.12, 10.1002/0471142727.mb1312s23.
-
Genomic characterization reveals a simple histone H4 acetylation codeProceedings of the National Academy of Sciences of USA 102:5501–5506.https://doi.org/10.1073/pnas.0500136102
-
Transcription of functionally related constitutive genes is not coordinatedNature Structural & Molecular Biology 18:27–34.https://doi.org/10.1038/nsmb.1934
-
Epigenetics in Saccharomyces cerevisiaeCold Spring Harbor Perspectives in Biology 5:.https://doi.org/10.1101/cshperspect.a017491
-
A novel yeast silencer: the 2um origin of Saccharomyces cerevisiae has HST3-, MIG1- and SIR-dependent silencing activityGenetics 162:59–71.
-
A new efficient gene disruption cassette for repeated use in budding yeastNucleic Acids Research 24:2519–2524.https://doi.org/10.1093/nar/24.13.2519
-
Genetic drift at expanding frontiers promotes gene segregationProceedings of the National Academy of Sciences of USA 104:19926–19930.https://doi.org/10.1073/pnas.0710150104
-
Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiaeMolecular and Cellular Biology 25:10060–10070.https://doi.org/10.1128/MCB.25.22.10060-10070.2005
-
Validating transcripts with probes and imaging technologyNature Methods 8:S12–S19.https://doi.org/10.1038/nmeth.1573
-
Role of NAD(+) in the deacetylase activity of the SIR2-like proteinsBiochemical and Biophysical Research Communications 278:685–690.https://doi.org/10.1006/bbrc.2000.3854
-
The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylasesProceedings of the National Academy of Sciences of USA 97:5807–5811.https://doi.org/10.1073/pnas.110148297
-
Histone gene multiplicity and position effect variegation in Drosophila melanogasterGenetics 105:327–344.
-
A pharmaco-epistasis strategy reveals a new cell size controlling pathway in yeastMolecular Systems Biology 9:707.https://doi.org/10.1038/msb.2013.60
-
Regulation of the nucleosome unwrapping rate controls DNA accessibilityNucleic Acids Research 40:10215–10227.https://doi.org/10.1093/nar/gks747
-
The establishment of gene silencing at single-cell resolutionNature Genetics 41:800–806.https://doi.org/10.1038/ng.402
-
Single molecule RNA FISH[Web log post]. https://sites.google.com/site/singlemoleculernafish/.
-
Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiaeGenetics 116:9–22.
-
Histone fold modifications control nucleosome unwrapping and disassemblyProceedings of the National Academy of Sciences of USA 108:12711–12716.https://doi.org/10.1073/pnas.1106264108
-
A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein familyProceedings of the National Academy of Sciences of USA 97:6658–6663.https://doi.org/10.1073/pnas.97.12.6658
-
Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridizationMolecular Biology of the Cell 9:3273–3297.https://doi.org/10.1091/mbc.9.12.3273
-
Control of cell type in yeast by the mating type locus: the α1-α2 hypothesisJournal of molecular Biology 147:357–372.https://doi.org/10.1016/0022-2836(81)90488-5
-
Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-riboseProceedings of the National Academy of Sciences of USA 97:14178–14182.https://doi.org/10.1073/pnas.250422697
-
Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: evidence for acetyl transfer from substrate to an NAD breakdown productProceedings of the National Academy of Sciences of USA 98:415–420.https://doi.org/10.1073/pnas.031563798
-
Acetylation of H3 K56 is required for RNA polymerase II transcript elongation through heterochromatin in yeastMolecular and Cellular Biology 30:1467–1477.https://doi.org/10.1128/MCB.01151-09
-
Acetylation in the globular core of histone H3 on Lysine-56 promotes chromatin disassembly during transcriptional activationProceedings of the National Academy of Sciences of USA 105:9000–9005.https://doi.org/10.1073/pnas.0800057105
-
HST3/HST4-dependent deacetylation of lysine 56 of histone H3 in silent chromatinMolecular Biology of the Cell 19:4993–5005.https://doi.org/10.1091/mbc.E08-05-0524
-
Imaging single mRNA molecules in yeastMethods in Enzymology 470:429–446.https://doi.org/10.1016/S0076-6879(10)70017-3
-
Single-RNA counting reveals alternative modes of gene expression in yeastNature Structural & Molecular Biology 15:1263–1271.https://doi.org/10.1038/nsmb.1514
Article and author information
Author details
Funding
National Institutes of Health (GM 031105)
- Jasper Rine
National Institutes of Health (5 T32 GM 7232-34)
- Anne E Dodson
National Science Foundation (DGE 1106400)
- Anne E Dodson
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Leonid Teytelman, Alexander van Oudenaarden and Abby Dernburg for their valuable guidance and provisions for the RNA FISH. We also thank Jack Wang for strain contributions, Oskar Hallatschek for use of the Zeiss Axio Zoom.V16, Liam Holt for use of the microfluidic platform and the Berkeley Stem Cell Center for use of the flow cytometer. We are grateful to members of the Rine laboratory for helpful discussions, especially Meru Sadhu, David Steakley, Sarah Bissonnette, Deborah Thurtle and Gavin Schlissel. We thank Doug Koshland and Georjana Barnes for critical review of the manuscript.
Copyright
© 2015, Dodson and Rine
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,518
- views
-
- 726
- downloads
-
- 82
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 82
- citations for umbrella DOI https://doi.org/10.7554/eLife.05007
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae
eLife 4:e05007.
https://doi.org/10.7554/eLife.05007




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}