Structural Biology: NMR illuminates the pathways to ALS
- Rutgers University, United States
Proteins can fold into many different conformations, and the conformation with the lowest energy is the most stable (Frauenfelder et al., 1991). Two techniques—X-ray crystallography and NMR spectroscopy—are routinely used to work out the structure of the protein in its ground state with atomic resolution. However, a given protein also spends time in other (higher-energy) states, and it has become clear over the past decade that these other states are involved in various biological processes, including protein folding, enzyme catalysis, protein-ligand interactions and protein allostery (Palmer, 2004; Sekhar and Kay, 2013; Tzeng and Kalodimos, 2013). However, as few of the proteins in a sample will be in any of these higher-energy states, most biophysical techniques are not able to detect them.
NMR spin relaxation is a powerful technique that can probe how proteins move over a wide range of timescales, from picoseconds to hours, with atomic resolution (Kay, 2011). Now, in eLife, Lewis Kay of the University of Toronto and co-workers—including Ashok Sekhar as first author—have used two complementary NMR spin relaxation methods to explore the different conformations of an enzyme that has been linked to amyotrophic lateral sclerosis (ALS), a devastating neurodegenerative disease (Sekhar et al., 2015).
Mutations in an enzyme called SOD1 have been identified as the main cause of the inherited form of ALS (Rosen et al., 1993). However, little is known about the cause of sporadic ALS, which has the same symptoms but appears to occur randomly throughout the population. The detailed molecular mechanisms of ALS remain to be clarified, although the inherited and sporadic forms of the disease are thought to share a common pathway. The detection of insoluble SOD1 in ALS patients suggests that mutations and modifications could lead to conformational changes in SOD1 that, in turn, increase the chances that it will misfold and form insoluble aggregates (Bosco et al., 2010).
The SOD1 enzyme can take on many different structural forms. The active form of the enzyme, known as Cu2Zn2SOD1S-S, exists as a dimer made up of two identical subunits. Each subunit is a β-barrel with eight strands, the most important of which are called the Zn loop and the electrostatic loop. Each subunit also contains a zinc (Zn) ion and a copper (Cu) ion. The Zn loop serves as the binding site for the Zn ion, while the electrostatic loop stabilizes the binding of both ions. There is also an intramolecular disulfide bond that further stabilizes the structure by anchoring the Zn loop to the β-barrel.
In contrast, the most immature and unstable form of the enzyme, apoSOD12SH, exists as a monomer and does not contain any metal ions or disulfide bonds. This form of the enzyme is thought to be the toxic species that causes ALS (Rotunno and Bosco, 2013), so there is a clear need to learn more about its structure and other properties. Sekhar et al.—who are at the University of Toronto and the University of Waterloo—found that the Zn and electrostatic loops were much more flexible in apoSOD12SH than in Cu2Zn2SOD1S-S. However, the β-barrel structures of both forms are very similar.
If a molecule switches between its ground and excited states around 200–2000 times per second, and more than ∼0.5% of the sample is in the excited state at any one time, a form of NMR called CPMG relaxation dispersion provides detailed information about the excited states (Sekhar and Kay, 2013). A different NMR technique called CEST can be used when the molecules switches between conformations around 20–300 times per second (Fawzi et al., 2011; Vallurupalli et al., 2012). Sekhar et al. demonstrated that a combination of CPMG and CEST can elucidate multiple exchange processes between different conformations. Moreover, despite the underlying complexity of the processes, they were able to determine the thermodynamic, kinetic and structural properties of four short-lived excited states that are in equilibrium with the ground state of apoSOD12SH (Figure 1). Measurements of chemical shift differences indicated that two of the excited states were actually native conformations of the Cu2Zn2SOD1S-S dimer (Figure 1A).
Figure 1
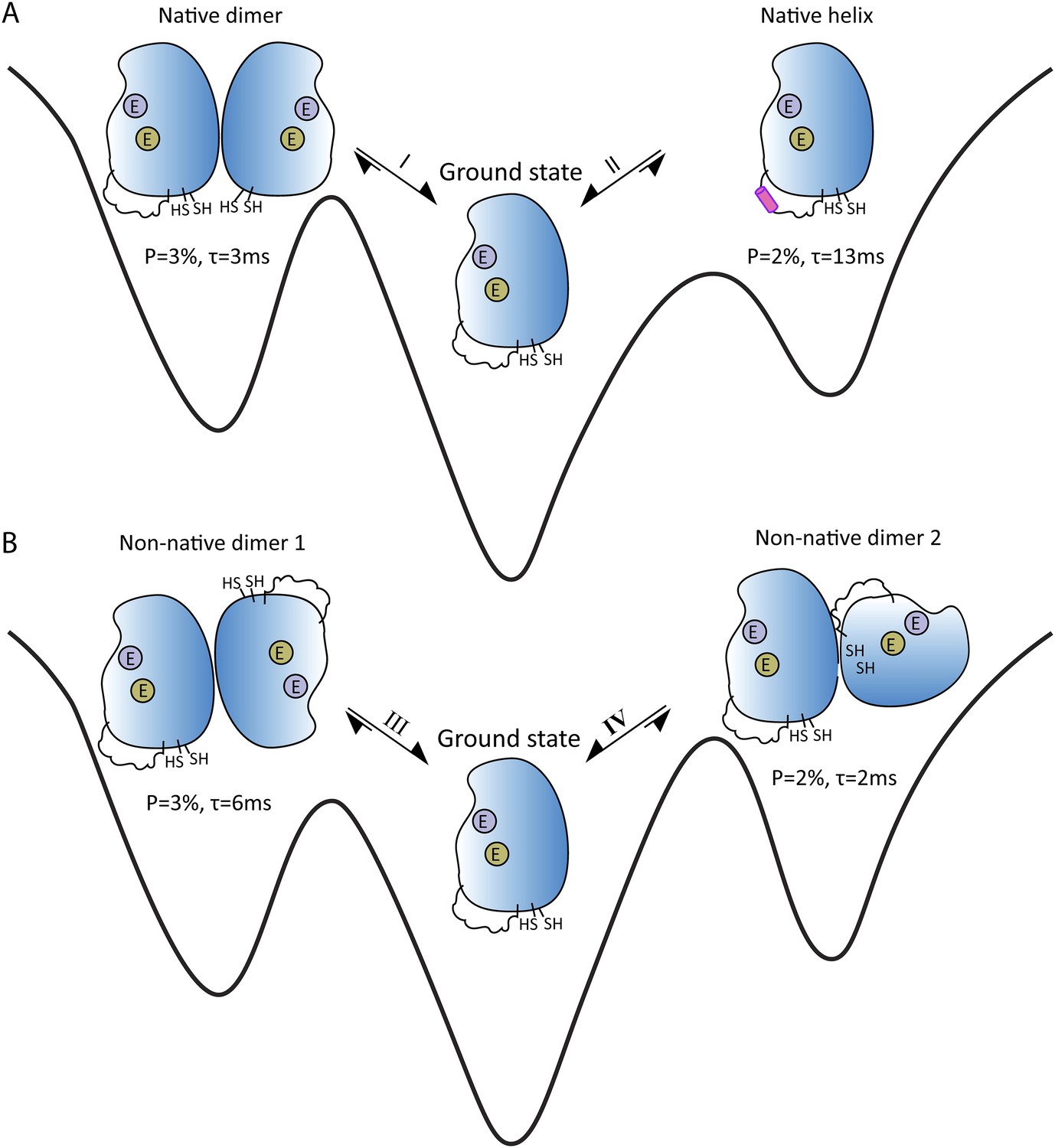
Energy landscape showing the four short-lived excited states that are in equilibrium with the ground state of apoSOD12SH, which is thought to be the form of the SOD1 enzyme that causes ALS.
(A) The ground state (center) is in equilibrium with two native (or working) conformations. Exchange process I leads to the formation of a dimer, with the changes being localized to the surface that forms the interface between the two SOD1 monomers in Cu2Zn2SOD1S-S (left); exchange process II folds the electrostatic loop within the enzyme to form a helix (pink). (B) The ground state (center) is also in equilibrium with two non-native conformations, both of which have aberrant dimer interfaces. These interfaces and the unstructured electrostatic loop in apoSOD12SH may act as sites for the formation of higher-order oligomers and aggregates that may have a role in ALS. The binding sites for metal ions are denoted by purple circles (Zn) and khaki circles (Cu); these sites are empty (denoted by E) for all these states. P is the percentage of enzymes in a state; τ is the lifetime of the state.
Of particular interest are the conformational exchanges between apoSOD12SH and two non-native oligomers (Figure 1B). If factors such as mutations or modifications shift the equilibrium towards these two states, they might serve as starting points for the formation of more complex oligomers that could have a role in ALS.
By showing how NMR spin relaxation methods can reveal such details, the approach developed by Sekhar, Kay and co-workers has the potential to assist in the design of therapeutic molecules that target these oligomers.
References
-
Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALSNature Neuroscience 13:1396–1403.https://doi.org/10.1038/nn.2660
-
The energy landscapes and motions of proteinsScience 254:1598–1603.https://doi.org/10.1126/science.1749933
-
NMR studies of protein structure and dynamics - a look backwards and forwardsJournal of Magnetic Resonance 213:492–494.https://doi.org/10.1016/j.jmr.2011.08.010
-
NMR characterization of the dynamics of biomacromoleculesChemical Reviews 104:3623–3640.https://doi.org/10.1021/cr030413t
-
An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesisFrontiers in Cellular Neuroscience 7:1–16.https://doi.org/10.3389/fncel.2013.00253
-
NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformersProceedings of the National Academy of Sciences of USA 110:12867–12874.https://doi.org/10.1073/pnas.1305688110
-
Allosteric inhibition through suppression of transient conformational statesNature Chemical Biology 9:462–465.https://doi.org/10.1038/nchembio.1250
-
Studying “invisible” excited protein states in slow exchange with a major state conformationJournal of the American Chemical Society 134:8148–8161.https://doi.org/10.1021/ja3001419
Article and author information
Author details
Publication history
Copyright
© 2015, Xie and Kalodimos
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,644
- views
-
- 233
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structural Biology: NMR illuminates the pathways to ALS
eLife 4:e08679.
https://doi.org/10.7554/eLife.08679




{kind=link}