Control of pili synthesis and putrescine homeostasis in Escherichia coli
- Department of Biological Sciences, The University of Texas at Dallas, United States
- Department of Urology, University of Texas Southwestern Medical Center, United States
eLife Assessment
This valuable study presents an interesting analysis of the role of the polyamine precursor putrescine in the pili-dependent surface motility of a laboratory strain of Escherichia coli. The overall data convincingly demonstrate a role in this case. This study presents interesting findings for those studying uropathogenic bacteria, and those studying bacterial polyamine function.
https://doi.org/10.7554/eLife.102439.3.sa0Significance of the findings:
Valuable: Findings that have theoretical or practical implications for a subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Convincing: Appropriate and validated methodology in line with current state-of-the-art
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
Polyamines are biologically ubiquitous cations that bind to nucleic acids, ribosomes, and phospholipids and, thereby, modulate numerous processes, including surface motility in Escherichia coli. We characterized the metabolic pathways that contribute to polyamine-dependent control of surface motility in the commonly used strain W3110 and the transcriptome of a mutant lacking a putrescine synthetic pathway that was required for surface motility. Genetic analysis showed that surface motility required type 1 pili, the simultaneous presence of two independent putrescine anabolic pathways, and modulation by putrescine transport and catabolism. An immunological assay for FimA—the major pili subunit, reverse transcription quantitative PCR of fimA, and transmission electron microscopy confirmed that pili synthesis required putrescine. Comparative RNAseq analysis of a wild type and ΔspeB mutant which exhibits impaired pili synthesis showed that the latter had fewer transcripts for pili structural genes and for fimB which codes for the phase variation recombinase that orients the fim operon promoter in the ON phase, although loss of speB did not affect the promoter orientation. Results from the RNAseq analysis also suggested (a) changes in transcripts for several transcription factor genes that affect fim operon expression, (b) compensatory mechanisms for low putrescine which implies a putrescine homeostatic network, and (c) decreased transcripts of genes for oxidative energy metabolism and iron transport which a previous genetic analysis suggests may be sufficient to account for the pili defect in putrescine synthesis mutants. We conclude that pili synthesis requires putrescine and putrescine concentration is controlled by a complex homeostatic network that includes the genes of oxidative energy metabolism.
Introduction
Polyamines are flexible aliphatic cations that are found in virtually all organisms. E. coli contains putrescine (1,4-diaminobutane) and lesser amounts of spermidine (N-(3-aminopropyl)–1,4-diaminobutane), and cadaverine (1,5-diaminopentane) (Tabor and Tabor, 1984). Both the pathways and enzymes of polyamine synthesis are redundant (Figure 1). Strains that lack eight of these genes do not contain detectable polyamines, grow slowly aerobically, and do not grow anaerobically (Chattopadhyay et al., 2009). Polyamines interact with nucleic acids and phospholipids, and can affect chromosome and ribosome structure, ribosome-mRNA interactions, and protein and nucleic acid elongation rates (Igarashi and Kashiwagi, 2000). Polyamines modulate, but are not required for, protein synthesis (Igarashi and Kashiwagi, 2015) and expression of hundreds of genes (Yoshida et al., 2004). A major mechanism of polyamine-dependent regulation is translational control of genes for 18 major transcription regulators: four of the seven σ factors – σ18 (FecI), σ24 (RpoE), σ38 (RpoS), and σ54 (RpoN); three histone-like DNA-binding proteins (Fis, H-NS, and StpA); and CpxR, Cya, Cra, and UvrY (Igarashi and Kashiwagi, 2015; Yoshida et al., 2004).
Figure 1
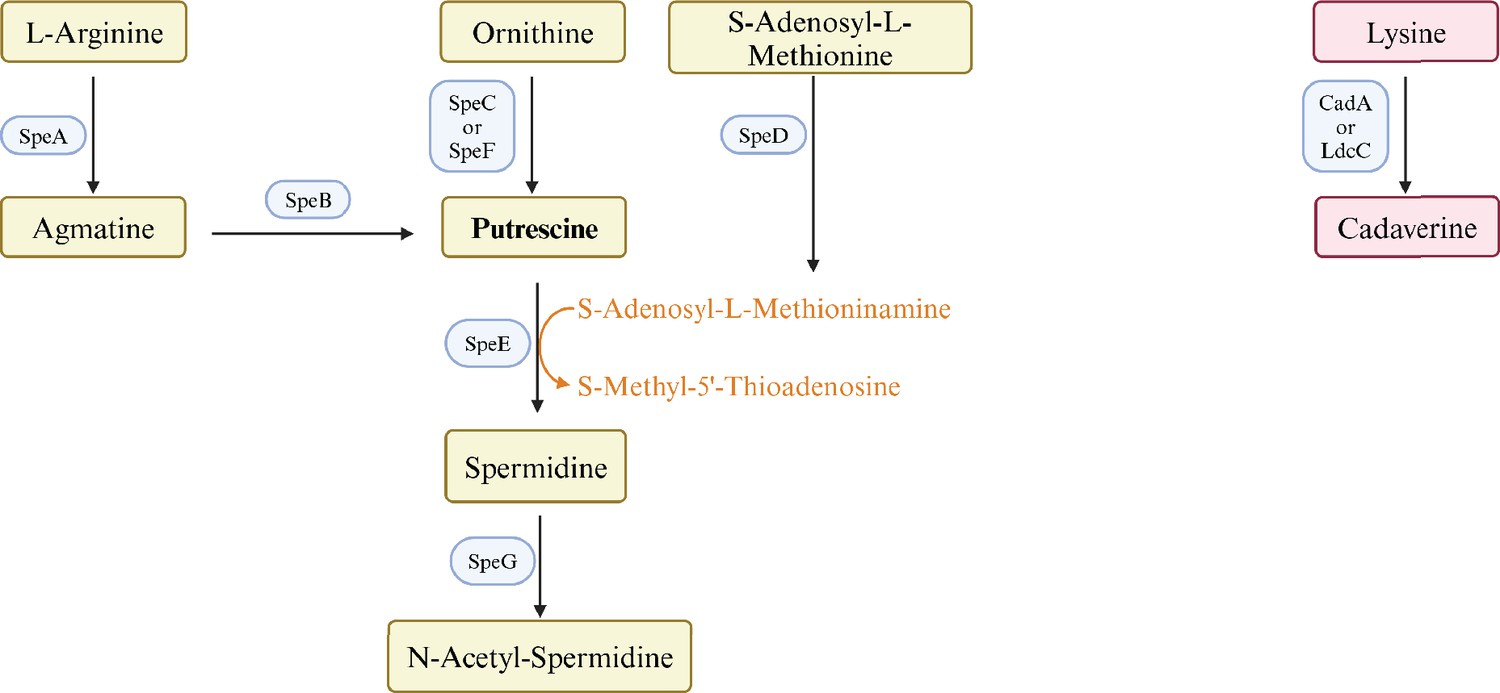
Pathways and enzymes of polyamine synthesis.
Polyamines control flagella-dependent surface motility of Proteus mirabilis and pili-dependent surface motility (PDSM) of E. coli: the latter has been reported to require the polyamine spermidine (Armbruster et al., 2013; Kurihara et al., 2009). While reconstructing putrescine-deficient strains to characterize the basis for the anaerobic growth defect, we noticed that a mutant unable to synthesize spermidine moved normally, while mutants lacking enzymes of putrescine synthesis moved poorly. We characterized the role of polyamines on E. coli surface motility and pili synthesis by genetic analysis, ELISA assays, RT-qPCR, electron microscopy, and RNAseq. We showed that PDSM and pili synthesis required two independent pathways of putrescine synthesis, but did not require spermidine, and was modulated by putrescine transport and catabolism. Results from an RNAseq analysis are consistent with multiple mechanisms of putrescine-dependent control, and, unexpectedly, suggest a putrescine homeostatic network that rewires metabolism to maintain intracellular putrescine.
Results
W3110 surface motility requires pili
An assessment of surface motility for E. coli K-12 and eight derivatives showed five strains covered a plate in 12–18 hr, while four did not traverse a plate in 36 hr (Ambagaspitiye et al., 2019). The slow-moving strains generated genetically stable fast-moving variants which suggests that the former were ancestral. We chose our lab strain of W3110 for further study because (a) it exhibited the least variability in spreading diameter and generated fewer fast-moving variants, and (b) we had previously analyzed this strain for the energy requirements for surface motility (Sudarshan et al., 2021). We refer to our slow-moving lab strain hereafter as W3110 but not all W3110 strains move similarly: the W3110 from the genetic stock center is a fast-moving variant (Ambagaspitiye et al., 2019).
Several results suggested that W3110 requires pili for surface motility. First, the deletion of fimA, which codes for the major type 1 pili subunit, abolished the oscillatory pattern and reduced, but did not eliminate, surface motility, but the deletion of fliC which encodes the major flagellum subunit did not affect surface motility (Figure 2A). Second, electron microscopy confirmed that W3110 isolated from surface motility plates possessed pili (Figure 2B). None of >500 cells expressed flagella. A mixed population of elongated (3–4 µm) and non-elongated (<2 µm) cells were visible, and pili were mostly associated with elongated cells. Finally, W3110 surface motility requires glucose (Sudarshan et al., 2021) which should prevent flagella synthesis via inhibition of cyclic AMP synthesis (Yokota and Gots, 1970). We confirmed that glucose suppressed swimming motility which implies suppression of flagella synthesis in W3110 during surface motility assays (Figure 2C). A W3110 derivative with deletions of both fliC and fimA still exhibited outward movement on a surface motility plate (Figure 2A), but electron microscopy showed the absence of appendages (Figure 2C). In summary, genetics, electron microscopy, and the glucose requirement show that W3110 surface motility requires pili.
Figure 2
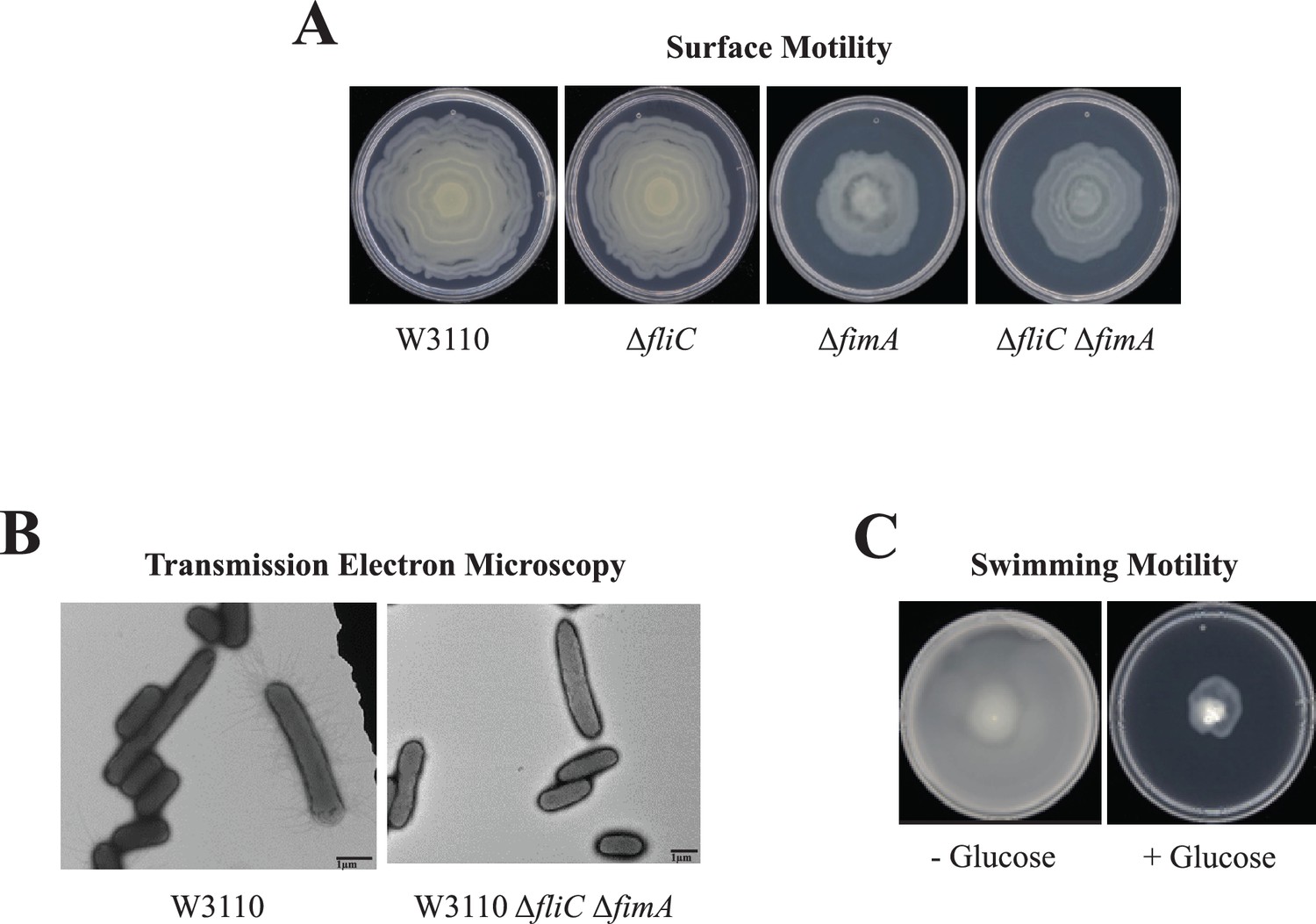
Surface motility of W3110.
(A) Surface motility of parental and derivative strains lacking the major subunits of the flagella (FliC), pili (FimA), or both. (B) Transmission electron microscopy (TEM) images of cells of W3110 and W3110 ΔfliC ΔfimA were taken from the movement’s edge directly from surface motility plates. (C) Swimming motility with and without 0.5% glucose.
Results from W3110 sequencing found an IS1 insertion in fimE which could explain its greater movement that is less variable and greater genetic stability. The FimB and FimE recombinases are the major components of phase variation that determine the orientation of the promoter that initiates transcription of the fimAICDFGH operon which codes for the pili structural proteins. The FimB recombinase favors the productive ON orientation, while the more active FimE recombinase favors the unproductive OFF orientation (Conway et al., 2023; Kelly et al., 2006; O’Gara and Dorman, 2000). The insertion in fimE is the likely basis for the relative stability of W3110.
PDSM required two independent putrescine synthesis pathways
We examined PDSM requirements for polyamine anabolic pathways. A major putrescine biosynthetic pathway is ornithine decarboxylation either by the constitutive SpeC or the low-pH inducible SpeF. TheΔspeC and ΔspeF mutants moved as well as the parental strain, but a ΔspeC ΔspeF double mutant moved less well (Figure 3). SpeA (arginine decarboxylase) and SpeB (agmatinase) catalyze an alternate two-step putrescine synthetic pathway (Figure 1; Tabor and Tabor, 1985): deletion of either gene reduced surface motility (Figure 3). The ΔspeA, ΔspeB, and ΔspeC ΔspeF strains grew as well as the parental strain and had normal swimming motility (Figure 3—figure supplement 1). A ∆speE mutant, which lacks the enzyme for spermidine synthesis, moved on a surface as well as the parental strain (Figure 3).
Figure 3 with 1 supplement see all
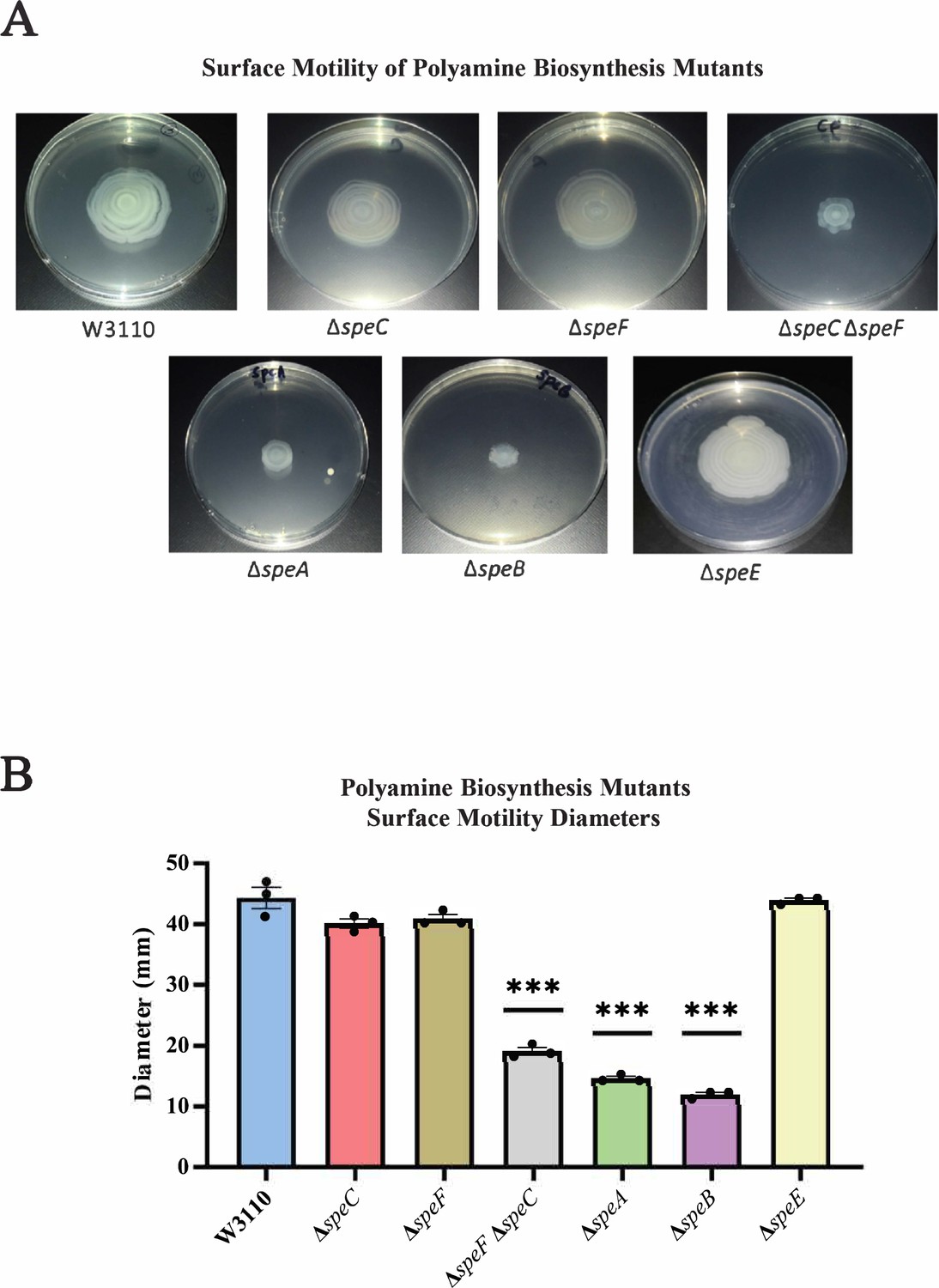
Genetics of surface motility.
(A) Pili-dependent surface motility (PDSM) of mutants with defects in polyamine anabolic genes. All assays were performed in triplicate, and representative images are shown. (B) Diameter of surface movement of polyamine mutants after 36 hr. Error bars represent standard deviations for three independent replicates. Statistical analysis was performed using the Dunnett test of significance: *p<0.05; **p<0.01; ***p<0.001. In this figure the ΔspeB mutant was IM26.
Supplemental putrescine at 1 mM restored the surface motility of a speB mutant (Figure 4A) and a speA mutant (not shown). Spermidine did not restore wild-type movement and, also, caused an unusual pattern of movement in the parental strain (Figure 4A). These exogenous concentrations of putrescine and spermidine had no effect on growth rates in a liquid motility medium (not shown). In summary, PDSM required the simultaneous presence of two independent pathways of putrescine synthesis but did not require spermidine.
Figure 4
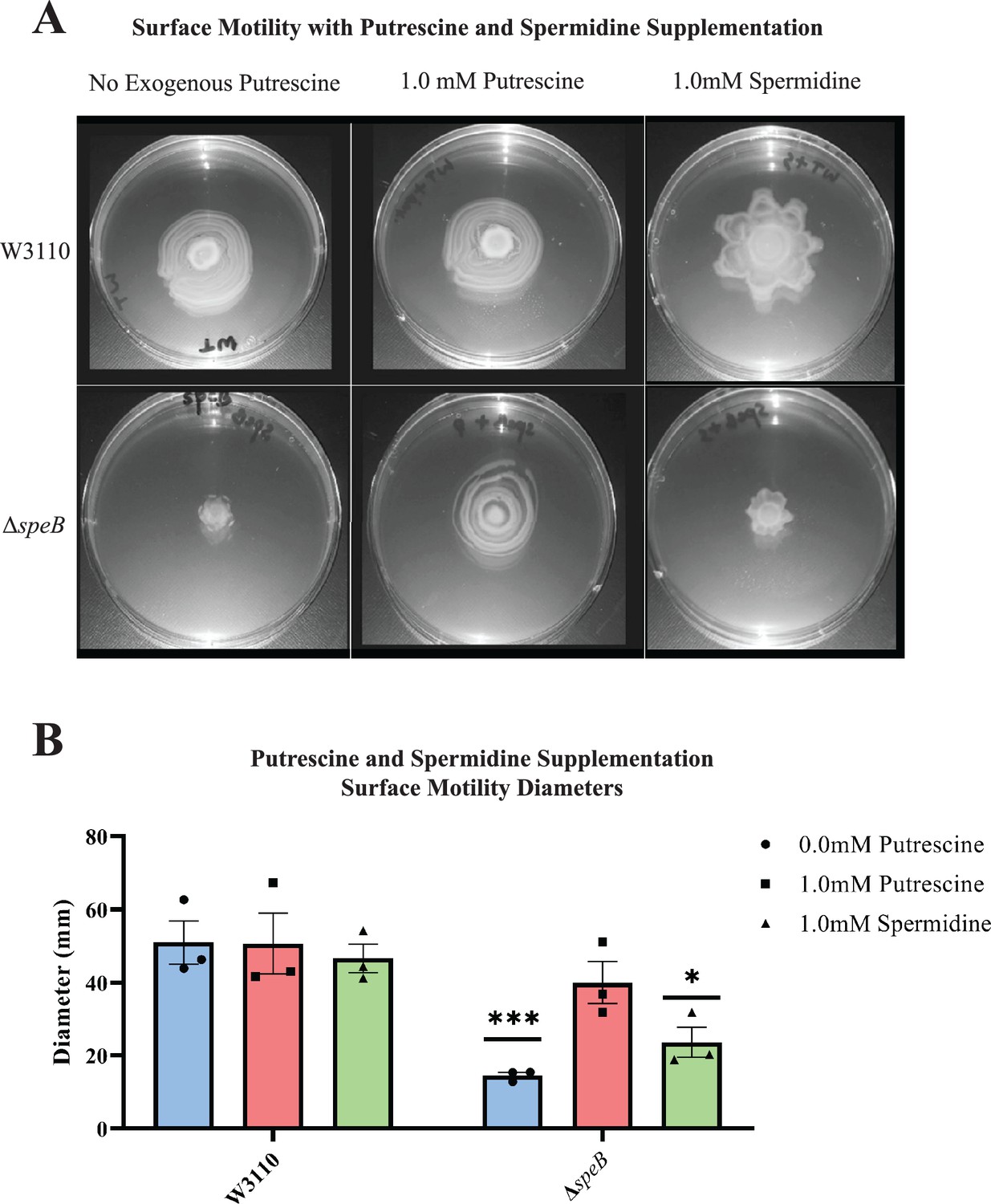
Nutritional supplementation of ΔspeB mutant.
(A) Putrescine and spermidine supplementation of wild-type and ΔspeB strains. (B) Diameter of surface movement of polyamine mutants after 36 hr. Error bars represent standard deviations for three independent replicates. Statistical analysis was performed using the Sadik test of significance: *p<0.05; **p<0.01; ***p<0.001. All assays were performed in triplicates and representative images are shown. In this figure the ΔspeB mutant was IM26.
Loss of putrescine transport systems affected surface motility
E. coli has several polyamine transport systems and one has been reported to be required for surface motility (Igarashi et al., 2001; Kurihara et al., 2011). Mutants lacking the PlaP and PotF putrescine transporters had 50% and 40% reduced surface motility, respectively, and lost the concentric ring pattern (Figure 5A). Loss of the PotE and PuuP transporters affected neither the movement diameter (Figure 5B) nor concentric ring formation (Figure 5A). We note that potF and plaP are more highly expressed than potE and puuP (85, 64, 17, and 6 counts per million transcripts, respectively) for W3110 grown in a liquid motility medium (Hogins et al., 2023a). We conclude that extracellular putrescine and its transport contribute to PDSM.
Figure 5
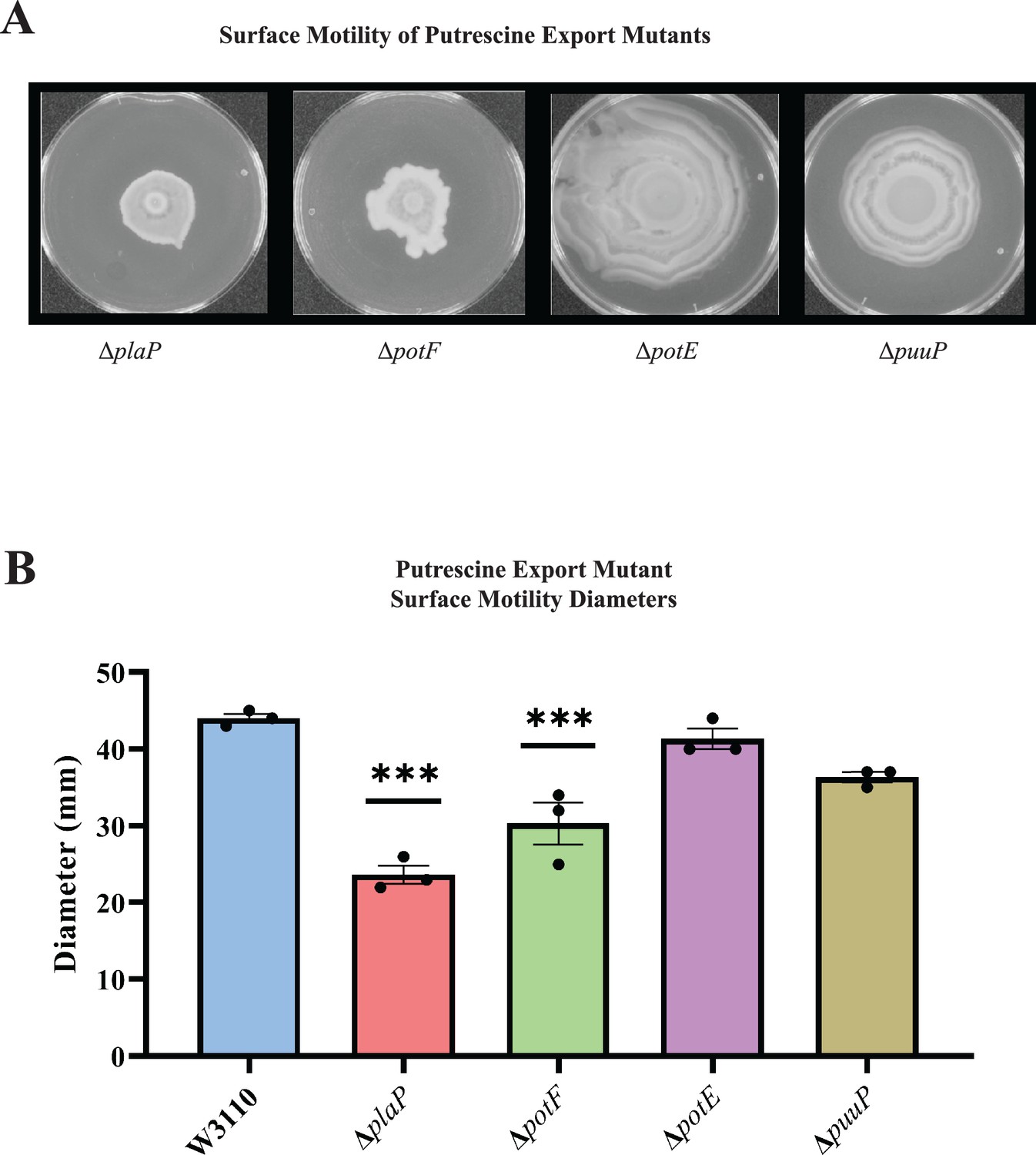
Putrescine transport and surface motility.
(A) Representative images of surface motility for strains defective in putrescine transport genes. (B) Diameter of surface movement of polyamine mutants after 36 hr. Error bars represent standard deviations for three independent replicates. Statistical analysis was performed using the Dunnett test of significance: *p<0.05; **p<0.01; ***p<0.001. All assays were performed in triplicate and representative images are shown.
Putrescine catabolism affected surface motility
Because putrescine catabolism modulates intracellular putrescine concentrations (Schneider et al., 2013; Schneider and Reitzer, 2012), we examined PDSM in putrescine catabolic mutants. The major putrescine catabolic pathway is initiated with putrescine glutamylation (Figure 6A, right column), and a second pathway initiates with putrescine deamination (Figure 6A, left column) (Schneider and Reitzer, 2012). Deletion of either puuA or patA which codes for the first enzymes of their respective pathways did not affect surface motility (Figure 6B). A double mutant moved less well which was unexpected because the proposed higher intracellular putrescine should have stimulated PDSM. Consistent with the stimulatory effect of putrescine, loss of puuR which codes for the repressor of PuuA-initiated pathway genes (Schneider and Reitzer, 2012), impaired PDSM (Figure 6B). One possible explanation to reconcile these seemingly contradictory results is that an optimal polyamine concentration stimulates PDSM, and a high concentration is inhibitory. The next section describes experiments to test this possibility.
Figure 6
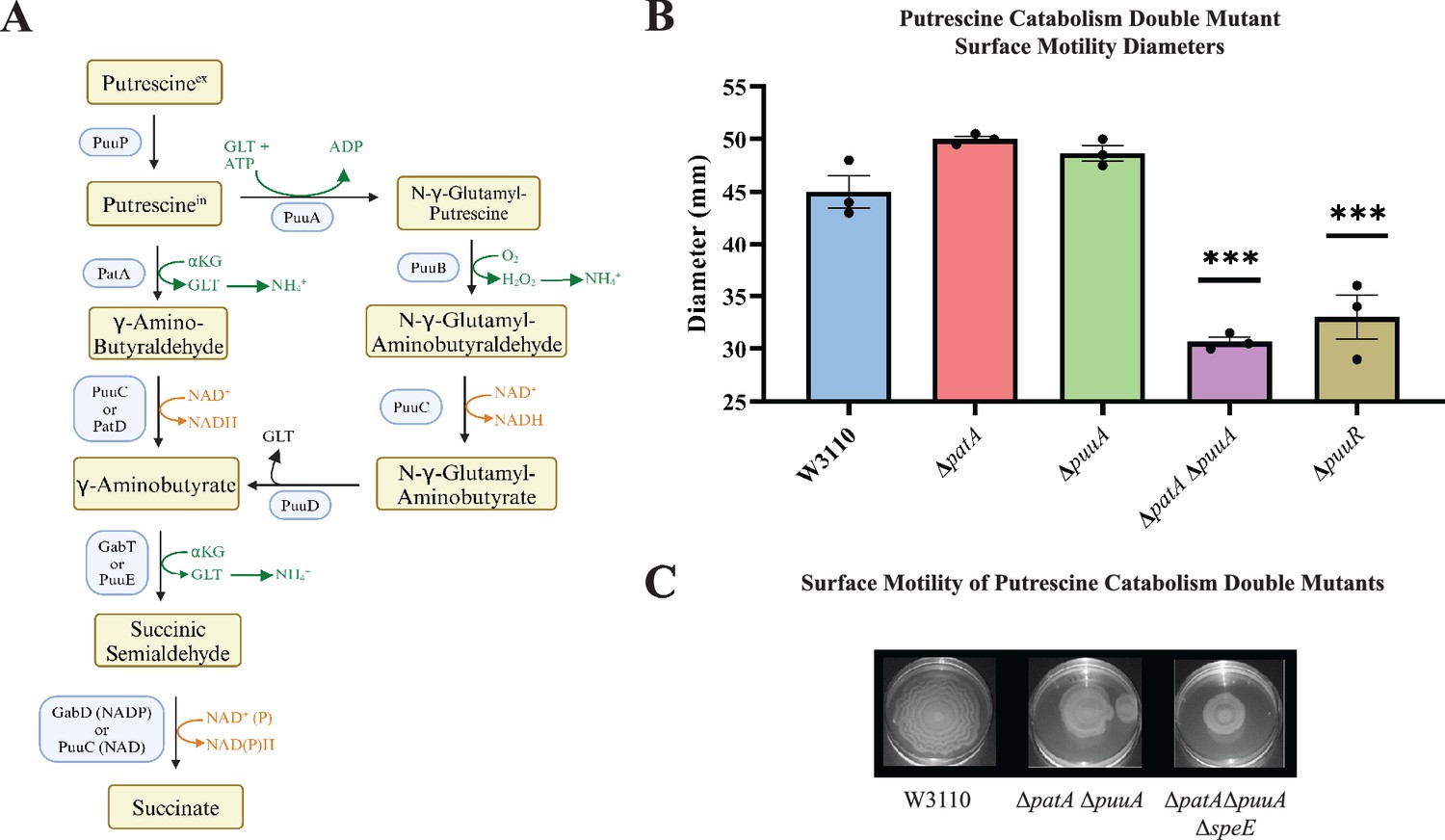
Putrescine catabolism and surface motility.
(A) Pathways and enzymes of putrescine catabolism. (B) Motility diameter of putrescine catabolic mutants after 36 hr. Error bars represent standard deviations for three independent replicates. Statistical analysis was performed using the Dunnett test of significance: *p<0.05; **p<0.01; ***p<0.001. All assays were performed in triplicates and representative images are shown. (C) Effect of loss of speE on the patA puuA catabolic double mutant.
High putrescine reduced the expression of pili genes
High putrescine inhibits the translation of some mRNAs, and high spermidine, a product of putrescine metabolism, inhibits growth (Fukuchi et al., 1995; Sakamoto et al., 2020). If the phenotype of the ΔpatA ΔpuuA double mutant results from spermidine toxicity, then loss of spermidine synthase (SpeE) should reverse the phenotype. This prediction was not met, which argues against spermidine toxicity (Figure 6C). For the speB mutant, 1 mM putrescine stimulated PDSM, and 4 mM putrescine was slightly inhibitory (Figure 7A and B). The concentric ring pattern was observed with 1 mM, but not 4 mM putrescine. RT-qPCR to test transcriptional regulation of pili expression by putrescine showed optimal fimA transcription at 1 mM putrescine in the speB mutant (Figure 7C). Indirect enzyme-linked immunosorbent assays (ELISAs) against FimA suggested that the level of FimA in the speB mutant at 1 mM putrescine was higher than with 4 mM putrescine, but the difference was not statistically significant (Figure 7D). Variation of supplemental putrescine did not affect FimA in the parental strain (Figure 7C and D). Transmission electron microscopy (TEM) showed that pili expression was highest for the speB mutant at 1 mM putrescine (Figure 8), moderate at 0.1- and 4 mM putrescine, and undetectable without putrescine. The cells grown with 4 mM putrescine were shorter and thinner, which suggests stress, possibly mimicking osmotic stress. From these results and those in the previous section, we conclude that pili expression requires an optimal putrescine concentration.
Figure 7
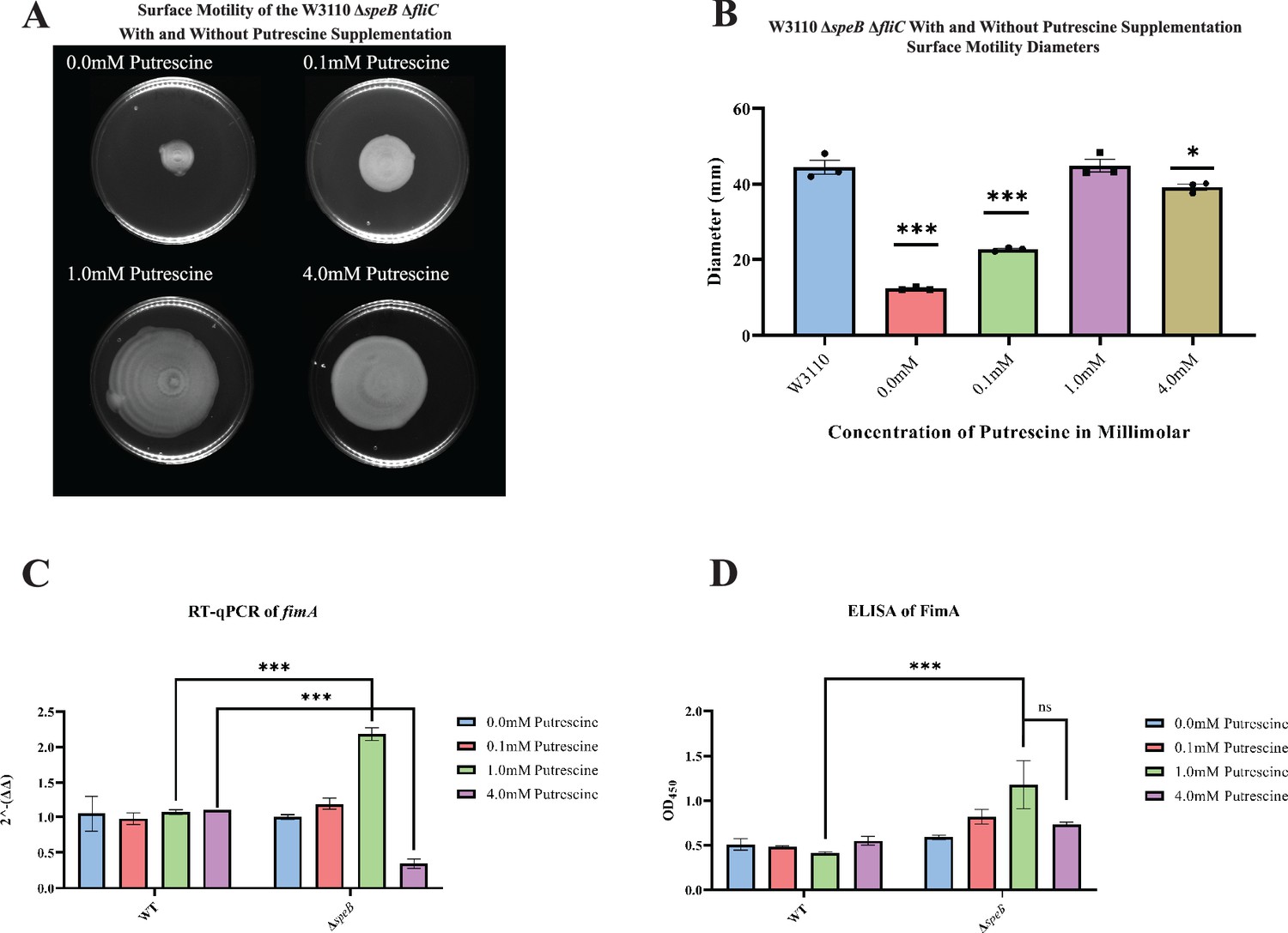
Surface motility and pili expression in W3110 and ΔspeB.
(A) W3110 ΔspeB surface motility with 0.0, 0.1, 1.0, and 4.0 mM exogenous putrescine. All assays were performed in triplicate. Representative images are shown. (B) Average diameter of three separate surface motility plates. The parental strain without putrescine is shown for reference. Significance was determined by comparing the diameters of the ΔspeB mutants in the different concentrations of putrescine compared to the parental W3110. One-way ANOVA was used with Dunnett hypothesis testing to determine p values, ***p<0.001. (C) Reverse transcriptase-quantitative PCR using primers targeting the fimA gene from cells grown in 0.0, 0.1, 1.0, and 4.0 mM of supplemented putrescine. Double deltas were generated by normalizing the parental and the ΔspeB mutant RNA libraries to rpoD t hen comparing fimA expression. Two-way ANOVA was used to determine significance followed by Sadik hypothesis testing. ***p<0.001. (D) Enzyme linked immunosorbent assays using antibodies targeting pili (FimA) from cells grown with 0.0, 0.1, 1.0, and 4.0 mM of supplemented putrescine. Two-way ANOVA was used to determine significance followed by FDR adjusting. ***p<0.001. In this figure the ΔspeB mutant was J15.
Figure 8
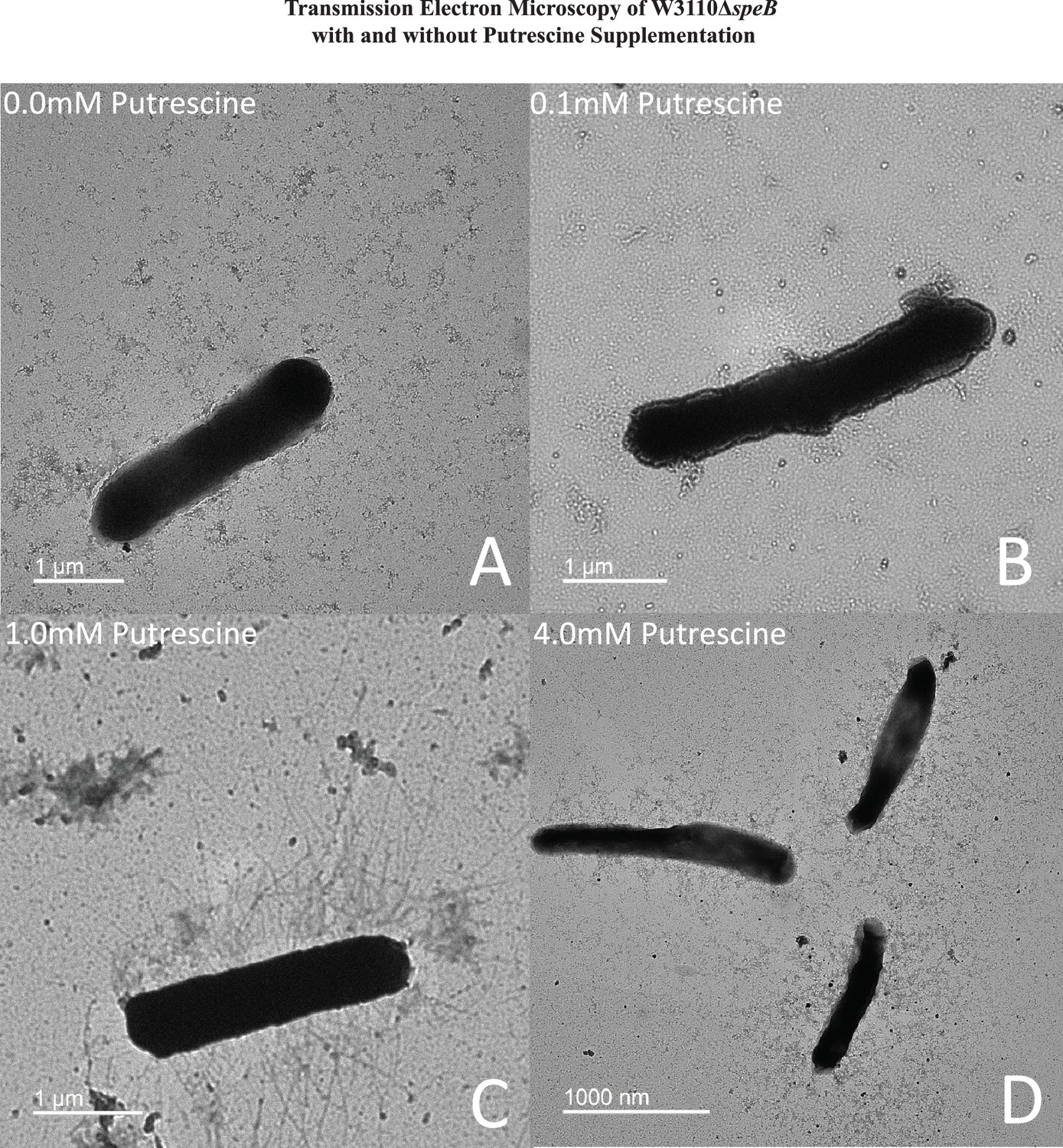
Transmission electron micrographs of ΔspeB mutant cells after surface motility.
Representative images are shown and cells were removed from motility plates with the following exogenous putrescine concentrations: (A) none, (B) 0.1 mM, (C) 1.0 mM, and (D) 4.0 mM. No pili were observed with 0 and 0.1 mM putrescine. Optimal pili production was observed with 1.0 mM putrescine. The bar represents one micron. In this figure the ΔspeB mutant was J15.
RNAseq analysis identifies fim operon expression and energy metabolism as targets of putrescine control
RNAseq analysis was used to further confirm or determine whether putrescine affected (a) transcription of the fim operon that codes for the pili structural genes, (b) fim operon promoter orientation, i.e., phase variation, or (c) energy metabolism: surface motility of W3110 has been shown to require glucose metabolism and oxidative phosphorylation (Sudarshan et al., 2021).
RNAseq analysis
We grew parental W3110 and its ΔspeB derivative with and without 1 mM putrescine. R2 values of pairwise comparisons showed that the transcriptome of the speB mutant grown without putrescine differed from the other transcriptomes which were similar (summarized in Table 1 and shown graphically in Supplementary file 1). A multidimensional scaling plot and a plot of the 100 most variable genes visualize these comparisons (Figure 9A and B). When compared to the parental strain, the speB mutant grown without putrescine had 310 downregulated genes and 159 upregulated genes with at least fourfold differential expression and FDR <0.05. Transcripts for the putrescine-induced puuAP and puuDRCBE operons, which specify genes of the major putrescine catabolic pathway, were reduced from 1.6- to 14-fold (FDR ≤0.02) in the speB mutant (Supplementary file 2). Because higher intracellular putrescine results in higher catabolic gene expression, these results imply lower intracellular putrescine in the ΔspeB mutant.
Figure 9
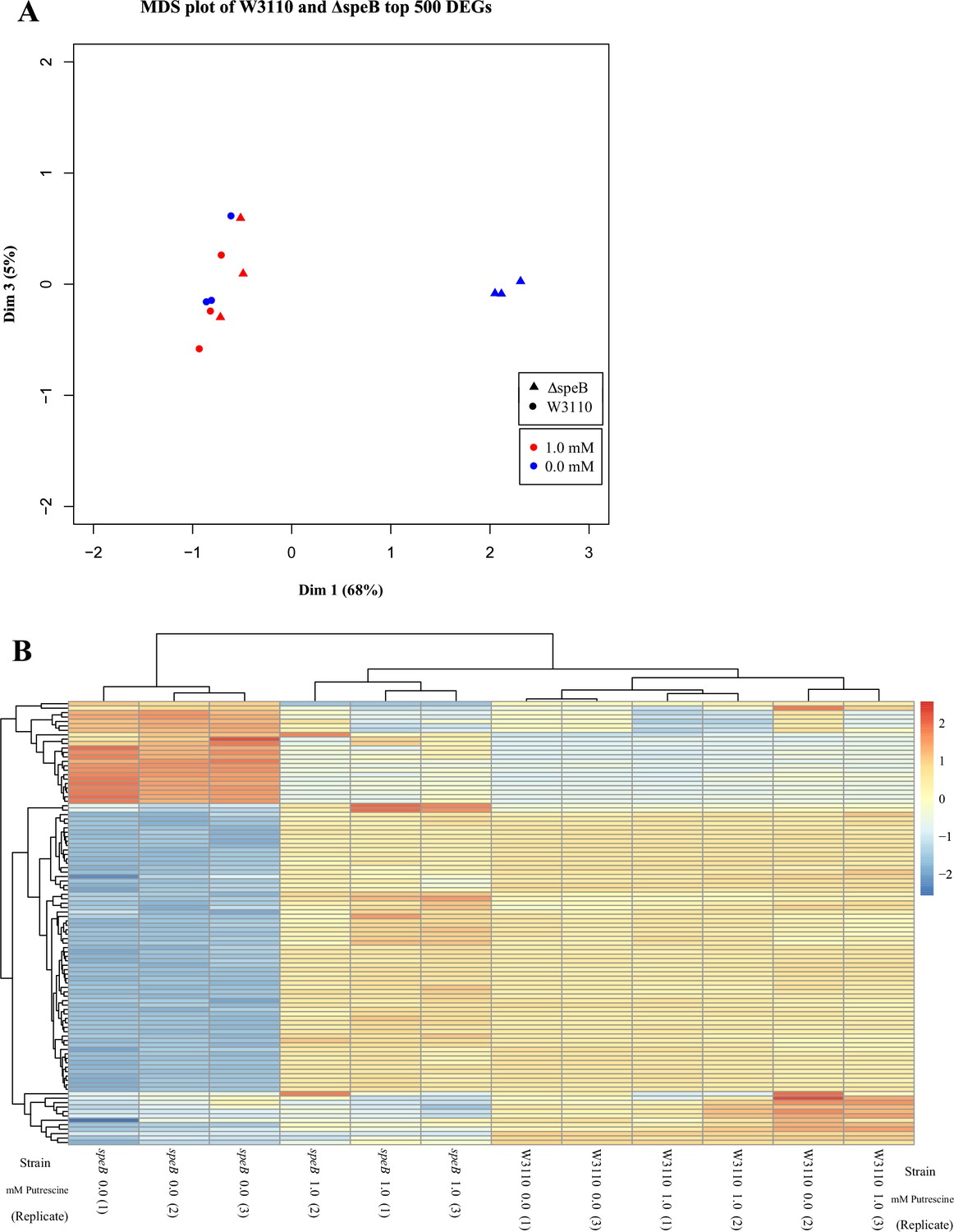
Visual representation of the results from transcriptomic sequencing of W3110 and the ΔspeB mutant’s gene expression in media with and without 1.0 mM putrescine.
(A) Multidimensional scaling plot of the parental and ΔspeB mutant transcriptomes. When grown with 1.0 mM putrescine (red), the ΔspeB mutant transcriptome (triangles) are nearly identical to the parental transcriptomes when grown without and with putrescine (blue and red circles, respectively). When grown without putrescine, the ΔspeB mutant transcriptome (blue triangles) is greatly skewed from transcriptomes of the parental strain and the ΔspeB mutant grown with putrescine. (B) Heatmap of the top 100 most variable genes further demonstrates the distinctiveness of the ΔspeB mutant grown without putrescine and the similarities of the ΔspeB mutant transcriptome when grown with 1.0 mM putrescine supplementation and the parental grown with or without putrescine. In this figure the ΔspeB mutant was J15.
Table 1
Pairwise statistical comparisons of transcriptomes.
Abbreviation: putr is putrescine.
| Strain 1 | Strain 2 | R2 |
|---|---|---|
| Wild-type (1 mM putr) | wild-type (0 mM putr) | 0.950 |
| Wild-type (1 mM putr) | ΔspeB (1 mM putr) | 0.954 |
| ΔspeB (0 mM putr) | ΔspeB (1 mM putr) | 0.820 |
| ΔspeB (0 mM putr) | wild-type (0 mM putr) | 0.796 |
| ΔspeB (0 mM putr) | wild-type (1 mM putr) | 0.786 |
Transcription of the fim operon is reduced in the ΔspeB mutant
Deletion of speB reduced transcripts for genes of the fimA operon (Figure 10A). Numerous transcription factors activate the fim operon (Schwan, 2011; Karp et al., 2018), and, of these, loss of speB reduced hns transcripts, and increased fis, lrp, and qseB transcripts, but had no effect on ihfA and ihfB (Figure 10B). Loss of hns, ihfA, and lrp impaired PDSM (Figure 10C and D), which confirms the requirement for their products for pili synthesis in our strain background.
Figure 10
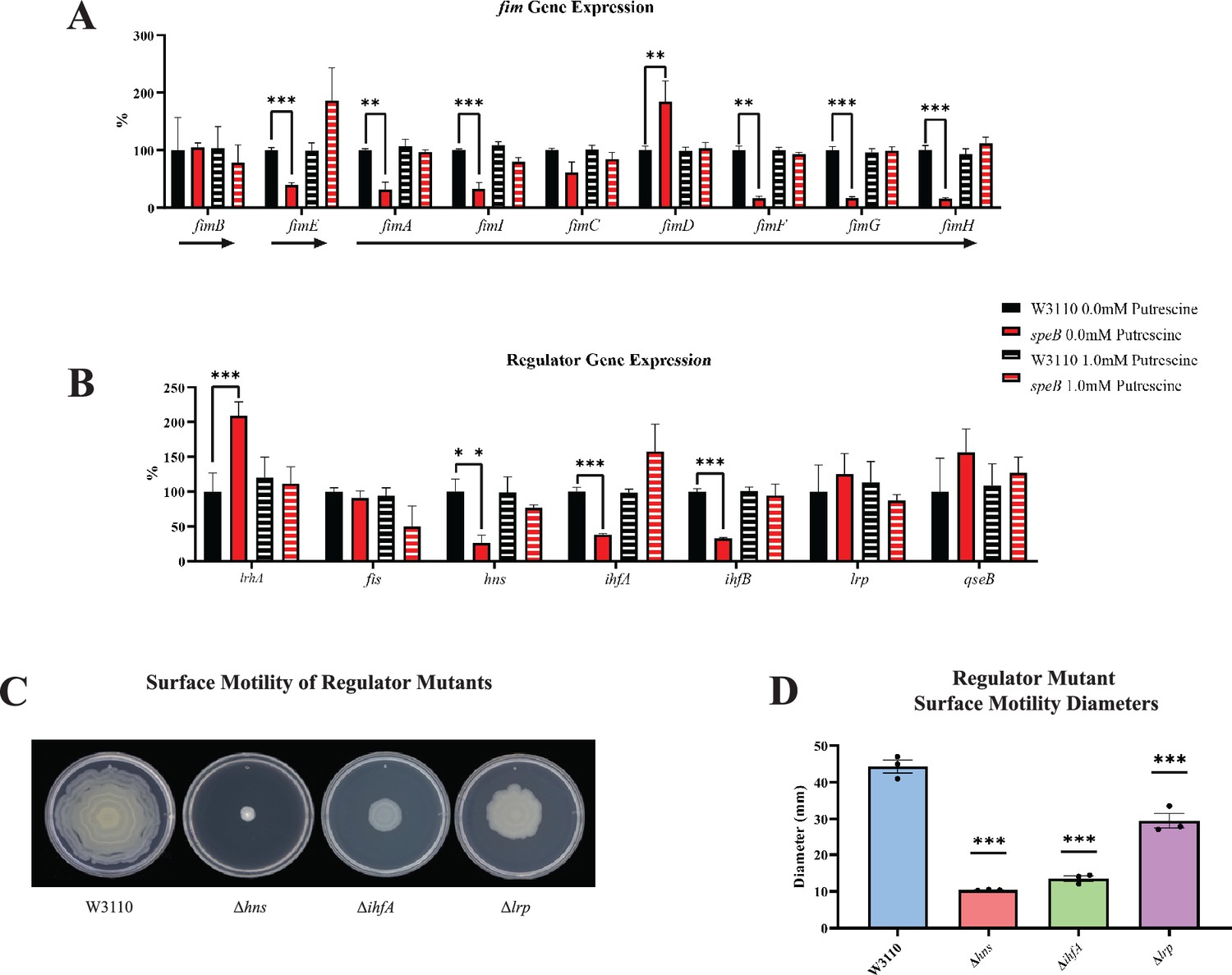
Transcriptomic sequencing of genes for the fim region and regulators that control fim gene expression.
(A) Expression of the fim genes in the parental W3110 and the speB mutant with and without putrescine supplementation. Values were calculated by dividing the individual replicates’ counts per million (CPM) value for each gene by the mean CPM of that gene in W3110 grown without putrescine and multiplying by 100 to yield the percent expression. Arrows below the genes signify the known operons: fimB and fimE belong to single gene operons, while fimAICDFGH belongs to one operon. One-way ANOVA was used to determine significance using Dunnett hypothesis testing. **p<0.01; ***p<0.001. (B) Expression of some regulators known to affect fim gene expression. Values were calculated as described in (A). ***p<0.001. (C) Surface motility of W3110 and three regulator mutants (Δhns, ΔihfA, and Δlrp). A gene found to be significantly different by this transcriptomic analysis (hns) was confirmed to be important in surface motility. In this figure the ΔspeB mutant was J15. (D) Diameter of surface movement of regulatory mutants after 36 hr. Error bars represent standard deviations for three independent replicates. Statistical analysis was performed using the Dunnett test of significance: *p<0.05; **p<0.01; ***p<0.001. All assays were performed in triplicate.
Putrescine does not control fim operon phase variation in W3110
The phase ON-favoring FimB and phase OFF-favoring FimE recombinases control the orientation of the fim operon promoter. Our version of W3110 has an insertion in fimE which means that FimB is the major factor that controls phase variation. Loss of speB reduced transcripts from fimB which could account for loss of motility. PCR analysis with specific primers can determine the fim operon promoter orientation (Figure 11A) and showed that loss of speB, loss of hns, or the presence of putrescine did not alter the ratio of ON to OFF (Figure 11B). This experiment determines the steady state phase ON to OFF ratio, which less FimB might not affect if FimB is in excess. In strains with FimE, the rate of phase ON orientation formation will be much slower, and even small changes in FimB activity could conceivably affect the switch orientation. We conclude that putrescine does not control phase variation, at least in our strain of W3110.
Figure 11

Phase variation in parental W3110, W3110 ΔspeB, W3110 Δhns, and MG1655.
(A) The diagram shows the genes for the FimB and FimE recombinases, the invertible fimS region which contains the promoter for the fim operon, and the fim operon which codes for the proteins of the type 1 pilus. Primer pairs 1–2 and 1–3 detect the fimS region in the phase OFF and ON orientations, respectively. The DNA sizes for phases OFF and ON are 884 and 394, respectively, for wild-type strains of E. coli, such as MG1655. Our lab strain of W3110 has an IS1 element insertion in fimE which increases the size of the amplified DNA fragment. MG1655 was analyzed as a control. Primer 1 is 5’-CCGCGATGCTTTCCTCTATG-3’; primer 2 is 5’-TAATGACGCCCTGAAATTGC-3’; and primer 3 is 5’-TGCTAACTGGAAAGGCGCTG-3’ (shown schematically). (B) Deletion of either speB or hns had no effect on fimS orientation. A possible explanation for the loss of pili or PDSM in the speB or hns mutants is locking the fimS switch in phase OFF. However, loss of either speB or hns had no effect on fimS orientation in W3110 (lanes 2–4), and putrescine did not alter fimS orientation of the W3110 ΔspeB mutant (lanes 6 and 7). We conclude that loss of speB in W3110 did not phase-lock fimS in phase OFF. Also note that W3110, which has an insertion in fimE, is not locked in phase ON.
Evidence for a putrescine homeostatic network
Altered transcript levels in the speB mutant suggest compensatory mechanisms for low putrescine (Supplementary file 2 for transcript differences for all genes, Table 2 for selected genes, and Figure 12 for a diagrammatic representation of transcript differences). The speB mutant had more transcripts from (a) genes of arginine and ornithine carboxylase, (b) all genes of ornithine and arginine synthesis from glutamate, and (c) genes for three separate putrescine transport systems; and fewer transcripts from the sap operon which codes for a putrescine exporter (Sugiyama et al., 2016) and rpmH and rpsT, whose products inhibit ornithine and arginine decarboxylase (Keseler et al., 2017). The net effect of these changes should be to increase intracellular putrescine (solid arrows in Figure 12). In other words, these changes are compensatory for low putrescine. In addition to these changes, the speB mutant had 60-fold and sevenfold more transcripts for mgtA and phoBR which code for the major magnesium transporter and the regulators of phosphate assimilation, respectively. These changes may also be compensatory and are discussed below.
Figure 12
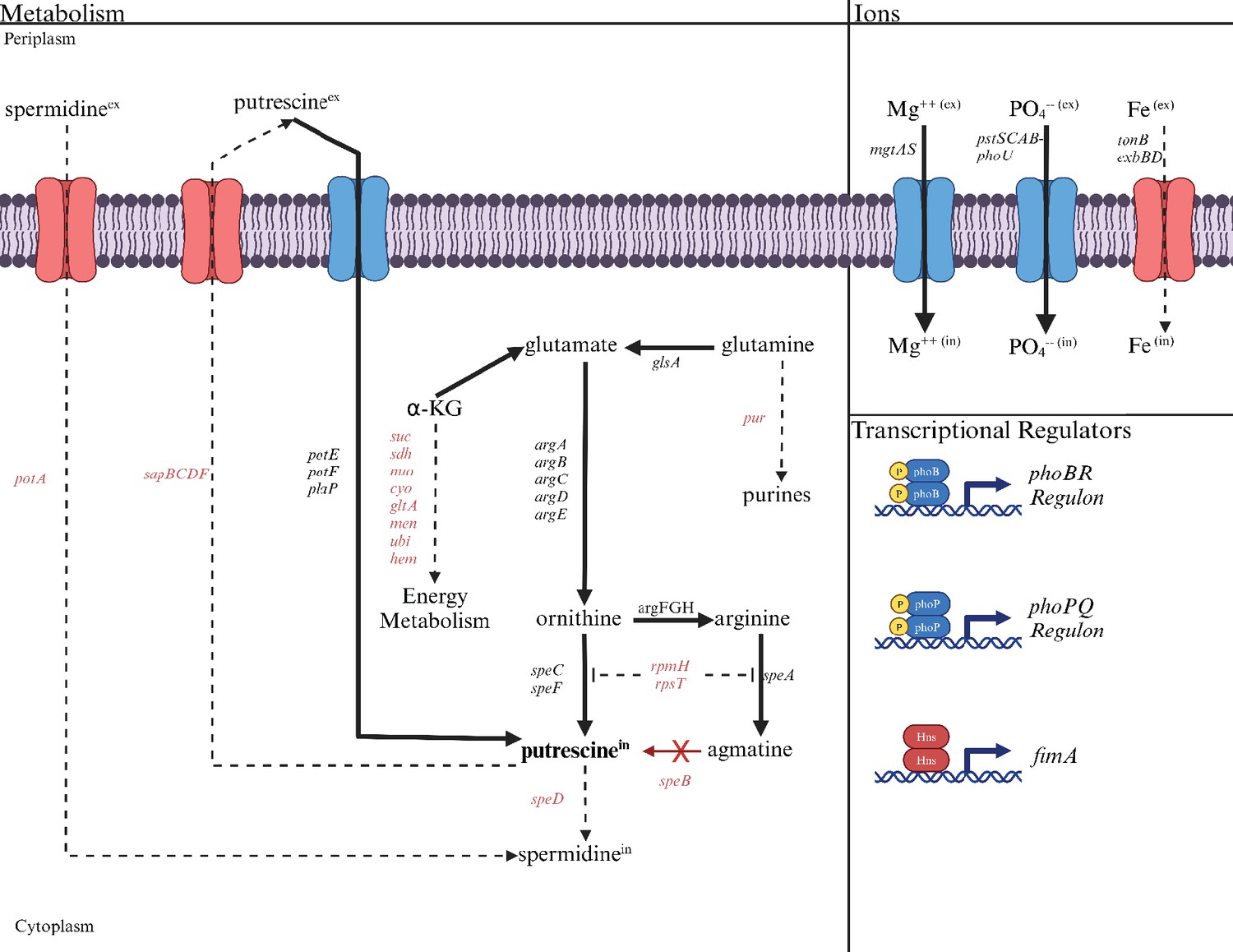
The deduced diversion of metabolism in a speB mutant away from energy metabolism toward putrescine synthesis compared to the parental strain.
The effects of low putrescine are shown. Genes and processes (transport and transcriptional regulators) in red have fewer transcripts, while those in black or blue have more. The dashed lines represent the proposed reduction in metabolic flux because of fewer transcripts from genes coding for the enzymes involved. Operons are shown except for the larger operons or regulons. For example, pur is meant to represent the unlinked genes that code for enzymes of purine synthesis. Table 2 or Supplementary file 2 should be consulted for specific genes and the quantitative change in transcripts.
Table 2
Differentially expressed genes that are proposed to contribute to putrescine homeostasis.
A positive number means more transcripts in the ΔspeB (lower putrescine) strain.
| Gene (function) | log2FC | FDR |
|---|---|---|
| Polyamine synthesis | ||
| speA (putrescine) | 0.99 | 0.003 |
| speC (putrescine) | 1.43 | 3E-4 |
| speD (spermidine) | –0.83 | 0.01 |
| speF (putrescine) | 1.05 | 0.007 |
| rpmH (inhibitor of SpeA and SpeC) | –2.77 | 2E-4 |
| rpsT (inhibitor of SpeA and SpeC) | –1.92 | 8E-4 |
| Polyamine transport | ||
| potABCD* (spermidine) | –2.17 | 3E-4 |
| potE (putrescine) | 2.07 | 2E-4 |
| potFGHI* (putrescine) | 1.41 | 4E-4 |
| plaP (putrescine) | 1.11 | 0.005 |
| sapBCDF* (putrescine export) | –1.18 | 8E-4 |
| Arginine synthesis | ||
| argA_2 (synthesis) | 1.77 | 0.001 |
| argB (synthesis) | 1.87 | 0.002 |
| argC (synthesis) | 2.86 | 0.005 |
| argD (synthesis) | 1.41 | 0.001 |
| argF (synthesis) | 1.63 | 0.01 |
| argG (synthesis) | 2.32 | 0.002 |
| argH (synthesis) | 0.97 | 0.001 |
| argI (synthesis) | 1.95 | 0.018 |
| argR (repressor of arginine regulon) | –1.35 | 4E-4 |
| Glutamate generation | ||
| glsA (glutamine degradation) | 2.16 | 5E-4 |
| glnG (regulation of ammonia assimilation) | –1.59 | 0.022 |
| TCA cycle/electron transport | ||
| acnA (TCA cycle) | –1.04 | 8E-4 |
| acnB (TCA cycle) | –1.96 | 2E-4 |
| fumA (TCA cycle) | –1.50 | 6E-4 |
| fumB (TCA cycle) | –1.49 | 0.003 |
| gltA (TCA cycle) | –1.64 | 2E-4 |
| icdA (TCA cycle) | –1.32 | 0.002 |
| sdhCDAB-sucABCD* (TCA cycle) | –3.88 | 5E-5 |
| nuo operon* (electron transport) | –0.82 | 0.004 |
| menFDHBCE* (electron transport) | –0.8 | 0.005 |
| ubiEJB* (electron transport) | –0.93 | 0.004 |
| cyoABCD* (electron transport) | –1.41 | 9E-4 |
| hemCD* (heme) | –2.05 | 2E-4 |
| Iron transport | ||
| exbBD* (transport of all iron chelates) | –2.33 | 4E-5 |
| tonB (transport of all iron chelates) | –1.9 | 4E-5 |
| entCEBA* (enterochelin iron) | –5.2 | 7E-6 |
| fecABCDE * (ferric citrate) | –4.4 | 3E-5 |
| fecIR (regulators, ferric citrate) | –4.4 | 6E-6 |
| feoABC* (ferrous iron) | –3.4 | 7E-4 |
| fur (regulator, iron assimilation) | –0.40 | 0.11 |
| Magnesium and phosphate transport | ||
| mgtA (magnesium) | 5.90 | 5E-6 |
| phoQ (regulator, magnesium) | 0.79 | 0.009 |
| pstSCAB* (phosphate) | 3.61 | 0.01 |
| phoBR* (regulator, phosphate assimilation) | 2.80 | 0.02 |
-
*
Values for transcripts of the first gene of the operon are given. Results for other genes of the operon are in Supplementary file 2.
Putrescine affects energy metabolism which could account for loss of motility
Loss of speB affected transcripts for genes of the major energy metabolism pathways (Table 2): fewer transcripts for genes coding for all citric acid cycle enzymes, NADH dehydrogenase I (the entire nuo operon), cytochrome oxidase (the cyo operon), and several genes of menaquinone (the menFDHBCE operon), ubiquinone, and heme synthesis. The speB mutant also had fewer transcripts for all genes of iron acquisition, which is consistent with diminished oxidative energy generation. A speB mutant had fewer transcripts for ptsH (Hpr of the phosphotransferase system of carbohydrate transport), ptsG (enzyme IIBC component of glucose transport), gltA (citrate synthase), sdhA (a succinate dehydrogenase subunit), and sucC (a succinyl-CoA synthetase subunit), and their loss has been shown to either impair or eliminate W3110 surface motility (Sudarshan et al., 2021). The cumulative effect of these differences could account for the motility defect in the speB mutant.
Discussion
Several aspects of polyamine metabolism affect PDSM in E. coli. Strains with loss of one or two polyamine synthesis genes ― speA and speB single mutants and a speC speF double mutant ― resulted in PDSM defects. This observation is remarkable because nine enzymes and several pathways are involved in polyamine synthesis. Our putrescine synthesis mutants had no growth defect which is not unexpected since deletions of eight biosynthetic genes were required to show a strong phenotype: impaired aerobic growth and elimination of anaerobic growth (Chattopadhyay et al., 2009). Strains with defects in the two most active putrescine transport systems had altered PDSM. The specific defects suggest the involvement of extracellular putrescine and arginine in intracellular putrescine homeostasis. The PDSM defect of putrescine transport mutants not only implicates extracellular putrescine but also suggests secretion of putrescine which is not added to the medium. The PDSM defect of speA and speB mutants implicates extracellular arginine because SpeA is mostly periplasmic (Tabor and Tabor, 1969). Defective putrescine catabolism also affected PDSM. The role of both putrescine synthesis and degradation suggests rapid adjustments to fluctuating putrescine concentrations and environmental conditions: putrescine catabolism is a core response to several stresses (Schneider et al., 2013).
Results from an RNAseq analysis showed that transcription of the fim operon was reduced in the speB mutant. Of the known transcription factors that control the fim operon, only the gene for H-NS had fewer transcripts. In contrast, transcripts for other relevant transcription factor genes were elevated in the speB mutant. Our results are consistent with H-NS mediating putrescine-dependent control, but we cannot exclude a general regulatory dysfunction mediated by altered nucleoprotein complexes at the fim operon promoter. The elevated transcripts for some transcription factors may be inhibitory.
The transcriptomic results showed that the speB mutant had more transcripts for genes whose products will maintain intracellular putrescine, which suggests compensatory mechanisms for low intracellular putrescine (Figure 11). The speB mutant also had more transcripts for magnesium and phosphate transport genes. The elevated transcripts for magnesium transport genes could compensate for low putrescine to the extent that magnesium can directly replace putrescine, e.g., binding to nucleotides and RNA (Miyamoto et al., 1993). Two recent studies observed an inverse correlation between intracellular magnesium and polyamines in Salmonella and proposed that either magnesium or the polyamines maintained an overall divalent cation homeostasis (Iwadate et al., 2023; Duprey and Groisman, 2020). The rationale for an increase in anionic phosphate assimilation is less clear, although the major phosphate permease transports metal-phosphates, including magnesium-phosphate (van Veen et al., 1994), which could help to compensate for low putrescine. Regardless of the rationale, the substantial changes in gene expression due to low putrescine suggest multiple compensatory mechanisms.
A major observation of the RNAseq analysis is that loss of speB lowered transcripts for many genes of energy metabolism and all genes of iron acquisition. This gene expression pattern suggests that low putrescine diverts metabolism away from energy generation and toward putrescine synthesis, i.e., a prioritization of putrescine synthesis over aerobic energy metabolism. Oxidative energy metabolism and putrescine synthesis compete for α-ketoglutarate and the metabolic diversion could also be a potential compensatory mechanism for low putrescine. Another way of looking at this relationship is that intracellular putrescine positively correlates with oxidative energy metabolism. Deletion of ptsH, ptsG, gltA, sdhA, and sucC have been shown to impair W3110 PDSM which suggests that a major, possibly the primary, effect of low putrescine is reduced energy generation.
In contrast to our results, a thorough and well-designed study showed that E. coli PDSM requires spermidine instead of putrescine (Kurihara et al., 2009). The differences between the studies were strain backgrounds, media (glucose-LB vs glucose-tryptone), and incubation temperature (33° vs 37°). We found that PDSM results for 37° incubations were highly variable and resulted in numerous fast-moving variants. Since growth at the higher temperature is faster, and faster growth is associated with higher putrescine (Tweeddale et al., 1998), we propose that growth at 37° results in an inhibitory putrescine concentration. In this case, spermidine would be stimulatory because of its known inhibition of the putrescine biosynthetic enzymes SpeA and SpeC (Applebaum et al., 1977; Wu and Morris, 1973) and subsequent reduction of intracellular putrescine to an optimal concentration. Regardless of the explanation, strain differences are not unexpected because of decades of strain passage for the strains employed with resulting laboratory evolution that can affect any of the numerous factors that control the intracellular putrescine concentration.
The transcript changes observed in our study were different from those in the only other systematic study of putrescine’s effect on E. coli gene expression (Yoshida et al., 2004). Both studies had almost identical experimental designs: 1.0- or 1.14 mM putrescine was added to a speB or speB speC mutant, respectively. Differences in strains, growth media (minimal media vs tryptone-containing media), and growth temperature (33° vs 37°) could account for the discrepancy in results. The most crucial difference could be the growth media: Igarashi and colleagues grew bacteria in a minimal medium, while we grew bacteria in an amino acid-containing medium that was required for surface motility. We propose that the slower growth in minimal medium, possibly correlated with higher guanosine tetraphosphate which is associated with slower growth, counters and obscures many growth-stimulatory putrescine effects. In contrast, the growth stimulation from the amino acids in our medium would not counter putrescine effects and would essentially allow the detection of effects caused solely by changes in putrescine concentrations. Regardless of the explanation, common results can be interpreted as core responses to putrescine limitation. Both studies observed that low putrescine increased transcripts for genes of arginine synthesis and magnesium transport, and reduced transcripts for the sdhCDAB-sucABCD operon which codes for three TCA cycle enzymes. The link between putrescine and arginine synthesis is probably mediated by the ArgR repressor because argR transcripts are 2.5-fold lower (FDR = 0.0004) in the speB mutant (Supplementary file 2).
Several results implicate putrescine in E. coli virulence during urinary tract infections. First, pili-dependent binding to the urothelium is an essential step for uropathogenic E. coli (Flores-Mireles et al., 2015). Second, putrescine is present in urine from infected, but not healthy, individuals (Puebla-Barragan et al., 2020; Satink et al., 1989). Third, urinary putrescine can result from uropathogenic E. coli growth in urine (Satink et al., 1989) and urothelial pathophysiology (Keay et al., 2014). Putrescine’s possible role in virulence is seemingly contradicted by the observation that a speB mutant outcompetes the parental CFT073 in the bladder of mice, i.e., less putrescine synthesis enhanced virulence (Alteri et al., 2009). A majority of uropathogenic E. coli is associated with phylogenetic group B2―including CFT073, whereas most E. coli lab strains, such as W3110, are group A. Group B2 strains lack genes for the major putrescine catabolic pathway and have significantly higher transcripts for genes coding for SpeA and SpeB (Hogins et al., 2023a). Both observations suggest that the cytoplasmic putrescine concentration is higher in group B2 strains. We propose that the putrescine concentration in group B2 strains is normally in the inhibitory range for pili synthesis, and that loss of SpeB reduces putrescine to a level that stimulates pili synthesis. Regardless of the explanation, putrescine control of pili synthesis and other processes in uropathogenic E. coli are worth examination.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Escherichia coli) | speB | PMID:16738554 | P1 transduction construction | |
| Gene (E. coli) | various, see strain list | PMID:16738554 | P1 transduction construction | |
| Strain, strain background (E. coli) | W3110 | other | Lab strain; history provided in Methods | |
| Genetic reagent (E. coli transducing virus) | P1 vir | other | Lab strain | |
| Antibody | anti-E. coli RpoD (rabbit polyclonal) | Cusabio | Cat #: CSB-PA360419XA01ENV RRID:AB_3678626 | 1:10,000 |
| Antibody | anti-E. coli FimA (rabbit polyclonal) | Cusabio | Cat #: CSB-PA361210ZA01ENV RRID:AB_3678627 | 1:10,000 |
| Antibody | Goat anti-rabbit IgG (H+L) HRP conjugated | Cusabio | Cat #: CSB-PA489724 RRID:AB_3678628 | 1:10,000 |
| Sequence-based reagent | FimA forward | This paper | PCR primers | ATGGTGGGACCGTTCACTTT |
| Sequence-based reagent | FimA reverse | This paper | PCR primers | GGCAACAGCGGCTTTAGATG |
| Sequence-based reagent | RpoD forward | This paper | PCR primers | TCGTGTTGAAGCAGAAGAAGCG |
| Sequence-based reagent | RpoD reverse | This paper | PCR primers | TCGTCATCGCCATCTTCTTCG |
| Sequence-based reagent | Phase variation test primer 1 | This paper | PCR primers | CCGCGATGCTTTCCTCTATG |
| Sequence-based reagent | Phase variation test primer 2 | This paper | PCR primers | TAATGACGCCCTGAAATTGC |
| Sequence-based reagent | Phase variation test primer 3 | This paper | PCR primers | TGCTAACTGGAAAGGCGCTG |
| Commercial assay or kit | TMB substrate kit | ThermoFisher | Cat #: 34021 | |
| Commercial assay or kit | Ligation sequencing DNA V14 | Oxford Nanopore Technologies | Cat #: SQK-LSK114 | |
| Commercial assay or kit | Rneasy Mini Kit | Qiagen | Cat #: 74104 | |
| Commercial assay or kit | RiboMinus Transcriptome Isolation Kit, bacteria | ThermoFisher | Cat #: K155004 | |
| Commercial assay or kit | RiboCop rRNA Depletion Kits for Bacteria | Lexogen | Cat #: 126.24 | |
| Commercial assay or kit | LunaScript RT Super | NEB | Cat #: E3010 | |
| Commercial assay or kit | PowerUp STBR Green Master Mix for qPCR | ThermoFisher | Cat #: A25777 | |
| Chemical compound, drug | Eiken Agar | Eiken Chemical Co., Ltd, Tokyo, Japan | Cat #: E-MJ00 | |
| Software, algorithm | bcl2fastq (v 4.2.4) | Illumina | RRID:SCR_015058 | |
| Software, algorithm | Guppy basecaller (v 6.5.7) | Oxford Nanopore Technologies | RRID:SCR_023196 | |
| Software, algorithm | Porechop (v 0.2.4) | Oxford Nanopore Technologies | RRID:SCR_016967 | |
| Software, algorithm | Flye (v 2.9.2) | PMID:27956617 | RRID:SCR_017016 | |
| Software, algorithm | Pilon (v 1.24) | https://doi.org.10.1371/journal.pone.0112963 | RRID:SCR_014731 | |
| Software, algorithm | Circulator (v 1.5.5) | PMID:26714481 | ||
| Software, algorithm | Prokka (v 1.14.6) | https://doi.org.10.1093/bioinformatics/btu153 | RRID:SCR_014732 | |
| Software, algorithm | QUAST (v 5.2.0) | PMID:23422339 | RRID:SCR_001228 | |
| Software, algorithm | CLC Genomics Workbench | Qiagen | RRID:SCR_011853 | |
| Software, algorithm | edgeR | PMID:19910308 | RRID:SCR_012802 |
Strains
All strains used for growth rate determinations and motility assays were derivatives of E. coli K-12 strain W3110 and are listed in Table 3. Great variations exist in standard lab strains, including W3110, from different labs. We initially tested nine strains derived from E. coli K-12, mostly from the Coli Genetic Stock Center (Ambagaspitiye et al., 2019), and chose our lab strain because of relative genetic stability and more quantitatively reproducible results. Our strain originated from the lab of Jon Beckwith in the 1960s via the lab of Boris Magasanik where it had been stored in a room temperature stab until the mid-1980s, at which time it was revived and frozen at –80 °C. To construct the mutant strains, the altered allele was obtained from the Keio collection in which the gene of interest had been deleted and replaced with an antibiotic-resistance gene (Baba et al., 2006). The marked deletion allele was transferred into W3110 by P1 transduction (Miller, 1972). The antibiotic gene was removed as described which generated an in-frame deletion (Datsenko and Wanner, 2000).
Table 3
Strains.
| Strain name | Genotype | Reference |
|---|---|---|
| BLS77 | W3110 ∆puuR::cat | Schneider and Reitzer, 2012 |
| BLS80 | W3110 ∆puuA::cat | Schneider and Reitzer, 2012 |
| BLS88 | W3110 ∆patA ∆puuA | Schneider and Reitzer, 2012 |
| CP2 | W3110 ∆patA | Schneider and Reitzer, 2012 |
| IM26 | W3110 ∆speB::kan fliC-lacZ | This study |
| IM27 | W3110 ∆speE::kan fliC-lacZ | This study |
| IM28 | W3110 ∆speF::cat fliC-lacZ | This study |
| IM29 | W3110 ∆speC::kan fliC-lacZ | This study |
| IM34 | W3110 ∆speC ∆speF::cat fliC-lacZ | This study |
| IM60 | W3110 ∆cadA::kan fliC-lacZ | This study |
| IM61 | W3110 ∆speA::kan | This study |
| IM62 | W3110 ∆patA ∆puuA ∆speE::kan | This study |
| IM63 | W3110 ∆potE::kan | This study |
| IM64 | W3110 ∆potF::kan | This study |
| IM65 | W3110 ∆plaP::kan | This study |
| IM66 | W3110 ∆puuP::kan | This study |
| J15 | W3110 ΔspeB ΔfliC::kan | This study |
| SA1 | W3110 ∆fliC::kan | This study |
| SA2 | W3110 ∆fimA::kan | This study |
| SA3 | W3110 ∆fliC ∆fimA | This study |
| SA4 | W3110 ∆hns::kan | This study |
| SA5 | W3110 ∆ihfA::kan | This study |
| SA6 | W3110 ∆lrp::kan | This study |
| W3110-LR referred to as W3110 | lacIqlacL8 | Lab strain |
Media and growth conditions
Request a detailed protocolFor P1 transductions and plasmid transformation experiments, cells were grown in standard LB liquid medium (1% tryptone, 1% NaCl, and 0.5% yeast extract, pH 7.0) at 37°C. Antibiotics were used for selection at concentrations of 25 µg/mL (both chloramphenicol and kanamycin). For growth analysis, bacteria were grown in a liquid motility medium (0.5% glucose, 1% tryptone, and 0.25% NaCl) which we refer to as GT medium. Starter cultures (typically 6–12 hr incubation) were grown in GT medium, harvested by centrifugation, washed twice with phosphate-buffered saline, and re-suspended in GT medium before inoculation. The cells were then grown at 37 °C in aerobic conditions (shaking at 240 rpm) and the turbidity was measured every 30 min. Cell growth was measured in Klett units using a Scienceware Klett colorimeter with a KS-54 filter. 100 Klett units represent an OD600 value of about 0.7.
Surface motility assay
Request a detailed protocolFor a standard surface motility assay, single colonies from fresh plates (streaked out from frozen stocks a day before) were inoculated in GT medium for 6 hr at 37 °C in aerobic conditions (shaking at 240 rpm). 30 ml of autoclaved GT medium with 0.45% agar (Eiken, Tokyo, Japan) was poured into a sterile polystyrene petri dish (100 mm × 15 mm) and allowed to solidify at room temperature for approximately 6 hr. Then, 1 µL of the pre-motility growth medium was inoculated at the center of the agar plate and incubated at 33°C for 36 hr. Each experiment was performed in triplicate and pictures were taken after 36 hr. Surface motility was extremely sensitive to humidity. Opening the incubator before 36 hr resulted in movement cessation. The surface motility assay was performed at 33 °C because results from incubations at 37°C were highly variable and the cultures more frequently generated genetically stable fast-moving variants.
Swim assay
Request a detailed protocolThe media and culturing for swimming motility are identical to that for surface motility, except that the plates contained 0.25% agar and were solidified at room temperature for about 1 hr before inoculation. Then, 1 µL of the culture was stabbed in the center of this swim agar plate and incubated at 33°C for 20 hr. Each experiment was performed in triplicate.
Transmission electron microscopy
Request a detailed protocolCells from surface motility plates were collected and fixed with 2.5% glutaraldehyde. Bacteria were absorbed onto Foamvar carbon-coated copper grids for 1 min. Grids were washed with distilled water and stained with 1% phosphotungstic acid for 30 s. Washed and stained grids were dried at 37 °C for 10 min. Samples were viewed on a JEOL 1200 EX transmission electron microscope at the University of Texas Southwestern Medical Center (Figure 2) and the University of Texas at Dallas (Figure 8).
ELISA analysis
Request a detailed protocolCells were grown overnight in 5 mL GT media followed by a 2 hr growth in GT media with 0.0, 0.1, 1.0, or 4.0 mM putrescine. Cells were then lysed using a 24-gauge needle. The cell lysate was diluted to a concentration of 1 mg/mL protein (based on A280) in pH 9.6 coating buffer (3 g Na2CO3, 6 g NaHCO3, 1000 mL distilled water) and then coated onto the walls of a 96-well plate by incubating overnight at 4 °C. The wells were rinsed three times with PBS and further blocked using coating buffer with 1% bovine albumin overnight at 4 °C. The following day, wells were rinsed three times with PBS, and primary antibodies to FimA or RpoD were added following supplier directions. After a 2 hr room temperature incubation, secondary antibodies conjugated to HRP were added and incubated for a further 2 hr. HRP was activated using the TMB substrate kit (Thermo-34021) following the provided protocol. Expression was read on a BioTek plate reader based on the provided kit protocol. Data was blanked to wells only treated with bovine albumin and then normalized to RpoD. Data was generated from technical and biological triplicates.
DNA isolation and genome assembly and annotation for W3110
Request a detailed protocolDNA was isolated based on previously established protocols (Hogins et al., 2023b). Short reads were sequenced at the SeqCenter (Pittsburgh, Pennsylvania) using Illumina tagmentation-based and PCR-based DNA prep and custom IDT indices targeting inserts of 280 bp without further fragmentation or size selection steps. The Illumina NovaSeq X Plus sequencer was run producing 2×151 paired-end reads. Demultiplexing, quality control, and adapter trimmer were performed with bcl-convert (v4.2.4). Total Illumina Reads (R1+R2): 4059078 with 553087045 bps >Q30.
Long reads were prepared using the PCR-free Oxford Nanopore Technologies ligation sequencing kit with the NEBNext companion module. No fragmentation or size selection was performed. Long-read libraries were performed using the R10.4.1 flow cell. The 400bps sequencing mode with a minimum read length of 200 bp was selected. Guppy (v6.5.7) was used at the super-accurate base calling, demultiplexing, and adapter removal. Total long reads: 53773 with 89.607% of bps >Q20.
To generate the completed genome, porechop (v0.2.4) was used to trim residual adapter sequences from long reads. Flye (v2.9.2) was used to generate the de novo assembly under the nano-hq model. Reads longer than the estimated N50 based on a genome size of 6Mbp initiated the assembly. Subsequent polishing using the short read data was performed using Pilon (v1.24) (RRID:SCR_014731) under default parameters. Long-read contigs with an average short-read coverage of 15 X or less were removed from the assembly. The assembled contig was confirmed to be circular via circulator (v1.5.5). Annotation was performed using prokka (v 1.14.6). Finally, statistics were recorded using QUAST (v5.2.0). The final genome contained 1 contig of 4750347 bp with a sequencing depth of 123.92 x. The N50 was 4750347. This genome can be accessed via the accession number CP165600 or via the BioProject PRJNA1142534 via NCBI. The completed genome has an estimated average nucleotide identity of 99.9694 to the W3110 genome deposited to NCBI (genome assembly ASM1024v1).
RNA isolation and quality control
Request a detailed protocolCells were grown for 2 hr in 1 mL GT media. 60 µL were then added to 1 mL of fresh GT, with or without 1 mM putrescine, and grown for another 2 hr. After growth, the cells were centrifuged, and frozen at –80 °C. The isolation and analysis protocol has been previously published (Hogins et al., 2023a). Cell pellets were thawed, resuspended in 0.7 mL of buffer RLT (Qiagen RNeasy kit), and mechanically lysed using a bead beater (FastPrep-24 Classic from MP Biomedical (RRID:SCR_013308)) set at the highest setting for three 45 s cycles with a 5 min rest period on ice between cycles. Cell lysates were used to isolate RNA using a Qiagen RNeasy mini kit. RNA was quantified, DNase treated, and re-quantified on a nanodrop. RNA that passed the first quality check was analyzed on RNA-IQ (Qubit). RNA with an IQ higher than 7.5 was used. RNA that passed both checks was run on an agarose gel to check for RNA integrity. RT-qPCR was used to ensure there was no DNA contamination. RNA libraries that passed all these quality checks were submitted to the genomics core facility at The University of Texas at Dallas for RNA sequencing. The core performed rRNA removal (RiboMinus Transcriptome Isolation Kit or RiboCop bacterial rRNA depletion—Lexogen), library preparation (Stranded Total RNA Prep—Illumina), and single-end Illumina sequencing.
Reverse transcription quantitative PCR
Request a detailed protocolRNA was isolated as described above. One microgram of RNA was reverse transcribed using LunaScript RT Super Mix (NEB E3010) and another microgram was subjected to the same reaction as the reverse transcribed RNA, except without reverse transcriptase (negative control) following manufacturer instructions. The cDNA was then diluted to 10 ng/µL for qPCR. PCR reactions were composed of 10 ng cDNA, 8 µL of nuclease-free water, 10 nM primers (rpoD: Forward: TCGTGTTGAAGCAGAAGAAGCG; Reverse: TCGTCATCGCCATCTTCTTCG) (fimA: Forward: ATGGTGGGACCGTTCACTTT; Reverse: GGCAACAGCGGCTTTAGATG), and PowerUp SYBR Green 2 X master mix for qPCR (ThermoFisher A25777) in a 20 µL reaction. A Quantstudio 3 qPCR machine was used to generate critical threshold (Ct) values. The Ct values were then analyzed using the 2-ΔΔCT method (Livak and Schmittgen, 2001) using rpoD as a library control.
RNAseq analysis
Request a detailed protocolTranscripts were aligned to the W3110 genome and CDS (CP165600) available on NCBI using the CLC genomics workbench (Qiagen version 22.00) for RNA-seq analysis. The gene expression counts generated as this output were used to perform differentially expressed gene (DEG) analysis using EdgeR (version 3.38.4) (RRID:SCR_012802) to analyze read counts (McCarthy et al., 2012; Robinson et al., 2010). DEGs had an adjusted p-value of less than 0.05.
Material availability statement
Request a detailed protocolAll strains are available upon request. All mutant alleles are readily accessible via the KEIO mutant collection (Baba et al., 2006).
Data availability
The raw RNA seq reads were deposited in GenBank: BioProject accession number PRJNA1126736. The genome was deposited in GenBank: BioProject accession number PRJNA1142534. The genome accession number is CP165600.
-
NCBI BioProjectID PRJNA1126736. Transcriptomic Sequencing of a speB mutant in Escherichia coli W3110.
-
NCBI BioProjectID PRJNA1142534. Escherichia coli str. K-12 substr. W3110 Genome sequencing.
-
NCBI NucleotideID CP165600. Escherichia coli str. K-12 substr. W3110 chromosome, complete genome.
References
-
Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collectionMolecular Systems Biology 2:e0050.https://doi.org/10.1038/msb4100050
-
Urinary tract infections: epidemiology, mechanisms of infection and treatment optionsNature Reviews. Microbiology 13:269–284.https://doi.org/10.1038/nrmicro3432
-
Decrease in cell viability due to the accumulation of spermidine in spermidine acetyltransferase-deficient mutant of Escherichia coliThe Journal of Biological Chemistry 270:18831–18835.https://doi.org/10.1074/jbc.270.32.18831
-
The distinct transcriptome of virulence-associated phylogenetic group B2 Escherichia coliMicrobiology Spectrum 11:e0208523.https://doi.org/10.1128/spectrum.02085-23
-
Genome sequences of seven clade B2 Escherichia coli strains associated with recurrent urinary tract infections in postmenopausal womenMicrobiology Resource Announcements 12:e0003523.https://doi.org/10.1128/mra.00035-23
-
Polyamines: mysterious modulators of cellular functionsBiochemical and Biophysical Research Communications 271:559–564.https://doi.org/10.1006/bbrc.2000.2601
-
Polyamine uptake systems in Escherichia coliResearch in Microbiology 152:271–278.https://doi.org/10.1016/s0923-2508(01)01198-6
-
Modulation of protein synthesis by polyaminesIUBMB Life 67:160–169.https://doi.org/10.1002/iub.1363
-
Evidence for bladder urothelial pathophysiology in functional bladder disordersBioMed Research International 2014:865463.https://doi.org/10.1155/2014/865463
-
The EcoCyc database: reflecting new knowledge about Escherichia coli K-12Nucleic Acids Research 45:D543–D550.https://doi.org/10.1093/nar/gkw1003
-
Dependence of swarming in Escherichia coli K-12 on spermidine and the spermidine importerFEMS Microbiology Letters 294:97–101.https://doi.org/10.1111/j.1574-6968.2009.01552.x
-
A novel putrescine importer required for type 1 pili-driven surface motility induced by extracellular putrescine in Escherichia coli K-12The Journal of Biological Chemistry 286:10185–10192.https://doi.org/10.1074/jbc.M110.176032
-
Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variationNucleic Acids Research 40:4288–4297.https://doi.org/10.1093/nar/gks042
-
Estimation of polyamine distribution and polyamine stimulation of protein synthesis in Escherichia coliArchives of Biochemistry and Biophysics 300:63–68.https://doi.org/10.1006/abbi.1993.1009
-
Cytotoxic mechanism of excess polyamines functions through translational repression of specific proteins encoded by polyamine modulonInternational Journal of Molecular Sciences 21:2406.https://doi.org/10.3390/ijms21072406
-
Microbial influences on urinary polyamine excretionClinica Chimica Acta; International Journal of Clinical Chemistry 179:305–314.https://doi.org/10.1016/0009-8981(89)90093-4
-
Pathway and enzyme redundancy in putrescine catabolism in Escherichia coliJournal of Bacteriology 194:4080–4088.https://doi.org/10.1128/JB.05063-11
-
Putrescine catabolism is a metabolic response to several stresses in Escherichia coliMolecular Microbiology 88:537–550.https://doi.org/10.1111/mmi.12207
-
Regulation of fim genes in uropathogenic Escherichia coliWorld Journal of Clinical Infectious Diseases 1:17–25.https://doi.org/10.5495/wjcid.v1.i1.17
-
A novel putrescine exporter sapBCDF of Escherichia coliThe Journal of Biological Chemistry 291:26343–26351.https://doi.org/10.1074/jbc.M116.762450
-
Partial separation of two pools of arginine in Escherichia coli; preferential use of exogenous rather than endogenous arginine for the biosynthesis of 1,4-diaminobutaneThe Journal of Biological Chemistry 244:6383–6387.
-
PolyaminesAnnual Review of Biochemistry 53:749–790.https://doi.org/10.1146/annurev.bi.53.070184.003533
-
Polyamines in microorganismsMicrobiological Reviews 49:81–99.https://doi.org/10.1128/mr.49.1.81-99.1985
-
Biosynthetic arginine decarboxylase from Escherichia coliJournal of Biological Chemistry 248:1687–1695.https://doi.org/10.1016/S0021-9258(19)44245-2
-
A unifying model for the role of polyamines in bacterial cell growth, the polyamine modulonThe Journal of Biological Chemistry 279:46008–46013.https://doi.org/10.1074/jbc.M404393200
Article and author information
Author details
Funding
Cain Foundation (The Felecia and John Cain Distinguished Chair in Women’s Health, in Honor of Philippe Zimmern, M.D.)
- Philippe E Zimmern
This work was funded through private donations. PEZ received endowment support from the Cain Foundation through the Felecia and John Cain Distinguished Chair in Women's Health in honor of Philippe E Zimmern, MD. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was funded through private donations. PEZ received endowment support from the Cain Foundation through the Felecia and John Cain Distinguished Chair in Women’s Health in honor of Philippe E Zimmern, MD. The authors declare no potential conflicts of interest.
The authors (JH, SH, GV, and LR) would like to thank Prof. Kelli Palmer of The University of Texas at Dallas for the use of her space, supplies, and tissue homogenizer. JH would like to further thank Prof. Palmer for the use of her CLC genomics program. The authors would like to thank the Genome Center at The University of Texas at Dallas for the services to support our research. The authors would like to thank the Olympus Discovery Center/Imaging Core facility at UT Dallas for providing equipment and support (Figure 8), and the UT Southwestern Medical Center for electron microscopy (Figure 2).
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.102439. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2024, Mehta, Hogins et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,059
- views
-
- 64
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 2
- citations for umbrella DOI https://doi.org/10.7554/eLife.102439
-
- 1
- citation for Version of Record https://doi.org/10.7554/eLife.102439.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Control of pili synthesis and putrescine homeostasis in Escherichia coli
eLife 13:RP102439.
https://doi.org/10.7554/eLife.102439.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}