TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1
- Walter and Eliza Hall Institute of Medical Research, Australia
- University of Melbourne, Australia
- Olivia Newton-John Cancer Research Institute, Australia
- La Trobe University, Australia
- Monash University, Australia
- Cornell University, United States
- SA Pathology, Australia
- University of Zurich, Switzerland
Figures
Figure 1
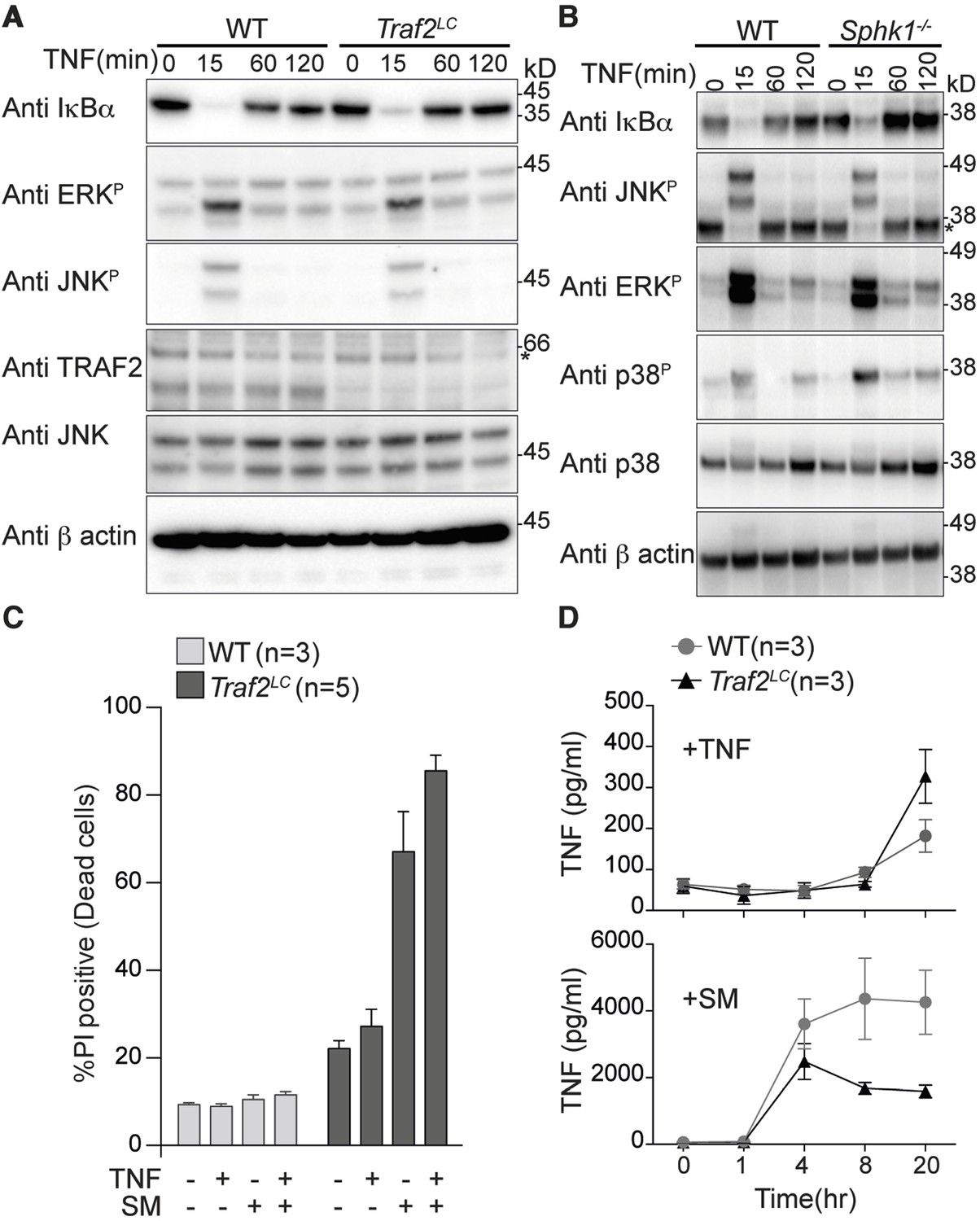
TRAF2 is not required for TNF-induced NF-κB and MAPK signalling in bone marrow derived macrophages.
(A and B) Western blot analysis of wild type (WT), Traf2lox/lox Lyz2-Cre (Traf2LC) and Sphk1-/- BMDMs treated with TNF (20 ng/ml) for the indicated times. (C) Flow cytometric analysis of WT, Traf2LCBMDMs treated with TNF (20 ng/ml) ± 100 nM Smac-mimetic (SM) for 24 hr and stained with Propidium Iodide (PI) and analysed by flow cytometry. (D) WT, Traf2LCmacrophages were treated with TNF (20 ng/ml) or Smac-mimetic (100 nM) for indicated time and supernatants analysed by TNF ELISA. Data are represented as mean ± SEM.
Figure 2
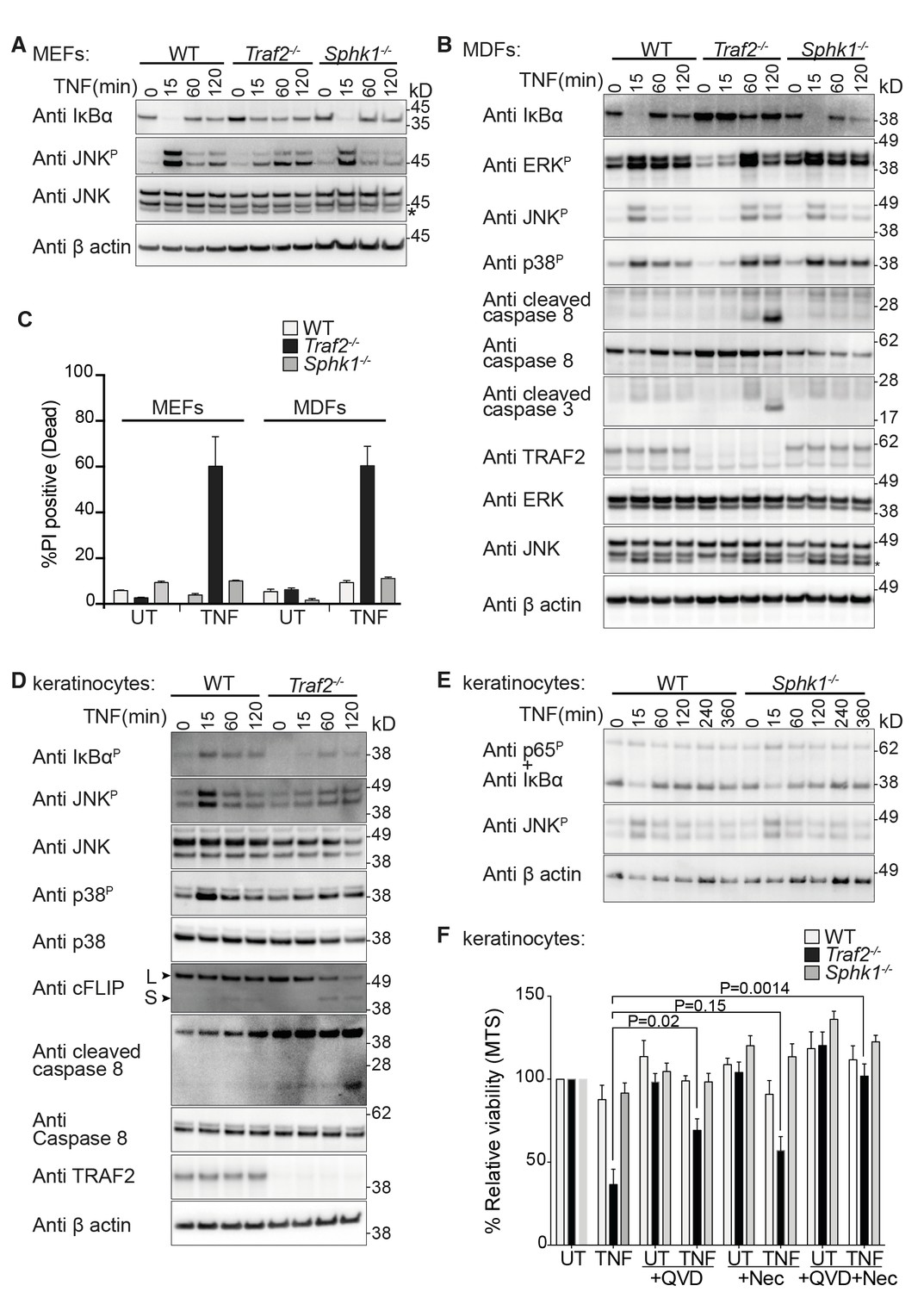
TRAF2 but not SPHK1 is required for TNF signalling in MEFs, MDFs and keratinocytes.
(A and B) Western blot analysis of wild-type (WT), Traf2-/- and Sphk1-/- immortalised MEFs and MDFs (as indicated) treated with TNF (100 ng/ml) for the indicated times. (C) FACS analysis of WT, Traf2-/- and Sphk1-/- MEFs and MDFs treated with TNF (100 ng/ml) for 24 hr and stained with Propidium Iodide (PI). n≥3 biological repeats. Data are represented as mean ± SEM. (D) Western blot analysis of WT and Traf2-/- keratinocytes treated with TNF (100 ng/ml) for the indicated times. (E) Western blot analysis of WT and Sphk1-/- keratinocytes treated with TNF (100 ng/ml) for the indicated times. (F) WT, Traf2-/- and Sphk1-/- keratinocytes were treated with TNF (100 ng/ml) in the absence or presence of Q-VD-OPh (QVD; 10 μM) and Necrostatin (Nec; 50 μM) as indicated for 24 hr and metabolically active cells were measured by MTS-PMS (MTS) assay. Biological repeats were WT and Traf2-/- (n=5) and Sphk1-/- (n=3). Data are represented as mean ± SEM. P values compare the indicated samples using a Students t-test.
Figure 3
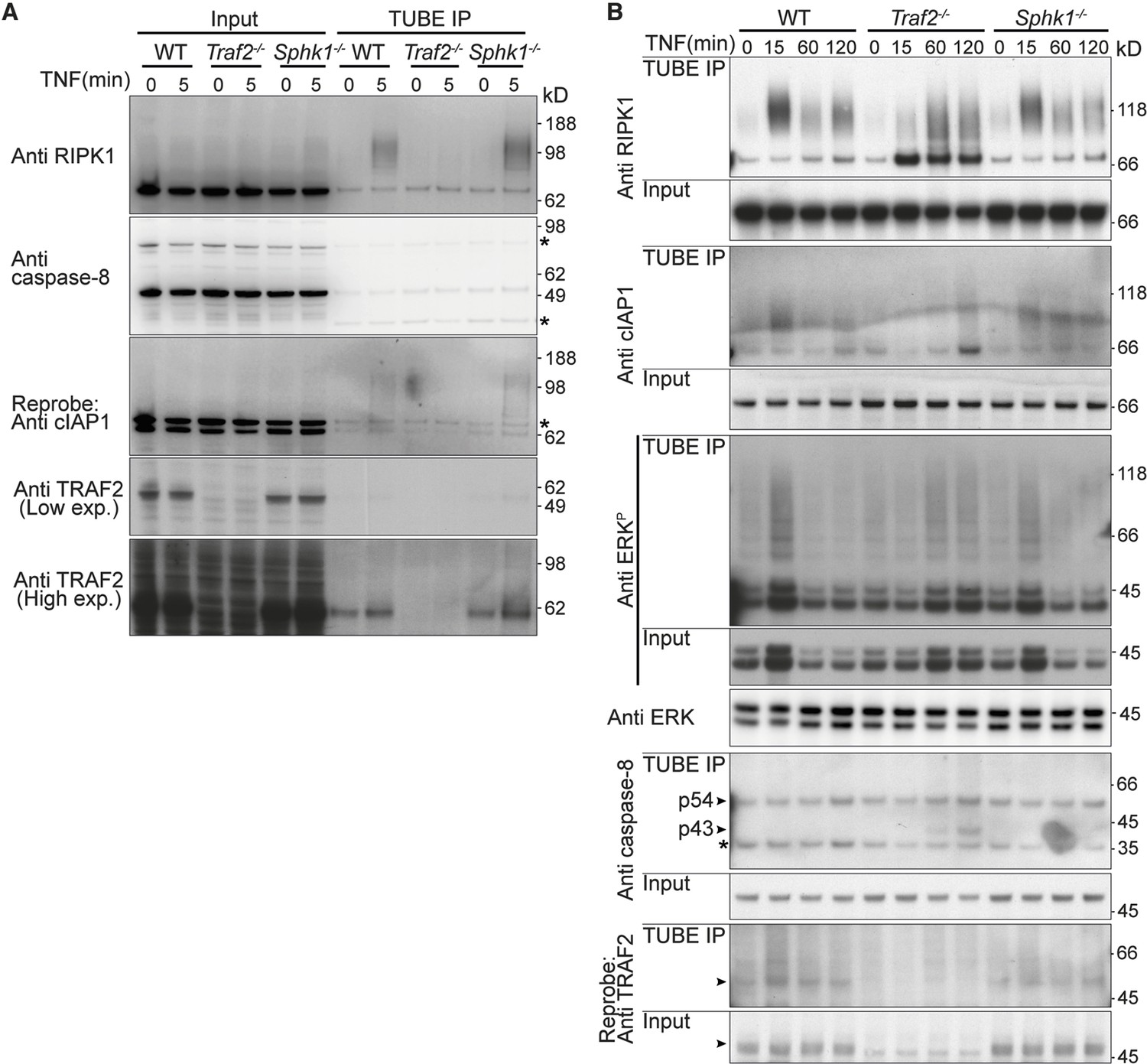
TRAF2 but not SPHK1 is required for rapid, TNF-induced, RIPK1 ubiquitylation.
(A and B) Wild-type, Traf2-/- and Sphk1-/- immortalised MDFs were treated with TNF (100 ng/ml) for the indicated times. Cell lysates were prepared and precipitated with ubiquitin-binding TUBE beads, then separated on an SDS-PAGE gel and Western blotted with the indicated antibodies.
Figure 4
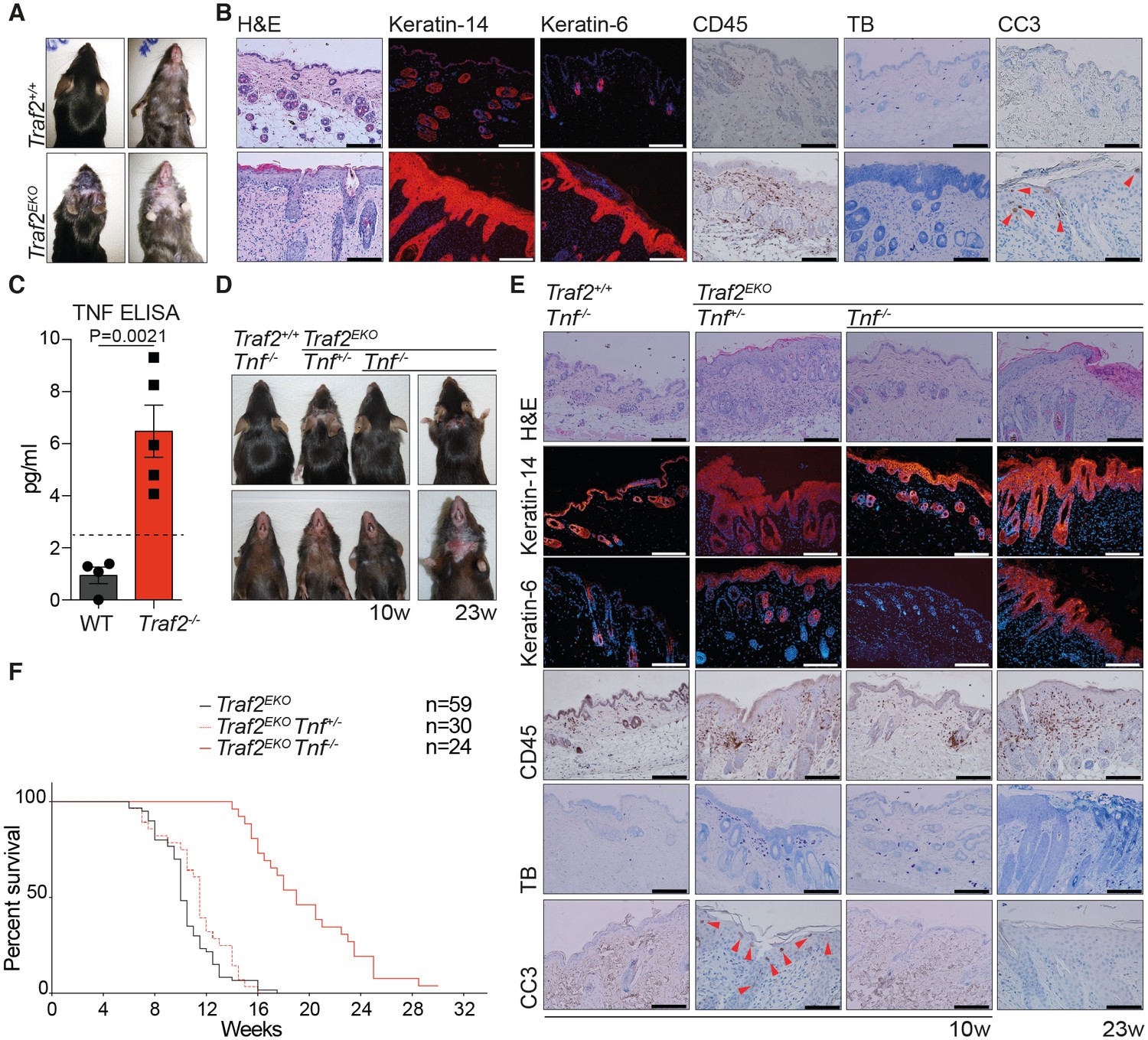
Loss of TRAF2 in keratinocytes causes epidermal hyperplasia and psoriasis-like skin inflammation.
(A, B, D and E) Representative images and skin sections of the indicated mice strains stained with haematoxylin/eosin (H&E), immuno-histochemistry for the pan leukocyte marker (CD45), cleaved caspase-3 (CC3), or toluidine blue (TB), or immuno-stained for Keratin-14 or Keratin-6 (in red) plus Hoechst (nuclei in blue). Scale bars = 100 μm. (C) The culture media of primary keratinocytes of the indicated genotypes at 80% confluency was replaced with serum-free media which was collected 48 hr later and analysed with TNF ELISA. Biological repeats WT (n=4) and Traf2-/- (n=5) are indicated. Data are represented as mean ± SEM. The dashed line indicates minimum detectable range of the ELISA. P values compare the indicated samples using a Students t-test. (F) Kaplan-Meier graph depicting survival of indicated mouse strains.
Figure 5
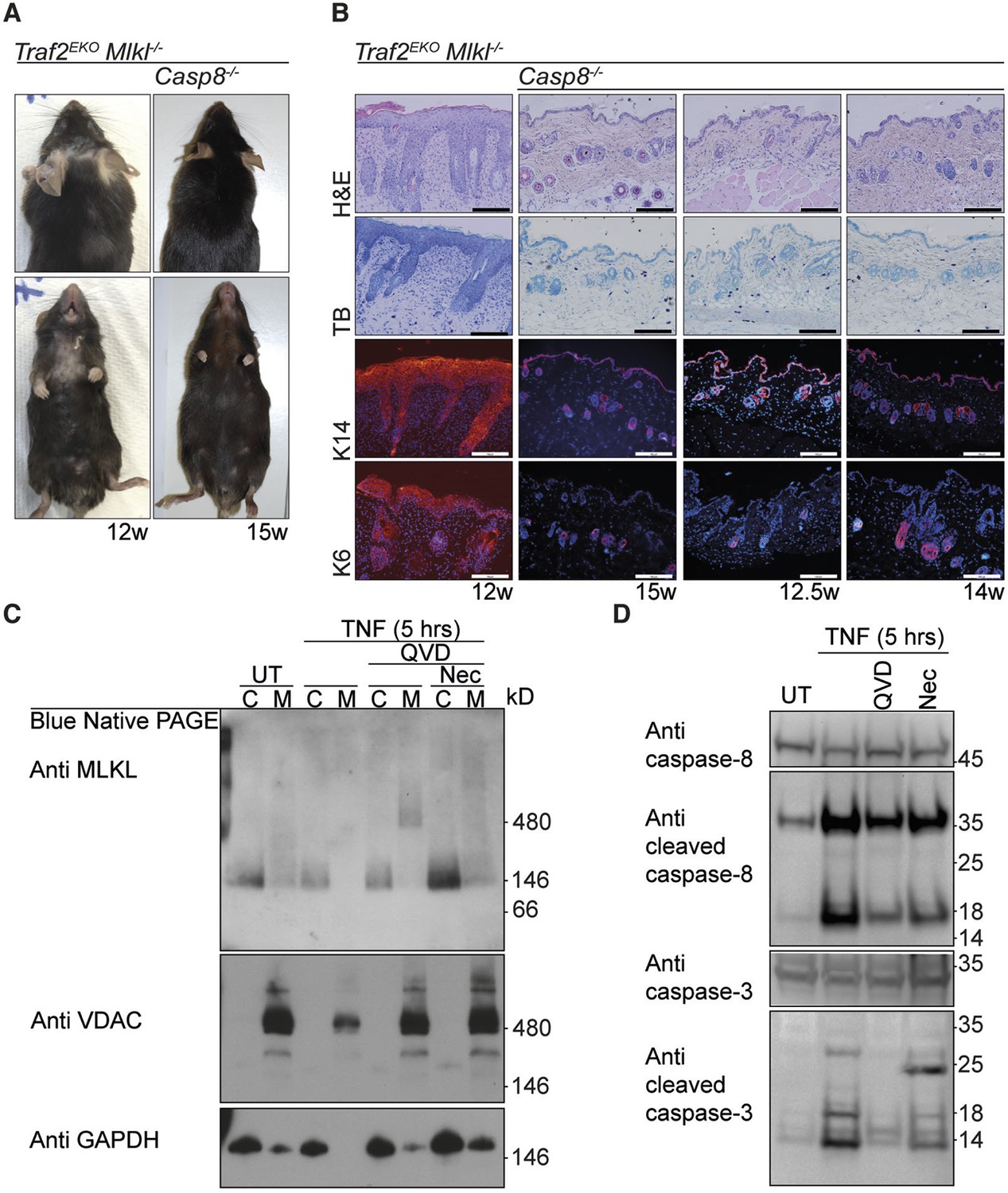
The early inflammation in Traf2EKO is caused by keratinocyte apoptosis.
(A and B) Representative images and skin sections of indicated mice stained with haematoxylin/eosin (H&E), toluidine blue (TB) or immuno-stained for Keratin-14 or Keratin-6 (in red) plus Hoechst (nuclei in blue). Scale bars = 100 μm. (C) Traf2-/- and Sphk1-/- keratinocytes were treated with TNF (100 ng/ml) for 5 hr in the absence or presence of QVD and/or Nec as indicated. Cytoplasmic (C) and Membrane (M) fractions were separated on a Blue Native PAGE gel and the Western blot probed with the indicated antibodies. (D) Traf2-/- keratinocytes were treated with TNF (100 ng/ml) for 5 hr in the absence or presence of QVD or Nec as indicated. The lysates were separated on an SDS-PAGE gel and the Western blot probed with the indicated antibodies.
Figure 6

Loss of TRAF2 in keratinocytes causes infiltration of neutrophils and IFNγ-producing CD4+ T cells to the skin.
(A-G) Single-cell suspensions from mouse ears with skin inflammation and healthy controls were prepared and the haematopoietic component (CD45.2+ and PI-) analysed by flow cytometry using the indicated markers. Representative contour plots for Ly6G and CD11b staining in the epidermis (A) and dermis (C) of mice with indicated genotype. Numbers indicate the proportion of CD11b+Ly6G+ neutrophils (boxed). Bar graph showing percentage infiltrated neutrophils to the epidermis (B) and dermis (D) n≥5, error bars are SEM. (C) Representative contour plots for TCRβ and CD4 staining. Numbers indicate the proportion of CD4+ T cells (boxed). (E) Representative contour plots for CD4 and TCRβ staining in the dermis of mice with indicated genotype. (F) Bar graph showing the percent of skin infiltrating CD4+ T cells, n≥8, error bars are SEM. (G) Bar graph showing percentage of infiltrating IFNγ-producing CD4+ T cells to the dermis of mice indicated genotype, n≥5, data are represented as mean ± SEM. Symbols in the graphs represent individual mice. All data are relative to the total CD45+ and PI- cells. P values compare the indicated samples using a Students t-test.
Figure 7
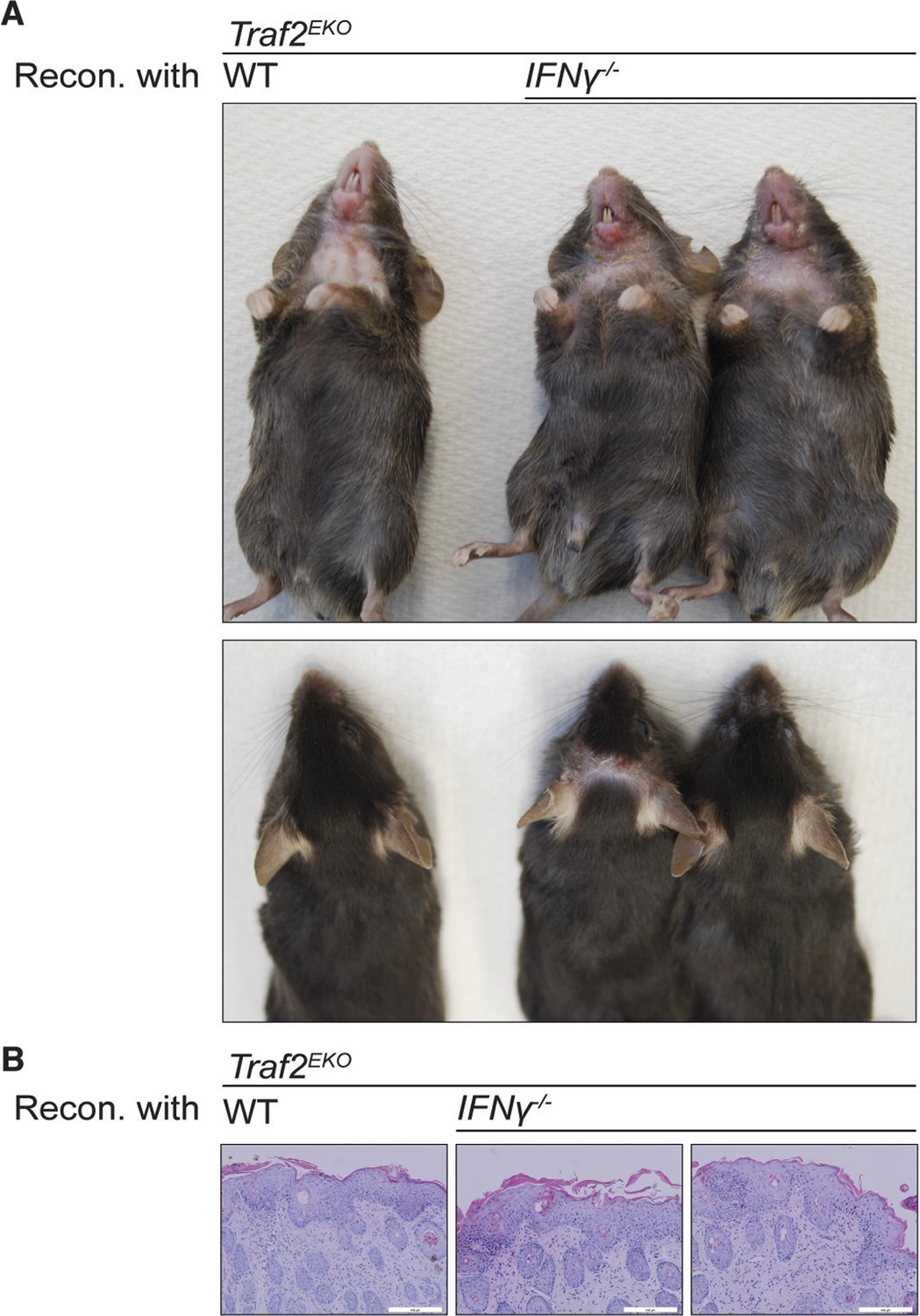
Reconstitution with IFNγ-deficient bone marrow does not prevent the inflammation in Traf2EKO mice.
(A,B) 6-week-old Traf2EKO mice were lethally irradiated and injected with wild-type (wt) or Ifnγ-/- bone marrow cells. After 4 weeks of reconstitution (Recon.), all mice, regardless of the origin of injected cells, started to develop inflammation similarly and needed to be sacrificed by 8 weeks after reconstitution. (A) Representative images. (B) H&E staining of skin sections. Scale bars = 100 μm.
Figure 8
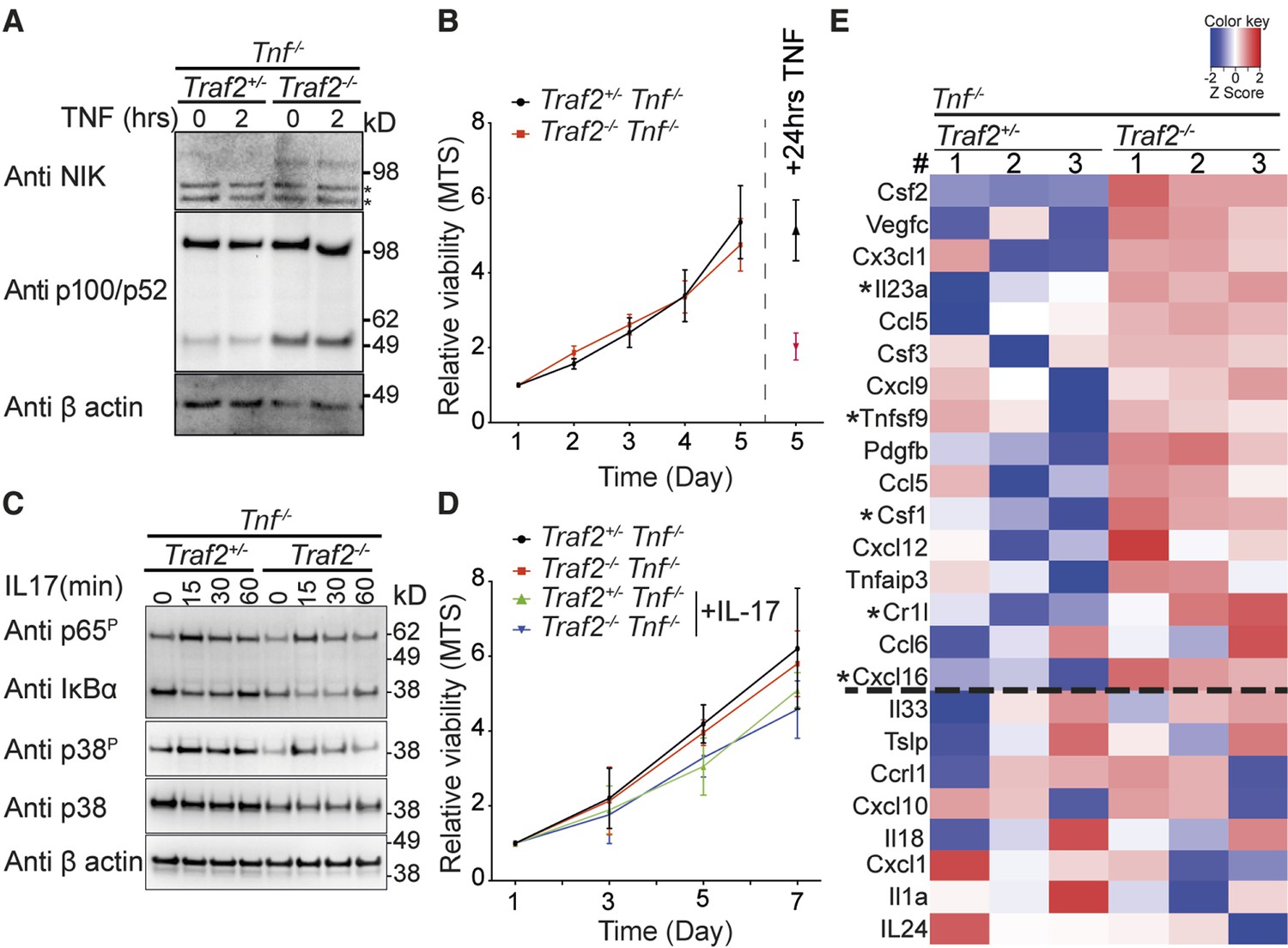
TRAF2 deletion causes constitutive activation of the non-canonical NF-κB transcription factor pathway.
(A) Tnf-/-Traf2+/- and Tnf-/-Traf2-/- keratinocytes were treated with or without TNF (100ng/ml) for the indicated times. The lysates were analysed as in Figure 1. Asterisks indicate non-specific bands. (B) The viability of keratinocytes of the indicated genotype was measured at indicated time points using an MTS assay. Viability following TNF treatment (100 ng/ml) on day 4 was used as a control. Data are represented as mean ± SEM, n=4 biological repeats. (C) Tnf-/-Traf2+/- and Tnf-/-Traf2-/- keratinocytes were treated with IL-17 (100 ng/ml) for the indicated times and lysates analysed as previously. (D) Viability of keratinocytes of indicated genotype ± IL-17 (100 ng/ml) was measured at indicated time points using MTS assay. Data are represented as mean ± SEM, n≥3 biological repeats. (E) Heat map depicting a selection from qPCR analysis of more than 600 inflammatory genes from Tnf-/-Traf2+/- and Tnf-/-Traf2-/- keratinocytes (3 mice for each genotype). Log expression values have been standardized to have mean 0 and standard deviation 1 for each row. Genes were ranked based on the fold change expression. Gens above the dashed line were highly elevated in Tnf-/-Traf2-/- keratinocytes. Asterisks indicate significant changes with P values <0.05.
-
Figure 8—source data 1
Complete zoom-able heat map of qPCR array.
Heat map depicting a selection from qPCR analysis of more than 600 inflammatory genes from Tnf-/-Traf2+/- and Tnf-/-Traf2-/- keratinocytes (3 mice for each genotype). Log expression values have been standardized to have mean 0 and standard deviation 1 for each row. Genes were ranked based on the fold change expression.
- https://doi.org/10.7554/eLife.10592.011
-
Figure 8—source data 2
Spreadsheet of result from qPCR array.
- https://doi.org/10.7554/eLife.10592.012
Figure 9
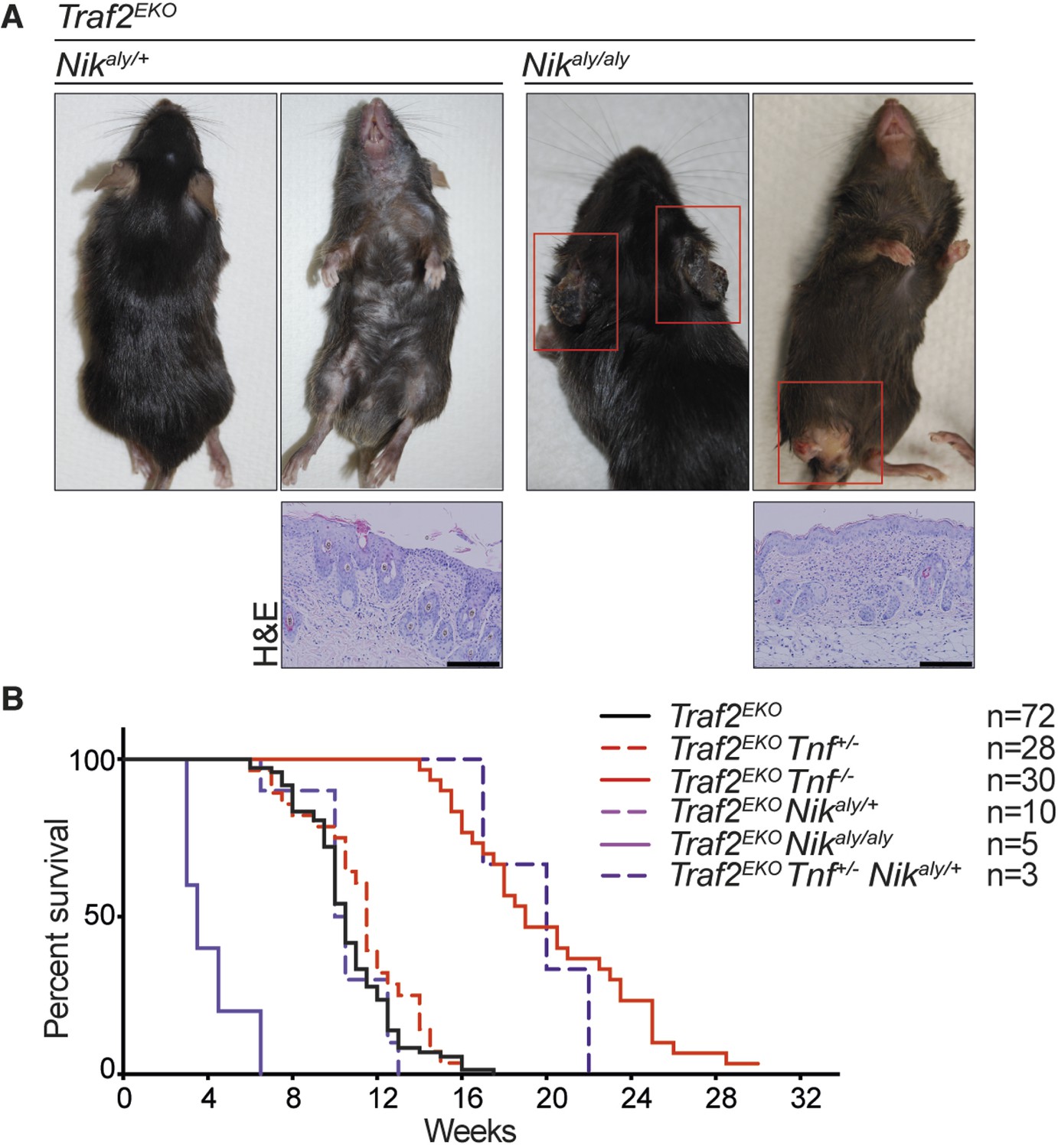
NIK mutation (aly) does not rescue the inflammation in Traf2EKO mice.
(A) Representative images and skin sections of mice with indicated genotypes. Scale bars = 100 μm. (B) Kaplan-Meier graph indicating the time that the mice with the skin lesions, needed to be sacrificed according to animal ethics regulations.
Figure 10
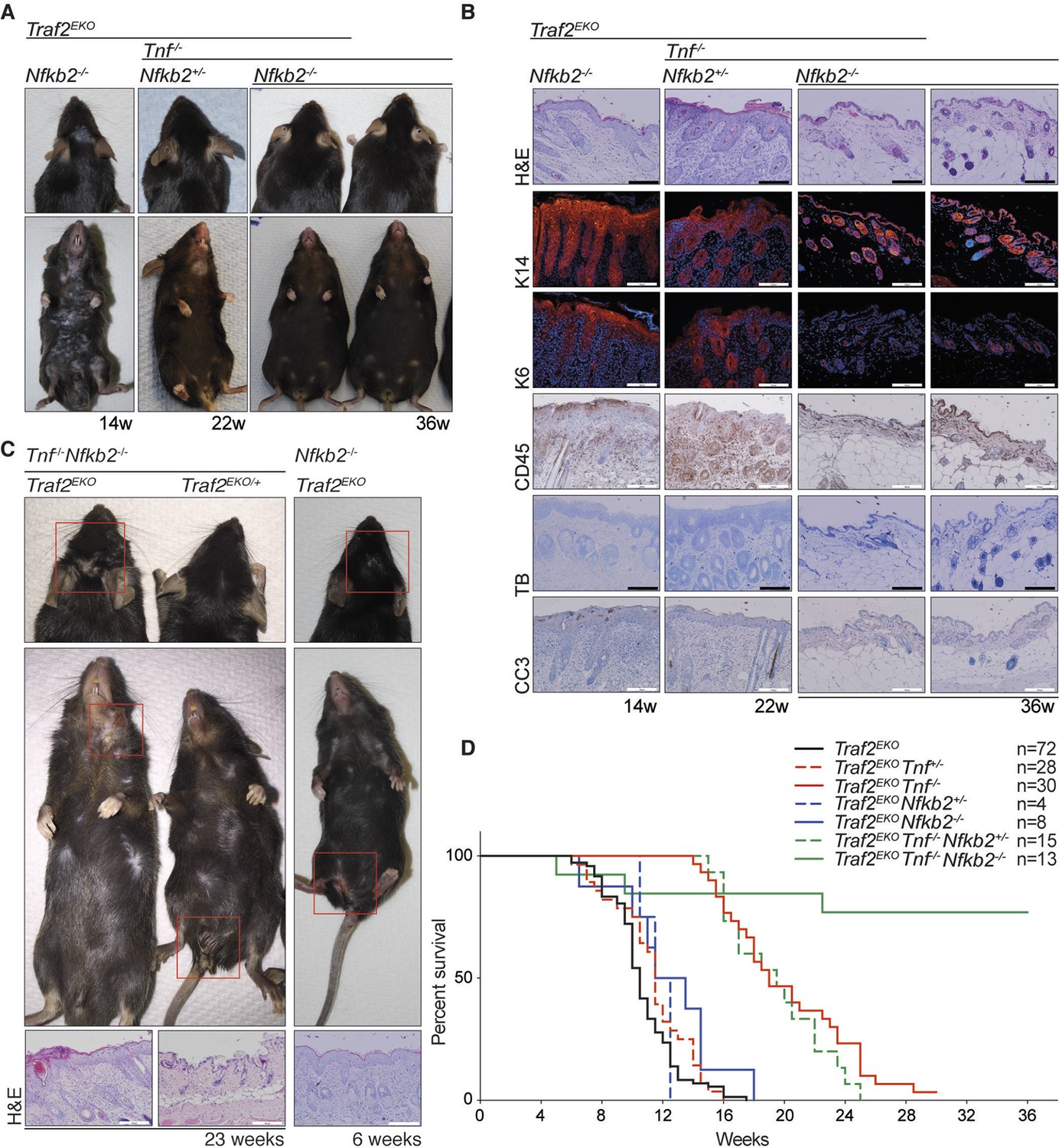
Depletion of both Tnf and Nfkb2 rescues the skin inflammation caused by loss of TRAF2 in keratinocytes.
(A-C) Representative images and skin sections of indicated mice stained with haematoxylin/eosin (H&E), immunostained for indicated epidermal differentiation markers (in red) and nuclei (Hoechst; in blue) and immunohistochemistry for pan leukocyte marker (CD45), cleaved caspase-3 (CC3) and toluidine blue (TB). Scale bars = 100 μm. (D) Kaplan-Meier graph depicting survival of indicated mouse strains.
Figure 11
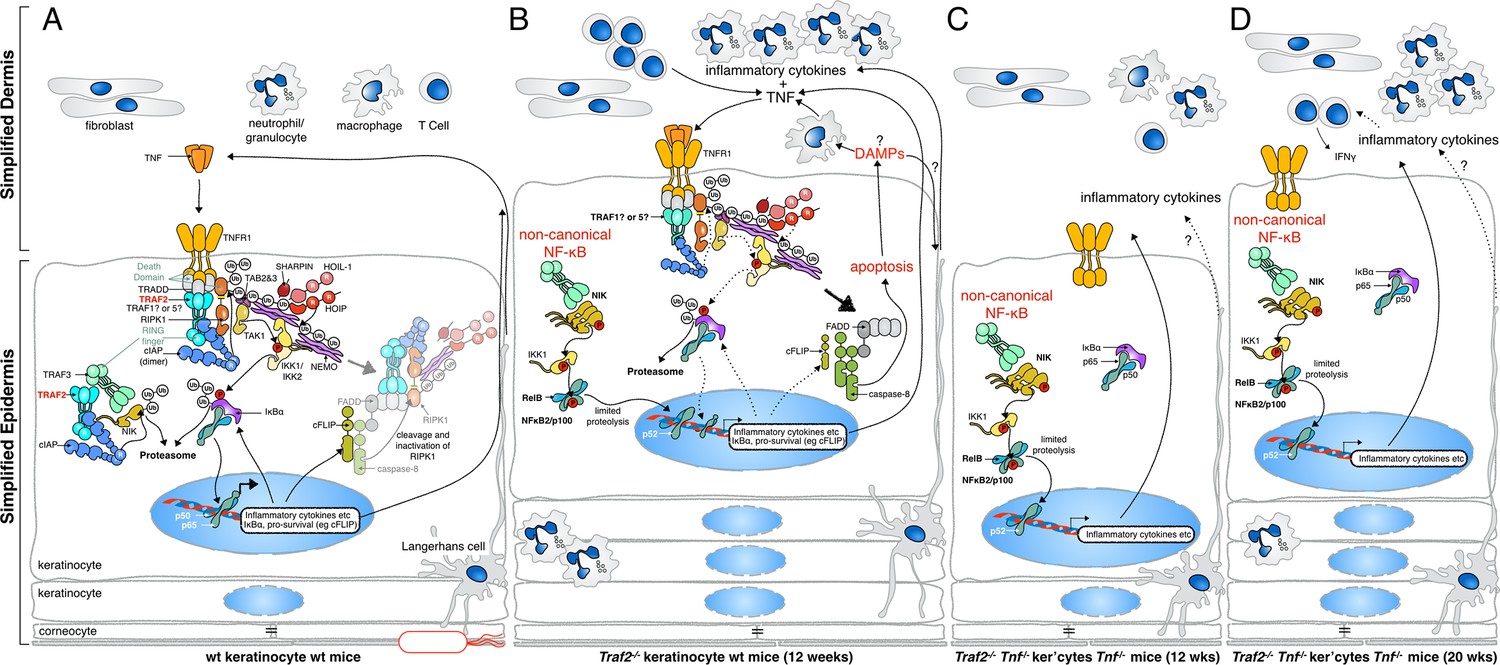
Proposed mechanisms by which loss of TRAF2 in keratinocytes causes a psoriasis-like skin inflammation.
(A) In normal wild-type skin TNF signalling might be activated by local penetration of bacteria into the epidermis. This will induce TNF, presumably by keratinocytes themselves or other resident cells such as Langerhans cells, and promotes canonical NF-κB signalling, inflammatory cytokine production and expression of prosurvival genes. If this response nullifies the threat, TNF signalling is turned off and homeostasis of the skin is maintained. (B) In the absence of TRAF2 in keratinocytes, TNF production can induce apoptotic cell death. This cell death, possibly because of the release of DAMPs, recruits neutrophils and other inflammatory cells to the skin and causes epidermal hyperplasia. Loss of TRAF2 also causes constitutive non-canonical NF-κB activation in viable keratinocytes, which increases the production and release of inflammatory cytokines, including TNF. Thus, TRAF2-deficient keratinocytes do not need to be stimulated by bacteria to produce TNF, and TNF-induced death sets up a potentially vicious cycle of inflammation. (C) Concomitant deletion of Tnf in mice with Traf2-deficient keratinocytes breaks this vicious cycle and prevents apoptosis of keratinocytes and early onset of the psoriasis-like phenotype. (D) In the absence of TNF, Traf2-deficient keratinocytes still produce inflammatory cytokines via the non-canonical NF-κB pathway that ultimately generate the same psoriasis-like phenotype in mice but with slower onset. These cytokines recruit inflammatory immune cells to the skin, including neutrophils and IFNγ+ T cells.
Author response image 1
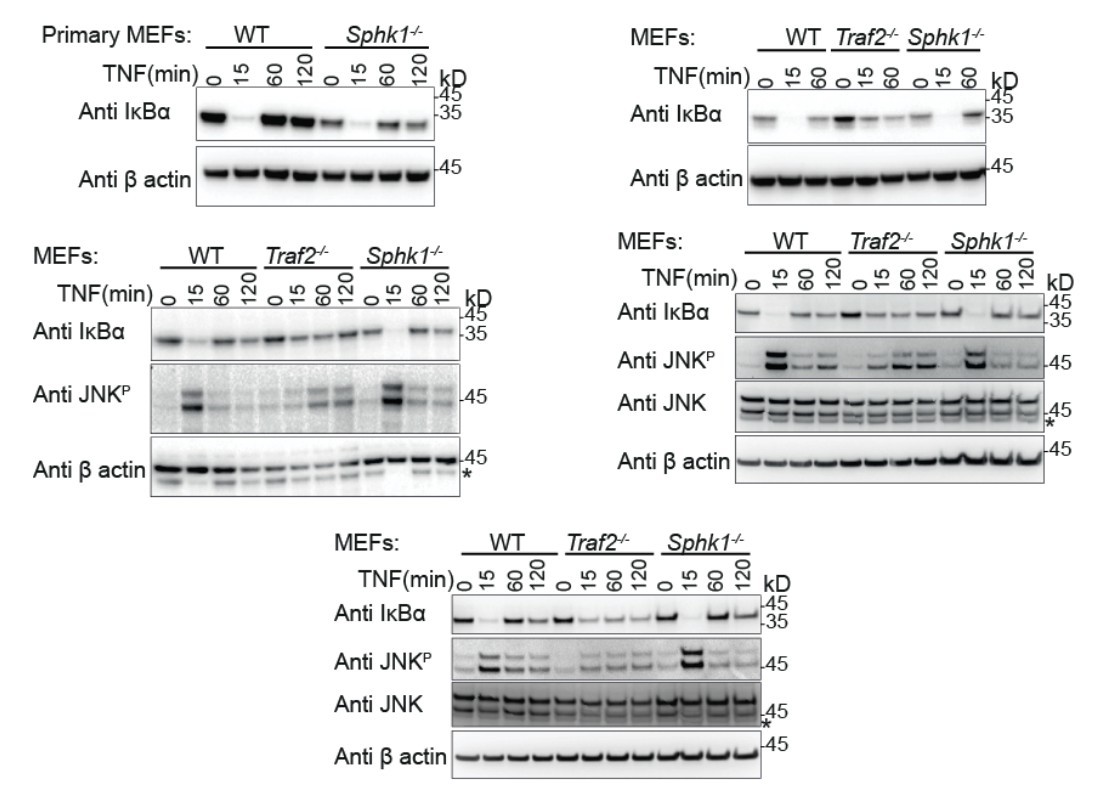
Primary (Top left) and transformed MEFs generated from the indicated knock-out mice stimulated with TNF for the indicated times and Western blotted with the indicated antibodies.
These indicate the reproducibility of the results that we have shown in our final manuscript.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1
eLife 4:e10592.
https://doi.org/10.7554/eLife.10592




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}