TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1
- Walter and Eliza Hall Institute of Medical Research, Australia
- University of Melbourne, Australia
- Olivia Newton-John Cancer Research Institute, Australia
- La Trobe University, Australia
- Monash University, Australia
- Cornell University, United States
- SA Pathology, Australia
- University of Zurich, Switzerland
Peer review process
This article was accepted for publication as part of eLife's original publishing model.
History
- Version of Record updated
- Version of Record published
- Accepted Manuscript published
- Accepted
- Received
Decision letter
-
Ivan DikicReviewing Editor; Goethe University Medical School, Germany
In the interests of transparency, eLife includes the editorial decision letter and accompanying author responses. A lightly edited version of the letter sent to the authors after peer review is shown, indicating the most substantive concerns; minor comments are not usually included.
Thank you for submitting your work entitled "TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of SPHK1" for peer review at eLife. Your submission has been favorably evaluated by Tadatsugu Taniguchi (Senior Editor) and three reviewers, one of whom is a member of our Board of Reviewing Editors.
The following individuals responsible for the peer review of your submission have agreed to reveal their identity: Manolis Pasparakis and Hao Wu (peer reviewers).
The reviewers have discussed the reviews with one another and the Reviewing editor has drafted this decision to help you prepare a revised submission.
The authors used genetic, cellular, immunological, and histological tools to investigate the roles of TRAF2 in inflammation and cell death. TRAF2 knockout does not affect TNF-induced canonical NF-κB or MAPK signaling, or cell death in macrophages. The authors show that TRAF2 is required for TNF signaling in fibroblasts and keratinocytes, but SPHK1 (sphingosine kinase 1) knockout cells did not show altered canonical NF-κB signaling, MAPK, or apoptotic cell death when compared to wild-type cells. Loss of TRAF2 leads to epidermal hyperplasia and psoriasis-like inflammation in mice, which can be delayed but not fully reversed by deletion of TNF. Deletion of TRAF2 activates the non-canonical NF-κB signaling pathway through NIK, and double deletion of NF-κB2 and TNF fully rescued inflammation induced by TRAF2 deletion. In addition, they conclude that in contrast to its previously suggested essential in regulating TRAF2 E3 ligase activity dependent TNF signalling, SPHK1 is not required for TNF-induced activation of NF-κB or MAPK and for resistance to TNF-induced cell death.
This is a very nice manuscript reporting clear and well-controlled experiments that reveal an important function of TRAF2 in the maintenance of skin homeostasis. Also, the data on the (lack of) function of SPHK1 in TNF signalling is very convincing. The authors should discuss more carefully the skin pathology developing in their mice comparing with human psoriasis in terms of histological findings, the immune infiltrates, gene expression as well as the genetic studies.
Reviewer #1 (Minor Comments):
1) The Materials and methods is missing information about how TUBE experiment was conducted, and source of RIPK1 and cIAP antibodies.
2) Figure 2 is missing blot for levels of anti ERK, JNK and p38 (A) and anti JNK (E).
3) The authors should be more consistent in all blots regarding time points when they compare effects of WT, Traf2-/- and Sphk1-/- cells.
4) In Figure 3A, why is there no observed caspase-8 ubiquitination in WT and Sphk1-/- cells upon TNF stimulation, since cIAPs and TRAF2 (Gonzalvez et al., 2012) should be active? The authors may consider discussing this issue.
5) Figure 3C: the authors should include also TUBE IP experiment with longer time points for Traf2-/- which would confirm their hypothesis about delayed NF-kB activation.
6) Figure 6: the authors should add FACS contour plot and corresponding bar diagram for CD11b+Ly6C+ infiltrating neutrophils in Traf2EKOTnf-/- as they did for other cell suspensions, and compare the data with results for CD4+ T cells.
7) The authors should focus in Discussion on experiments with IL-17 and discuss potential other signals for activation of non-canonical NF-kB independent of TNF.
8) Figure 10C: Kaplan-Meier graph does not depict final result with a survival of Traf2EKOTnf-/-Nfkb2-/- mice for up to 36 weeks. Instead, it is showing only survival percentage for other 6 genotypes. Since this survival curve is one of the most crucial results, the authors should add it.
9) The authors describe that loss of Traf2 mostly induced lesions in anterior parts of the body, while animals lacking active TRAF2 and functional non-canonical NF-kB signaling were more susceptible to develop lesions in ventral and posterior parts. They should discuss possible reasons for these differences.
10) In the last Results paragraph, the authors twice talk about the phenotype of Traf2EKO/+Tnf-/-Nfkb2-/- which isn't presented nowhere on figures. Please, provide figures showing these results or correct that paragraph.
Reviewer #2 (Minor Comments):
In Figures 1A and 2A, the β actin blots are not equalized among the lanes. It probably will not change the conclusions, but loading the same amount of samples to each lane will make the figures look more convincing.
Reviewer #3 (Minor Comments):
The authors state that the triple mutant mice (TRAF2EKO, Tnf-/-, Nfkb2-/-) 'occasionally' developed skin lesions but do not indicate how many mice have been analysed and how many developed lesions. Please clearly state the number of mice analysed for each genotype. It would also be helpful to clearly state the age of mice shown in all figures.
There is a wrong citation in the Discussion "Several mouse models of skin inflammation have been rescued by depletion of TNF (Gerlach et al., 2011; Nenci et al., 2006; Pasparakis, Schmidt-Supprian, and Rajewsky, 2002)." The citation Pasparakis, Schmidt-Supprian, and Rajewsky, 2002 is not the right one, the correct one is Pasparakis, Courtois et al., 2002.
Figure 6D in the subsection “Psoriasis-like inflammation in Traf2EKO mice is characterised by skin infiltration of neutrophils and IFNγ producing CD4+ T cells in” referring to the data on IFNγ+ T cells should be Figure 6E.
[Editors' note: further revisions were requested prior to acceptance, as described below.]
Thank you for resubmitting your work entitled "TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine Kinase 1" for further consideration at eLife. Your revised article has been favorably evaluated by Tadatsugu Taniguchi (Senior Editor), a Reviewing Editor, and one reviewer. The manuscript has been improved but there are some remaining issues that need to be addressed before acceptance, as outlined below.
The inflammatory skin lesions developing in mice with keratinocyte-specific TRAF2 knockout can be described as psoriasis-like, but it is not correct to write that these mice develop psoriasis, as in the following instances in the manuscript:
In the subsection “Psoriasis-like inflammation in Traf2EKO mice is characterised by skin infiltration of neutrophils and IFNγ producing CD4+ T cells in”: "However, both wild type and Ifng-/-chimeras developed skin inflammation to the same extent indicating that IFNγ is not a major contributor to the psoriasis (Figure 7)."
In the subsection “Blocking non-canonical NF-κB signalling and depletion of TNF together prevent inflammation caused by TRAF2 deficiency in keratinocytes”: "Contrary to our hypothesis, deficiency in Map3k14 or Nfkb2 did not prevent the psoriasis in Traf2EKO mice (Figure 9 and 10)."
Discussion: "Unfortunately, we were unable to age these mice more than 15 weeks as they developed lymphadenopathy and could therefore not examine the effect of Mlkl-/-Casp8-/- on the late onset psoriasis."
Discussion: "While this does not exclude a role for IL-17 in the psoriasis observed in Traf2EKO or Traf2EKOTnf-/- mice we hypothesized that Traf2EKOTnf-/- keratinocytes produced other factors to recruit inflammatory cells to the skin."
In Figure 11, the schematic model gives the impression that in the absence of TNF the inflammatory skin lesions are induced by M-CSF and IL-23 released by keratinocytes and IFNγ produced by T cells. As there is no data in the paper supporting causal function of these cytokines, it would be more appropriate not to refer to specific cytokines or to at least clearly describe in the scheme and the legend that these cytokines may be implicated but their functional role remains to be experimentally addressed.
https://doi.org/10.7554/eLife.10592.017Author response
[…] This is a very nice manuscript reporting clear and well-controlled experiments that reveal an important function of TRAF2 in the maintenance of skin homeostasis. Also, the data on the (lack of) function of SPHK1 in TNF signalling is very convincing. The authors should discuss more carefully the skin pathology developing in their mice comparing with human psoriasis in terms of histological findings, the immune infiltrates, gene expression as well as the genetic studies.
Reviewer #1 (Minor Comments):
1) The Materials and methods is missing information about how TUBE experiment was conducted, and source of RIPK1 and cIAP antibodies.
Thanks for picking this up, we have now included the full information for the method and the source of the antibodies (subsection “Immuno precipitation of ubiquitin-conjugates (TUBE IP)”).
2) Figure 2 is missing blot for levels of anti ERK, JNK and p38 (A) and anti JNK (E).
We repeated this experiment using the same time points as before and tested with a full panel of antibodies and the appropriate loading controls (new Figure 2A and Author response image 1). As before, IκBα degradation is complete in Sphk1-/- and wild type MEFs but incomplete in Traf2-/- MEFs (n=1 primary MEF, n=4; comprising 3 biologically independent transformed MEF lines, one of which was tested twice). Likewise, JNK phosphorylation is maximal in wild type and Sphk1-/- MEFs and is reduced and delayed in Traf2-/- MEFs (n=3). In the previous figure we had included phospho-p38 and phospho-ERK blots, however, in repeating experiments with different sources of MEFs for the revision, we observed variations in these responses. Therefore we cannot draw conclusions based on this data and have removed them. This fits with our poor experience with different biological MEF lines and is the reason that we prefer to use the far more reliable dermal fibroblasts (e.g. Figure 3).
Author response image 1
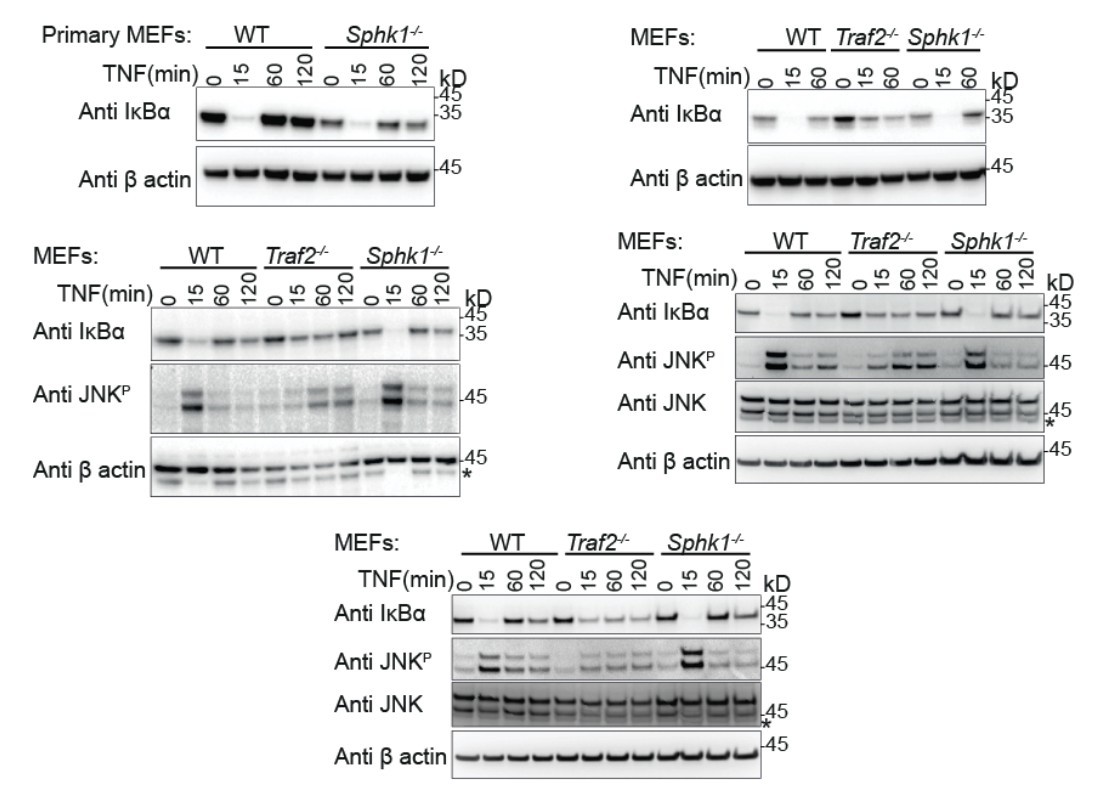
Primary (Top left) and transformed MEFs generated from the indicated knock-out mice stimulated with TNF for the indicated times and Western blotted with the indicated antibodies.
These indicate the reproducibility of the results that we have shown in our final manuscript.
3) The authors should be more consistent in all blots regarding time points when they compare effects of WT, Traf2-/- and Sphk1-/- cells.
In Figure 2A, B, D and E we analyse 15, 60 and 120 minutes following TNF stimulation, which are well established time points that allow us to monitor activation and the return to baseline. In Figure 2E we also include time points of 4 and 6 hr because keratinocytes are a particularly important cell type in the study and we thought it relevant to investigate a prolonged response. In Figure 3A, using the more involved TUBE protocol in MDFs we examined the early time
point of 5 minutes because ubiquitylation occurs as an early during the signalling cascade. When we saw a defect in ubiquitylation in Traf2-/- cells we extended the analysis to 60 minutes (old Figure 3B) because that is where we started to observe NF-κB activation in Traf2-/- MDFs (Figure 2B). Because we saw a
delayed RIPK1 ubiquitylation in Traf2-/-cells (old Figure 3B) this led us to question whether Sphk1-/- cells might also activate ubiquitylation aberrantly and therefore we performed a more detailed analysis of 15, 30, 60, 90 and 120 minutes in these cells (old Figure 3C).
In response to the reviewer's comment we have now analysed the 0, 15, 60 and 120 minute time points following TNF stimulation of Traf2-/- and Sphk1-/- MDFs and compared to wild type MDFs in the same experiment (New Figure 3B). The reviewers will appreciate that these new results are identical with the old results and entirely consistent with the other data that we show in Figure 2B (IκBα degradation, caspase-8 activation and ERK phosphorylation). Therefore we have removed the old Figure 3B and C and replaced these panels with our new Figure 3B.
4) In Figure 3A, why is there no observed caspase-8 ubiquitination in WT and Sphk1-/- cells upon TNF stimulation, since cIAPs and TRAF2 (Gonzalvez et al., 2012) should be active? The authors may consider discussing this issue.
In Gonzalvez et althe authors use TRAIL or the Fas activating antibody Jo2 in HCT116 Bax-/- cells to examine ubiquitylation of caspase-8, which they observed most strongly 1 hr post stimulation. In Figure 3A we analysed 5 minutes post TNF stimulation in MDFs. In our new experiment (new Figure 3B), we extended our analysis to 120 minutes and also do not see signs of an ubiquitylated form of caspase-8 in wild type and Sphk1-/- cells. However we think the completely different treatments and cell types used in these two works preclude us from drawing any conclusion or adding anything productive to the Discussion.
5) Figure 3C: the authors should include also TUBE IP experiment with longer time points for Traf2-/- which would confirm their hypothesis about delayed NF-kB activation.
As requested we have repeated this experiment (new Figure 3B), which repeats and extends our previous observation that ubiquitylation of RIPK1 and phosphorylation of ERK is delayed and weaker in Traf2-/-MDFs compared to wild type or Sphk1-/- cells.
6) Figure 6: the authors should add FACS contour plot and corresponding bar diagram for CD11b+Ly6C+ infiltrating neutrophils in Traf2EKOTnf-/- as they did for other cell suspensions, and compare the data with results for CD4+ T cells.
We have added the requested data quantifying CD11b Ly6G neutrophils from the epidermis of Traf2EKOTnf-/- mice (new panel in Figure 6A and new data in 6B). We have also now added flow cytometry plots to examine and quantify recruitment of neutrophils into the dermis of Traf2EKOand Traf2EKOTnf-/- mice compared with wild type dermis (New Figure 6C and 6D).
7) The authors should focus in Discussion on experiments with IL-17 and discuss potential other signals for activation of non-canonical NF-kB independent of TNF.
In the Discussion section, we have added discussion on the clinical results with secukinumab and ixekizumab and the implications and limitations of our in vitroexperiments.
"IL-17 plays an important role in inflammatory diseases and monoclonal antibodies targeting IL-17 such as secukinumab and ixekizumab are performing well in psoriasis patients (Tse, 2013). […] However, in our experiments IL-17 did not increase Traf2EKO/EKOTnf-/- keratinocyte proliferation compared to Traf2EKO/+Tnf-/- cells in vitroand loss of TRAF2 had no impact on IL-17 mediated activation of NF-κB and p38 in keratinocytes. While this does not exclude a role for IL-17 in the psoriasis observed in Traf2EKOor Traf2EKOTnf-/- mice we hypothesized that Traf2EKOTnf-/- keratinocytes produced other factors to recruit inflammatory cells to the skin."
Likewise we have included a discussion on the implications of our results regarding non-canonical NF-κB signalling and psoriasis:
"This observation might be relevant to human psoriasis because it has been reported that the levels of TWEAK and its receptor Fn-14 are elevated in human psoriatic lesions (Cheng et al., 2015) and it is well established that TWEAK/Fn14 signalling induces degradation of TRAF2 and activation of non-canonical NF-κB signalling (Saitoh et al., 2003; Vince et al., 2008). Thus, our results suggest that inhibition of non-canonical NF-κB signalling might be beneficial in chronic inflammatory disease together with TNF inhibitions."
8) Figure 10C: Kaplan-Meier graph does not depict final result with a survival of Traf2EKOTnf-/-Nfkb2-/- mice for up to 36 weeks. Instead, it is showing only survival percentage for other 6 genotypes. Since this survival curve is one of the most crucial results, the authors should add it.
Our apologies. The data was there but it was incorrectly colour coded (pink instead of blue) and was in an unfortunate position so that it gave the impression of demarking the graph boundary. We have now changed the format of this graph so that it is clear which survival line corresponds to which mouse strain. We have also substantially increased the numbers of the strains that are most directly relevant, namely Traf2-/-Tnf-/-Nfkb2-/- (from 4 to 13) and Traf2-/-Tnf-/-Nfkb2-/+ (from 9 to 15). These new data also incorporate three Traf2-/-Tnf-/-Nfkb2-/- mice that die of other causes than psoriasis. These causes are most likely due to their immune deficiency and also occur in Tnf-/-Nfkb2-/- and Traf2EKO/EKONik(Map3K14)aly/aly mice (Figures 9A and 10C).
9) The authors describe that loss of Traf2 mostly induced lesions in anterior parts of the body, while animals lacking active TRAF2 and functional non-canonical NF-kB signaling were more susceptible to develop lesions in ventral and posterior parts. They should discuss possible reasons for these differences.
We note that we also see the same lesions developing in Traf2EKO/EKONfkb2-/-Tnf-/- and Tnf-/-Nfkb2-/- mice (Figure 10D). NIK and non-canonical NF-κB signalling play an important role in immune responses and in establishing the immune system. Given the location of the lesions in Traf2EKO/EKONfkb2-/- and Traf2EKO/EKONfkb2-/-Tnf-/- mice, we think it likely that these lesions are connected with a failure to correctly regulate responses to bacteria in these locations. This is consistent with the other report that fails to generate complete Traf2-/-Map3k14-/- or Traf2-/-Map3k14-/-Tnf-/- mice (Lin, PNAS 2011) that we have cited.
10) In the last Results paragraph, the authors twice talk about the phenotype of Traf2EKO/+Tnf-/-Nfkb2-/- which isn't presented nowhere on figures. Please, provide figures showing these results or correct that paragraph.
These mice were shown in Figure 11, which has now been merged with Figure 10. We have provided the appropriate figure reference (Figure 10C) for the statement concerning these mice in the text. One of the references to this strain of mice, in the last sentence of the Results section, was however a typo. It should have referred to Traf2EKO/EKOTnf-/-Nfkb2-/-and we have now fixed this error.
Reviewer #2 (Minor Comments):
In Figures 1A and 2A, the β actin blots are not equalized among the lanes. It probably will not change the conclusions, but loading the same amount of samples to each lane will make the figures look more convincing.
In Figure 1A the actin signal was burnt out due to over-exposure. We have replaced this loading control with a shorter exposure time.
We repeated the experiment in Figure 2A and the new experiment has loading controls for JNK and a more representative actin blot.
Reviewer #3 (Minor Comments):
The authors state that the triple mutant mice (TRAF2EKO, Tnf-/-, Nfkb2-/-) 'occasionally' developed skin lesions but do not indicate how many mice have been analysed and how many developed lesions. Please clearly state the number of mice analysed for each genotype. It would also be helpful to clearly state the age of mice shown in all figures.
We have discussed and included pictures of these mice and provided the pertinent information about the age of the mice:
"Three out of 13 of the Traf2EKOTnf-/-Nfkb2-/-mice developed skin lesions at an early age around the mouth and anus (Figure 10C and 10D). This is likely to be due to the susceptibility of NFκB2 deficient mice to opportunistic infections because both Tnf-/-Nfkb2-/-and Traf2EKO/EKONfkb2-/- mice succumbed to such infections (Figure 10C and Shinkura et al., 1996; Yin et al., 2001)."
There is a wrong citation in the Discussion "Several mouse models of skin inflammation have been rescued by depletion of TNF (Gerlach et al., 2011; Nenci et al., 2006; Pasparakis, Schmidt-Supprian, and Rajewsky, 2002)." The citation Pasparakis, Schmidt-Supprian, and Rajewsky, 2002 is not the right one, the correct one is Pasparakis, Courtois et al., 2002.
Thank you for this correction.
Figure 6D in the subsection “Psoriasis-like inflammation in Traf2EKO mice is characterised by skin infiltration of neutrophils and IFNγ producing CD4+ T cells in” referring to the data on IFNγ+ T cells should be Figure 6E.
The whole figure has changed and we have endeavoured to cite the new figure correctly.
[Editors' note: further revisions were requested prior to acceptance, as described below.]
The inflammatory skin lesions developing in mice with keratinocyte-specific TRAF2 knockout can be described as psoriasis-like, but it is not correct to write that these mice develop psoriasis, as in the following instances in the manuscript:
In the subsection “Psoriasis-like inflammation in Traf2EKO mice is characterised by skin infiltration of neutrophils and IFNγ producing CD4+ T cells in”: "However, both wild type and Ifng-/- chimeras developed skin inflammation to the same extent indicating that IFNγ is not a major contributor to the psoriasis (Figure 7)."
In the subsection “Blocking non-canonical NF-κB signalling and depletion of TNF together prevent inflammation caused by TRAF2 deficiency in keratinocytes”: "Contrary to our hypothesis, deficiency in Map3k14 or Nfkb2 did not prevent the psoriasis in Traf2EKO mice (Figures 9 and 10)."
Discussion: "Unfortunately, we were unable to age these mice more than 15 weeks as they developed lymphadenopathy and could therefore not examine the effect of Mlkl-/-Casp8-/- on the late onset psoriasis."Discussion: "While this does not exclude a role for IL-17 in the psoriasis observed in Traf2EKO or Traf2EKOTnf-/- mice we hypothesized that Traf2EKOTnf-/- keratinocytes produced other factors to recruit inflammatory cells to the skin."
Thanks for pointing out this. We have changed all the above items to the appropriate and relevant terms to the phenotype of Traf2EKOmice, which are consistent with the title and rest of the manuscript.
In Figure 11, the schematic model gives the impression that in the absence of TNF the inflammatory skin lesions are induced by M-CSF and IL-23 released by keratinocytes and IFNγ produced by T cells. As there is no data in the paper supporting causal function of these cytokines, it would be more appropriate not to refer to specific cytokines or to at least clearly describe in the scheme and the legend that these cytokines may be implicated but their functional role remains to be experimentally addressed.
Now we have changed and improved Figure 11 and its legend addressing the mentioned issues. We have changed the tile of the figure to the “Proposed mechanisms by which loss of TRAF2 in keratinocytes causes a psoriasis-like skin inflammation.” We have changed the specific cytokines to a general term of “inflammatory cytokines”.
https://doi.org/10.7554/eLife.10592.018Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1
eLife 4:e10592.
https://doi.org/10.7554/eLife.10592




{kind=link}