Viral-mediated Pou5f1 (Oct4) overexpression and inhibition of Notch signaling synergistically induce neurogenic competence in mammalian Müller glia
- Department of Neuroscience, Johns Hopkins University School of Medicine, United States
- Department of Ophthalmology and Visual Sciences, University of Michigan School of Medicine, United States
- Department of Cell and Developmental Biology, University of Michigan School of Medicine, United States
- Michigan Neuroscience Institute, University of Michigan School of Medicine, United States
- Department of Ophthalmology, Johns Hopkins University School of Medicine, United States
- Department of Neurology, Johns Hopkins University School of Medicine, United States
- Institute for Cell Engineering, Johns Hopkins University School of Medicine, United States
- Kavli Neuroscience Discovery Institute, Johns Hopkins University School of Medicine, United States
eLife Assessment
This manuscript demonstrates that Oct4 overexpression synergizes with Notch inhibition (Rbpj knockout) to promote the conversion of adult murine Müller glia (MG) into bipolar cells. These findings are important as the authors used rigorous genetic lineage tracing (GLAST-CreER; Sun-GFP) to confirm that neurogenesis indeed originates from MGs, addressing a key issue in the field. The single-cell multiomic analyses are compelling, and while functional studies of MG-derived bipolar cells would strengthen the conclusions, they are beyond the scope of this study.
https://doi.org/10.7554/eLife.106450.3.sa0Significance of the findings:
Important: Findings that have theoretical or practical implications beyond a single subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Compelling: Evidence that features methods, data and analyses more rigorous than the current state-of-the-art
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
Retinal Müller glia in cold-blooded vertebrates can reprogram into neurogenic progenitors to replace neurons lost to injury, but mammals lack this ability. While recent studies have shown that transgenic overexpression of neurogenic bHLH factors and glial-specific disruption of NFI family transcription factors and Notch signaling induce neurogenic competence in mammalian Müller glia, induction of neurogenesis in wildtype glia has thus far proven elusive. Here, we report that viral-mediated overexpression of the pluripotency factor Pou5f1 (Oct4) induces transdifferentiation of mouse Müller glia into bipolar neurons, and synergistically stimulates glial-derived neurogenesis in parallel with Notch loss of function. Single-cell multiomic analysis shows that Pou5f1 overexpression leads to widespread changes in gene expression and chromatin accessibility, inducing activity of both the neurogenic transcription factor Rfx4 and the Yamanaka factors Sox2 and Klf4. This study demonstrates that viral-mediated overexpression of Pou5f1 induces neurogenic competence in adult mouse Müller glia, identifying mechanisms that could be used in cell-based therapies for treating retinal dystrophies.
Introduction
The homeodomain factor Pou5f1 (Oct4) is a central component of transcriptional regulatory networks that maintain pluripotency in embryonic stem (ES) cells. Along with Sox2, Klf4, and Myc, Pou5f1 is one of the Yamanaka factors sufficient to induce the formation of induced pluripotent stem cells from virtually all somatic cell types (Takahashi and Yamanaka, 2006). In contrast to these other factors, Pou5f1 is selectively expressed in ES cells and essentially absent from somatic cells, including adult stem cells from every tissue examined. The two clear exceptions are found in gonadal stem cells (Takashima et al., 2013; Uhlen et al., 2010) and during neural crest formation, where Pou5f1 expression is essential for the ability of these cells to generate diverse cell types (Morrison and Brickman, 2006; Zalc et al., 2021). This has raised the question of whether Pou5f1 overexpression in vivo, alone or in combination with other pluripotency factors, may confer some level of multipotency on somatic cells in the central nervous system.
Sustained overexpression of the Yamanaka factors Pou5f1, Sox2, Klf4, and Myc induces teratoma formation in many tissues (Abad et al., 2013; Ohnishi et al., 2014). Transient or low-level Yamanaka factor overexpression, in contrast, has been reported to induce cellular rejuvenation in both neurons and other cell types, although this occurs without clear induction of proliferation or conferring multipotency (Ocampo et al., 2016; Wang et al., 2021a). However, several studies have directly investigated whether viral-mediated overexpression in CNS neural stem or glial cells of Pou5f1 alone, or in combination with subsets of other pluripotency factors, can induce multipotency and neurogenesis. Results from these studies are mixed, with Pou5f1 overexpression in neural stem cells reported to either not induce neurogenesis (Asadi et al., 2015; Dehghan et al., 2015; Sim et al., 2011), to promote formation of oligodendrocyte progenitors (OPCs) (Yu et al., 2021), or to promote remyelination in committed OPCs (Dehghan et al., 2016). Two studies reported that viral-mediated Pou5f1 overexpression, either alone (Niu et al., 2013) or in combination with other Yamanaka factors (Gao et al., 2016), could directly convert reactive astrocytes into neurons. However, these studies used the GFAP minipromoter to drive gene expression, which has been reported to show insert-dependent ectopic insert-dependent silencing in glia and activation in neurons (Wang et al., 2021b). This limitation makes these findings difficult to interpret, as additional methods of validating cell lineage were not used.
Two previous studies have reported injury-dependent induction of Pou5f1 in retinal Müller glial cells in zebrafish and mice. In zebrafish, where injury induces Müller glia to undergo conversion to neurogenic progenitors (Wan and Goldman, 2016), it was reported that Pou5f1 is essential for glial reprogramming to occur (Sharma et al., 2019). In mice, where neurogenic competence is rapidly and actively suppressed following injury, it was reported that Pou5f1 expression is transiently induced, but fully repressed within 24 hr following injury (Reyes-Aguirre and Lamas, 2016). This raises the possibility that Pou5f1 expression may be sufficient to confer neurogenic competence on retinal Müller glial cells. In this study, we assessed whether AAV-mediated overexpression of Pou5f1 could induce reprogramming of adult mouse Müller glia. While we did not detect any expression of Pou5f1 in either zebrafish or mouse Müller glia following injury, we used both genetic cell lineage and single-cell omics analyses to unambiguously demonstrate that viral-mediated overexpression of Pou5f1 selectively induces the formation of bipolar neurons from control GlastCreER;Rosa26LSL-Sun1-GFP mouse Müller glia without inducing substantial levels of proliferation. We further showed that Pou5f1 overexpression synergistically enhances existing levels of Müller glia-derived neurogenesis in the absence of Notch signaling. Single-cell multiomic analysis demonstrated that Pou5f1 cooperates with Sox2 and Klf4, which are constitutively expressed in Müller glia, to broadly alter the gene regulatory landscape and induce expression of genes such as the neurogenic transcription factor Rfx4. This study demonstrates that substantial levels of in situ glia-to-neuron reprogramming in mammalian retina can be induced by viral overexpression of a Yamanaka factor.
Results
Pou5f1 and Nanog are not detectably expressed in Müller glia or Müller glia-derived progenitor cells in either zebrafish or mice
To examine the expression of Pou5f1 and other pluripotency-associated genes in Müller glia, we first analyzed previously published gene expression data from zebrafish and mouse retina (Hoang et al., 2020; Le et al., 2024). We first analyzed both single-cell RNA-seq data obtained from wildtype retinas and bulk RNA-seq data obtained from FACS-isolated Müller glia carrying glial-specific GFP reporter lines. Consistent with previous findings (Nelson et al., 2011; Surzenko et al., 2013), in mice we detected expression of Sox2 and Klf4 in resting and both N-methyl-D-aspartate (NMDA)-treated and light-damaged Müller glia, along with injury-induced expression of Myc, which is lost as activated glia progressively return to a resting state by 72 hr post-injury. Likewise, in zebrafish we observed expression of sox2 and myca/b (but not klf4) in both resting and injured Müller glia (Figure 1—figure supplement 1A, B). However, we did not detect expression of either Pou5f1 or its direct target Nanog in either species in any treatment condition (Loh et al., 2006). Bulk RNA-seq data obtained from purified Müller glia detected very low levels of pou5f3v(Oct4), but not Nanog, expression in some injured zebrafish Muller glia, but neither Pou5f1 nor Nanog was detected in either resting or injured mouse Müller glia (Figure 1—figure supplement 1A, B). In addition, we did not observe any induction of Pou5f1 or Nanog in snRNA-seq data obtained from mouse Müller glia following selective loss of function of Rbpj alone or in combination with Nfia/b/x (Le et al., 2024; Figure 1—figure supplement 1C). These results indicate that neither NMDA excitotoxicity, light damage, or induction of neurogenic competence by loss of function of Nfia/b/x and/or Rbpj induces detectable levels of Pou5f1 or Nanog in neurogenic mammalian Müller glia.
AAV-mediated overexpression of Pou5f1 induces conversion of control GlastCreER;Rosa26LSL-Sun1-GFP mouse Müller glia to bipolar-like cells
To determine whether Pou5f1 overexpression could reprogram mammalian Müller glia into a neurogenic state, we administered intraperitoneal tamoxifen injections into P21 GlastCreER;Rosa26LSL-Sun1-GFP mice, thereby irreversibly labeling Müller glia and their progeny with nuclear envelope-targeted Rosa26LSL-Sun1-GFP under control of the ubiquitous CAG promoter (de Melo et al., 2012; Hoang et al., 2020; Figure 1A, Figure 1—figure supplement 2a). One week following the final tamoxifen dose, we performed intravitreal injections of either Gfap-Pou5f1-mCherry or Gfap-mCherry control 7m8 AAV (Figure 1B, Figure 1—figure supplement 2b). While expression of other neurogenic bHLH factors driven by the GFAP promoter were rapidly silenced in Müller glia and activated in amacrine and retinal ganglion cells, Gfap-Pou5f1-mCherry remained selectively expressed in Müller glia. However, Pou5f1 expression alone did not induce detectable levels of Müller glia-derived neurogenesis in the uninjured retina (Le et al., 2022).
Figure 1 with 3 supplements see all
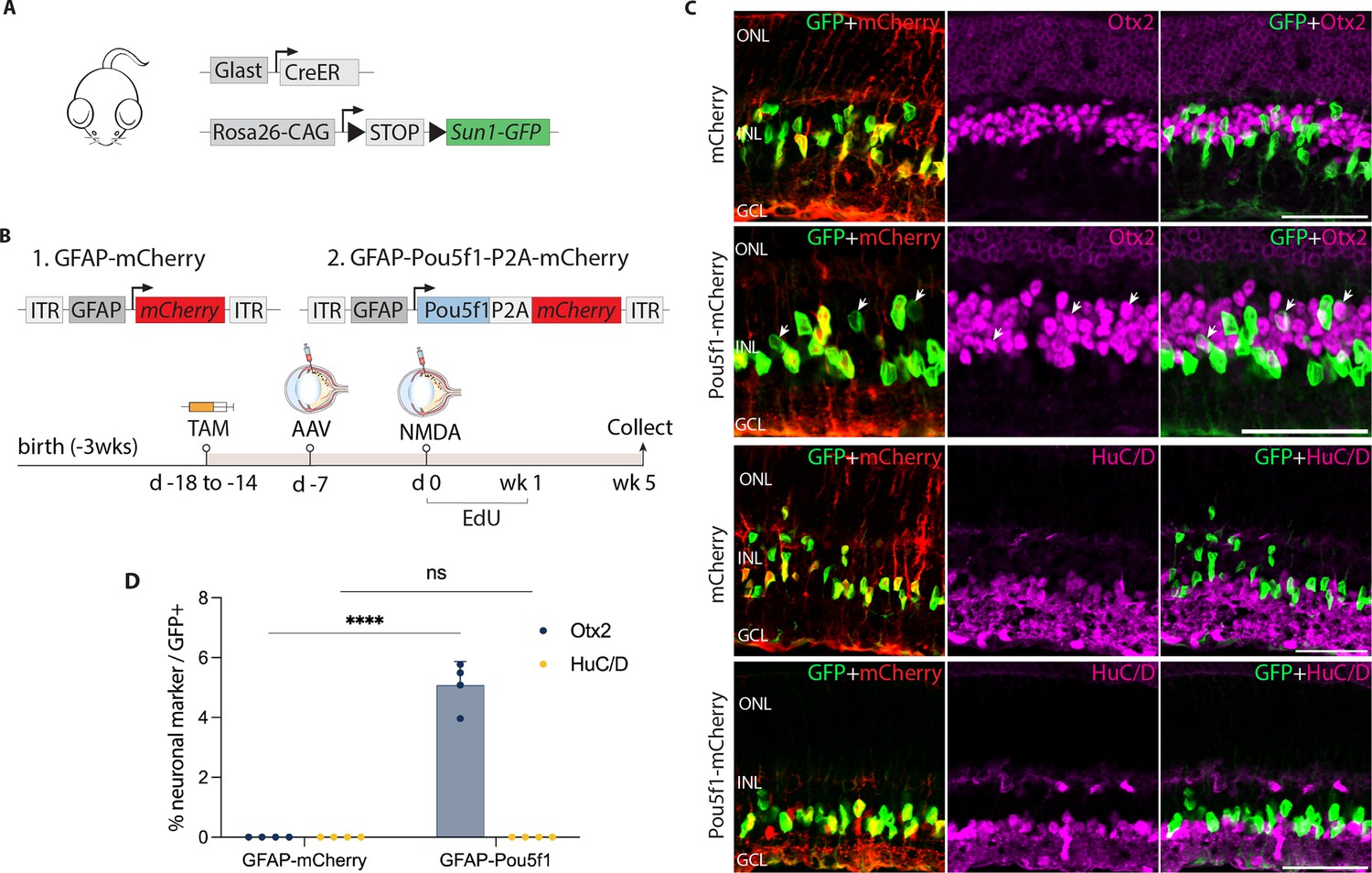
AAV-mediated overexpression of Pou5f1 induces neurogenesis in control GlastCreER;Rosa26LSL-Sun1-GFP Müller glia following N-methyl-D-aspartate (NMDA)-induced excitotoxicity.
(A) Schematic of the transgenic construct used to specifically label Müller glia with Sun1-GFP expression. (B) Schematic of the GFAP AAV constructs and experimental workflow. (C) Representative images of retinas immunolabeled for GFP, mCherry, Otx2, and HuC/D. White arrowheads indicate GFP-positive Müller glia-derived neurons expressing neuronal markers Otx2 or HuC/D. (D) Quantification of mean percentage ± SD of GFP-positive Müller glia-derived neurons expressing either Otx2 or HuC/D. Significance was determined via two-way ANOVA with Tukey’s multiple comparison test: ****p < 0.0001. Each data point was calculated from an individual retina. TAM, tamoxifen; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar = 50 μm.
Since injury is often required to promote Müller glia-derived neurogenesis following Ascl1 overexpression or loss of function of Nfia/b/x (Hoang et al., 2020; Jorstad et al., 2017), we hypothesized that this might also be so for Müller glia overexpressing Pou5f1. To test this, we injected NMDA 1 week post-viral injection. We then administered 5-ethynyl-2′-deoxyuridine (EdU) to label proliferating cells. Retinas were harvested and analyzed at 1, 2, and 5 weeks post-injury (Figure 1—figure supplement 2b, Figure 1B). Immunostaining analysis revealed enriched mCherry expression in GlastCreER;Rosa26LSL-Sun1-GFP Müller glia in both control mCherry and Pou5f1-mCherry-infected retinas at all timepoints (Figure 1C, Figure 1—figure supplement 2c–l). In addition, Pou5f1 expression was detected in GFP-positive Müller glia in Pou5f1-mCherry-infected retinas but not in mCherry-infected GlastCreER;Rosa26LSL-Sun1-GFP retina (Figure 1—figure supplement 2c, d, Figure 1—figure supplement 3). While most GFP-positive cells infected with Pou5f1-mCherry expressed the Müller glial marker Sox2 (Figure 1—figure supplement 2e, f, Figure 1—figure supplement 3b), a subset of GFP-positive cells lost Sox2 expression from 2 weeks post-injury onward (Figure 1—figure supplement 2f, Figure 1—figure supplement 3b, yellow arrows), which is consistent with glia-to-neuron conversion.
We observed selective expression of the neurogenic bHLH factor Ascl1 in Pou5f1-mCherry-infected GFP-positive Müller glia at 1 week post-injury (Figure 1—figure supplement 2g), with the number of Ascl1-positive cells declining somewhat by 2 and 5 weeks post-injury (Figure 1—figure supplement 2h, Figure 1—figure supplement 3c). While at 1 week post-injury, we did not observe any significant Müller glia-derived neurogenesis (Figure 1—figure supplement 2i, m), by 2 weeks post-injury we observed 1.1% of GFP-positive cells colocalized with the bipolar cell marker Otx2 in Pou5f1-mCherry-infected retinas (Figure 1—figure supplement 2j, m), and this increased to 5.1% of GFP-positive cells at 5 weeks post-injury (Figure 1A, D). We found that Müller glia-derived Otx2+ bipolar-like cells lost mCherry reporter expression, likely due to reduced activity of the Gfap-Pou5f1-mCherry construct in neurons (Le et al., 2022). Immunostaining with additional cell-specific markers revealed that a subset of these GFP-positive cells expressed the cone bipolar markers Scgn and Cabp5 (Figure 1—figure supplement 3d, e), but not the rod and cone ON bipolar cell marker Isl1 or the rod marker Nrl (Figure 1—figure supplement 3f, g). Furthermore, we did not observe expression of the amacrine cell marker HuC/D in GFP-positive cells (Figure 1C, D), which was observed in neurons generated from neurogenic mouse Müller glia deficient in either Rbpj or Nfia/b/x (Hoang et al., 2020; Le et al., 2024). No EdU/GFP-positive cells were detected at either 1 week (Figure 1—figure supplement 2k) or 5 weeks post-injury (Figure 1—figure supplement 3h). However, at 2 weeks post-injury, we observed a very limited number of GFP-positive cells incorporating EdU in retinas infected with both mCherry control and Pou5f1-mCherry, with modest EdU incorporation but significantly increased by Pou5f1 overexpression (Figure 1—figure supplement 2l, n). These findings indicate that Pou5f1 overexpression induces a very low level of proliferation in Müller glia in the second week following NMDA injury. However, the absence of EdU/Otx2/GFP-positive cells at both 2 and 5 weeks post-injury suggests that bipolar cell generation at this stage was, at least in part, driven by direct transdifferentiation.
Pou5f1 overexpression synergistically enhances glial-derived neurogenesis induced by genetic disruption of Notch signaling
In our previous study, we demonstrated that disrupting Notch signaling in adult mouse Müller glia – either by selective deletion of the transcriptional mediator Rbpj or by combined disruption of Notch1 and Notch2 receptors – induces transdifferentiation into bipolar and amacrine-like cells (Le et al., 2024). To further determine the effects of Pou5f1 overexpression in the absence of Notch signaling, we used GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP mice to genetically disrupt Rbpj function (Figure 2A, Figure 2—figure supplement 1a). We then performed intravitreal injection of either control or Pou5f1 overexpression constructs followed by NMDA and EdU injection as performed for GlastCreER;Rosa26LSL-Sun1-GFP mice. Retinas were collected at 1, 2, and 5 weeks following NMDA treatment (Figure 2B, Figure 2—figure supplement 1b). Immunostaining confirmed expression of Pou5f1 protein in GFP-positive Müller glia of Pou5f1-mCherry-infected retinas, but not in age-matched mCherry-infected controls (Figure 2—figure supplement 1c, d, Figure 2—figure supplement 2a). We observed that many GFP-positive cells in Pou5f1-mCherry-infected retinas did not express the Müller glial markers Sox2 (Figure 2—figure supplement 1e, f) or Sox9 (Figure 2—figure supplement 2b, yellow arrows), suggesting that they have lost their glial identity.
Figure 2 with 4 supplements see all
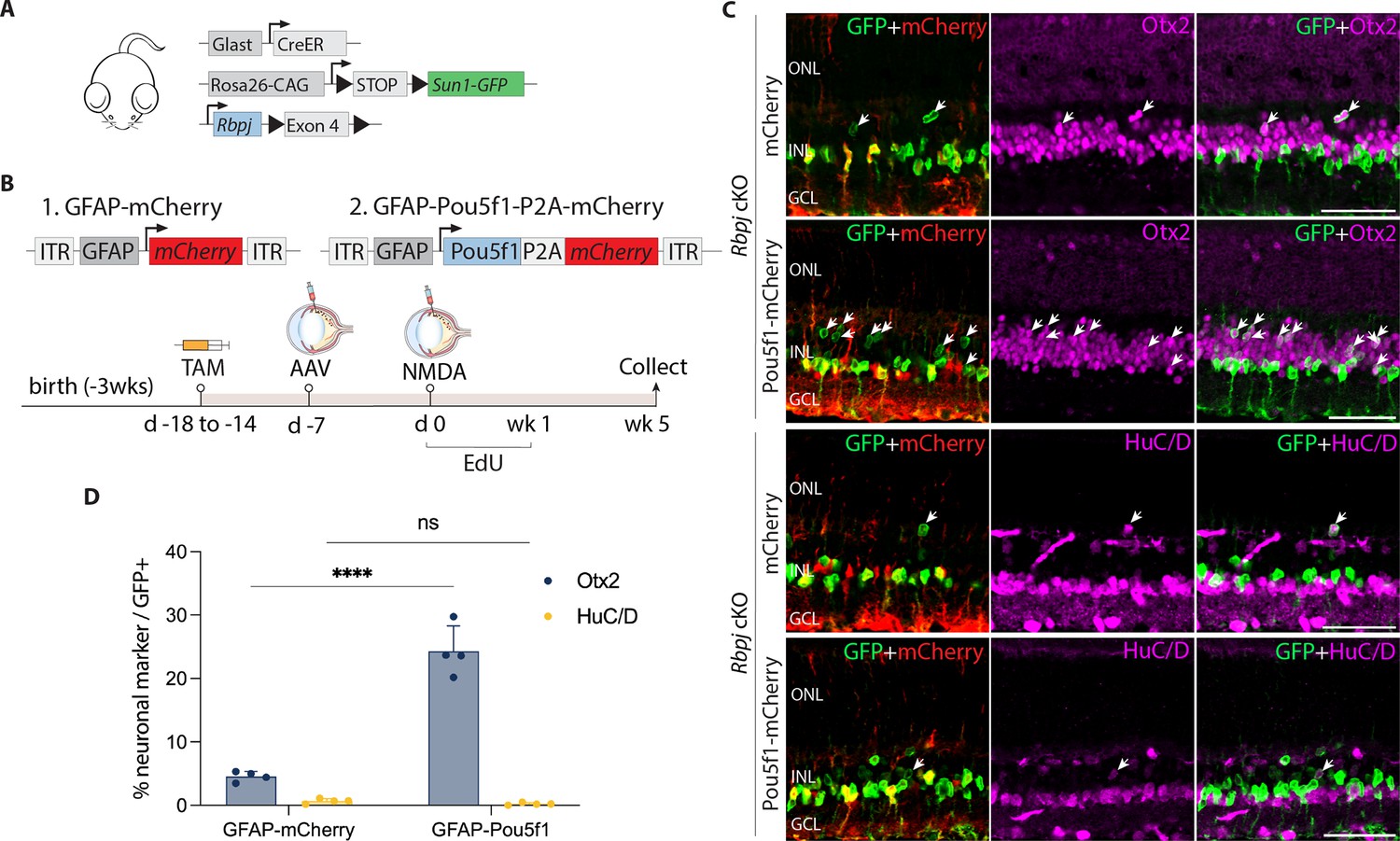
Pou5f1 overexpression enhances neurogenesis in Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia.
(A) Schematic of the transgenic constructs used to induce loss of function of Rbpj specifically in Müller glia. (B) Schematic of the GFAP AAV constructs and experimental workflow. (C) Representative images of retinas immunolabeled for GFP, mCherry, Otx2, and HuC/D. White arrowheads indicate GFP-positive Müller glia-derived neurons expressing neuronal markers Otx2 or HuC/D. (D) Quantification of mean percentage ± SD of GFP-positive Müller glia-derived neurons expressing either Otx2 or HuC/D. Significance was determined via two-way ANOVA with Tukey’s multiple comparison test: ****p < 0.0001. TAM, tamoxifen; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar = 50 μm.
At 1 and 2 weeks post retinal injury, we observed selective expression of the neurogenic bHLH factor Ascl1 in both control mCherry and Pou5f1-mCherry-infected GFP-positive Rbpj-deficient Müller glia (Figure 2—figure supplement 1g, h). Time course analysis showed that Müller glia-derived cells expressing the bipolar cell marker Otx2 appeared as early as 1 week following NMDA injury (Figure 2—figure supplement 1i–k). However, we did not observe a significant change in the proportion of GFP-positive Rbpj-deficient Müller glia expressing the amacrine cell marker HuC/D. By 5 weeks after NMDA injury, we observed 4.5% of GFP-positive cells in retinas of mCherry-infected controls expressed Otx2, while 0.7% expressed HuC/D in Pou5f1-mCherry-infected retinas (Figure 2C, D). These levels of neurogenesis are consistent with previous findings in uninfected Rbpj-deficient Müller glia (Le et al., 2024). In contrast, we observed 24.3% of GFP-positive cells expressing Otx2 in Pou5f1-mCherry-infected retinas, representing a significant (p < 0.0001) increase relative to mCherry control. Unlike in control (GlastCreER;Rosa26LSL-Sun1-GFP) retinas, Pou5f1-expressing, Rbpj-deficient Müller glia showed no EdU incorporation at either 1, 2, or 5 weeks following infection (Figure 2—figure supplement 1l, m, Figure 2—figure supplement 2c). EdU labeling instead colocalized with the microglia marker Iba1. The absence of Müller glia proliferation indicates that the observed neurogenesis is due largely to direct transdifferentiation of Müller glia into retinal neuron-like cells, rather than through dedifferentiation to a proliferative neuronal progenitor-like state.
We have previously shown that Müller glia-specific Notch1/2 double loss of function mutants phenocopy neurogenesis seen in Rbpj mutants (Le et al., 2024). We then examined the effect of Pou5f1 overexpression on GlastCreER;Notch1lox/lox;Notch2lox/lox;Rosa26LSL-Sun1-GFP mice using the same protocol previously described for control and Rbpj-deficient Müller glia (Figure 2—figure supplement 3a, b). We observed 15.8% of GFP+ Müller glia colocalized with Otx2 in Pou5f1-mCherry-infected retinas compared to 6.6% GFP/Otx2+ cells in samples infected with control mCherry vector (Figure 2—figure supplement 3c, d). As expected, infection with Pou5f1-mCherry vector induced both Pou5f1 (Figure 2—figure supplement 3e) and Ascl1 (Figure 2—figure supplement 3f) expression in Notch1/2-deficient Müller glia.
Finally, we tested whether Pou5f1 overexpression could enhance Müller glia-derived neurogenesis induced by loss of function of the transcription factors Nfia, Nfib, and Nfix (Hoang et al., 2020). To test this, we injected GlastCreER;Nfialox/lox;Nfiblox/lox;Nfixlox/lox;Rosa26LSL-Sun1-GFP mice with both mCherry and Pou5f1-mCherry vectors as previously described, then analyzed the retinas 5 weeks after NMDA injury (Figure 2—figure supplement 4a, b). We observed limited coexpression of mCherry and GFP following infection with either construct (Figure 2—figure supplement 4c), indicating that NFI factors may be required for appropriate glial-specific expression of the GFAP minipromoter construct, as suggested by previous findings in astrocytes (Cebolla and Vallejo, 2006).
Pou5f1 overexpression promotes expression of reactive glial markers and neurogenic bHLH factors in Rbpj-deficient Müller glia
To gain insight into the mechanism by which Pou5f1 stimulates neurogenesis in Rbpj-deficient Müller glia, we conducted single-cell (sc)RNA-seq analysis of GFP-positive cells FACS-isolated from retinas of GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP mice infected with either GFAP-mCherry control or GFAP-Pou5f1-mCherry virus. Mice were tamoxifen- and NMDA-treated as described in Figure 2, and GFP-positive cells were isolated at 8 weeks following NMDA treatment (Figure 3A). We profiled 12,143 GFP+ cells from control-infected and 14,612 GFP+ cells from Pou5f1-mCherry-infected retinas (Figure 3B).
Figure 3 with 1 supplement see all
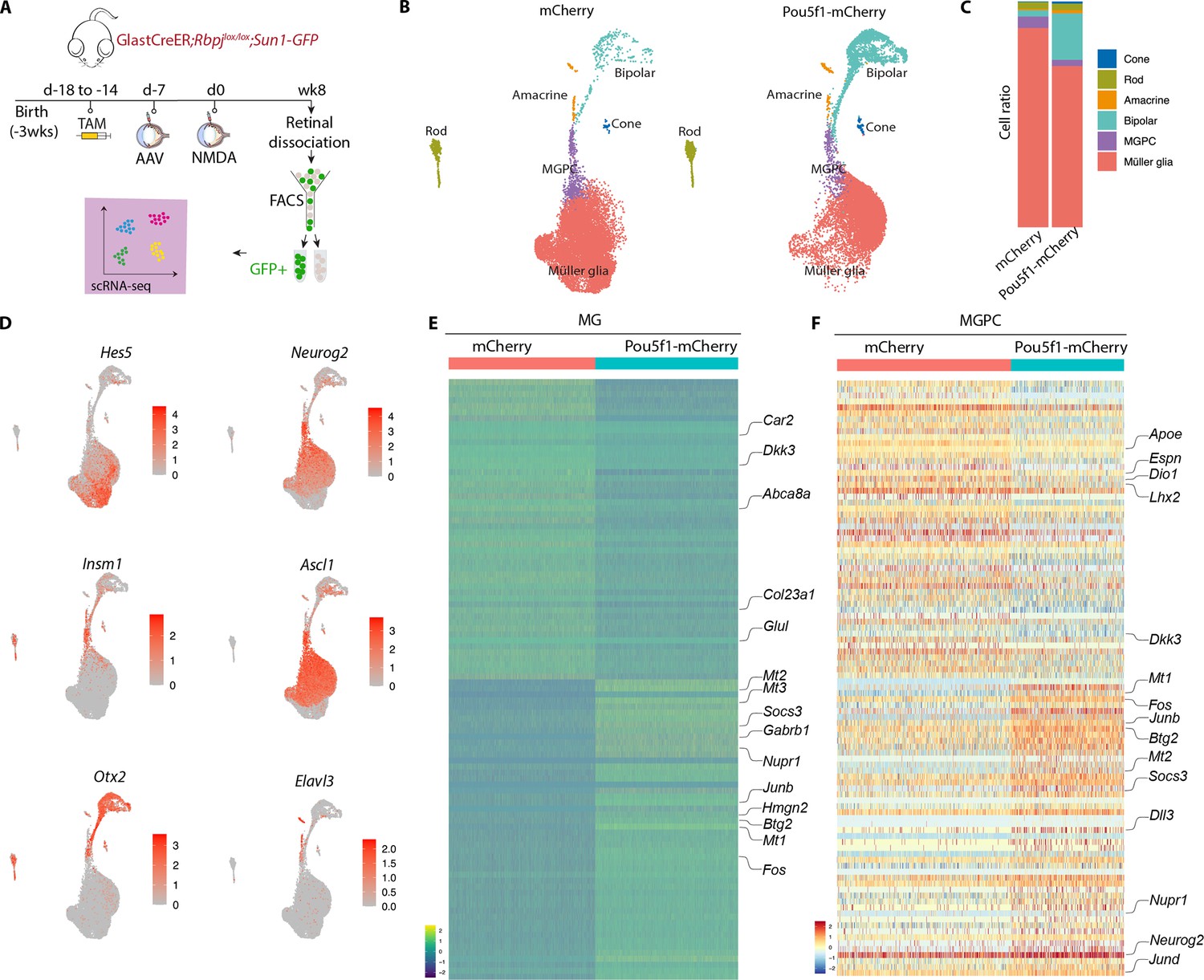
Single-cell RNA-sequencing (scRNA-seq) analysis of Müller glia and Müller glia-derived neurons from Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia following Pou5f1 overexpression.
(A) Schematic of the scRNA-seq experimental pipeline. (B) UMAP plot showing the clustering of GFP-positive cells from Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia retinas infected with GFAP-mCherry and GFAP-Pou5f1-mCherry AAV constructs. (C) Stacked bar plots showing the proportion of cells in each cluster across two sample groups. (D) Feature plots highlighting the cluster of Müller glia (Hes5), neurogenic Müller glia-derived progenitor cells (MGPCs) (Neurog2, Insm1, Ascl1), bipolar cells (Otx2), and amacrine cells (Elavl3). (E) Heatmap showing the expression of top differentially expressed genes (DEGs) from the Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia cell cluster from retinas infected with GFAP-mCherry and GFAP-Pou5f1-mCherry AAV constructs. (F) Heatmap showing the expression of top DEGs for MGPC cell cluster from retinas infected with GFAP-mCherry and GFAP-Pou5f1-mCherry AAV constructs.
In both samples, we observed a clear differentiation trajectory connecting Müller glia, neurogenic Müller glia-derived progenitor cells (MGPCs), and differentiating amacrine and bipolar cells (Figure 3B). Pou5f1-overexpressing samples consistently show substantially higher fractions of bipolar cells (20.5% vs. 2.7 %), and a smaller fraction of Müller glia (71.4% vs. 88.2 %), consistent with immunohistochemical data (Figure 3C). Small numbers of contaminating rod and cone photoreceptors were also observed, as previously reported (Hoang et al., 2020), but no immature photoreceptor-like cells were observed, indicating that these represent contaminating mature cells that were not excluded by FACS analysis.
Selective molecular markers could readily distinguish Müller glia (Glul, Apoe, Vim, Clu, Rlbp1) from neurogenic MGPCs (Neurog2, Ascl1, Sox4, Hes6, Dll1), and clearly identify differentiating bipolar (Otx2, Pcp2, Pcp4, Car10, Gabrb3) and amacrine-like cells (Nrxn1, Tfap2b, Nrg1, Snap25) (Figure 3D–F, Figure 3—figure supplement 1a–c, Supplementary file 1). In both MGs and MGPCs, we observed substantial differences in gene expression between samples infected with control and Pou5f1-mCherry samples. Pou5f1-infected Müller glia selectively downregulated genes specific to resting Müller glia (Car2, Dkk3, Glul, Col23a1, Abca8a), and upregulated genes enriched in reactive glia (Gfap, Socs3, Nupr1, Mt1/2/3, Fos, Jun, Klf6) (Figure 3E, F, Figure 3—figure supplement 1, Supplementary file 1). In MGPCs, Pou5f1 overexpression also downregulated both transcription factors (Lhx2, Hopx) and other genes (Apoe, Dio1, Il33, Espn) that are enriched in resting Müller glia, and upregulated many other genes that are selectively expressed in activated glia (Mt1/2/3, Fos, Jun, Nupr1, Socs3). In addition, Pou5f1-infected MGPCs expressed consistently higher levels of neurogenic bHLH factors (Ascl1, Neurog2, Hes6), bipolar cell specification (Otx2), or genes broadly associated with neurogenesis (Insm1, Dll3, Mybl1). Pou5f1-infected MGPCs also expressed genes (Gadd45a, Btg2) enriched in late-stage neurogenic progenitors (Clark et al., 2019; Figure 3E, F, Supplementary file 1). Together, these findings imply that Pou5f1 enhances bipolar cell formation in Rbpj-deficient Müller glia potentially by repressing expression of genes such as Lhx2 that promote quiescence (de Melo et al., 2018; de Melo et al., 2016), increasing expression of genes that broadly promote neurogenesis, and selectively increasing expression of the bipolar cell-promoting factor Otx2 (Chan et al., 2020; Wang et al., 2014).
Single-cell multiomic analysis identifies targets of Pou5f1 in control and Rbpj-deficient Müller glia
To further investigate the mechanism by which Pou5f1 promotes neurogenesis, we conducted simultaneous single-nucleus (sn)RNA-seq and ATAC-seq on GFP-positive cells from both control and Rbpj-deficient Müller glia infected with either control or Pou5f1-mCherry vectors at 8 weeks following NMDA injection (Figure 4A). UMAP analysis of integrated snRNA/ATAC-seq profiles was then used to identify cell clusters. Müller glia from control and Rbpj-deficient animals form two separate Müller glia clusters (Figure 4B, C). Control Pou5f1-expressing Müller glia gave rise to both small numbers of neurogenic MGPCs (Figure 4D), in line with the small numbers of Ascl1-positive cells observed (Figure 1—figure supplement 2g, h, Figure 3—figure supplement 1b), as well as mature bipolar cells, consistent with histological findings (Figure 1C, D). Pou5f1 overexpression substantially enhances bipolar cell generation from Rbpj-deficient MGPC (Figure 4D, Figure 4—figure supplement 1). Small numbers of rod and cone photoreceptors were also detected in this analysis, but their proportions did not substantially change between treatment conditions and clearly defined immature transitional states were identified, and therefore likely represent contaminating native cells.
Figure 4 with 1 supplement see all
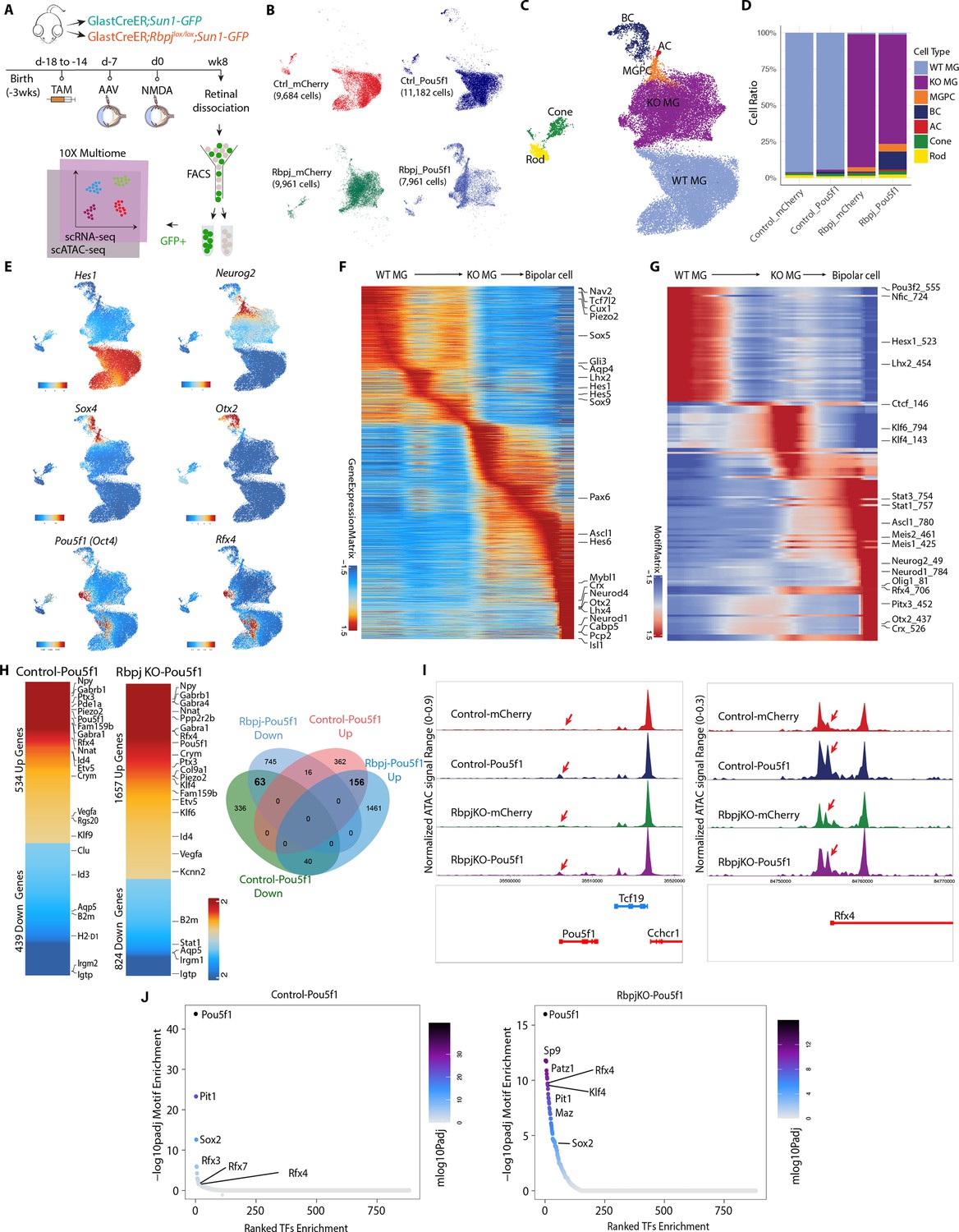
Integrated snRNA/scATAC-seq analysis of control GlastCreER;Rosa26LSL-Sun1-GFP and Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia following Pou5f1 overexpression.
(A) Schematic of the multiomic scRNA/ATAC-seq experimental pipeline. (B) UMAP plot of multiomic datasets showing the clustering of GFP+ cells from control GlastCreER;Rosa26LSL-Sun1-GFP and Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia from GFAP-mCherry and GFAP-Pou5f1-mCherry AAV infected retinas. (C) UMAP plot showing the identity of cell clusters determined by marker gene expression. (D) Stacked bar plots represent the proportion of cells in each cluster across different sample groups. (E) Feature plots highlighting the cluster of Müller glia (Hes1), neurogenic Müller glia-derived progenitor cells (MGPCs) (Neurog2, Sox4), bipolar cells (Otx2), Pou5f1- and Rfx4-expressing cells. (F) Heatmap showing expression of differentially expressed genes (DEGs) along the neurogenesis trajectory. (G) Heatmap showing differential motif activity along the neurogenesis trajectory. (H) Heatmaps of DEGs differentially expressed between Pou5f1 and mCherry control samples in the control GlastCreER;Rosa26LSL-Sun1-GFP and Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia clusters. Venn diagram showing unique and common DEGs between Pou5f1 and mCherry control in the control GlastCreER;Rosa26LSL-Sun1-GFP and Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia. (I) Increased chromatin accessibility regions associated with the Pou5f1 and Rfx4 loci observed following Pou5f1 overexpression in both control GlastCreER;Rosa26LSL-Sun1-GFP and Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia. (J) Top-ranked enriched motifs in chromatin regions showing increased accessibility following Pou5f1 overexpression in both control GlastCreER;Rosa26LSL-Sun1-GFP and Rbpj-deficient GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP Müller glia cell clusters.
Pou5f1 overexpression leads to significant changes in gene expression in both control and Rbpj-deficient Müller glia. The transition from resting to neurogenic MGPCs to bipolar cells is associated with downregulation of genes that are specifically expressed in resting Müller glia, including Aqp4, Hes1, Tcf7l2, and Lhx2 (Figure 4E, F, Figure 4—figure supplement 1a–c, Supplementary file 2). Similarly, motif analysis showed that chromatin accessibility is reduced for target motifs for transcription factors that maintain glial quiescence such as Nfia/b/x and Lhx2 (Figure 4G, Supplementary file 2). MGPCs likewise show increased accessibility at Stat1/3 motifs, as well as target motifs for neurogenic transcription factors such as Ascl1, Neurog2, and Neurod1 (Figure 4G), consistent with previous studies of MGPCs generated from Nfia/b/x and Rbpj-deficient Müller glia (Hoang et al., 2020; Le et al., 2024).
Pou5f1 overexpression in both control and Rbpj-deficient Müller glia clusters, respectively, induces upregulation of 534 and 1657 genes, while, respectively, downregulating 439 and 824 genes (Figure 4H). A total of 156 genes are upregulated in both control and Rbpj-deficient Müller glia, while 63 genes are downregulated. In both control and Rbpj-deficient Müller glia, Pou5f1 upregulates multiple classes of ion channels, including mechanosensitive (Piezo2), TRP (Trpm1, Trpm3), and chloride channels (Clca3a1, Clca3a2), as well as GABA receptors (Gabra4, Gabrb1), cyclic nucleotide phosphodiesterases (Pde8b, Pde11a), and the neuropeptide Npy. Pou5f1 overexpression also selectively downregulates multiple inflammation-associated genes (B2m, H2-D1), small GTPases (Gbp2, Igtp, Irgm2, Rab13, Rgs20, Tgtp2), along with the glycolytic enzyme Gapdh and many ribosomal subunits (Figure 4H, Supplementary file 3). This comprises a functionally diverse range of genes that are often not prominently expressed in retinal progenitors, and whose role in controlling neurogenic competence remains to be investigated.
Pou5f1 also differentially regulates multiple transcription factors in both control and Rbpj-deficient Müller glia. As expected, Pou5f1 mRNA is strongly upregulated (Figure 4E, H), with increased chromatin accessibility also being observed at the endogenous Pou5f1 promoter, indicating that Pou5f1 regulates its self-expression (Figure 4I, Supplementary file 4). Similarly, Pou5f1 overexpression strongly upregulates the transcription factor Rfx4 and accessibility at its endogenous promoter (Figure 4E, H, I, Supplementary file 4). Previous studies showed that overexpression of Rfx4 is sufficient to induce neurogenesis when overexpressed in human ES cells (Choi et al., 2024; Joung et al., 2024). However, Rfx4 expression is not detectably expressed in developing mouse retinas (Clark et al., 2019), indicating that Pou5f1-induced neurogenesis may involve mechanisms beyond simply inducing dedifferentiation to a retinal progenitor-like state.
Several other transcription factors are only differentially regulated following Pou5f1 overexpression in Rbpj-deficient Müller glia. One notable upregulated transcription factor is Klf4, which cooperates with Pou5f1 to induce pluripotency (Takahashi and Yamanaka, 2006). In contrast, Stat1, which inhibits neurogenesis induced by Ascl1 overexpression in Müller glia (Jorstad et al., 2020), is selectively downregulated in Rbpj-deficient Müller glia (Supplementary file 3).
Analysis of changes in chromatin accessibility induced by Pou5f1 reveals that the consensus motif for Pou5f1 and its paralogue Pit1 (Pou1f1) show the strongest increase in accessibility, followed by Rfx family motifs (Figure 4J, Supplementary file 5). Both control and Rbpj-deficient Müller glia show increased accessibility associated with Sox2, which directly interacts with Pou5f1 and cooperates to induce pluripotency (Rizzino, 2013). While Pou5f1 does not significantly upregulate the pluripotency factor Klf4 in control Müller glia, it robustly induces Klf4 expression Rbpj-deficient Müller glia, and likewise selectively induces accessibility at Klf4 consensus motif sites (Figure 4J). In addition, Rbpj-deficient Müller glia also show smaller but still significant increases in accessibility at target sites for the proneural factor Neurog2 and the neurogenic factor Insm1 (Lyu et al., 2021), identifying additional mechanisms by which Pou5f1 might synergistically enhance neurogenesis (Figure 4J, Supplementary file 5).
Discussion
In this study, we demonstrate that AAV-mediated overexpression of Pou5f1 is sufficient to induce neurogenic competence in Müller glia in adult mice, and to synergistically enhance neurogenesis induced by genetic disruption of Notch signaling. While Pou5f1 overexpression induces very low levels of proliferation, Pou5f1-induced neurogenesis appears to mostly result from direct glia-to-bipolar cell transdifferentiation by simultaneously upregulating expression of neurogenic bHLH factors and the bipolar cell-promoting factor Otx2. In addition, Pou5f1 upregulates expression of the neurogenic transcription factor Rfx4, which is not expressed during retinal development. Pou5f1 exerts reprogramming effects by broadly increasing chromatin accessibility at predicted regulatory sites associated with these genes.
While we did not detect endogenous Pou5f1 expression in either injured or neurogenic mouse Müller glia, the fact that these cells express robust levels of Sox2, Klf4, and Myc implies that they may already be prime to reprogramming induced by Pou5f1. Our findings differ from two previous reports that initially motivated us to carry out this study. A previous study in zebrafish showed weak levels of the Pou5f1 homolog pou5f3 in resting glia, with these levels increasing dramatically following injury (Sharma et al., 2019). It remains unclear why we were unable to detect more than trace levels of pou5f3 expression in zebrafish Müller glia in any of the many bulk and single-cell RNA-seq samples we analyzed (Hoang et al., 2020). We also did not detect the previously reported low levels of injury-induced Pou5f1 expression in mouse Müller glia (Reyes-Aguirre and Lamas, 2016). Similar discrepancies in observed cellular patterns of Pou5f1 expression have been observed in many other tissues (Lengner et al., 2008), and further work will be needed to clarify these discrepancies.
This study represents a clear instance in which viral-mediated expression coupled with genetic cell lineage analysis was effectively used to induce the conversion of adult mammalian Müller glia into retinal neurons in situ. A similar approach has been also successfully used to generate bipolar neurons from both immature (Pollak et al., 2013) and very recently also in mature, Müller glia by overexpression of the neurogenic bHLH factors Ascl1 and Atoh1 (Pavlou et al., 2024). Other studies reported efficient conversion of Müller glia to photoreceptors using a complex cocktail of overexpressed factors, including Ctnnb1, Otx2, Crx, and Nrl (Yao et al., 2018), and efficient conversion of Müller glia to retinal ganglion cells following overexpression of Atoh7 and Pou4f2 (Xiao et al., 2021). However, these studies both used GFAP minipromoters to both drive expression of these constructs and to infer cell lineage. Multiple studies have shown that GFAP minipromoter specificity is heavily influenced by insert sequences present in AAVs, which can result in both silencing of expression in glia and ectopic activation of expression in neurons (Le et al., 2022; Wang et al., 2021b). As a result, it cannot be concluded that these represent bona fide glia-to-neuron conversion in vivo. Similar claims have been made about knockdown of the RNA binding protein Ptbp1, which was variously claimed to directly convert Müller glia to either retinal ganglion cells or photoreceptors, but has been debunked using genetic cell lineage analysis (Hoang et al., 2022; Xie et al., 2022). In this study, the combination of prospective cell lineage analysis using the GlastCreER;Rosa26LSL-Sun1-GFP transgene before infection with GFAP-Pou5f1-mCherry vector, in combination with single-cell RNA-sequencing (scRNA-seq) analysis of infected cells, rules out any potentially confounding factors resulting from the GFAP minipromoter.
Our finding that Pou5f1 can synergistically enhance neurogenesis induced by genetic disruption of Notch signaling raises the question of whether this might be more broadly applicable to other models of induced neurogenesis in mammalian Müller glia. The mechanism by which Pou5f1 induces glial-derived neurogenesis appears complex, and to differ fundamentally from that seen following either overexpression of Ascl1 or loss of function of Nfia/b/x and/or Rbpj (Hoang et al., 2020; Jorstad et al., 2017; Le et al., 2024). In these cases, Müller glia broadly upregulate proneural genes and/or downregulate Notch signaling. Pou5f1 instead induces expression of the neurogenic transcription factor Rfx4, which is not expressed in developing retina. While this may represent a parallel pathway to neurogenic competence that in part accounts for synergistic induction of neurogenesis seen in Rbpj-deficient Müller glia, confirmation of the neurogenic activity of Rfx4 in Müller glia requires direct experimental confirmation.
Other mechanisms enhancing Pou5f1-dependent neurogenesis in the Rbpj-deficient Müller glia may include enhanced induction of Klf4 expression and increased accessibility at target sites for neurogenic transcription factors such as Ascl1. While Ascl1 overexpression induces Müller glia-derived neurogenesis in adult mice, this occurs only following treatment with HDAC inhibitors (Jorstad et al., 2017), even though Ascl1 is itself a pioneer factor (Păun et al., 2023). Pou5f1 overexpression broadly increases the accessibility of putative cis-regulatory sequences associated with both Neurog2 and Insm1, identifying another potential mechanism of synergy. Transient overexpression of Pou5f1 could potentially further enhance the neurogenic function of Ascl1 and enhance glial-derived neurogenesis more broadly. This may also be the case for pathways reported to induce glial reprogramming in other CNS regions, such as Sox2 overexpression (Niu et al., 2015). Since Pou5f1 overexpression only weakly and transiently induces proliferation in adult Müller glia, this reduces the potential oncogenic risk, and may ultimately prove to be relevant for the design of cell-based regenerative therapies for treating retinal dystrophies.
Methods
Mice
Mice were raised and housed in a climate-controlled pathogen-free facility on a 14/10 hr light/dark cycle. Mice used in this study were GlastCreER;Rosa26LSL-Sun1-GFP, which were generated by crossing the GlastCreER (JAX#012586) and Rosa26LSL-Sun1-GFP (JAX#021039) lines developed by Dr. Jeremy Nathans at Johns Hopkins (de Melo et al., 2012; Mo et al., 2015), and were obtained from his group. GlastCreER;Rbpjlox/lox;Rosa26LSL-Sun1-GFP mice were generated by crossing GlastCreER;Sun1GFP with conditional Rbpjlox/lox mice (JAX #034200).
GlastCreER;Notch1lox/lox;Notch2lox/lox;Rosa26LSL-Sun1-GFP mice were obtained by crossing GlastCreER;Notch1lox/lox;Rosa26LSL-Sun1-GFP and GlastCreER;Notch2lox/lox;Rosa26LSL-Sun1-GFP mice. GlastCreER;Nfia/b/xlox/lox;Rosa26LSL-Sun1-GFP mice were generated by crossing Nfialox/lox (see below); Nfiblox/lox (Hsu et al., 2011), and Nfixlox/lox (Campbell et al., 2008) mice to GlastCreER;Rosa26LSL-Sun1-GFP mice. Nfialox/lox mice were generated in the Roswell Park Gene Targeting and Transgenic Shared Resource using heterozygous targeted ES cells from EUCOMM project 38437 (KOMP). All mice used in this study contain both the GlastCreER and Rosa26LSL-Sun1-GFP transgenes, allowing visualization of both Müller glia and Müller glia-derived neurons. Maintenance and experimental procedures performed on mice were in accordance under protocol MO22M22 approved by the Institutional Animal Care and Use Committee (IACUC) at the Johns Hopkins School of Medicine.
Intraperitoneal tamoxifen injection
To induce Cre recombination, animals at ~3 weeks of age were intraperitoneally injected with tamoxifen (Sigma-Aldrich, #H6278-50mg) in corn oil (Sigma-Aldrich, #C8267-500ML) at 1.5 mg/dose for 5 consecutive days.
NMDA treatment
Adult mice were anesthetized with isoflurane inhalation. Two microliters of 100 mM NMDA in PBS were intravitreally injected using a microsyringe with a 33 G blunt-ended needle.
EdU treatment
At 1 week post tamoxifen induction, mice were administered EdU (Abcam, #ab146186) via i.p. injections (150 μl of 5 mg/ml in PBS) and drinking water (0.3 mg/ml) for 7 consecutive days.
Cloning, production, and intravitreal injection of adeno-associated virus
The Addgene #50473 construct, which contains a GFAP promoter, was used in this study. The EGFP sequence was replaced by the mCherry sequence. The P2A ribosomal self-cleaving peptide is used to simultaneously express Pou5f1 5′ to the mCherry reporter as a single polypeptide, which is then cleaved to generate Pou5f1 and mCherry. The coding sequence of Pou5f1 was synthesized by GeneWiz. AAV constructs were packaged into AAV2.7m8 by Boston Children’s Hospital Viral Core. Following tamoxifen induction, 1-month-old GlastCreER;Rosa26LSL-Sun1-GFP mice were intravitreally injected with GFAP AAV constructs using a microsyringe with a 33 G blunt-ended needle. The titers and injection volume for each construct are listed in Supplementary file 6.
Immunohistochemistry and imaging
Collection and immunohistochemical analysis of retinas were performed as described previously (Hoang et al., 2020). Mouse eye globes were fixed in 4% paraformaldehyde (ElectronMicroscopySciences, #15710) for 4 hr at 4°C. Retinas were dissected in 1× PBS and incubated in 30% sucrose overnight at 4°C. Retinas were then embedded in OCT (VWR, #95057-838), cryosectioned at 16 μm thickness, and stored at −20°C. Sections were dried for 30 min in a 37°C incubator and washed 3 × 5 min with 0.1% Triton X-100 in PBS (PBST). EdU labeling was performed by using Click-iT EdU kit (Thermo Fisher, #C10340, #C10636) following the manufacturer’s instructions. Sections were then incubated in blocking buffer (10% Horse Serum (Thermo Fisher, #26050070), 0.4% Triton X-100 in 1× PBS) for 2 hr at room temperature (RT) and then incubated with primary antibodies in the blocking buffer overnight at 4°C. Primary antibodies used are listed in Appendix 1—key resources table.
Sections were washed 4 × 5 min with PBST to remove excess primary antibodies and were incubated in secondary antibodies in blocking buffer for 2 hr at RT. Sections were then counterstained with DAPI in PBST, washed 4 × 5 min in PBST and mounted with ProLong Gold Antifade Mountant (Invitrogen, #P36935) under coverslips (VWR, #48404-453), air-dried, and stored at 4°C. Fluorescent images were captured using a Zeiss LSM 700 confocal microscope. Z-stack images were collected for all sections. Colocalization of markers scored only if observed in individual Z-stack images. Secondary antibodies used are listed in Appendix 1—key resources table.
Cell quantification and statistical analysis
Otx2/GFP- and HuC/D/GFP-positive cells were counted and divided by the total number of GFP-positive cells from a single random whole section per retina. Each data point in the bar graphs was calculated from an individual retina. All cell quantification data were graphed and analyzed using GraphPad Prism 10. Two-way ANOVA was used for analysis between three or more samples of multiple groups. All results are presented as mean ± SD.
Retinal cell dissociation
Retinas were dissected in fresh ice-cold PBS and retinal cells were dissociated using an optimized protocol as previously described (Fadl et al., 2020). Each sample contains a minimum of four retinas from four animals of both sexes. Dissociated cells were resuspended in ice-cold HBAG Buffer containing Hibernate A (BrainBits, #HALF500), B-27 supplement (Thermo Fisher, #17504044), and Glutamax (Thermo Fisher, #35050061).
scRNA-seq library preparation
ScRNA-seq libraries were prepared using dissociated retinal cells using the 10X Genomics Chromium Single Cell 3′ Reagents Kit v3.1 (10X Genomics, Pleasanton, CA). Libraries were constructed following the manufacturer’s instructions and were sequenced using Illumina NextSeq. Sequencing data were processed through the Cell Ranger 7.0.1 pipeline (10X Genomics) using default parameters.
Single-cell Multiome ATAC + GEX sequencing library preparation
ScATAC-seq and scRNA-seq libraries were prepared using FACS-isolated GFP-positive cells using the 10X Genomic Chromium Next GEM Single Cell Multiome ATAC + Gene Expression kit following the manufacturer’s instructions. Briefly, cells were spun down at 500 × g for 5 min, resuspended in 100 μl of ice-cold 0.1× Lysis Buffer, lysed by pipette-mixing four times, and incubated on ice for 4 min total. Cells were washed with 0.5 ml of ice-cold Wash Buffer and spun down at 500 × g for 5 min at 4°C. Nuclei pellets were resuspended in 10–15 μl Nuclei Buffer and counted using Trypan blue. Resuspended cell nuclei (10–15k) were utilized for transposition and loaded into the 10 Genomics Chromium Single Cell system. ATAC libraries were amplified with 10 PCR cycles and were sequenced on Illumina NovaSeq with ~200 million reads per library. RNA libraries were amplified from cDNA with 14 PCR cycles and were sequenced on Illumina NovaSeq 6000.
scRNA-seq data analysis
ScRNA-seq data were pre-processed using Cellranger v7.1.0 using a custom mouse genome (version mm10) with mCherry and Sun1GFP sequences included. The cell-by-genes count matrices were further analyzed using Seurat V5.1.0 (Hao et al., 2024). Cells with RNA counts less than 1000 or greater than 25,000, and number of genes less than 500 or greater than 6000 were filtered out as low-quality cells. Cells with a mitochondrial fraction of greater than 10% were removed. For data visualization, UMAP was generated using the first 20 dimensions. Müller glia and MGPC cell clusters were subsetted for further analysis. Differential gene expression of mCherry control and Pou5f1-mCherry groups was performed using the ‘FindAllMarkers’ function.
Single-cell multiomic analysis
Raw scRNA-seq and scATAC-seq data were processed with the Cell Ranger software version 2.0.2 specifically using cellranger-arc pipeline for formatting reads, demultiplexing samples, genomic alignment, and generating the cell-by-gene count matrix and the fragments files. Both the cell-by-gene count matrix and the fragments files were the final output from the cellranger-arc pipeline, and were used for all downstream analysis.
ScRNA-seq and scATAC-seq were analyzed using Archr version 1.0.2. EnsDb.Mmusculus.v79 and BSgenome.Mmusculus.UCSC.mm10 were used for annotations. The arrow file for each sample was created using the cell-by-gene count matrix and the fragments files output from the cellranger-arc pipeline. To filter poor quality cells, Gex_nGenes greater than 500 and less than 5000 were kept, Gex_nUMI greater than 1000 and less than 15,000 were kept. TSSEnrichment greater than 10 and nFrags greater than 5000 were kept, while other cells were deemed of poor quality and were filtered out. Gene expression values from paired scATAC-seq and scRNA-seq multi modal assay were added to the arrow files using the ‘addGeneExpressionMatrix’ function. The LSI dimensionality reduction for both scATAC and scRNASeq was computed and combined using the ‘addCombinedDims’ function. UMAP embedding is calculated using ‘addUMAP’ function and clusters are identified from the reduced dimensions. Marker features were used to annotate clusters.
To identify peaks, ‘addReproduciblePeakSet’ function was used to get insertions from coverage files, call peaks, and merge peaks and this step internally used macs software. Then the ‘addPeakMatrix’ function was called to independently compute counts for each peak per cell. To link peaks to genes, ‘getPeak2GeneLinks’ function was called. The ‘getMarkerFeatures’ function was used to get the differential gene expressions and the differential peaks expressions using the GeneExpressionMatrix and PeakMatrix calculated, respectively. Peaks with motifs were annotated using the cisbp motif set using ‘annotatePeaks.pl’ from HOMMER version 4.11 to associate the peaks with nearby genes. The full code used for the analysis is available at https://github.com/SherineAwad/OCT4scRNA-ATAC, copy archived at Awad, 2025.
Appendix 1
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | Chicken anti-GFP (chicken polyclonal) | ThermoFisher | A10262, RRID:AB_2534023 | IF (1:400) |
| Antibody | Rabbit anti-RFP (rabbit polyclonal) | Abcam | ab124754, RRID:AB_10971665 | IF (1:400) |
| Antibody | Goat anti-RFP (goat polyclonal) | Rockland | 200-101-379, RRID:AB_2744552 | IF (1:400) |
| Antibody | Goat anti-Otx2 (goat polyclonal) | R&D Systems | AF1979, RRID:AB_2157172 | IF (1:200) |
| Antibody | Mouse anti-HuC/D (mouse monoclonal) | ThermoFisher | A-21271, RRID:AB_221448 | IF (1:200) |
| Antibody | Mouse anti-NeuN (mouse monoclonal) | Sigma-Aldrich | MAB377, RRID:AB_2298772 | IF (1:200) |
| Antibody | Mouse anti-Oct3/4 (mouse monoclonal) | Santa Cruz | sc-5279, RRID:AB_628051 | IF (1:200) |
| Antibody | Rabbit anti-Sox9 (mouse monoclonal) | Sigma-Aldrich | AB5535, RRID:AB_2239761 | IF (1:400) |
| Antibody | Goat anti-Nrl (goat polyclonal) | R&D Systems | AF2945, RRID:AB_2155098 | IF (1:400) |
| Antibody | Rabbit anti-Scgn (rabbit polyclonal) | Biovendor Laboratory Medicine | RD181120100, RRID:AB_2034060 | IF (1:400) |
| Antibody | Rabbit anti-Ascl1 (rabbit monoclonal) | Abcam | ab211327, RRID:AB_2924270 | IF (1:400) |
| Antibody | Rabbit anti-CaBP5 (rabbit monoclonal) | SynapticSystems | 475 002, RRID:AB_2924962 | IF (1:400) |
| Antibody | Goat anti-Sox2 (goat polyclonal) | R&D Systems | AF2018, RRID:AB_355110 | IF (1:400) |
| Antibody | Mouse anti-Isl1 (mouse monoclonal) | DSHB | 40.2D6, RRID:AB_528315 | IF (1:400) |
| Antibody | Donkey anti-Chicken 488 (donkey polyclonal) | Sigma-Aldrich | SAB4600031, RRID:AB_2721061 | IF (1:400) |
| Antibody | Donkey anti-Rabbit 568 (donkey polyclonal) | ThermoFisher | A-10042, RRID:AB_2757564 | IF (1:400) |
| Antibody | Donkey anti-Goat 568 (donkey polyclonal) | ThermoFisher | A11057, RRID:AB_2534104 | IF (1:400) |
| Antibody | Donkey anti-Mouse 568 (donkey polyclonal) | ThermoFisher | A10037, RRID:AB_2534013 | IF (1:400) |
| Antibody | Donkey anti-Rat 568 (donkey polyclonal) | ThermoFisher | A78946, RRID:AB_2910653 | IF (1:400) |
| Antibody | Donkey anti-Goat 633 (donkey polyclonal) | ThermoFisher | A-21082, RRID:AB_2535739 | IF (1:400) |
| Antibody | Donkey anti-Rabbit 647 (donkey polyclonal) | ThermoFisher | A-31573, RRID:AB_2536183 | IF (1:400) |
| Antibody | Donkey anti-Mouse 647 (donkey polyclonal) | ThermoFisher | A-31571, RRID:AB_162542 | IF (1:400) |
| Commercial assay or kit | 10 x scRNaseq 3′ v3.1 | 10 X Genomics | 1000268 | |
| Commercial assay or kit | 10 x Multiome ATAC +GEX | 10 X Genomics | 1000283 | |
| Commercial assay or kit | Click-iT EdU Alexa Fluor 647 | ThermoFisher | C10340 | |
| Commercial assay or kit | Click-iT EdU Pacific Blue | ThermoFisher | C10418 | |
| Software, algorithm | ImageJ/Fiji | https://imagej.net/software/fiji/ | RRID:SCR_002285 | |
| Software, algorithm | Adobe Illustrator | http://www.adobe.com | RRID:SCR_010279 | v.26.5 |
| Software, algorithm | GraphPad Prism | https://www.graphpad.com/ | RRID:SCR_002798 | v.10 |
| Software, algorithm | Cell Ranger | 10 X Genomics | RRID:SCR_017344 | v.2.0.2 |
| Software, algorithm | ArchR | https://github.com/GreenleafLab/ArchR | RRID:SCR_020982 | v.1.0.2 |
| Software, algorithm | Seurat | https://github.com/satijalab/seurat | RRID:SCR_007322 | v.5.1.0 |
Data availability
All scRNA-seq, snRNA-seq, and scATAC-seq data described in this study are available at Gene Expression Omnibus under accession number GSE277390.
-
NCBI Gene Expression OmnibusID GSE277390. Oct4 overexpression and suppression of Notch signaling synergistically induce neurogenic competence in mammalian Muller glia.
References
-
SoftwareOCT4scRNA-ATAC, version swh:1:rev:87d62136cced3355c36c8aadffb3d91b795e5075Software Heritage.
-
The transcription factor Nfix is essential for normal brain developmentBMC Developmental Biology 8:52.https://doi.org/10.1186/1471-213X-8-52
-
RFX4 is an intrinsic factor for neuronal differentiation through induction of proneural genes POU3F2 and NEUROD1Cellular and Molecular Life Sciences 81:99.https://doi.org/10.1007/s00018-024-05129-y
-
Lhx2 is an essential factor for retinal gliogenesis and notch signalingThe Journal of Neuroscience 36:2391–2405.https://doi.org/10.1523/JNEUROSCI.3145-15.2016
-
An optimized protocol for retina single-cell RNA sequencingMolecular Vision 26:705–717.
-
Dictionary learning for integrative, multimodal and scalable single-cell analysisNature Biotechnology 42:293–304.https://doi.org/10.1038/s41587-023-01767-y
-
Ectopic insert-dependent neuronal expression of GFAP promoter-driven AAV constructs in adult mouse retinaFrontiers in Cell and Developmental Biology 10:914386.https://doi.org/10.3389/fcell.2022.914386
-
In vivo reprogramming of astrocytes to neuroblasts in the adult brainNature Cell Biology 15:1164–1175.https://doi.org/10.1038/ncb2843
-
Regulation of pluripotency in male germline stem cells by Dmrt1Genes & Development 27:1949–1958.https://doi.org/10.1101/gad.220194.113
-
Towards a knowledge-based human protein atlasNature Biotechnology 28:1248–1250.https://doi.org/10.1038/nbt1210-1248
-
Retina regeneration in zebrafishCurrent Opinion in Genetics & Development 40:41–47.https://doi.org/10.1016/j.gde.2016.05.009
-
In vivo regeneration of ganglion cells for vision restoration in mammalian retinasFrontiers in Cell and Developmental Biology 9:755544.https://doi.org/10.3389/fcell.2021.755544
Article and author information
Author details
Funding
National Eye Institute (R01EY031685)
- Seth Blackshaw
Research to Prevent Blindness (Stein Innovation Award)
- Seth Blackshaw
Alcon Research Institute (Young Investigator Award)
- Thanh Hoang
The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Acknowledgements
This study was supported by an award from the National Eye Institute (R01EY031685) to SB, a Stein Innovation Award from Research to Prevent Blindness to SB, and a Young Investigator Award from Alcon Research Institute to TH.
Ethics
Maintenance and experimental procedures performed on mice were in accordance under protocol MO22M22 approved by the Institutional Animal Care and Use Committee (IACUC) at the Johns Hopkins School of Medicine.
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.106450. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2025, Le et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,841
- views
-
- 105
- downloads
-
- 12
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 9
- citations for umbrella DOI https://doi.org/10.7554/eLife.106450
-
- 3
- citations for Reviewed Preprint v2 https://doi.org/10.7554/eLife.106450.2
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Viral-mediated Pou5f1 (Oct4) overexpression and inhibition of Notch signaling synergistically induce neurogenic competence in mammalian Müller glia
eLife 14:RP106450.
https://doi.org/10.7554/eLife.106450.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}