Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps
- University of Illinois at Urbana-Champaign, United States
- University of California San Francisco School of Medicine, United States
Figures
Figure 1

Visual summary of advanced MDFF methodology.
A graphic table illustrating MDFF refinement of a model of carbon monoxide dehydrogenase using a high-resolution map. The map represents an open conformation while the initial search model was obtained through crystallography of a closed conformation. This search model was independently fitted, using direct MDFF, to individual members of a set of maps obtained by applying Gaussian blurs of various half-widths (, first column) to the experimental density. These maps are visualized as a 3D surface in the second column, while the resulting MDFF potentials are represented in cross-section in the third column. Notice the increase in number of contiguous density regions as increases. This increase in contiguity is manifested in the lowering of high barriers (red) for small values to low or flat energy profiles (blue) for larger values, as observed in the potential cross-sections. Reduced barrier heights allow the structure to explore the conformational space freely during fitting. The structure after 500 ps of fitting, shown in red, is superimposed on the known target structure, shown in blue, in the fourth column. The time evolution of RMSD with respect to the target during fitting is shown in the fifth column. The RMSD plots show that direct fitting to lower resolution maps requires fewer time steps to reach convergence. In fact, the structure never becomes less deviated than the initial 7-Å RMSD from the target in the direct MDFF of the highest-resolution map (i.e. in the absence of Gaussian blurring). The inset shows refinements of the same structure through cMDFF and ReMDFF employing the same set of maps. A clear improvement over direct MDFF is apparent, with convergence to within 1.7 Å and 1.0 Å of the target achieved within 1000 and 100 ps for cMDFF and ReMDFF respectively.
Figure 2 with 8 supplements
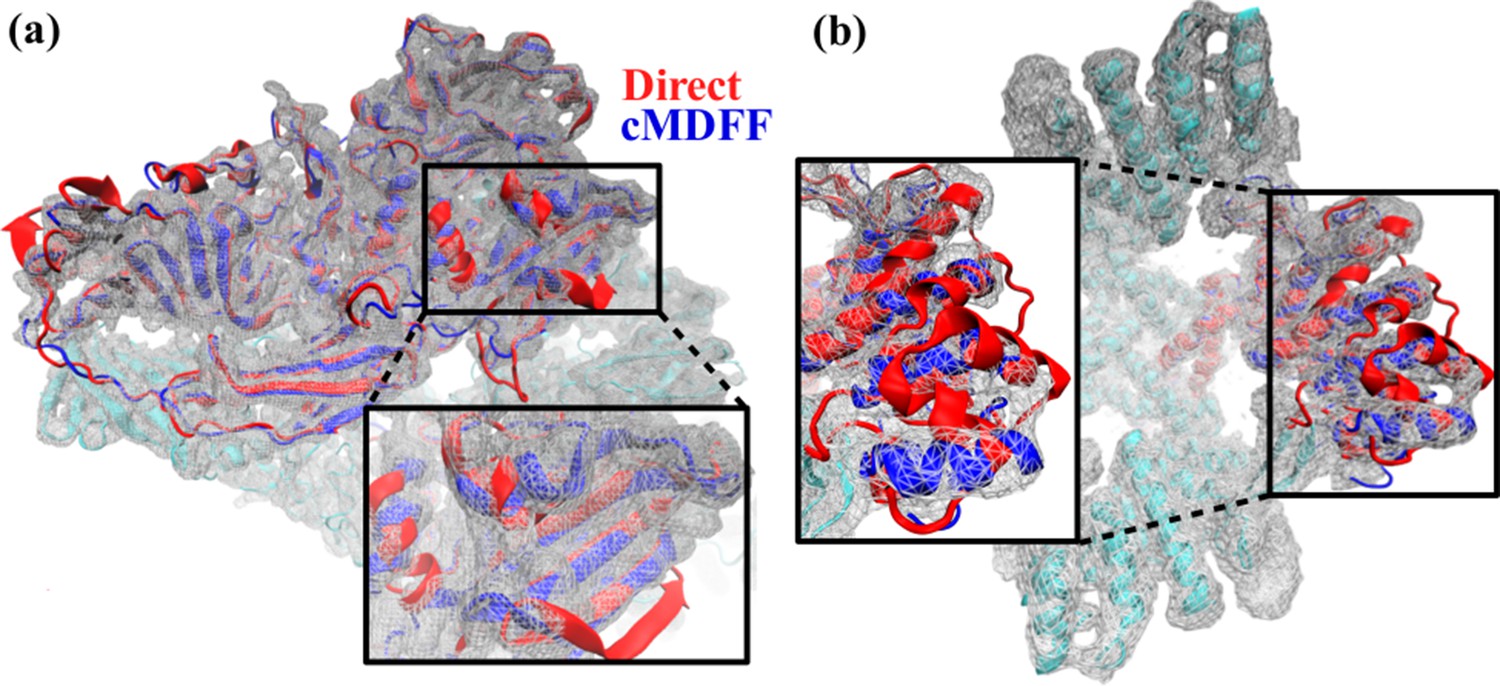
Comparison between cMDFF and direct MDFF fitted models.
Models of (a) β-galactosidase and (b) TRPV1, obtained from cMDFF (blue) and direct MDFF (red) fitting simulations are superimposed. The cMDFF-fitted models fit well into the high-resolution maps (grey) of each molecule, whereas the direct MDFF models have become trapped in local minima that result in portions of the models protruding from the maps. ReMDFF-fitted models are almost identical to those from cMDFF and are therefore not shown.
Figure 2—figure supplement 1
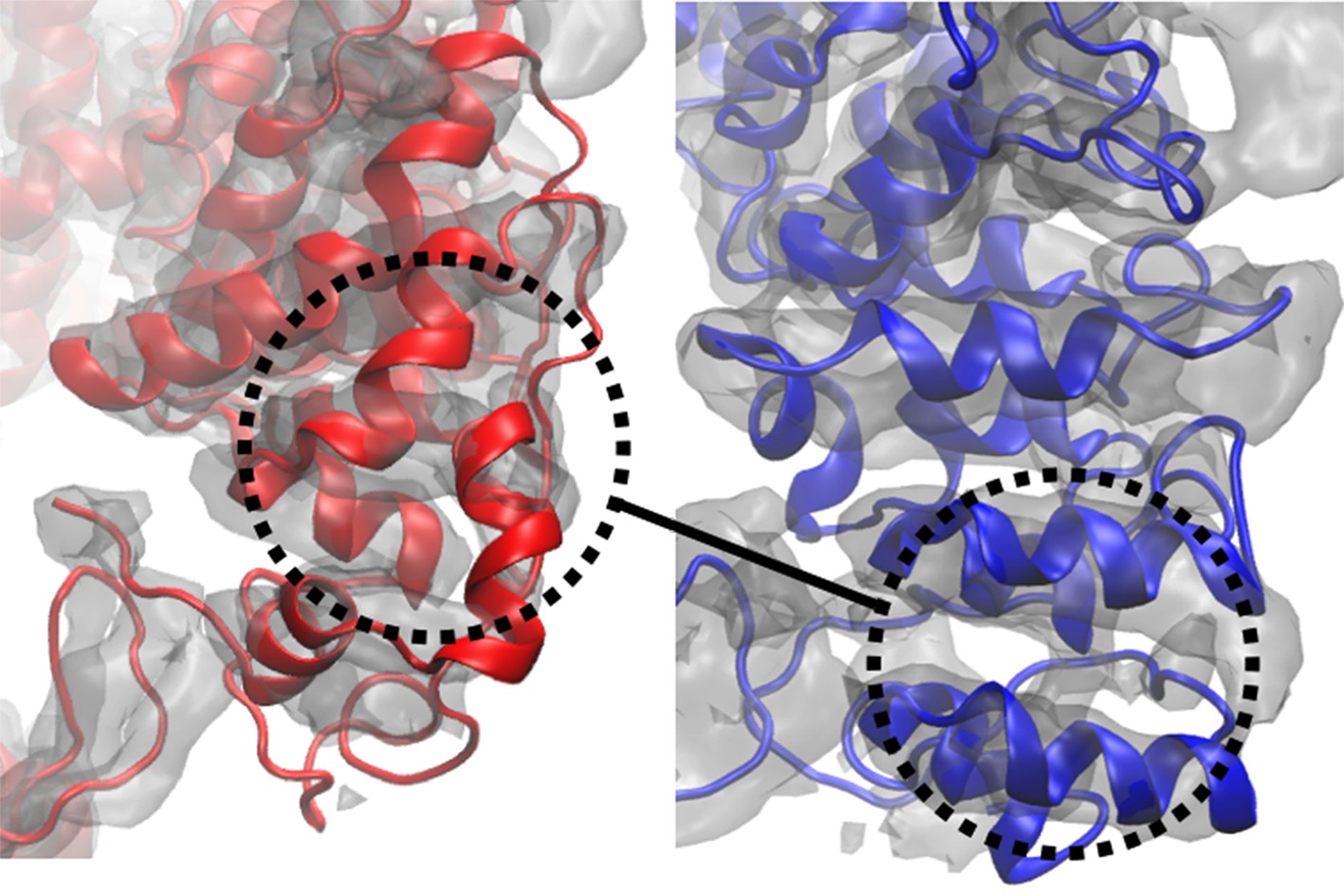
Global cross-correlation as a measure of fit.
The blue and red structures represent the same region of a segment of TRPV1 that have been fitted differently into the density map shown. The global cross-correlations of the structural region shown in each case are 0.728 (red) and 0.723 (blue). However, the blue structure is clearly better fitted than the red structure, as reflected in RMSDs from the published structure of 6.2 Å (red) and 2.3 Å (blue). Although the case described is an extreme one, it shows that global cross-correlation, as a measure of fit, can be misleading, particularly in regards to local correspondence of residues to the map.
Figure 2—figure supplement 2
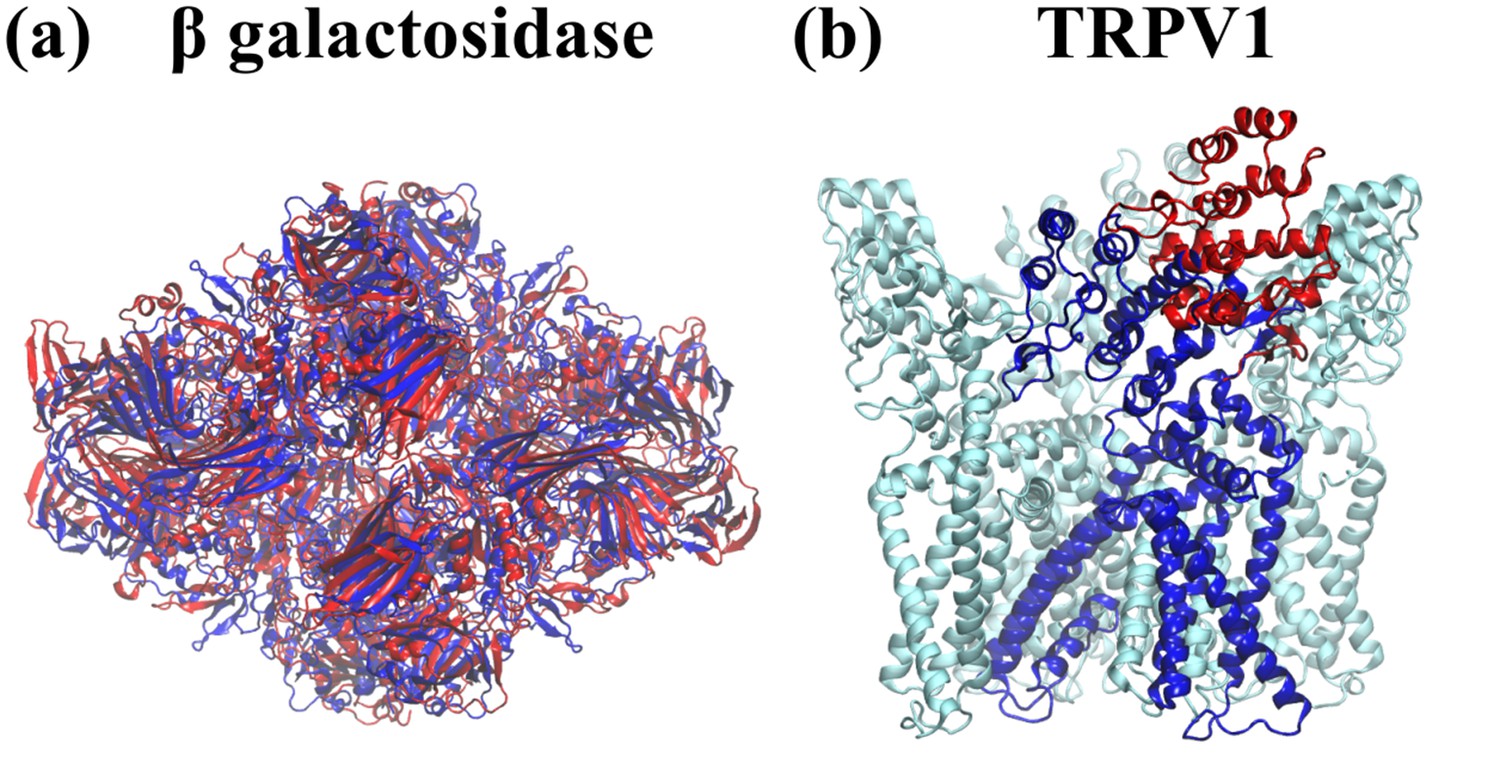
Comparison of initial models to target (published) models.
For the purpose of testing cMDFF on (a) β-galactosidase and (b) TRPV1, the published models (blue) were distorted to provide the initial models (red) for fitting. In the case of TRPV1, the distortion was applied to only one subunit.
Figure 2—figure supplement 3
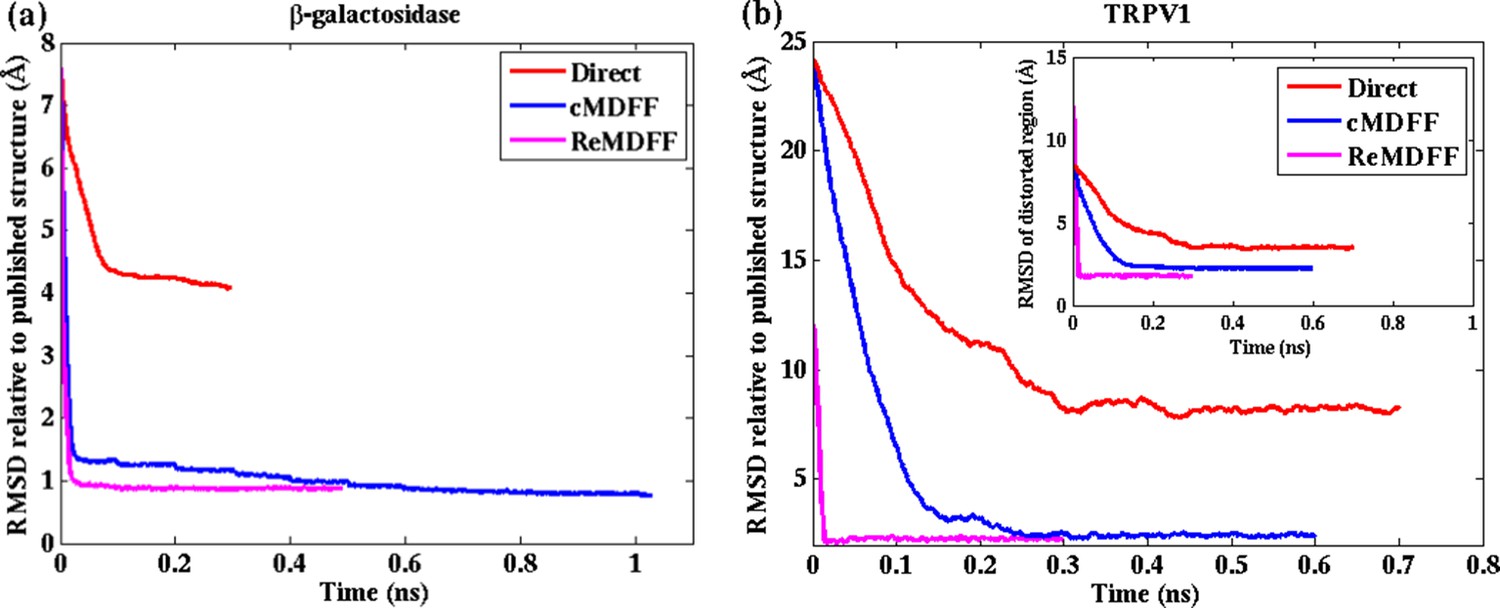
Convergence of cMDFF, ReMDFF, and direct MDFF simulations.
RMSD over simulation time is plotted for the cMDFF, ReMDFF, and direct MDFF simulations of (a) β-galactosidase and (b) TRPV1 monomer. RMSD is calculated with respect to the published models (PDB 3J7H for 3.2-Å resolution and PDB 5A1A for 2.2-Å resolution). For ReMDFF, the plot contains data from a single, best-fit, replica. (inset) Same as (b) but now for the TRPV1 tetramer. For both β-galactosidase, and TRPV1 monomer and tetramer, cMDFF and ReMDFF outperformed direct MDFF in terms of both efficiency and fitting accuracy, as reflected in Tables 1 and Supplementary file 1A.
Figure 2—figure supplement 4
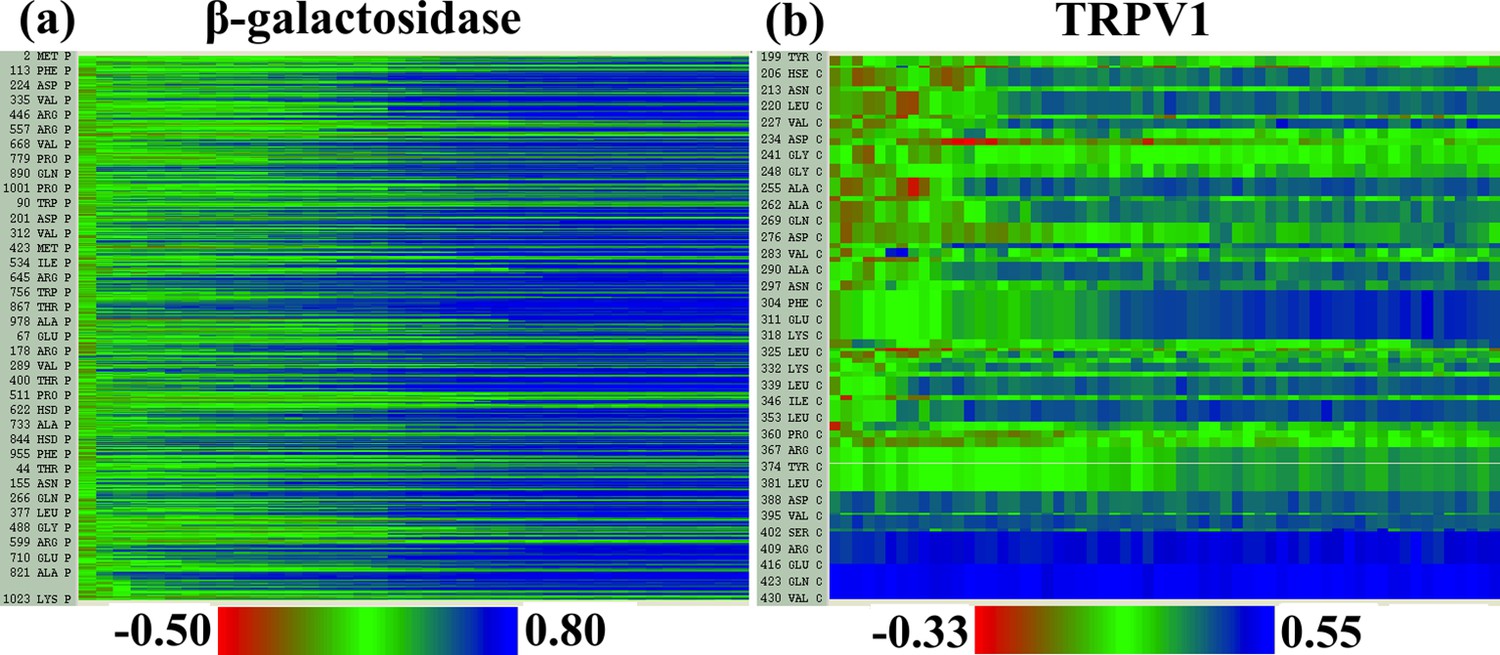
Local cross-correlations during cMDFF.
Local cross-correlations of residues within the fitted regions of (a) β-galactosidase and (b) TRPV1 plotted over the course of the cMDFF fitting show improvement over the successive MDFF refinement steps.
Figure 2—figure supplement 5
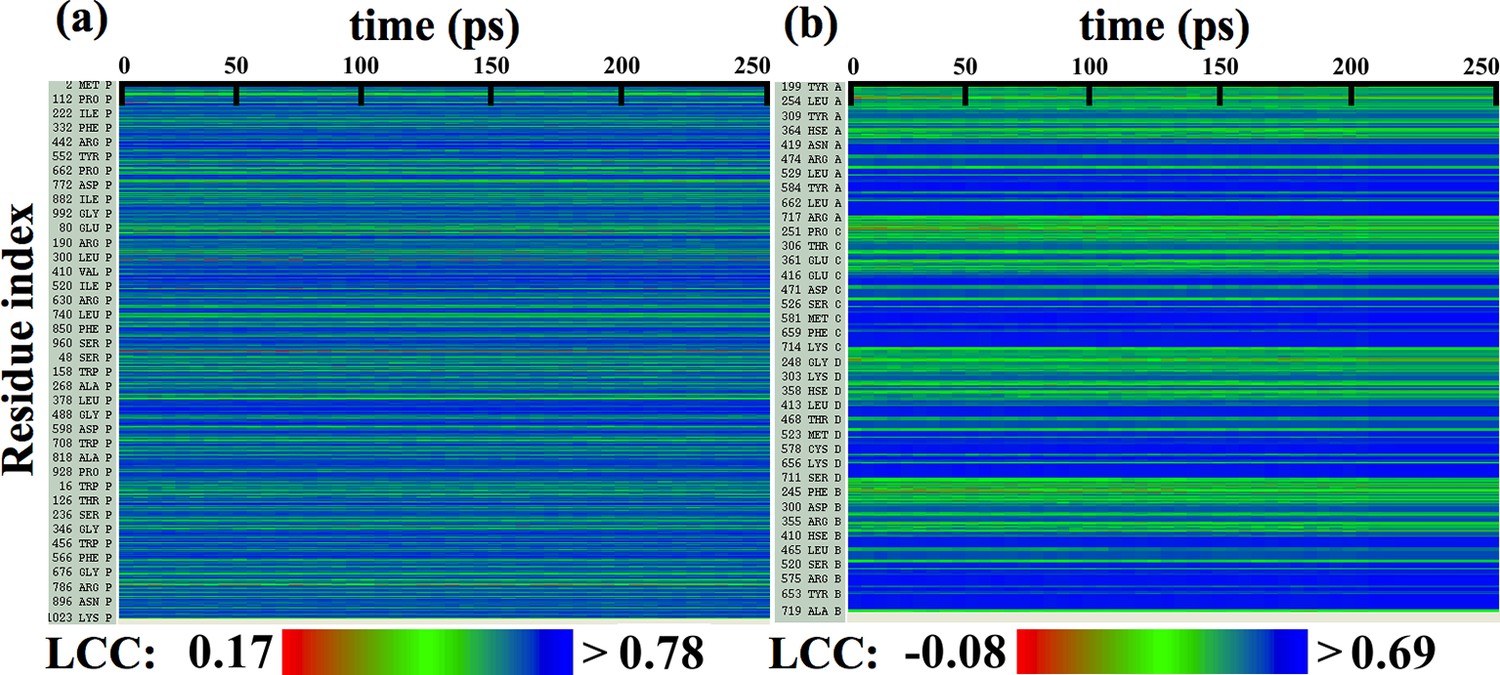
Local cross-correlations during direct MDFF to refine de novo structures.
Local cross-correlations of residues within the fitted regions of (a) β-galactosidase and (b) TRPV1 plotted over the course of direct MDFF of the respective de novo structures show little change. The large-scale structure of the starting de novo models are already well-fitted within the maps, increases in fit and overall structure quality of the refined de novo structures over the starting structures are thus due to local, sporadic improvements.
Figure 2—figure supplement 6
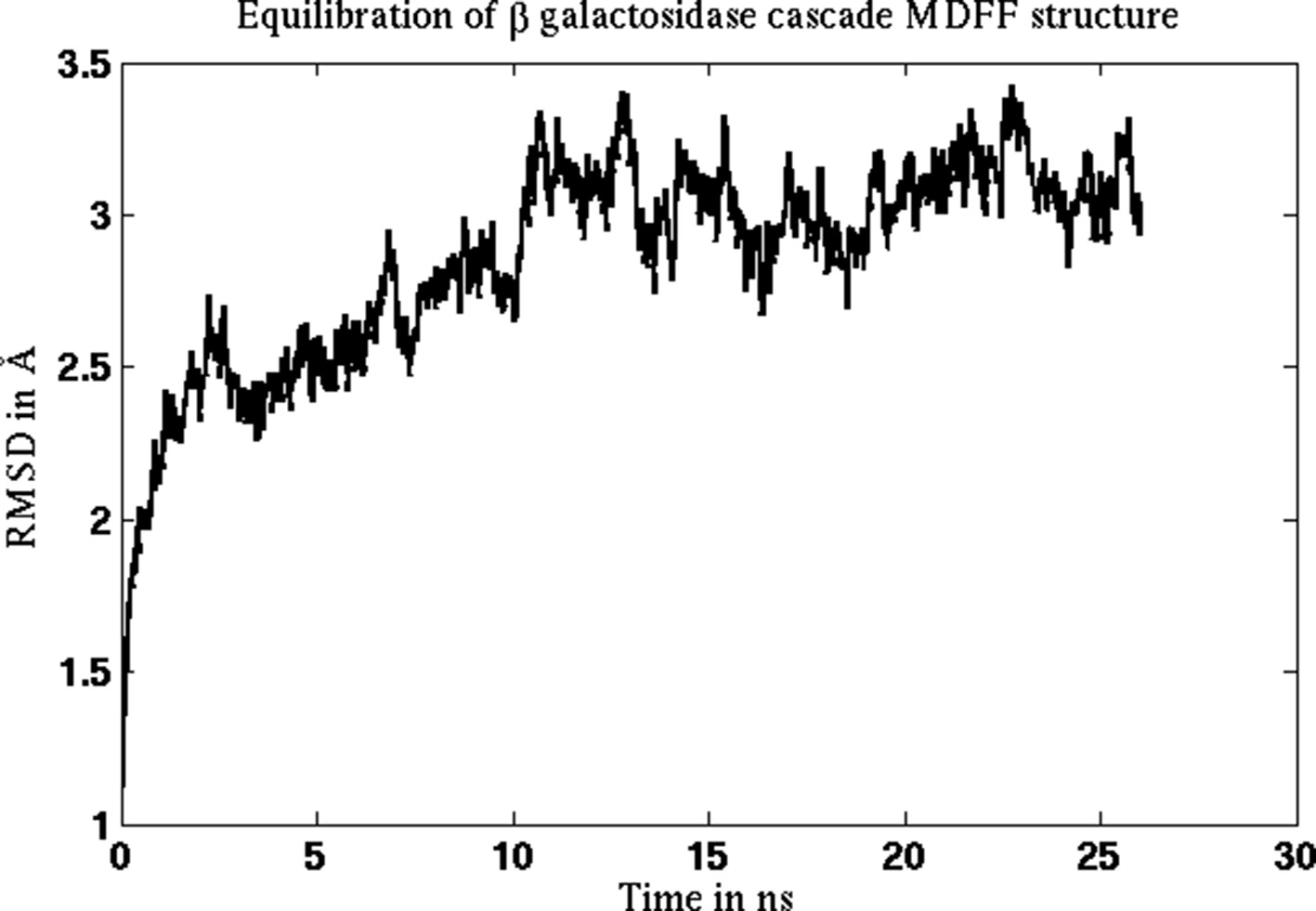
Equilibration of cMDFF-refined model of β-galactosidase.
The model resulting from a cMDFF fitting of β-galactosidase to the 3.2-Å map is subject to an equilibration MD simulation. The RMSD plot of the structure shows that it converges within 10 ns to an RMSD value of 3 Å.
Figure 2—figure supplement 7
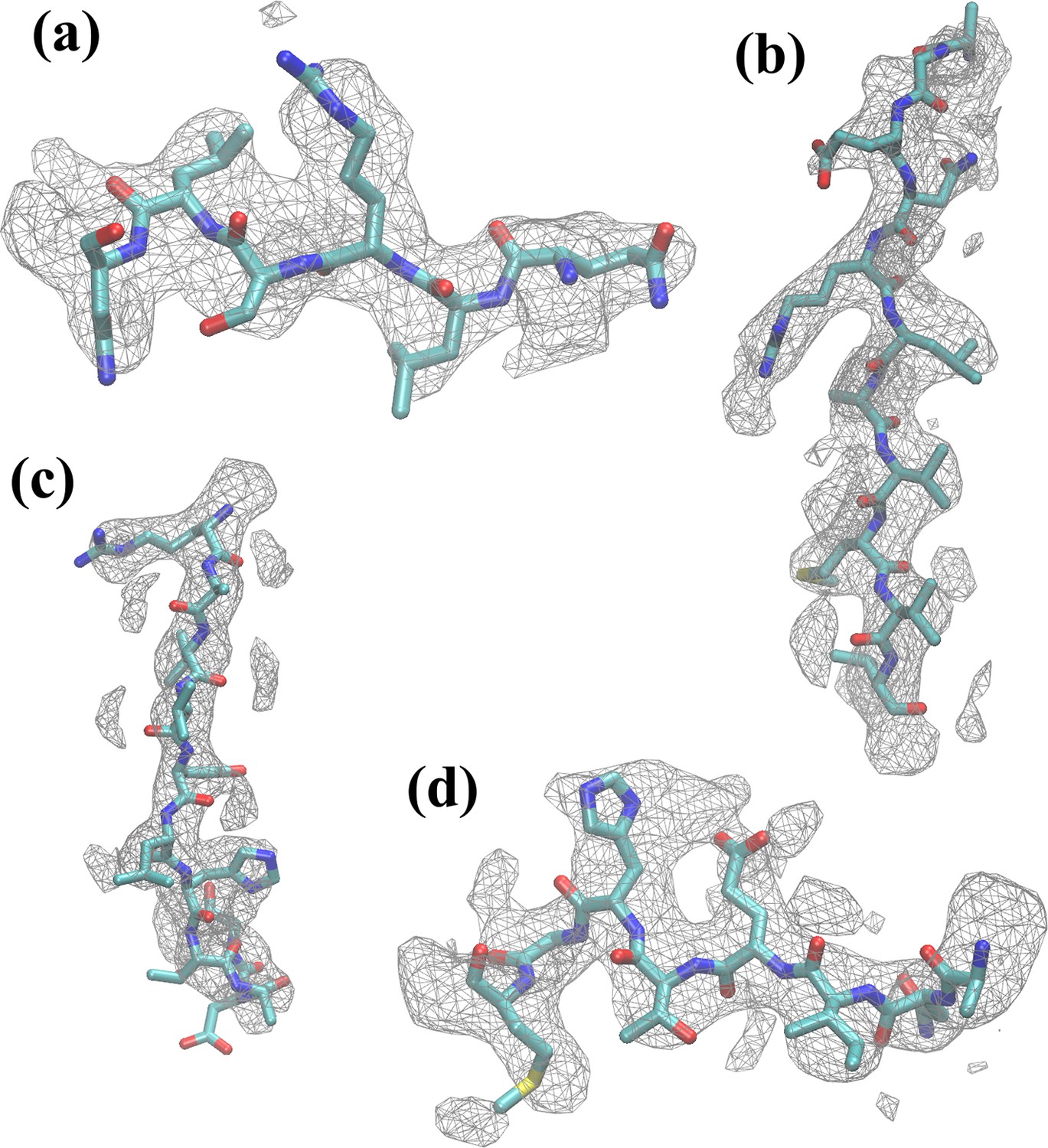
Residues of β-galactosidase fitted within density map.
Several examples of residue segments, consisting of residues (a) 50–55, (b) 179–189, (c) 310–320, and (d) 413–420, are shown within the corresponding map regions. In general, both backbone and sidechains were found to have fitted well after MDFF refinement.
Figure 2—figure supplement 8
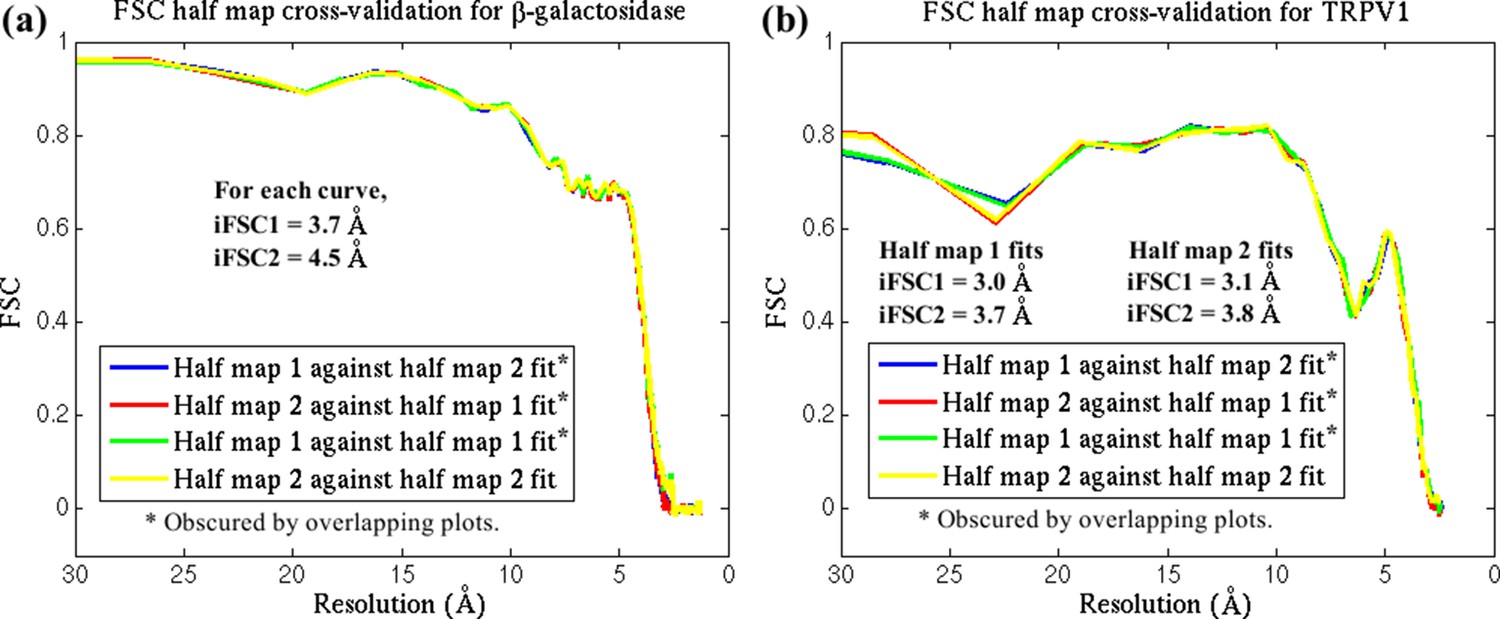
FSC cross-validation plots.
The reported structures for (a) β-galactosidase and (b) TRPV1 were each fitted by direct MDFF against two half-maps, labelled 1 and 2, from their respective EM data. Simulated maps were generated from the resulting structures, with labels corresponding to the half-maps used in the fitting. FSC plots of the simulated maps against the half-maps are so similar that they superimpose on one another. In addition, the differences in iFSCs between the various plots are negligible. These results demonstrate that the MDFF method, with the parameters used in the present study, does not overfit the structure.
Figure 3

Models colored by local resolution, square of RMSF, and B-factor.
The published models of (a) β-galactosidase (PDB 5A1A) and (b) TRPV1 (PDB 3J5P) are colored by the local EM map resolutions, the per-residue mean square fluctuations (RMSF) during MDFF simulation, and published B-factors. Comparison of these figures shows qualitative agreement between local resolution, RMSF, and B-factor. In fact, the local resolutions and B-factors correlate linearly with RMSF of a fitted model both in the presence as well as absence of the EM map (more details in Figure 4).
Figure 4 with 4 supplements
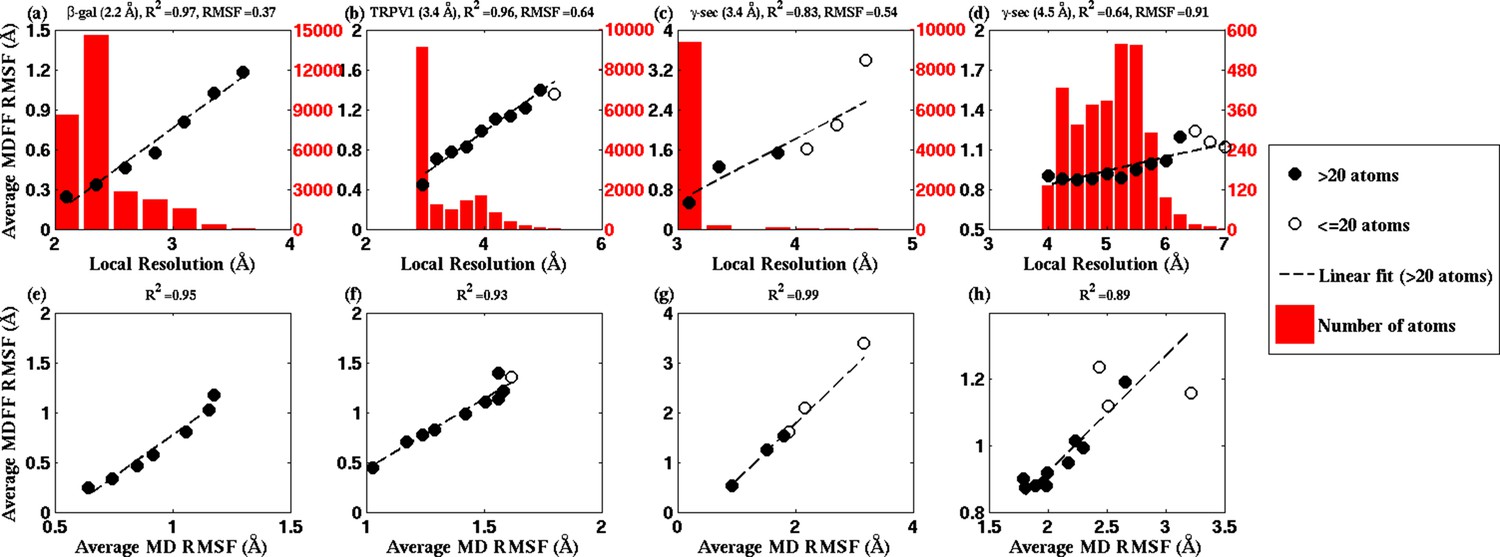
RMSF vs. local resolution plots for various simulations.
For each test case shown, atoms in the MDFF-refined structure are classified by local resolution of the map regions they are fitted into. The average RMSF value of atoms (during MDFF simulation) in each resolution bin is calculated and plotted against the local resolution in the cases of (a) β-galactosidase (β-gal) at 2.2 Å, (b) TRPV1 at 3.4 Å, γ-secretase (γ-sec) at (c) 3.4 Å and (d) 4.5 Å resolution, and proteasome (see Figure 4—figure supplement 3). The numbers of atoms in the resolution bins are displayed as a histogram (in red) spanning a system-specific range of resolutions. The lowest resolution bins contained low (<20) populations and visual inspection consistently revealed the atoms to be on the edges of the density or were otherwise located inside map noise, and were therefore ignored during further analysis. A clear linear correlation between RMSF and local resolution can be found in each case, such that applying a linear fit produces the high value shown in each graph heading. Also displayed in each heading is an overall RMSF, averaged over all atoms in the system. The overall RMSF reflects the conformational variety of structures that fit within the map, and is found to correspond to the map resolution such that higher resolutions produce lower RMSFs. The second row of plots show that the RMSF during MDFF simulation also linearly correlates with RMSF during unbiased MD simulations of (e) β-gal, (f) TRPV1 and (g,h) γ-sec, establishing that fluctuations during MDFF reflect the inherent flexibility of a system.
Figure 4—figure supplement 1
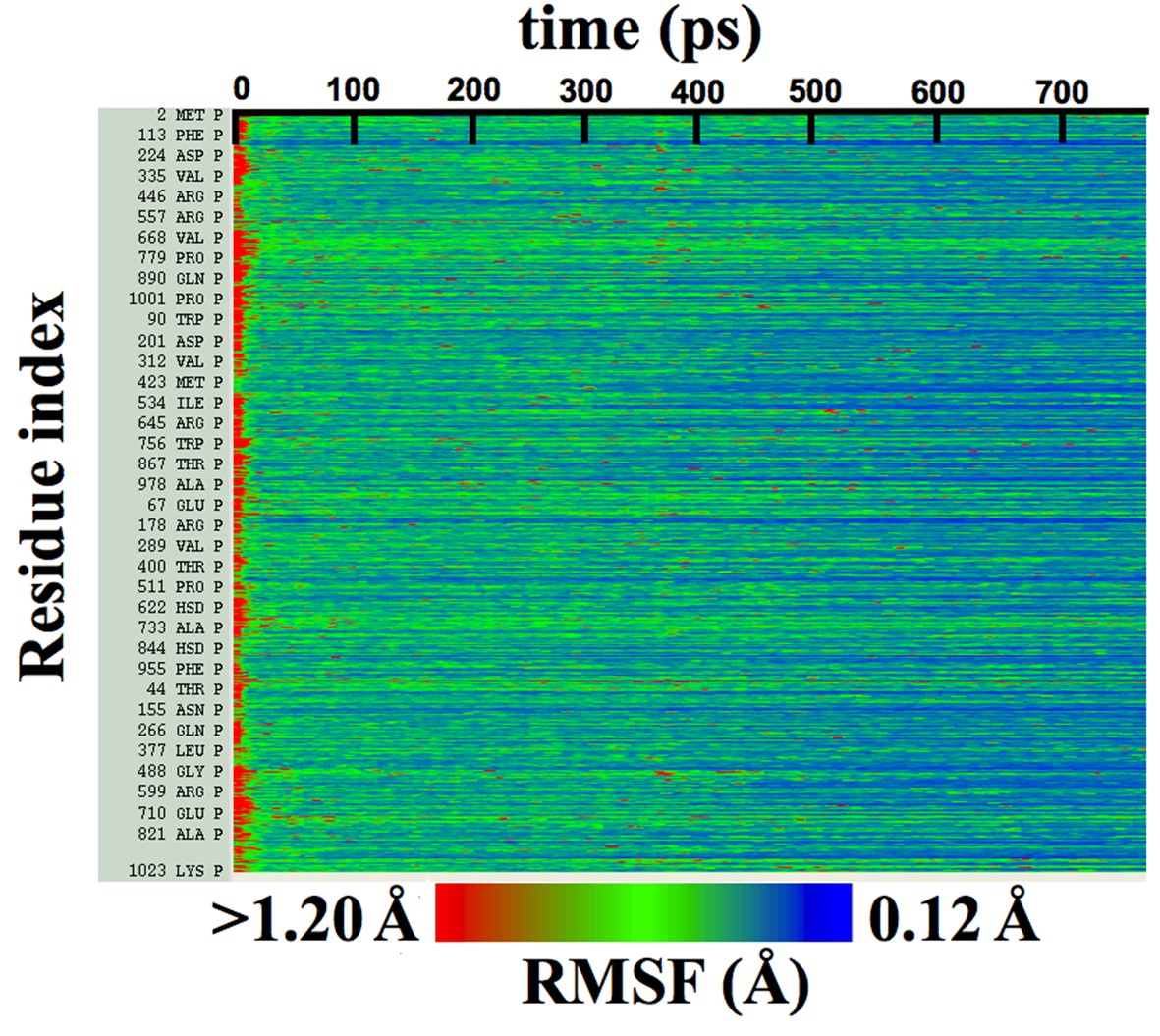
Per-residue RMSFs over β-galactosidase cMDFF fitting.
Residue RMSFs as a function of progress of cMDFF fitting show a general trend of decrease as the structure becomes better fit.
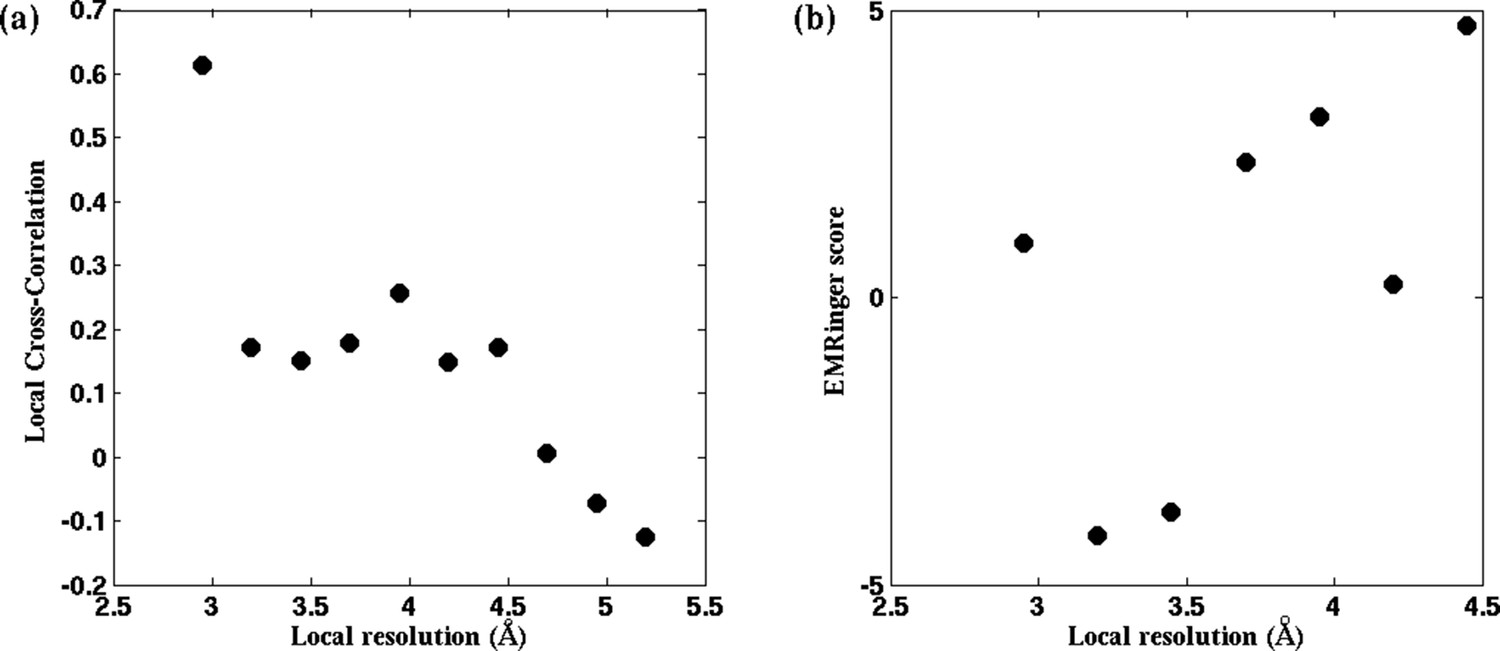
Figure 4—figure supplement 2
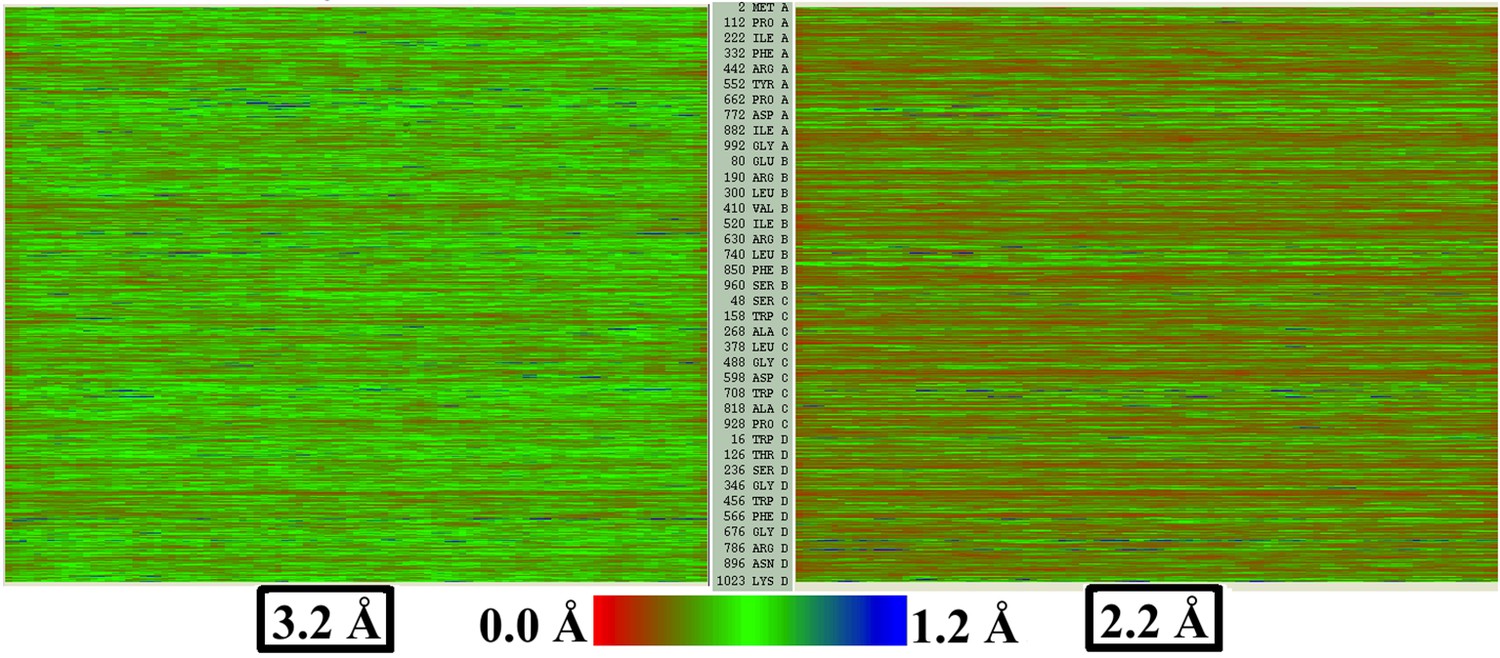
EMRinger score and LCC do not predict local resolution in TRPV1.
Published models corresponding to the 2.2-Å and 3.2-Å maps of β-galactosidase are fitted to their respective maps using direct MDFF. The RMSF values of all the residues along the protein sequence are plotted showing those from the 2.2-Å map are lesser than those from 3.2-Å map.
Figure 4—figure supplement 3
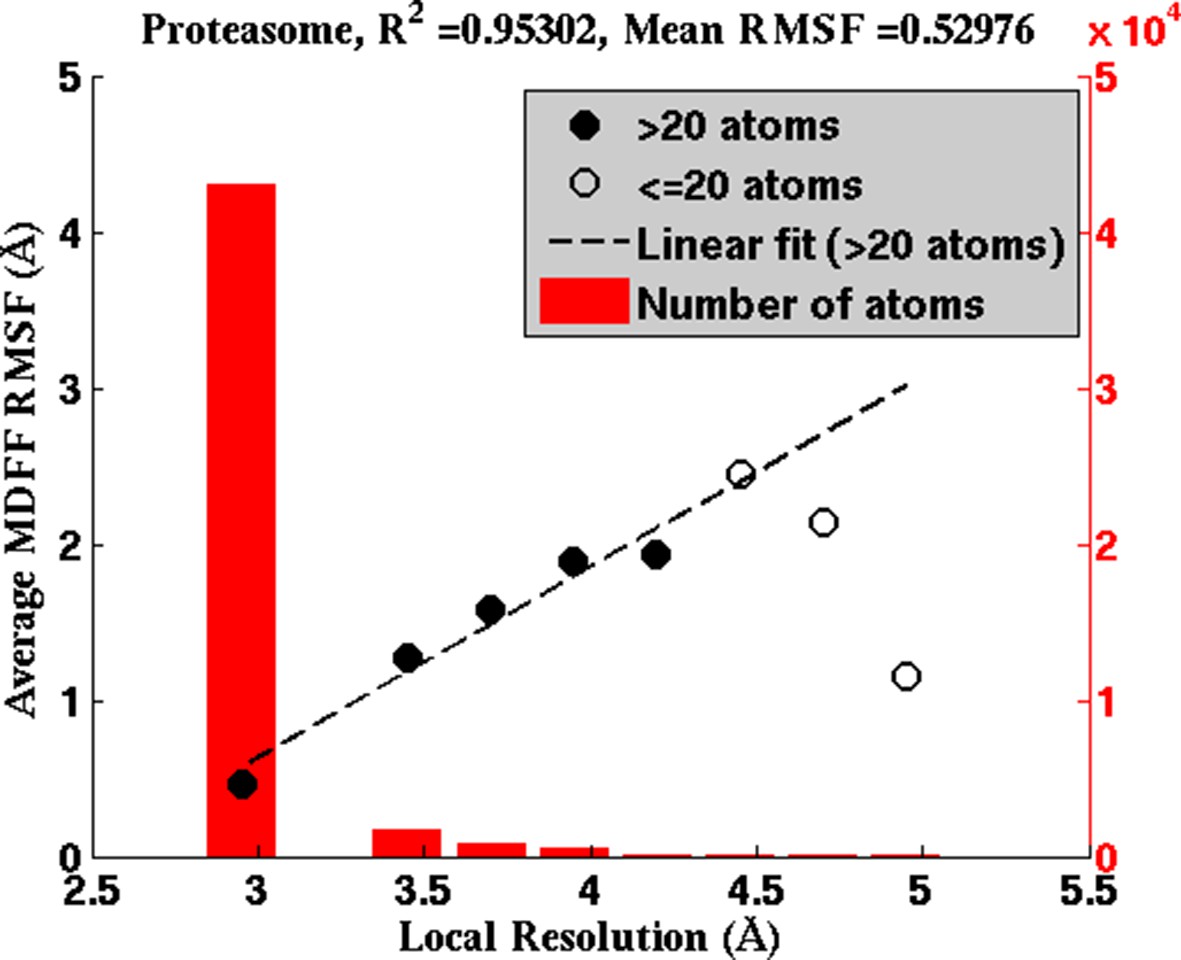
Average RMSF vs. local resolution during MDFF simulation of proteasome.
In the proteasome test case, the average RMSF of atoms corresponding to each local resolution, determined by ResMap, correlates linearly with the resolution. The same correlation was observed in all test cases considered (see Results).
Figure 4—figure supplement 4
RMSF values of individual residues during direct MDFF of published β-galactosidase models.
(a) Local cross-correlation and (b) EMRinger scores obtained from residues of a fitted model of TRPV1 do not exhibit one-to-one correspondence to local map resolutions.
Figure 5 with 4 supplements
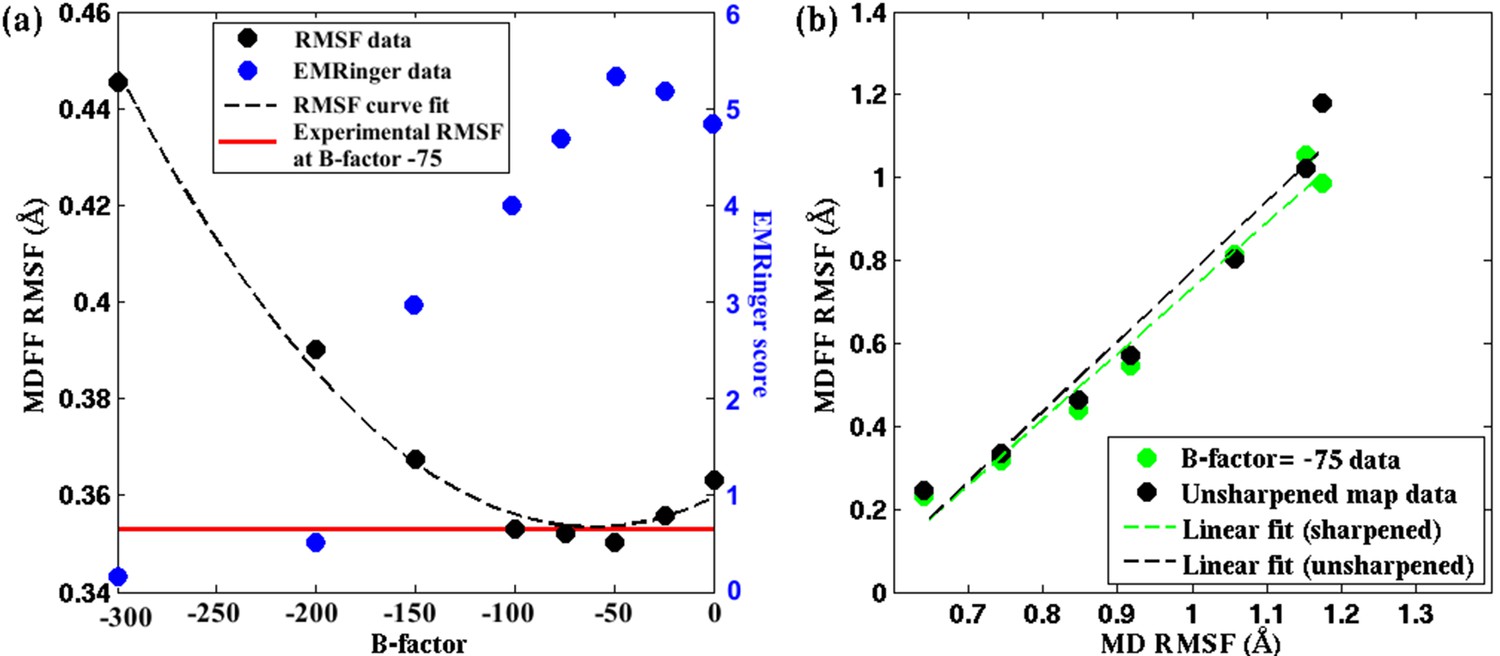
Effect of map sharpening on residue flexibility of β-galactosidase.
(a) Overall RMSF of a fitted 2.2Å β-galactosidase structure (PDB 5A1A) during direct MDFF fitting as a function of the B-factor of the fitting map exhibits a parabolic trend. Guinier analysis identifies a B-factor of −75 as optimal, for which the corresponding RMSF (shown in red) coincides with the minimum of the trend line. EMRinger scores (shown in blue) of the same structures show a negative parabolic trend, with the peak coinciding with the minimum of the RMSF plot. (b) The linear relationships between local RMSF during MDFF and during unbiased MD for the unsharpened map and optimally sharpened map are compared. While the linear relationship is preserved in both cases, RMSFs in the sharpened case are slightly lower than in the unsharpened case.
Figure 5—figure supplement 1

Effect of map sharpening on residue flexibility in TRPV1.
RMSF values of a fitted TRPV1 structure during MDFF fitting as a function of the B-factor of the fitting map exhibits a parabolic trend. This trend is prsented for (a) the whole protein, (b) the soluble region, and (c) the transmembrane domain. Guinier analysis identifies a B-factor of −100 as optimal for the whole protein as well as the transmembrane domain; the soluble region is characterized by a B-factor of −150. These B-factors are in close agreement with those representing a minimal RMSF: −100 (whole protein), −150 to −200 (soluble region), −100 to −125 (transmembrane domain).
Figure 5—figure supplement 2
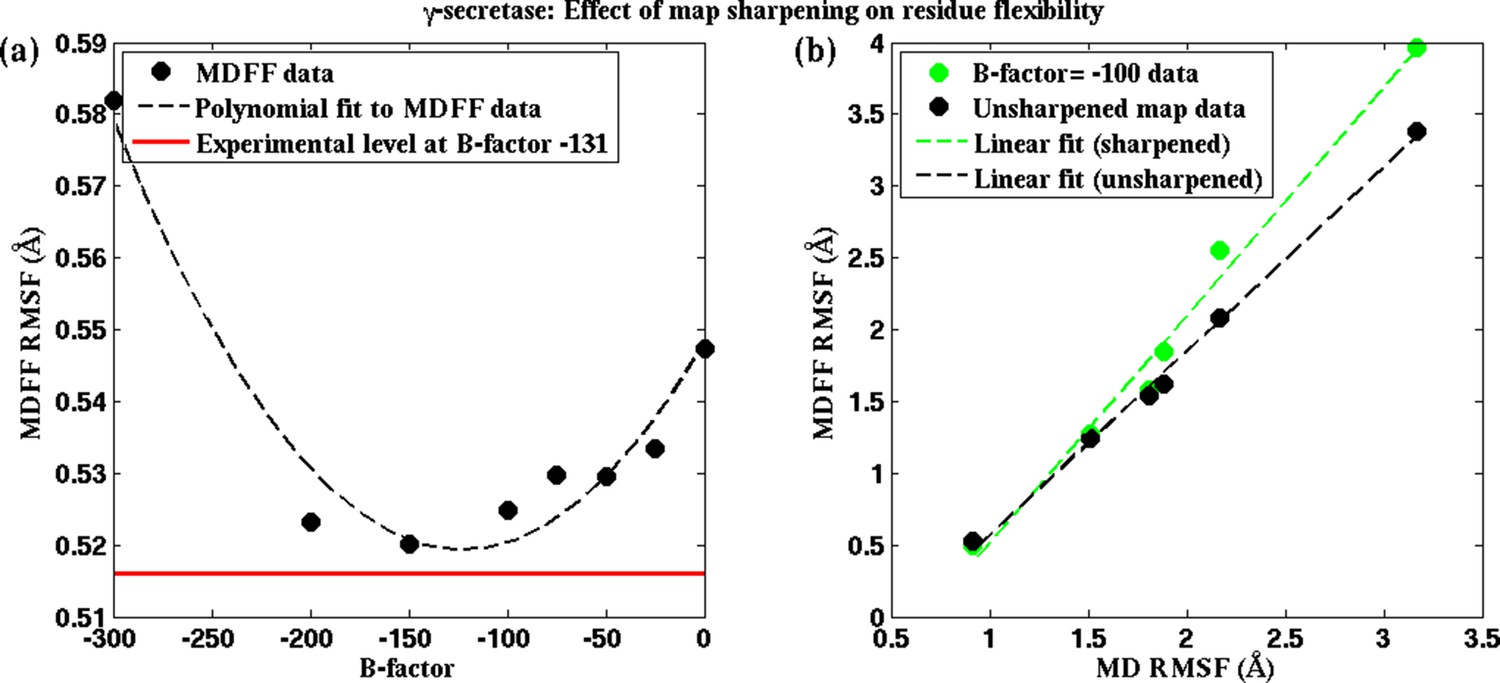
Effect of map sharpening on residue flexibility in γ-secretase.
(a) Overall RMSF of a fitted γ-secretase structure during MDFF fitting plotted as a function of the B-factor of the fitting map forms a parabola. Guinier analysis of map sharpening identifies a B-factor of −131 as optimal. The corresponding RMSF (shown in red) lies close to the minimum of the parabola. (b) The linear relationships between local RMSF during MDFF and during unbiased MD for the unsharpened map and the sharpened map of B-factor −100 are compared. For all but the first resolution bin, RMSF for the sharpened map is higher than that of the unsharpened map. However, the first resolution bin contains about 98% of atoms in the structure, so that atoms in the other bins are outliers, which fall into map regions of non-optimal local resolution.
Figure 5—figure supplement 3
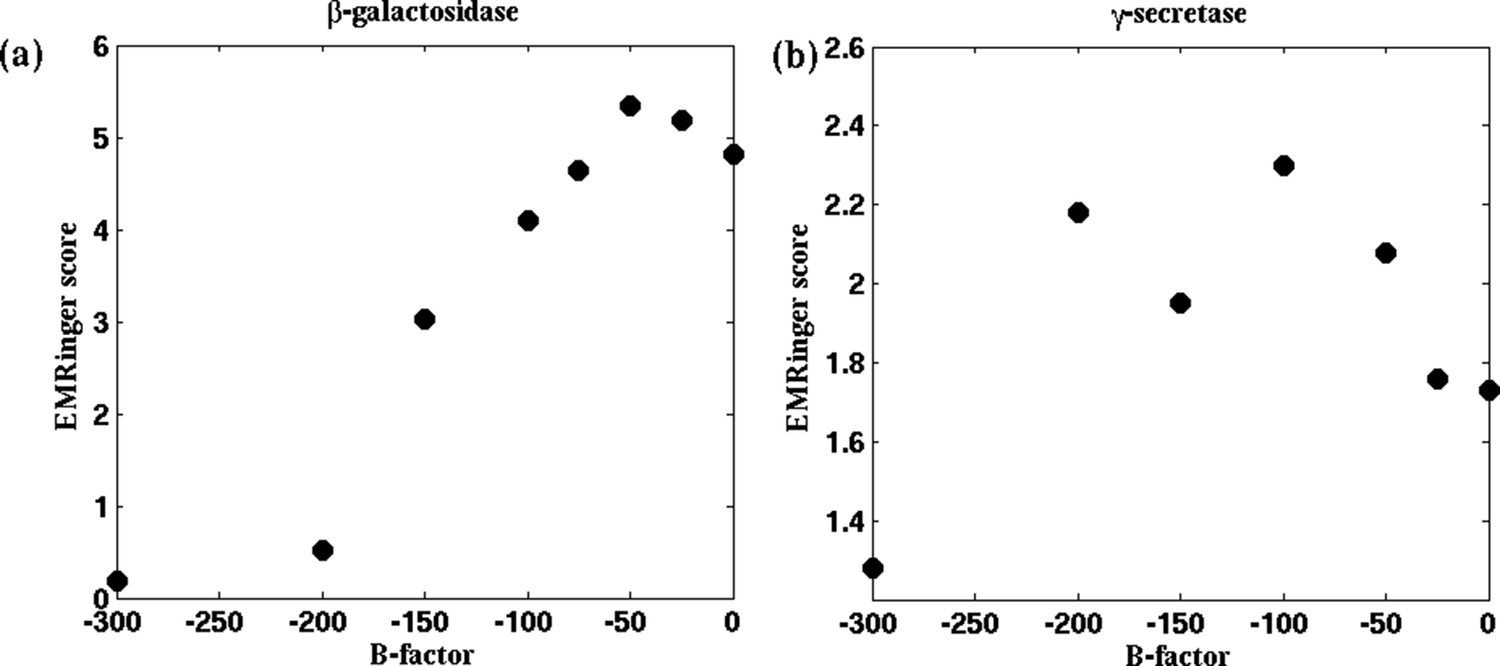
EMRinger scores as a function of B-factor.
For the cases of (a) β-galactosidase and (b) γ-secretase, the reported model was fitted to maps sharpened with various B-factors. EMRinger scores for the fitted model/map pairs are plotted against the corresponding B-factors. The maxima of the plots, at B-factors −50 and −100 for β-galactosidase and γ-secretase respectively, correspond with the RMSF minima in Figures 4a and 4—figure supplement 1a.
Figure 5—figure supplement 4
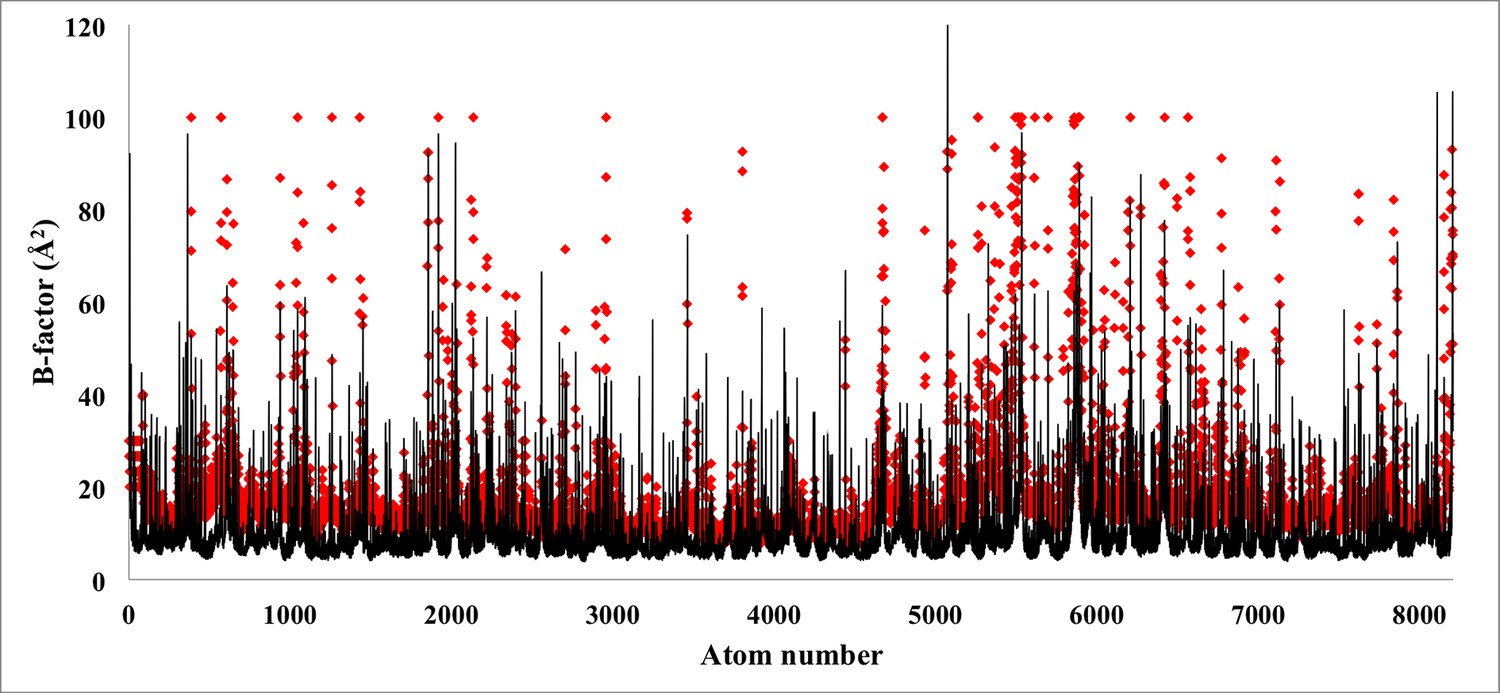
Atom-by-atom B-factor for a β-galactosidase monomer.
The B-factor, measured for each atom employing the relationship 8π2/3(RMSF)2 employing RMSF values from Figure 3 (black line), are fairly comparable to the B-factors reported experimentally (red rhombus). An overall cross-correlation of 55% is found between these two sets of B-factors.
Videos
Video 1
cMDFF Refinement of TRPV1.
https://doi.org/10.7554/eLife.16105.004Tables
Table 1
β-galactosidase MDFF results. cMDFF and ReMDFF provide better fitted structures than direct MDFF according to various criteria. It is noteworthy that all structures refined by any form of MDFF display an improved MolProbity (Chen et al., 2010) score compared to the original de novo structure.
| Structure | RMSD(Å) | EMRinger | iFSC1(Å) | iFSC2(Å) | MolProb. | GCC |
|---|---|---|---|---|---|---|
| de novo (Bartesaghi et al., 2014) | 0.0 | 2.25 | 4.03 | 5.00 | 3.14 | 0.67 |
| Refined de novo | 0.6 | 4.23 | 4.19 | 5.20 | 1.23 | 0.68 |
| Initial | 7.7 | 0.24 | 0.14 | 0.15 | 1.49 | 0.48 |
| Direct MDFF | 3.7 | 2.31 | 2.11 | 2.74 | 1.38 | 0.56 |
| cMDFF | 0.7 | 3.16 | 4.22 | 5.22 | 1.37 | 0.67 |
| ReMDFF | 0.9 | 3.45 | 3.76 | 4.66 | 1.13 | 0.67 |
Table 2
Structure quality indicators for β-galactosidase structures. β-galactosidase structures investigated in the present study were uploaded to the MolProbity server (http://molprobity.biochem.duke.edu) to extract the quantities presented below. The results show that the cMDFF- and ReMDFF-refined structures not only exhibit good measures of fit, but also improve the clash score and rotamer geometries, relative to the de novo and initial structures, while incurring only a small expense in Ramachandran statistics, bad angles, and Cβ deviations.
| de novo (Bartesaghi et al., 2014) | Refined de novo | Initial | Direct MDFF | cMDFF | ReMDFF | |
|---|---|---|---|---|---|---|
| Clashscore | 53.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Poor rotamers (%) | 11.6 | 3.8 | 4.2 | 3.0 | 4.4 | 1.37 |
| Favored rotamers (%) | 67.4 | 90.8 | 87.8 | 92.1 | 89.8 | 95.3 |
| Ramachandran outliers (%) | 0.2 | 0.7 | 2.7 | 3.0 | 1.6 | 2.7 |
| Ramachandran favored (%) | 97.4 | 95.8 | 91.1 | 91.1 | 94.4 | 90.9 |
| MolProbity | 3.14 | 1.23 | 1.49 | 1.38 | 1.37 | 1.13 |
| C deviations (%) | 0.0 | 0.05 | 4.92 | 0.18 | 0.29 | 0.39 |
| Bad bonds (%) | 0.09 | 0.04 | 3.61 | 0.02 | 0.01 | 0.03 |
| Bad angles (%) | 0.03 | 0.60 | 3.98 | 0.63 | 0.49 | 0.37 |
| RMS distance (Å) | 0.007 (0.025%) | 0.019 (0%) | 0.035 (0.237%) | 0.022 (0%) | 0.019 (0%) | 0.021 (0%) |
| RMS angle (degrees) | 1.1 (0.009%) | 2.2 (0.009%) | 3.6 (1.177%) | 2.4 (0.103%) | 2.1 (0.018%) | 2.3 (0.085%) |
| Cis prolines (%) | 8.06 | 8.06 | 6.45 | 6.45 | 6.45 | 8.06 |
| Cis non-prolines (%) | 1.15 | 1.15 | 0.0 | 0.0 | 1.15 | 0.0 |
Table 3
Performance and cost results for ReMDFF of carbon monoxide dehydrogenase on Amazon Web Services (AWS) Elastic Compute Cloud (EC2) platform. Costs are incurred on a per-hour basis, with a 1 hr minimum.
| Instance type | CPU | Performance (ns/day) | Time (hours) | Simulation cost ($) |
|---|---|---|---|---|
| c3.8xlarge | 30 | 5.88 | 0.41 | 1.68 |
| c3.4xlarge | 12 | 3.33 | 0.72 | 0.84 |
| c3.2xlarge | 6 | 1.35 | 1.78 | 0.84 |
Additional files
-
Supplementary file 1
Supplementary tables.
(A) TRPV1 MDFF Results. (B) Structure quality indicators for TRPV1 structures. (C) MDFF for the TRPV1 TM region. (D) Structure quality indicators for γ-secretase. (E) Measures of fit for MDFF refinements of β-galactosidase prepared initially at 1000 K. (F) Structural quality indicators for MDFF-refined β-galactosidase prepared initially at 1000 K.
- https://doi.org/10.7554/eLife.16105.028
-
Supplementary file 2
Initial and refined structures.
- https://doi.org/10.7554/eLife.16105.029
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps
eLife 5:e16105.
https://doi.org/10.7554/eLife.16105




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}