Transcription factor TFCP2L1 patterns cells in the mouse kidney collecting ducts
- Columbia University, United States
- Max Delbruck Center for Molecular Medicine, Germany
- Tongji University, China
- Institute for European Expertise in Physiology, France
- University of Washington, United States
- Fudan University, China
- University of Illinois at Chicago, United States
Abstract
Although most nephron segments contain one type of epithelial cell, the collecting ducts consists of at least two: intercalated (IC) and principal (PC) cells, which regulate acid-base and salt-water homeostasis, respectively. In adult kidneys, these cells are organized in rosettes suggesting functional interactions. Genetic studies in mouse revealed that transcription factor Tfcp2l1 coordinates IC and PC development. Tfcp2l1 induces the expression of IC specific genes, including specific H+-ATPase subunits and Jag1. Jag1 in turn, initiates Notch signaling in PCs but inhibits Notch signaling in ICs. Tfcp2l1 inactivation deletes ICs, whereas Jag1 inactivation results in the forfeiture of discrete IC and PC identities. Thus, Tfcp2l1 is a critical regulator of IC-PC patterning, acting cell-autonomously in ICs, and non-cell-autonomously in PCs. As a result, Tfcp2l1 regulates the diversification of cell types which is the central characteristic of 'salt and pepper' epithelia and distinguishes the collecting duct from all other nephron segments.
https://doi.org/10.7554/eLife.24265.001Introduction
Kidney development starts from two embryonic structures, the metanephric mesenchyme (MM) and the ureteric bud (UB) (Little and McMahon, 2012). The Metanephric Mesenchyme gives rise to tubules organized in segments, each with a single type of epithelial cell. The UB, in contrast, gives rise to tubules containing mixtures of two different paradigmatic cell types called principal cells (PC) and intercalated cells (IC). These two cell types have different functions and they express different sets of signature proteins. PCs control water and electrolyte balance and express ion-channels such as ROMK, ENaC and Aquaporins 2–4 (Sasaki et al., 1995). ICs, on the other hand, control acid-base balance (Gluck et al., 1982) and contribute to immune defense (Paragas et al., 2014; Azroyan et al., 2015). ICs express vacuolar H+--ATPase, including kidney specific subunits (D-B subunits), bicarbonate conversion enzymes (carbonic anhydrase Ca2 and Ca12), bicarbonate transporters, AE1 (Slc4a1), AE4 (Slc4a4) and pendrin (Slc26a4). Nonetheless, despite their many differences, ICs and PCs function coordinately: for example, the absorption of Na+ by PCs creates a transepithelial electrical gradient which stimulates H+ secretion by ICs (Chang et al., 1996; DuBose and Caflisch, 1985).
The underlying mechanisms that allow ICs and PCs to coordinate their activities have been difficult to identify because their genesis from progenitors and their ultimate relatedness to one another has been uncertain. This is because the collecting duct is thought to be populated by many subtypes of ICs, including α-IC and β-IC, non-α-β-IC, and various mixed cell types (Schwartz et al., 1985) interspersed among PCs. Yet the definition of these various ICs and their relationship to PCs has been called into question as one cell type may convert into another in response to environmental challenges and recently reproduced in genetic models (Aigner et al., 1995; Al-Awqati and Schwartz, 2004; Bagnis et al., 2001; Wu et al., 2013; Trepiccione et al., 2016). Moreover, the knockout of the IC specific transcription factor, Foxi1 exhibited a cell type that co-expressed mixtures of IC (Ca2) and PC (Aqp2) proteins, implying that Foxi1 restricted the otherwise facile interconversions of ICs to PCs (Blomqvist et al., 2004). Consequently, while many distinct cellular phenotypes are known to populate the collecting duct, the underlying logic that coordinates these cell types has not been uncovered.
A clue to the mechanisms that coordinate ICs and PCs was suggested by their stereotyped spatial patterning. Immunofluorescence analysis found rosette-like structures in the adult collecting duct, a pattern reminiscent of tissues governed by Notch mediated lateral inhibition (Blanpain et al., 2006; Kiernan, 2013; Noah et al., 2013). In fact, recent studies have shown that manipulation of Notch signaling modifies the ratio of PCs and ICs (Jeong et al., 2009; Guo et al., 2015; Grimm et al., 2015; Nam et al., 2015) suggesting that not only Foxi1 but also components of the Notch pathway are critical to determine cell type. However, the developmental context for these regulators is currently indeterminate, in part because of incomplete description of the developmental origin of IC and PC.
Here we show that IC-PC coordination is under control of a poorly studied transcription factor called Tfcp2l1. We found that Tfcp2l1 induces the initial formation of a cellular intermediate which we call the ‘double positive’ mixed IC-PC cell. Thereafter Tfcp2l1 regulates the formation of discrete ICs and PCs by both cell-autonomous and cell non-autonomous mechanisms. The latter includes the regulation of the Jag1-Notch1 pathway in rosettes composed of ICs and PCs. These data indicate that UB tubules are patterned by Notch dependent interactions of neighboring cells rather than demarcated in nephron segments controlled by Notch signaling (Costantini and Kopan, 2010).
In sum, coordinate development of ICs and PCs is linked by Tfcp2l1 acting late in gestation in progenitors of the collecting duct. This mechanism explains the apparent reciprocal relationship in the relative abundance of ICs and PCs in the adult collecting duct (Jeong et al., 2009; Guo et al., 2015) as well as their physiologic linkage. We suggest that coordination between ICs and PCs by Tfcp2l1 is critical for homeostasis, since these cells co-regulate the balance of electrolytes, acid-base, and water.
Results
Expression of Tfcp2l1 in the development of the distal nephron
Tfcp2l1 (also known as LBP-9 or CRTR-1) is a nuclear transcription factor and a member of the CP2 subfamily of the LSF/Grainyhead family (Kokoszynska et al., 2008; Yamaguchi et al., 2005; Aue et al., 2015; Werth et al., 2010; Walentin et al., 2015; Traylor-Knowles et al., 2010). Tfcp2l1 has been implicated in the maintenance of pluripotency networks of ES cells where it is targeted by both LIF (Dunn et al., 2014; Martello et al., 2013; Ye et al., 2013) and Wnt (Yamaguchi et al., 2005). In addition, Tfcp2l1 is implicated in the development of arborizing epithelial trees, including the collecting ducts (Yamaguchi et al., 2006; Paragas et al., 2014). In fact, Tfcp2l1 was detected at E11 in the primordium of the collecting ducts (the Wolffian Duct and the Ureteric Bud; data not shown), and then throughout its arborized Krt8+ stalks at E15-E18 (Figure 1A, Figure 1—figure supplement 1) when Tfcp2l1 appeared to localize exclusively to the nucleus. In adult collecting ducts (P60), Tfcp2l1 was prominent in both PCs (Krt8+) and ICs (Atp6v1b1+; abbreviated Atp6b1) (Figure 1B), but demonstrated greater immunoreactivity in ICs than in PCs (Figure 1C). Tfcp2l1 was also expressed in the Thick Limb of Henle and connecting segments of the nephron (data not shown), but its most persistent location was the collecting duct system.
Figure 1 with 1 supplement see all

Tfcp2l1 is a nuclear protein expressed in the collecting ducts.
(A) Immunofluorescence detection of Tfcp2l1 (green) in stalks of ureteric-collecting ducts at E15 and at E18. Nuclear localization was prominent at E18. The ducts were identified by the uniform expression of Krt8 (red). Bars = 5 µm. (B) In adult collecting ducts,Tfcp2l1 (green) was expressed by both Intercalated Cells (IC), identified by immunodetection of Atp6v1b1, abbreviated Atp6b1 (red), and Principal Cells (PC) identified by immunodetection of Aqp2 (blue). Z-stack projection. Bar = 10 µm. (C) Quantification of Tfcp2l1 immunoflourescence in adult collecting ducts normalized per measurement area. Atp6b1+ IC cells expressed higher levels of Tfcp2l1 than did Aqp2+ PC cells.
Identification of IC-PC ‘double positive’ progenitors
In order to identify the cellular targets of Tfcp2l1, the development of collecting duct epithelia was characterized using signature proteins found in adult PCs and ICs. While the E13 and E15 collecting duct demonstrated homogeneous expression of proteins typical of PCs, there were rare cells co-expressing IC proteins (not shown). By E18 these cells became abundant and we designated them ‘double-positive’ progenitors, because they co-expressed IC and PC proteins (e.g. IC proteins: Foxi1+, Atp6b1+; together with PC proteins: Calb1+, Krt8+: Figure 2A, Figure 2—figure supplement 1A,B). The ‘double positive’ phenotype was spatially restricted, because neighboring cells expressed PC but not IC proteins (Figure 2—figure supplement 1A,B). The ‘double-positive’ progenitors were also transient; by birth, IC and PC proteins were generally not co-expressed (Figure 2—figure supplement 1C, Figure 3, Figure 3—figure supplement 2; four independent mice for each immunofluorescence analysis) except in rare collecting duct cells (Figure 2—figure supplement 2).
Figure 2 with 2 supplements see all

‘Double positive’ progenitors populate the E18 collecting duct.
(A) IC proteins were expressed at E18 in presumptive PC cells. Co-expression of Atp6b1 (typical of ICs, red) and Calbindin (typical of PCs, blue) is shown in Tfcp2l1+ cells in the cortical region of the collecting duct. (white arrows) Bar = 10 µm. (B) Lineage of ICs and PCs was detected with genetic reporters. HoxB7-Cre;mTmG (green) marked every cell in the collecting duct including AQP2+ PC cells (red-yellow) and Atp6b1+ IC cells (blue-green). Atp6b1-Cre;mTmG labeled every IC cell (endogenous Atp6b1+; blue-green), as well as some Aqp2+ PCs (white arrows, yellow). Atp6b1-Cre;mTmG-negative PC cells are also found (Aqp2+, Atp6b1-, Atp6b1-Cre;mTmG-, red) (Bars= top 5 µm, bottom 20 µm).
Figure 3 with 2 supplements see all

Tfcp2l1 is necessary for the development of ICs.
(A) Structure of mouse Tfcp2l1 gene showing LoxP sites flanking the DNA-Binding CP2 domain (exon 3 and 4). (B) Control (Tfcp2l1f/f) and Tfcp2l1 knockout kidneys (Cdh16-Cre;Tfcp2l1f/f) were analyzed for PC (Aqp2 red) and IC (Atp6b1 blue) proteins. Note that the deletion of Tfcp2l1 replaced the normal patterning of IC and PC cells with a monotonous array of PC like cells (Aqp2+). Z-stack reconstruction. Bars = 10 µm.
To further analyze the origin of ICs and PCs, we crossed cre-reporter (mTmG; Muzumdar et al., 2007) with cre-drivers that become active at different developmental stages of collecting duct development. We used HoxB7-Cre, which becomes active before the appearance of ‘double positive’ cells, and Atp6b1-Cre, which is active only after the expression of Atp6b1 in ICs between E15-E18 (Miller et al., 2009). All ICs and PCs were labeled in HoxB7-Cre/mTmG adult kidneys (P60; Figure 2B) confirming that ICs and PCs developed from a HoxB7+ ureteric progenitor cell. Surprisingly, Atp6b1-Cre/mTmG labeled not only Atp6b1+ ICs (74% of GFP-labeled cells were ICs) but also a subset of Aqp2+ PCs (24% of GFP-labeled cells were Aqp2+ cells expressing low levels of Atp6b1). In addition, rare (2%) GFP-labeled cells expressed equivalent levels of Aqp2 and Atp6b1, typical of ‘double positive’ cells (n = 80 ducts in four independent mice were inspected for each genetic label). Therefore, all adult ICs and PCs derive from HoxB7+ progenitors and all adult ICs and at least some PCs derive from HoxB7+ progenitors that subsequently express Atp6b1+.
Tfcp2l1 is required for epithelial patterning
To determine whether Tfcp2l1 is important for the development of ‘double-positive’ progenitors or for later stages of IC and PC development, we created a floxed allele flanking the CP2 domain (Figure 3a), and we used a set of cre-drivers to inactivate Tfcp2l1 in a stage- and cell-specific manner. EIIA-Cre was used to delete Tfcp2l1 early in development (Lakso et al., 1996), the Cdh16-Cre driver was used to inactivate Tfcp2l1 throughout the distal nephron and collecting duct before ICs develop (Shao et al., 2002), and Atp6b1-Cre was used for cell-specific deletion in maturing ICs (Miller et al., 2009). In each case, the efficiency of Tfcp2l1 inactivation was confirmed by immunostaining (Figure 3B). EIIA-Cre;Tfcp2l1f/f knockouts died quickly after birth when IC maturation was still ongoing (similar to published gene-trap; Yamaguchi et al., 2006), but both Cdh16-Cre and Atp6b1-Cre;Tfcp2l1f/f knockouts survived to adulthood (P60).
Tfcp2l1 deletion with EIIA-Cre or with Cdh16-Cre produced grossly normal kidneys at birth (Figure 3—figure supplement 1), except that all ‘double positive’ progenitors and all mature ICs were deleted, as measured by their signature proteins, Foxi1, Ca2, and Atp6b1 which for example were reduced by ~89% (4.8 ± 1.9 vs 41.5 ± 15.1 Atp6b1+ cells per kidney section; n = 3 independent mice; p=0.0014; Figure 3B; Figure 3—figure supplement 2). In contrast, PC cell numbers and proteins demonstrated variable responses and were either mildly downregulated (e.g. 78 ± 23.6 vs 112.5 ± 22.6 Aqp2+ cells per kidney section; n = 3 independent mice; p=0.04; Figure 3B) or mildly upregulated (e.g. Krt8; Figure 3—figure supplement 2A). PCs also remained correctly polarized, displaying apical ROMK and basolateral Cdh1 and Aqp4, indicating that Tfcp2l1 did not regulate epithelial polarity in PCs (not shown). Hence, Tfcp2l1 was critical to initiate the expression of IC signature genes which were first expressed in ‘double positive’ progenitors.
In contrast to the deletion of Tfcp2l1 by EIIA-Cre and Cdh16-Cre drivers which act before the appearance of IC cells, we found that deletion of Tfcp2l1 with Atp6b1Cre resulted in a normal number and distribution of ICs and PCs at P14. By P60, however, there were fewer than half the number of ICs (14 ± 5.4 vs. 29 ± 13 Atp6b1+ cells per section or as a percentage of collecting duct cells; n = 3 independent kidneys each; p=0.01; Figure 4A). In fact, the surviving ICs demonstrated only faint Atp6b1 staining which was co-expressed with PC protein Krt8. When we introduced the floxed mTmG Reporter with Tfcp2l1-flox and Atp6b1-Cre, and focused our analysis on GFP+ cells by measuring endogenous markers in single cells by spot imaging, we found that GFP+Tfcp2l1+ cells demonstrated IC (Atp6b1>>Aqp2) or PC (Aqp2>Atp6b1) phenotypes, but GFP+ Tfcp2l1-deleted cells appeared strikingly similar to PCs (Aqp2>Atp6b1; Figure 4B) or double positive cells (Atp6b1≈Aqp2).
Figure 4
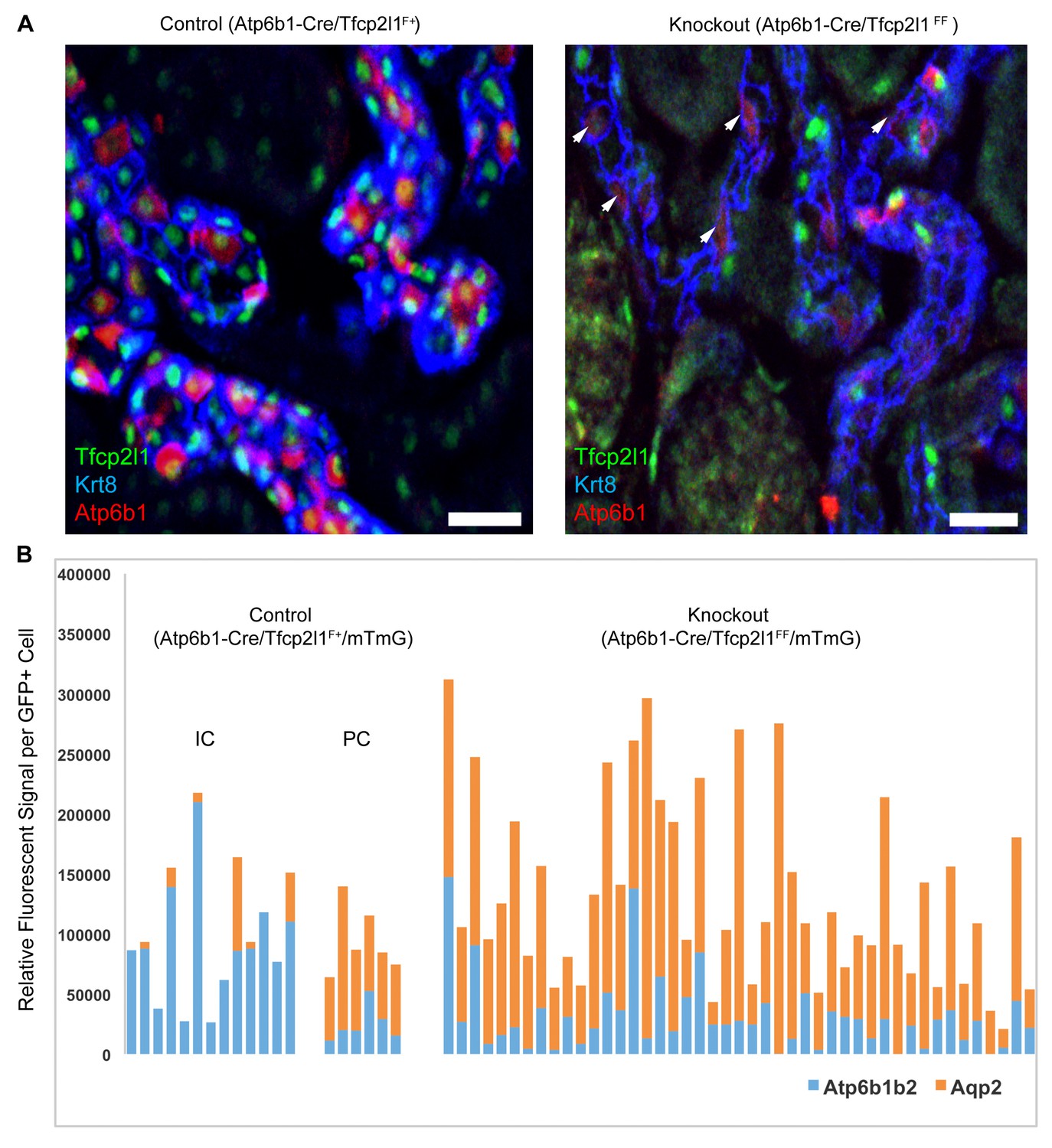
IC specific deletion of Tfcp2l1 (green) by Atp6b1Cre results in loss of IC cells.
(A) Deletion of Tfcp2l1 resulted in the widespread loss of IC and PC patterning. Only residual expression of Atp6b1 (red) in Krt8+ (blue) PCs was detected in cortical collecting ducts (white arrows; n = 3 independent mice; Bars = 25 µm). (B) Cell fate analysis of Tfcp2l1 knockout IC cells using genetic reporter (Atp6b1-Cre;mTmG). We analyzed single GFP+ cells in Control (Atp6b1-Cre;Tfcp2l1f/+;mTmG) and in Knockout (Atp6b1-Cre;Tfcp2l1f/f;mTmG) collecting ducts by spot imaging. In control kidneys, GFP+ cells were ICs or PCs (e.g. GFP+ ICs: Atp6b1>Aqp2 and GFP+ PCs: Aqp2>Atp6b1), or expressed both markers in variable ratios (Atp6b1≈Aqp2). In contrast, in knockout kidneys, the majority of GFP+ cells appeared to be PC-like or double positives cell types (compare knockout with wild type profiles). (n = 20 GFP+ Control and n = 45 Tfcp2l1 deleted GFP+ cells from representative images; n = 4 independent kidneys).
Taken together, Tfcp2l1 was critical in the primary development of ‘double positive’ progenitors and the long-term maintenance of mature ICs which are otherwise capable of assuming a mixed or even a PC cell phenotype. In contrast, Tfcp2l1 was not likely to mediate IC development by regulating their survival, because we failed to find the apoptosis marker activated caspase 3 in any of our models (data not shown).
Identification of Tfcp2l1 target genes
To identify transcriptional targets of Tfcp2l1 that might explain IC regulation, we used an integrative approach that combined high-throughput data from two independent assays: (1) We found 843 differentially expressed genes in a comparison of EIIA-Cre;Tfcp2l1f/f knockout and Tfcp2l1f/f control P1 mouse kidneys (Affymetrix Microarrays analyzed by limma, n = 3, p<0.05). According to GUDMAP, the comprehensive genitourinary development database (Harding et al., 2011, www.gudmap.org), our analysis showed that 62% of downregulated genes were expressed either by UB, cortical or medullary collecting ducts or by the TALH including multiple subunits of the H+ATPase complex (Atp6v0d2; Atp6b1; Atp6v1c2) and IC specific Foxi1, Ca12, Ca2, Aqp6 and Oxgr1 (Figure 5A, Figure 5—figure supplements 1 and 2; Supplementary file 1); (2) We found 6564 Tfcp2l1-bound genomic regions in P1 mouse kidney using ChIP-Seq (Figure 5A; Supplementary file 2). Comparing the two datasets we found that Tfcp2l1 bound both up and down regulated genes at the TSS (Figure 5A,B). In fact, in a global analysis of all Tfcp2l1 peaks, 30% accumulated near the TSS within known promoters (Figure 5C). In addition these peaks demonstrated the established Tfcp2l1 motif (Supplementary file 4) (Chen et al., 2008). Moreover, cell type specific actions of Tfcp2l1 were suggested in a comparison with Tfcp2l1 ChIP-Seq from ES (Chen et al., 2008) and from Grhl2 ChIP-Seq in kidney (Werth et al., 2010) which revealed little overlap with Tfcp2l1 ChIP-Seq in kidney (Figure 5D). In contrast, a random set of genes that were not modulated by the Tfcp2l failed to show TSS enrichment (Figure 5B).
Figure 5 with 2 supplements see all
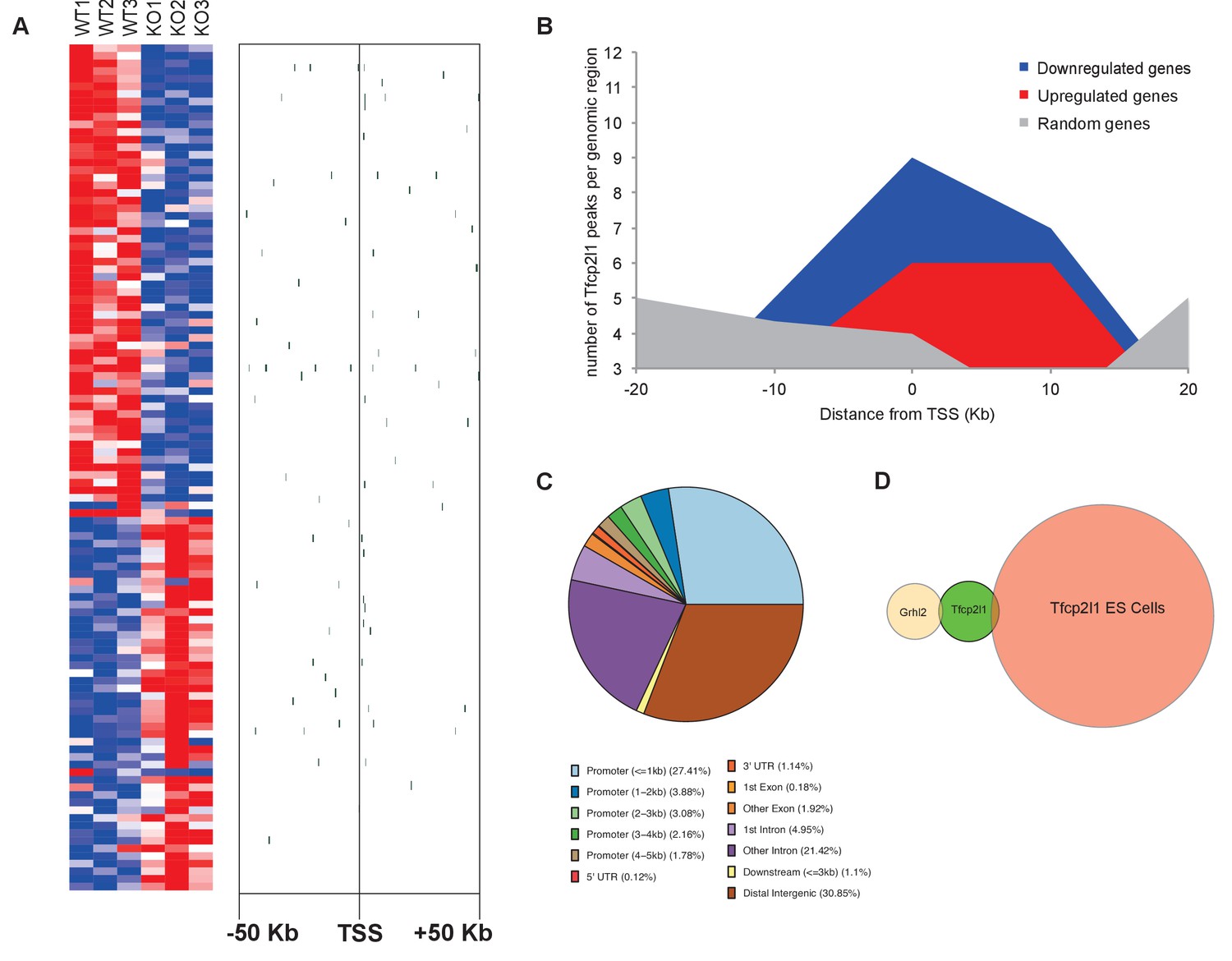
Identification of Tfcp2l1 targets.
(A) Identification of Tfcp2l1 targets. Integration of knockout and Tfcp2l1 ChIP-seq gene expression data obtained from P1 kidneys. Most of the genes significantly up or down regulated by Tfcp2l1 (WT-Tfcp2l1f/f vs KO-EIIACre;Tfcp2l1f/f) demonstrated binding peak(s) mapping between +50 KB to −50 KB relative to the TSS for each gene. (B) Tfcp2l1 peaks of both up and down regulated genes were enriched at the TSS in comparison with a random set of Tfcp2l1 independent genes. (C) Genome wide annotation of Tfcp2l1 peaks revealed that ~27% of peaks were within 1 kb of the TSS and 38% located within 10 kb from TSS. (D) Cell type specificity of Tfcp2l1 ChIP peaks. Comparison of Tfcp2l1 ChIP peaks in different models. P1 kidney (our study, Green) is compared with Tfcp2l1 peaks identified in ES Cells (Chen et al., 2008, Red) and with Grhl2 peaks identified in E18 kidney (Werth et al., 2010), Tan color). Note the limited overlap between these datasets.
The comparison between the two datasets also allowed us to identify the critical transcriptional targets of Tfcp2l1. Since Tfcp2l1 is known as an activator (To et al., 2010), we sought genes that were significantly downregulated in knockouts and contained a Tfcp2l1-DNA interaction site ±50 KB from the gene’s transcriptional start site. Using this approach, we found that Tfcp2l1 positively regulated expression of many IC genes including subunits B1 and D2 of the V-ATPase complex, Oxgr1, Ca12, Slc4a1, Aqp6 and IC-specific transcription factor Foxi1 (Figure 5, Figure 5—figure supplement 2). In contrast, no known PC markers were represented in the downregulated gene set.
It has also been reported that Tfcp2l1 is a repressor (Rodda et al., 2001). Consequently we analyzed genes that were upregulated in the knockouts and that demonstrated a Tfcp2l1-DNA interaction site ±50 KB from the gene’s TSS. None of these genes were associated with a particular function or a cell-type in the distal nephron.
Taken together, Tfcp2l1 directly controls genes that are critical for the function of IC cells (differentiation genes, V-ATPase, Oxgr1 etc).
Tfcp2l1 regulates Jag1 and Notch signaling in the collecting duct
The analysis of our two datasets demonstrated significant over-representation of components of the Notch pathway suggesting that Tfcp2l1 might regulate Notch signaling in ICs or in PCs.
We analyzed the Notch pathway using well characterized antibodies for Notch Ligands (Jag1, Jag2, Dll1, Dll4), Receptors (Notch1-4) and Notch Signaling States (‘ON’: characterized by detection of cleaved Notch and its target, nuclear Hes1, and ‘OFF’: characterized by detection of uncleaved Notch). We found that Jag1 marked the development of ICs: Jag1 and Atp6b1 co-localize with Ktr8 in IC-PC ‘double positive cells’ at E18 (Figure 6A) and in the adult Jag1 was specifically expressed by a subset of ICs called Pendrin+ β-ICs (Figure 6B). The localization of Jag1 was specific because, α-ICs and PCs were Jag1- (Figure 6C). In contrast, activated Notch1 (NICD; Val1744) and activated nuclear Hes1 were found in PCs (Jag1-,Atp6b1-,Aqp2+) immediately surrounding the Jag1+ β-ICs (Figure 7). Inactive, uncleaved Notch1 (extracellular domain) was detected in other locations such as the basal membrane of some ICs, identified as Krt8-, Jag1-α-ICs (Figure 8 and additional data, not shown). Therefore, the expression of Jag1 and Notch activity correlated with the development of cell types: PCs (‘Notch ON’: Jag1-, Cleaved Notch1), β-IC (‘Notch OFF’ Jag1+, Notch1-), and α-IC (‘Notch OFF’ Jag1-, Uncleaved Notch1).
Figure 6
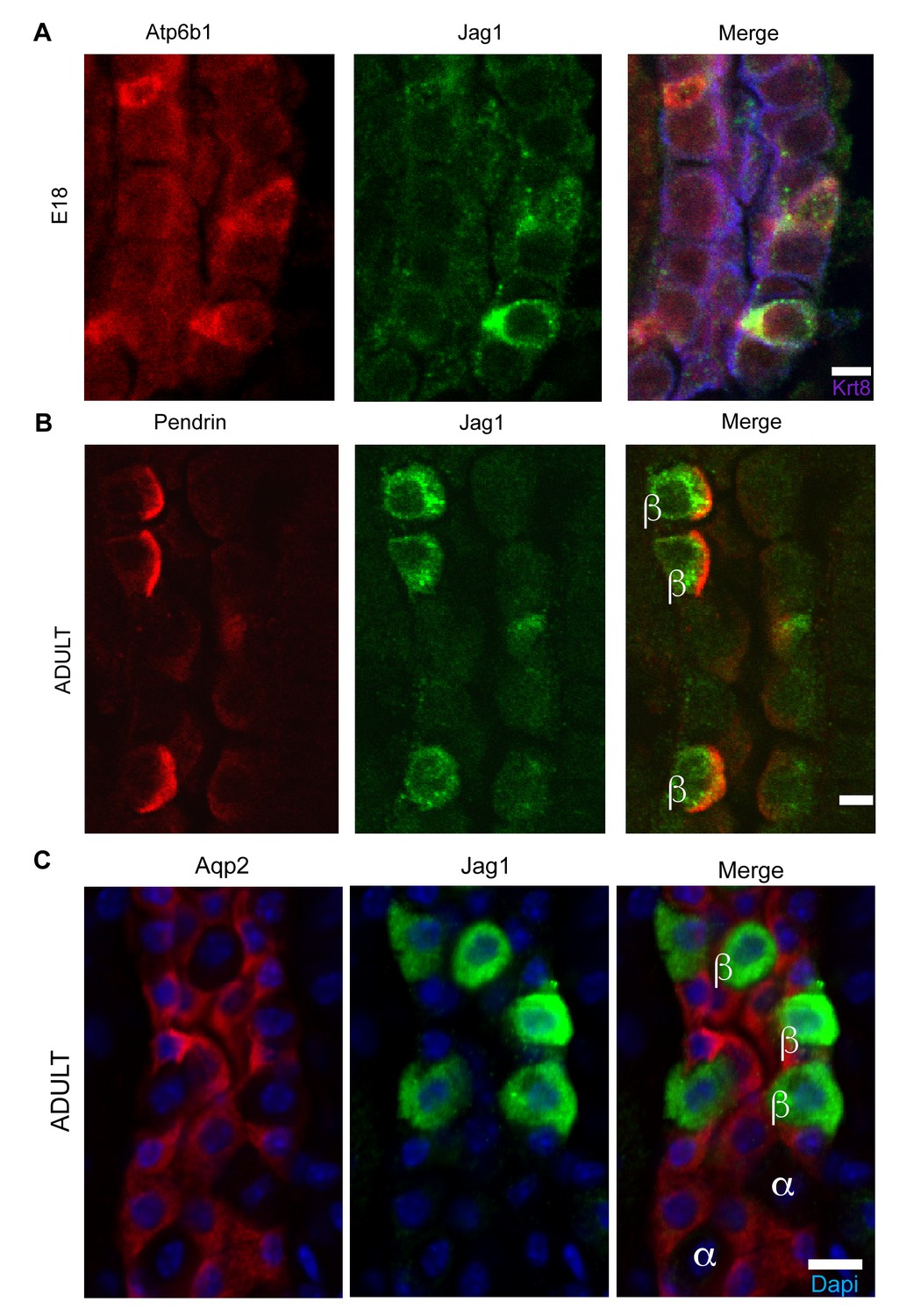
Jag1 is a novel early marker of developing (E18) and adult (P60) IC cells and activated Notch is found in PCs
(A) Jag1 (green) co-expressed with Atp6b1 (red) at the first appearance of ‘double positive’ cells. Krt8 (purple) is expressed by all cells at this stage. (B) In the adult kidney, Jag1 (green) is specifically expressed in a subset of IC cells called Pendrin+ β-ICs (red) (P60), but C not in other collecting duct cell types including Aqp2+ PC and Aqp2- α-ICs. Nuclei, blue (A,B) Bars = 5 µm. (C) Bar = 10 µm.
Figure 7
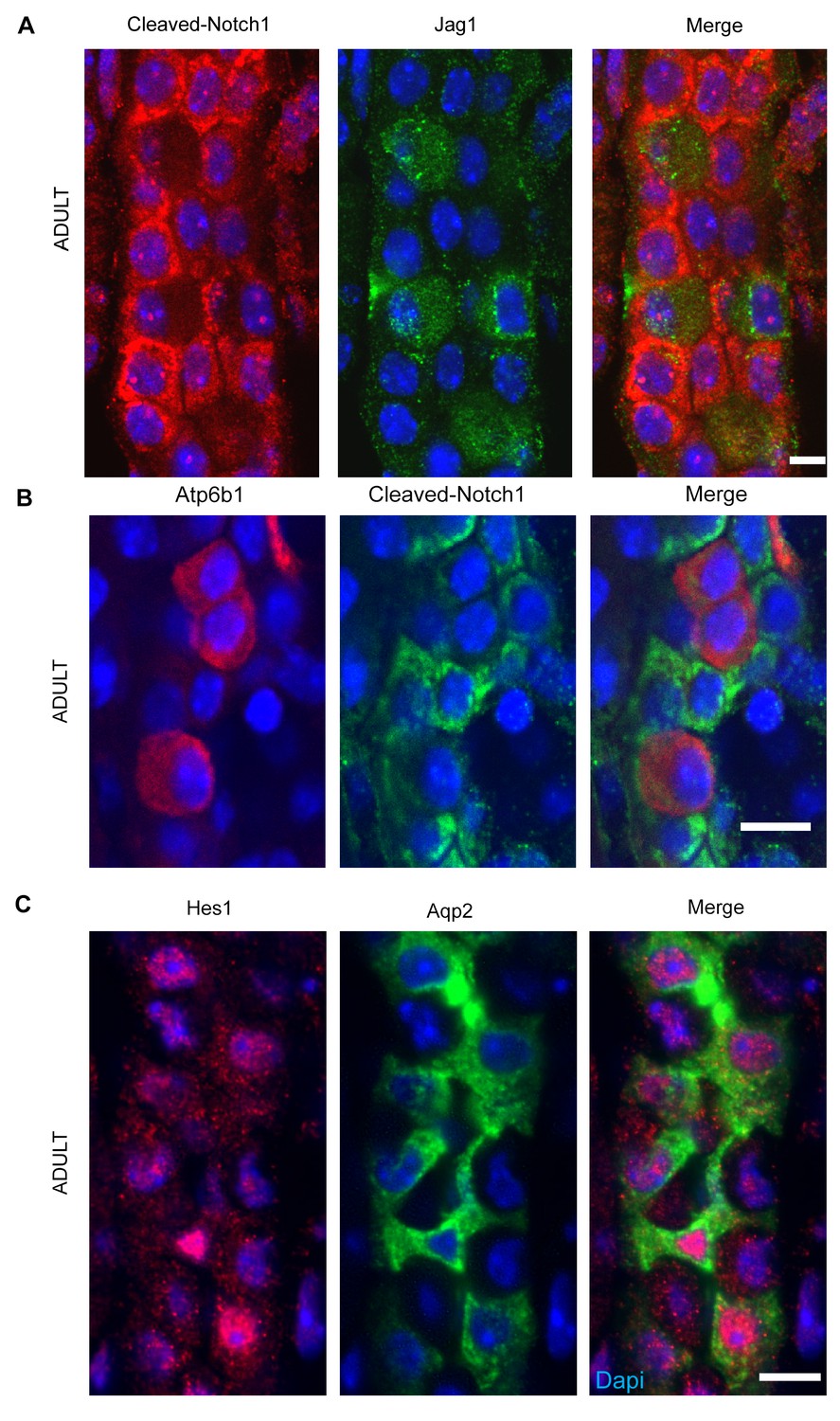
(A,B) Jag1-Notch1 signaling in the collecting duct. Activated Notch is found in PC cells adjacent to Jag1+Atp6b1+β-ICs. (C) Consistently, Notch target gene Hes1 (red) was detected in Aqp2+ PC (green). A, C Bar = 10 µm, B Bar = 5 µm.
Figure 8 with 1 supplement see all
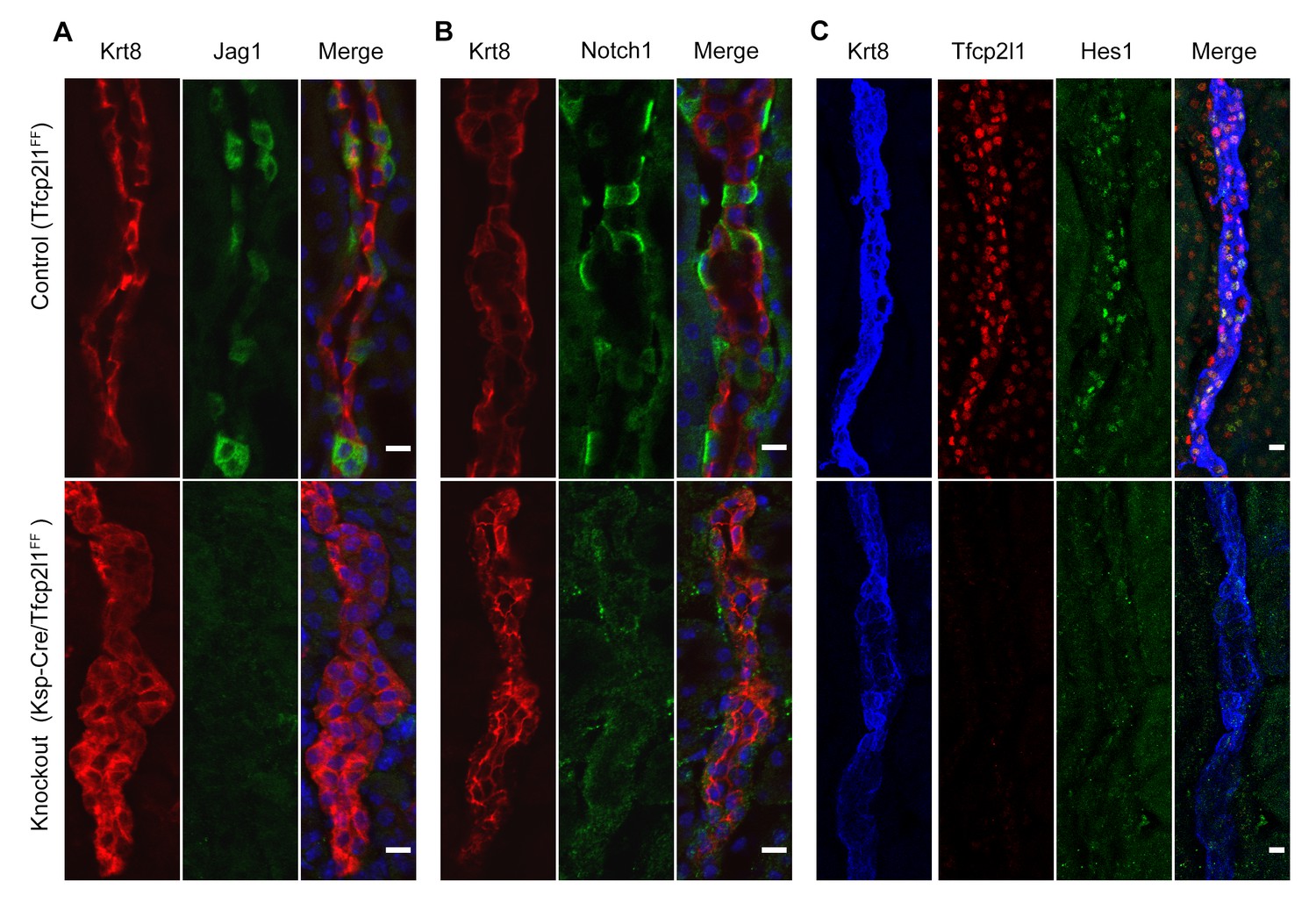
Tfcp2l1 is required for Notch signaling in collecting ducts.
Tfcp2l1 knockout (Cdh16-Cre;Tfcp2l1f/f) results in A depletion of Jag1 (green) from Krt8-β-ICs (red) B. depletion of cell surface (inactive) Notch1 (green) from Krt8- α-ICs (red) C depletion of nuclear Hes (green) from Krt8+ PCs. (n = 4 independent mice for each immunodetection; Bars = 10 µm).
To determine whether Tfcp2l1 regulated the distinctive patterning of Jag1 and Notch1 expression, we examined Cdh16-Cre;Tfcp2l1f/f kidneys and found nearly complete deletion of Jag1 in ICs and nuclear Hes1 in PCs. In addition, Notch staining was deleted in the Tfcp2l1 knockout collecting duct (Figure 8; n = 4 independent mice). These experiments suggested that Jag1 and Notch1 signaling were dependent on Tfcp2l1.
To determine whether Tfcp2l1 might directly control Notch signaling, we examined ChIP analysis and found that Tfcp2l1 directly bound the promoter of 18 Notch associated genes including Jag1 (-904, -510), Hes1 (-56202, -1823, 50028), Hey1 (-127494, -336, 138049) and Hey2 (-212) (Supplementary file 3). To broaden this analysis, we examined Tfcp2l1-bound genomic regions for the enrichment of transcription factor motifs adjacent to the Tfcp2l1 binding site (PWMErich using top 1000 Tfcp2l1-bound regions, Supplementary file 4). We found significant enrichment of motifs known to be targets of Notch signaling within ±20 bp from the Tfcp2l1 peak center, including Hes- and Tfap-, Atoh-, and Tcf-families. We confirmed that promoters of IC-specific genes bound Tfcp2l1, Hes1 and Foxi1 by using Tfcp2l1 ChIP followed by Hes1 or Foxi1 ChIP (Figure 8—figure supplement 1). Considering that Hes1 is a well-known repressor, these observations suggest a functional feedback loop from the Notch pathway to a subset of genes regulated by Tfcp2l1, potentially explaining why some IC genes are expressed at low levels in Tfcp2l1+Hes1+ PCs.
Jag1 is required for cellular patterning and differentiation of the collecting ducts
To analyze whether Notch signaling regulates the patterning of IC and PC proteins, we studied a conditional deletion of the Notch interacting domain exon 4 of Jag-1 (Kiernan et al., 2006). We deleted Jag1 with either Cdh16-Cre or Atp6b1-Cre-drivers and the deletions were confirmed at P60 by immunostaining (Figure 9—figure supplement 1). Cdh16-Cre;Jag1f/f did not phenocopy Cdh16-Cre;Tfcp2l1f/f; in fact, ICs signature proteins were not only preserved in the Jag1 knockout but were now widely co-expressed with PC proteins (e.g. Tfcp2l1+, Atp6b1+ with Aqp2+ or Tfcp2l1+, Atp6b1+ with Krt8+ or Tfcp2l1+, Foxi1+ with Aqp2+; Figure 9; Figure 9—figure supplement 2; n = 4 independent mice). Tubular structural failure was also found: tubules contained luminal debris (Figure 9—figure supplement 2) and collecting duct diameter was enlarged 28% in Cdh16-Cre;Jag1f/f (Aqp4 staining facilitated the measurements: 30.1 ± 3.5 µm vs 23.5 ± 2 µm, SEM, n = 4 independent mice, p=2×10−14, Figure 9—figure supplement 3). Krt8 was downregulated in these tubules, perhaps contributing to the structural failure. At later stages (P90) we observed hydronephrosis (3/4 mice), similar to another collecting duct-specific Notch-inactivation model (Jeong et al., 2009). These data suggest that while expression of IC proteins are ‘upstream’ of Jag1, their relative level in specific cells of the collecting duct is controlled by Jag1 signaling.
Figure 9 with 4 supplements see all
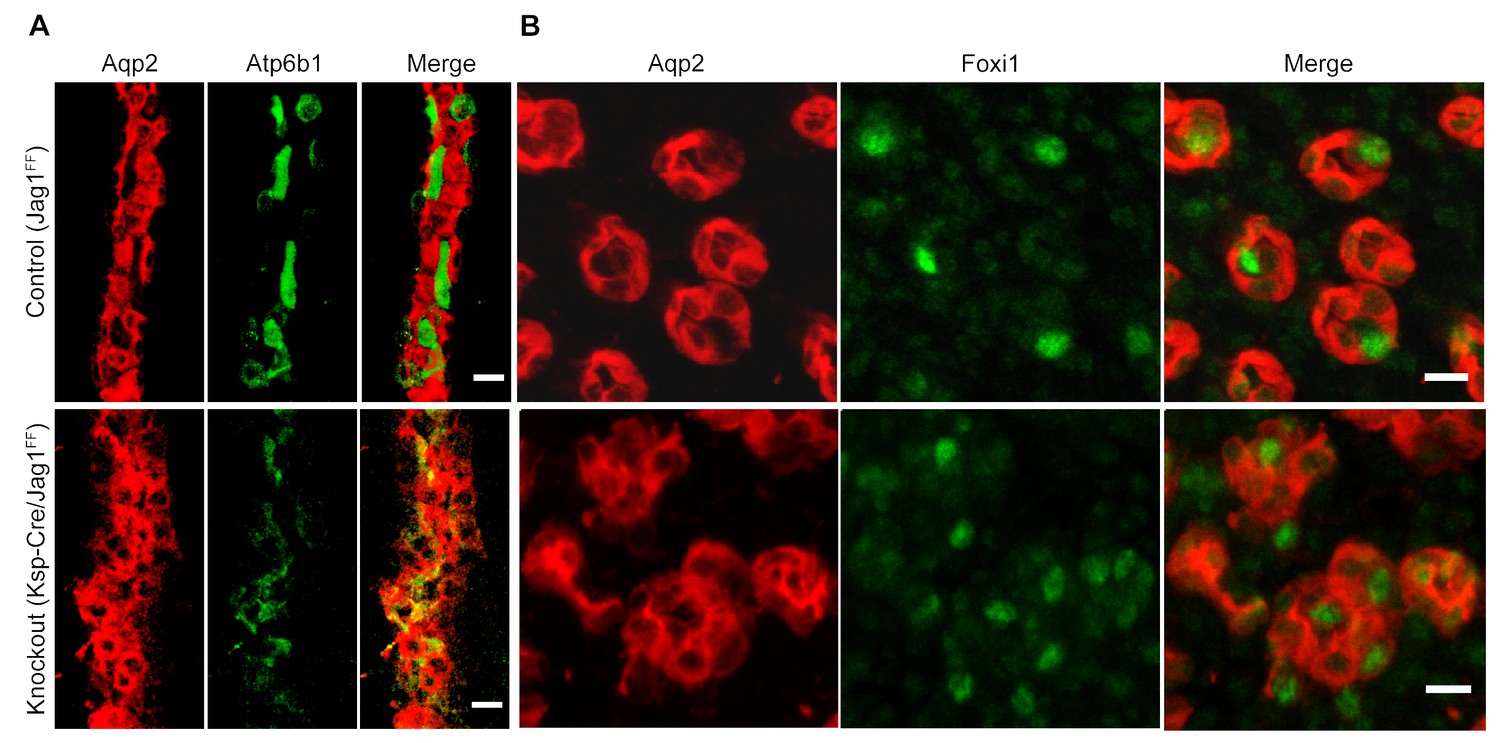
Jag1 regulates the distribution of IC proteins in the collecting ducts.
Knockout of Jag1 (Jag1f/f;Cdh16-Cre) resulted in diffuse expression of IC proteins A Atp6b1 and B Foxi1. Expression of these proteins overlapped with Aqp2 creating ‘double positive’ cells. (n = 4 independent mice; Bars = 10 µm).
To dissect the role of Jag-Notch in collecting cell development at higher resolution, we generated an IC Atp6b1-Cre;Jag1f/f;mTmG mouse and analyzed the GFP-labeled knockout cells. We found an increase in ‘double positive cells’ (from 2% to 12%, p=0.004) and a decrease in Aqp2+ PC cells (24% to 12%; p=4.7E−06; n = 3 independent mice; Figure 9—figure supplement 4). Similar data were obtained when we acutely interrupted Notch signaling by treating explanted E15 mouse kidneys with the gamma-secretase inhibitor Compound E (100 nM, 48 hr, n = 6 independent cultures of mouse kidneys). The treatment generated striking ‘double positive’ cells at the tips of the Ureteric Bud (Foxi1+, Atp6b1+ and Krt8+; Figure 10). In sum, inactivation of Jag1 disrupted IC and PC identity and arrested cells expressing inappropriate levels of IC and PC proteins.
Figure 10

Manipulation of Notch signaling in vivo.
(A) Inhibition of Notch signaling upregulates Foxi1 and promotes IC cell differentiation. Acute inhibition of Notch signaling in E15 kidneys with Compound E (48 hr) resulted in the differentiation of IC cells at the tips of the UB/Collecting Ducts. Note that the IC cells demonstrated a ‘double-positive’ phenotype with the co-expression of PC (Krt8+ blue) and IC (Foxi1+ red, Atp6b1+ green) proteins. (B) High power of Compound E treated kidneys (n = 6 independent mouse kidney cultures). (A) Bars = 50 µm and (B) Bar = 10 µm.
Discussion
While most segments of the kidney have one type of epithelia, multiple cell types are found in the collecting ducts including a diverse population of ICs, PCs and ‘double-positive’ progenitor cells. While these cells differ in structure, gene expression and function, they cooperate to perform physiologic functions. For example, Na+ absorption by PCs via ENaC stimulates H+ excretion by ICs, and conversely the blockade of Na+ absorption in PCs by amiloride or by mutation of a subunit of the epithelial sodium channel results in the suppression of H+ secretion creating an ‘electrogenic’ renal tubular acidosis (Chang et al., 1996; DuBose and Caflisch, 1985). Conversely, the deletion of bicarbonate transporters in ICs reduces the expression of Na+ channels in PCs (Pech et al., 2010). The physical interactions between ICs and PCs not only promotes functional coupling of these cells by trans-epithelial gradients, but additionally permit juxtacrine signaling which regulates acid-base excretion (e.g. CXCL12::CXCR4 (Schwartz et al., 2015).
ICs and PCs may serve opposing but linked functions such as K+ excretion (PC) and K+ absorption (IC) (Park et al., 2012; Kim et al., 2016). They may also serve independent roles which cannot be mutually compensated. For example, the decrease in PCs after Li treatment results in polyuria (Christensen et al., 2004) since PCs regulate water balance, while the decrease in ICs by Tfcp2l1 deletion enhances kidney infection since ICs mediate urinary acidification and the production of a number of antimicrobials including Lcn2/NGAL (Shohl and Janney, 1917; Paragas et al., 2014). In sum, appropriate numbers of PCs and ICs are required for both physiologically linked and physiologically independent functions. Here we suggest that the integration of IC and PC populations is mediated by Tfcp2l1 and Jag1 and is reflected in their ‘rosette’ like architecture (e.g. Figure 3; Figure 3—figure supplement 2B).
Tfcp2l1 and Jag1 act on a primordial cell type which appears in the collecting duct between E15-E18 and expresses both IC and PC proteins (Figure 2; Figure 2—figure supplements 1 and 2). Descendants of these cells (tagged by HoxB7-Cre or Atp6b1-Cre;mTmG; Figure 2B) generated distinct ICs and PCs, yet even as canonical adult cells, they continued to express low levels of each other’s signature proteins (Figure 2—figure supplement 2; Figure 4B), and remain plastic enough to change their phenotypes from PC to IC or IC to PC in vitro (Fejes-Tóth and Náray-Fejes-Tóth, 1992) and in vivo (Park et al., 2012; Trepiccione et al., 2013). In this setting the deletion of either Tfcp2l1 (after it had already induced Atp6b1, Figure 4) or Jag1 (Figure 9) demonstrated that cells initially destined to become ICs can fail to achieve their adult phenotype and demonstrate ‘double positive’ or even PC characteristics. In sum, the most likely explanation for our data is that collecting duct cells have the potential to choose either IC or PC fate and that the canonical cell types probably represent the extremes of a spectrum regulated by Tfcp2l1 and Jag1.
Tfcp2l1 appears to act on the IC-PC progenitors by both cell autonomous and cell non-autonomous mechanisms. Its cell autonomous activities were demonstrated both by an interaction of Tcfcp2l1 with IC specific genes (Figure 5; Figure 5—figure supplements 1 and 2) as well as loss of their expression in the Tfcp2l1 knockout (Figure 3; Figure 3—figure supplement 2; Figure 4). These cell autonomous activities may explain the absence of all stages of IC development in a cell population destined to express PC-like characteristics. These data are reminiscent of the deletion of Foxi1 (Blomqvist et al., 2004) which lies downstream of Tfcp2l1.
The cell non-autonomous role of Tfcp2l1 was evidenced by a direct interaction of Tfcp2l1 and the Jag1 promoter (quantified in Supplemental Table 3) and by the loss of Jag1 expression in Tfcp2l1 knockouts (Figure 8). Hence, Tfcp2l1 induced Jag1 which we propose mediates cell-non-autonomous signaling from Jag1+ICs to Notch1+ PCs.
Notch signaling is known to control cell fate decisions and to pattern many different organs through lateral-inhibition (Afelik and Jensen, 2013; Martinez Arias et al., 2002). In an initially homogeneous cell population, some cells start to express more Notch ligand which activates Notch expressing neighboring cells. Notch activation downregulates the ligand, which in turn leads to reduced Notch signaling in the ligand expressing neighbor, locking the two neighboring cells into a Notch OFF-state (ligand-expressing) or a Notch ON-state (Notch expressing). Notch activation initiates gamma-secretase-mediated cleavage of Notch-receptor permitting the Notch intracellular domain (NICD) to translocate to the nucleus where it acts as a transcriptional co-activator with RBPJ. NICD-RBPJ complex regulates a set of BHLH transcription factors including Hey1, Hey2, Hes1, Hes5.
Notch-mediated generation of multiple cell types has been previously identified in the zebrafish pronephric duct (Liu et al., 2007; Ma and Jiang, 2007) and in the Drosophila Malpighian tubule (Wan et al., 2000) which are both phylogenetically related to the collecting duct of the mammalian nephron, as well as in functionally related cells in the frog skin (Quigley et al., 2011). Many components of the Notch signaling pathway were expressed in the embryonic mammalian collecting duct. Before E18 there was faint expression of Jag1 and Notch1 throughout the collecting duct (data not shown), but by E18 there was clear excess expression of Jag1 in a subset of duct cells which displayed IC and PC proteins. Surrounding these IC progenitors were PCs with activated nuclear Hes1, a pattern which became distinct after birth (Figure 6; Figure 7). In sum, the classical lateral-inhibition paradigm of Notch signaling appears to be relevant to patterning collecting duct cell types.
‘Notch-ON’ signaling may be relevant to cell fate choices because while Hes1 targets are not known in detail, TF binding site overrepresentation analysis (PWMEnrich; Thomas-Chollier et al., 2011) of Tfcp2l1 bound peaks demonstrated neighboring Tfcp2l1 and Hes1 binding sites, as well as combinatorial binding of Tfcp2l1-Foxi1-Hes1 (Figure 8—figure supplement 1) raising the possibility that Hes1 signaling in PCs may negatively modulate Tfcp2l1 and Foxi1 targets. Hes1 might repress IC genes in PC cells by enhancing chromatin modification enzymes since the inactivation of histone methyltransferase Dot1l appeared to convert PCs into ICs (Xiao et al., 2016). In sum, Notch signaling might antagonize Tfcp2l1 in PC cells where Notch activates Hes1.
‘Notch-OFF’ in IC cells may result from the basolateral localization of Notch (perhaps making it sterically inaccessible to ligands), or perhaps as a result of the expression of NUMB in some ICs (not shown; McGill et al., 2009). The ‘Notch-OFF’ state is likely to be critical for IC function because it may link Tfcp2l1 to Foxi1. While Tfcp2l1 might stimulate the expression of Foxi1 directly (Tfcp2l1 bound Foxi1 at +50 kb from the TSS), the upregulation of Foxi1 appeared to be a direct consequence of Notch-OFF because activated Notch signaling negatively regulated Foxi1 (Guo et al., 2015) and conversely Notch inactivation models (Jag1 knockout and pharmacologic inhibition) generated diffuse expression of Foxi1 (Figure 9, Figure 10). In addition, we found that Foxi1 bound to the Jag1 promoter (Figure 8—figure supplement 1), consistent with observations made in the endolymphatic duct cells of Foxi1 knockouts where Jag1 was absent (Hulander et al., 2003). Hence, while Tfcp2l1 and Foxi1 may act independently and synergistically to upregulate the IC phenotype (Blomqvist et al., 2004; Vidarsson et al., 2009), we expect that a positive regulatory loop between Foxi1 and Jag1 maintains high levels of Jag1 expression in ICs, but conversely inhibits Foxi1 in neighboring PCs. Hence, the induction of Jag1 by Tfcp2l1 suppressed IC gene expression in PCs.
Our data provide a developmental explanation for experiments demonstrating that the balance of cell types in the adult collecting duct can be modified by Notch signaling. For example, conditional inactivation of Mib1 (Jeong et al., 2009; Nam et al., 2015), a E3 ubiquitin-ligase that positively regulates Notch signaling by regulating Notch ligands, resulted in more ICs as did the inactivation of a cell surface protease, Adam10 (Guo et al., 2015), which regulates the cleavage and activation of Notch proteins. Conversely, overexpression of Notch intracellular domain ICD directed collecting duct cells towards a PC fate (Jeong et al., 2009). While these data were observed in the adult, we suggest that each of these experimental manipulations may have acted at the embryonic IC-PC ‘double positive’ stage. In other words, rather than a binary choices between canonical IC and PC phenotypes, a large body of literature may be explained by the notion that the manipulation of Notch signaling caused a failure to choose among competing programs of gene expression, producing the ‘double positive’ phenotype.
Our model has subtle differences with classical demonstrations of Notch signaling. In other developing organs, including the inner ear and intestine, classical negative feedback loops act in a paracrine manner between neighboring cells to inhibit the genes which induced the Notch ligands (e.g. Ngn1 induces expression of Dll1, but Notch activation inhibits Ngn1; Atoh1 activates Dll1/4, but Dll1/4 suppresses Atoh1 [Morrison et al., 1999; Kazanjian et al., 2010]). In contrast, Tfcp2l1 expression was inhibited only by two fold in Hes1+ PCs (IC>PC). While this differential expression might contribute to cell speciation by driving higher levels of Jag1 expression in ICs, we note that Foxi1, which is downstream of Tfcp2l1, is not only critical for Jag1 expression, but it is reciprocally inhibited by Notch signaling (Figures 9 and 10). Consequently, bothTfcp2l1 and Foxi1, rather than either alone, may mediate the Notch signaling.
A second distinction with the classical models was the response of PCs to the loss of Hes1. In other developing organs, loss of Hes1 signaling resulted in a failure of tissue development (e.g. spinous skin cells; Blanpain et al., 2006). In contrast, Tfcp2l1 deletion and the failure of Hes1 signaling did not block PC development, indicating that Hes1 may play an inhibitory role in the collecting duct by preventing IC development in cells otherwise competent to become PCs. Rather than Hes1, additional Notch targets such as Elf5 may mediate Notch dependent PC development (Grassmeyer et al., 2017).
The plasticity of collecting duct cells was identified in the adult (Schwartz et al., 1985); here we show that this plasticity reflects a developmental program that may remain active long after birth and is likely to permit adaptations to environmental cues. Tfcp2l1 was found in a screen for transcription factors which induce epithelialization (Barasch et al., 1999; Schmidt-Ott et al., 2007); Werth and Leete, Unpublished). However its key role lays in regulating the plasticity of collecting duct progenitors (HoxB7+, Atp6b1+, Jag1+ cells; Model Figure 11). Tfcp2l1 provides essential directions for the initiation and refinement of this cell type via cell autonomous and non-cell autonomous mechanisms. As a consequence, it regulates the cellular diversity, which is the principal feature that distinguishes the collecting duct from other segments of the nephron.
Figure 11
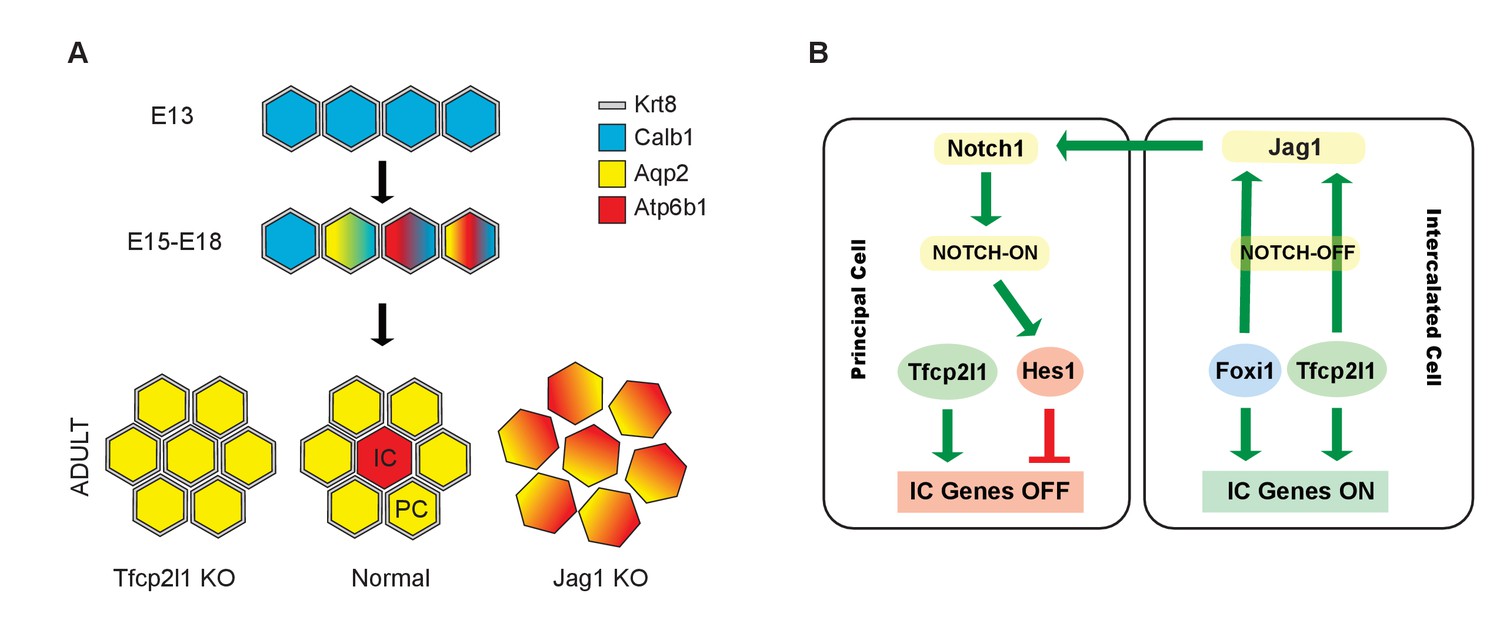
Models.
(A) Development of cellular diversity in the collecting duct. Initially, we found a monotonous expression of PC proteins at E13, and then a transitional stage (E15–E18) characterized by the appearance of ‘double-positive’ cells. After birth, these cells achieved distinct identities and assumed rosette-like patterning. Deletion of Tfcp2l1 resulted in a monotonic cell type expressing PC cell, but not IC cell markers. Inactivation of Jag1 in contrast resulted in the loss of cell identity and patterning, increasing the number of ‘double-positive’ cells typified by E18 collecting ducts. (B) Proposed model of cell-autonomous and non-cell-autonomous actions of Tfcp2l1. Tfcp2l1 induces the expression of IC genes, including Jag1. Expression of Jag1 triggers Notch signaling in adjacent PC cells. Jag1 signaling results in activation of Hes1 (Notch-ON) in PCs and conversely Foxi1 expression in ICs (Notch-OFF). We envision that the combination of Tfcp2l1 with either Foxi1 or Hes1 drives cell identity. Maintenance of this circuit may depend on an excess of Tfcp2l1 in ICs (Figure 1) as well as the expression of Foxi1 which is known to induce Jag1. Conversely, Jag1 suppression of Foxi1 (Figures 8 and 9) demonstrates a negative feedback loop in neighboring PCs.
Materials and methods
Animal models Tfcp2l1 conditional knockouts were generated by flanking exons 3, 4 and 5 (~1.8 kb) with LoxP sites. These exons included the CP2 conserved functional domain and their deletion created an open reading frame shift. The targeting construct was generated by using a modified NCI BAC recombineering protocol. Briefly, mouse (C57) Tfcp2l1 BAC clone (RP24-291G6, CHORI, Oakland, California) was recombineered using Loxp-Neomycin-Loxp (LNL) cassette with homologous arms upstream of exon 3 and Frt-Neomycin-Frt-Loxp (FNFL) cassette downstream of exon5. The Tfcp2l1 -targeting BAC DNA was electroporated into mouse KV1 ES cells (generated by Columbia University Transgenic Facility), followed by neomycin selection. Neomycin-resistant ES clones were screened using PCR and Southern Blots and selected clones were microinjected into C57B6 blastocysts to generate chimeras. The Tfcp2l1 chimeric mice were subsequently mated with EIIa-flpe C57B6 female mice for both germline transmission and flpe-based removal of the Frt-floxed neomycin cassette, which generated F1 Tfcp2l1-floxed mice. Cre mediated deletion was confirmed at the genomic level using PCR, at the RNA level using quantitative Real-Time PCR and at protein level using immunohistochemistry. Tfcp2l1 knockout and wild type kidneys were assayed using Affymetrix Mouse Genome 430.2 microarrays.
EIIA-Cre (RRID:IMSR_JAX:003724), Cdh16-Cre (RRID:IMSR_JAX:012237) and Jag1f/f (RRID:IMSR_JAX:010618) were purchased from the Jackson Labs, Rosa26mTmG (RRID:IMSR_JAX:007576) was kindly provided by C Mendelsohn and Atp6b1-Cre was a gift from R Nelson, Utah. Rosa26mTmG genetic reporters express membrane-bound green fluorescein protein (GFP) upon Cre-mediated recombination (Muzumdar et al., 2007). GFP was detected with anti-GFP post-antigen retrieval. All rodents experiments were approved by Columbia University IACUC.
Chromatin immunoprecipitation
Request a detailed protocolAs described (Werth et al., 2010), P1 mouse kidneys were cut into pieces and then sieved to single cells in DMEM. Chromatin was crosslinked and fragmented for 15 min (30 s stroke/30 s pause) using a Fisher Sonicator 450 (power 8; on 10 s and off 20 s for 6 min) to a fragment size of 100–500 bp. DNA concentration was measured by NanoDrop and then subjected to Tfcp2l1 ChIP with anti-human Tfcp2l1 (RRID:AB_2202564) or goat IgG control (Jackson Immunoresearch). The ChIP antibody was extensively validated in ChIP-PCR assays. In brief, we used conservative Tfcp2l1 binding site at Krt7 promoter to validate the performance of the Tfcp2l1 antibody in a ChIP PCR assay. A ChIP Library was prepared according to the Illumina ChiPseq library preparation kit using size selection (150–250 bp). The library was sequenced on Illumina Genome Analyzer IIx (San Diego, California). Sequences were aligned to mm9 genome assembly using Bowtie (Langmead et al., 2009) and binding sites were detected using MACS (Zhang et al., 2008). We used GREAT (McLean et al., 2010) for gene association/enrichment analysis. For motif analysis we used PWMEnrich (http://www.bioconductor.org/packages/release/bioc/html/PWMEnrich.html)
RNA extraction and cDNA synthesis, Affymetrix Arrays
Request a detailed protocolWe isolated RNA using Qiagen RNAeasy micro kit, analyzed the isolate by Bioanalyzer (Agilent, Santa Clara, California), and synthesized cDNA using kits from Applied Biosystems. Preparation of templates and hybridization on Affymetrix Arrays was performed at the Columbia Genome Center. Differential gene expression was identified using R Limma (https://bioconductor.org/packages/limma). Induced genes were validated by QPCR using Sybr Green (Applied Biosystems, Forster City, California) and relative levels of mRNA expression were normalized to β-actin mRNA. Primer pairs were designed using Primer3 software (Whitehead Institute, MIT) and validated by product size (Supplementary file 5). P-values cut-offs (0.05 and 0.01) were chosen to generate datasets of comparable sizes.
Our data has been deposited with GEO, Accession Number GSE87769 (and Super Series: Accession Number GSE85325, GSE87744, GSE87752). Tfcp2l1 ChIP-Seq data from ES cells was obtained from GEO, Accession Number GSE11431. Grhl2 ChIP-Seq data from the kidney was obtained from GEO, Accession Number GSE24295.
In situ hybridization and immunocytochemistry
Request a detailed protocolTissues were prepared by intracardial perfusion with PBS followed by 1% PFA/PBS, 30% Sucrose/PBS, OCT (Tissue-Tek) and cryosectioned at 50 µm. Blocking and staining was performed in PBS/3% BSA/0.2% Triton X-100 solution. Controls and experimental samples were always placed on the same slide, stained in parallel and imaged with the same parameters using confocal Zeiss LSM510 Confocal Microscope (Columbia Core Facilities). We used confocal microscopy to reconstruct whole cells from the image stacks and estimated the amount of protein per cell by measuring total fluorescent signal. Antibodies are listed in Supplementary file 5. Data were analyzed using Fiji (ImageJ). Digoxigenin-labeled antisense riboprobes were generated from cDNAs using reverse primers containing a 5’ T7 polymerase promoter sequence (Supplementary file 5; Schmidt-Ott et al., 2005).
Data availability
-
Identification of Tfcp2l1 target genes in the mouse kidneyPublicly available at the NCBI Gene Expression Omnibus (accession no: GSE87769).
-
Tfcp2l1 controls cellular patterning of the collecting duct.Publicly available at the NCBI Gene Expression Omnibus (accession no: GSE85325).
-
RNA-Seq Gene Expression Analysis in Rat Metanephric Mesenchyme overexpressing Transcription Factor Tfcp2l1Publicly available at the NCBI Gene Expression Omnibus (accession no: GSE87744).
-
Genome wide map of Tfcp2l1 binding sites from mouse kidneyPublicly available at the NCBI Gene Expression Omnibus (accession no: GSE87752).
-
Mapping of transcription factor binding sites in mouse embryonic stem cellsPublicly available at the NCBI Gene Expression Omnibus (accession no: GSE11431).
-
Gene expression in epithelial and non-epithelial cells of renal originPublicly available at the NCBI Gene Expression Omnibus (accession no: GSE24295).
References
-
Notch signaling in the pancreas: patterning and cell fate specificationWiley Interdisciplinary Reviews: Developmental Biology 2:531–544.https://doi.org/10.1002/wdev.99
-
Transitional differentiation patterns of principal and intercalated cells during renal collecting duct developmentEpithelial Cell Biology 4:121–130.
-
A fork in the road of cell differentiation in the kidney tubuleJournal of Clinical Investigation 113:1528–1530.https://doi.org/10.1172/JCI22029
-
A Grainyhead-Like 2/Ovo-Like 2 pathway regulates renal epithelial barrier function and lumen expansionJournal of the American Society of Nephrology 26:2704–2715.https://doi.org/10.1681/ASN.2014080759
-
Remodeling the cellular profile of collecting ducts by chronic carbonic anhydrase inhibitionAmerican Journal of Physiology. Renal Physiology 280:F437–448.
-
Canonical notch signaling functions as a commitment switch in the epidermal lineageGenes & Development 20:3022–3035.https://doi.org/10.1101/gad.1477606
-
Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1Journal of Clinical Investigation 113:1560–1570.https://doi.org/10.1172/JCI20665
-
The proton translocating ATPase responsible for urinary acidificationThe Journal of Biological Chemistry 257:9230–9233.
-
Integrated compensatory network is activated in the absence of NCC phosphorylationJournal of Clinical Investigation 125:2136–2150.https://doi.org/10.1172/JCI78558
-
Adam10 mediates the choice between principal cells and intercalated cells in the kidneyJournal of the American Society of Nephrology 26:149–159.https://doi.org/10.1681/ASN.2013070764
-
The GUDMAP database--an online resource for genitourinary researchDevelopment 138:2845–2853.https://doi.org/10.1242/dev.063594
-
Inactivation of notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in miceJournal of Clinical Investigation 119:3290–3300.https://doi.org/10.1172/JCI38416
-
Notch signaling during cell fate determination in the inner earSeminars in Cell & Developmental Biology 24:470–479.https://doi.org/10.1016/j.semcdb.2013.04.002
-
Aquaporin 2-labeled cells differentiate to intercalated cells in response to potassium depletionHistochemistry and Cell Biology 145:17–24.https://doi.org/10.1007/s00418-015-1372-9
-
Mammalian kidney development: principles, progress, and projectionsCold Spring Harbor Perspectives in Biology 4:a008300.https://doi.org/10.1101/cshperspect.a008300
-
CSL-independent notch signalling: a checkpoint in cell fate decisions during development?Current Opinion in Genetics & Development 12:524–533.https://doi.org/10.1016/S0959-437X(02)00336-2
-
Numb regulates post-endocytic trafficking and degradation of Notch1Journal of Biological Chemistry 284:26427–26438.https://doi.org/10.1074/jbc.M109.014845
-
GREAT improves functional interpretation of cis-regulatory regionsNature Biotechnology 28:495–501.https://doi.org/10.1038/nbt.1630
-
Expression of Delta1 and Serrate1 (Jagged1) in the mouse inner earMechanisms of Development 84:169–172.https://doi.org/10.1016/S0925-4773(99)00066-0
-
Altered response of pendrin-positive intercalated cells in the kidney of Hoxb7-Cre;Mib1f/f miceHistology and Histopathology 30:751–762.https://doi.org/10.14670/HH-30.751
-
Notch in the intestine: regulation of homeostasis and pathogenesisAnnual Review of Physiology 75:263–288.https://doi.org/10.1146/annurev-physiol-030212-183741
-
α-Intercalated cells defend the urinary system from bacterial infectionJournal of Clinical Investigation 124:2963–2976.https://doi.org/10.1172/JCI71630
-
Proposed mechanism in the change of cellular composition in the outer medullary collecting duct during potassium homeostasisHistology and Histopathology 27:1559–1577.https://doi.org/10.14670/HH-27.1559
-
Pendrin modulates ENaC function by changing luminal HCO3-Journal of the American Society of Nephrology 21:1928–1941.https://doi.org/10.1681/ASN.2009121257
-
CRTR-1, a developmentally regulated transcriptional repressor related to the CP2 family of transcription factorsJournal of Biological Chemistry 276:3324–3332.https://doi.org/10.1074/jbc.M008167200
-
Water channels in the kidney collecting ductKidney International 48:1082–1087.https://doi.org/10.1038/ki.1995.391
-
Novel regulators of kidney development from the tips of the ureteric budJournal of the American Society of Nephrology 16:1993–2002.https://doi.org/10.1681/ASN.2004121127
-
SDF1 induction by acidosis from principal cells regulates intercalated cell subtype distributionJournal of Clinical Investigation 125:4365–4374.https://doi.org/10.1172/JCI80225
-
Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tractJournal of the American Society of Nephrology 13:1837–1846.https://doi.org/10.1097/01.ASN.0000016444.90348.50
-
The growth of Bacillus coli in urine at varying hydrogen ion concentrationsThe Journal of Urology 1:211–229.
-
RSAT 2011: regulatory sequence analysis toolsNucleic Acids Research 39:W86–W91.https://doi.org/10.1093/nar/gkr377
-
A fate-mapping approach reveals the composite origin of the connecting tubule and alerts on "single-cell"-specific KO model of the distal nephronAmerican Journal of Physiology - Renal Physiology 311:F901–F906.https://doi.org/10.1152/ajprenal.00286.2016
-
Aqp2-expressing cells give rise to renal intercalated cellsJournal of the American Society of Nephrology 24:243–252.https://doi.org/10.1681/ASN.2012080866
-
Dot1l deficiency leads to increased intercalated cells and upregulation of V-ATPase B1 in miceExperimental Cell Research 344:167–175.https://doi.org/10.1016/j.yexcr.2015.09.014
-
Model-based analysis of ChIP-Seq (MACS)Genome Biology 9:R137.https://doi.org/10.1186/gb-2008-9-9-r137
Article and author information
Author details
Funding
National Institutes of Health (RO1DK073462)
- Jonathan Barasch
March of Dimes Foundation (Research Grant)
- Jonathan Barasch
National Institutes of Health (RO1DK092684)
- Jonathan Barasch
National Institutes of Health (U54DK104309)
- Jonathan Barasch
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Ethics
Animal experimentation: All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Columbia. Protocol # AC-AAAH7404.
Copyright
© 2017, Werth et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,535
- views
-
- 559
- downloads
-
- 75
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 75
- citations for umbrella DOI https://doi.org/10.7554/eLife.24265
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Transcription factor TFCP2L1 patterns cells in the mouse kidney collecting ducts
eLife 6:e24265.
https://doi.org/10.7554/eLife.24265




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}