IRF4 haploinsufficiency in a family with Whipple’s disease
- INSERM U1163, France
- Paris Descartes University, France
- Sidra Medicine, Qatar
- The Rockefeller University, United States
- University Aix-Marseille, URMITE, UM63, CNRS 7278, IRD 198, France
- Garvan Institute of Medical Research, Australia
- University of New South Wales, Australia
- Memorial Sloan Kettering Cancer Center, United States
- Institut Pasteur, France
- CNRS UMR2000, France
- Cochin Hospital, France
- Weill Cornell Graduate School of Medical Sciences, United States
- Assistance Publique-Hôpitaux de Paris, Necker Hospital for Sick Children, France
- Howard Hughes Medical Institute, United States
Peer review process
This article was accepted for publication as part of eLife's original publishing model.
History
- Version of Record published
- Accepted Manuscript published
- Accepted
- Received
Decision letter
-
Jos WM van der MeerReviewing Editor; Radboud University Medical Centre, Netherlands
In the interests of transparency, eLife includes the editorial decision letter and accompanying author responses. A lightly edited version of the letter sent to the authors after peer review is shown, indicating the most substantive concerns; minor comments are not usually included.
Thank you for submitting your article "IRF4 haploinsufficiency in a family with Whipple's disease" for consideration by eLife . Your article has been favorably evaluated by Harry Dietz (Senior Editor) and three reviewers, one of whom, Jos WM van der Meer (Reviewer #1), is a member of our Board of Reviewing Editors. The following individual involved in review of your submission has agreed to reveal their identity: Don Vinh (Reviewer #3).
The reviewers have discussed the reviews with one another and the Reviewing Editor has drafted this decision to help you prepare a revised submission.
Summary:
This paper represents a solid piece of clinical investigation. The investigators describe a rare loss-of-function mutation in IRF4 , leading to an autosomal dominant probably selective immunodeficiency in the defense against Tropheryma Whipplei .
The role of this variant/gene in disease is supported by the authors:
a) The authors show that IRF4 has evolutionary constraint against variation in the human population;
b) The affected residue is highly conserved and in silico prediction programs predict pathogenicity;
c) The authors provide additional experimental evidence for pathogenicity by introducing the identified variant (and others observed in the human population) in an overexpression system;
d) Use an EMSA assay to give clues about the pathophysiological consequences of the variant; and finally show RNAseq data with and w/o in vitro stimulations.
Essential revisions:
1) The work is convincing that this mutation underlies the susceptibility in this family. That being said, we come to the major weakness of the paper, i.e., the lack of confirmation in either a second family or in (some) sporadic cases. The authors have investigated transcription in a series of unrelated patients with Whipple's disease (Figure 4). They do not provide much information on these patients. The least they could have done was sequencing of the entire IRF4 gene in these patients. In addition, the authors should take the effort to use Matchmaker exchange to increase the chances of identifying other cases.
2) The findings they present in fact show that not only WD is associated with the mutation, but also (long-term?) carrier status. This point is rather prominent, but not discussed until pretty late in the Discussion. The possibility that the defect merely leads to (asymptomatic) infection of the GI tract should be discussed more clearly. It is possible that a second hit is necessary to lead to symptomatic infection.
Since T. whipplei primarily enters through the GI tract, the question is relevant which cells in the intestine express IRF4 . This could be added to the Discussion.
3) The claim that IRF4 haploinsufficiency is the mutational mechanism is not clear enough. Haploinsufficiency, per se, requires that the gene product (RNA and/or protein) is present in only 50%. The authors show even higher RNA levels for IRF4 variants and no difference for protein levels, which would argue against HI. The term "functional haploinsufficiency" might be applied to indicate that the protein derived from one allele has no function, but this would need to be distinguished from a hypomorphic or dominant-negative allele, both of which are also defined by loss-of-function. It is questionable whether overexpression experiments are conclusive for LoF variants and at all allow insights into haploinsufficiency. What about IRF4 deletions (and clear cut stop-gains or frameshift mutations), in controls and cases (with other diseases)?
https://doi.org/10.7554/eLife.32340.035Author response
Essential revisions:
1) The work is convincing that this mutation underlies the susceptibility in this family. That being said, we come to the major weakness of the paper, i.e., the lack of confirmation in either a second family or in (some) sporadic cases.
It is true that we describe a single kindred with WD due to IRF4 haploinsufficiency. However, this kindred is multiplex and large, and includes four distantly related patients. The first genetic etiology of primary immunodeficiencies has often been found in a single kindred, and even in a single patient (see Casanova JL et al., J. Exp. Med., 2014). The three previously reported inborn errors of human IRFs were each documented in a single patient: autosomal dominant IRF3 deficiency causing herpes simplex encephalitis (Andersen LL et al., 2015), autosomal recessive IRF7 deficiency causing life-threatening influenza (Ciancanelli M. et al., 2015), and autosomal recessive IRF8 deficiency causing combined immunodeficiency (Hambleton S. et al., New Eng. J. Med., 2011). In this context, our multiplex kindred with WD and autosomal dominant IRF4 deficiency provides a sufficient level of causality. It also provides an “anchor gene”, as other genetic etiologies of WD discovered in the future are likely to be related to IRF4 . Indeed, a hallmark of the human genetic basis of infectious diseases to date is the high level of physiological homogeneity despite enormous genetic heterogeneity (see Casanova, 2015a and b). The manuscript has been revised accordingly (Discussion, first paragraph).
The authors have investigated transcription in a series of unrelated patients with Whipple's disease (Figure 4). They do not provide much information on these patients. The least they could have done was sequencing of the entire IRF4 gene in these patients.
We thank the editors and referees for this helpful suggestion. We sequenced the coding and non-coding exons of IRF4 , and the flanking intron regions, in 25 other unrelated patients with Whipple’s disease (WD). These findings are described in revised Figure 4. No rare or private IRF4 variants were identified. We also searched for copy number variations (CNVs) encompassing parts of IRF4 , but we found none. In addition, we amplified the full-length IRF4 cDNA from EBV-B cell lines from the other 25 unrelated patients with WD. The structure of the cDNA was found to be intact in all these patients, ruling out regulatory mutations disrupting splicing of theIRF4 mRNA (see Author response image 1).
Author response image 1

Electrophoresis of the full-length IRF4 cDNA from EBV-B cell lines.
EBV-B cell lines from 25 patients diagnosed with Whipple’s disease (WD1 to WD25, WT/WT at all coding exons of IRF4) and from three patients (P1, P2 and P3), two asymptomatic heterozygous relatives (HET1 and HET2), four homozygous WT relatives (WT1 to WT4) and two healthy controls (C1 and C2).
Finally, we analyzed IRF4 protein levels by western blotting on EBV-B cell lines from the other 25 unrelated patients with WD. We found that IRF4 levels were normal, strongly suggesting that these patients have no regulatory mutation decreasing the levels of IRF4 transcript and protein (see Author response image 2). The manuscript has been revised accordingly (subsection “IRF4 mRNA levels in transfected HEK293T cells”; subsection “IRF4 qPCR”; Figure 4 legend).
Author response image 2
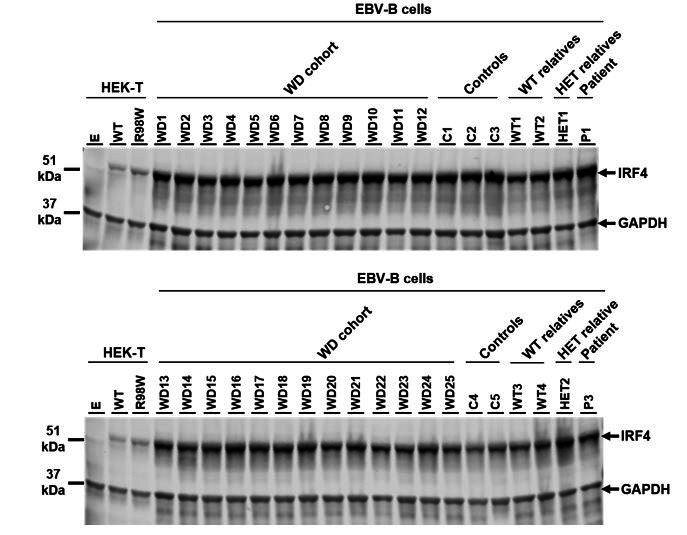
IRF4 protein levels in EBV-B cell lines.
Total lysates of EBV-B cells from five healthy controls (C1 to C5), four homozygous WT relatives (WT1 to WT4), two asymptomatic heterozygous relatives (HET1 and HET2), two patients (P1 and P3) and 25 other patients diagnosed with Whipple’s disease (WD1 to WD25, WT/WT at all coding exons of IRF4 ). Protein extracts from HEK293T cells transfected with empty vector (E), WT or R98W plasmids were used as controls for the specific band corresponding to IRF4.
In addition, the authors should take the effort to use Matchmaker exchange to increase the chances of identifying other cases.
We thank the editors and reviewers for this suggestion. We used Matchmaker exchange and found no matches, for either the gene (IRF4 ) or the clinical phenotype (WD). This was not completely unexpected, as WD is not even suspected to be a genetic disorder, and IRF4 was not reported as a disease-causing gene.
2) The findings they present in fact show that not only WD is associated with the mutation, but also (long-term?) carrier status. This point is rather prominent, but not discussed until pretty late in the Discussion. The possibility that the defect merely leads to (asymptomatic) infection of the GI tract should be discussed more clearly. It is possible that a second hit is necessary to lead to symptomatic infection.
We thank the editors and reviewers for this important suggestion. We have revised our manuscript accordingly (Discussion, last paragraph).
Since T. whipplei primarily enters through the GI tract, the question is relevant which cells in the intestine express IRF4. This could be added to the Discussion.
We thank the editors and reviewers for this helpful suggestion. We have revised our manuscript accordingly (Discussion, last paragraph).
3) The claim that IRF4 haploinsufficiency is the mutational mechanism is not clear enough. Haploinsufficiency, per se, requires that the gene product (RNA and/or protein) is present in only 50%. The authors show even higher RNA levels for IRF4 variants and no difference for protein levels, which would argue against HI. The term "functional haploinsufficiency" might be applied to indicate that the protein derived from one allele has no function, but this would need to be distinguished from a hypomorphic or dominant-negative allele, both of which are also defined by loss-of-function. It is questionable whether overexpression experiments are conclusive for LoF variants and at all allow insights into haploinsufficiency. What about IRF4 deletions (and clear cut stop-gains or frameshift mutations), in controls and cases (with other diseases)?
We thank the editors and referees for raising this key question, which we have addressed experimentally in great depth.
We began by testing in more detail the hypothesis of negative dominance for the R98W allele. We first repeated the dominance negative (DN) assay using an (ISRE )3 promoter, testing a broader range of experimental conditions. We did not detect any negative dominance (revised Figure 3—figure supplement 1A), consistent with our original findings.
We then performed additional experiments using another type of promoter. IRF4 has been described to bind to promoters containing AP-1-IRF composite elements (AICE ) with a high affinity. IRF4 binds to the AP-1 complex in this way (Peng Li et al., Nature, 2012). We, therefore, assessed the ability of mutant IRF4 proteins to induce transcription from an AICE motif-containing promoter. As for the (ISRE )3 promoter, both R98W and R98A-C99A failed to activate the AICE motif-containing promoter (revised Figure 2D). R98W is, therefore, loss-of-function (LOF) for both (ISRE )3 and AICE motif-containing promoters. In this new context, we assessed the possible negative dominance of R98W using the AICE motif-containing promoter. We found no DN effect, even with high proportions of the mutant (revised Figure 3—figure supplement 1B). The R98W mutant allele is, therefore, LOF, as opposed to hypomorphic. Moreover, R98W is not DN, even when overexpressed, in any of the conditions tested. The R98W mutant allele being LOF but not DN, by definition, autosomal dominant IRF4 deficiency must operate by haploinsufficiency.
We then investigated the mechanism of haploinsufficiency in more detail. We found that IRF4 protein levels in the nuclei of EBV-B cells, where they would be expected to exert their effects on transcription, were similar in R98W heterozygous and WT homozygous individuals (revised Figure 4—figure supplement 1C). Similar results were obtained with nuclei from primary CD4+ T cells stimulated with activating anti-CD2/CD3/CD28 monoclonal antibody-coated beads (revised Figure 5C). Moreover, we have shown, by the TA cloning of amplified IRF4 cDNAs from EBV-B cells, that cells from symptomatic and asymptomatic heterozygous individuals produce about 50% WT IRF4 mRNA and 50% mutant IRF4 mRNA (revised Figure 4B). Finally, we have shown that the overexpression of IRF4 R98W or WT plasmids in HEK293T cells results in similar levels of IRF4 protein in the nucleus (revised Figure 3B). Together, these results indicate that IRF4 haploinsufficiency does not operate by loss-of-expression, but by LOF.
Finally, and importantly, we completed the functional characterization of all non-synonymous variants from the gnomAD database (revised Figure 3—figure supplement 4) or our in-house WES database (revised Figure 3—figure supplement 5). We had tested 39 of the 156 known variants by the time of our initial submission. For this revision, we recently tested a total of 153 variants. We did not test an essential splice variant (c.4032T>C) and two variants present only in a non-canonical transcript predicted to undergo nonsense-mediated decay (p.L406P and p.R407W).
The five stop-gain or frameshift variants predicted to be LOF in the gnomAD database were not detectable on western blots following the transfection of HEK293T cells with the corresponding plasmids (the epitope of the antibody used being located at the C-terminus of IRF4; revised Figure 3—figure supplement 2). We also performed RT-qPCR and amplified the full-length IRF4 cDNA from HEK293T cells transfected with the corresponding plasmids. We found that the IRF4 mRNAwas present and intact, confirming that the cells had been correctly transfected with the corresponding plasmids (see Author response image 3 and Author response image 4).
Author response image 3
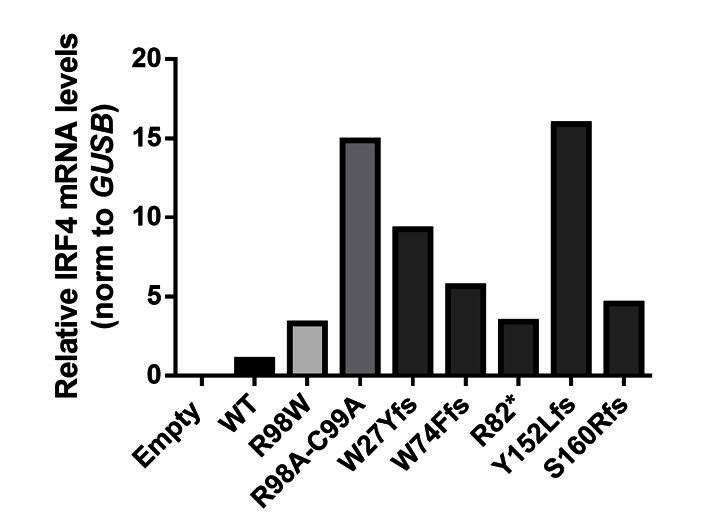
IRF4 mRNA levels in transfected HEK293T cells.
Total RNA extracted from HEK293T cells transfected with E, WT, R98W, R98A-C99A or the five predicted LOF variant plasmids, was subjected to RT-qPCR for total IRF4 . Data are displayed as 2ΔΔCt after normalization against endogenous GUS control gene expression (ΔCt) and the WT value (ΔΔCt).
Author response image 4
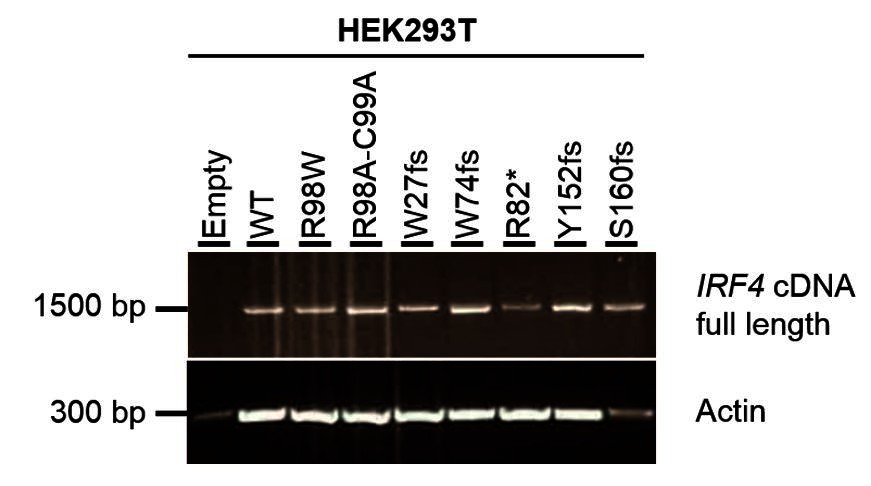
Electrophoresis of full-length IRF4 cDNA from transfected HEK293T cells.
Total cDNA extracted from HEK293T cells transfected with E, WT, R98W, R98A-C99A or the five predicted LOF variant plasmids. A partial actin B cDNA was used as a loading control.
Overall, we showed that the five stop-gain or frameshift variants predicted to be LOF in the gnomAD database were LOF in functional tests in our overexpression system. All these variants are individually (MAF<6x10-6 ) and collectively (MAF <2x10-5 ) extremely rare, and we have no access to additional information for these individuals (a single heterozygote per variant) from the gnomAD database.
We also showed that, of the 148 non-synonymous coding variants tested, only one in-frame deletion (p.E46del) of one amino acid previously described in the gnomAD database was hypomorphic, and that only one in-frame deletion (p.G279-H280 del) of two amino acids found in our in-house WES database was LOF. The E46del is extremely rare in the general population (MAF= 9x10-6 ), and we have no access to additional information for the carrier of this IRF4 variant in the gnomAD database. G279_H280 del has not been reported in public databases. The individual carrying this mutation does not suffer from WD and belongs to a family in which the allele does not segregate with the clinical phenotype.
Overall, the cumulative frequency of these seven LOF (n =6) or hypomorphic (n =1) variants (from a pool of 153 variants) is <4x10-5 , fully consistent with the frequency of WD (occurring only in adults chronically infected with Tw). The other 146 alleles behaved like the WT allele. These new findings confirm that IRF4 is intolerant to LOF, and even hypomorphic variants in the heterozygous state. They support our initial conclusion that autosomal dominant IRF4 deficiency underlies WD in this kindred through haploinsufficiency.
These new studies have resulted in several changes to the revised manuscript. The main text has been revised throughout. The following figures have also been modified:
- Revised Figure 3C: data are now shown as dose responses.
- Revised Figures 5A, 5B and 5C: data are no longer normalized relative to the mean value for controls.
- Revised Figure 2: as all variants have been tested, all dots are now in black.
- Revised Figure 3—figure supplement 2: we have added the new variants tested.
- Revised Figure 3—figure supplement 4: we have added the new variants tested.
- Revised Figure 4B: the data are now represented as a table.
Figures S3B and S3C, which showed the 39 variants previously tested have been removed, as 153 variants have now been tested, and the corresponding information is provided in revised Figure 2—source data 1.
Revised Figure 3D has been added. The previous Figures 3D and 3E are now revised Figures 3E and 3F, respectively.
https://doi.org/10.7554/eLife.32340.036Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
IRF4 haploinsufficiency in a family with Whipple’s disease
eLife 7:e32340.
https://doi.org/10.7554/eLife.32340




{kind=link}
{kind=link}
{kind=link}
{kind=link}