IRF4 haploinsufficiency in a family with Whipple’s disease
- INSERM U1163, France
- Paris Descartes University, France
- Sidra Medicine, Qatar
- The Rockefeller University, United States
- University Aix-Marseille, URMITE, UM63, CNRS 7278, IRD 198, France
- Garvan Institute of Medical Research, Australia
- University of New South Wales, Australia
- Memorial Sloan Kettering Cancer Center, United States
- Institut Pasteur, France
- CNRS UMR2000, France
- Cochin Hospital, France
- Weill Cornell Graduate School of Medical Sciences, United States
- Assistance Publique-Hôpitaux de Paris, Necker Hospital for Sick Children, France
- Howard Hughes Medical Institute, United States
Abstract
Most humans are exposed to Tropheryma whipplei (Tw). Whipple’s disease (WD) strikes only a small minority of individuals infected with Tw (<0.01%), whereas asymptomatic chronic carriage is more common (<25%). We studied a multiplex kindred, containing four WD patients and five healthy Tw chronic carriers. We hypothesized that WD displays autosomal dominant (AD) inheritance, with age-dependent incomplete penetrance. We identified a single very rare non-synonymous mutation in the four patients: the private R98W variant of IRF4, a transcription factor involved in immunity. The five Tw carriers were younger, and also heterozygous for R98W. We found that R98W was loss-of-function, modified the transcriptome of heterozygous leukocytes following Tw stimulation, and was not dominant-negative. We also found that only six of the other 153 known non-synonymous IRF4 variants were loss-of-function. Finally, we found that IRF4 had evolved under purifying selection. AD IRF4 deficiency can underlie WD by haploinsufficiency, with age-dependent incomplete penetrance.
https://doi.org/10.7554/eLife.32340.001eLife digest
In 1907, George Hoyt Whipple – an American pathologist working at Johns Hopkins University in Baltimore – described a new inflammatory disease that affects the intestine. Patients with this condition, now known as Whipple’s disease, experience diarrhea, weight loss, and abdominal and joint pain. The disease is rare; it affects about one in a million people, mostly those over the age of 50 who are of European descent.
Later it was discovered that bacteria called Tropheryma whipplei cause Whipple’s disease and that antibiotics can cure it. These bacteria are widespread and yet only a small minority of individuals infected with T. whipplei goes on to develop Whipple’s disease. In some populations, over 50% of individuals have been infected with the bacteria at some point in their lives, but most will get rid of the infection. This raised the question: when exposed to the same microbe, why do some individuals develop a severe disease, while others remain unharmed?
From the 1950s onwards, scientists identified a few families with multiple members who have developed Whipple’s disease. These observations suggested that human genes may play a role in determining whether a person infected with T. whipplei becomes ill. Rare genetic mutations that affect the immune system have also been linked to the development of life-threatening cases of influenza or tuberculosis.
Now, Guérin et al. report that, in one French family, an extremely rare mutation in the gene that codes for a protein called IRF4 may contribute to the development of Whipple’s disease. This family had four members with Whipple’s disease, all of whom had one copy of the gene with this rare mutation and one normal copy of the gene. The IRF4 protein acts like a switch that turns on and off some genes involved in the body’s response to infection. In patients with this mutation, the IRF4 protein does not work as it should.
Guérin et al. suggest that Whipple’s disease may be caused by specific genetic mutations affecting the immune system in subjects infected by T. whipplei. More studies are needed to see if other genetic mutations also contribute to other cases of Whipple’s disease. Such studies may help physicians to better understand why some people become sick after T. whipplei infections while others do not. They may also help physicians to diagnose the disease, and even lead to better treatments.
https://doi.org/10.7554/eLife.32340.037Introduction
Whipple’s disease (WD) was first described as an intestinal inflammatory disease by George H. Whipple in 1907 (Whipple, 1907). Its infectious origin was suspected in 1961 (Yardley and Hendrix, 1961), and the causal microbe, Tropheryma whipplei (Tw), a Gram-positive actinomycete, was detected by PCR in 1992 (Relman et al., 1992), and cultured in 2000 (Raoult et al., 2000). Tw is probably transmitted between humans via the oro-oral or feco-oral routes. WD is a chronic condition with a late onset (mean age at onset: 55 years) (Braubach et al., 2017) affecting multiple organs. The clinical manifestations of classical WD are arthralgia, diarrhea, abdominal pain, and weight loss (Dobbins, 1987; Durand et al., 1997; Fleming et al., 1988; Mahnel et al., 2005; Maizel et al., 1970). However, about 25% of WD patients display no gastrointestinal or osteoarticular symptoms, instead presenting with cardiac and/or neurological manifestations (Durand et al., 1997; Fenollar et al., 2014, Fenollar et al., 2001; Gubler et al., 1999; Schneider et al., 2008). WD is fatal if left untreated, and relapses occur in 2% to 33% of treated cases, even after prolonged appropriate antibiotic treatment (Lagier et al., 2011; Marumganti and Murphy, 2008). WD is rare and has been estimated to affect about one in a million individuals (Dobbins, 1981, 1987; Fenollar et al., 2007). However, about two thousand cases have been reported in at least nine countries worldwide, mostly in North America and Western Europe (Bakkali et al., 2008; Desnues et al., 2010; Fenollar et al., 2008a; Lagier et al., 2010; Puéchal, 2016; Schneider et al., 2008). Chronic asymptomatic carriage of Tw is common in the general population, and this bacterium has been detected in feces, saliva, and intestinal mucosae. The prevalence of Tw carriage in the feces has been estimated at 2% to 11% for the general population, but can reach 26% in sewer workers and 37% in relatives of patients and carriers (Amsler et al., 2003; Ehrbar et al., 1999; Fenollar et al., 2014, Fenollar et al., 2007; Maibach et al., 2002; Rolain et al., 2007; Schneider et al., 2008; Street et al., 1999).
Seroprevalence for specific antibodies against Tw in the general population varies from 50% in France to 70% in Senegal (Fenollar et al., 2009, Fenollar et al., 2014; Raoult et al., 2000; Schneider et al., 2008). At least 75% of infected individuals clear Tw primary infections, but a minority (<25%) become asymptomatic carriers, a very small proportion of whom develop WD (<0.01%) (Fenollar et al., 2008b). Tw infection is therefore, necessary but not sufficient, for WD development, and it is unclear whether prolonged asymptomatic carriage necessarily precedes WD. The hypothesis that WD results from the emergence of a more pathogenic clonal strain of Tw was not supported by bacterial genotyping (Li et al., 2008). WD mostly affects individuals of European origin, but does not seem to be favored by specific environments. WD is typically sporadic, but six multiplex kindreds have been reported, with cases often diagnosed years apart, suggesting a possible genetic component (Durand et al., 1997; Fenollar et al., 2007; Ponz de Leon et al., 2006). WD patients are not prone to other severe infections (Marth et al., 2016). Moreover, WD has never been reported in patients with conventional primary immunodeficiencies (PIDs) (Picard et al., 2018). This situation is reminiscent of other sporadic severe infections, such as herpes simplex virus-1 encephalitis, severe influenza, recurrent rhinovirus infection, severe varicella zoster disease, trypanosomiasis, invasive staphylococcal disease, and viral infections of the brainstem, which are caused by single-gene inborn errors of immunity in some patients (Andersen et al., 2015; Casanova, 2015a, Casanova, 2015b; Ciancanelli et al., 2015; Israel et al., 2017; Lamborn et al., 2017; Zhang et al., 2015; Ogunjimi et al., 2017; Vanhollebeke et al., 2006; Zhang et al., 2018). We therefore hypothesized that WD might be due to monogenic inborn errors of immunity to Tw, with age-dependent incomplete penetrance.
Results
A multiplex kindred with WD
We investigated four related patients diagnosed with WD (P1, P2, P3, and P4) with a mean age at diagnosis of 58 years. They belong to a large non-consanguineous French kindred (Figure 1A). The proband (P1), a 69-year-old woman, presented with right knee arthritis in 2011, after recurrent episodes of arthritis of the right knee since 1980. Tw was detected in the synovial fluid by PCR and culture, but not in saliva, feces, or small intestine tissue. Treatment with doxycycline and hydroxychloroquine was effective. At last follow-up, in 2016, P1 was well and Tw PCR on saliva and feces was negative. P2, a second cousin of P1, is a 76-year-old woman with classical WD diagnosed at 37 years of age in 1978 by periodic acid–Schiff (PAS) staining of a small intestine biopsy specimen. She was treated with sulfamethoxazole/trimethoprim. At last follow-up, in 2016, Tw PCR on saliva and feces was positive. P3, the father of P1, is a 92-year-old man with classical WD diagnosed at 62 years of age, in 1987, based on positive PAS staining of a small intestine biopsy specimen. Long-term sulfamethoxazole/trimethoprim treatment led to complete clinical and bacteriological remission. P4, the brother of P2, is a 70-year-old man who consulted in 2015 for arthralgia of the knees and right ulna-carpal joints. PCR and culture did not detect Tw in saliva and feces, but serological tests for Tw were positive. Treatment with methotrexate and steroids was initiated before antibiotics, the effect of which is currently being evaluated. All four patients are otherwise healthy. Saliva and/or feces samples from 18 other members of the family were tested for Tw (Figure 1A; Figure 1—source data 1). Five individuals are chronic carriers (mean age: 55 years) and 13 tested negative (mean age: 38 years). Nine additional relatives could not be tested. The distribution of WD in this kindred was suggestive of an AD trait with incomplete clinical penetrance.
Figure 1 with 1 supplement see all
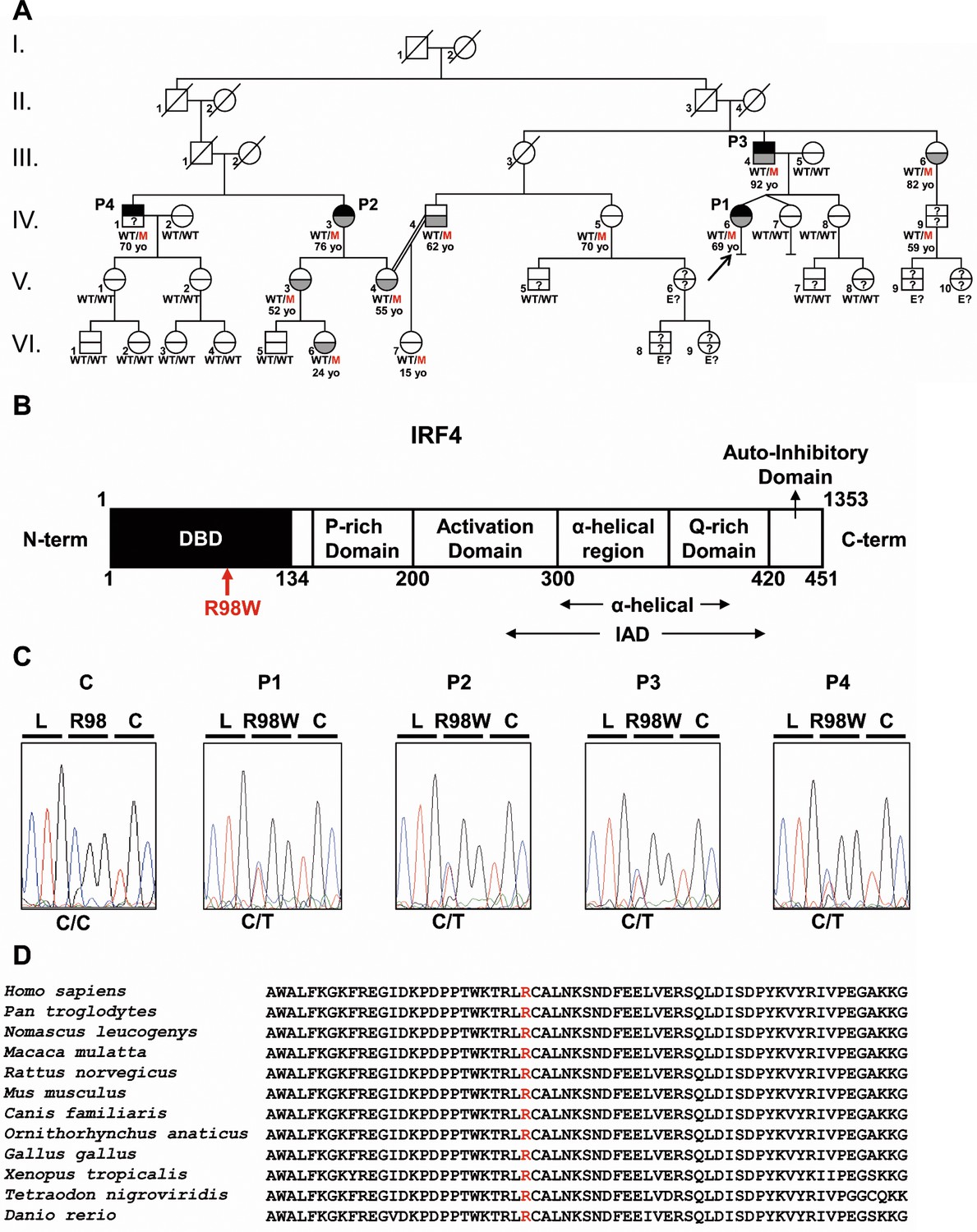
Autosomal dominant IRF4 deficiency.
(A) Pedigree of the kindred, with allele segregation. Generations are designated by Roman numerals (I, II, III, IV, V and VI), and each individual is designated by an Arabic numeral (from left to right). Each symbol is divided into two parts: the upper part indicates clinical status for WD (black: affected, white: healthy, ‘?': not known); the lower part indicates whether Tw was identified by PCR (in saliva, blood, feces or joint fluid) or by PAS staining on bowel biopsy specimens (gray: Tw-positive, white: Tw-negative, ‘?': not tested). Whipple’s disease patients are indicated as P1, P2, P3, and P4; the proband is indicated with an arrow. Genotype status and age (for IRF4-heterozygous individuals) are reported below the symbols. Individuals whose genetic status could not be evaluated are indicated by the symbol ‘E?”. (B) Schematic representation of the IRF4 protein, showing the DNA-binding domain (DBD), P-rich domain, activation domain, α-helical domain, Q-rich domain, IRF association domain (IAD), and auto-inhibitory domain. The R98W substitution is indicated in red. (C) Electropherogram of IRF4 genomic DNA sequences from a healthy unrelated control (C) and the patients (P1, P2, P3, P4). The R98W IRF4 mutation leads to the replacement of an arginine with a tryptophan residue in position 98 (exon 3, c.292 C > T). The corresponding amino acids are represented above each electropherogram. (D) Alignment of the R98W amino acid in the DBD domain of IRF4 in humans and 11 other animal species. R98 is indicated in red.
-
Figure 1—source data 1
Kindred information summary.
For each subject, Tw carriage status, IRF4 genotype, clinical status and date of birth (DOB) are reported. NA: not available; Pos: positive; Neg: negative; Tw: Tropheryma whipplei; WD: Whipple’s disease; E?: genotype not assessed.
- https://doi.org/10.7554/eLife.32340.004
-
Figure 1—source data 2
Non-synonymous variants within the linkage regions found in WES data from patients.
- https://doi.org/10.7554/eLife.32340.005
A private heterozygous missense IRF4 variant segregates with WD
We analyzed the familial segregation of WD by genome-wide linkage (GWL), using information from both genome-wide single-nucleotide polymorphism (SNP) microarrays and whole-exome sequencing (WES) (Belkadi et al., 2016). Multipoint linkage analysis was performed under an AD model, with a very rare disease-causing allele (<10−5) and age-dependent incomplete penetrance. Twelve chromosomal regions linked to WD were identified on chromosomes 1 (x3), 2, 3, 6, 7, 8, 10, 11, 12 and 17, with a LOD score close (>1.90) to the maximum expected value (1.95) (Figure 1—figure supplement 1A). These regions covered 27.18 Mb and included 263 protein-coding genes. WES data analysis for these 263 genes identified 54 heterozygous non-synonymous coding variants common to all four WD patients (Figure 1—source data 2). Only one, a variant of the interferon regulatory factor (IRF) four gene encoding a transcription factor from the IRF family (Ikushima et al., 2013), located in a 200 kb linked region on chromosome 6 (Figure 1—figure supplement 1A and B), was very rare, and was even found to be private [not found in the gnomAD database, http://gnomad.broadinstitute.org, or in our in-house WES database (HGID)], whereas all other variants had a frequency >0.001, which is inconsistent with the frequency of WD and our hypothesis of a very rare (<10−5) deleterious heterozygous allele. The variant is a c.292 C > T substitution in exon 3 of IRF4, replacing the arginine residue in position 98 with a tryptophan residue (R98W) (Figure 1A, B and C). IRF4 is a transcription factor with an important pleiotropic role in innate and adaptive immunity, at least in a few strains of inbred mice (Shaffer et al., 2009). Mice heterozygous for a null Irf4 mutation have not been studied, but homozygous null mice have various T- and B-cell abnormalities and are susceptible to both Leishmania and lymphocytic choriomeningitis virus (Klein et al., 2006; Lohoff et al., 2002; Mittrücker et al., 1997; Suzuki et al., 2004; Tamura et al., 2005; Tominaga et al., 2003). We confirmed the IRF4 R98W mutation by Sanger sequencing genomic DNA from the blood of the four WD patients (Figure 1C). Thirteen relatives of the WD patients were WT/WT at the IRF4 locus, and 10 of these relatives (77%) tested negative for Tw carriage. Eight other relatives were heterozygous for the IRF4 R98W mutation, five of whom (62.5%) were Tw carriers (mean age: 55 years) (Figure 1A; Figure 1—source data 1). Overall, 12 individuals from the kindred, including the four patients, the five chronic carriers of Tw, two non-carriers of Tw and one relative not tested for Tw, were heterozygous for IRF4 R98W (Figure 1A; Figure 1—source data 1). The familial segregation of the IRF4 R98W allele was therefore consistent with an AD pattern of WD inheritance with incomplete clinical penetrance. Chronic Tw carriage also followed an AD mode of inheritance.
R98W is predicted to be loss-of-function, unlike most other IRF4 variants
The R98 residue in the DNA-binding domain (DBD) of IRF4 is highly conserved in the 12 species for which IRF4 has been sequenced (Figure 1B and D). It has been suggested that this residue is essential for IRF4 DNA-binding activity, because the R98A-C99A double mutant is loss-of-function (LOF) (Brass et al., 1999; Escalante et al., 2002). The R98W mutation is predicted to be damaging by multiple programs (Kircher et al., 2014); it has a combined annotation–dependent depletion score (CADD) score of R98W (26.5), well above the mutation significance cutoff (MSC) of IRF4 (11.125) (Figure 2) (Itan et al., 2016; Kircher et al., 2014). The R98W variant was not present in the gnomAD database or our in-house HGID database of more than 4,000 WES from patients with various infectious diseases. The mutant allele was not found in the sequences for the CEPH-HGDP panel of 1,052 controls from 52 ethnic groups, or in 100 French controls, confirming that this variant was very rare, probably private to this kindred. Therefore, the minor allele frequency (MAF) of this private allele is <4 × 10−6. Moreover, the IRF4 gene has a gene damage index (GDI) of 2.85, a neutrality index score of 0.15 (Itan et al., 2015), and a purifying selection f parameter of 0.32 (among the <10% of genes in the genome subject to the greatest constraint; Figure 2—figure supplement 1), strongly suggesting that IRF4 has evolved under purifying selection (i.e. strong evolutionary constraints) (Eilertson et al., 2012). Biologically disruptive heterozygous mutations of IRF4 are therefore likely to have clinical effects. We identified 156 other high-confidence heterozygous non-synonymous coding or splice variants of IRF4 (Figure 2—source data 1) in public (gnomAD: 153 variants, all with MAF <0.009) and HGID (three variants) databases: 147 were missense variants (two of which were also found in the homozygous state: p.S149N and p.A370V), four were frameshift indels leading to premature stop codons (p.W27YfsTer50, p.W74GfsTer28, p.Y152LfsTer60, and p.S160RfsTer11), three were in-frame indels (p.E46del, p.G279_H280del, and p.S435del), one was a nonsense variant (p.R82*), one was an essential splice variant (c.4032T > C), and two were missense variants found only in a non-canonical transcript predicted to undergo nonsense-mediated decay (p.L406P and p.R407W). Up to 150 of the 156 variants are predicted to be benign, whereas only six were predicted to be potentially LOF according to the gnomAD database classification (the four frameshift indels, the nonsense variant, and the essential splice variant). Comparison of the CADD score and MAF of these IRF4 variants showed R98W to have the second highest CADD score of the four variants with a MAF <4 × 10−6 (Figure 2). These findings suggest that the private heterozygous IRF4 variant of this kindred is biochemically deleterious, unlike most other rare (MAF <0.009) non-synonymous variants in the general population, 150 of 156 of which were predicted to be benign (Lek et al., 2016).
Figure 2 with 1 supplement see all

Analysis in silico of IRF4 variants.
Minor allele frequency (MAF) and combined annotation–dependent depletion (CADD) score of all coding variants previously reported in a public database (gnomAD) (http://gnomad.broadinstitute.org) and in our in-house (HGID) database. The dotted line corresponds to the mutation significance cutoff (MSC) with 95% confidence interval. The R98W variant is shown as a red square.
-
Figure 2—source data 1
156 non-synonymous heterozygous coding or splice variants reported in the gnomAD or HGID databases.
†: non-canonical transcript predicted to undergo nonsense mediated decay.
- https://doi.org/10.7554/eLife.32340.008
R98W is LOF, unlike most other previously observed IRF4 variants
We first characterized IRF4 R98W production and function in vitro, in an overexpression system. We assessed the effect of the IRF4 R98W mutation on IRF4 levels by transiently expressing WT or mutant R98W in HEK293T cells. IRF4 R98A-C99A, which is LOF for DNA binding (Brass et al., 1999), was included as a negative control. In total cell extracts, mutant IRF4 proteins were more abundant than the WT protein and had the expected molecular weight (MW) of 51 kDa, as shown by western blotting (Figure 3A). The R98 residue has been shown to be located in a nuclear localization signal, the complete disruption of which results in a loss of IRF4 retention in the nucleus (Lau et al., 2000). We therefore analyzed the subcellular distribution of IRF4 WT and R98W proteins, in total, cytoplasmic, and nuclear extracts from transiently transfected HEK293T cells. The R98W mutant was more abundant than the WT protein in total cell and cytoplasmic extracts, but these proteins were similarly abundant in nuclear extracts (Figure 3B). We performed luciferase reporter assays to assess the ability of the mutant IRF4 protein to induce transcription from interferon-stimulated response element (ISRE) motif-containing promoters. Unlike the WT protein, both R98W and R98A-C99A failed to activate the (ISRE)3 promoter (Figure 3C). We also assessed the ability of IRF4 to induce transcription from an (AP-1)-IRF composite element (AICE) motif-containing promoter (Li et al., 2012). Both R98W and R98A-C99A failed to activate the AICE promoter (Figure 3D). Moreover, we observed no dominant-negative effect of the IRF4 R98W protein, with either the ISRE or AICE motif-containing promoter (Figure 3—figure supplement 1). We assessed the ability of R98W to bind DNA, in an electrophoretic mobility shift assay (EMSA) (Figure 3E and F). Signal specificity was assessed by analyzing both supershift with an IRF4-specific antibody and by competition with an unlabeled competitor probe. The R98W mutation abolished IRF4 binding to the ISRE cis element (Figure 3E), and binding of the IRF4-PU.1 complex to interferon composite elements (EICEs) containing both IRF4 and PU.1 recognition motifs (Brass et al., 1999) (Figure 3F). The R98W allele of IRF4 is therefore LOF for both DNA binding and the induction of transcription. We then tested 153 of the other 156 IRF4 variants: 150 variants previously described in the gnomAD database and three variants found in the HGID database. The essential splice variant and the two variants present only in a non-canonical transcript were not tested. All the variants tested were normally expressed (two with a higher MW), except for the five predicted LOF variants tested (the epitope of the antibody being at the C-terminus of IRF4) (Figure 3—figure supplements 2; 3). Transfection with the five predicted LOF plasmids was efficient, as assessed by cDNA amplification and sequencing (data not shown). When tested for (ISRE)3 promoter activation, the five stop-gain or frameshift variants predicted to be LOF in the gnomAD database (very rare variants, MAF <6 × 10−6) were found to be LOF. We also showed that among all the non-synonymous coding variants tested, only one rare very rare (MAF 9 × 10−6) inframe deletion of one amino acid (E46del) reported in the gnomAD database, was hypomorphic, and another variant from our in-house WES database (G279_H280 del, private to one family) was LOF (Figure 3—figure supplements 4; 5). The cumulative frequency of these seven LOF (n = 6) or hypomorphic (n = 1) variants was <4 × 10−5, fully consistent with the frequency of WD (occurring only in adults chronically infected with Tw). Overall, our data show that the R98W IRF4 allele is LOF, like only six other very rare non-synonymous IRF4 coding variants of the 153 variants tested. Moreover, R98W is not dominant negative.
Figure 3 with 5 supplements see all
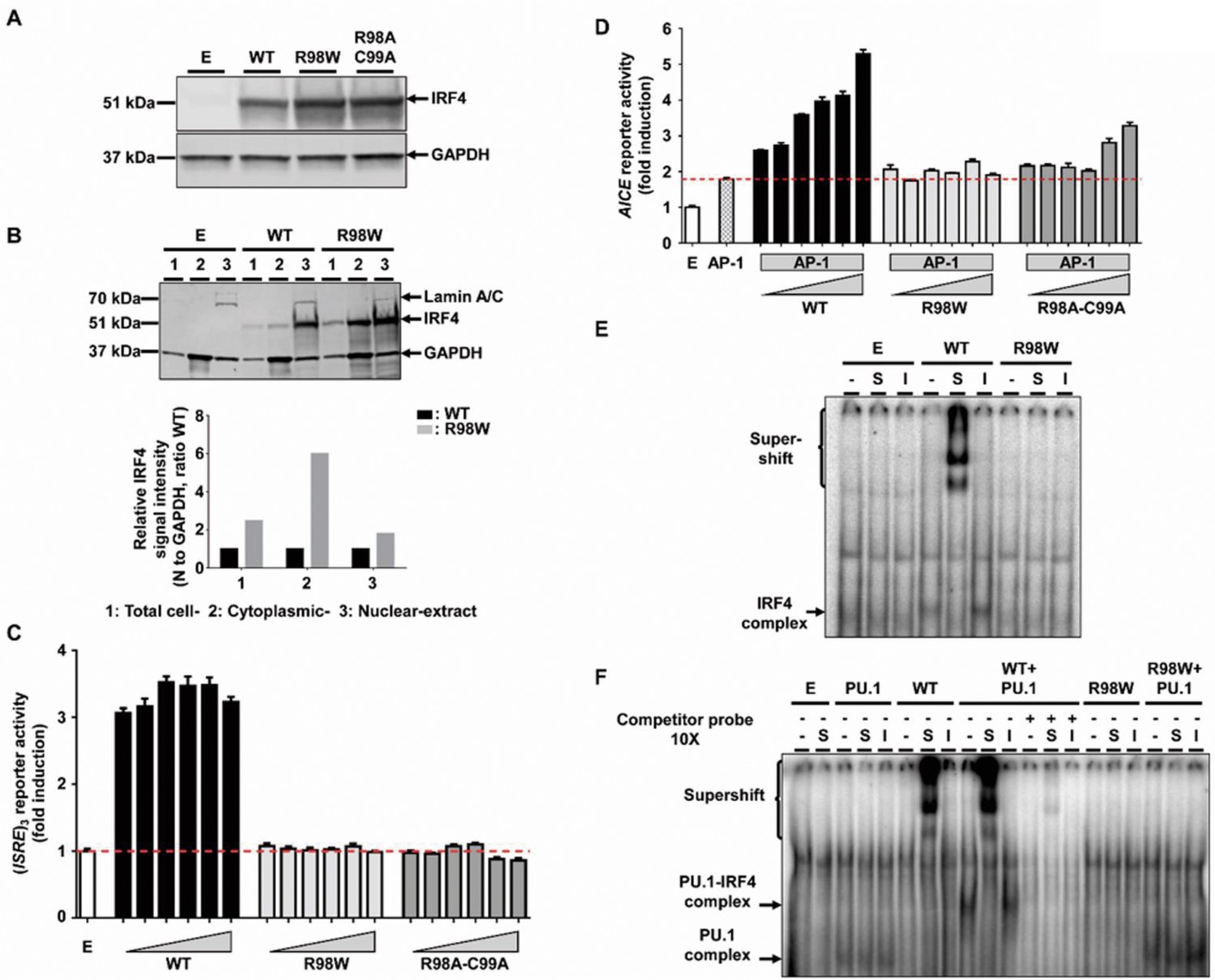
Molecular characterization of the R98W IRF4 mutation (loss of DNA binding).
(A) HEK293T cells were transfected with the pcDNA3.1 empty vector (E) or plasmids encoding IRF4 WT, IRF4 R98W or IRF4 R98A-C99A. Total cell extracts were subjected to western blotting; the upper panel shows IRF4 levels and the lower panel shows the levels of GAPDH, used as a loading control. The results shown are representative of three independent experiments. (B) (upper panel) HEK293T cells were transfected with the pcDNA3.1 empty vector (E) or plasmids encoding IRF4 WT or IRF4 R98W . Total cell (1), cytoplasmic (2) and nuclear (3) extracts were subjected to western blotting. Lamin A/C and GAPDH were used as loading controls. (lower panel) IRF4 signal intensity for R98W-transfected cells and WT-transfected cells, in various cell compartments (total, cytoplasmic and nuclear), normalized against the GAPDH signal, as shown by western blotting. The results shown are representative of three independent experiments. (C) Luciferase activity of HEK293T cells cotransfected with an (ISRE)3 reporter plasmid plus the pcDNA3.1 empty vector (E, 100 ng) and various amounts of plasmids encoding IRF4 WT or IRF4 R98W or IRF4 R98A/C99A (6.25, 12.5, 25, 50, 75 and 100 ng). Results are shown as the fold induction of activity relative to E-transfected cells. The red dotted line indicates mean activity for E-transfected cells. The mean and standard error of three experiments are shown. (D) Luciferase activity of HEK293T cells cotransfected with an AICE reporter plasmid plus the pcDNA3.1 empty vector (E, 100 ng) and/or constant amounts of plasmids encoding BATF WT and JUN WT (25 ng each, AP-1) and/or various amounts of plasmids encoding IRF4 WT or IRF4 R98W or IRF4 R98A/C99A (6.25, 12.5, 18.8, 25, 37.5 and 50 ng). Results are shown as the fold induction of activity relative to E-transfected cells. The red dotted line indicates mean activity for AP-1-transfected cells. The mean and standard error of two experiments are shown. (E) Electrophoretic mobility shift assay (EMSA) with nuclear extracts of HEK293T cells transfected with the pcDNA3.1 empty vector (E), or plasmids encoding IRF4 WT or IRF4 R98W. Extracts were incubated with a 32P-labeled ISRE probe. Extracts were incubated with a specific anti-IRF4 antibody (S) to detect DNA-protein complex supershift, with an isotype control antibody (I) to demonstrate the specificity of the complex, and with no antibody (-), as a control. The results shown are representative of three independent experiments. (F) EMSA of nuclear extracts of HEK293T cells transfected with the pcDNA3.1 empty vector (E), or plasmids encoding PU.1, IRF4 WT, or IRF4 R98W, or cotransfected with PU.1 and IRF4 WT or PU.1 and IRF4 R98W plasmids. Extracts were incubated with a 32P-labeled λB probe (EICE). Extracts were incubated with a specific anti-IRF4 antibody (S) to detect DNA-protein complex supershift, with an isotype control antibody (I) to demonstrate the IRF4 specificity of the complex and with no antibody (-), as a control. Experiments in the presence of excess non-radioactive probe (cold probe) demonstrated the probe specificity of the complexes. The results shown are representative of three independent experiments.
AD IRF4 deficiency phenotypes in heterozygous EBV-B cells
We investigated the cellular phenotype of heterozygosity for the R98W allele in EBV-transformed B-cell lines (EBV-B cells) from patients. We performed reverse transcription-quantitative polymerase chain reaction (RT-qPCR) on EBV-B cells from P1, P3, two healthy heterozygous relatives (IRF4 WT/R98W), four healthy IRF4-WT homozygous relatives, and seven healthy unrelated controls. We also investigated 25 unrelated WD patients with Tw carriage. We sequenced all IRF4 coding exons for these patients, who were found to be WT. They were also found to have an intact IRF4 cDNA structure and normal IRF4 protein levels in EBV-B cells (data not shown). Cells from individuals heterozygous for the R98W mutation (patients and healthy carriers) had higher IRF4 mRNA levels than those from WT homozygous relatives, unrelated WD cohort patients and EBV-B cells from healthy unrelated controls (Figure 4A). We compared the relative abundances of WT and R98W IRF4 mRNA in EBV-B cells from heterozygous carriers of the mutation, by performing TA-cloning experiments on P1, P3, one healthy heterozygous relative, one relative homozygous for WT IRF4, and two previously tested healthy unrelated controls. In heterozygous carriers of the mutation (patients and healthy relatives), the R98W mutation was present in 48.1–60% of the total IRF4 mRNA, whereas the rest was WT (Figure 4B). We evaluated the levels and distribution of IRF4 protein by western blotting on EBV-B cells from P1, P2, P3, one healthy heterozygous relative, three healthy homozygous WT relatives and five unrelated healthy individuals. As in transfected HEK293T cells, IRF4 protein levels were high both in total cell extract and even more so in cytoplasmic extracts of EBV-B cells from heterozygous carriers (Figure 4—figure supplement 1). By contrast, IRF4 protein levels in EBV-B cell nuclei were similar in heterozygous carriers and controls (Figure 4—figure supplement 1). As IRF4 is a transcription factor, we then analyzed the steady-state transcriptome of EBV-B cells from three healthy homozygous WT relatives and three WT/R98W heterozygotes (P1, P3, VI.6). We identified 37 protein-coding genes as differentially expressed between subjects heterozygous for IRF4 and those homozygous WT for IRF4 (18 upregulated and 19 downregulated; data not shown). We identified no marked pathway enrichment based on these genes. EBV-B cells from individuals heterozygous for IRF4 had a detectable phenotype, in terms of IRF4 production and function, consistent with AD IRF4 deficiency underlying WD.
Figure 4 with 1 supplement see all

IRF4 mRNA levels in EBV-B cells.
(A) Total RNA extracted from healthy unrelated controls (n = 7; IRF4 WT/WT), patients diagnosed with Whipple’s disease (n = 25; WT/WT for all coding exons of IRF4) not related to this kindred, healthy homozygous WT relatives (n = 4, IRF4 WT/WT), patients with monoallelic IRF4 mutations (n = 2; IRF4 WT/R98W) and asymptomatic heterozygous relatives with monoallelic IRF4 mutations (n = 2; IRF4 WT/R98W) was subjected to RT-qPCR for total IRF4. Data are displayed as 2-ΔΔCt after normalization according to endogenous GUSB control gene expression (ΔCt) and the mean of controls (ΔΔCt). The results shown are the mean ± SD of three independent experiments. (B) Calculated frequency (%) of each mRNA (WT and R98W allele) obtained by the TA-cloning of cDNA generated from EBV-B cells from healthy unrelated controls (n = 2), healthy homozygous WT relatives (n = 1), patients with monoallelic IRF4 mutations (n = 2) and asymptomatic heterozygous relatives with monoallelic IRF4 mutations (n = 1).
AD IRF4 deficiency phenotypes in heterozygous leukocytes
We assessed IRF4 levels in peripheral blood mononuclear cells (PBMCs) from healthy controls (Figure 5—figure supplement 1). IRF4 were also expressed in CD4+ T cells, particularly after stimulation with activating anti-CD2/CD3/CD28 monoclonal antibody-coated (mAb-coated) beads (data not shown). We therefore assessed the IRF4 protein expression profile in CD4+ T cells from four healthy unrelated controls, P1 and P3, with and without (non-stimulated, NS) stimulation with activating anti-CD2/CD3/CD28 mAb-coated beads. The results were consistent with those for transfected HEK293T and EBV-B cells, as IRF4 levels were higher in activated CD4+ T cells from P1 and P3 than in controls, both for total cell extracts, and even more so for the cytoplasmic compartment (Figure 5A and B). By contrast, IRF4 levels in the nucleus were similar and, possibly, even slightly lower in patients than in controls (Figure 5C). We also investigated peripheral myeloid (Figure 5—figure supplements 1–4) and lymphoid blood cell subsets in patients (Figure 5—figure supplements 5–7; Figure 5—source data 1), which display an apparently normal development compared to healthy controls’ cells. Then, we checked for transcriptomic differences associated with genotype and/or infection, by investigating the transcriptomes of PBMCs from six IRF4-heterozygous individuals (three patients, P1-P3; and three healthy relatives, HET1-HET3) and six IRF4 WT-homozygous individuals (four healthy relatives, WT1-WT4; and two healthy unrelated controls, C1-C2) with and without in vitro infection for 24 hr with Tw, or Mycobacterium bovis-Bacillus Calmette-Guerin (BCG), which, like Tw, belongs to phylum Actinobacteria. We performed unsupervised hierarchical clustering of the differentially expressed (DE) transcripts (infected versus uninfected) to analyze the overall responsiveness of PBMCs from individual subjects to BCG and Tw infections in vitro. Heterozygous individuals clearly clustered separately from homozygous WT individuals (Figure 6A), revealing a correlation between genotype and response to infection. Overall, we found that 402 transcripts from 193 unique genes were responsive to BCG infection (Figure 6—source data 1), and 119 transcripts from 29 unique genes were responsive to Tw infection (Figure 6—source data 2) in homozygous WT subjects, according to the criteria described in the Materials and methods. Due to the small number of Tw-responsive transcripts linked to unique genes, we were unable to detect any pathway enrichment for this specific condition. However, we identified 24 canonical pathways as enriched after the exposure of PBMCs to BCG. We ranked these pathways according to the difference in mean z-score between homozygous WT and heterozygous subjects (Figure 6B). The top 10 pathways included the interferon signaling network, the Th1 pathway network, the HMGB1 signaling network, the p38 MAPK signaling network, the NF-κB signaling network, the dendritic cell maturation network and the network responsible for producing nitric oxide and reactive oxygen species. These pathways were highly ranked mostly due to IFNG and STAT1, which were strongly downregulated in IRF4 heterozygotes, particularly in P1, P2 and P3, relative to WT homozygotes. IRF4 is predicted to bind the promoter regions of 47% of the genes identified in the BCG study (91 of 193 genes), including those of IFNG and STAT1. Subjects heterozygous for IRF4 also had lower levels of LTA expression, and lower levels of IL2RA expression were observed specifically in patients (GSE102862). These data suggest a general impairment of the T-cell response in subjects heterozygous for IRF4 upon BCG infection in vitro. Moreover, the lower levels of CD80 expression suggest a possible impairment of myeloid and/or antigen-presenting cell function upon BCG infection in patients, but not in healthy heterozygous or homozygous WT subjects (GSE102862). Peripheral leukocytes from IRF4-heterozygous individuals therefore had a phenotype in terms of IRF4 production and function.
Figure 5 with 7 supplements see all
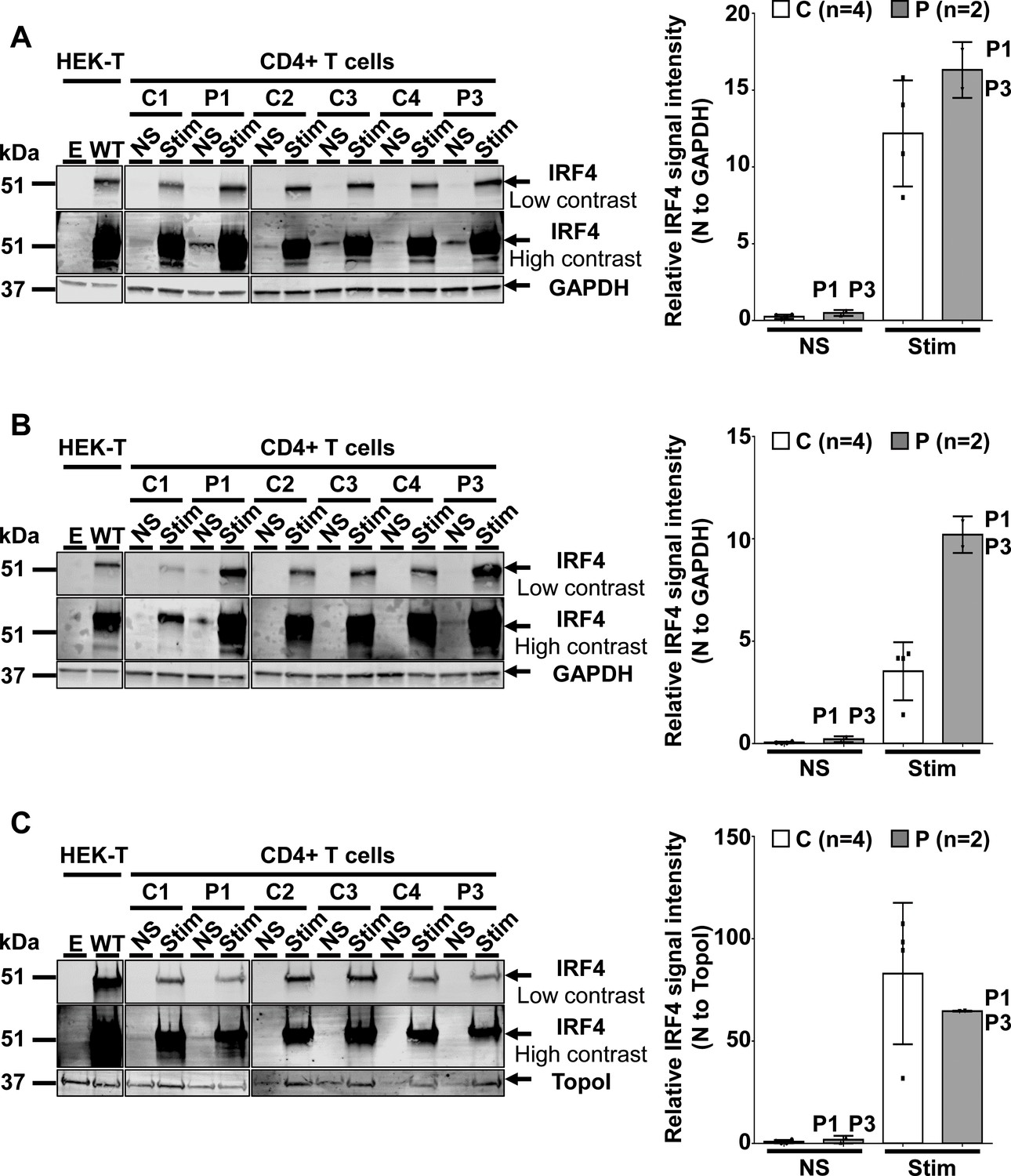
IRF4 protein levels in CD4+ T cells.
(A–C) (Left) Total-cell (A), cytoplasmic (B) and nuclear (C) extracts from CD4+ T cells from four healthy unrelated controls (C1 to C4) and two patients (P1 and P3) stimulated with activating anti-CD2/CD3/CD28 monoclonal antibody-coated beads (Stim) or left unstimulated (NS). Protein extracts from HEK293T cells transfected with the pcDNA3.1 empty vector (E) or plasmids encoding IRF4 WT plasmids were used as controls for the specific band corresponding to IRF4. (Right) Representation of IRF4 signal intensity for each individual, obtained by western blotting, with normalization against the GAPDH signal (total, cytoplasmic extracts) or the topoisomerase I signal (nuclear extracts).
-
Figure 5—source data 1
Immunophenotyping of patients (P1, P2 and P3) and a WT homozygous relative.
All subjects had normal numbers and percentages of T, B, and NK cells for age.
- https://doi.org/10.7554/eLife.32340.029
Figure 6
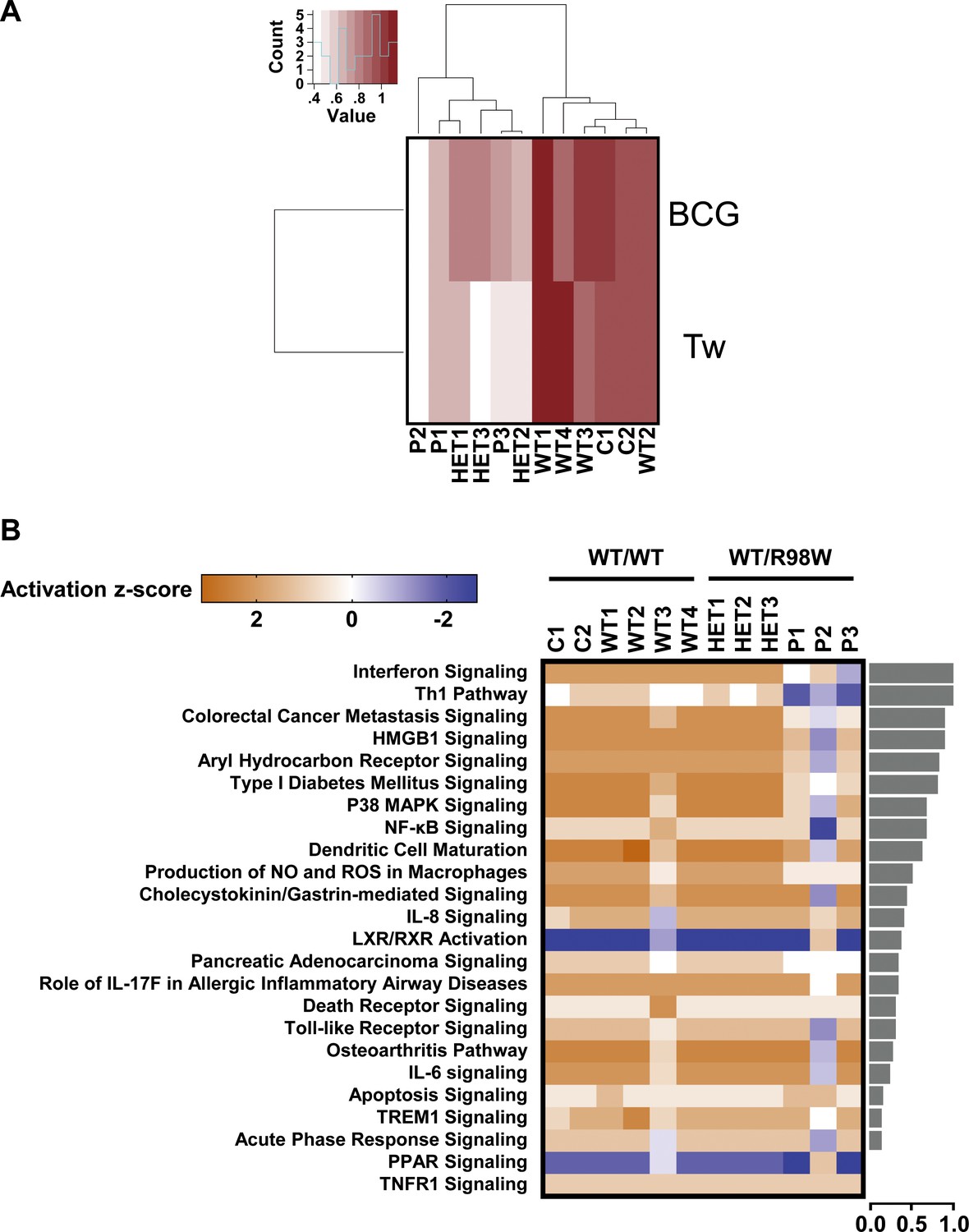
Overall transcriptional responsiveness of PBMCs following in vitro exposure to Tw and BCG and pathway activity analysis for genes responsive to BCG exposure.
(A) The overall responsiveness of individual subjects following stimulation with BCG and Tw, relative to non-stimulated conditions (along the horizontal axis) is shown as a heatmap. For each individual and each stimulus, overall responsiveness was assessed on the basis of normalized counts of differentially expressed transcripts, as described in the corresponding Materials and methods section. Subjects were grouped by unsupervised hierarchical clustering. (B) Enriched canonical pathways were ranked according to differences in mean activation z-score between genotypes (WT/WT individuals vs. WT/R98W individuals). The activation z-scores for each individual and pathway are shown as heat maps. Pathways predicted to be activated are depicted in orange, pathways predicted to be inhibited are depicted in blue. A lack of prediction concerning activation is depicted in white. Individuals are presented in columns, pathways in rows. The pathways are ranked from most different between genotypes (at the top of the list) to the least different (at the bottom). The differences in mean activation z-scores between WT/WT and WT/R98W individuals for each pathway are depicted as bars to the right of the heat maps (the direction of difference is not shown). The Ingenuity Pathway Analysis (IPA) tool was used to generate a list of the most significant canonical pathways and their respective activation z-scores.
-
Figure 6—source data 1
Differentially expressed (DE) genes found to be responsive to BCG in homozygous WT subjects using the criteria described in the Materials and methods section.
Genes are grouped by up- or down- regulation and ranked in alphabetical order.
- https://doi.org/10.7554/eLife.32340.027
-
Figure 6—source data 2
Differentially expressed (DE) genes found to be responsive to Tw in homozygous WT subjects using the criteria described in the Materials and methods section.
Genes are grouped by up- or down- regulation and ranked in alphabetical order.
- https://doi.org/10.7554/eLife.32340.028
Discussion
WD was initially described as an inflammatory disease (Whipple, 1907) but was subsequently shown to be infectious (Raoult et al., 2000; Relman et al., 1992; Yardley and Hendrix, 1961). We provide evidence that WD is also a genetic disorder. We show here that, in a large multiplex kindred, heterozygosity for the private, LOF R98W mutation of IRF4 underlies an AD form of WD with incomplete penetrance. The causal relationship between IRF4 genotype and WD was demonstrated as follows. First, the IRF4 R98W mutation is the only non-synonymous rare variant segregating with WD in this kindred. Second, the mutation was demonstrated experimentally to be LOF, unlike 146 of 153 other non-synonymous coding IRF4 variants in the general population. Only seven of the 153 non-synonymous coding variants identified (including the five predicted to be LOF in the gnomAD database) were found to be LOF (n = 6) or hypomorphic (n = 1) and they were all extremely rare (cumulative MAF <4 × 10−5). Moreover, IRF4 has evolved under purifying selection, suggesting that deleterious heterozygous variants of this gene entail fitness costs (Barreiro and Quintana-Murci, 2010; Quintana-Murci and Clark, 2013; Rieux-Laucat and Casanova, 2014). Third, EBV-B cells and activated CD4+ T cells heterozygous for IRF4 R98W have a distinctive phenotype, particularly for IRF4 expression in the cytoplasm. This mutation also has a strong functional impact on gene expression in IRF4 R98W-heterozygous PBMCs stimulated with BCG or Tw. These findings unequivocally show that heterozygosity for the R98W allele of IRF4 is the genetic etiology of WD in this kindred. Although we did not find any IRF4 mutations in a pilot cohort of 25 patients with sporadic WD, these and other patients may also develop WD due to other inborn errors of immunity, possibly related to IRF4, as suggested by the apparent genetic heterogeneity and physiological homogeneity underlying severe infectious diseases (Andersen et al., 2015; Casanova, 2015a; Casanova, 2015b; Ciancanelli et al., 2015; Israel et al., 2017; Lamborn et al., 2017; Ogunjimi et al., 2017; Vanhollebeke et al., 2006; Zhang et al., 2018; Zhang et al., 2015). This observation therefore extends our model, in which life-threatening infectious diseases striking otherwise healthy individuals during primary infection can result from single-gene inborn errors of immunity.
In this kindred with AD IRF4 deficiency, haploinsufficiency was identified as the key mechanism, although IRF4 protein levels in the cytoplasmic compartment were higher in patients with the mutation than in wild-type homozygotes. The protein was not more abundant in the nucleus, where IRF4 exerts its effects on transcription. Moreover, half of the IRF4 mRNA in EBV-B cells from heterozygous subjects is WT. The total amount of IRF4 mRNA was higher in the EBV-B cells of heterozygous subjects, but the total amount of IRF4 protein in the nuclear compartment of heterozygous EBV-B and activated CD4+ T cells was similar to that in WT homozygous cells. These data suggest that no more than half the IRF4 protein in the nucleus is WT in heterozygous cells. In addition, not only is IRF4 subject to purifying selection, but also the R98W mutation is itself LOF, with no detectable dominant-negative effect at cell level. Haploinsufficiency is an increasingly recognized mechanism underlying AD inborn errors of immunity (Afzali et al., 2017; Rieux-Laucat and Casanova, 2014). It is commonly due to loss-of-expression alleles, contrasting with the negative dominance typically exerted by expressed proteins, but many mutations are known to cause haploinsufficiency without actually preventing protein production (Afzali et al., 2017; Pérez de Diego et al., 2010; Rieux-Laucat and Casanova, 2014). Haploinsufficiency in this kindred is not due to loss-of-expression of IRF4. Instead, it results from a lack of activity of the R98W IRF4 proteins present in the nucleus.
Incomplete penetrance is common in conditions resulting from haploinsufficiency. In this kindred, incomplete penetrance may result from a lack of Tw infection (in heterozygous individuals IV.5 and VI.7), or a lack of WD development in infected individuals (in heterozygous individuals III.6, IV.4, V.3, V.4, VI.6). All five chronic carriers of Tw were heterozygous for the IRF4 R98W mutation, suggesting that AD IRF4 deficiency also favors the development of chronic Tw carriage. The five asymptomatic carriers were 24 to 82 years old, whereas the four patients were 69 to 92 years old. The impact of IRF4 R98W may therefore increase with age, initially facilitating chronic carriage in Tw-infected individuals, and subsequently predisposing chronic carriers to the development of WD. We cannot exclude the possibility that a modifier allele at another locus contributes to the development of WD in infected heterozygous individuals with IRF4 mutations. Future studies will attempt to define the cellular basis of WD in individuals with IRF4 mutations. The apparently normal development of all peripheral myeloid and lymphoid blood cell subsets studied in patients, and the selective predisposition of these individuals to WD suggest that the disease mechanism is subtle and specifically affects protective immunity to Tw and that it may act in the gastrointestinal (GI) tract. Interestingly, the data of several public databases indicate that IRF4 RNA is expressed in the stomach, colon, and small intestine (https://www.gtexportal.org, http://biogps.org), and that the IRF4 protein is expressed in glandular cells from the stomach, duodenum, small intestine, and rectum (https://www.proteinatlas.org). In addition, a recent analysis of human intestinal macrophage subsets (Bujko et al., 2018) showed IRF4 RNA to be expressed in human intestinal myeloid resident cells. Further studies of GI-tract-resident cells, including myeloid and lymphoid cells in particular, should make it possible to decipher the molecular and cellular mechanisms by which human IRF4 haploinsufficiency underlies WD upon infection by Tw.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
| Gene (Human) | IRF4 (NM_002460.3) | This paper | Vector backbone: pcDNA 3.1D/V5-His-TOPO vector (Thermo Fisher Scientific) | |
| Gene (Human) | PU.1 (NM_001080547.1) | This paper | Vector backbone: pcDNA 3.1D/V5-His-TOPO vector (Thermo Fisher Scientific) | |
| Gene (Human) | BATF (NM_006399.3) | OriGene | RC207104 | |
| Gene (Human) | JUN (NM_002228.3) | OriGene | RC209804 | |
| Strain (Tropheryma whipplei), strain background (DIG APD 25) | Tw | This paper | NCBI taxon: 2039 | Obtained from ‘Research Unit of Infectious and Tropical Emerging Diseases, University Aix-Marseille, URMITE, UM63, CNRS 7278, IRD 198, 13005 Marseille, France, EU’. Strain isolated from mesenteric lymph node (29/01/09). |
| Strain (Mycobacterium bovis-Bacillus Calmette-Guerin) , strain background (pasteur) | BCG | doi: 10.1084/jem.20021769 | NCBI taxon: 33892 | |
| Cell line (Human) | HEK293T | ATCC | CRL-3216 | |
| Cell line (Human) | EBV-B cells | This paper | For each individual, purified B cells were immortalized with EBV in the laboratory | |
| Transfected construct (PGL4.10[luc2]) | (ISRE)3 reporter plasmid, | This paper, backbone: Promega | #E6651 | Obtained from ‘Department of Biotechnology and Food Engineering, Technion-Israel Institute of Technology’ |
| Transfected construct (PGL4.10[luc2]) | AICE reporter plasmid | This paper, backbone: Promega | #E6651 | Generated by metabion international ag |
| Transfected construct (pRL-SV40 vector) | pRL-SV40 vector | Promega | #E2231 | |
| Biological sample (Human) | Patients' blood samples | This paper | ||
| Biological sample (Human) | Controls' blood samples | This paper | ||
| Antibody | anti-IRF4 | Santa Cruz | M-17 | Dilution: 1/1000 |
| Antibody | anti-GAPDH | Santa Cruz | FL-335 | Dilution: 1/1000 |
| Antibody | anti-topoisomerase I | Santa Cruz | C-21 | Dilution: 1/1000 |
| Antibody | anti-lamin A/C | Santa Cruz | H-110 | Dilution: 1/1000 |
| Antibody | anti-CD11b | Miltenyi Biotec | # 130-110-611 | Fluorochrome: PE |
| Antibody | anti-CD86 | Miltenyi Biotec | #130-094-877 | Fluorochrome: PE |
| Antibody | anti-CD206 | Miltenyi Biotec | #130-099-732 | Fluorochrome: PE |
| Antibody | anti-CD209 | Miltenyi Biotec | #130-109-589 | Fluorochrome: PE |
| Antibody | anti-HLADR | Miltenyi Biotec | #130-111-789 | Fluorochrome: PE |
| Antibody | anti-CD20 | BD biosciences | Fluorochrome: PE; clone H1 | |
| Antibody | anti-CD10 | BD biosciences | Fluorochrome: APC, clone HI10a | |
| Antibody | anti-CD27 | BD biosciences | Fluorochrome: PerCP-Cy5.5; clone L128 | |
| Antibody | anti-IgM | Miltenyi | Clone PJ2-22H3 | |
| Antibody | anti-IgG | BD biosciences | Fluorochrome: BV605; clone G18-145 | |
| Antibody | anti-IgA | Miltenyi | Clone IS11-8E10 | |
| Antibody | anti-CD4 | eBioscience | Fluorochrome: Pacific blue; clone OKT4 | |
| Antibody | anti-CD45RA | BD biosciences | Fluorochrome: PerCP-Cy5.5, clone HI100 | |
| Antibody | anti-CCR7 | Sony | Fluorochrome: FITC; clone G043H7 | |
| Recombinant DNA reagent | pcDNA 3.1D/V5-His-TOPO vector | Thermo Fisher Scientific | #K4900-01 | |
| Sequence-based reagent | IRF4-specific primer | Thermo Fisher Scientific | #Hs01056533_m1 | |
| Sequence-based reagent | GUSB | Thermo Fisher Scientific | #4326320E | |
| Peptide, recombinant protein | rhGM-CSF | R and D System | #CAA26822 | |
| Peptide, recombinant protein | rhM-CSF | R and D System | #NP_757350 | |
| Peptide, recombinant protein | IFN-γ | Boehringer Ingelheim | Imukin | |
| Peptide, recombinant protein | rhIL4 | R and D System | #P05112 | |
| Commercial assay or kit | Lipofectamine LTX kit | Thermo Fisher Scientific | #15338100 | |
| Commercial assay or kit | Dual-Luciferase 1000 assay system kit | Promega | #E1980 | |
| Commercial assay or kit | ZR RNA Microprep kit | Zymo research | #R1061 | |
| Commercial assay or kit | High-Capacity RNA-to-cDNA kit | Thermo Fisher Scientific | #R4387406 | |
| Commercial assay or kit | TOPO TA cloning kit | Thermo Fisher Scientific | #K450001 | |
| Commercial assay or kit | directional TOPO expression kit | Thermo Fisher Scientific | #K4900-01 | |
| Commercial assay or kit | QuikChangeII XL Site-Directed Mutagenesis Kit | Agilent Technologies | #200522 | |
| Commercial assay or kit | LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | #L34957 | |
| Chemical compound, drug | Tris | MP biomedicals | #11TRIS01KG | |
| Chemical compound, drug | HCl | Sigma | #H1758 | |
| Chemical compound, drug | NaCl | Sigma | #S3014 | |
| Chemical compound, drug | Triton X-100 | Sigma | #T8532 | |
| Chemical compound, drug | EDTA | MP biomedicals | #11EDTA05M1 | |
| Chemical compound, drug | protease inhibitors Complete | Roche | #04693116001 | |
| Chemical compound, drug | Phosphatase inhibitor cocktail | Roche | #04906837001 | |
| Chemical compound, drug | DTT | Thermo Fisher Scientific | #20290 | |
| Chemical compound, drug | pepstatin A | Sigma | #P4265 | |
| Chemical compound, drug | leupeptin | Sigma | #L2884 | |
| Chemical compound, drug | antipain | Sigma | #A6191 | |
| Chemical compound, drug | Hepes | Sigma | #H3375 | |
| Chemical compound, drug | KCl | Sigma | #P9333 | |
| Chemical compound, drug | EGTA | Amresco | #0732 | |
| Chemical compound, drug | NP40 | Sigma | #N6507 | |
| Chemical compound, drug | NaF | Sigma | #S7920 | |
| Chemical compound, drug | PMSF | Sigma | #P7626 | |
| Chemical compound, drug | MgCl2 | Sigma | #M8266 | |
| Chemical compound, drug | Klenow fragment | NEB | #M0210S | |
| Chemical compound, drug | d-ATP-32P | PerkinElmer | #BLU012H250UC | |
| Chemical compound, drug | TBE migration buffer | Euromedex | #ET020-B | |
| Chemical compound, drug | acrylamide/bis-acrylamide 37.5:1 | Sigma | #A7168 | |
| Software, algorithm | affy R package | Gautier et al., 2004; Irizarry et al., 2003 | ||
| Software, algorithm | IPA software | Alsina et al., 2014 | ||
| Software, algorithm | Microsoft Excel | Microsoft | ||
| Software, algorithm | GraphPad Prism V7.0 | GraphPad | ||
| Software, algorithm | Image studio | Licor |
Patients and family
Request a detailed protocolAll members of the multiplex kindred studied, the pedigree of which is shown in Figure 1A, live in France and are of French descent. Informed consent was obtained from all family members, and the study was approved by the national ethics committee.
Patient 1 (P1, proband) was born in 1948 and presented arthritis of the right knee in 2011, after recurrent episodes of arthritis of this joint associated with effusion since 1980. Tropheryma whipplei (Tw) was detected in synovial fluid by PCR and culture in 2011, but was not detected by PCR in saliva, feces, and small-bowel biopsy specimens. Physical examination revealed a large effusion of the right knee, limiting mobility. The fluid aspirated from this joint contained 4,000 erythrocytes/mm3 and 8,800 leukocytes/mm3, but no crystals or evidence of microbes. Synovial hypertrophy of the right knee and a narrowing of the right internal femoro-tibial joint were detected on MRI. X-ray showed an extension of the right femoro-tibial joint and erosion of the posterior part of the femoro-tibial joint. However, erythrocyte sedimentation rate (ESR) (3 mm/h) and C-reactive protein (CRP) (1.8 mg/l) determinations gave negative results. P1 received methotrexate (15 mg/week) for 4 months, without remission. Antibiotic treatment with doxycycline (200 mg/day) was then immediately initiated. The arthralgia resolved, but right knee effusion persisted. Hydroxychloroquine was therefore added to the treatment regimen. At last follow-up, in 2016, the patient was well.
P2, a second cousin of P1, was born in 1941 and was diagnosed with classical WD and digestive problems in 1978, based on positive periodic acid–Schiff (PAS) staining of a small intestine biopsy specimen. She was treated with sulfamethoxazole/trimethoprim. At last follow-up, in 2016, Tw PCR was positive for the saliva and feces.
P3, the father of P1, was born in 1925 and was diagnosed with classical WD in 1987 on the basis of positive PAS staining of a small intestine biopsy specimen. Clinical manifestations included diarrhea, abdominal pain and weight loss. P3 displayed no extraintestinal manifestations. He was successfully treated with sulfamethoxazole/trimethoprim, with complete clinical and bacteriological remission.
P4, the brother of P2, was born in 1947 and sought medical advice in 2015 for arthralgia affecting the knees and right ulna-carpal joints. The other joints were unaffected. A culture of the joint fluid was negative for bacteria, but Tw was not sought. Tw was not detected in the saliva and feces by PCR or culture, but serological tests for Tw were positive. The fluid aspirated from the right knee contained 4,800 erythrocytes/mm3 and 10,900 leukocytes/mm3 (91% neutrophils and 9% lymphocytes) without crystals. Blood tests revealed an ESR of 30 mm/h and a CRP concentration of 50 mg/l, with no rheumatoid factor, anti-cyclic citrullinated peptide antibodies (anti-CCP) or anti-nuclear antibodies. An X-ray revealed a narrowing of the joint space in the knees and vertebral hyperostosis was visible. The joints of the hands were unaffected. The patient was treated with anti-inflammatory drugs, without success. Treatment with methotrexate and steroids was introduced, followed by antibiotics, the effect of which is currently being evaluated.
Saliva and/or feces samples from 18 other members of the family were checked for the presence of Tw, by a PCR specifically targeting T. whipplei, as previously described (Figure 1A, Figure 1—source data 1) (Edouard et al., 2012). Five individuals were found to be chronic carriers (mean age: 55 years) and 13 were not (mean age: 38 years). Testing was not possible for nine other relatives. The overall distribution of WD in this kindred was suggestive of an AD trait with incomplete penetrance.
Genome-wide analysis
Request a detailed protocolGenome-wide linkage analysis was performed by combining genome-wide array and whole-exome sequencing (WES) data (Belkadi et al., 2016). In total, nine family members were genotyped with the Genome-Wide Human SNP Array 6.0. Genotype calling was achieved with the Affymetrix Power Tools Software Package (http://www.affymetrix.com/estore/partners_programs/programs/developer/tools/powertools.affx). SNPs were selected with population-based filters (Purcell et al., 2007), resulting in the use of 905,420 SNPs for linkage analysis. WES was performed as described in the corresponding section, in four family members, P1, P2, P3 and P4. In total, 64,348 WES variants were retained after application of the following filtering criteria: genotype quality (GQ) >40, minor read ratio (MRR) >0.3, individual depth (DP) >20 x, retaining only diallelic variants with an existing RS number and a call rate of 100%. Parametric multipoint linkage analysis was performed with the Merlin program (Abecasis et al., 2002), using the combined set of 960,267 variants. We assumed an AD mode of inheritance, with a frequency of the deleterious allele of 10−5 and a penetrance varying with age (0.8 above the age of 65 years, and 0.02 below this threshold). Data for the family and for Europeans from the 1000 Genomes project were used to estimate allele frequencies and to define linkage clusters, with an r2 threshold of 0.4.
The method used for WES has been described elsewhere (Bogunovic et al., 2012; Byun et al., 2010). Briefly, genomic DNA extracted from the patients’ blood cells was sheared with a Covaris S2 Ultrasonicator (Covaris). An adapter-ligated library was prepared with the Paired-End Sample Prep kit V1 (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (71 Mb version - Agilent Technologies). Paired-end sequencing was performed on an Illumina Genome Analyzer IIx (Illumina), generating 72- or 100-base reads. We used a BWA-MEM aligner (Li and Durbin, 2009) to align the sequences with the human genome reference sequence (hg19 build). Downstream processing was carried out with the Genome analysis toolkit (GATK) (McKenna et al., 2010) SAMtools (Li et al., 2009), and Picard Tools (http://picard.sourceforge.net). Substitution calls were made with a GATK UnifiedGenotyper, whereas indel calls were made with a SomaticIndelDetectorV2. All calls with a read coverage <2 x and a Phredscaled SNP quality <20 were filtered out. Single-nucleotide variants (SNV) were filtered on the basis of dbSNP135 (http://www.ncbi.nlm.nih.gov/SNP/) and 1000 Genomes (http:browser.1000genomes.org/index.html) data. All variants were annotated with ANNOVAR (Wang et al., 2010). All IRF4 mutations identified by WES were confirmed by Sanger sequencing.
Tw detection
Request a detailed protocolPCR and serological tests for Tw were performed as previously described (Fenollar et al., 2009).
Cell culture and subpopulation separation
Request a detailed protocolPBMCs were isolated by Ficoll-Hypaque density centrifugation (GE Healthcare) from cytopheresis or whole-blood samples obtained from healthy volunteers and patients, respectively. PBMCs and EBV-B cells (purified B cells were immortalized with EBV in the laboratory) were cultured in RPMI medium supplemented with 10% FBS, whereas HEK293T cells (ATCC; CRL-3216) were cultured in DMEM medium supplemented with 10% FBS. Subsets were separated by MACS, using magnetic beads conjugated with the appropriate antibody (Miltenyi Biotec) according to the manufacturer’s protocol. All cells used in this study were tested for mycoplasma contamination and found to be negative.
Site-directed mutagenesis and transient transfection
Request a detailed protocolThe full-length cDNA of IRF4 and PU.1 was inserted into the pcDNA 3.1D/V5-His-TOPO vector with the directional TOPO expression kit (Thermo Fisher Scientific). BATF anf JUN were obtained from Origene companie (#RC207104 and #RC209804, respectively). Constructs carrying mutant alleles were generated from this plasmid by mutagenesis with a site-directed mutagenesis kit (QuikChangeII XL; Agilent Technologies), according to the manufacturer’s instructions. HEK293T cells were transiently transfected with the various constructs, using the Lipofectamine LTX kit (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions.
Cell lysis and western blotting
Request a detailed protocolTotal protein extracts were prepared by mixing cells with lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Triton X-100, and 2 mM EDTA) supplemented with protease inhibitors (Complete, Roche) and phosphatase inhibitor cocktail (PhoStop, Roche), 0.1 mM dithiothreitol DTT (Life Technologies), 10 µg/ml pepstatin A (Sigma, #P4265), 10 µg/ml leupeptin (Sigma, #L2884), 10 µg/ml antipain dihydrochloride (Sigma, #A6191) and incubating for 40 min on ice. A two-step extraction was performed to separate the cytoplasmic and nuclear content of the cells; cells were first mixed with a membrane lysis buffer (10 mM Hepes pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.05 % NP40, 25 mM NaF supplemented with 1 mM PMSF, 1 mM DTT, 10 µg/ml leupeptin, 10 µg/ml aprotinin) and incubated for 30 min on ice. The lysate was centrifuged at 10,000 x g. The supernatant, corresponding to the cytoplasm-enriched fraction, was collected and the nuclear pellet was mixed with nuclear lysis buffer (20 mM Hepes pH 7.9, 0.4 M NaCl, 1,mM EDTA, 1, mM EGTA, 25% glycerol supplemented with 1 mM PMSF, 1 mM DTT, 10 µg/ml leupeptin, 10 µg/ml aprotinin). Equal amounts of protein, according to a Bradford protein assay (BioRad, Hercules, CA), were resolved by SDS-PAGE in a Criterion TGX 10% precast gel (Biorad) and transferred to a low-fluorescence PVDF membrane. Membranes were probed with unconjugated antibody: anti-IRF4 (Santa Cruz, M-17) antibody was used at a dilution of 1:1000 and antibodies against GAPDH (Santa Cruz, FL-335), topoisomerase I (Santa Cruz, C-21), and/or lamin A/C (Santa Cruz, H-110) were used as loading controls. The appropriate HRP-conjugated or infrared dye (IRDye)-conjugated secondary antibodies were incubated with the membrane for the detection of antibody binding by the ChemiDoc MP (Biorad) or Licor Odyssey CLx system (Li-Cor, Lincoln, NE), respectively.
EMSA
Request a detailed protocolDouble-stranded unlabeled oligonucleotides (cold probes) were generated by annealing in TE buffer (pH 7.9) supplemented with 33.3 mM NaCl and 0.67 mM MgCl2. The annealing conditions were 100°C for 5 min, followed by cooling overnight at room temperature. After centrifugation at 3000 x g at 4°C for 30 min, the pellet was suspended in water. We labeled 0.1 µg of cold probe in Klenow buffer supplemented with 9.99 mM dNTP without ATP, 10 U Klenow fragment (NEB) and 50 µCi d-ATP-32P, at 37°C for 60 min. Labeled probes were purified on Illustra MicroSpin G-25 Columns (GE Healthcare Life Sciences), according to the manufacturer’s protocol. We incubated 10 µg of nuclear protein lysate on ice for 30 min with a 32P-labeled (a-dATP) ISRE probe (5′ – gat cGG GAA AGG GAA ACC GAA ACT GAA-3′) designed on the basis of the ISG15 promoter or the λB probe (5′- gat cGC TCT TTA TTT TCC TTC ACT TTG GTT AC-3′) described by Brass et al. in 1999 (Brass et al., 1999). For supershift assays, nuclear protein lysates were incubated for 30 min on ice with 2 µg of anti-IRF4 (Santa Cruz, M-17) antibody or anti-goat Ig (Santa Cruz) antibody. Protein/oligonucleotide mixtures were then subjected to electrophoresis in 12.5% acrylamide/bis-acrylamide 37.5:1 gels in 0.5% TBE migration buffer for 80 min at 200 mA. Gels were dried on Whatman paper at 80°C for 30 min and placed in a phosphor-screen cassette for 5 days. Radioactivity levels were analyzed with the Fluorescent Image Analyzer FLA-3000 system (Fujifilm).
Luciferase reporter assays
Request a detailed protocolThe (ISRE)3 reporter plasmid (pGL4.10[luc2] backbone, Promega #E6651), which contains three repeats of the ISRE sequence separated by spacers, was kindly provided by Prof. Aviva Azriel (Department of Biotechnology and Food Engineering, Technion-Israel Institute of Technology). The AICE reporter plasmid (pGL4.10[luc2] backbone, Promega #E6651) contains part of the IL23R promoter (−254 to −216). HEK293T cells were transiently transfected with the (ISRE)3 reporter plasmid (100 ng/well on a 96-well plate), the pRL-SV40 vector (Promega # E2231, 40 ng/well) and a IRF4 WT or mutant pcDNA 3.1D/V5-His-TOPO plasmid (Invitrogen #K4900-01, 25 ng/well or the amount indicated, made up to 100 ng with empty plasmid), with the Lipofectamine LTX kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. For the AICE assay, we used the same protocol, but with the addition of BATF and JUN expression plasmids (25 ng/well each). Cells were used 24 hr after transfection for the ISRE assay and 48 hr after transfection for the AICE assay, with the Dual-Luciferase 1000 assay system kit (Promega #E1980), according to the manufacturer’s protocol. Signal intensity was determined with a Victor X4 plate reader (Perkin Elmer). Experiments were performed in triplicate and reporter activity is expressed as fold-induction relative to cells transfected with the empty vector. Negative dominance was assessed by performing the same protocol with the following modifications: (ISRE)3 reporter plasmid (100 ng/well for a 96-well plate), pRL-SV40 vector (40 ng/well), +IRF4+ WT and mutant plasmids were used to cotransfect cells, with a constant amount of WT plasmid (25 ng/well) but various amounts of mutant plasmid (25 ng/well alone made up to 100 ng with empty plasmid or with the indicated amount, made up to 100 ng with empty plasmid) amounting to a total of 240 ng/well. For the AICE assay, the same protocol was used, but with the addition of BATF and JUN expression plasmids (25 ng/well each).
Microarrays
Request a detailed protocolFor the microarray analysis of PBMCs, cells from six IRF4-heterozygous individuals (three patients, P1-P3; and three asymptomatic relatives, HET1-HET3) and six IRF4 WT-homozygous individuals (four healthy relatives, WT1-WT4; and two healthy unrelated controls, C1-C2) were dispensed into a 96-well plate at a density of 200,000 cells/well and were infected in vitro with live Tw at a multiplicity of infection (MOI) of 1, or with live BCG (M. bovis-BCG, Pasteur substrain) at a MOI of 20, or were left uninfected (mock). Two wells per condition were combined 24 hr post-infection for total RNA isolation with the ZR RNA Microprep kit (Zymo Research). For the microarray on EBV-B cells, we used 400,000 cells from three IRF4-heterozygous mutation carriers and three WT individuals from the kindred for total RNA isolation with the ZR RNA Microprep kit (Zymo Research). Microarray experiments on both PBMCs and EBV-B cells were performed with the Affymetrix Human Gene 2.0 ST Array. Raw expression data were normalized by the robust multi-array average expression (RMA) method implemented in the affy R package (Gautier et al., 2004; Irizarry et al., 2003). Normalized expression data were processed as previously described (Alsina et al., 2014) and briefly summarized here. First, fold-changes (FC) in expression between mock-infected and BCG-infected or Tw-infected conditions were calculated for each individual separately. For each set of conditions, transcripts were further filtered based on a minimal 1.5 FC in expression (up- or downregulation). In a final stage, transcripts satisfying the previous filters in at least four of the six homozygous WT individuals for each in vitro infection condition were retained for downstream analysis. We counted the differentially expressed (DE) transcripts affected by stimulation in samples from all subjects for each stimulus, and determined the mean counts for these DE transcripts in all homozygotes. The mean values obtained were then used to normalize the counts of DE transcripts, yielding an overall transcriptional responsiveness for each individual separately, and for each stimulus. This overall responsiveness of subjects to either Tw or BCG is shown as a heatmap, and individual subjects were grouped by unsupervised hierarchical clustering. Responsive transcripts were further analyzed with Ingenuity Pathway Analysis (IPA) Software, Version 28820210 (QIAGEN) (Alsina et al., 2014) for functional interpretation. In brief, the FC values for each individual and treatment were used as input data for the identification of canonical pathway enrichment (z-score cut-off set at 0.1). The activation z-score values calculated for the identified pathways were exported from IPA and used to calculate mean values and differences between WT homozygotes and heterozygotes, and for graphical representation, with Microsoft Excel and GraphPad Prism Version 7.0, respectively. The direction of the difference was not considered further. Negative mean difference values were converted into positive values before the ranking of the canonical pathways according to the difference between the genotypes. The microarray data used in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database, under accession number GSE102862.
IRF4 qPCR
Request a detailed protocolTotal RNA was prepared from the EBV-B cells of individuals heterozygous for IRF4 mutations (two patients and two asymptomatic relatives), and healthy homozygous WT relatives (n = 4). We also included samples from unrelated individuals (seven healthy controls and 25 patients with Tw carriage; all IRF4 coding exons for each individual were sequenced and shown to be WT). Moreover, the 25 WD patients included in this experiment were found to have an intact IRF4 cDNA structure and normal levels of IRF4 protein production in EBV-B cells. RNA was prepared from 500,000 cells with the ZR RNA Microprep kit (Zymo Research), according to the manufacturer’s instructions. A mixture of random octamers and oligo dT-16 was used, with the MuLV reverse transcriptase (High-Capacity RNA-to-cDNA kit, Thermo Fisher Scientific), to generate cDNA. Quantitative real-time PCR was performed with the TaqMan Universal PCR Master Mix (Thermo Fisher Scientific), the IRF4-specific primer (Hs01056533_m1, Thermo Fisher Scientific) and the endogenous human β-glucuronidase (GUSB) as a control (4326320E, Thermo Fisher Scientific). Data were analyzed by the ΔΔCt method, with normalization against GUSB.
IRF4 TA-cloning
Request a detailed protocolThe full-length cDNA generated from the EBV-B cells of IRF4-heterozygous and WT homozygous individuals was used for the PCR amplification of exon 3 of IRF4. The products obtained were cloned with the TOPO TA cloning kit (pCR2.1-TOPO TA vector, Thermo Fisher Scientific), according to the manufacturer’s instructions. They were then used to transform chemically competent bacteria, and 100 clones per individual were Sanger-sequenced with M13 primers (forward and reverse).
CD4+ T-cell stimulation by activating anti-CD2/CD3/CD28 mAb-coated beads
Request a detailed protocolIsolated CD4+ T cells from patients (P1 and P3) and from healthy unrelated controls (C1-C4) were either left unstimulated (NS) or stimulated with activating anti-CD2/CD3/CD28 mAb-coated beads (Miltenyi Biotec) for 24 hr in RPMI medium supplemented with 10% FBS (24-well plate).
Differentiation and activation of in vitro monocyte-derived macrophages (MDMs)
Request a detailed protocolAfter isolation, monocytes from P1 or two healthy unrelated controls were plated (24-well plate; 600,000 cells/well) in RPMI medium supplemented with 40% human serum (M1-like) or 10% FBS (M2-like). Differentiation cytokines (R and D Systems) were immediately added: 0.5 ng/ml rhGM-CSF (M1-like) or 20 ng/ml rhM-CSF (M2-like). Every 3 days, we replaced 30% of the medium with fresh complete medium supplemented with the appropriate cytokines. After 14 days of differentiation, cells were left unstimulated (NS) or were activated by incubation with 2.5 ng/ml IFN-γ (M1-like) or 50 ng/ml rh-IL-4 (M2-like) for 48 hr.
MDM surface marker expression
Request a detailed protocolDifferentiated/activated MDMs were detached by treatment with trypsin (1.6 µg/ml) in PBS. Cells were treated with Fc receptor blocking agent (Miltenyi Biotec) and Aqua Dead Cell Stain kit (Thermo Fisher Scientific) for 1 hr. They were then washed and stained for 1 hr at room temperature with appropriate antibodies (see Figure 5—figure supplement 4) and appropriate isotype controls (BD biosciences). Samples were analyzed on a Beckman Coulter Gallios flow cytometer.
Percentage of memory B cells
Request a detailed protocolPBMCs from healthy unrelated controls and patients (P1, P2 and P3) were stained with antibodies against CD20, CD10, and CD27, and IgM, IgD, IgG, or IgA. The percentages of transitional (CD20+ CD10+ CD27-), naïve (CD20+ CD10- CD27-) and memory (CD20+ CD10- CD27+) B cells were determined by flow cytometry. We then assessed the IgM/IgD or IgG or IgA expression of the memory B cells, to determine the extent of Ig isotype switching in the memory compartment.
Ex vivo naïve and effector/memory CD4+ T-cell stimulation
Request a detailed protocolCD4+ T cells were isolated as previously described (Ma et al., 2012). Briefly, cells were labeled with anti-CD4, anti-CD45RA, and anti-CCR7 antibodies, and naive (defined as CD45RA+ CCR7+ CD4+) T cells or effector/memory T cells (defined as CD45RA- CCR7± CD4+) were isolated (>98% purity) with a FACS Aria cell sorter (BD Biosciences). Purified naive or effector/memory CD4+ T cells were cultured with T-cell activation and expansion beads (anti-CD2/CD3/CD28; Miltenyi Biotec) for 5 days. Culture supernatants were then used to assess the secretion of IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17A, IL-17F, IFNγ, and TNFα in a cytometric bead array (BD biosciences), and the secretion of IL-22, by ELISA (Peprotech).
In vitro differentiation of naïve CD4+ T cells
Request a detailed protocolNaïve CD4+ T cells (CD45RA+ CCR7+) were isolated (>98% purity) from healthy unrelated controls or patients, with a FACS Aria sorter (BD Biosciences). They were cultured under polarizing conditions, as previously described (Ma et al., 2016). Briefly, cells were cultured with T-cell activation and expansion beads (anti-CD2/CD3/CD28; Miltenyi Biotec) alone or under Th1 (IL-12 [20 ng/ml; R and D Systems]) or Th17 (TGFβ, IL-1β [20 ng/ml; Peprotech], IL-6 [50 ng/ml; PeproTech], IL-21 [50 ng/ml; PeproTech], IL-23 [20 ng/ml; eBioscience], anti-IL-4 [5 μg/ml], and anti-IFN-γ [5 μg/ml; eBioscience]) polarizing conditions. After 5 days, culture supernatants were used to assess the secretion of the cytokines indicated, by ELISA (IL-22), or with a cytometric bead array (all other cytokines).
Data availability
-
IRF4 haploinsufficiency in a family with Whipple’s diseasePublicly available at the NCBI Gene Expression Omnibus (accession no: GSE102862).
References
-
Functional IRF3 deficiency in a patient with herpes simplex encephalitisThe Journal of Experimental Medicine 212:1371–1379.https://doi.org/10.1084/jem.20142274
-
Acquired resistance to trimethoprim-sulfamethoxazole during Whipple disease and expression of the causative target geneThe Journal of Infectious Diseases 198:101–108.https://doi.org/10.1086/588706
-
From evolutionary genetics to human immunology: how selection shapes host defence genesNature Reviews Genetics 11:17–30.https://doi.org/10.1038/nrg2698
-
Transcriptional and functional profiling defines human small intestinal macrophage subsetsThe Journal of Experimental Medicine 215:441–458.https://doi.org/10.1084/jem.20170057
-
Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcomaThe Journal of Experimental Medicine 207:2307–2312.https://doi.org/10.1084/jem.20101597
-
New insights into Whipple's disease and Tropheryma whipplei infectionsMicrobes and Infection 12:1102–1110.https://doi.org/10.1016/j.micinf.2010.08.001
-
Is there an immune deficit in Whipple's disease?Digestive Diseases and Sciences 26:247–252.https://doi.org/10.1007/BF01391638
-
Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine InterneMedicine 76:170–184.
-
The rise of Tropheryma whipplei: a 12-year retrospective study of PCR diagnoses in our reference centerJournal of Clinical Microbiology 50:3917–3920.https://doi.org/10.1128/JCM.01517-12
-
SnIPRE: selection inference using a Poisson random effects modelPLoS Computational Biology 8:e1002806.https://doi.org/10.1371/journal.pcbi.1002806
-
Crystal structure of PU.1/IRF-4/DNA ternary complexMolecular Cell 10:1097–1105.https://doi.org/10.1016/S1097-2765(02)00703-7
-
A paradoxical Tropheryma whipplei western blot differentiates patients with whipple disease from asymptomatic carriersClinical Infectious Diseases 49:717–723.https://doi.org/10.1086/604717
-
Tropheryma whipplei and Whipple's diseaseJournal of Infection 69:103–112.https://doi.org/10.1016/j.jinf.2014.05.008
-
Prevalence of asymptomatic Tropheryma whipplei carriage among humans and nonhuman primatesThe Journal of Infectious Diseases 197:880–887.https://doi.org/10.1086/528693
-
Whipple endocarditis without overt gastrointestinal disease: report of four casesAnnals of Internal Medicine 131:112–116.https://doi.org/10.7326/0003-4819-131-2-199907200-00007
-
The IRF family transcription factors at the interface of innate and adaptive immune responsesCold Spring Harbor Symposia on Quantitative Biology 78:105–116.https://doi.org/10.1101/sqb.2013.78.020321
-
Failure and relapse after treatment with trimethoprim/sulfamethoxazole in classic Whipple's diseaseJournal of Antimicrobial Chemotherapy 65:2005–2012.https://doi.org/10.1093/jac/dkq263
-
Evidence of lifetime susceptibility to Tropheryma whipplei in patients with Whipple's diseaseJournal of Antimicrobial Chemotherapy 66:1188–1189.https://doi.org/10.1093/jac/dkr032
-
Recurrent rhinovirus infections in a child with inherited MDA5 deficiencyThe Journal of Experimental Medicine 214:1949–1972.https://doi.org/10.1084/jem.20161759
-
The Sequence Alignment/Map format and SAMtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
Unique and shared signaling pathways cooperate to regulate the differentiation of human CD4+ T cells into distinct effector subsetsThe Journal of Experimental Medicine 213:1589–1608.https://doi.org/10.1084/jem.20151467
-
Immunosuppressive therapy in Whipple's disease patients is associated with the appearance of gastrointestinal manifestationsThe American Journal of Gastroenterology 100:1167–1173.https://doi.org/10.1111/j.1572-0241.2005.40128.x
-
Detection of Tropheryma whipplei DNA in feces by PCR using a target capture methodJournal of Clinical Microbiology 40:2466–2471.https://doi.org/10.1128/JCM.40.7.2466-2471.2002
-
Whipple's disease: a review of 19 patients from one hospital and a review of the literature since 1950Medicine 49:175–205.
-
Tropheryma whipplei infection and Whipple's diseaseThe Lancet Infectious Diseases 16:e13–e22.https://doi.org/10.1016/S1473-3099(15)00537-X
-
Whipple's disease: neurological relapse presenting as headache for two yearsJournal of General Internal Medicine 23:2131–2133.https://doi.org/10.1007/s11606-008-0776-9
-
Inborn errors in RNA polymerase III underlie severe varicella zoster virus infectionsJournal of Clinical Investigation 127:3543–3556.https://doi.org/10.1172/JCI92280
-
Whipple's disease in a father-son pairInternal and Emergency Medicine 1:254–256.https://doi.org/10.1007/BF02934754
-
PLINK: a tool set for whole-genome association and population-based linkage analysesThe American Journal of Human Genetics 81:559–575.https://doi.org/10.1086/519795
-
Population genetic tools for dissecting innate immunity in humansNature Reviews Immunology 13:280–293.https://doi.org/10.1038/nri3421
-
Cultivation of the bacillus of Whipple's diseaseNew England Journal of Medicine 342:620–625.https://doi.org/10.1056/NEJM200003023420903
-
Identification of the uncultured bacillus of Whipple's diseaseNew England Journal of Medicine 327:293–301.https://doi.org/10.1056/NEJM199207303270501
-
Whipple's disease: new aspects of pathogenesis and treatmentThe Lancet Infectious Diseases 8:179–190.https://doi.org/10.1016/S1473-3099(08)70042-2
-
IRF4: Immunity. Malignancy! Therapy?Clinical Cancer Research 15:2954–2961.https://doi.org/10.1158/1078-0432.CCR-08-1845
-
IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversityThe Journal of Immunology 174:2573–2581.https://doi.org/10.4049/jimmunol.174.5.2573
-
Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4International Immunology 15:1–10.https://doi.org/10.1093/intimm/dxg001
-
Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-INew England Journal of Medicine 355:2752–2756.https://doi.org/10.1056/NEJMoa063265
-
A hitherto undescribed disease characterized anatomically by deposits of fat and fattyacids in the intestinal and mesentericlymphatic tissuesBulletin of the Johns Hopkins Hospital 18:382.
-
Combined electron and light microscopy in Whipple's disease. Demonstration of "bacillary bodies" in the intestineBulletin of the Johns Hopkins Hospital 109:80–98.
-
Genomics is rapidly advancing precision medicine for immunological disordersNature Immunology 16:1001–1004.https://doi.org/10.1038/ni.3275
Article and author information
Author details
Jacinta Bustamante

Jean-Laurent Casanova
Funding
National Institutes of Health (5R01AI089970-02)
Jean-Laurent Casanova
National Health and Medical Research Council (1042925)
Cindy S Ma Stuart G Tangye
European Research Council (281297)
Lluis Quintana-Murci
Seventh Framework Programme (FP/2007-2013)
Lluis Quintana-Murci
Agence Nationale de la Recherche (ANR-IFNPHOX (ANR-13-ISV3-001-01))
Jacinta Bustamante
Agence Nationale de la Recherche (ANR-10-LABX-62-IBEID)
Lluis Quintana-Murci Laurent Abel Jean-Laurent Casanova
Agence Nationale de la Recherche (ANR-GENMSMD (ANR-16-CE17-0005-01))
Jacinta Bustamante
Agence Nationale de la Recherche (ANR-14-CE14-0008-02)
Lluis Quintana-Murci
Agence Nationale de la Recherche (ANR-14-CE14-0007-02)
Lluis Quintana-Murci
National Health and Medical Research Council (1113904)
Stuart G Tangye Cindy S Ma
Agence Nationale de la Recherche (NKIR-ANR-13-PDOC-0025-01)
Vivien Béziat
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank the patients and their families for participating in the study. We thank Yelena Nemirovskaya, Tatiana Kochetkov, Lahouari Amar, Cécile Patissier, Céline Desvallées, Dominick Papandrea, Mark Woollett, and Amy Gall for technical and secretarial assistance and all members of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. We acknowledge the use of the biological resources of the Imagine Institute DNA biobank (BB-33–00065). AG was supported by ANR-IFNPHOX (ANR-13-ISV3-0001-01), ANR-GENMSMD (ANR-16-CE17-0005-01) and the Imagine Institute. The Laboratory of Human Genetics of Infectious Diseases is supported in part by grants from the St. Giles Foundation, The Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, and the European Research Council (ERC), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French National Research Agency (ANR) under the ‘Investments for the future’ program (grant number ANR-10-IAHU-01), ANR-IFNPHOX (ANR-13-ISV3-0001-01, to JB), ANR-GENMSMD (ANR-16-CE17-0005-01, to JB). Research in the Quintana-Murci laboratory was supported by the Pasteur Institute, the Centre National de la Recherche Scientifique (CNRS), the French Government’s Investissement d’Avenir program, (ANR-10-LABX-62-IBEID), IEIHSEER (ANR-14-CE14-0008-02) and TBPATHGEN (ANR-14-CE14-0007-02), and the European Union’s Seventh Framework Program (FP/2007–2013)/ERC Grant Agreement No. 281297. SGT and CSM are supported by grants and fellowship awarded by the National Health and Medical Research Council of Australia (1113904, 1042925) and the Office of Health and Medical Research of the New South Wales State Government. TN and LW are supported by Australian Postgraduate Research Awards from the University of NSW.
Ethics
Informed consent was obtained from all family members, and the study was approved by the national ethics committee.
Copyright
© 2018, Guérin et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,401
- views
-
- 568
- downloads
-
- 65
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 65
- citations for umbrella DOI https://doi.org/10.7554/eLife.32340
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
IRF4 haploinsufficiency in a family with Whipple’s disease
eLife 7:e32340.
https://doi.org/10.7554/eLife.32340




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}