An assay for de novo kinetochore assembly reveals a key role for the CENP-T pathway in budding yeast
- Howard Hughes Medical Institute, Fred Hutchinson Cancer Research Center, United States
- University of Washington, United States
Abstract
Chromosome segregation depends on the kinetochore, the machine that establishes force-bearing attachments between DNA and spindle microtubules. Kinetochores are formed every cell cycle via a highly regulated process that requires coordinated assembly of multiple subcomplexes on specialized chromatin. To elucidate the underlying mechanisms, we developed an assay to assemble kinetochores de novo using centromeric DNA and budding yeast extracts. Assembly is enhanced by mitotic phosphorylation of the Dsn1 kinetochore protein and generates kinetochores capable of binding microtubules. We used this assay to investigate why kinetochores recruit the microtubule-binding Ndc80 complex via two receptors: the Mis12 complex and CENP-T. Although the CENP-T pathway is non-essential in yeast, we demonstrate that it becomes essential for viability and Ndc80c recruitment when the Mis12 pathway is crippled by defects in Dsn1 phosphorylation. Assembling kinetochores de novo in yeast extracts provides a powerful and genetically tractable method to elucidate critical regulatory events in the future.
https://doi.org/10.7554/eLife.37819.001Introduction
Chromosomes must be accurately segregated to daughter cells during cell division to avoid aneuploidy, a hallmark of birth defects and cancers (Pfau and Amon, 2012). Faithful segregation relies on the attachment of chromosomes to spindle microtubules via the kinetochore, a conserved protein complex that assembles at centromeres (Yamagishi et al., 2014; Musacchio and Desai, 2017). Kinetochores must track dynamically growing and shrinking microtubule tips, monitor for erroneous kinetochore-microtubule attachments, and serve as the platform for the spindle assembly checkpoint (Biggins, 2013; Joglekar and Kukreja, 2017). To carry out these many functions, the kinetochore is a highly regulated, megadalton protein structure composed of many subcomplexes (Figure 1A). Although these subcomplexes must faithfully assemble onto the centromere every cell cycle, the underlying mechanisms that regulate kinetochore assembly are not well understood.
Figure 1 with 2 supplements see all
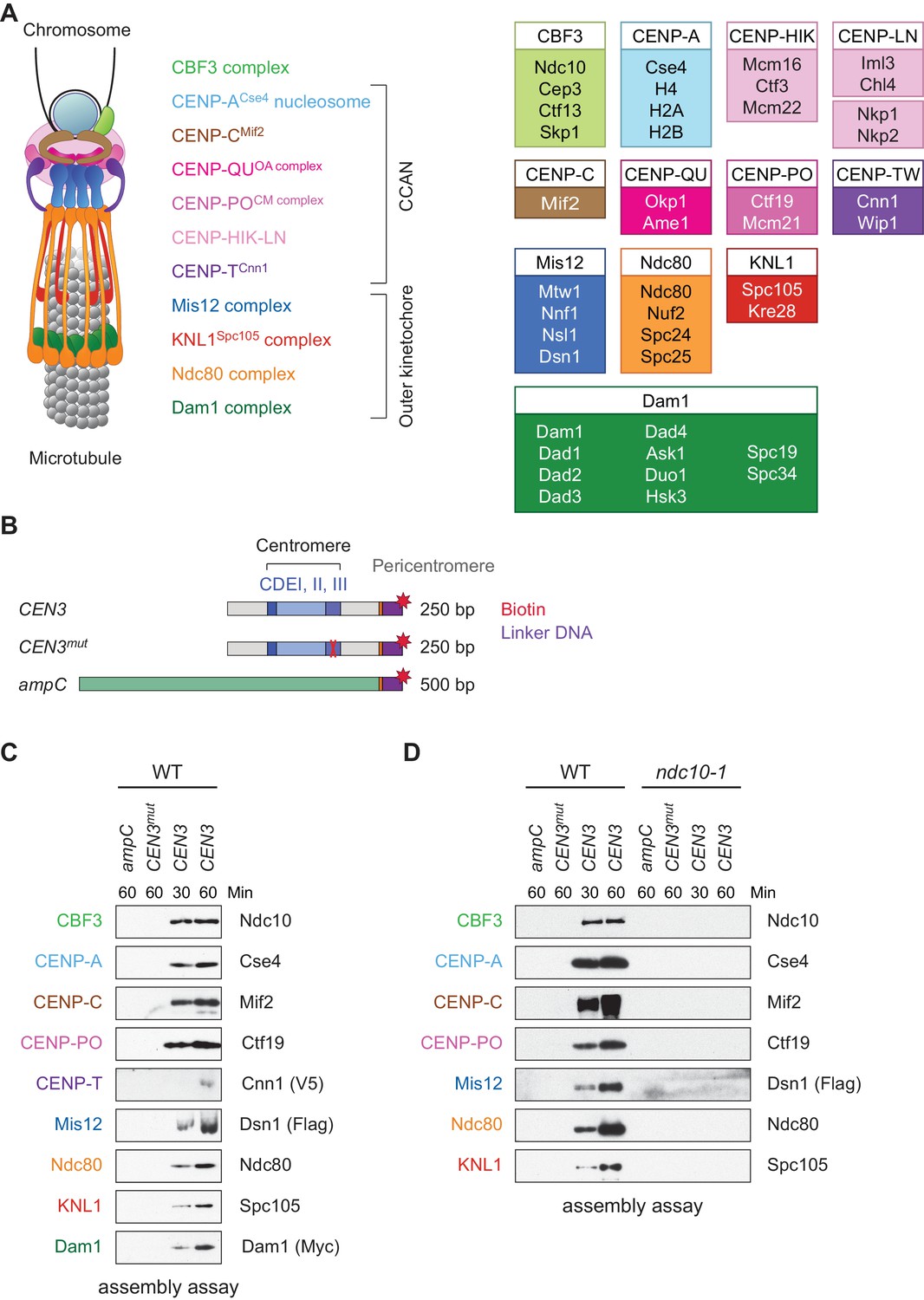
Assembly of kinetochores de novo on a centromere DNA template.
(A) A schematic of the budding yeast kinetochore. Inner kinetochore subcomplexes assemble onto centromeres, serving as the platform for outer kinetochore recruitment. The listed subcomplexes are ordered based on physical interactions, and the yeast proteins in each kinetochore subcomplex are shown on the right. (B) DNA templates for the assembly assay. The templates include 500 bp from the E. coli ampC gene that encodes for β-lactamase (green) as a negative control, the 117 bp chromosome III centromere (CEN3), or a mutant CEN3 (CEN3mut) containing three point mutations in the CBF3 binding site (red ‘X’). The three Centromere-Determining Elements (CDEs) are indicated and ~70 bp of flanking pericentromeric DNA on either side is shown (grey). The DNA templates also contain linker DNA (purple) before the biotinylation (red star) at the 3’ end of the centromere. (C) Kinetochores assembled in vitro are centromere-specific and span the entire kinetochore. The indicated DNA templates were incubated in WT whole cell extracts prepared from a CNN1-3V5 DSN1-3Flag DAM1-9myc strain (SBY17228) for the indicated time (min). DNA-bound proteins were analyzed by immunoblotting with the indicated antibodies. Extracts are shown in Figure 1—figure supplement 1. (D) Kinetochore assembly is inhibited in an ndc10-1 temperature sensitive mutant. Extracts from a DSN1-6His-3Flag strain (SBY8253) or a DSN1-6His-3Flag ndc10-1 strain (SBY8361) shifted to the non-permissive temperature were used for assembly assays. DNA-bound proteins were analyzed by immunoblotting with the indicated antibodies. Extracts in Figure 1—figure supplement 2.
Kinetochores are built on a specialized centromeric chromatin structure, in which canonical histone H3 is replaced with a centromere-specific variant, CENP-A (Earnshaw and Rothfield, 1985; Salmon and Bloom, 2017). Most eukaryotes have complex ‘regional’ centromeres composed of repetitive DNA stretches with interspersed CENP-A- and H3-containing nucleosomes (Blower et al., 2002). The constitutive centromere-associated network (CCAN) binds to centromeric chromatin to form the inner kinetochore and serve as the scaffold for outer kinetochore assembly, which mediates microtubule attachment (Foltz et al., 2006). The Ndc80 complex (‘Ndc80c’) is a key microtubule-binding site within the kinetochore, because it directly mediates attachment and recruits additional attachment factors, such as the Ska complex in vertebrates and its functional ortholog Dam1 in fungi (Cheeseman et al., 2006; DeLuca et al., 2005; Maure et al., 2011; Zhang et al., 2017; Lampert et al., 2013). Interestingly, there are two parallel kinetochore receptors for Ndc80c: the Mis12 complex (‘Mis12c’) and CENP-T (Maskell et al., 2010; Malvezzi et al., 2013; Schleiffer et al., 2012; Gascoigne et al., 2011; Nishino et al., 2013; Nishino et al., 2012). Mis12c interacts with the KNL1 and Ndc80 complexes to create a larger network called KMN (KNL1-Mis12-Ndc80) (Cheeseman et al., 2006). CENP-T, a histone-fold domain containing protein, recruits Ndc80c via the same interaction surface on the Ndc80 complex that binds Mis12c (Nishino et al., 2013; Schleiffer et al., 2012; Malvezzi et al., 2013; Hori et al., 2008; Dimitrova et al., 2016). Although Mis12c and CENP-T each contribute to Ndc80c recruitment in vivo (Malvezzi et al., 2013; Gascoigne et al., 2011), it has been unclear why cells employ two competing receptors for Ndc80c and whether the CENP-T protein functions as a histone at centromeres.
Substantial progress in understanding kinetochore assembly has been made using reconstitution systems in vitro. For example, pre-formed nucleosomal arrays incubated in Xenopus egg extracts assemble microtubule-binding elements that allowed the identification of events required to initiate kinetochore assembly (Guse et al., 2011). Furthermore, the binding selectivity of some kinetochore proteins for CENP-A nucleosomes (over H3 nucleosomes) was recently determined by reconstituting the entire linkage between the CENP-A nucleosome and KMN (Weir et al., 2016). To identify additional events that regulate kinetochore assembly, we set out to develop a reconstitution system that combines the strengths of these previously developed methods with the added ability to genetically manipulate the system and maintain post-translational modifications. To do this, we used budding yeast because they have a simple ‘point’ centromere that is defined by a ~ 125 bp specific DNA sequence and a single microtubule attachment site per chromosome (Winey et al., 1995; Biggins, 2013). The kinetochore subcomplexes and functions are largely conserved, including the specialized chromatin structure containing CENP-ACse4 that serves as the platform for kinetochore assembly. Additionally, dual pathways for Ndc80 recruitment are used in yeast, although the CENP-T ortholog, Cnn1, is not essential for viability (Schleiffer et al., 2012; Bock et al., 2012). However, CENP-TCnn1 contributes to kinetochore function because mutants display increased chromosome loss, and the tethering of CENP-TCnn1 imparts partial stability to acentric minichromosomes via Ndc80 recruitment (Malvezzi et al., 2013; Bock et al., 2012). In yeast, CENP-TCnn1 localization to kinetochores peaks in mitosis as a result of phosphoregulation (Schleiffer et al., 2012; Bock et al., 2012), but it is unclear why its recruitment is cell cycle regulated and whether this affects the Mis12 pathway of Ndc80 recruitment.
Here, we develop a cell-free method to assemble complete kinetochores de novo using centromeric DNA and yeast extracts. We demonstrate that this assay has the same basic requirements as kinetochore assembly in vivo, including the need for the CENP-ACse4 chaperone HJURPScm3, suggesting the formation of a centromeric nucleosome. Conserved mitotic phosphorylation events of the Mis12 complex enhance kinetochore assembly, revealing that the assay is sensitive to key post-translational modifications. Furthermore, this method generates kinetochores that exhibit microtubule-binding activity and employ both Ndc80 recruitment pathways. We applied this assay to identify the requirements for CENP-TCnn1 assembly and find that it requires all other inner kinetochore subcomplexes, suggesting it does not have independent DNA-binding activity. Furthermore, we discovered that the CENP-TCnn1 pathway is required for Ndc80 recruitment and cell viability when the Mis12 pathway is impaired by defects in conserved mitotic phosphoregulation (Akiyoshi et al., 2013a; Kim and Yu, 2015; Yang et al., 2008). Taken together, we have established a kinetochore assembly assay that identifies a critical function for the yeast CENP-TCnn1 pathway and that provides a powerful method to identify other key regulatory events required for kinetochore assembly and function in the future.
Results
Development of a method to assemble kinetochores de novo
Because we had previously identified conditions to purify functional kinetochores from yeast cells (Akiyoshi et al., 2010), we reasoned that these extracts might be permissive for de novo kinetochore assembly. To test this, we linked the chromosome III centromere (117 bp) and ~70 bp of pericentromeric DNA on each side (referred to as ‘CEN3’; Figure 1B) to beads via a biotin tag and incubated it in whole cell yeast extracts in the presence of excess non-specific competitive DNA. As negative controls, we used a template (CEN3mut) with mutations in the centromere determining element III (CDEIII) region of DNA that abolishes kinetochore assembly in vivo (Sorger et al., 1995; Lechner and Carbon, 1991) as well as a 500 bp DNA template from within the E. coli ampC gene. We optimized the extract conditions for assembly in vitro by altering the lysis buffer and method, most notably switching from potassium chloride to potassium glutamate, a salt utilized in other reconstitution assays (Seki and Diffley, 2000; Heller et al., 2011). The assembly reaction was initially performed using an extract prepared from asynchronously growing wildtype (WT) cells and analyzed by immunoblotting against representative components of most kinetochore subcomplexes. Within 30 min of assembly, every protein assayed bound specifically to centromeric DNA (Figure 1C). Inner kinetochore components are generally saturated within 30 min, while outer kinetochore proteins require longer to reach saturation. To compare the efficiency of assembly in various mutants and conditions, we analyzed assembly on CEN3 DNA at two time points hereafter.
To further analyze the composition of the assembled particles, we performed mass spectrometry. We detected 39 out of 49 core kinetochore proteins at higher coverage levels on CEN3 DNA relative to either ampC DNA or CEN3mut DNA (Table 1). In support of centromeric nucleosome assembly, CENP-ACse4 was specifically enriched on CEN3 DNA. Importantly, we detected components from all known kinetochore subcomplexes on CEN3 DNA, including the CENP-TCnn1 protein. The only proteins that were not detected are small proteins that are components of subcomplexes that were otherwise detected in the MS (for example, Dad2 in the Dam1 complex). Together, these data suggest that all kinetochore complexes assemble on centromeric DNA under the conditions we developed.
Table 1
Components from each of the core subcomplexes are detected on assembled kinetochores.
Kinetochores were assembled on ampC, CEN3mut, or CEN3 DNA from an asynchronous WT DSN1-3Flag (SBY14441) extract and analyzed by LC/MS/MS mass spectrometry. The table indicates the human ortholog (if applicable) of each yeast protein, the percent coverage, and the number of unique and total peptides detected from each assembly. We included the only detected microtubule-associated protein.
| Table 1. WT assembled kinetochores | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ampC | ampC | ampC | CEN3mut | CEN3mut | CEN3mut | CEN3 | CEN3 | CEN3 | ||||
| Subcomplex | Yeast Protein | Human Protein | % Coverage | Unique Peptides | Total Peptides | % Coverage | Unique Peptides | Total Peptides | % Coverage | Unique Peptides | Total Peptides | |
| CPC | Ipl1 | Aurora B | Not present | Not present | 23.7 | 8 | 10 | |||||
| Sli15 | INCENP | 7 | 2 | 2 | 14.8 | 6 | 7 | 64.3 | 54 | 113 | ||
| Bir1 | Survivin | 15.1 | 10 | 11 | 22.6 | 14 | 17 | 59.2 | 70 | 177 | ||
| Nbl1 | Borealin | Not present | Not present | 76.7 | 6 | 11 | ||||||
| CCAN | Cbf1 | Cbf1 | Not present | 62.7 | 30 | 77 | 59.3 | 29 | 60 | |||
| Cbf3 | Ndc10 | 11.4 | 7 | 8 | 32.9 | 23 | 26 | 63.2 | 78 | 194 | ||
| Cep3 | 3.5 | 1 | 2 | 15.3 | 8 | 9 | 34.2 | 25 | 76 | |||
| Ctf13 | 2.7 | 1 | 1 | 18.4 | 5 | 5 | 46 | 22 | 38 | |||
| Skp1 | 28.4 | 3 | 3 | 19.6 | 2 | 2 | 41.8 | 12 | 24 | |||
| Nucleosome | Cse4 | CENP-A | 13.1 | 3 | 3 | 24.5 | 6 | 8 | 49.8 | 10 | 31 | |
| Hta2 | H2A | 35.6 | 7 | 20 | 35.6 | 5 | 13 | 35.6 | 6 | 19 | ||
| Htb2 | H2B | 45 | 8 | 18 | 39.7 | 7 | 29 | 39.7 | 7 | 29 | ||
| Hht1 | H3 | 5.1 | 1 | 1 | 5.1 | 1 | 1 | Not present | ||||
| Hhf1 | H4 | 45.6 | 7 | 11 | 56.3 | 8 | 19 | 46.6 | 8 | 13 | ||
| Nucleosome | Psh1 | 3.9 | 1 | 1 | Not present | Not present | ||||||
| Associated | Scm3 | HJURP | Not present | 6.3 | 1 | 1 | 28.3 | 8 | 10 | |||
| Mif2 | Mif2 | CENP-C | Not present | 9.7 | 4 | 4 | 58.7 | 29 | 39 | |||
| OA | Okp1 | CENP-Q | Not present | 20.9 | 7 | 9 | 42.6 | 21 | 34 | |||
| Ame1 | CENP-U | Not present | 20.4 | 5 | 6 | 61.4 | 22 | 41 | ||||
| CM | Ctf19 | CENP-P | Not present | 6.8 | 2 | 2 | 44.7 | 21 | 31 | |||
| Mcm21 | CENP-O | Not present | 10.3 | 3 | 4 | 65.8 | 26 | 39 | ||||
| Iml3 | Iml3 | CENP-L | Not present | Not present | 60.8 | 13 | 19 | |||||
| Chl4 | CENP-N | Not present | 7.9 | 3 | 3 | 37.1 | 16 | 19 | ||||
| Nkp1 | Not present | 26.9 | 4 | 6 | 57.6 | 17 | 28 | |||||
| Nkp2 | Not present | 15 | 2 | 4 | 55.6 | 7 | 10 | |||||
| Ctf3 | Mcm16 | CENP-H | Not present | 22.7 | 2 | 3 | 48.6 | 7 | 11 | |||
| Ctf3 | CENP-I | Not present | 5.3 | 3 | 3 | 23.7 | 17 | 27 | ||||
| Mcm22 | CENP-K | Not present | 25.5 | 3 | 4 | 81.6 | 18 | 27 | ||||
| Cnn1 | Cnn1 | CENP-T | Not present | Not present | 45.7 | 13 | 18 | |||||
| Wip1 | CENP-W | Not present | Not present | 39.3 | 2 | 2 | ||||||
| Mhf1 | CENP-S | 48.9 | 4 | 4 | 48.9 | 3 | 7 | 40 | 2 | 5 | ||
| Mhf2 | CENP-X | 43.8 | 4 | 8 | 47.5 | 4 | 6 | 28.8 | 3 | 4 | ||
| Outer Kt | Mtw1 | Mtw1 | Mis12 | Not present | Not present | 22.8 | 4 | 4 | ||||
| Nnf1 | PMF1 | Not present | Not present | 13.9 | 2 | 2 | ||||||
| Nsl1 | Nsl1 | Not present | Not present | 24.1 | 3 | 3 | ||||||
| Dsn1 | Dsn1 | Not present | Not present | 7.3 | 2 | 2 | ||||||
| Ndc80 | Ndc80 | HEC1 | Not present | Not present | 28.4 | 15 | 16 | |||||
| Nuf2 | NUF2 | Not present | Not present | 32.8 | 12 | 13 | ||||||
| Spc24 | SPC24 | Not present | Not present | 57.3 | 8 | 8 | ||||||
| Spc25 | SPC25 | Not present | Not present | 27.1 | 5 | 5 | ||||||
| Spc105 | Spc105 | KNL1 | Not present | Not present | 5 | 3 | 3 | |||||
| Kre28 | Zwint1 | Not present | Not present | Not present | ||||||||
| Dam1 | Dam1 | Not present | Not present | 10.8 | 2 | 2 | ||||||
| Dad1 | 26.6 | 1 | 1 | 26.6 | 1 | 2 | 26.6 | 1 | 1 | |||
| Dad3 | Not present | Not present | Not present | |||||||||
| Ask1 | Not present | Not present | 8.2 | 1 | 1 | |||||||
| Duo1 | Not present | Not present | 7.3 | 1 | 1 | |||||||
| Hsk3 | 15.9 | 1 | 1 | 15.9 | 1 | 1 | 15.9 | 1 | 1 | |||
| Spc19 | Not present | Not present | 8.5 | 1 | 1 | |||||||
| Spc34 | Not present | Not present | 4.7 | 1 | 2 | |||||||
| Dad2 | Not present | Not present | Not present | |||||||||
| Dad4 | Not present | Not present | Not present | |||||||||
| MAPs | Stu2 | CHTOG | 3.5 | 2 | 2 | Not present | Not present | |||||
We next asked whether the assembly assay reflected requirements in vivo. Kinetochore assembly is initiated by the binding of the CBF3 complex to CDEIII, which facilitates the deposition of CENP-ACse4 (Poddar et al., 2004; Camahort et al., 2007). All kinetochore proteins except Cbf1, which binds directly to CDEI, require the Ndc10 component of the CBF3 complex for their localization in vivo (He et al., 2000). We therefore tested the requirement for CBF3 by performing the assembly assay with extracts prepared from WT cells and an ndc10-1 temperature sensitive mutant. Similar to the negative controls, the assembly reaction was completely inhibited on the CEN3 DNA in the ndc10-1 extracts (Figure 1D). Together, these data indicate that the assembly reaction is initiated by CBF3, consistent with the requirements for assembly in vivo.
Kinetochores assemble on a single CENP-A nucleosome
Kinetochore assembly in vivo requires a CENP-A nucleosome, so we tested whether CENP-ACse4 requires its chaperone HJURPScm3 for deposition (Camahort et al., 2007; Shivaraju et al., 2011; Stoler et al., 2007). To do this, we generated cells containing an auxin-inducible degron (AID) allele of SCM3, scm3-AID, which targets the protein for proteasomal degradation when the TIR1 F-box protein and the hormone auxin are present (Nishimura et al., 2009). Although we could not detect HJURPScm3 protein in extracts due to low intracellular levels, we concluded that the protein was degraded because the cells were inviable when plated on auxin (Figure 2—figure supplement 1). We prepared extracts from scm3-AID strains (with or without TIR1) treated with auxin and performed the assembly assay (Figure 2A). As expected for the most upstream protein in the assembly pathway, Ndc10 associated with CEN3 DNA in the presence or absence of HJURPScm3. However, CENP-ACse4 and all other CCAN components assayed no longer associated with CEN3 when HJURPScm3 was depleted (Figure 2A). This strict requirement for CENP-ACse4 recruitment by its chaperone suggests that CENP-ACse4 is forming a functional nucleosome in vitro.
Figure 2 with 3 supplements see all

Assembled kinetochore particles contain a single, chaperone-dependent CENP-ACse4 nucleosome.
(A) Degradation of HJURPScm3 blocks assembly of the kinetochore beginning with CENP-ACse4. A DSN1-3Flag scm3-EGFP-AID strain (SBY16440) and a DSN1-3Flag scm3-EGFP-AID OsTIR1-myc strain (SBY16438) were treated with auxin and the extracts were used for assembly assays. DNA-bound proteins were analyzed by immunoblotting for the indicated proteins. Extracts in Figure 2—figure supplement 2. (B) Assembly on a centromeric DNA template of only 180 bp is similar to a 250 bp template. Extract from a DSN1-3Flag CNN1-3V5 DAM1-9myc (SBY17228) strain was used for assembly assays with the indicated DNA templates. DNA-bound proteins were analyzed by immunoblotting with the indicated antibodies. Extracts in Figure 2—figure supplement 3.
Centromeric nucleosomes can be detected in the surrounding pericentromeric region in vivo (Coffman et al., 2011; Lawrimore et al., 2011; Wisniewski et al., 2014), leading to debate about whether a single CENP-ACse4 nucleosome is sufficient for kinetochore assembly (Aravamudhan et al., 2013; Furuyama and Biggins, 2007; Wisniewski et al., 2014). We therefore performed the assembly assay using a shorter 180 bp template that cannot form more than one octameric nucleosome. CENP-ACse4 levels were similar on both templates, and the entire kinetochore formed in both cases (Figure 2B). Together, these data suggest that a single, well-positioned centromeric nucleosome is sufficient for kinetochore assembly in the absence of surrounding pericentromeric DNA.
Assembly in vitro is regulated by the cell cycle and phosphorylation
Kinetochore assembly is regulated during the cell cycle and occurs during S phase in budding yeast (Kitamura et al., 2007; Pearson et al., 2004), although it isn’t clear whether this reflects a requirement for active DNA replication or another S phase event. During mitosis, there are dynamic changes in kinetochore composition and CENP-TCnn1 levels at kinetochores peak due to phosphoregulation (Schleiffer et al., 2012; Bock et al., 2012; Dhatchinamoorthy et al., 2017). To test whether the assembly assay is subject to cell cycle regulation, WT cells were grown asynchronously or arrested in G1, S phase, or mitosis, and the extracts were used for in vitro assembly assays. Assembly is least efficient in extracts from cells arrested in G1 and most efficient in S phase and mitosis (Figure 3A), consistent with cell cycle regulation that occurs in vivo. As expected, the CENP-TCnn1 pathway is noticeably enhanced in kinetochores assembled from mitotic extracts. The cellular levels of some proteins, particularly CENP-TCnn1 and CENP-CMif2, are different in the various arrests and it is not clear whether this is due to changes in expression level or solubility. We also note that there appears to be a preference for the assembly of slower-migrating forms of CENP-CMif2 during S phase and mitosis, which may reflect post-translational modifications.
Figure 3 with 1 supplement see all
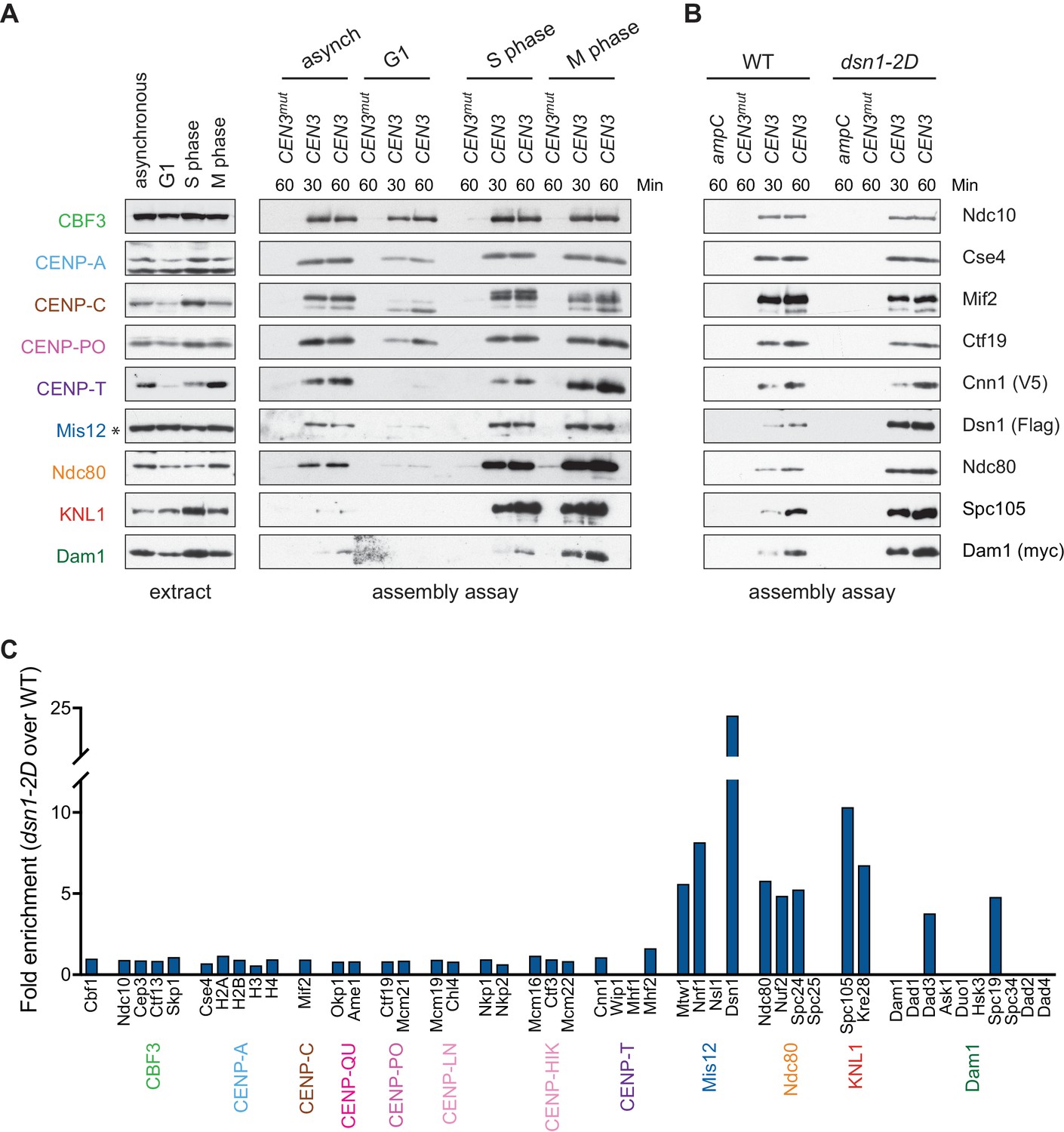
Kinetochore assembly in vitro is regulated by the cell cycle and phosphorylation.
(A) Assembly in vitro is most efficient in extracts made from mitotically arrested cells. Kinetochores were assembled using extract from WT cells (DSN1-3Flag CNN1-3V5 DAM1-9myc (SBY17227)) that were either asynchronously growing or arrested in G1 (with alpha factor), S phase (with hydroxyurea), or early mitosis (with benomyl). Diluted whole cell extracts (left) and DNA-bound proteins (right) were analyzed by immunoblotting with the indicated antibodies. (B) Outer kinetochore assembly is enhanced in dsn1-2D extracts. Assembly assays were performed using extracts prepared from benomyl-arrested DSN1-3Flag CNN1-3V5 DAM1-9myc (SBY17228) and dsn1-2D-3Flag CNN1-3V5 DAM1-9myc (SBY17234) strains on the indicated DNA templates. DNA-bound proteins were analyzed by immunoblotting with the indicated antibodies. Extracts in Figure 3—figure supplement 1. (C) dsn1-2D enhances the assembly of most outer kinetochore proteins by at least 5-fold. Assembly assays were performed using extracts from DSN1-3Flag (SBY14441) and dsn1-2D-3Flag (SBY14151) on CEN3 DNA. Assembled proteins were labeled with tandem mass tags and analyzed by quantitative mass spectrometry. For each protein, the relative abundance in dsn1-2D assembled kinetochores was divided by the relative abundance in WT to calculate the fold enrichment in the dsn1-2D assembled kinetochores.
A conserved mitotic phosphorylation event that promotes kinetochore assembly is Aurora B-mediated phosphorylation of Dsn1, which promotes the interaction between Mis12c and the inner kinetochore protein CENP-CMif2 (Akiyoshi et al., 2013a; Kim and Yu, 2015; Yang et al., 2008; Dimitrova et al., 2016; Petrovic et al., 2016). To test the effects of this phosphorylation on kinetochore assembly in vitro, we made extracts made from a dsn1-S240D, S250D (dsn1-2D) phosphomimetic mutant (Akiyoshi et al., 2013a). While the innermost CCAN proteins were present at equivalent levels, the dsn1-2D assembled kinetochores showed a strong enrichment for outer kinetochore proteins beginning with the Mis12 complex itself when assayed by immunoblotting and mass spectrometry (Figure 3B and Table 2). To directly quantify the difference between WT and dsn1-2D assembly reactions, we performed quantitative mass spectrometry (qMS) using tandem mass tag labeling (McAlister et al., 2014). Although the qMS data does not allow us to analyze the stoichiometry of components within one sample, we were able to compare relative protein levels between WT and dsn1-2D assembled kinetochores. Similar to the immunoblot analysis, there was a strong enrichment of outer kinetochore proteins (3- to 7-fold enrichment) in the dsn1-2D assembled kinetochores while the CCAN levels were similar to WT assembled kinetochores (Figure 3C). Together, these data indicate that our assembly assay in vitro reflects requirements known for assembly in vivo and is sensitive to critical post-translational modifications.
Table 2
Outer kinetochore assembly is enhanced by Dsn1 phosphorylation.
Kinetochores were assembled on the indicated DNA templates from an asynchronous dsn1-2D-3Flag (SBY14151) extract and analyzed by mass spectrometry as in Table 1. We included the detected microtubule-associated proteins.
| Table 2. dsn1-2D assembled kinetochores | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ampC | ampC | ampC | CEN3mut | CEN3mut | CEN3mut | CEN3 | CEN3 | CEN3 | ||||
| Subcomplex | Yeast Protein | Human Protein | % Coverage | Unique Peptides | Total Peptides | % Coverage | Unique Peptides | Total Peptides | % Coverage | Unique Peptides | Total Peptides | |
| CPC | Ipl1 | Aurora B | Not present | Not present | 24.5 | 9 | 15 | |||||
| Sli15 | INCENP | 11 | 4 | 5 | 21.9 | 10 | 11 | 62.6 | 61 | 221 | ||
| Bir1 | Survivin | 20.2 | 13 | 15 | 25.5 | 17 | 20 | 64.3 | 71 | 257 | ||
| Nbl1 | Borealin | 23.3 | 1 | 1 | 21.9 | 1 | 1 | 61.6 | 7 | 19 | ||
| CCAN | Cbf1 | Cbf1 | Not present | 65 | 27 | 53 | 59.3 | 30 | 92 | |||
| Cbf3 | Ndc10 | 21.2 | 2 | 14 | 24.5 | 19 | 22 | 58.9 | 68 | 563 | ||
| Cep3 | 20.6 | 8 | 9 | 15.3 | 8 | 10 | 34.5 | 21 | 111 | |||
| Ctf13 | 2.7 | 1 | 1 | 12.3 | 5 | 5 | 40.6 | 21 | 71 | |||
| Skp1 | 8.8 | 1 | 2 | 22.2 | 3 | 3 | 44.3 | 11 | 32 | |||
| Nucleosome | Cse4 | CENP-A | 20.5 | 5 | 5 | 14 | 4 | 6 | 49.3 | 10 | 34 | |
| Hta2 | H2A | 35.6 | 5 | 13 | 35.6 | 7 | 20 | 35.6 | 5 | 34 | ||
| Htb2 | H2B | 39.7 | 7 | 18 | 39.7 | 7 | 31 | 31.3 | 6 | 29 | ||
| Hht1 | H3 | Not present | 5.1 | 1 | 1 | Not present | ||||||
| Hhf1 | H4 | 56.3 | 9 | 11 | 56.3 | 8 | 16 | 55.3 | 8 | 22 | ||
| Nucleosome Associated | Psh1 | 7.4 | 2 | 2 | Not present | Not present | ||||||
| Scm3 | HJURP | Not present | Not present | 30.5 | 8 | 17 | ||||||
| Mif2 | Mif2 | CENP-C | Not present | 13.7 | 5 | 5 | 55.7 | 27 | 54 | |||
| OA | Okp1 | CENP-Q | Not present | 22.7 | 8 | 9 | 43.6 | 21 | 50 | |||
| Ame1 | CENP-U | Not present | 28.7 | 5 | 5 | 54.9 | 19 | 43 | ||||
| CM | Ctf19 | CENP-P | Not present | 9.8 | 3 | 3 | 42.3 | 17 | 41 | |||
| Mcm21 | CENP-O | Not present | 25.8 | 7 | 8 | 48.6 | 23 | 42 | ||||
| Iml3 | Iml3 | CENP-L | Not present | 15.5 | 3 | 3 | 60.8 | 13 | 28 | |||
| Chl4 | CENP-N | Not present | 7.2 | 3 | 3 | 29 | 12 | 21 | ||||
| Nkp1 | Not present | 26.5 | 4 | 6 | 58 | 18 | 35 | |||||
| Nkp2 | Not present | 15 | 2 | 3 | 35.9 | 5 | 14 | |||||
| Ctf3 | Mcm16 | CENP-H | 14.9 | 1 | 1 | 19.9 | 2 | 2 | 44.8 | 6 | 12 | |
| Ctf3 | CENP-I | Not present | 4 | 2 | 2 | 13.9 | 12 | 19 | ||||
| Mcm22 | CENP-K | Not present | 14.6 | 2 | 2 | 74.9 | 17 | 34 | ||||
| Cnn1 | Cnn1 | CENP-T | Not present | Not present | 27.1 | 8 | 11 | |||||
| Wip1 | CENP-W | Not present | Not present | 21.1 | 2 | 2 | ||||||
| Mhf1 | CENP-S | 48.9 | 3 | 4 | 48.9 | 4 | 9 | 21.1 | 2 | 2 | ||
| Mhf2 | CENP-X | 62.5 | 7 | 8 | 47.5 | 4 | 4 | 28.8 | 2 | 3 | ||
| Outer KT | Mtw1 | Mtw1 | Mis12 | Not present | 18.7 | 4 | 4 | 48.1 | 13 | 21 | ||
| Nnf1 | PMF1 | Not present | 16.9 | 2 | 2 | 30.3 | 10 | 13 | ||||
| Nsl1 | Nsl1 | Not present | 15.3 | 2 | 2 | 69 | 15 | 21 | ||||
| Dsn1 | Dsn1 | Not present | 13.4 | 4 | 4 | 39.2 | 21 | 31 | ||||
| Ndc80 | Ndc80 | HEC1 | Not present | 18.5 | 8 | 9 | 56.4 | 37 | 63 | |||
| Nuf2 | NUF2 | Not present | 15.7 | 6 | 6 | 51 | 27 | 42 | ||||
| Spc24 | SPC24 | Not present | 38.5 | 4 | 5 | 63.4 | 12 | 26 | ||||
| Spc25 | SPC25 | Not present | 8.1 | 1 | 1 | 37.6 | 8 | 10 | ||||
| Spc105 | Spc105 | KNL1 | Not present | 3.1 | 2 | 2 | 45.9 | 41 | 60 | |||
| Kre28 | Zwint1 | Not present | 3.6 | 1 | 1 | 7 | 2 | 2 | ||||
| Dam1 | Dam1 | Not present | Not present | 21.6 | 5 | 6 | ||||||
| Dad1 | 26.6 | 1 | 1 | 26.6 | 1 | 1 | 37.2 | 2 | 4 | |||
| Dad3 | Not present | Not present | 29.8 | 2 | 3 | |||||||
| Ask1 | Not present | Not present | 21.2 | 3 | 4 | |||||||
| Duo1 | Not present | Not present | 18.2 | 4 | 4 | |||||||
| Hsk3 | 15.9 | 1 | 1 | 15.9 | 1 | 1 | 15.9 | 1 | 1 | |||
| Spc19 | Not present | Not present | 30.3 | 4 | 6 | |||||||
| Spc34 | Not present | Not present | 31.5 | 6 | 11 | |||||||
| Dad2 | Not present | Not present | Not present | |||||||||
| Dad4 | Not present | Not present | Not present | |||||||||
| MAPs | Stu2 | CHTOG | Not present | Not present | 33.6 | 23 | 31 | |||||
| Bim1 | Not present | Not present | 17.7 | 4 | 5 | |||||||
| Slk19 | Not present | Not present | 1.6 | 1 | 1 | |||||||
| Bik1 | Not present | Not present | 10 | 3 | 4 | |||||||
Assembled kinetochores are capable of binding microtubules
One of the most fundamental activities of the kinetochore is microtubule binding, so we next tested whether the assembled kinetochores are competent to attach to microtubules. We assembled kinetochores in extracts made from dsn1-2D cells, as well as extracts depleted of the major microtubule-binding component Ndc80 as a control (Cheeseman et al., 2006; DeLuca et al., 2005). Because the Chromosomal Passenger Complex (CPC) can mediate microtubule binding when bound to centromeric DNA in vitro (Sandall et al., 2006), we also performed the experiment in extracts depleted of INCENPSli15, the CPC scaffold protein (Jeyaprakash et al., 2007; Carmena et al., 2012). Although the AID-tagged proteins were significantly depleted from cells after auxin addition (Figure 4A), low levels of Ndc80 remained that were capable of assembling in vitro (Figure 4B). However, the residual levels were not sufficient for the proteins to perform their recruitment functions since Dam1 was absent in the ndc80-AID assembled kinetochores and Aurora BIpl1 was absent from the sli15-AID assembled kinetochores (Jeyaprakash et al., 2007; Klein et al., 2006; Lampert et al., 2013). We incubated the bead-bound assembled kinetochores with either taxol-stabilized microtubules or free tubulin as a negative control. The bound proteins were eluted from the beads and copurifying tubulin was analyzed by fluorescence. Microtubules bound much more robustly than free tubulin to the assembled dsn1-2D kinetochores (Figure 4B). Although the single mutants did not significantly alter microtubule binding, the double ndc80-AID sli15-AID mutant kinetochores were not able to bind microtubules. Thus, kinetochores assembled in vitro are capable of binding to microtubules through the established microtubule-binding interfaces.
Figure 4 with 1 supplement see all
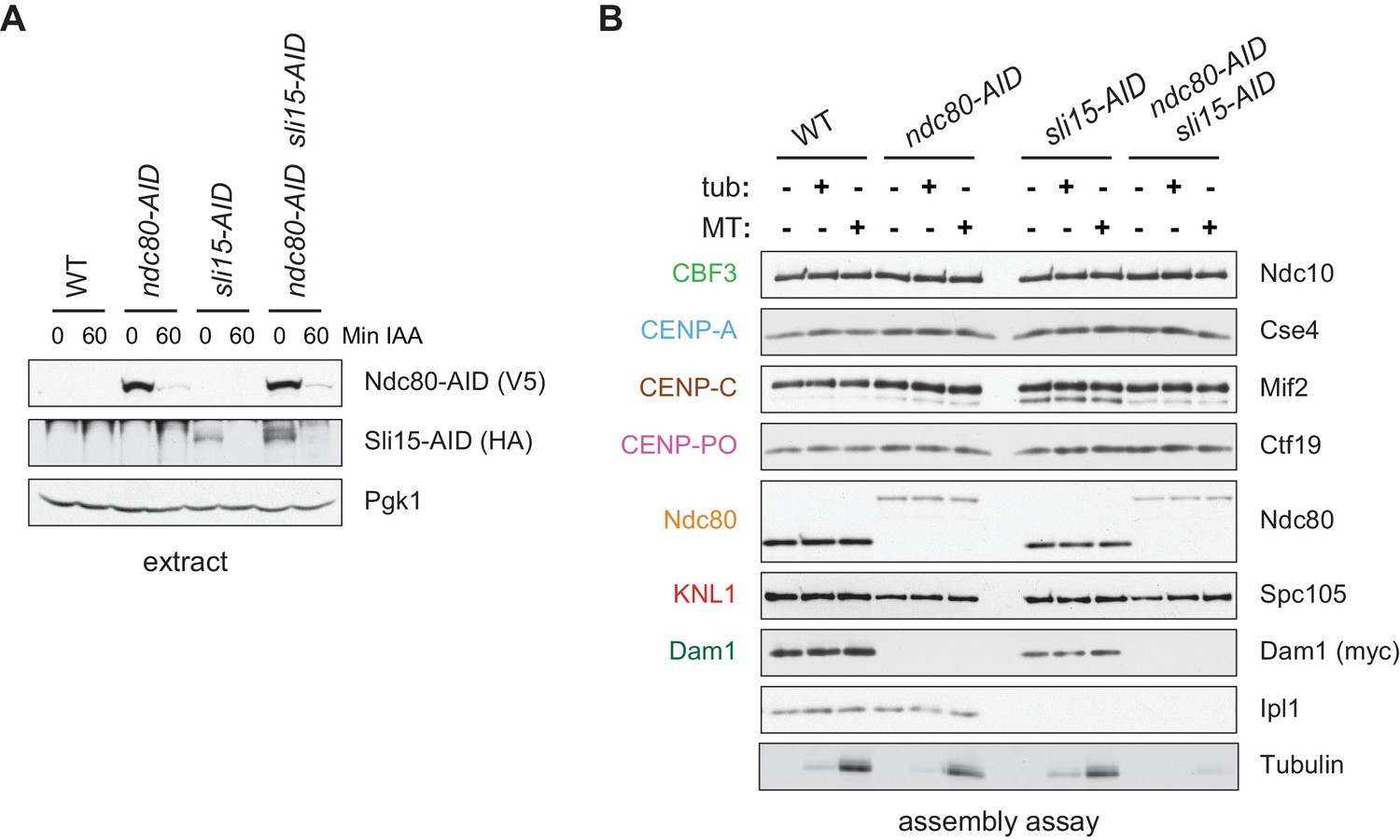
Assembled kinetochore particles bind to microtubules.
(A) The Ndc80-3V5-AID and Sli15-3HA-AID proteins are degraded after one hour of auxin treatment as determined by immunoblotting of whole cell extracts. (B) Assembled kinetochores bind microtubules but not free tubulin. Assembly assays were performed using extracts from the following strains: dsn1-2D-3Flag DAM1-9myc OsTIR1 (SBY14343), dsn1-2D-3Flag DAM1-9myc OsTIR1 ndc80-3V5-AID (SBY14336), dsn1-2D-3Flag DAM1-9myc OsTIR1 sli15-3HA-AID (SBY14890), and dsn1-2D-3Flag DAM1-9myc OsTIR1 ndc80-3V5-AID sli15-3HA-AID (SBY17238). All strains were arrested in benomyl and treated with auxin before harvesting. The assembled kinetochores were then incubated with buffer, free tubulin, or taxol-stabilized microtubules. The free tubulin and the microtubules contained alexa-647-labeled tubulin. DNA-bound proteins were analyzed by immunoblotting with the indicated antibodies, and the tubulin and microtubules were analyzed by fluorescence imaging. The Ndc80-3V5-AID protein migrates slower than untagged Ndc80. Extracts and tubulin input in Figure 4—figure supplement 1.
CENP-TCnn1 requires all CCAN proteins for kinetochore localization
A conserved feature of kinetochore assembly is the recruitment of Ndc80 via two complexes: Mis12c and CENP-TCnn1. The mechanism of CENP-TCnn1 recruitment to the kinetochore has been controversial. CENP-TCnn1 and its partner CENP-W have histone fold domains (HFD) and can tetramerize with two additional HFD proteins, CENP-S/X, to form a nucleosome-like structure in vitro (Nishino et al., 2012; Takeuchi et al., 2014; Schleiffer et al., 2012). The human proteins require their heterotetramerization and DNA-binding capabilities to assemble a functional kinetochore in vivo (Nishino et al., 2012), leading to the model that CENP-TCnn1 forms a unique centromeric chromatin structure. However, CENP-TCnn1 localization to kinetochores requires other CCAN proteins, suggesting it may not directly bind to centromeric DNA (Carroll et al., 2010; Basilico et al., 2014; Samejima et al., 2015; Suzuki et al., 2015; Pekgöz Altunkaya et al., 2016; Logsdon et al., 2015).
To address these issues, we used our DNA-based method to analyze the requirements for CENP-TCnn1 recruitment. The CCAN is composed of distinct subcomplexes and the physical interactions between them have been mapped using co-immunoprecipitation experiments (Schleiffer et al., 2012; Pekgöz Altunkaya et al., 2016) (Figure 1A). To map the CENP-TCnn1 assembly pathway on centromeric DNA, we performed the assembly assay from cells containing a representative mutant of each conserved CCAN subcomplex that had been arrested in mitosis. CENP-TCnn1 is absent or severely reduced in all inner kinetochore mutants tested (Figure 5A–C), indicating that CENP-TCnn1 does not have independent DNA-binding properties in our assay conditions. In addition, CENP-TCnn1 appears to be the most distal component of the CCAN because every other subcomplex is required for its kinetochore localization (Figure 5D) (Pekgöz Altunkaya et al., 2016).
Figure 5
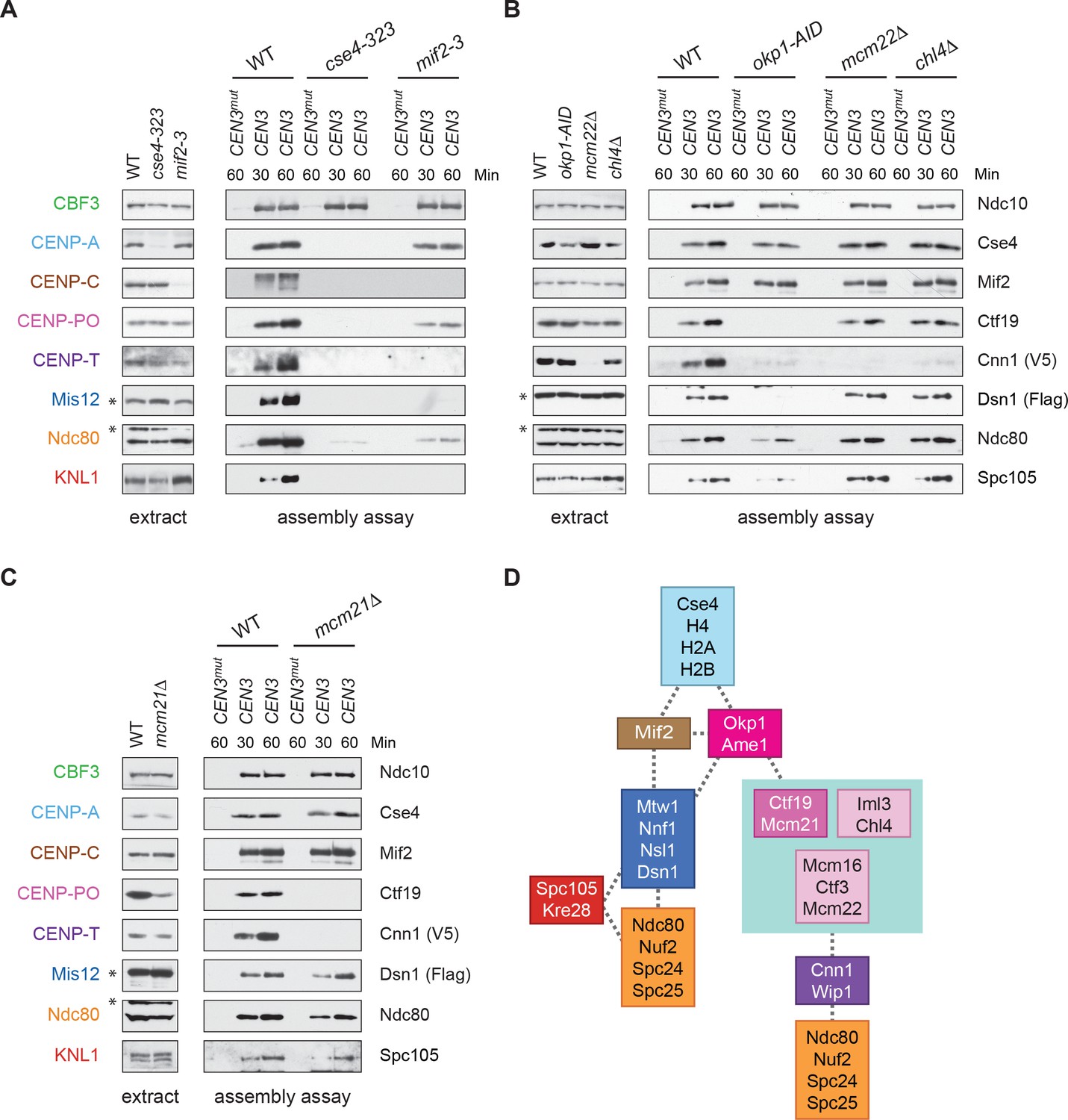
CENP-TCnn1 localization to the kinetochore requires the CCAN.
(A–C) CENP-TCnn1 assembly occurs downstream of all other inner kinetochore components. Assembly assays were performed on the indicated DNA templates using extracts prepared from cells arrested in benomyl. The strains used in (A) were also shifted to the non-permissive temperature for three hours before harvesting: DSN1-3Flag CNN1-3V5 (SBY17230), DSN1-3Flag CNN1-3V5 cse4-323 (SBY17770), and DSN1-3Flag CNN1-3V5 mif2-3 (SBY17603). The strains used in (B) were treated with auxin for three hours before harvesting: DSN1-3Flag CNN1-3V5 (SBY17230), DSN1-3Flag CNN1-3V5 okp1-3V5-AID OsTIR1 (SBY17764), DSN1-3Flag CNN1-3V5 mcm22Δ (SBY17460), and DSN1-3Flag CNN1-3V5 chl4Δ (SBY17607). The strains used in (C) were benomyl treated only: DSN1-3Flag CNN1-3V5 (SBY17230) and DSN1-3Flag CNN1-3V5 mcm21Δ (SBY18304). (D) A schematic distinguishing the proteins involved in the CENP-TCnn1 and Mis12 recruitment pathways.
Kinetochore assembly in vitro utilizes both pathways to Ndc80 recruitment
It has been unclear how CENP-TCnn1 facilitates kinetochore assembly in yeast, because Ndc80 levels are not noticeably reduced by the loss of CENP-TCnn1in vivo (Bock et al., 2012; Schleiffer et al., 2012). We therefore performed the assembly assay in a cnn1Δ extract and found that Ndc80 and KNL1Spc105 levels are slightly reduced, suggesting that CENP-TCnn1 contributes to Ndc80 recruitment (Figure 6). We next compared this to the role of Mis12c in Ndc80 recruitment by performing the assay in an extract from which dsn1-AID had been degraded. Here, Ndc80 assembly is considerably reduced but not absent. To test whether the residual Ndc80 is due to CENP-TCnn1, we analyzed assembly from a cnn1Δ dsn1-AID double mutant and found that Ndc80 recruitment is abolished. Together, these data show that the de novo assay uses both pathways of assembly and that CENP-TCnn1 contributes to Ndc80 recruitment independently of the Mis12 complex.
Figure 6 with 1 supplement see all
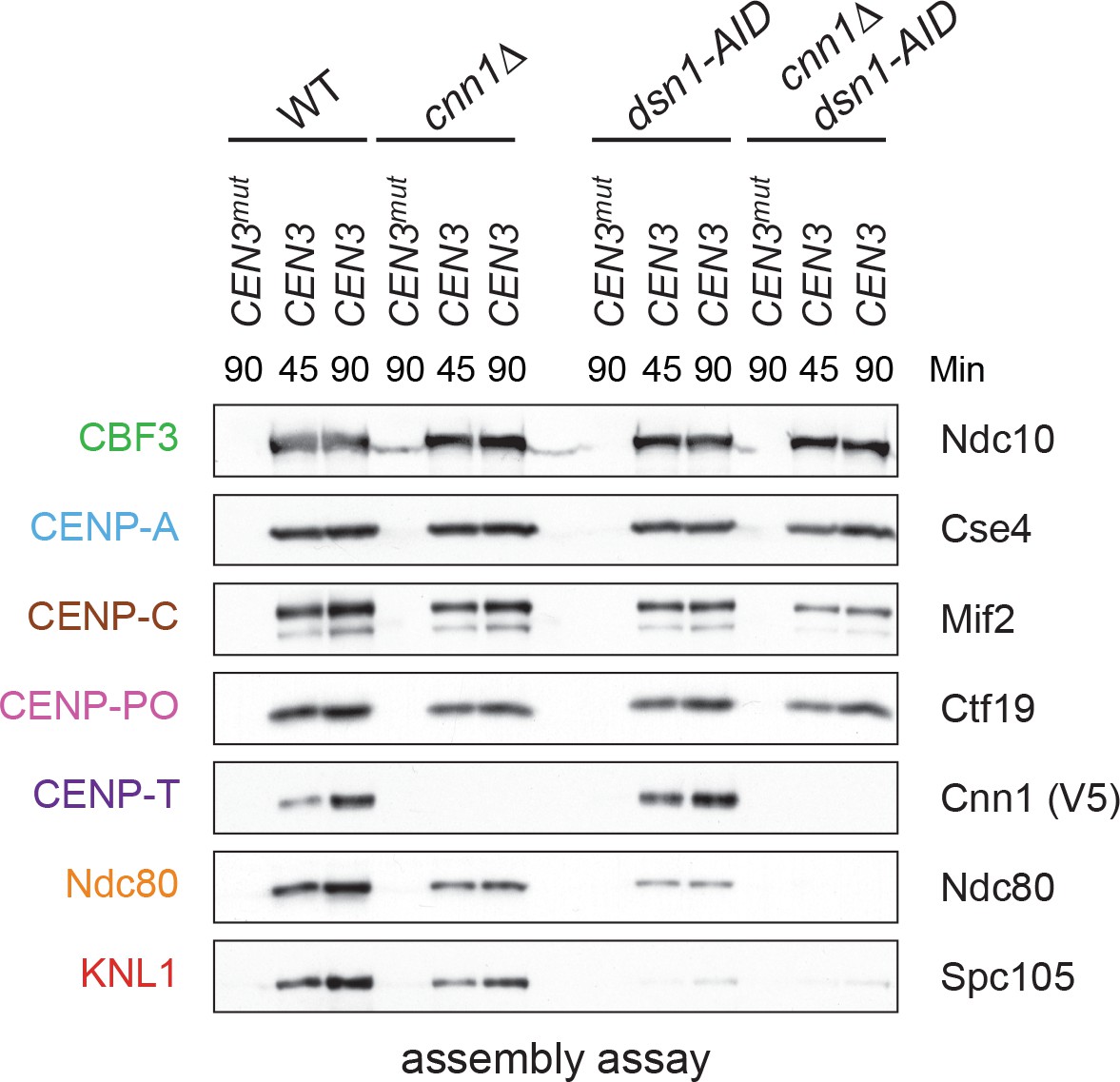
Kinetochore assembly utilizes two pathways for Ndc80 recruitment.
Dsn1 and CENP-TCnn1 both contribute to Ndc80 recruitment. Kinetochores were assembled using extract from WT, dsn1-AID, cnn1Δ, or dsn1-AID cnn1Δ double mutant cells that were arrested in benomyl and treated with auxin: DSN1-3Flag Cnn1-3V5 OsTIR1 (SBY17548), DSN1-3Flag cnn1Δ OsTIR1 (SBY17546), dsn1-3HA-AID Cnn1-3V5 OsTIR1 (SBY17544), and dsn1-3HA-AID cnn1Δ OsTIR1 (SBY17380). Extracts in Figure 6—figure supplement 1.
CENP-TCnn1 is essential for Ndc80 recruitment when Mis12c lacks mitotic phosphorylation
Although CENP-TCnn1 contributes to Ndc80 recruitment, Mis12c is clearly the major Ndc80 receptor in budding yeast (Schleiffer et al., 2012; Bock et al., 2012). Consistent with this, Mis12c is essential for viability while CENP-TCnn1 is non-essential, leading to the question of why yeast have retained the CENP-TCnn1 pathway. We hypothesized that CENP-TCnn1 may compensate for downregulation of the Mis12c pathway when the conserved Aurora B sites on Dsn1 are dephosphorylated. Although yeast cells lacking the Aurora B phosphorylation sites (dsn1-2A) are inviable due to low Dsn1 protein expression, mutating an additional Cdk1 site (serine 264) restores protein levels and viability (dsn1-3A) (Akiyoshi et al., 2013a; Akiyoshi et al., 2013b). Because dsn1-3A weakens the interaction between Mis12c and CENP-CMif2, we postulated that this linkage is not essential due to compensation by the CENP-TCnn1 pathway. Consistent with this, there was synthetic lethality between dsn1-3A and cnn1Δ (Figure 7A), indicating that CENP-TCnn1 becomes essential when the Mis12 pathway is misregulated. This result is similar to previous reports indicating that mutants in CENP-T/W exacerbate mutations in CENP-CMif2 (Schleiffer et al., 2012; Hornung et al., 2014). To further test this, we crossed dsn1-3A to additional mutants in the CENP-TCnn1 pathway (deletions of CENP-KMcm22 and CENP-NChl4). These deletions are also synthetically lethal with dsn1-3A (Figure 7A), indicating that the entire CENP-TCnn1 pathway is essential for viability when the interaction between Dsn1 and CENP-CMif2 is crippled by a lack of Aurora B phosphorylation. These data are consistent with previous observations that a CENP-TCnn1 deletion has synthetic phenotypes with a CENP-CMif2 mutant that cannot bind to Mis12c and with a temperature sensitive allele of the Mis12 complex component NNF1 (Bock et al., 2012; Hornung et al., 2014).
Figure 7 with 3 supplements see all

Cells require CENP-TCnn1 when the Mis12 pathway is impaired.
(A) The CENP-TCnn1 pathway is required for viability when the Mis12c assembly pathway is compromised. Dsn1-3A is synthetic lethal with cnn1Δ and deletions of other genes (MCM22 and CHL4) in the CENP-TCnn1 recruitment pathway. A dsn1-3A strain (SBY14170) was crossed to cnn1Δ (SBY13386), mcm22Δ (SBY6997), and chl4Δ (SBY8788). The meiotic products (tetrads) of the resulting diploids are oriented left to right, haploid spores were genotyped, and double mutants are indicated with circles. (B) A dsn1-3A mcm22-AID double mutant is lethal when treated with auxin. Serial dilutions of the following yeast strains were plated on the indicated media: WT (SBY3), dsn1-3A-3Flag (SBY14170), mcm22-3HA-AID OsTIR1 (SBY17982), and dsn1-3A-3Flag mcm22-3HA-AID OsTIR1 (SBY18171). (C) The CENP-TCnn1 pathway recruits Ndc80 when Mis12 complex assembly is compromised. Assembly was performed with extracts from HU-arrested strains that were treated with auxin: DSN1-3Flag OsTIR1 (SBY14131), DSN1-3Flag mcm22-3HA-AID OsTIR1 (SBY18044), dsn1-3A-3Flag OsTIR1 (SBY14169), and dsn1-3A-3Flag mcm22-3HA-AID OsTIR1 (SBY18034). Extracts in Figure 7—figure supplement 2. (D) WT (SBY18498) and dsn1-3A mcm22-3HA osTIR1 (SBY18324) cells containing MTW1-3GFP were released from G1 and kinetochores were analyzed by fluorescence microscopy during metaphase. The percentage of cells containing mono-lobed, bi-lobed or scattered kinetochores was quantified and a representative picture of the bi-lobed and scattered categories is shown above the graph. The p value for the difference between WT and the double mutant for bi-lobed kinetochores is 0.04 and for scattered kinetochores is 0.036. (E) The sequential order of kinetochore subcomplex recruitment to the DNA, as determined from our data and from (Pekgöz Altunkaya et al., 2016). Dotted lines indicate physical interactions.
We hypothesized that the synthetic lethality exhibited in the double mutant strain is due to a defect in Ndc80 recruitment. To test this, we attempted to generate a conditional double mutant strain to analyze Ndc80 assembly. However, a cnn1-AID allele was hypomorphic and exhibited synthetic lethality with dsn1-3A even in the absence of auxin (data not shown). We therefore generated an mcm22-AID allele that blocks the centromere recruitment of CENP-TCnn1 (Figure 7—figure supplements 1 and 2). The mcm22-AID dsn1-3A double mutant was inviable when auxin was added (Figure 7B). To analyze Ndc80 recruitment, we performed the assembly assay from extracts made from the single and the double mutant cells treated with auxin. Because we expected the double mutant to be deficient for Ndc80 recruitment, we reasoned that the mitotic checkpoint might be compromised and therefore arrested cells in S phase rather than mitosis. As expected, the dsn1-3A extracts showed a considerable decrease in Ndc80 assembly as well as the other KMN components, Dsn1 and KNL1Spc105, and that Ndc80 recruitment was further reduced in the dsn1-3A mcm22-AID double mutant (Figure 7C and Figure 7—figure supplement 3). Despite this reduction in Ndc80, KNL1Spc105 assembly appears unaffected by CENP-KMcm22 degradation, suggesting that the CENP-TCnn1 pathway may specifically recruit Ndc80 and not the full KMN network. To determine how the defect in Ndc80 recruitment we detected in vitro affects kinetochore function in vivo, we analyzed the distribution of kinetochores in the dsn1-3A mcm22-AID mutant. WT and dsn1-3A mcm22-AID cells containing Mtw1-3GFP were released from G1 and analyzed for kinetochore distribution when they were in metaphase. While the majority of WT cells exhibited a normal bi-lobed distribution of kinetochores, the kinetochores were scattered in the double mutant cells indicating a defect in establishing normal kinetochore-microtubule attachments (Figure 7D). Together, our data suggest that the CENP-TCnn1 pathway is required to recruit critical levels of Ndc80 complex to kinetochores to mediate proper kinetochore-microtubule attachments when the centromere recruitment of the Mis12c is compromised by a defect in Aurora B phosphorylation.
Discussion
Kinetochores can be assembled de novo
We developed an assay using centromeric DNA and whole cell yeast extracts to assemble kinetochores de novo. Although similar assays incubating yeast centromeric DNA in extracts were previously developed, none achieved assembly of the outer kinetochore (Ohkuni and Kitagawa, 2011; Sandall et al., 2006; Sorger et al., 1994). By altering the extract conditions, we were able to assemble all known kinetochore subcomplexes on a centromeric DNA template. Outer kinetochore assembly was dramatically enhanced when the extracts were made from cells expressing a conserved phospho-mimetic mutant that promotes kinetochore assembly in vivo (Akiyoshi et al., 2013a; Kim and Yu, 2015; Yang et al., 2008; Dimitrova et al., 2016; Petrovic et al., 2016). In addition, the assembly assay utilizes both conserved pathways for Ndc80 recruitment, and the assembled kinetochores are competent to attach to microtubules in vitro. In the future, it will be important to fully characterize the microtubule binding mode of the assembled kinetochores using biophysical assays.
A number of criteria indicate that the assay we developed reflects kinetochore assembly de novo. First, the assembly of all kinetochore proteins depends on the CBF3 complex, which is required to initiate assembly in vivo (Poddar et al., 2004). Second, CENP-A association with the template requires its chaperone (Camahort et al., 2007; Shivaraju et al., 2011; Stoler et al., 2007). Histone H2A, H2B, and H4 are also present, suggesting that a centromeric nucleosome forms on the DNA. We found that DNA templates capable of wrapping a single centromeric nucleosome efficiently assemble kinetochores, consistent with recent work showing that KMN can link to a single centromeric nucleosome (Weir et al., 2016). In addition, these data demonstrate that pericentromeric chromatin is not required for kinetochore assembly in vitro, although it contributes to kinetochore function in vivo (Bloom, 2014). Third, outer kinetochore protein recruitment depends on the inner kinetochore proteins (Gascoigne and Cheeseman, 2011; Hara and Fukagawa, 2018). Fourth, kinetochore assembly is more efficient in extracts made from cells arrested in S phase or mitosis. Although it was previously shown that yeast kinetochores assemble during S phase, it was not clear if assembly required DNA replication (Kitamura et al., 2007; Pearson et al., 2004). Because replication cannot occur in extracts without sequential kinase treatment (Heller et al., 2011), our assay also shows that active DNA replication is not a strict requirement for kinetochore assembly.
CENP-TCnn1 localization to kinetochores requires the CCAN
Kinetochores recruit the microtubule-binding complex Ndc80 through both the Mis12 complex and CENP-TCnn1 (Nishino et al., 2013; Schleiffer et al., 2012; Malvezzi et al., 2013; Hori et al., 2008). The position of CENP-TCnn1 within the kinetochore has been unclear because it can form a nucleosome-like structure with CENP-W/S/X in vitro (Nishino et al., 2012). However, the CENP-TCnn1 and CENP-ACse4 DNA-binding sites overlap in yeast (Pekgöz Altunkaya et al., 2016), suggesting that CENP-TCnn1 may not directly contact the centromere. We found that all CCAN subcomplexes analyzed are required for CENP-TCnn1 kinetochore localization in vitro, which is generally consistent with work analyzing its localization in human cells (Carroll et al., 2010; Basilico et al., 2014; Samejima et al., 2015; Suzuki et al., 2015; Pekgöz Altunkaya et al., 2016; Logsdon et al., 2015). However, an OA mutant that is defective in the recruitment of other CCAN components does retain some CENP-TCnn1in vivo, suggesting that CENP-TCnn1 localization requirements in vivo may be more complex than revealed by our assay (Thapa et al., 2015; Schmitzberger et al., 2017). Regardless, these data are consistent with the conclusion that CENP-TCnn1 does not have intrinsic DNA binding activity under our assay conditions. In addition, we did not detect CENP-S/XMhf1/2 specifically binding to centromeric DNA, suggesting they are not yeast kinetochore components.
It was previously known that CENP-ACse4 recruits CENP-CMif2 and OA, but the relative order of CCAN components downstream of these complexes was unclear. We therefore combined our data with known physical interactions of each subcomplex to map the order of the pathway from CENP-ACse4 to CENP-TCnn1 (Figure 7D) (Pekgöz Altunkaya et al., 2016). We propose that the OA complex is the bifurcation point of the Mis12c and CENP-TCnn1 assembly pathways, because the CENP-QOkp1 mutant perturbed the assembly of both KMN and CENP-POCtf19-Mcm21, while the CCAN subcomplexes downstream of CENP-CMif2 and OA specifically altered only the CENP-TCnn1 pathway. The CENP-TCnn1 recruitment pathway is therefore comprised of the CENP-PO (CM complex), CENP-HIK, and CENP-LN complexes. We note that all of the non-essential, conserved yeast kinetochore proteins are specific to the CENP-T pathway, providing an explanation for why the yeast kinetochore contains both essential and non-essential proteins.
Functions of the CENP-TCnn1 pathway in budding yeast
In human cells, the CENP-T pathway recruits Ndc80 both directly and indirectly. The CENP-T protein directly binds to two Ndc80 complexes and recruits a third via a phospho-regulated interaction with a Mis12 complex that is also bound to an Ndc80 complex (Huis In 't Veld et al., 2016; Rago et al., 2015). CENP-T knockdown in human cells results in severely decreased Mis12 and KNL1 complexes at kinetochores (Gascoigne et al., 2011; Kim and Yu, 2015). In contrast, we did not find evidence for the recruitment of KMN by CENP-TCnn1 in budding yeast, consistent with data showing that recombinant CENP-TCnn1 does not interact with recombinant Mis12 complex in vitro (Schleiffer et al., 2012). The CCAN mutants we assayed that specifically inhibit the CENP-TCnn1 pathway did not alter Mis12 or KNL1Spc105 assembly. A lack of linkage between CENP-TCnn1 and the Mis12 complex in yeast may also explain why CENP-TCnn1 is non-essential and does not contribute to spindle checkpoint signaling (Schleiffer et al., 2012; Bock et al., 2012). It will be important to further analyze the relationship between the yeast Mis12 complex and CENP-TCnn1 in the future.
The Mis12 pathway is responsible for the majority of Ndc80 recruitment in yeast, so it is surprising that yeast cells are viable when the conserved phosphorylation sites that promote Mis12c localization are mutated (Akiyoshi et al., 2013a; Kim and Yu, 2015; Yang et al., 2008; Dimitrova et al., 2016; Petrovic et al., 2016). Here, we discovered these cells are viable because they use the CENP-TCnn1 pathway to assemble a functional kinetochore. When the CENP-TCnn1 pathway is eliminated in cells lacking Dsn1 phosphorylation, Ndc80 levels are significantly reduced and kinetochores are defective in making normal attachments to microtubules in vivo. A deletion of CENP-TCnn1 has synthetic phenotypes with two other mutants that cripple Mis12c assembly: a CENP-CMif2 truncation lacking its Mis12c binding site and a temperature sensitive allele of NNF1 in the Mis12 complex (Bock et al., 2012; Hornung et al., 2014). Taken together, these data suggest that the CENP-TCnn1 assembly pathway is required to recruit critical levels of Ndc80 when the function of the Mis12c pathway is reduced. CENP-TCnn1 kinetochore levels peak at anaphase (Bock et al., 2012; Dhatchinamoorthy et al., 2017), which is the time when Aurora B-mediated phosphorylation of kinetochore proteins is reversed by phosphatase activity. Therefore, the anaphase enrichment of CENP-TCnn1 might not only increase the load-bearing potential of kinetochore-microtubule attachments by recruiting more Ndc80, but also reinforce kinetochore stability when Aurora B-mediated phosphorylation of Dsn1 is removed. In addition, switching to an Ndc80-recruiting pathway that does not recruit KMN may also help silence the spindle assembly checkpoint, as KNL1 is the critical scaffold for the SAC.
The development of a kinetochore assembly assay de novo has helped to define the two pathways that assemble Ndc80 at kinetochores. Our assay is complementary to a previously developed assembly method using preassembled chromatin templates and frog egg extracts (Guse et al., 2011), but provides the advantage of being genetically tractable. In the future, our assembly assay will be useful for directly examining the role of other post-translational modifications in kinetochore assembly. In addition, it will provide a method to assess the biophysical and structural properties of each Ndc80 recruitment pathway to better understand how cells maintain kinetochore-microtubule attachments to ultimately ensure accurate chromosome segregation.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (S. cerevisiae) | See supplementary file 1 | |||
| Strain, strain background (Saccharomyces cerevisiae) | W303 | |||
| Genetic reagent (S. cerevisiae) | See supplementary file 1 | |||
| Antibody | anti-Ndc10 (rabbit polyclonal) | Desai lab | OD1 | (1:5,000) |
| Antibody | anti-Cse4 (rabbit polyclonal) | Biggins lab | 9536 | (1:500) |
| Antibody | anti-Mif2 (rabbit polyclonal) | Desai lab | OD2 | (1:6,000) |
| Antibody | anti-Ctf19 (rabbit polyclonal) | Desai lab | OD10 | (1:15,000) |
| Antibody | anti-Ndc80 (rabbit polyclonal) | Desai lab | OD4 | (1:10,000) |
| Antibody | anti-Spc105 (rabbit polyclonal) | Biggins lab | PAC4065 | (1:10,000) |
| Antibody | anti-Ipl1 (rabbit polyclonal) | Desai lab | OD121 | (1:300) |
| Antibody | anti-HA (mouse monoclonal) | Roche | 12AC5, Catalog #1-583-816 | (1:10,000) |
| Antibody | anti-V5 (mouse monoclonal) | Invitrogen | Catalog #R960-25 | (1:5,000) |
| Antibody | anti-Flag (mouse monoclonal) | Sigma-Aldrich | Catalog #F3165 | (1:3,000) |
| Antibody | anti-Myc (mouse monoclonal) | Covance | 9E10, Catalog #MMS-150R | (1:10,000) |
| Antibody | anti-mouse secondary (goat monoclonal) | GE Healthcare BioSciences | NA931 | (1:10,000) |
| Antibody | anti-rabbit secondary (goat monoclonal) | GE Healthcare BioSciences | NA934 | (1:10,000) |
| Recombinant DNA reagent | See supplementary file 2 | |||
| Sequence-based reagent | See supplementary file 3 | |||
| Chemical compound, drug | α-factor | United Biochemical Research Inc. | 10 mg/mL | |
| Chemical compound, drug | hydroxyurea | Sigma | H8627 | 0.2M |
| Chemical compound, drug | benomyl | Sigma | 381586–25G | 60 mg/mL |
| Chemical compound, drug | indole-3-acetic acid (IAA) | Sigma | I3750-5G-A | 500 mM |
| Other | Dynabeads M-280 Streptadivin | Invitrogen | 112-05D |
Yeast strain construction
Request a detailed protocolThe Saccharomyces cerevisiae strains used in this study are listed in Supplementary file 1. Standard genetic crosses and media were used to generate and grow yeast (Sherman et al., 1974). Gene deletions, AID alleles, and epitope tagged alleles (3Flag, 9myc, 3 HA, and 3V5) were constructed at the endogenous loci by standard PCR-based integration as described in (Longtine et al., 1998) and confirmed by PCR. DSN1-3Flag, dsn1-2D-3Flag, and dsn1-3A-3Flag were generated by PCR amplification of part of the DSN1 gene, the Flag tags, and URA3 using primers SB4570 and SB4571 on plasmids pSB1113, pSB1115, and pSB1142, respectively. The PCR products were transformed into yeast, and the transformants were confirmed by sequencing. The plasmids and primers used to generate strains are listed in Supplementary file 2 and 3.
Yeast methods
Request a detailed protocolAll liquid cultures were grown in yeast peptone dextrose rich (YPD) media. Cells were arrested in G1 or S phase by adding either 10 μg/mL α-factor in DMSO or 0.2M hydroxyurea, respectively, to log phase cells in liquid culture for three hours until at least 90% of the cells were shmoos (α-factor) or large-budded (hydroxyurea). To arrest cells in mitosis, log phase cultures were diluted 1:1 with liquid media containing 60 μg/mL benomyl and grown for another three hours until at least 90% of cells were large-budded.
Temperature sensitive alleles were inactivated by diluting log phase cultures 1:1 with 37°C liquid media and shifting the cultures to 37°C for 2 hr (ndc10-1) or 3 hr (cse4-323 and mif2-3) before harvesting. For cse4-323 and mif2-3, the added 37°C media included 60 μg/mL benomyl.
For strains with auxin inducible degron (AID) alleles, all cultures used in the experiment were treated with 500 μM indole-3-acetic acid (IAA, dissolved in DMSO) for the final 60 min of growth (scm3-AID, ndc80-AID, sli15-AID, and dsn1-AID) as described in (Nishimura et al., 2009; Miller et al., 2016). For the experiment that included okp1-AID (Figure 5C), all log phase cultures were diluted 1:1 with media containing benomyl and IAA such that the final concentrations were 30 μg/mL benomyl and 500 μM IAA. After two hours, another 150 μM IAA was added, and cultures were harvested after one more hour. For the experiment in Figure 7B, 0.2M hydroxyurea and 500 μM IAA was added to log phase liquid cultures. After two hours, another 150 μM IAA was added, and cultures were harvested after one more hour. For the analysis of kinetochore distribution (Figure 7D), cells were arrested in G1 with alpha factor for three hours and IAA was added during the final hour. The cells were washed and released into media with IAA and harvested after 100 min (when cells were in metaphase) for microscopy analysis. At least 200 cells were analyzed in duplicate biological replicates.
Growth assays were performed by diluting log phase cultures to OD600 ~ 1.0 from which a 1:5 serial dilution series was made. This series was plated on YPD plates that were top-plated with either DMSO or 500 μM IAA plates and incubated at 23°C.
Preparation of DNA templates, Dynabeads, and competitive DNA
Request a detailed protocolPlasmid pSB963 was used to generate the ampC and CEN3 DNA templates and pSB972 was used to generate the CEN3mut template used in this study. DNA templates were generated by PCR using a 5’ primer with pericentromeric homology upstream of the centromere and a biotinylated 3’ primer with linker DNA, an EcoRI restriction site, and pericentromeric homology downstream of the centromere. The latter primer was ordered from Invitrogen with a 5’ biotinylation. Sequences of the primers used to PCR amplify the DNA templates are listed in Supplementary file 3.
The PCR product was purified using the Qiagen PCR Purification Kit and conjugated to Streptadivin-coated Dynabeads (M-280 Streptavidin, Invitrogen) for 2.5 hr at room temperature, using 1 M NaCl, 5 mM Tris-HCl (pH7.5), and 0.5 mM EDTA as the binding and washing buffer. Per 1 mg (100 μL) of beads, we conjugated 1.98 μg/mg of the 180 bp templates, 2.75 μg/mg of the 250 bp centromeric templates, or 5.5 μg/mg of the 500 bp ampC template to have equivalent numbers of templates on beads. After washing three times, the beads were stored in 10 mM HEPES-KOH and 1 mM EDTA at 4°C until use. Competitive DNA was made by sonicating 5 μg/mL salmon sperm DNA in dH2O. The sonicated salmon sperm DNA was stored at −20°C in between uses.
Kinetochore assembly assay
Request a detailed protocolFor a standard kinetochore assembly in vitro, cells were grown in 600 mL of liquid YPD media to log phase and harvested by centrifugation. All subsequent steps were performed on ice with 4°C buffers. Cells were washed once with dH2O with 0.2 mM PMSF, then once with Buffer L (25 mM HEPES pH 7.6, 2 mM MgCl2, 0.1 mM EDTA pH 7.6, 0.5 mM EGTA pH 7.6, 0.1 % NP-40, 175 mM K-Glutamate, and 15% Glycerol) supplemented with protease inhibitors (10 μg/ml leupeptin, 10 μg/ml pepstatin, 10 μg/ml chymostatin, 0.2 mM PMSF), and 2 mM DTT. Cells were resuspended in Buffer L according to the following calculation: (OD of culture) x (number of mL of culture harvested)=number of μL of Buffer L. This suspension was then snap frozen in liquid nitrogen by pipetting drops directly into liquid nitrogen. These dots were then lysed using a Freezer/Mill (SPEX SamplePrep), using 10 rounds that consisted of 2 min of bombarding the dots at 10 cycles per second, then cooling for 2 min. The subsequent powder was thawed on ice and clarified by centrifugation at 16,100 g for 30 min at 4°C. The resulting soluble whole cell extracts (WCE) generally have a concentration of 50–70 mg/mL. The dots, powder, and WCE were stored at −80°C if needed. 5 μL of WCE were saved in a sodium dodecyl sulfate (SDS) buffer for immunoblot analysis.
Typically, 750 μL of whole cell extract was incubated on ice for 15 min with 24.75 μg sonicated salmon sperm DNA (30-fold excess competitive DNA relative to the DNA template on beads). Then, 30 μL of beads pre-conjugated with DNA were added, and the reaction was rotated constantly at room temperature for 30–90 min. The reaction was stopped on ice by addition of 3–5 times the reaction volume of Buffer L. The beads were then washed once with 1 mL Buffer L supplemented with 33 μg/mL of competitive DNA, then three more times with 1 mL Buffer L. Bound proteins were eluted by resuspending the beads in 75 μL of SDS buffer, boiling the beads at 100°C for 3 min, and collecting the supernatant. Samples were stored at −20°C. Bound proteins were examined by immunoblotting, described below. All experiments were repeated two or more times as biological replicates to verify reproducibility and a representative result is reported.
Mass spectrometry
Request a detailed protocolFollowing the standard assembly protocol and washes, assembled kinetochores were washed twice with 1 mL of 50 mM HEPES pH 8, then resuspended in ~60 μL of 0.2% RapiGest SF Surfactant (Waters) in 50 mM HEPES pH 8. Proteins were eluted by gentle agitation at room temperature for 30 min. A small portion of the eluate was added to SDS buffer and analyzed by SDS-PAGE and immunoblotting and/or silver staining. The remaining sample was snap frozen in liquid nitrogen and sent to the Taplin Mass Spectrometry Facility for LC/MS/MS analysis, or to Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School (TCMP@HMS) for TMT labeling and MS3 analysis.
Bulk microtubule-binding assay
Request a detailed protocolMicrotubules were polymerized at 37°C for 15 min using a 1:50 mixture of Alexa-647-labeled and unlabeled bovine tubulin in polymerization buffer [BRB80 (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8), 1 mM GTP, 5.7% (v/v) DMSO, and an additional 4 mM MgCl2]. The polymerization was stopped with the addition of BRB80 and 10 μM taxol. Microtubules were sheared by pulling them through a 27 1/2G needle 10 times, and then pelleted by room temperature centrifugation for 10 min at 170,000 g. Polymerized microtubules were resuspended in BRB80 with 10 μM taxol to approximately 14.4 μM, based on the initial amount of tubulin. Serial dilutions of both the polymerized microtubules and the equivalent amount of initial tubulin mixture were run on an SDS-PAGE gel and analyzed by fluorescence imaging with a Typhoon Trio (GE Healthcare). The amount of tubulin that successfully polymerized was estimated to ensure that comparable amounts of free tubulin and microtubules were introduced to the assembled kinetochores. Assembled and washed kinetochores on beads were resuspended in room temperature Buffer L with 0.9 mg/mL κ-casein, 20 uM taxol, and either ~5 nM tubulin or polymerized microtubules. This reaction was incubated at room temperature with constant rotation for 45 min, then washed twice with room temperature Buffer L, resuspended in SDS buffer, and eluted by boiling. Bound tubulin or microtubules were detected by fluorescence imaging.
Whole cell extracts for AID degradation
Request a detailed protocolWhole cell extracts for immunoblotting were made by freezing cells in liquid nitrogen and resuspending in SDS buffer. Cells were lysed using glass beads and a beadbeater (Biospec Products), then clarified by centrifugation at 16,100 g for 5 min at 4°C.
Immunological methods
Request a detailed protocolWhole cell extract or samples were prepared as described above and separated by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane (BioRad) and standard immunoblotting was performed. Primary and secondary antibodies were used as described in (Miller et al., 2016). Additionally, α-Ndc10, α-Mif2, and α-Ipl1 were generous gifts from Arshad Desai and were used as follows: α-Ndc10 (OD1) 1:5,000; α-Mif2 (OD2) 1:6,000; and α-Ipl1 (OD121) 1:300. We also used α-Cse4 (9536) 1:500 (Pinsky et al., 2003). HRP conjugated secondary antibodies were detected with Pierce enhanced chemiluminescent (ECL) substrate and SuperSignal West Dura and Femto ECL (ThermoFisher Scientific). Note that the immunoblot exposures vary to best represent differences across extracts or assembly samples. The levels of proteins in input extracts and assembly samples can therefore not be directly compared.
Data availability
All data generated during this study are included in the manuscript and supplementary data.
References
-
Centromeric heterochromatin: the primordial segregation machineAnnual Review of Genetics 48:457–484.https://doi.org/10.1146/annurev-genet-120213-092033
-
Conserved organization of centromeric chromatin in flies and humansDevelopmental Cell 2:319–330.https://doi.org/10.1016/S1534-5807(02)00135-1
-
Cnn1 inhibits the interactions between the KMN complexes of the yeast kinetochoreNature Cell Biology 14:614–624.https://doi.org/10.1038/ncb2495
-
The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosisNature Reviews Molecular Cell Biology 13:789–803.https://doi.org/10.1038/nrm3474
-
Dual recognition of CENP-A nucleosomes is required for centromere assemblyThe Journal of Cell Biology 189:1143–1155.https://doi.org/10.1083/jcb.201001013
-
CENP-A exceeds microtubule attachment sites in centromere clusters of both budding and fission yeastThe Journal of Cell Biology 195:563–572.https://doi.org/10.1083/jcb.201106078
-
Hec1 and Nuf2 are core components of the kinetochore outer plate essential for organizing microtubule attachment sitesMolecular Biology of the Cell 16:519–531.https://doi.org/10.1091/mbc.e04-09-0852
-
Structural plasticity of the living kinetochoreThe Journal of Cell Biology 216:3551–3570.https://doi.org/10.1083/jcb.201703152
-
The human CENP-A centromeric nucleosome-associated complexNature Cell Biology 8:458–469.https://doi.org/10.1038/ncb1397
-
Kinetochore assembly: if you build it, they will comeCurrent Opinion in Cell Biology 23:102–108.https://doi.org/10.1016/j.ceb.2010.07.007
-
Kinetochore assembly and disassembly during mitotic entry and exitCurrent Opinion in Cell Biology 52:73–81.https://doi.org/10.1016/j.ceb.2018.02.005
-
A cooperative mechanism drives budding yeast kinetochore assembly downstream of CENP-AThe Journal of Cell Biology 206:509–524.https://doi.org/10.1083/jcb.201403081
-
How kinetochore architecture shapes the mechanisms of its functionCurrent Biology 27:R816–R824.https://doi.org/10.1016/j.cub.2017.06.012
-
Multiple assembly mechanisms anchor the KMN spindle checkpoint platform at human mitotic kinetochoresThe Journal of Cell Biology 208:181–196.https://doi.org/10.1083/jcb.201407074
-
Kinetochore microtubule interaction during S phase in Saccharomyces cerevisiaeGenes & Development 21:3319–3330.https://doi.org/10.1101/gad.449407
-
Molecular requirements for the formation of a kinetochore-microtubule interface by Dam1 and Ndc80 complexesThe Journal of Cell Biology 200:21–30.https://doi.org/10.1083/jcb.201210091
-
Point centromeres contain more than a single centromere-specific Cse4 (CENP-A) nucleosomeThe Journal of Cell Biology 195:573–582.https://doi.org/10.1083/jcb.201106036
-
Both tails and the centromere targeting domain of CENP-A are required for centromere establishmentThe Journal of Cell Biology 208:521–531.https://doi.org/10.1083/jcb.201412011
-
Molecular architecture and assembly of the yeast kinetochore MIND complexThe Journal of Cell Biology 190:823–834.https://doi.org/10.1083/jcb.201002059
-
CENP-T provides a structural platform for outer kinetochore assemblyThe EMBO Journal 32:424–436.https://doi.org/10.1038/emboj.2012.348
-
Whole-proteome genetic analysis of dependencies in assembly of a vertebrate kinetochoreThe Journal of Cell Biology 211:1141–1156.https://doi.org/10.1083/jcb.201508072
-
CENP-T proteins are conserved centromere receptors of the Ndc80 complexNature Cell Biology 14:604–613.https://doi.org/10.1038/ncb2493
-
BookMethods in Yeast GeneticsCold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
-
Scm3 is a centromeric nucleosome assembly factorJournal of Biological Chemistry 286:12016–12023.https://doi.org/10.1074/jbc.M110.183640
-
Factors required for the binding of reassembled yeast kinetochores to microtubules in vitroThe Journal of Cell Biology 127:995–1008.https://doi.org/10.1083/jcb.127.4.995
-
A quantitative description of Ndc80 complex linkage to human kinetochoresNature Communications 6:8161.https://doi.org/10.1038/ncomms9161
-
The centromeric nucleosome-like CENP-T-W-S-X complex induces positive supercoils into DNANucleic Acids Research 42:1644–1655.https://doi.org/10.1093/nar/gkt1124
-
Three-dimensional ultrastructural analysis of the Saccharomyces cerevisiae mitotic spindleThe Journal of Cell Biology 129:1601–1615.https://doi.org/10.1083/jcb.129.6.1601
-
Kinetochore composition and its function: lessons from yeastsFEMS Microbiology Reviews 38:185–200.https://doi.org/10.1111/1574-6976.12049
-
Phosphorylation of HsMis13 by Aurora B kinase is essential for assembly of functional kinetochoreJournal of Biological Chemistry 283:26726–26736.https://doi.org/10.1074/jbc.M804207200
Article and author information
Author details
Funding
National Institutes of Health (5R01GM064386-17)
- Sue Biggins
Howard Hughes Medical Institute (Investigator Award)
- Sue Biggins
National Institutes of Health (Chromosome metabolism training grant)
- Jackie Lang
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are grateful to Arshad Desai for generously providing many of the antibodies used in this study, and to Erman Akbay for assisting with mass spectrometry data analysis. We thank Phil Gafken and Yuko Ogata from the Fred Hutch Proteomics Facility, Ross Tomaino from the Taplin Mass Spectrometry Facility, and Ryan Kunz from TCMP@HMS for providing experimental advice and performing mass spectrometry. We thank all members of the Biggins lab for strains and insightful discussions. Finally, we thank the Biggins lab, Chip Asbury, Josh Larson and Toshi Tsukiyama for critical comments on the manuscript. The authors declare no competing financial interests.
Copyright
© 2018, Lang et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,063
- views
-
- 411
- downloads
-
- 69
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 69
- citations for umbrella DOI https://doi.org/10.7554/eLife.37819
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
An assay for de novo kinetochore assembly reveals a key role for the CENP-T pathway in budding yeast
eLife 7:e37819.
https://doi.org/10.7554/eLife.37819




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}