Adaptation to constant light requires Fic-mediated AMPylation of BiP to protect against reversible photoreceptor degeneration
- UT Southwestern Medical Center, United States
- Howard Hughes Medical Institute, United States
Abstract
In response to environmental, developmental, and pathological stressors, cells engage homeostatic pathways to maintain their function. Among these pathways, the Unfolded Protein Response protects cells from the accumulation of misfolded proteins in the ER. Depending on ER stress levels, the ER-resident Fic protein catalyzes AMPylation or de-AMPylation of BiP, the major ER chaperone and regulator of the Unfolded Protein Response. This work elucidates the importance of the reversible AMPylation of BiP in maintaining the Drosophila visual system in response to stress. After 72 hr of constant light, photoreceptors of fic-null and AMPylation-resistant BiPT366A mutants, but not wild-type flies, display loss of synaptic function, disintegration of rhabdomeres, and excessive activation of ER stress reporters. Strikingly, this phenotype is reversible: photoreceptors regain their structure and function within 72 hr once returned to a standard light:dark cycle. These findings show that Fic-mediated AMPylation of BiP is required for neurons to adapt to transient stress demands.
https://doi.org/10.7554/eLife.38752.001Introduction
Post-translational modifications (PTMs) of proteins are important for rapid responses to environmental challenges of cells. One such PTM is AMPylation, the reversible addition of adenosine monophosphate (AMP) to hydroxyl groups (also known as adenylylation) (Kingdon et al., 1967; Brown et al., 1971; Woolery et al., 2014; Casey and Orth, 2018). AMPylation is catalyzed by at least two protein families, among them the conserved Fic-domain proteins (Harms et al., 2016; Casey and Orth, 2018). Eukaryotic Fic, an ER-resident type-II membrane protein (Rahman et al., 2012), AMPylates BiP (GRP78), a highly conserved and ubiquitous ER chaperone (Ham et al., 2014; Preissler et al., 2015). Working together with a multitude of associated quality control proteins, BiP is critical for the translocation, folding, and secretion of proteins from the ER as well as for aiding in the clearing of misfolded ER aggregates and degradation of membrane-associated proteins (Hendershot et al., 1988; Kozutsumi et al., 1988; Meunier et al., 2002). BiP is both a mediator and transcriptional target of the Unfolded Protein Response (UPR), a coordinated cell signaling pathway that is activated during times of high misfolded protein levels in the ER. Like many protein chaperones, BiP depends on its ATPase activity to undergo a conformational change to bind to its substrates (Gaut and Hendershot, 1993). AMPylation locks BiP into a state resembling the ATP-bound conformation with high substrate off-rates, thereby inhibiting its chaperone function (Preissler et al., 2017b; Wieteska et al., 2017).
In agreement with this PTM’s inhibitory role, BiP AMPylation levels are linked to protein homeostasis (Ham et al., 2014). Reduction of ER protein load promotes Fic-mediated AMPylation of BiP, whereas Fic catalyzes the deAMPylation of BiP under elevated ER stress conditions (Ham et al., 2014; Preissler et al., 2015). This switch in Fic’s activity is linked to a key regulatory salt bridge in eukaryotic Fic. Mutations in Fic that disrupt this salt bridge result in an overactive AMPylator that lacks deAMPylation activity (Casey et al., 2017; Preissler et al., 2017a). Together, these studies suggest a model in which BiP is AMPylated in times of low ER stress, creating a reserve pool of inactive BiP that can be readily activated to respond to changes of ER homeostasis (Figure 1A). This reserve pool of BiP is proposed to act as a buffer to attenuate or shorten the need for a more dramatic activation of the transcriptional and translational arms of the UPR (Casey et al., 2017; Preissler et al., 2017a; Wieteska et al., 2017). However, the physiological importance of endogenous Fic-mediated AMPylation remains unclear.
Figure 1 with 3 supplements see all

BiP is a target of Fic AMPylation and deAMPylation in vivo.
(A) BiP AMPylation during times of low ER stress reserves a portion of the chaperone to allow for a rapid, deAMPylation-driven, response to high ER stress (Casey et al., 2017; Preissler et al., 2017a). (B) Bar graphs show the percentage of null mutant BiPG0102/y males rescued by the indicated genomic BiPWT, BiPT366A or BiPT518A genomic transgene (Tg) relative to sibling controls. N = 3 biological replicas. At least 50 flies scored for each replica. Bar graphs show means ± Standard Deviation (SD). (C) Bar graphs show the percentage of viable flies of the indicated wild-type or fic30C genotypes expressing the overactive FicE247G under the ubiquitous Da-Gal4 driver relative to sibling controls. Among the indicated genomic BiP transgenes, only BiPT366A provides partial rescue of lethality in the BiP+/+ background and near complete rescue in a BiPG0102 null background. N = 3 biological replicas. At least 100 total flies scored for each replica. Bar graphs show means ± SD.
-
Figure 1—source data 1
Relates to Figure 1B and C.
Quantification of fly survival.
- https://doi.org/10.7554/eLife.38752.006
In the fruit fly, Drosophila melanogaster, we previously demonstrated that fic-null mutants harbor a defect in visual signaling, as assessed by electroretinogram (ERG). The well-characterized Drosophila visual system has proven a valuable model for many fields, such as neuroscience (Borycz et al., 2002; Sugie et al., 2015), cell signaling (Dolph et al., 1993; Scott et al., 1995), protein trafficking (Lee et al., 2003; Akbar et al., 2009), and neurodegeneration (Leonard et al., 1992; Johnson et al., 2002; Ryoo et al., 2007). The specialized photoreceptor cells possess tightly packed microvilli-like membranes, termed rhabdomeres, that endow remarkable sensitivity to minute changes in light conditions (Montell, 2012). The ability to maintain this sensitivity is critical for flight behavior, foraging, and escape from predators. Thus, under a wide range of conditions, photoreceptors must maintain their light detection cascade, which requires the constant production, trafficking, and degradation of proteins through the endomembrane system (Colley et al., 1995; Kiselev et al., 2000; Rosenbaum et al., 2006).
We postulated that as a regulator of proteostasis and the UPR, BiP must be tightly regulated for proper maintenance of vision in the fly. Here we demonstrate that mutants lacking AMPylation of BiP at a specific residue, Thr366, possess the same ERG defects as fic-null animals, implicating BiP as the target of Fic necessary for visual signaling. We go on to find that application of an eye-specific stress, constant light, leads to exaggerated electrophysiology defects and the loss of photoreceptor rhabdomeres, akin to neurodegeneration. However, these defects exhibit a remarkable and unique reversibility: structural and functional phenotypes observed in light-stressed fic-null and AMPylation-resistant BiPT366A mutants are reversed after the flies are returned to a standard light/dark cycle. In addition, we identify changes in the regulation of UPR during constant light stress in these mutants, implicating dysregulation of ER homeostasis as a probable cause of the inability to adapt to altered light conditions.
Results
BiPT366A rescues over-expression of constitutively active AMPylating FicE247G
To test the hypothesis that BiP is a critical target of Fic AMPylation in vivo (Figure 1A), we designed and generated transgenes expressing wild-type and AMPylation-resistant FLAG-tagged BiP proteins under control of the endogenous BiP promoter (Figure 1—figure supplement 1A). BiP null fly mutants die early in development; this lethality is rescued by including a copy of the genomic transgene expressing FLAG-BiPWT or the AMPylation-resistant FLAG-BiPT366A mutant (Figure 1B). We will refer to these rescued flies as BiPWT or BiPT366A, respectively. In contrast, a BiP transgene mutated at a second reported AMPylation site (Preissler et al., 2015; Casey et al., 2017), BiPT518A, did not rescue BiP-/- lethality. As fic null mutants that lack BiP AMPylation are viable, the lethality of the BiPT518A mutant is not likely to be due to the loss of AMPylation (Casey et al., 2017). Instead, these observations indicate an essential role for Thr518 in BiP’s chaperone activity. Consistent with this notion, the equivalent residue, Thr538, in the S. cerevisiae BiP homolog Kar2 is required for survival under heat stress even though yeast lack both Fic domain proteins and BiP AMPylation (Figure 1—figure supplement 1B).
Previously, we reported that over-expression of the constitutively active AMPylating FicE247G was lethal in a fic-null fly background (fic30C) because it lacks the essential deAMPylation activity (Casey et al., 2017). We tested whether flies expressing the AMPylation-resistant BiPT366A could survive this lethality. Consistent with previous findings, we observe over-expression of the mutant UAS-FicE247G transgene using the ubiquitous Da-Gal4 driver was lethal in an otherwise fic-null animal (Figure 1C). Lethality of the constitutively active AMPylating FicE247G was suppressed in flies expressing the AMPylation-resistant BiPT366A but not BiPWT (Figure 1C). These results indicate that Thr366 of BiP is a physiologically relevant AMPylation target in vivo.
The UPR protects eyes from overactive AMPylation
To test the interaction between Fic-mediated AMPylation and the UPR, we employed an eye-specific Fic gain-of-function model. Eye-specific expression of the constitutively active AMPylating UAS-FicE247G transgene using a LongGMR-Gal4 driver in otherwise fic-null animals results in a severe rough-eye defect (Casey et al., 2017). However, in a fic heterozygous background, eye-specific expression of constitutively active AMPylating FicE247G yields only a mildly rough eye (Figure 2A). We used this intermediate phenotype to assess genetic interactions between FicE247G and components of the UPR with a candidate-based targeted RNAi screen (Figure 2—figure supplement 1). FicE247G-induced eye roughness was significantly enhanced by knockdown of the key UPR components Perk, Atf4, and Ire1 (Figure 2C–E and Figure 2—figure supplement 1), but not ATF6 (Figure 2B). Furthermore, Xbp1 knockdown in conjunction with FicE247G expression was lethal (Figure 2F). By contrast, knockdown of these UPR genes in the absence of FicE247G did not influence eye phenotype or fly survival (Figure 2A’–F’). These genetic interactions suggest a role for UPR signaling in mitigating cellular stress imposed by expressing the constitutively active AMPylating FicE247G in the eye.
Figure 2 with 1 supplement see all
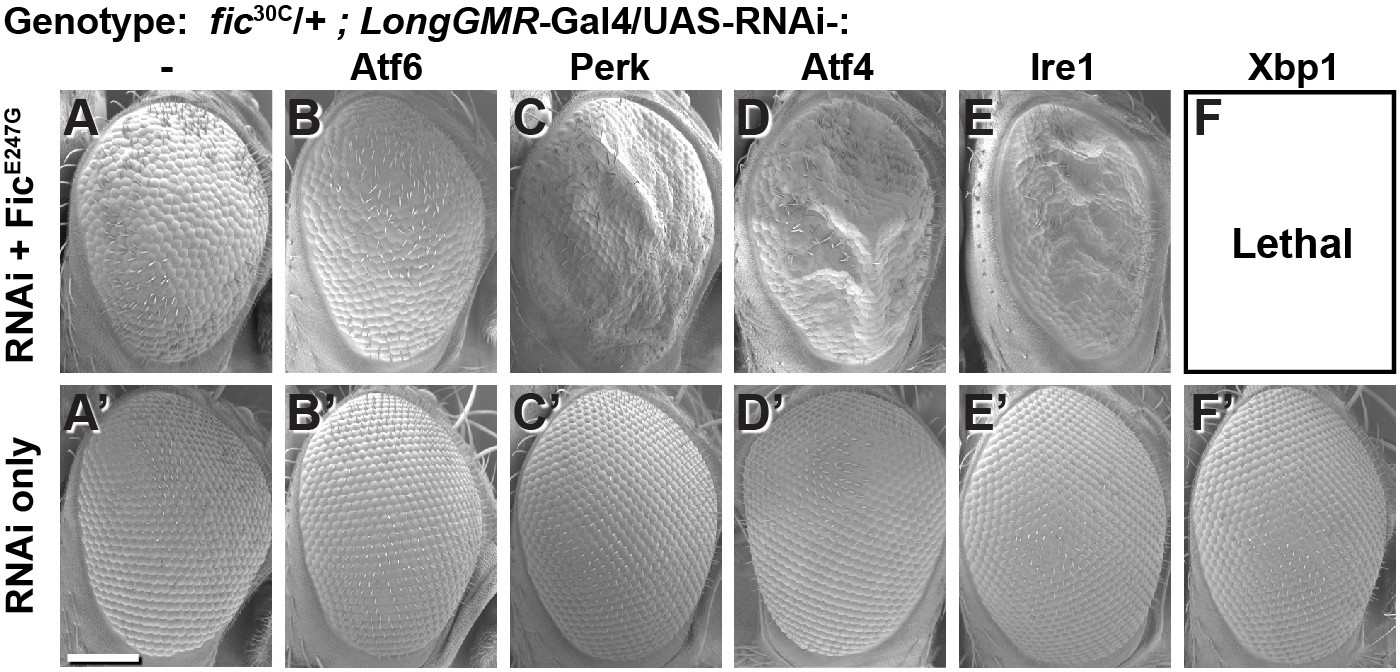
Genetic interactions between Fic and UPR genes.
Representative SEM images of heterozygote mutant fic30C/+ eyes expressing the indicated UAS-RNAi transgenes with (A–F) or without (A’–F’) UAS-FicE247G under longGMR-Gal4 control. See Figure 2—figure supplement 1 for quantification. Scale bar: 100 µM.
AMPylation of BiP is necessary for maintaining vision
The findings that BiP is a target of Fic in vivo and that silencing UPR pathway components enhances the severity of the constitutively active AMPylating FicE247G-induced rough-eye phenotype prompted us to assay the physiological effects of cellular stress in flies lacking BiP AMPylation. To do this we utilized flies that are either null for fic (fic30C) or express the AMPylation-resistant BiPT366A instead of wild-type BiP. By using this strategy, we are able to discern BiP AMPylation-specific changes from other potential changes that are due to as-yet-unknown targets of Fic AMPylation.
As previously reported in ERG recordings, fic-null flies display a reduction of the initial response (termed the ON Transient, Figure 3A) to a light pulse compared to wild-type controls. Interestingly, BiPT366A, but not BiPWT flies, exhibited the same defect in ON Transients as fic30C mutants, consistent with BiP being the primary target of Fic AMPylation required for proper visual neurotransmission (Figure 1—figure supplement 2). Of note, we used an eye-specific RNAi construct against white to minimize any effect of the mini-white gene used as a marker in these BiP transgenes. When we compared ERG traces of fic30C and BiPT366A flies in white+ (red eyed) backgrounds, the reductions in ON transients were no longer detectable (Figure 1—figure supplement 3). This is likely due to the previously established protective effect provided by the red pigment in white+ flies. Indeed, a similar white-dependent phenotype has been reported for photoreceptor synaptic plasticity after prolonged light exposure (Sugie et al., 2015; Damulewicz et al., 2017), consistent with previous observations that a functional white gene masks some degenerative phenotypes in the retina (Lee and Montell, 2004; Soukup et al., 2013). Therefore, we used white-eyed flies to test whether AMPylation may play a role in this type of photoreceptor plasticity, which is induced by rearing flies in uninterrupted low light for 72 hr (Sugie et al., 2015; Damulewicz et al., 2017).
Figure 3 with 3 supplements see all
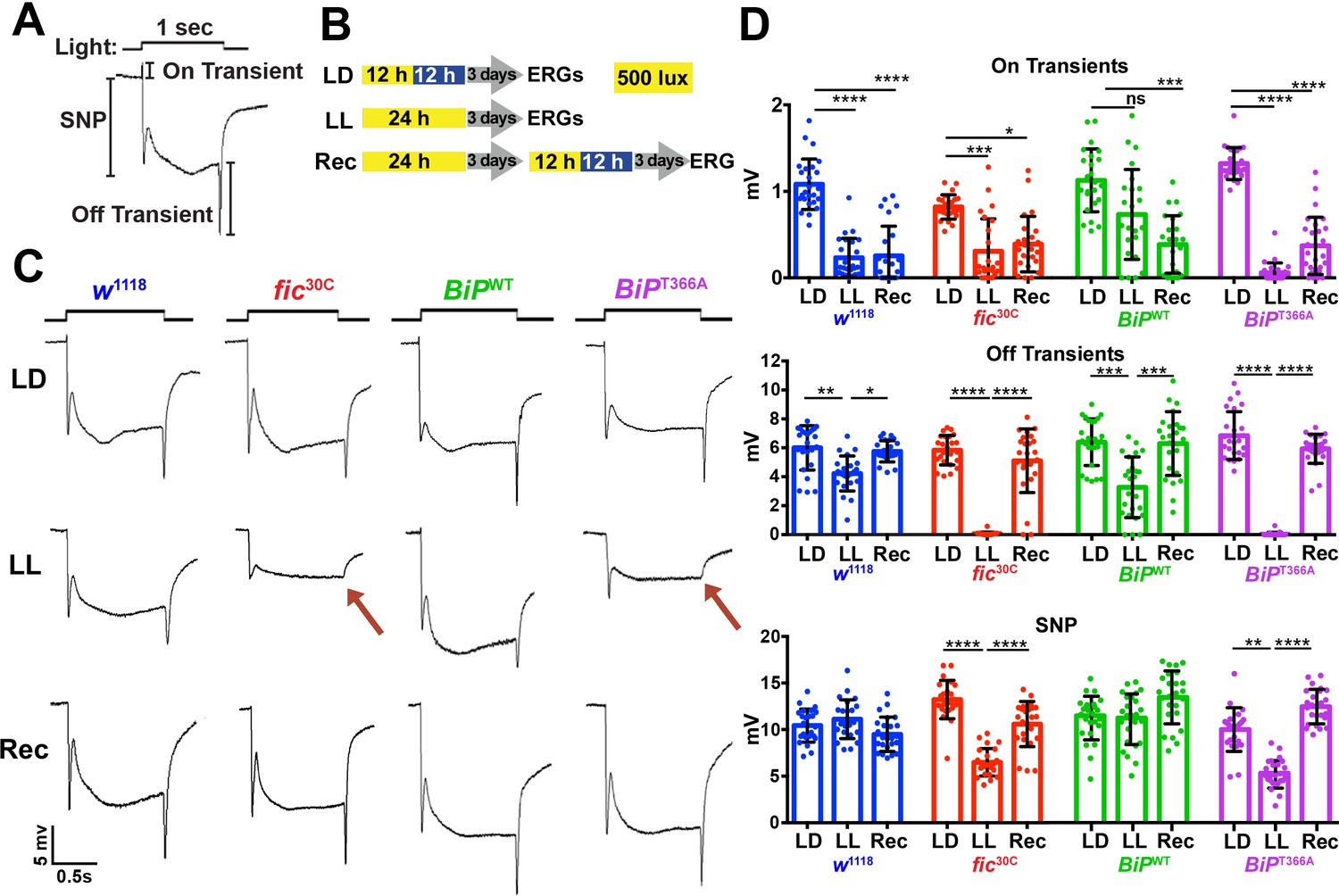
Fic-mediated AMPylation of BiP is required for photoreceptor maintenance.
(A) A representative ERG trace in response to a 1 s light pulse displaying the sustained negative potential (SNP), representing the depolarization within photoreceptor neurons, and the ON and OFF transients, reflecting post-synaptic activity of lamina neurons. (B) Representation of the different light treatments of flies before ERG recordings: 3 days of 12 hr light (500 lux) and 12 hr dark (LD), 3 days of continuous light (LL) or 3 days of continuous light followed by 3 days of LD (Rec). 1 s light pulses were performed at 4 s intervals. (C) Representative traces from w1118, fic30C, BiPWT and BiPT366A flies. Under LL, fic30C and BiPT366A mutants lose ON and OFF transients (red arrows) and have reduced SNPs. The changes are reversed after 3 days of recovery (Rec). (D) Quantification of key components of ERGs shown in panel C. Bar graphs show means ± SD. ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05; n = 24 flies for each genotype/condition, pooled from three independent biological replicas.
-
Figure 3—source data 1
Relates to Figure 3D.
Quantification of ERG components with LD, LL, and Recovery.
- https://doi.org/10.7554/eLife.38752.015
We conducted ERG recordings under different light conditions with four fly lines, w1118, fic30C, BiPWT and BiPT366A (Figure 3B). Compared to age-matched siblings reared under the standard 12 hr Light:12 hr Dark (LD) treatment, fic30C and BiPT366A flies reared for three days under continuous light (LL) at 500 lux exhibited severe ERG defects. This included reduction in the sustained negative potential (SNP), a measure of photoreceptor activation, and loss of ON and OFF transients, which reflect synaptic transmission to downstream L1/L2 lamina neurons (Figure 3C and D). Wild-type controls maintained healthy OFF transients following LL, but ON transients were reduced, reflecting the sensitivity of this component to various light conditions (Figure 3D). To test for behavioral consequences, we assayed w1118 and fic30C flies after 72 hr of LD or LL treatment for light-induced startle behavior using single-fly activity chambers (Ni et al., 2017). Following a 5 min light pulse, LD-reared fic30C flies exhibited a startle response indistinguishable from control w1118 flies, whereas fic30C flies, but not w1118 flies, failed to respond to the light pulse after 72 hr of LL (Figure 3—figure supplement 1). Thus, Fic-mediated AMPylation is required to maintain vision acuity under LL conditions.
We next designed experiments to test whether these functional ERG changes in flies lacking AMPylation reflected light-induced neurodegeneration or a failure to adapt to constant stimulation. First, we asked if the LL-induced ERG defects of BiPT366A and fic30C flies were reversible. We reared mutant and control flies for 72 hr in LL followed by 72 hr of recovery in LD (referred to as ‘Rec’; Figure 3B). This recovery period was sufficient to restore both healthy OFF transients and SNPs in BiPT366A and fic30C flies (Figure 3C and D). Second, we asked if the intensity of the light would exaggerate the defects of BiPT366A and fic30C flies. Exposure of mutant or control flies with 5000 lux, instead of 500 lux, did not alter the severity of ERG defects, indicating the changes were not simply a reflection of the increased amount of total light exposure during LL treatment (Figure 3—figure supplement 2). Third, we asked if prolonging the LL stress would alter the reversibility of these defects. Mutant flies reared under LL for ten days retained the capability to recover healthy ERG traces after only three days on LD, indicating that photoreceptors are not dying but maintained during prolonged light stress (Figure 3—figure supplement 3). Together, these data support a model for a dysregulated adaptive response, rather than phototoxicity, inducing the reversible loss of OFF transients and reduced SNPs.
Constant light induces severe but reversible morphological defects in AMPylation mutants
To determine if the underlying eye substructures were being altered in these AMPylation deficient mutants, we performed TEM on ultrathin transverse eye sections. Under LD conditions, fic30C and BiPT366A mutant and wild-type controls appeared indistinguishable (Figure 4A). However, following 72 hr of LL (500 lux), fic30C and BiPT366A mutants, but not w1118 and BiPWT controls, displayed severe defects in the integrity of rhabdomeres, the microvilli-like membrane structures that house the phototransduction cascade (Figure 4B). After a three-day recovery at LD, the rhabdomeres were nearly restored in both AMPylation-deficient mutants (Figure 4C). To quantify these structural changes in large cohorts of flies, we assessed flies for the presence of wild-type ‘deep pseudopupils’ (DPP) (Figure 4D). Visualization of the DPP affords an assessment of rhabdomere structural integrity in living flies (Franceschini and Kirschfeld, 1971). Consistent with the TEM data, 3 days of LL caused loss of DPP in fic30C and BiPT366A, and DPPs returned after a 3 day recovery (Figure 4D). This suggests that proper regulation of BiP through AMPylation is required for maintaining both function and structure of photoreceptor cells.
Figure 4
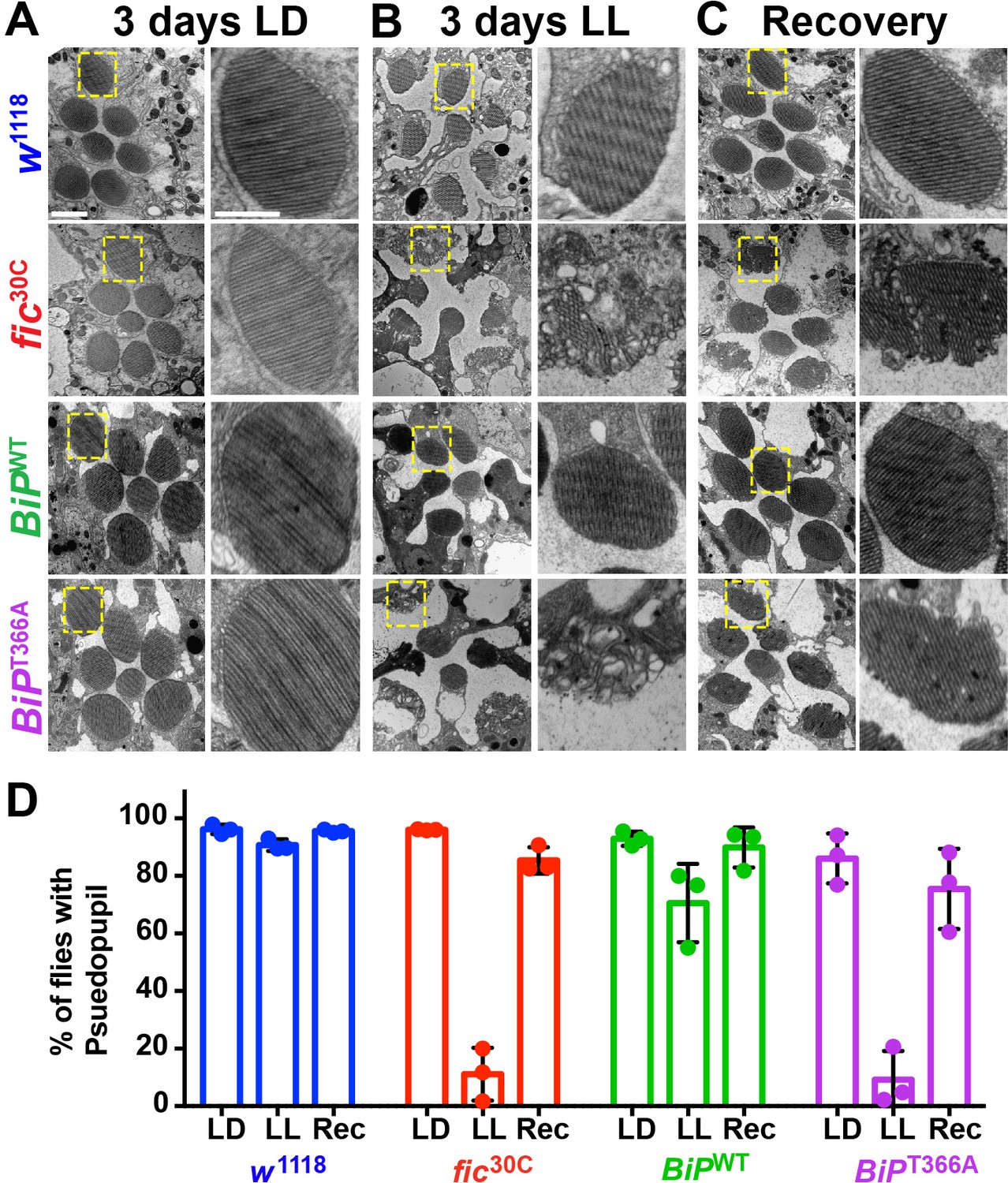
AMPylation of BiP is required for maintaining structural plasticity in the retina.
(A–C) Representative TEM images of retina thin sections from the indicated genotypes with either standard LD (A), the stress-inducing LL (B) or recovery treatment (C). Scale bars: 1 µM. Yellow boxes indicate rhabdomeres shown in high magnification images. High magnification scale bars: 0.5 µM. (D) Percentages of flies with intact deep pseudopupil following LD, LL and Rec. N = 3 independent biological replicas with approximately 50 flies scored per genotype per replica. Bar graphs show means ± SD.
-
Figure 4—source data 1
Relates to Figure 4D.
Scoring of flies for deep pseudopupil defects.
- https://doi.org/10.7554/eLife.38752.020
Fic is required for ER homeostasis in the visual system during constant light
Given the unique role of Fic in both AMPylating and deAMPylating BiP to modulate its chaperone activity and maintaining ER homeostasis, we evaluated fic30C flies for changes in the UPR under LD, LL, and Rec conditions. First, we performed immunohistochemistry against BiP, a transcriptional target of the UPR, which is upregulated during states of ER stress (Gardner et al., 2013; Ham et al., 2014). After 3 days of LL, sections of fic30C revealed increased levels of BiP in retinas and in the lamina neuropils where photoreceptor axons form synapses onto lamina neurons. BiP levels returned to control levels following three days of recovery (Figure 5A and B). To further assess UPR signaling in these tissues, we utilized a sensor for Ire1-mediated Xbp1 splicing (Sone et al., 2013) and an Atf4 translational reporter which serves as a proxy for Perk-mediated phosphorylation of eIF-2a (Kang et al., 2015). In wild-type flies, Xbp1-GFP was slightly induced in the lamina after 24 hr of LL in wild-type flies and the signal decreased over time (Figure 5C, top row, and Figure 5D). However, in fic30C flies, the Xbp1-GFP signal in the lamina continued to increase after 48 hr of LL and remained elevated after 72 hr (Figure bottom row, and Figure 5D). In the retina, control flies showed little to no increase of Xbp1-GFP levels, while fic30C flies showed a significant transient increase after one and two days LL. With the Atf4-DsRed reporter, we observed a significant increase of signal in both the retina and lamina of wild-type flies after one day, but no difference in fic30C mutants at one or two days LL when compared to LD controls (Figure 5E and F). However, by three days of LL, Atf4-DsRed reporter activity in the wild-type flies returned to control levels, while the fic30C mutants showed a significant increase in both the retina and lamina neuropil (Figure 5E and F). These changes in UPR signaling were reversible as each of the reporters returned to near control intensity after 72 hr of LD recovery (Figure 5A,C and E, last columns). The elevated UPR response in fic30C mutants correlated with the timing of the observed severe defects in the integrity of rhabdomeres (Figure 4B). Together, these data identify a role for Fic-mediated BiP AMPylation in regulating ER stress during homeostatic responses of the visual system.
Figure 5
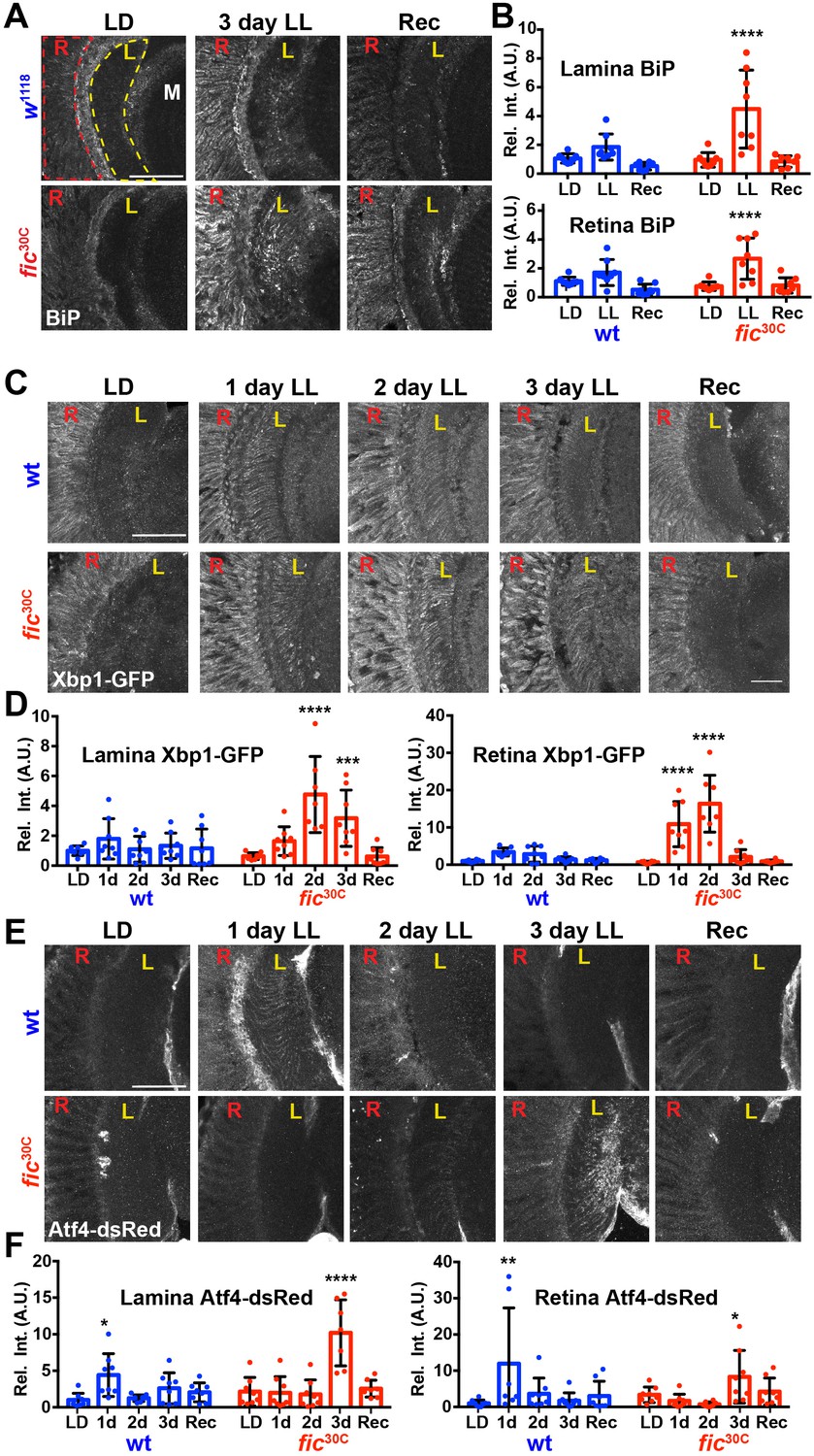
ER homeostasis is disturbed in fic mutants during prolonged light stimulation.
(A) Representative images of BiP immunohistochemistry in sections of w1118 and fic30C flies following 3 days LD, LL or Recovery treatments. (B) Quantification of BiP fluorescence intensity, normalized to wild-type LD controls, in the lamina neuropil and retina from two independent experiments. (C) Representative images of a Xbp1-GFP splicing reporter in either a Fic wild-type or the null fic30C background following LD, 1 day LL, 2 day LL, 3 day LL, and Recovery conditions. (D) Quantification of GFP fluorescence intensity, normalized to wild-type LD controls, in the lamina neuropil and retina from two independent experiments. (E) Representative images of an Atf4-dsRed reporter in either a wild-type or fic30C background following LD, 1 day LL, 2 day LL, 3 day LL, and Recovery conditions. (F) Quantification of Atf4-dsRed intensity, normalized to wild-type LD controls, in the lamina neuropil and retina from two independent experiments. For all experiments, n = 8 flies per genotype/condition, with exceptions of outliers falling three standard deviations outside the mean. Bar graphs show means ± SD. For all experiments, significance is indicated for treatment compared to the LD condition for the corresponding genotype. ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05. All scale bars: 50 µM.
-
Figure 5—source data 1
Relates to Figures 5B, D and F.
Quantification of integrated intensity of BiP (5B), Xbp1-GFP (5D), and Atf4-DsRed (5F).
- https://doi.org/10.7554/eLife.38752.022
Discussion
Here we demonstrate that BiP is a critical in-vivo target of Fic-mediated AMPylation, as an AMPylation-resistant BiP blocks lethality caused by over-expressed constitutively active AMPylating FicE247G and recapitulates fic-null vision defects. This work also sheds light on the physiological connection between AMPylation/deAMPylation of BiP and the UPR. We observe genetic interactions with the constitutively active AMPylating FicE247G and the UPR sensors Ire1 and Perk as well as their effectors, perhaps due to the critical role of BiP as both a regulator (Bertolotti et al., 2000; Shen et al., 2005; Carrara et al., 2015; Amin-Wetzel et al., 2017) and downstream transcriptional target of the UPR (Kozutsumi et al., 1988; Ham et al., 2014). Indeed, we hypothesize that unregulated FicE247G, in the absence of deAMPylation activity, AMPylates excess BiP, excluding it from its normal chaperone role and leading to cell death (Casey et al., 2017; Truttmann et al., 2017). In support of this hypothesis, the developmental defects due to excessive unregulated AMPylation are suppressed by AMPylation-resistant BiPT366A. Furthermore, the enhancement of the rough-eye FicE247G phenotype by knockdown of the Ire1 and PERK pathways suggest a protective role for the UPR, perhaps through the known effects on translation by Ire1-mediated decay of mRNA, Xbp1-driven transcription or Perk-mediated phosphorylation of eIF-2a (Gardner et al., 2013).
Our work supports an in-vivo requirement for Fic-mediated AMPylation of BiPT366 in the context of long-term adaptation to prolonged light exposure. BiPT366A replacement mutants phenocopy fic-null flies in both the light-induced blindness and the unexpected recovery from this phenotype. We observe changes in the ON/OFF transients reflecting defects in transmission to the downstream L1/L2 neurons and also a decreased magnitude in the SNP of photoreceptors. Functional changes in the SNP component are mirrored in the structural changes of photoreceptor rhabdomeres. Rhabdomere appearance of AMPylation-deficient flies was reminiscent of retinal degeneration mutants (Smith et al., 1991; Ryoo et al., 2007), however the remarkable recovery of the tissue structure in three days of LD is unlike any reported retinal degeneration phenotype. It is worth emphasizing that both rhabdomere structure and synaptic transmission recover, as these are not necessarily linked. For example, some mutant alleles of ninaE, encoding the R1-6 rhodopsin, or rdgC, encoding a rhodopsin phosphatase, maintain synaptic transmission despite substantial rhabdomere degeneration (Webel et al., 2000; Lee and Montell, 2001). Together, our findings demonstrate a seminal role for Fic-mediated AMPylation of BiP in vivo: enabling photoreceptors to adapt and maintain both structural and functional integrity during periods of prolonged stress due to uninterrupted light stimulation. The exact mechanism through which these defects in fic mutants arise remains undetermined, but previous studies have demonstrated a requirement for maintaining normal ER folding and trafficking of transmembrane visual signaling components such as Rhodopsin (Colley et al., 1995; Rosenbaum et al., 2006). We hypothesize this intense demand for proper ER stress regulation sensitizes the eye to otherwise mild defects in fic mutants, and the additional demands posed by the stress of constant light stimulation.
We also observed that loss of BiP AMPylation deregulates, but does not block, the UPR to this physiological stress. This observation supports previous claims that AMPylation and deAMPylation of BiP aids in maintaining ER homeostasis (Figure 1A) by establishing a reserve pool of BiP that can readily be deAMPylated in response to acute ER insults (Ham et al., 2014; Casey et al., 2017; Preissler et al., 2017a). This regulation would allow for fine-tuning of the UPR response under specific contexts, comparable to findings in C. elegans in which fic-1 and hsp3 (a BiP homologue) mutants are sensitive to bacterial infection (Truttmann et al., 2016). We speculate that the eye requires tight control of BiP activity, to facilitate adaptation of the vision signaling cascade. Under standard LD conditions, only slight differences are observed, presumably because ER stress is low (Figure 5C and D, first column). However, exposure to constant light results in ER stress and UPR signaling which wild-type flies can clear, presumably because there is a reserve pool of AMPylated BiP to rapidly respond to the stress. In fic mutants, we speculate, loss of the reserve BiP results in the UPR dysregulation revealed by the IreI and Perk activity reporters (Figure 5C and D) as a larger proportion of BiP would be previously engaged and not able to respond to the extra stress. Additionally, the prolonged UPR response in the eyes with dysregulated AMPylation may result in increased expression of UPR-regulated proteins while blocking production of the visual signaling components necessary for adapting to transient stress. It is unknown whether the ratio of AMPylated to unmodified BiP directly regulates activation of the UPR sensors, given the multiple levels of feedback on the system (Gardner et al., 2013). It remains to be determined whether the effects on UPR signaling are the direct cause of the visual system defects or if the phenotypes in fic and BiPT366A mutants are primarily due to changes in protein folding and secretion. We also cannot rule out the influence of AMPylation on other ER processes that involve BiP, such as ER-associated degradation (Hegde et al., 2006).
BiP expression is subject to multiple levels of feedback regulation and is induced when the UPR is activated (Kozutsumi et al., 1988; Ma and Hendershot, 2003). However, in a negative feedback loop, BiP also inhibits activation of the UPR sensors Ire1, Perk, and Atf6, through direct binding (Bertolotti et al., 2000; Shen et al., 2005). It remains unknown how AMPylation affects the interactions of BiP with these proteins in vivo; however, in-vitro work suggests that AMPylation of BiP abolishes its inhibitory effect on Ire1 dimerization and activation (Amin-Wetzel et al., 2017). We speculate that the loss of BiP AMPylation/deAMPylation cycle in a fic null affects both the ability of BiP to quickly respond to misfolded protein aggregates and to regulate UPR activation. Future studies on the dynamic role of reversible BiP AMPylation and its interaction with downstream UPR sensors should provide unique insight into neuronal plasticity and regeneration.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Drosophila melanogaster) | fic | NA | FLYB:FBgn0263278 | |
| Gene (D. melanogaster) | Hsc3-70 | NA | FLYB:FBgn0001218 | |
| Genetic reagent (D. melanogaster) | Da-Gal4 | Bloomington Drosophila Stock Center | BDSC:55851; FLYB:FBst0055851; RRID: BDSC_55851 | |
| Genetic reagent (D. melanogaster) | LongGMR-Gal4 | Bloomington Drosophila Stock Center | BDSC:8121; FLYB:FBst0008121; RRID:BDSC_8121 | |
| Genetic reagent (D. melanogaster) | W[1118] | Bloomington Drosophila Stock Center | BDSC:3605; FLYB:FBst0003605; RRID:BDSC_3605 | |
| Genetic reagent (D. melanogaster) | BiP[G0102]/FM7c | Bloomington Drosophila Stock Center | BDSC:11815; FLYB:FBal0098203; RRID:BDSC_11815 | |
| Genetic reagent (D. melanogaster) | UAS[Scer]-Xbp1- GFP.hg | Bloomington Drosophila Stock Center | BDSC:60731; FLYB:FBst0060731; RRID:BDSC_60731 | Sone et al. (2013) |
| Genetic reagent (D. melanogaster) | dsRed.crc(ATF4). 5'UTR.tub | DOI: 10.1371/journal.pone. 0126795; PMID:25978358 | FLYB: FBal0304834 | Gift from Don Ryoo, NYU. Tubulin promoter and ATF4 5'UTR drive DsRed expression (Flybase FBal0304834) |
| Genetic reagent (D. melanogaster) | genomic 3xFLAG- BiP[WT] | This paper | pAttb_gen3xFLAG-BiP[WT] inserted in AttP landing site at 89E11 | |
| Genetic reagent (D. melanogaster) | genomic 3xFLAG- BiP[T366A] | This paper | pAttb_gen3xFLAG-BiP[T366A] inserted in AttP landing site at 89E11 | |
| Genetic reagent (D. melanogaster) | genomic 3xFLAG- BiP[T518A] | This paper | pAttb_gen3xFLAG-BiP[T518A] inserted in AttP landing site at 89E11 | |
| Genetic reagent (D. melanogaster) | GMR-dsRNA[white] | This paper | pAttb_GMR-dsRNA[white] inserted in AttP landing site at 43A1 | |
| Genetic reagent (D. melanogaster) | fic[30C] | DOI: 10.1074/jbc.M117. 799296; PMID:29089387 | Casey et al. (2017) | |
| Genetic reagent (D. melanogaster) | UAS[Scer]-V5- Fic[E247G] | DOI: 10.1074/jbc.M117. 799296; PMID:29089387 | Casey et al. (2017) | |
| Genetic reagent (D. melanogaster) | UPR and ER protein RNAi lines screened are contained in Supplemental Table 1 | |||
| Genetic reagent (Saccharomyces cerevisiae) | KAR2::KAN | GE Healthcare Life Sciences | SGD:S000003571 | gene replacement generated using PCR-based gene deletion strategy yielding start- to stop-codon deletion |
| Strain, strain background (Saccharomyces cerevisiae) | BY4741 MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | DOI: 10.1002/(SICI)1097- 0061(19980130)14:2 < 115:: AID-YEA204 > 3.0.CO;2–2; PMID: 9483801 | GenBank: JRIS00000000.1 | |
| Antibody | anti-Hsc70-3 (BiP) (Guinea Pig polyclonal) | DOI: 10.1038/sj.emboj. 7601477; PMID:17170705 | FLYB: FBgn0001218; RRID: AB_2569409 | Gift from Don Ryoo, NYU (1:2000 IHC, 1:8000 WB) |
| Antibody | anti-RFP (Rabbit polyclonal) | Rockland | Rockland:600-401-379; RRID:AB_2209751 | (1:1000 IHC) |
| Antibody | anti-GFP (Chicken polyclonal) | ThermoFisher Scientific | ThermoFisher Scientific:A10262; RRID: AB_2534023 | (1:1000 IHC) |
| Antibody | anti-Flag (mouse monoclonal) | Sigma | Sigma:F-3165; RRID:AB_259529 | (1:2000 WB) |
| Antibody | anti-Actin (mouse monoclonal) | Developmental Studies Hybridoma Bank | DSHB:JLA20; RRID: AB_528068 | 1:2000 (WB) |
| Antibody | Alexa 488- or 568- secondaries | Molecular Probes | (1:1000 IHC) | |
| Antibody | LICOR 800 or 700- secondaries | LICOR Biosciences | (1:20,000 WB) | |
| Recombinant DNA reagent | pAttb_gen3xFLAG- BiP[WT] | This paper | PCR in multiple steps from genomic DNA {sequence location = X: 11,801,696..11,807,117 [-]}. Cloned into modified pAttb vector | |
| Recombinant DNA reagent | pAttb_gen3xFLAG- BiP[T366A] | This paper | Progenitors: pAttb_gen3xFLAG- BiP[WT]. Mutated sequence synthesized with Geneblock (IDT) | |
| Recombinant DNA reagent | pAttb_gen3xFLAG- BiP[T518A] | This paper | Progenitors: pAttb_gen3xFLAG- BiP[WT]. Mutated sequence synthesized with Geneblock (IDT) | |
| Recombinant DNA reagent | pAttb_GMR- dsRNA[white] | This paper | Progenitors: pUASt_dsRNA[white] (gift from Dean Smith, UT Southwestern, PMID: 11804566). GMR sequence: Geneblock (IDT) | |
| Recombinant DNA reagent | pKAR2:LEU2 | This paper | Cloned from amplification of endogenous KAR2 with primers (see below) | |
| Recombinant DNA reagent | pKAR2[T386A]: LEU2 | This paper | Progenitor: pKAR2:LEU2. Site directed mutagenesis used to make mutation | |
| Recombinant DNA reagent | pKAR2[T538A]: LEU2 | This paper | Progenitor: pKAR2:LEU2. Site directed mutagenesis used to make mutation | |
| Sequence-based reagent | 5’-GCATCCGCGGATACT CTCGTACCCTGCCGC-3’ | This paper | Cloning for pKAR2:LEU2 | |
| Sequence-based reagent | 5’-ATGCGAGCTCCGTAT ATACTCAGTATAATC-3’ | This paper | Cloning for pKAR2:LEU2 | |
| Sequence-based reagent | 5’-GGTTGGTGGTTCTG CTAGAATTCCAAAGGT CCAACAATTGTTAGAA TCATACTTTGATGG-3’ | This paper | Mutagenesis primer for pKAR2[T386A]:LEU2 | |
| Sequence-based reagent | 5’-ACCTTTGGAATTCT AGCAGAACCACCAAC CAAAACGATATCATCA ACATCCTTCTTTTCC-3’. | This paper | Mutagenesis primer for pKAR2[T386A]:LEU2 | |
| Sequence-based reagent | 5’-AGATAAGGGAGCTGG TAAATCCGAATCTATCAC CATCACTAACG-3’ | This paper | Mutagenesis primer for pKAR2[T538A]:LEU2 | |
| Sequence-based reagent | 5’-GGATTTACCAGCTCC CTTATCTGTGGCAGACA CCTTCAGAATACC-3’. | This paper | Mutagenesis primer for pKAR2[T538A]:LEU2 | |
| Chemical compound, drug | VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | Vector Laboratories: H-1200 | |
| Software, algorithm | Adobe Photoshop | Adobe | RRID:SCR_014199 | |
| Software, algorithm | ImageJ | NIH | RRID:SCR_003070 |
Fly stocks and genetics
View detailed protocolBloomington Stock Center provided w1118 (BS# 3605), BiPG0102/FM7c (BS#11815), Da-Gal4 (BS#55850), LongGMR-Gal4 (BS#8121) stocks. The fic30C allele and UAS-FicE247G flies was previously described (Casey et al., 2017). Lines used in the RNAi screen are described in Figure 2—figure supplement 1 and were obtained from Bloomington Stock Center and the Vienna Drosophila Resource Center (Dietzl et al., 2007). The Atf45’UTR-dsRed (Kang et al., 2015) and the Xbp1-GFP (Sone et al., 2013) lines were a gift from Dr. Don Ryoo (NYU) and were recombined with the fic30C allele. We generated the p[gen3xFLAG-BiPWT]AttP-89E11, p[gen3xFLAG-BiPT366A]AttP-89E11, p[gen3xFLAG-BiPT518A]AttP-89E11 and p[GMR-dsRNAwhite] alleles using the Phi30C integrase strategy (Venken et al., 2006). p[GMR-dsRNAwhite] was recombined with the fic30C allele and white-eyed candidates were screened for the fic allele by PCR. BiPG0102;; p[gen3xFLAG-BiPWT]AttP-89E11 and BiPG0102;;p[gen3xFLAG-BiPT366A]AttP-89E11 stocks were made by crossing males harboring the genomic transgene to BiPG0102/FM7c female flies. Surviving males were backcrossed to BiPG0102/FM7c female flies, and stable stocks were established from the resulting progeny. None of the rare escaping BiPG0102; ; p[gen3xFLAG-BiPT518A]AttP-89E11 male flies were fertile. The LongGMR-Gal4,UASScer-V5-FicE247G-attP-B3/TM6B,Hu and Da-Gal4,UASScer-V5-FicE247G-attP-3B/TM6B,hu stocks were made using standard Drosophila recombination and crossed into w1118 and w1118; fic30C backgrounds.
List of fly strains and stocks
Request a detailed protocolw1118 (BS#3605)
OreR
; fic30C
w1118; fic30C
w1118; p[GMR-dsRNAwhite]
w1118; fic30C, p[GMR-dsRNAwhite]
w1118; fic30C; LongGMR-Gal4,UASScer-V5-FicE247G- attP-3B/TM6B,hu
w1118; fic30C/CyO; Da-Gal4,UASScer-V5-FicE247G- attP-3B/TM6B,hu
BiPG0102/FM7c (BS#11815)
w1118;; p[gen3xFLAG-BiPWT]AttP-89E11
w1118;; p[gen3xFLAG-BiPT366A]AttP-89E11
w1118;; p[gen3xFLAG-BiPT518A]AttP-89E11
w1118; fic30C; p[gen3xFLAG-BiPWT]AttP-89E11
w1118; fic30C; p[gen3xFLAG-BiPT366A]AttP-89E11
w1118; fic30C; p[gen3xFLAG-BiPT518A]AttP-89E11
BiPG0102; ; p[gen3xFLAG-BiPWT]AttP-89E11
BiPG0102; ; p[gen3xFLAG-BiPT366A]AttP-89E11
BiPG0102; p[GMR-dsRNAwhite]; p[gen3xFLAG-BiPWT]AttP-89E11
BiPG0102; p[GMR-dsRNAwhite]; p[gen3xFLAG-BiPT366A]AttP-89E11
w1118; p[tub-Atf45’UTR-dsRed]/p[GMR-dsRNAwhite]
w1118; fic30C,p[tub-Atf45’UTR-dsRed]/fic30C,p[GMR-dsRNAwhite]
w1118; p[GMR-dsRNAwhite]; p[Da-Gal4]/p[UASScer-Xbp1-GFP.hg]
w1118; fic30C,p[GMR-dsRNAwhite]; p[Da-Gal4]/p[UASScer-Xbp1-GFP.hg]
Generation of genomic BiP transgenes
Request a detailed protocolPCR-amplified BiP genomic sequences were cloned into a pAttB vector and a 3X-FLAG tag was inserted after the N-terminal signal sequence. To create the T366A and T518A mutations, gBlocks (IDT, Coralville, IA) for the mutant sequences were synthesized and subcloned into the pAttB_genomic BiP vector via NEB HiFi Assembly Kit (NEB, Ipswich, MA). These constructs were sequence-verified and injected into embryos (BestGene, Chino Hills, CA) for insertion at the 89E11 landing site. Expression levels of FLAG-BiP transgenes were determined with western blotting. In brief, fly heads were homogenized in lysis buffer (10% SDS, 6M urea, and 50 mM Tris-HCl, pH 6.8 + 10% DTT), sonicated for 5 min, boiled for 2 min, and centrifuged for 10 min at 10,000 g to remove debris. 10 µL were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blots were probed with anti-BiP (1:8000, gift from Dr. Don Ryoo, NYU, NY), anti-FLAG (1:2000 M2- F3165, Millipore Sigma, St. Louis, MO) and anti-Actin (1:4000, JLA-20, DSHB, Iowa City, IA) and detected using IRdye-labeled antibodies and an Odyssey scanner (LI-COR Biosciences, Lincoln, NE).
Generation of GMR_dsRNAwhite transgenes
View detailed protocolTo make the eye-targeted dsRNA constructs against the white gene, the dsRNA sequence was obtained from a pAttb-UASS.cer-dsRNAwhite vector (Kalidas and Smith, 2002) (a gift from Dr. Dean Smith, UT Southwestern Medical Center, TX) and the UASS.cer-Hsp40 promotor sequence was replaced with a 5X-GMR promotor sequence, synthesized as a gBlock (IDT) and cloned with NEB HiFi Assembly Kit (NEB).
Fly rearing conditions
Request a detailed protocolAll flies were reared on standard molasses fly food, under room temperature conditions. For light treatments, flies were collected within one to two days of enclosing, and placed in 5 cm diameter vials containing normal food, with no more than 25 flies, and placed at either LD (lights ON 8am/lights OFF 8pm) or LL. ERGs, head dissections and behavior assays were performed between 1pm and 4pm. The same intensity white LED light source was used for both conditions and flies were kept the same distance away from the light source, which amounted to approximately 500 lux. LD and LL treatments were done at 25°C. For the UPR and FicE247G rough-eye interaction experiments, all flies were raised at 28°C.
Survival analysis of flies expressing genomic BiP construct
Request a detailed protocolBiPG0102/FM7c female virgin flies were crossed to males with either gen3xFLAG-BiPWT, gen3xFLAG-BiPT366A or gen3xFLAG-BiPT518A. The number of surviving non-FM7c male flies was scored by presence or lack of the Bar eye marker. Percent of expected was calculated from the actual number or recovered flies of the relevant genotypes compared the expected Mendelian number [# observed flies/ #expected flies]. Crosses were repeated three times (n = 3). Total number of flies scored was at least 100 for each BiP variant.
Survival analysis of flies expressing BiP variants in a Da-Gal4, UAS-FicE247G background
Request a detailed protocolC-terminally V5-His6-tagged UAS-FicE247G (Casey et al., 2017) was expressed via the ubiquitous Da-Gal4 driver in fic30C/CyO heterozygous flies. These flies were crossed to w-; fic30C (controls), w-; fic30C; gen3xFLAGBiPWT, w-; fic30C; gen3xFLAGT366A, w-; fic30C; gen3xFLAG-BiPT518A, or BiPG0102; fic30C; gen3xFLAGT366A. Offspring were scored and the number of adults homozygous for fic30C with the Da-Gal4, UAS-FicE247G allele and were compared to the number of fic30C heterozygous sibling controls. Percent of expected was calculated from the actual number or recovered flies of the relevant genotypes compared the expected Mendelian number [# observed flies/ #expected flies]. Crosses were repeated three times (n = 3). Total number of flies scored was at least 100 for each BiP variant each repeat.
Electroretinograms
Request a detailed protocolERGs were recorded as previously described (Montell, 2012). Glass electrodes filled with 2M NaCl were placed in the fly thorax and surface of the corneal lens (recording). A computer-controlled LED light source (MC1500; Schott, Mainz, Germany) was pulsed for 1 s at 4 s intervals. The resulting ERG traces were collected by an electrometer (IE-210; Warner Instruments, Hamden, CT), digitized with a Digidata 1440A and MiniDigi 1B system (Molecular Devices, San Jose, CA), and recorded using Clampex 10.2 (Molecular Devices) and quantified with Clampfit software (Molecular Devices). Flies were assayed in batches of eight to ten, and resulting quantifications are pooled from three independent biological repeats.
Deep pseudopupil analysis
Request a detailed protocolFlies were anesthetized on CO2 and aligned with one eye facing up. Using a stereoscopic dissection microscope, each fly was scored for presence or loss of the deep pseudopupil (Franceschini and Kirschfeld, 1971), and the percentage of flies with intact pseudopupils was calculated. For each genotype/treatment, over 50 flies were scored per replica and three biological replicas were performed (n = 3).
Light-startle behavior assay
Request a detailed protocolAssay was adapted from a previously described method (Ni et al., 2017). After 72 hr of LD or LL treatment, 16 flies per genotype were collected at the same time each morning and placed into individual Drosophila Assay Monitoring (DAM) chambers (TriKinetics Inc, Waltham, MA). The DAM monitors were placed into a dark incubator. Two hours later, a 500 lux light was turned on by a timer for five minutes. Data was collected with DAMSystem3.0 and DAMFileScan11.0 (TriKinetics Inc). The resulting data was exported to Microsoft Excel and graphed in GraphPad Prism. Three replica experiments were averaged and plotted as Time (min) vs Average activity per 2 min. bin (n = 3). The change in response to light was calculated for each light pulse as [mean beam breaks for 10 min. post-pulse] – [mean beam breaks for 10 min. pre-pulse].
Scanning electron microscopy
Request a detailed protocolSEMs of fly eyes were obtained as previously described (Wolff, 2011). Eyes were fixed in 2% paraformaldehyde, 2% glutaraldehyde, 0.2% Tween 20, and 0.1 M cacodylate buffer, pH 7.4, for 2 hr. Samples were washed four times with increasing ethanol (25–100%) for 12 hr each followed by a series of hexamethyldisilazane washes (25–100% in ethanol) for one hour each. Flies were air dried for 24 hr, mounted on SEM stubs, and the bodies were coated in fast-drying silver paint. Flies were sputter coated with a gold/pallidum mixture for 60 s and imaged at 900X magnification, with extra high tension set at 3.0 kV on a scanning electron microscope (Sigma SEM; Carl Zeiss, Germany). Ten flies per genotype were mounted and three were imaged (n = 3). Blinding of the samples’ identity to the user acquiring the images was performed.
Transmission electron microscopy
Request a detailed protocolTEMs of retina sections were performed as previously described (Jenny, 2011; Rahman et al., 2012). Briefly, 550 nm sections were cut and stained with toluidine blue to confirm orientation and section depth. Blocks were subsequently thin-sectioned at 70 nm with a diamond knife (Diatome, Hatfield, PA) on a Leica Ultracut six ultramicrotome (Leica Microsystems, Wetzlar, Germany) and collected onto formvar-coated, glow-discharged copper grids, post-stained with 2% aqueous uranyl acetate and lead citrate. Images were acquired on a Tecnai G2 spirit transmission electron microscope (FEI) equipped with a LaB6 source using a voltage of 120 kV. Blinding of the samples to the technicians performing the processing and the user acquiring the images was performed. Two fly heads per genotype/condition and at least three thin sections per sample were examined (n = 2). Samples were unmasked after the images were processed.
Immunohistochemistry for BiP and UPR reporters
Request a detailed protocolFly heads were dissected in HL3 hemolymph-like solution, fixed for four hours in ice-cold 4% para-formaldehyde in filtered PBS, washed overnight in 25% (wt/vol) sucrose in phosphate buffer (pH 7.4), embedded in Optimal Cutting Temperature compound (EMS, Hatfield, PA), frozen in dry ice and sectioned at 20 μm thickness on a cryostat microtome (CM 1950, Leica Microsystems, Wetzlar, Germany). Sections were probed overnight with primary antibodies against Drosophila BiP (1:2000, Gift from Don Ryoo (Ryoo et al., 2007), GFP (1:1000, A10262, ThermoFisher Scientific, Waltham, MA) or RFP (1:1000, 600-401-379, Rockland, Limerick, PA). Secondary antibodies were labeled with Alexa488-conjugated Goat anti-Chicken (Molecular Probes, P/N# A-11039), Alexa488-conjugated Goat anti-Guinea Pig (Molecular Probes, P/N# A-11073), or Alexa568-conjugated Goat anti-Rabbit (Molecular Probes, P/N# A-11011). Alexa 647-conjugated phallodin was also added to label Actin for identifying structures. Images were captured with an oil-immersion 63 × NA−1.4 lens on an inverted confocal microscope (LSM710, Carl Zeiss). For each genotype and light rearing conditions, immunohistochemistry experiments were performed in two biological replicas with new sets of flies, using identical acquisition settings. Blinding of the samples to the user acquiring the images was performed when appropriate.
Quantification of fluorescence staining
Request a detailed protocolFluorescence images were quantified using ImageJ (NIH) adapting previous methods (Nandi et al., 2017). For each antibody, a threshold was determined, removing the lowest 10% of signal in LD control samples (to reduce variation from low level background signals). This same threshold was applied, and a mask was created for every image in a batch of staining. Within a 1 µm optical slice, the retina and lamina regions were selected manually using an Actin stain and assigned as Regions of Interest. The integrated pixel intensity per unit area was measured within this selected area, redirecting to the threshold mask. In each fly, four sequential optical slices were quantified and averaged. For each genotype and treatment, four flies were quantified from two independent biological replicas for a total of eight flies. Data was normalized to the wild-type LD control for each replica. Outliers of greater than three standard deviations were omitted from the analysis.
Yeast plasmids and strains
Request a detailed protocolYeast genetic techniques were performed by standard procedures described previously. (Sherman et al., 1981). All strains were cultured in either rich (YPD: 1% yeast extract, 2% peptone, and 2% glucose) or complete synthetic minimal (CSM) media lacking appropriate amino acids with 2% glucose. Yeast were grown to log phase, serially diluted, and spotted onto agar plates to assay fitness and temperature sensitivity as previously described (Tran et al., 2007).
DNA fragments of KAR2 was generated by PCR amplification of the endogenous KAR2 gene using the primers 5’-GCATCCGCGGATACTCTCGTACCCTGCCGC-3’ and 5’-ATGCGAGCTCCGTATATACTCAGTATAATC-3’. Plasmid pKAR2:LEU2 and pKAR2:URA3 were generated by subcloning genomic DNA fragments containing promoter and coding sequence of KAR2 into the SacI and SacII sites of pRS315 and pRS316, respectively. pKAR2T386A:LEU2 was generated by site directed mutagenesis of pKAR2:LEU2 using the primers 5’-GGTTGGTGGTTCTGCTAGAATTCCAAAGGTCCAACAATTGTTAGAATCATACTTTGATGG-3’ and 5’-ACCTTTGGAATTCTAGCAGAACCACCAACCAAAACGATATCATCAACATCCTTCTTTTCC-3’. pKAR2T538A:LEU2 was generated by site directed mutagenesis of pKAR2:LEU2 using the primers 5’-AGATAAGGGAGCTGGTAAATCCGAATCTATCACCATCACTAACG-3’ and 5’-GGATTTACCAGCTCCCTTATCTGTGGCAGACACCTTCAGAATACC-3’.
ACY008 yeast (mat A kar2::KAN his3∆0 leu2∆0 LYS met15∆0 ura3∆0 pKAR2:URA) were obtained by sporulation and dissection of KAR2 heterozygous null yeast (Mata/mat@ KAR2::KAN/KAR2 his3∆0/his3∆0 leu2∆0/leu2∆0 LYS/lys MET/met15∆0 ura3∆0/ura3∆0) (GE) transformed with pKAR2:URA. Standard plasmid shuffle techniques with 5-FOA(Zymo) were utilized to obtain ACY016 (mat A kar2::KAN his3∆0 leu2∆0 LYS met15∆0 ura3∆0 pKAR2:LEU2) ACY017(mat A kar2::KAN his3∆0 leu2∆0 LYS met15∆0 ura3∆0 pKAR2T386A:LEU2), and ACY020(mat A kar2::KAN his3∆0 leu2∆0 LYS met15∆0 ura3∆0 pKAR2T538A:LEU2)
Statistics
Statistics were performed using GraphPad Prism 7. Normality of data distribution was determined using D'Agostino’s and Pearson’s normality test. For the genetic analysis in Figure 1 and the ERG measurements in Figure 1—figure supplement 2, Figure 1—figure supplement 3, Figure 3—figure supplement 2, Figure 3—figure supplement 3, significance was determined using one-way ANOVA, followed by Tukey’s multiple comparisons tests. Statistical significance for non-parametric data, including the ERGs with light treatment quantifications in Figure 2, was determined by Kruskal-Wallis tests followed by multiple comparisons testing with Dunn's correction. For the image quantification data in Figure 4, significance was determined by two-way ANOVA followed by multiple comparisons with Benjamini-Krieger-Yekutieli’s False Discovery Rate correction. All tests were two-sided with no experimental matching. RStudio (version 1.1.442, 2018, RStudio, Inc.) was used for Fisher’s Exact Tests for the eye interaction screen, with Bonferroni’s multiple comparison method to determine significance (Figure 2—figure supplement 1). Tests were two-sided. When possible, blinding of sample identities was performed for image acquisition and fluorescence intensity quantification. Sample sizes for ERG assays, EM experiments, fluorescence quantifications and fly genetic analysis were determined based from previous experience (Rahman et al., 2012; Stenesen et al., 2015; Nandi et al., 2017).
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
The SM protein Car/Vps33A regulates SNARE-mediated trafficking to Lysosomes and lysosome-related organellesMolecular Biology of the Cell 20:1705–1714.https://doi.org/10.1091/mbc.e08-03-0282
-
tan and ebony genes regulate a novel pathway for transmitter metabolism at fly photoreceptor terminalsThe Journal of Neuroscience 22:10549–10557.https://doi.org/10.1523/JNEUROSCI.22-24-10549.2002
-
Fic-mediated deAMPylation is not dependent on homodimerization and rescues toxic AMPylation in fliesJournal of Biological Chemistry 292:21193–21204.https://doi.org/10.1074/jbc.M117.799296
-
Enzymes involved in AMPylation and deAMPylationChemical Reviews 118:1199–1215.https://doi.org/10.1021/acs.chemrev.7b00145
-
Cryptochrome is a regulator of synaptic plasticity in the visual system of Drosophila melanogasterFrontiers in Molecular Neuroscience 10:165.https://doi.org/10.3389/fnmol.2017.00165
-
Endoplasmic reticulum stress sensing in the unfolded protein responseCold Spring Harbor Perspectives in Biology 5:a013169.https://doi.org/10.1101/cshperspect.a013169
-
Mutations within the nucleotide binding site of immunoglobulin-binding protein inhibit ATPase activity and interfere with release of immunoglobulin heavy chainThe Journal of Biological Chemistry 268:7248–7255.
-
Unfolded protein response-regulated Drosophila fic (dFic) protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasisJournal of Biological Chemistry 289:36059–36069.https://doi.org/10.1074/jbc.M114.612515
-
Biological diversity and molecular plasticity of FIC domain proteinsAnnual Review of Microbiology 70:341–360.https://doi.org/10.1146/annurev-micro-102215-095245
-
Preparation of adult Drosophila eyes for thin sectioning and microscopic analysisJournal of Visualized Experiments e2959.https://doi.org/10.3791/2959
-
Degeneration of photoreceptors in rhodopsin mutants of DrosophilaJournal of Neurobiology 23:605–626.https://doi.org/10.1002/neu.480230602
-
Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stressJournal of Biological Chemistry 278:34864–34873.https://doi.org/10.1074/jbc.M301107200
-
A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteinsMolecular Biology of the Cell 13:4456–4469.https://doi.org/10.1091/mbc.e02-05-0311
-
Drosophila visual transductionTrends in Neurosciences 35:356–363.https://doi.org/10.1016/j.tins.2012.03.004
-
FICD acts bifunctionally to AMPylate and de-AMPylate the endoplasmic reticulum chaperone BiPNature Structural & Molecular Biology 24:23–29.https://doi.org/10.1038/nsmb.3337
-
Stable binding of ATF6 to BiP in the endoplasmic reticulum stress responseMolecular and Cellular Biology 25:921–932.https://doi.org/10.1128/MCB.25.3.921-932.2005
-
Role of asparagine-linked oligosaccharides in rhodopsin maturation and association with its molecular chaperone, NinaAJournal of Biological Chemistry 275:24752–24759.https://doi.org/10.1074/jbc.M002668200
-
Preparation of Drosophila eye specimens for scanning electron microscopyCold Spring Harbor Protocols 2011:1383–1385.https://doi.org/10.1101/pdb.prot066506
-
AMPylation of Rho GTPases subverts multiple host signaling processesJournal of Biological Chemistry 289:32977–32988.https://doi.org/10.1074/jbc.M114.601310
Article and author information
Author details
Funding
National Institute of General Medical Sciences (R01GM120196)
- Helmut Krämer
National Eye Institute (RO1EY010199)
- Helmut Krämer
Howard Hughes Medical Institute
- Kim Orth
Welch Foundation (I-1561)
- Kim Orth
Once Upon A Time Foundation
- Kim Orth
National Science Foundation (1000176311)
- Andrew T Moehlman
National Institute of General Medical Sciences (RO1GM115188)
- Kim Orth
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Drs. Eric Olson and Joe Takahashi and the members of the Krämer and Orth labs for discussion and technical assistance. We thank the Bloomington Stock Center (NIH P40OD018537) and the Vienna Drosophila Resource Center (VDRC, www.vdrc.at) for flies and the Molecular and Cellular Imaging Facility at the University of Texas Southwestern Medical center for help with electron microscopy (NIH S10 OD020103-01). KO is a Burroughs Welcome Investigator in Pathogenesis of Infectious Disease, a Beckman Young Investigator, and a W W Caruth, Jr., Biomedical Scholar and has an Earl A Forsythe Chair in Biomedical Science.
Copyright
© 2018, Moehlman et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,191
- views
-
- 244
- downloads
-
- 50
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 50
- citations for umbrella DOI https://doi.org/10.7554/eLife.38752
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Adaptation to constant light requires Fic-mediated AMPylation of BiP to protect against reversible photoreceptor degeneration
eLife 7:e38752.
https://doi.org/10.7554/eLife.38752




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}