Expanded genetic screening in Caenorhabditis elegans identifies new regulators and an inhibitory role for NAD+ in axon regeneration
- University of California, San Diego, United States
- University of California, San Diego, School of Medicine, United States
Abstract
The mechanisms underlying axon regeneration in mature neurons are relevant to the understanding of normal nervous system maintenance and for developing therapeutic strategies for injury. Here, we report novel pathways in axon regeneration, identified by extending our previous function-based screen using the C. elegans mechanosensory neuron axotomy model. We identify an unexpected role of the nicotinamide adenine dinucleotide (NAD+) synthesizing enzyme, NMAT-2/NMNAT, in axon regeneration. NMAT-2 inhibits axon regrowth via cell-autonomous and non-autonomous mechanisms. NMAT-2 enzymatic activity is required to repress regrowth. Further, we find differential requirements for proteins in membrane contact site, components and regulators of the extracellular matrix, membrane trafficking, microtubule and actin cytoskeleton, the conserved Kelch-domain protein IVNS-1, and the orphan transporter MFSD-6 in axon regrowth. Identification of these new pathways expands our understanding of the molecular basis of axonal injury response and regeneration.
https://doi.org/10.7554/eLife.39756.001Introduction
Axon regeneration after injury is an important and conserved biological process in many animals, involving a large number of genes and pathways (He and Jin, 2016; Mahar and Cavalli, 2018; Tedeschi and Bradke, 2017). Upon axonal injury, distal axon segments degenerate and segments proximal to the cell body remain alive and can in certain cases regenerate (Chen et al., 2007; McQuarrie and Grafstein, 1973; Neumann and Woolf, 1999). Axon regeneration after injury requires rapid sealing of the damaged plasma membrane (PM) and subsequent formation of growth cones, leading to regrowth and extension from damaged proximal axons. These cellular changes involve numerous molecular pathways, starting with rapid calcium influx at injury sites (Ghosh-Roy et al., 2010; Rishal and Fainzilber, 2014; Wolf et al., 2001), retrograde injury signaling, transcriptional reprogramming to re-structuring of the cytoskeleton and re-organization of the extracellular matrix (ECM) (Blanquie and Bradke, 2018). In the adult mammalian central nervous system (CNS), axon regeneration is limited, due to the combination of a repressive glial environment and a lower intrinsic growth capacity of CNS neurons (He and Jin, 2016). The lack of axonal regrowth after CNS injuries, therefore, impairs functional recovery.
Many approaches have been proposed and tested to promote axon regeneration over the past decades (David and Aguayo, 1981; He and Jin, 2016; Park et al., 2008). Yet, mechanistic understanding of how damaged axons regenerate in a permissive environment remains fragmented. Since the discovery of functional axon regeneration in the nematode Caenorhabditis elegans (Yanik et al., 2004), several function-based genetic screens have revealed conserved axon regeneration genes and pathways, notably the highly conserved MAPKKK DLK-1 signaling cascade (Yan et al., 2009; Chen et al., 2011; Hammarlund et al., 2009; Nix et al., 2014). We previously reported a distinct set of genes identified from a genetic screen of 654 genes in mechanosensory axon regeneration (Chen et al., 2011). For example, regulators of microtubule (MT) dynamics play a rate-limiting role in axon regrowth, consistent with findings from other animal models (Bradke et al., 2012; Hur et al., 2012). Additional studies reveal other conserved pathways include the RNA-binding protein CELF/UNC-75 (Chen et al., 2016a), the miRNA and piRNA pathway (Kim et al., 2018; Zou et al., 2013), the fusogen EFF-1 (Ghosh-Roy et al., 2010; Neumann et al., 2015), and the apoptotic pathway (Pinan-Lucarre et al., 2012). Importantly, the findings from C. elegans are echoed from similar screening in mammalian neurons (Sekine et al., 2018; Zou et al., 2015).
Here, we report our analysis of 613 additional new genes using the C. elegans mechanosensory axon regeneration assay. We find new gene classes with inhibitory roles in axon regrowth, such as the NAD+ salvage pathway and the conserved Kelch-domain protein IVNS-1. We also find several permissive factors, such as A Disintegrin and Metalloprotease with Thrombospondin repeats (ADAMTS) proteins, a Rab GTPase RAB-8, and the membrane transporter MFSD-6. We show that the endoplasmic reticulum (ER)-PM contact site protein Extended Synaptotagmin (ESYT-2) is sensitive to axonal injury, and that Junctophilin (JPH-1) inhibits axon-axon fusion. Our studies of genes encoding lipid or phospholipid metabolic enzymes indicate extensive functional redundancy. This expanded screen reinforces several themes from the previous study, such as the inhibitory role of ECM components and the permissive role of MT stabilization (Chen et al., 2011). Together, our findings highlight the molecular complexity of axon regeneration and provide the genetic framework for a more comprehensive understanding of axon regeneration.
Results
We screened 613 additional genes representing nine classes of protein function and structure, selected based on their sequence conservation and the availability of viable genetic mutants with normal axon development (Figure 1A,B; Figure 1—source data 1). We tested genetic null or strong loss-of-function mutations in each gene for effects on mechanosensory PLM (Posterior Lateral Microtubule) axon regeneration. In the PLM axon regrowth model, we sever the axon ~50 μm distal from the cell body in the fourth larval (L4) stage using a femtosecond laser and measure axon regrowth 24 hr post-axotomy in at least 10 animals per strain (Wu et al., 2007). From these 613 genes, we identified 49 genes promoting PLM regrowth (i.e. showing reduced regrowth in loss-of-function mutants) and 34 genes inhibiting regrowth (i.e. increased regrowth in loss-of-function mutants) (Tables 1 and 2; Figure 1—source data 1). As in our previous screen, genes affecting axon regrowth are found across all functional and structural classes tested (Figure 1B). The percentage of genes having positive or negative effects on regrowth was similar to that reported in our previous screen (Chen et al., 2011) (Figure 1—figure supplement 1), suggesting this screen remains far from saturated. The combined analyses of >1200 genes reinforce the conclusion that regenerative axon regrowth requires many genetic pathways, most of which are not involved in developmental axon outgrowth or guidance. Below, we first focus on a set of genes with previously uncharacterized roles in axon regeneration and then summarize common themes from the expanded screen.
Figure 1 with 2 supplements see all
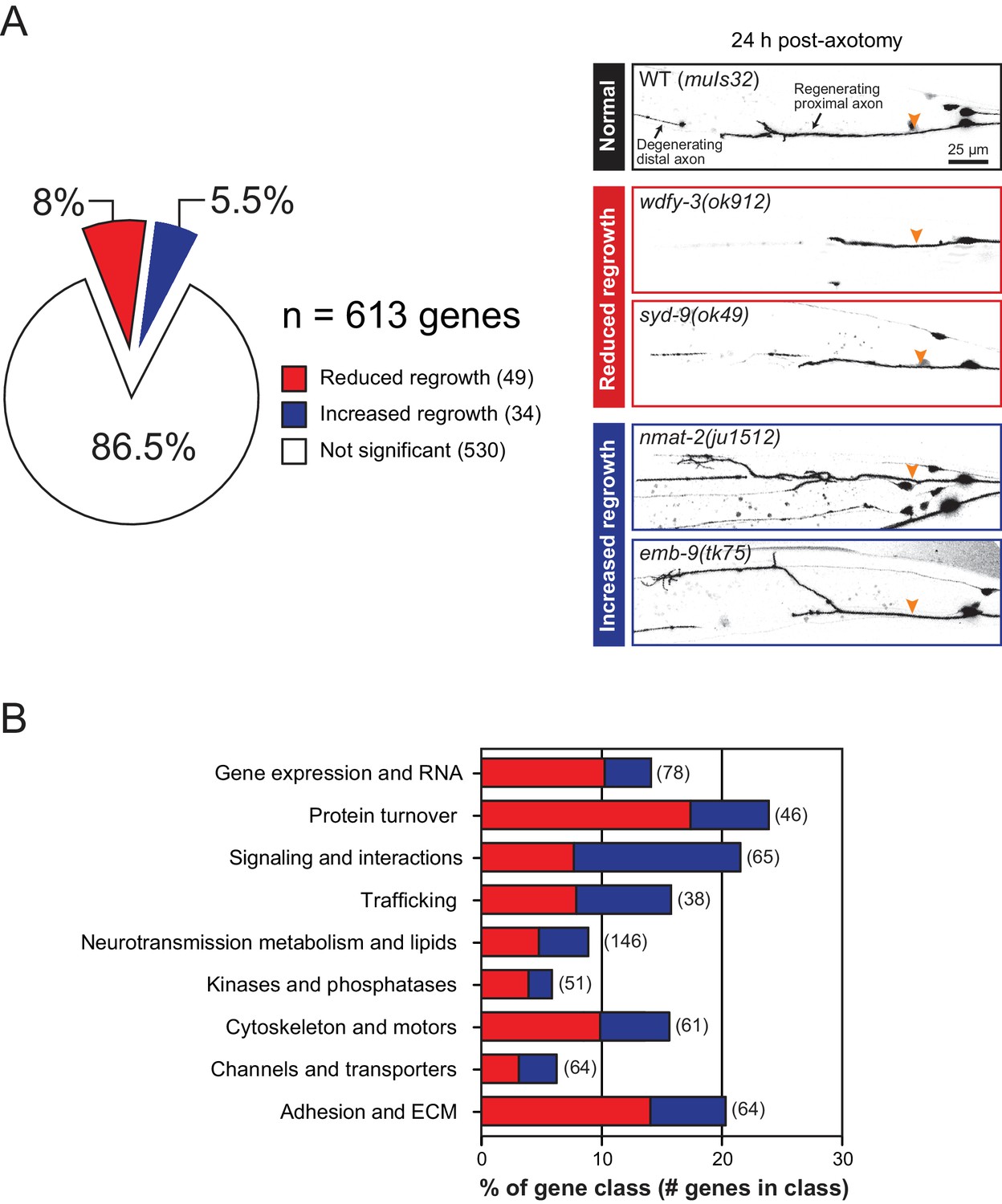
Overview and results of expanded axon regrowth screen.
(A) Pie chart showing fraction of genes screened displaying significantly reduced or increased regrowth at 24 hr. Right: representative inverted grayscale images of PLM 24 hr post-axotomy in wild type (black box), and mutants with reduced (red boxes) or increased regrowth (blue boxes). Orange arrowhead, site of axotomy. (B) Distribution of reduced/increased regrowth mutants among nine functional or structural gene classes, shown as percentage of genes in each class. See Figure 1—source data 1 for lists of genes in each class.
-
Figure 1—source data 1
List of screened genes, reference alleles, and the functional categories.
- https://doi.org/10.7554/eLife.39756.006
Table 1
Mutants displaying reduced PLM regrowth
https://doi.org/10.7554/eLife.39756.007| Gene name | Mutations | Normalized regrowth (24 hr) | N | P value | Molecular function | Closest human Genea |
|---|---|---|---|---|---|---|
| A. Cell Adhesion and ECM | ||||||
| adt-1 | cn30 | 0.75 | 21 | ** | ADAM metalloprotease | ADAMTS3 |
| adt-3 | ok923 | 0.71 | 46 | ** | ADAM metalloprotease | ADAMTS2 |
| C05D9.7 | ok2931 | 0.60 | 28 | *** | Unknown | N/A |
| dpy-10 | e128 | 0.67 | 28 | ** | 5FMC ribosome biogenesis complex | PELP1 |
| F35G2.1 | ok1669 | 0.68 | 27 | *** | Quiescin sulfhydryl oxidase | QSOX1 |
| gly-2 | tm839 | 0.69 | 16 | * | Mannosyl-glycoprotein N-acetylglucosaminyltransferases | MGAT5 |
| osm-11 | rt142 | 0.72 | 13 | ** | Secreted protein | N/A |
| zig-1 | ok784 | 0.68 | 12 | * | Basigin | BSG |
| zig-3 | ok1476 | 0.73 | 18 | ** | Kazal type serine peptidase inhibitor domain | KAZALD1 |
| B. Channels and transporters | ||||||
| abts-1 | ok1566 | 0.74 | 34 | *** | Anion exchange protein | SLC4A7 |
| mfsd-6 | ju833 | 0.68 | 15 | ** | Major facilitator | MFSD6 |
| C. Cytoskeleton and motors | ||||||
| fli-1 | ky535 | 0.61 | 27 | *** | Actin remodeling protein | FLII |
| mec-12 | e1605 | 0.73 | 15 | * | Tubulin α−3 chain | TUBA1C |
| mec-17 | ok2109 | 0.55 | 14 | ** | α-Tubulin N-acetyltransferase | ATAT1 |
| tba-9 | ok1858 | 0.70 | 24 | ** | α-Tubulin | TUBA3 |
| vab-10 | e698 | 0.78 | 11 | * | Spectraplakin | DST |
| D. Protein kinases and phosphatases | ||||||
| plk-1 | or683ts | 0.59 | 13 | ** | Polo like kinase | PLK1 |
| svh-2 | tm737 | 0.68 | 30 | *** | Receptor Tyrosine kinase | MET |
| E. Neurotransmission, metabolism, and lipid | ||||||
| cept-2 | ok3135 | 0.68 | 27 | * | Choline/ethanolamine phosphotransferase | CEPT1 |
| cpr-1 | ok1344 | 0.60 | 32 | *** | Cysteine proteinase | CTSB |
| dhhc-11 | gk1105 | 0.75 | 32 | *** | Palmitoyltransferase | ZDHHC11 |
| eat-3 | tm1107 | 0.74 | 16 | ** | Mitochondrial dynamin like GTPase | OPA1 |
| npr-20 | ok2575 | 0.49 | 44 | *** | G-protein coupled receptor | CCKBR/TRHR |
| ptps-1 | tm1984 | 0.55 | 27 | *** | 6-pyruvoyl tetrahydrobiopterin synthase | PTS |
| supr-1 | ju1118 | 0.78 | 30 | ** | Unknown | N/A |
| F. Trafficking | ||||||
| jph-1 | ok2823 | 0.77 | 14 | ** | Junctophilin | JPH1 |
| rab-8 | tm2526 | 0.77 | 28 | ** | Ras GTPase | RAB8B |
| rsef-1 | ok1356 | 0.66 | 17 | * | Endosomal Rab family GTPase | RASEF |
| G. Signaling and interactions | ||||||
| osm-7 | tm2256 | 0.57 | 28 | ** | Unknown | N/A |
| par-2 | or373 | 0.63 | 47 | *** | C3HC4-type RING-finger | TRIM |
| rgl-1 | ok1921 | 0.74 | 30 | ** | Ral guanine nucleotide dissociation stimulator | RGL1 |
| wdfy-3 | ok912 | 0.52 | 17 | *** | WD40 and FYVE domain | WDFY3 |
| wdr-23 | tm1817 | 0.66 | 30 | *** | DDB1 and CUL4 associated factor | DCAF11 |
| H. Protein turnover, proteases, cell death | ||||||
| brap-2 | ok1492 | 0.61 | 62 | *** | BRCA1-associated protein; zinc ion binding activity | BRAP |
| cdc-48.1 | tm544 | 0.69 | 19 | ** | Transitional ER ATPase homolog | VCP |
| ced-9 | n1950 | 0.73 | 16 | * | Cell-death inhibitor Bcl-2 homolog | BCL2 |
| dnj-23 | tm7102 | 0.69 | 32 | ** | DNaJ domain (prokaryotic heat shock protein) | DNAJC9 |
| fbxc-50 | tm5154 | 0.73 | 12 | * | F-box protein | N/A |
| math-33 | ok2974 | 0.64 | 12 | *** | Ubiquitin-specific protease | USP7 |
| skr-5 | ok3068 | 0.69 | 12 | * | S-phase kinase associated protein | SKP1 |
| tep-1 | tm3720 | 0.53 | 36 | *** | ThiolEster containing Protein; endopeptidase inhibitor activity | CD109 |
| I. Gene expression and RNA regulation | ||||||
| mec-8 | e398 | 0.23 | 16 | *** | RNA binding protein, mRNA processing factor | RBPMS |
| rict-1 | mg360 | 0.58 | 26 | *** | Subunit of TORC2 | RICTOR |
| rtcb-1 b | gk451 | 0.58 | 25 | *** | tRNA-splicing ligase RtcB homolog | RTCB |
| skn-1 | ok2315 | 0.78 | 10 | * | Basic leucine zipper protein | NFE2 |
| smg-3 | r930 | 0.68 | 28 | *** | Nonsense mediated mRNA decay regulator | UPF2 |
| syd-9 | ju49 | 0.47 | 15 | *** | Zinc finger E-box binding homeobox | ZEB1 |
| tdp-1 | ok803 | 0.70 | 36 | *** | TAR DNA-binding protein | TARDBP/TDP-43 |
| wdr-5.1 | ok1417 | 0.70 | 26 | *** | WD repeat-containing protein | WDR5 |
-
Genes are classified in nine functional or structural classes. Mutations are genetic or predicted molecular nulls, or partial loss-of-function. Normalized regrowth is relative to matched same-day controls or to pooled controls. Significant levels (*p<0.05; **p<0.01; ***p<0.001) based on Student’s t-test.
a Closest human gene based on BLASTP score in Wormbase WS263; Ensembl/HGNC symbol.
-
brtcb-1(gk451) mutant reported to show increased regrowth in the C. elegans motor neurons (Kosmaczewski et al., 2015).
Table 2
Mutants displaying increased PLM regrowth
https://doi.org/10.7554/eLife.39756.008| Gene name | Mutations | Normalized regrowth (24 hr) | N | P value | Molecular function | Closest human Genea |
|---|---|---|---|---|---|---|
| A. Cell adhesion and ECM | ||||||
| emb-9 | tk75 b | 1.37 | 28 | *** | Collagen type IV α3 chain | COL4A3 |
| epi-1 | gm121 | 1.33 | 36 | *** | Laminin | LAMA |
| mig-17 | k174 | 1.24 | 37 | *** | ADAM metalloprotease | ADAMTS5 |
| ZC116.3 | ok1618 | 1.40 | 26 | *** | Cubilin | CUBN |
| B. Channels and transporters | ||||||
| lgc-12 | ok3546 | 1.33 | 26 | ** | Serotonin receptor 3E | HTR3E |
| tmc-1 | ok1859 | 1.31 | 30 | ** | Transmembrane channel-like protein | TMC1 |
| C. Cytoskeleton and motors | ||||||
| ivns-1 | ok3171 | 1.31 | 18 | ** | Actin-binding; splicing | IVNS1ABP |
| twf-2 | ok3564 | 1.33 | 39 | ** | Twinfilin actin binding protein | TWF |
| nud-1 | ok552 | 1.30 | 25 | ** | Nuclear distribution C, Dynein complex regulator | NUDC |
| tba-7 | gk787939 | 1.45 | 15 | *** | α-tubulin | TUBA |
| E. Neurotransmission, metabolism, and lipid | ||||||
| nmat-2 | tm2905 | 1.55 | 38 | *** | Nicotinamide mononucleotide adenylyltransferase | NMNAT1 |
| qns-1 | ju1563 | 1.13 | 40 | * | NAD + synthetase | NADSYN1 |
| mgl-1 | tm1811 | 1.24 | 28 | ** | Glutamate metabotropic receptor | GRM3 |
| mgl-3 | tm1766 | 1.32 | 22 | ** | Glutamate metabotropic receptor | GRM6 |
| npr-25 | ok2008 | 1.27 | 26 | ** | Coagulation factor II thrombin receptor | F2RL2 |
| ucr-2.3 | ok3073 | 1.41 | 24 | *** | Ubiquinol-cytochrome C reductase core protein | UQCRC2 |
| F. Trafficking | ||||||
| nex-1 c | gk148 | 1.38 | 27 | *** | Annexin | ANXA13 |
| nex-2 d | ok764 | 1.23 | 30 | ** | Annexin | ANXA7 |
| snb-6 | tm5195 | 1.30 | 38 | ** | Vesicle associated membrane protein | VAMP1 |
| G. Signaling and interactions | ||||||
| drag-1 | tm3773 | 1.52 | 26 | ** | Repulsive guidance molecule BMP co-receptor | RGMB |
| ect-2 | ku427 | 1.31 | 31 | ** | RhoGEF | ECT2 |
| lin-2 | e1309 | 1.31 | 27 | *** | Membrane associated guanylate kinase | CASK |
| magi-1 | zh66 | 1.40 | 29 | *** | Membrane associated guanylate kinase | MAGI2 |
| prmt-5 | gk357 | 1.26 | 24 | ** | Protein arginine N-methyltransferase | PRMT5 |
| rap-1 | tm861 | 1.33 | 10 | *** | Ras small GTPase | RAP1 |
| smz-1 | ok3576 | 1.39 | 13 | * | PDZ domain-containing protein | N/A |
| trxr-1 | tm2047 | 1.3 | 31 | * | Thioredoxin reductase | TXNRD2 |
| H. Protein turnover, proteases, cell death | ||||||
| natb-1 | ju1405 | 1.29 | 14 | * | N(α)-acetyltransferase 20 | NAA20 |
| rnf-5 | tm794 | 1.28 | 15 | * | Ring finger protein | RNF5 |
| ulp-5 | tm3063 | 1.22 | 30 | *** | SUMO specific peptidase | SENP7 |
| I. Gene expression and RNA regulation | ||||||
| csr-1 | fj54 | 1.24 | 38 | *** | Argonaute | AGO1 |
| hda-6 | tm3436 | 1.29 | 42 | *** | Histone deacetylase | HDAC6 |
| elpc-3 | ok2452 | 1.31 | 26 | *** | Elongator acetyltransferase complex subunit | ELP3 |
-
Genes are classified in nine functional or structural classes. Mutations are genetic or predicted molecular nulls, or partial loss-of-function. Normalized regrowth is relative to matched same-day controls or to pooled controls. Significant levels (*p<0.05; **p<0.01; ***p<0.001) based on Student’s t-test.
a Closest human gene based on BLASTP score in Wormbase WS263; Ensembl/HGNC symbol.
-
b emb-9(tk75) mutant reported to be a gain-of-function allele that makes stable EMB-9/Type IV collagen (Kubota et al., 2012).
cnex-1(gk148) mutant reported to show reduced regrowth in the C. elegans motor neurons (Nix et al., 2014).
-
d nex-2(bas4) mutant reported to show normal regrowth in the C. elegans motor neurons (Nix et al., 2014).
The conserved enzyme NMNAT inhibits axon regeneration
Among genes with significant inhibitory effects on axon regrowth, we identified NMAT-2, a member of the nicotinamide mononucleotide adenylyltransferase (NMNAT) enzyme family (Figure 2A,B). NMNAT enzymes catalyze a vital step in NAD+ biosynthesis and confer neuroprotection in several injury models of flies and mice (Gerdts et al., 2016). In mammalian neurons increasing NMNAT activity protects against Wallerian degeneration and axon degradation following trophic factor withdrawal (Mack et al., 2001; Vohra et al., 2010). In C. elegans, overexpression of NMAT-2/NMNAT protects against neuronal degeneration caused by the toxic mutant ion channel MEC-4(d) (Calixto et al., 2012), but does not protect against distal axon degeneration after laser axotomy (Nichols et al., 2016). We found that PLM regrowth was enhanced in two independent nmat-2 null (0) mutants, tm2905 and ju1512 (Figure 2B,C). A null mutation of NMAT-1, a close paralog, did not affect PLM regrowth (Figure 2C). nmat-2(0) adult animals are sterile, while nmat-1(0) are fertile, indicating that these two NMNATs may have distinct tissue- or cell-type-specific roles.
Figure 2 with 1 supplement see all
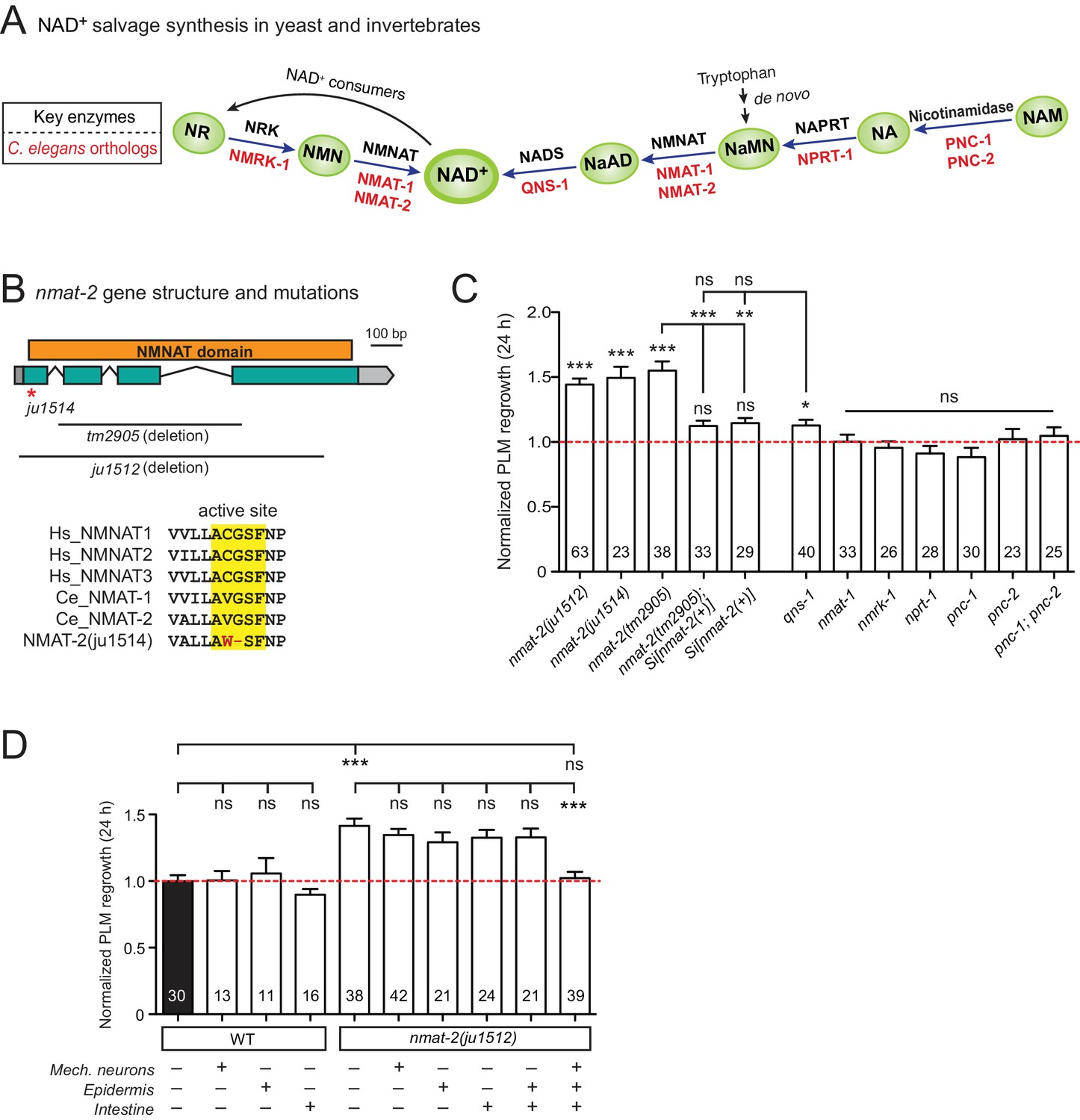
NMNAT/NMAT-2 inhibits PLM axon regrowth via its catalytic domain.
(A) Overview of NAD+ salvage biosynthesis pathway. Top, key enzymes; Bottom, C. elegans orthologs (Shaye and Greenwald, 2011). (B) Top, nmat-2 gene structure and mutant alleles. NMAT-2 contains an NMNAT domain. nmat-2(ju1514) point mutation and nmat-2(ju1512) deletion alleles were generated using CRISPR-Cas9 genome editing. Bottom, sequence alignment of the active site of NMNAT domain of C. elegans NMAT-2 (accession number: NP_492480.1; amino acids 4–14) with human NMNAT1–3 (NP_073624.2, NP_055854, NP_001307441) and C. elegans NMAT-1 (NP_510010.2). Sequences were analyzed using Clustal Omega. (C) Normalized regrowth 24 hr post-axotomy in mutants lacking genes encoding enzymes in the NAD+ biosynthesis pathway. Statistics, Student’s t-test with same day controls. For the statistical test of transgene analysis, one-way ANOVA followed by Tukey’s multiple comparison test. (D) PLM axon regrowth 24 hr post-axotomy in transgenic animals expressing nmat-2(+) driven by tissue-specific promoters for mechanosensory neurons (Pmec-4), epidermis (Pcol-12) or intestine (Pmtl-2) in a nmat-2(ju1512) background. One-way ANOVA followed by Tukey’s multiple comparison test. Data are shown as mean ± SEM. n, number of animals shown within columns. ns, not significant; *p<0.05; **p<0.01; ***p<0.001.
-
Figure 2—source data 1
Each data point in Figure 2C,D.
- https://doi.org/10.7554/eLife.39756.011
To address whether the observed effects of nmat-2(0) are related to NAD+ synthesis, we examined loss-of-function mutants of other enzymes in the invertebrate NAD+ salvage synthesis pathway (Figure 2A), including the glutamine-dependent NAD+ synthase QNS-1, nicotinamide riboside kinase (NRK) NMRK-1, nicotinate phosphoribosyltransferase (NAPRT) NPRT-1, nicotinamidase PNC-1 and PNC-2 (Magni et al., 1999; Vrablik et al., 2009). Among these, only qns-1(0) mutants showed marginally increased axon regrowth (Figure 2C). NMAT-2 and QNS-1 catalyze the terminal steps of the NAD+ salvage pathway. Like nmat-2(0), qns-1(0) mutants are sterile (Wang et al., 2015) (this work), while other single mutants are fertile, suggesting that NMAT-2 and QNS-1 define essential steps in the biosynthesis of NAD+. To address whether sterility of the animals might contribute to the observed effects on axon regrowth, we cultured animals on 5’fluoro-2’ deoxyuridine (FUdR) and found that neither wild type or nmat-1(0) grown in FUdR showed increased PLM regrowth (Figure 2—figure supplement 1). Additionally, we have previously reported that sterile animals following germline ablation do not affect PLM regrowth (Kim et al., 2018). Thus, we conclude that NMAT-2’s role in axon regrowth is independent of animal fertility.
We next focused on NMAT-2 to define the role of NAD+ pathway in axon regeneration. Using CRISPR genome editing, we generated a single copy transgene expressing nmat-2(+) under its endogenous promoter (juSi347). This transgene fully rescued the sterility of nmat-2(0) and restored the increased axon regrowth in nmat-2(0) mutants to wild-type levels (Figure 2C), confirming that the increased axon regrowth is due to loss of NMAT-2 function. We then asked in which tissues NMAT-2 acts to inhibit axon regeneration using transgenic expression of NMAT-2 in the epidermis, intestine, or mechanosensory neurons (Figure 2—source data 1). Transgenic expression of NMAT-2 in individual tissues was not able to restore axon regeneration in nmat-2(0) to normal (Figure 2D). Interestingly, the combined expression of NMAT-2 in all three tissues restored normal axon regeneration (Figure 2D), and also partially rescued sterility. We conclude that NMAT-2 may act in both neuronal and non-neuronal cells to inhibit axon regeneration.
In addition to their enzymatic roles, several NMNAT proteins function as molecular chaperones, including Drosophila NMNAT, mouse NMNAT2, and human NMNAT3 (Ali et al., 2016; Zhai et al., 2006; Zhai et al., 2008). We therefore tested whether the enzymatic properties of NMAT-2 are required for inhibition of axon regeneration. Using CRISPR genome editing, we mutated the active site motif involved in ATP recognition (Zhang et al., 2002) (Figure 2B). This mutant nmat-2(ju1514) displayed sterility and enhanced regrowth of PLM neurons (Figure 2C), indistinguishable from nmat-2(0) mutants. Therefore, the role of NMAT-2 in axon regeneration likely requires its enzymatic activity. Here, we infer that the enhanced axon regeneration in nmat-2(0) reflects sustained low levels of NAD+.
The neuroprotective effect of NMNAT is cell-autonomous in Drosophila and in mice (Gilley et al., 2013; Wen et al., 2011). Our finding that NMAT-2 inhibits axon regrowth via several tissues suggests that NMNAT may function via distinct mechanisms for neuroprotection vs. axon regeneration. The PLM axon is adjacent to the intestine and is enveloped by the surrounding epidermis (Emtage et al., 2004). Speculatively, NAD+ might activate inhibitory factors in neurons and in surrounding tissues, which act together to repress the axon regenerative response; some of these factors might regulate cell-cell interaction and signal transduction. In Drosophila, lack of NMNAT also led to enhanced sensory axon regeneration (Chen et al., 2016b). Together, these data suggest conserved roles of NMNAT in axon regeneration. Future work will be required to dissect specific mechanisms by which NMNAT inhibits axon regeneration.
Differential roles and functional redundancy of ER-PM contact site components in axon regeneration
Membrane contact sites (MCSs) are regions where membranes from two organelles or an organelle and the PM are held together by protein tethers, most of which are conserved from yeast to mammals (Phillips and Voeltz, 2016; Saheki et al., 2016). MCSs can coordinate activities such as calcium entry or lipid transfer between membranes. Calcium entry via voltage-gated Ca2+ channels in the PM is critical for PLM axon regeneration (Ghosh-Roy et al., 2010). Additionally, MCSs between the PM and ER might be involved in lipid addition to the PM during rapid extension of regrowing axons (Hausott and Klimaschewski, 2016). We therefore examined mutants affecting conserved ER-PM MCS components such as Junctophilin, Extended synaptotagmin (E-Syt), Anoctamins, and OxySterol Binding Proteins (OSBP).
Junctophilins are multi-pass transmembrane proteins that are localized to ER-PM contacts in excitable cells, where they couple PM- and ER-localized calcium channels (Landstrom et al., 2014). JPH-1 is the sole Junctophilin in C. elegans (Yoshida et al., 2001) (Figure 3A). We observed that jph-1(ok2823) mutants, likely null, exhibited a significantly increased rate of reconnection or fusion between the regrowing axon and distal fragment (Figure 3B). Axons that did not reconnect in jph-1 mutants exhibited reduced axon regeneration, compared to controls (Figure 3C). As reconnected axons were not measured for regrowth analysis, the reduced regrowth in jph-1 mutants might be due to an overrepresentation of poorly growing axons. Axon-axon fusion requires the fusogen EFF-1 (Ghosh-Roy et al., 2010; Pérez-Vargas et al., 2014) and a phosphoserine-mediated apoptotic cell engulfment pathway (Neumann et al., 2015). We analyzed eff-1; jph-1 double mutants and found that the enhanced reconnection in jph-1 was greatly reduced (Figure 3B). Drosophila Junctophilin-like molecule functions in apoptotic cell removal (Gronski et al., 2009). These observations suggest JPH-1-mediated contacts may restrict axon-axon fusion, dependent on eff-1.
Figure 3 with 1 supplement see all
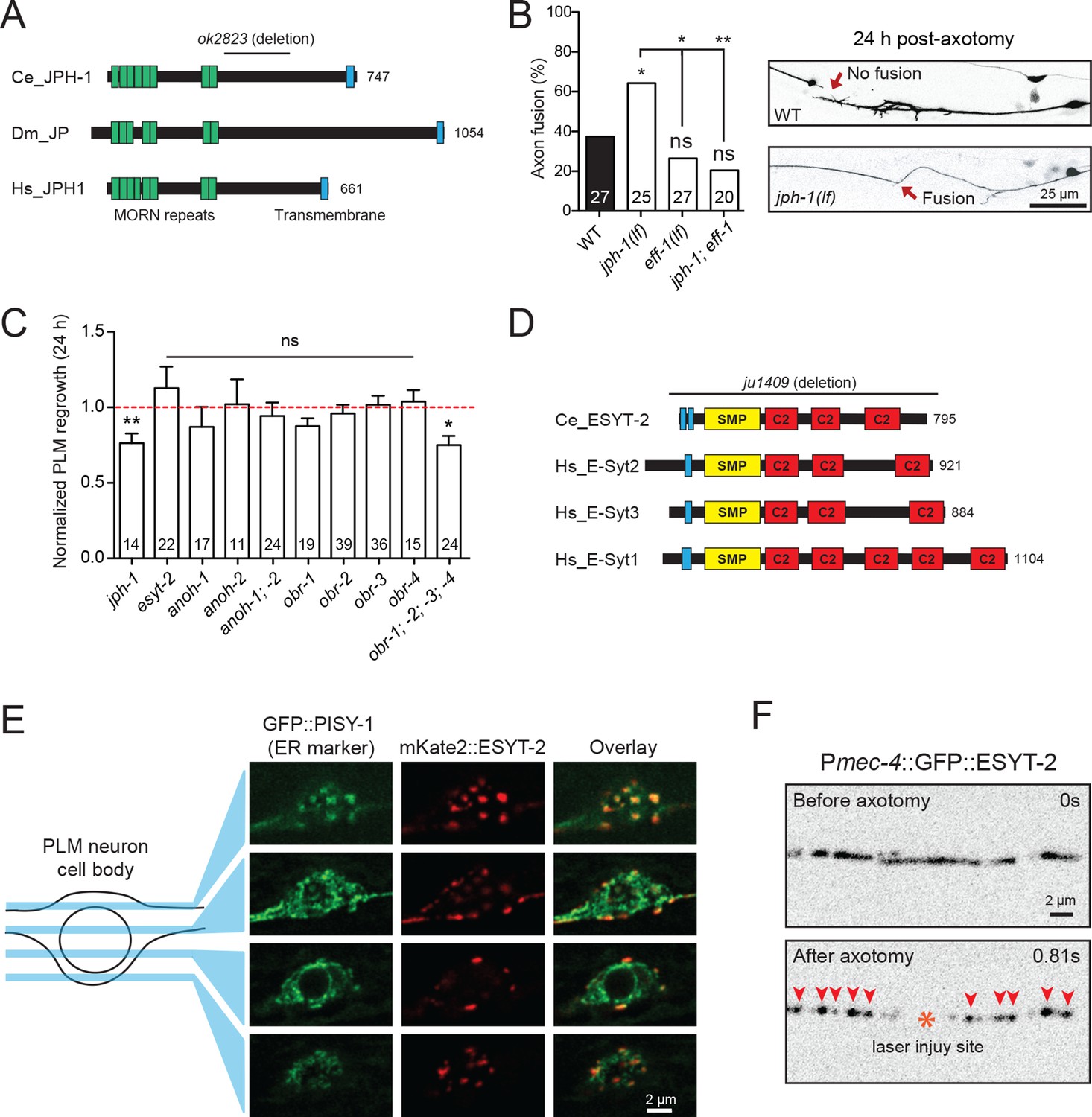
Select ER-PM membrane contact site proteins are required for axon regeneration and are sensitive to injury.
(A) Junctophilin-1 protein structure. From top to bottom: C. elegans JPH-1 (NP_492193.2), its Drosophila ortholog JP (NP_523525.2), and human ortholog JPH1 (NP_065698.1). Junctophilins contain N-terminal MORN (Membrane Occupation and Recognition Nexus) repeats (green) and a C-terminal transmembrane domain (blue). C. elegans deletion allele is indicated above (ok2823). (B) Percentage of axons that exhibit fusion between the regrowing axon and distal fragment 24 hr post-axotomy. Upper image shows a regrowing axon that has not fused with the distal fragment in a wild-type animal. Lower image shows fusion between the regrowing axon and the distal fragment in a jph-1(ok2823) animal. Fisher’s exact test. *p<0.05; **p<0.01. (C) Normalized regrowth 24 hr post-axotomy in mutants lacking selected genes encoding ER-PM MCS proteins. Data are shown as mean ± SEM. n, number of animals shown within columns. Student’s t-test with same day controls. ns, not significant; *p<0.05; **p<0.01. (D) E-Syt protein structure. From top to bottom: C. elegans ESYT-2 and its human orthologs E-Syt2, E-Syt3, and E-Syt1 (NP_065779.1, NP_114119.2, NP_056107.1, respectively). Amino acid length is indicated to the right of each protein. E-Syt proteins contain N-terminal hydrophobic regions (blue), SMP (Synaptotagmin-like Mitochondrial and lipid-binding Protein) domains (yellow), and C-terminal C2 domains (red). C. elegans deletion allele is indicated above (ju1409). (E) Images of the PLM cell body and surrounding neurites. Left, GFP::PISY-1 ER marker; Middle, mKate2::ESYT-2 driven by the mec-4 promoter; Right, Image overlays. Images show single slices taken at 1 μm intervals. (F) Representative inverted grayscale images of GFP::ESYT-2 in the axon of the PLM neuron before and immediately after axotomy (upper and lower panels, respectively). Site of laser axotomy indicated by asterisk; puncta indicated by arrowheads.
-
Figure 3—source data 1
Each data point in Figure 3C.
- https://doi.org/10.7554/eLife.39756.014
Extended synaptotagmins (E-Syt) are a family of proteins containing multiple C2 domains (Figure 3D) that have been shown to tether the ER to the PM (Giordano et al., 2013) and are implicated in membrane lipid transfer (Saheki et al., 2016; Yu et al., 2016). ESYT-2 is the sole E-Syt in C. elegans and is most closely related to human E-Syt2 and E-Syt3 (Figure 3D). We found that esyt-2 showed wide expression in the nervous system (Figure 3—figure supplement 1). In the mechanosensory neuron cell body, full-length GFP-ESYT-2 showed a punctate pattern, colocalizing with an ER marker PISY-1 (Rolls et al., 2002) at the peripheral ER (Figure 3E). In uninjured axons, ESYT-2 was distributed intermittently (Figure 3F; upper panel). Strikingly, upon axon injury, axonal ESYT-2 condensed into small puncta almost immediately (<1 s) (Figure 3F; lower panel). As axon injury triggers a rapid rise in axonal calcium (Ghosh-Roy et al., 2010), we speculate that the injury-induced Ca2+ transient triggers ESYT-2 relocalization to axonal ER-PM contact sites. This is consistent with the observation that vertebrate E-Syt1 can localize to ER-PM contact sites following an increase in cytosolic calcium (Giordano et al., 2013; Idevall-Hagren et al., 2015). We generated esyt-2 null mutants by genome editing (Figure 3D). These mutant animals were indistinguishable from wild-type animals in growth rate, body morphology, and exhibited normal axon development and regrowth (Figure 3C). Thus, while ESYT-2 undergoes temporal changes in response to axon injury, it does not appear to be essential for axon regrowth.
The Anoctamin protein family function as tethers at ER-PM contact sites in yeast (Manford et al., 2012; Wolf et al., 2012). C. elegans has two orthologs, ANOH-1 and ANOH-2. ANOH-1 is expressed in mechanosensory neurons and acts together with the apoptotic factor CED-7 to promote phosphatidylserine exposure in the removal of necrotic cells (Li et al., 2015). ced-7(0) reduces PLM axon regrowth (Neumann et al., 2015). However, we found that loss of function in anoh-1 or anoh-2, or the anoh-1; anoh-2 double mutant, did not affect PLM axon regeneration (Figure 3C).
The eukaryotic OSBP and OSBP-related (ORP) family of MCS-localized lipid transfer proteins includes multiple members. ORP5/8 act as tethers at ER-PM MCSs where they mediate PI4P/Phosphatidylserine counter-transport, while OSBP and the other ORPs function at different MCSs (Chung et al., 2015). We tested the four C. elegans homologs individually as well as a quadruple mutant. Each obr single mutant displayed normal regeneration, and the quadruple mutant displayed a significant decrease in axon regrowth (Figure 3C). While the expression pattern and action site of these OBR proteins remain to be determined, our finding is consistent with the known redundancy within the OBR family (Kobuna et al., 2010).
Altogether, the above analysis echoes a recent study in yeast where elimination of multiple MCS components did not impair ER-PM sterol exchange (Quon et al., 2018), highlighting the challenge to tease apart the functional redundancy of MCS proteins in biological processes.
Lipid metabolic enzymes likely have extensive functional redundancy in axon regrowth
Lipids are essential components of membranes and regulate many biological functions including energy storage and lipid signaling. In C. elegans, the majority of triglyceride is obtained from the diet, and lipogenesis accounts for less than 10% of stored body fat (Srinivasan, 2015). Lipolysis is required for cellular uptake or release of fatty acids and glycerol (Zechner et al., 2012). Classical ‘neutral’ lipolysis involves at least three different lipases: ATGL (adipose triglyceride lipase), HSL (hormone sensitive lipase), and MGL (monoglyceride lipase). ATGL requires a coactivator protein, CGI-58/ABHD5. C. elegans encodes a single ATGL (ATGL-1), three CGI-58/ABHD5 (ABHD-5.2, ABHD-5.3, and LID-1), a single HSL (HOSL-1), but lacks MGL by sequence homology (Zechner et al., 2012). We tested single mutants for all these genes and double or triple mutants for ABHD (α/β hydrolase domain) genes and observed no detectable effects in PLM axon regrowth (Figure 4A).
Figure 4

PLM axon regeneration involves membrane lipid biosynthesis pathway.
(A) Normalized PLM axon regrowth 24 hr post-axotomy in mutants affecting neutral lipolysis. (B) Normalized PLM axon regrowth 24 hr post-axotomy in mutants affecting acid lipolysis. (C) Overview of C.elegans Kennedy pathway for de novo biosynthesis of PE and PC, the major phospholipids in the PM. (D) Normalized PLM axon regrowth 24 hr post-axotomy in mutants lacking select genes encoding enzymes in the Kennedy pathway. Data are shown as mean ±SEM. n, number of animals shown within columns. Student’s t-test with same day controls. ns, not significant; *p<0.05.
-
Figure 4—source data 1
Each data point in Figure 4A,B,D.
- https://doi.org/10.7554/eLife.39756.016
Triglycerides can also be hydrolyzed through autophagy-mediated degradation of lipid droplets by some lysosomal acid lipases, termed lipophagy or ‘acid’ lipolysis (Singh et al., 2009). C. elegans lysosomal lipases (LIPL-1, LIPL-3, and LIPL-4), autophagy proteins (LGG-1 and LGG-2), and transcription factors (HLH-30/TFEB and MXL-3/MXL) act in lipophagy (Folick et al., 2015; O'Rourke and Ruvkun, 2013). Two nuclear hormone receptors NHR-49/PPARα and NHR-80/HNF4α are reported to regulate LIPL-4 (Folick et al., 2015). We found that single mutants for all these genes showed normal PLM regrowth (Figure 4B), suggesting that lipolysis may not play an essential role in PLM axon regeneration.
The Kennedy pathway synthesizes the most abundant phospholipids in eukaryotic membranes, phosphatidylcholine (PC) and phosphatidylethanolamine (PE) (Gibellini and Smith, 2010), and involves conserved enzymes catalyzing a series of consecutive reactions (Figure 4C). Of all mutants affecting individual enzymes in the Kennedy pathway, we found that cept-2 null mutants showed a significant reduction in axon regrowth (Figure 4D). In testing functional redundancy between cept-1 and cept-2, we found double mutants to be embryonic or larval lethal (data not shown), preventing further analysis. Definitive conclusions will require tissue-specific and temporal manipulation of this pathway. Overall, our analysis suggests that the Kennedy pathway may affect axon regeneration.
The conserved NS1A-BP ortholog IVNS-1 inhibits axon regrowth
Among other conserved proteins, we identified the BTB-Kelch family protein IVNS-1 (Influenza Virus NS1A binding protein/NS1A-BP) as an inhibitor of axon regrowth. BTB/POZ (Broad-Complex, Tramtrack, and Bric-a-Brac/Poxvirus and Zinc finger) domain and Kelch repeats function in a wide variety of biological processes including gene expression, protein ubiquitination, and cytoskeleton binding (Dhanoa et al., 2013). Human NS1A-BP was originally identified based on interaction with the influenza A virus via its Kelch domain (Wolff et al., 1998) (Figure 5A) and was later found to interact with actin filaments (Perconti et al., 2007) and RNA binding proteins, including heterogeneous nuclear ribonucleoprotein and splicing factors (hnRNPs) and RNA helicase (Tsai et al., 2013). C. elegans IVNS-1 has the same overall domain organization as NS1A-BP (Figure 5A).
Figure 5
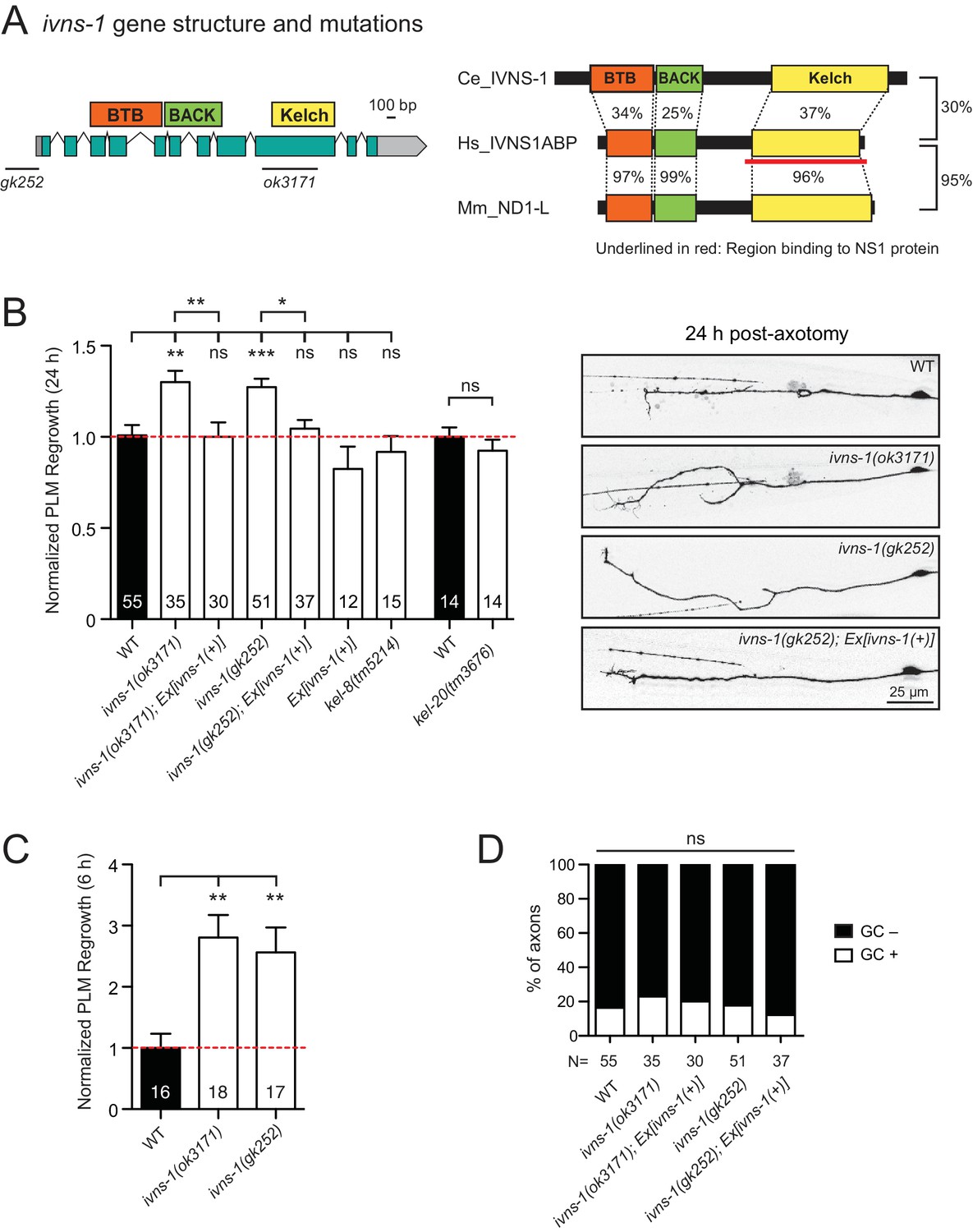
The Kelch-domain protein IVNS-1 inhibits axon regeneration.
(A) ivns-1 gene structure. Left: Loss-of-function alleles are indicated below (gk252 and ok3171). Right: Alignment of the C. elegans IVNS-1 (NP_510109.1) with its human ortholog IVNS1ABP (NP_006460.1) and mouse ortholog ND1-L (NP_473443.2). Number indicates percentage identity of protein sequences. Sequences were analyzed using Clustal Omega. (B) Normalized PLM axon regrowth 24 hr post-axotomy in mutants of Kelch-domain proteins. Data are shown as mean ±SEM. n, number of animals shown within columns. Student’s t-test with same day controls. ns, not significant; *p<0.05; **p<0.01; ***p<0.001. Right: representative inverted grayscale images of PLM 24 hr post-axotomy. Scale bar, 25 μm. (C) Normalized PLM axon regrowth 6 hr post-axotomy. Data are shown as mean ±SEM. One-way ANOVA followed by Tukey’s multiple comparison test. n, number of animals shown within columns. **p<0.01. (D) Percentage of axons with growth cones (GCs) 6 hr post-axotomy. n, Number of animals shown below columns. Fisher’s exact test. ns, not significant.
-
Figure 5—source data 1
Each data point in Figure 5B,C.
- https://doi.org/10.7554/eLife.39756.018
We analyzed two independent ivns-1 mutants (gk252 and ok3171) and observed increased axon regrowth, which was restored to control levels following transgenic expression of ivns-1 driven by its own promoter (Figure 5A,B). ivns-1 mutants showed increased regrowth as early as 6 hr post-injury (Figure 5C), while growth cone formation in ivns-1 mutants was normal (Figure 5D). The effects of ivns-1 on axon regrowth appeared to be unique, as mutants in two other BTB-Kelch proteins kel-8 and kel-20 displayed normal regrowth (Figure 5B). Whether the function of IVNS-1 involves actin cytoskeleton or RNA regulation remains to be determined.
Overview of common themes
Complex roles of basement membrane ECM and ADAMTSs
ECM plays diverse roles in axon regeneration (Barros et al., 2011). In C. elegans, neuronal processes are closely associated with basement membrane (BM) (White et al., 1976), which is a thin, specialized ECM adjacent to epithelial tissues (Jayadev and Sherwood, 2017). We previously reported that BM components SPON-1/F-spondin and PXN-2/Peroxidasin inhibit axon regrowth (Chen et al., 2011; Gotenstein et al., 2010). We further analyzed mutants of essential BM structural components and found that a loss-of-function mutant (gm121) of EPI-1/Laminin α and a gain-of-function mutant (tk75) of EMB-9/Type IV collagen (Kubota et al., 2012) both showed enhanced regrowth (Figure 1—figure supplement 2A), supporting defined roles of specific BM components in axon regrowth.
ADAMTS proteins are secreted metalloproteases and act as key ECM remodeling enzymes (Tang, 2001). In the mammalian nervous system, ADAMTS4 promotes axon regeneration and recovery after spinal cord injury, by digesting chondroitin sulfate proteoglycans (CSPGs), which are known to be prominent inhibitory components of the glial scar (Tauchi et al., 2012). In C. elegans, multiple chondroitin proteoglycans are expressed, but are not sulfated (Olson et al., 2006). The PLM axon is enveloped by the surrounding epidermis (Emtage et al., 2004) and is not in direct contact with the BM after embryogenesis. However, regrowing PLM axons may come in contact with the BM during regrowth. We tested null mutants in all five ADAMTS homologs and found that ADT-1 and ADT-3 promote and MIG-17 inhibits PLM axon regrowth (Figure 1—figure supplement 2A). These results suggest opposing roles for ADAMTS family members in PLM axon regrowth. ADT-1 and ADT-3 may normally degrade inhibitory BM such that their deficiency leads to elevated BM and impairs axonal regrowth. In contrast, MIG-17 may degrade permissive BM (for example, Type IV collagen) such that its deficiency leads to elevated stable Type IV collagen and enhances axonal regrowth. Together, these data indicate the complex roles of ECM components and ADAMTSs.
Permissive role of Rab GTPase RAB-8 and inhibitory role of annexin proteins NEX-1 and NEX-2 in axon regeneration
We previously showed that genes implicated in endocytosis of synaptic vesicles (e.g., unc-57/endophilin) or membrane trafficking (rsef-1/RASEF) are required for axon regeneration (Chen et al., 2011). Here, we tested additional membrane-trafficking factors, especially the Rab small GTPases. Trafficking of secretory vesicles from the Golgi is partly regulated by Rab8 (Stenmark, 2009), and trafficking of recycling endosomes is regulated by Rab11 (Ascaño et al., 2009). Lack of Rab8 results in decreased neurite outgrowth in embryonic hippocampal neurons (Huber et al., 1995). C. elegans RAB-8 has been implicated in membrane trafficking in ciliated neurons (Kaplan et al., 2010). We found that RAB-8 was required for PLM axon regrowth, whereas RAB-11.2 showed no impact (Figure 1—figure supplement 2B), suggesting that post-Golgi vesicle trafficking, rather than endosome recycling, may be important for axon regrowth.
The Annexins are calcium-dependent phospholipid-binding proteins (Monastyrskaya et al., 2007) with a wide variety of roles in membrane biology (Mirsaeidi et al., 2016) and plasma membrane repair/resealing (Boye and Nylandsted, 2016). C. elegans has four Annexins (NEX-1/–2/−3/–4) (Daigle and Creutz, 1999); and NEX-1 was shown to promote GABAergic motor neuron regeneration (Nix et al., 2014). We found that NEX-1 and NEX-2 have an inhibitory role on PLM regrowth, whereas NEX-3 and NEX-4 have no impact (Figure 1—figure supplement 2B). These results suggest cell-type-dependent roles of Annexins in regrowth.
Further evidence for permissive roles of the MT cytoskeleton in axon regeneration
Precise regulation of MT dynamics is a critical factor in axon regrowth (Blanquie and Bradke, 2018; Tang and Chisholm, 2016). Our previous studies identified EFA-6 as an intrinsic inhibitor of regrowth by acting as an axonal MT-destabilizing factor (Chen et al., 2015; Chen et al., 2011). We and others have also reported that MT post-translational modifications have differential roles in axon regeneration (Cho and Cavalli, 2012; Ghosh-Roy et al., 2012). MT stabilization is linked to acetylation of α-tubulins (Janke and Montagnac, 2017) and has been shown to improve regrowth; for example, pharmacological MT stabilization by Paclitaxel or Epothilone B promotes axon regrowth in multiple models (Chen et al., 2011; Ruschel et al., 2015; Sengottuvel et al., 2011). Here, we tested two α-tubulin acetyltransferases, MEC-17 and ATAT-2, which acetylate the α-tubulin MEC-12 that is enriched in mechanosensory neurons (Akella et al., 2010). We found that MEC-17, but not ATAT-2, was required for normal axon regrowth (Figure 1—figure supplement 2C). mec-17; atat-2 double mutants showed reduced axon regrowth resembling the mec-17 single mutant (Figure 1—figure supplement 2C), suggesting that MEC-17-dependent acetylated MTs are permissive for axon regrowth. In addition, the HDAC orthologs HDA-3 and HDA-6 inhibit axon regrowth (Chen et al., 2011) (Table 2). HDAC family proteins, which can deacetylate MTs and other targets, have been shown to be involved in mammalian axon regeneration (Cho and Cavalli, 2012). Overall, our results support a pro-regenerative role for acetylated MTs in axon regrowth.
An increasing notion is that isotypes of tubulins influence MT composition and stability (Tang and Jin, 2018). PLM axons contain predominantly unusual 15 MT filaments made of MEC-7/β-tubulin and MEC-12/α-tubulin, and also express multiple tubulin isotypes (Kaletsky et al., 2018; Lockhead et al., 2016) that likely contribute to 11 protofilaments. We found that loss of function in mec-12 or tba-9 resulted in reduced regrowth (Figure 1—figure supplement 2D). In contrast, loss of function in tba-7 showed enhanced regrowth. A recent study that examined the neurite growth of mechanosensory neurons has proposed that TBA-7/ α -tubulin likely functions as a destabilizing factor for MTs (Zheng et al., 2017). Our observation is consistent with this proposal, and supports the general role of stabilized MTs in promoting axon regrowth.
Roles for actin filament regulators in axon regeneration
Growth cone formation is an important initial stage of axon regeneration and involves extensive remodeling of actin filaments (Gomez and Letourneau, 2014). Actin-binding proteins can promote actin filament assembly or disassembly, for example, Gelsolin severs actin filaments to promote disassembly (Klaavuniemi et al., 2008; Liu et al., 2010), while Twinfilin binds to the ADP-actin monomers and prevents their assembly into filaments (Moseley et al., 2006; Palmgren et al., 2002). Of the three Gelsolin-related proteins in C. elegans, gsnl-1 and viln-1 null mutants showed normal regrowth while partial loss-of-function mutants of fli-1 displayed reduced PLM axon regrowth (Figure 1—figure supplement 2E). In contrast, lack of the Twinfilin homolog TWF-2 increased axon regrowth. Although both Gelsolin and Twinfilin can promote actin filament disassembly, they may have differential roles in regenerating C. elegans axons.
Novel ion channels and transporters involved in PLM axon regeneration
Neuronal activity plays a significant role in axon regeneration in vertebrates and invertebrates (Chen et al., 2011; Ghosh-Roy et al., 2010; Lim et al., 2016; Tedeschi et al., 2016). Our prior screen tested 54 genes encoding channels and transporters and overall was consistent with neuronal excitability promoting PLM regrowth. Here, we examined an additional 58 channel or transporter genes (Figure 1—source data 1). We found several new genes in which loss-of-function mutation results in enhanced regeneration, including the sodium-sensitive channel tmc-1 (Chatzigeorgiou et al., 2013) and an acetylcholine receptor alpha subunit (CHRNA6) lgc-12 (Cohen et al., 2014) (Table 2). Additionally, we found that MFSD-6, a member of the Major Facilitator Superfamily Domain (MFSD) family, promotes PLM regrowth (Figure 1—figure supplement 2F). MFSD family proteins have 10–12 transmembrane regions (Yan, 2015); some mediate nutrient transport across the blood-brain barrier (Ceder et al., 2017; Perland et al., 2017), but most are of unknown function. C. elegans MFSD-6 was previously identified as a regulator of motor circuit activity and mfsd-6(0) mutants are resistant to inhibitors of cholinesterase (McCulloch et al., 2017). mfsd-6 is expressed in most neurons, including mechanosensory neurons (Ogurusu et al., 2015). Loss-of-function mutants of mfsd-6 exhibited reduced axon regrowth, which was rescued by expressing wild type mfsd-6 under a pan-neuronal promoter (Figure 1—figure supplement 2F). As mechanosensory neurons are not thought to be cholinergic, yet other mutants of cholinergic signaling (e.g. cha-1/ChAT, unc-17/VChAT) are defective in axon regrowth (Chen et al., 2011), these data suggest a possible neuronal, but cell non-autonomous, role for acetylcholine signaling to be permissive for PLM regrowth.
Discussion
Functional screening for axon regeneration phenotypes is a powerful approach to identify novel regulators of axon regrowth after injury. C. elegans PLM axons exhibit robust response to injury, and therefore allow efficient screening of positive and negative regulators of regrowth. In this work we have nearly doubled the number of genes tested using genetic mutations and the PLM regeneration assay, taking the total number of genes screened to 1267. We expanded some gene classes previously analyzed in depth (e.g. kinases, ECM components, ion channels, and transporters) and have also specifically targeted several pathways not addressed in our earlier screen, such as NAD+ biosynthesis, MCS components, lipid metabolism, and actin regulators. Interestingly, both MCS components and lipid metabolism tested display a high degree of genetic redundancy, such that single mutants only occasionally display regeneration defects, and compound mutant strains are required to assess functional requirements. Nevertheless, our findings suggest that ER-PM contact sites may be regulated by axon injury and that phospholipid synthesis may be critical for axon regeneration. Further work will be required to define whether these pathways play a role in lipid addition to the regrowing axon membrane or a more general signaling role.
Several axon regeneration screens have now been reported in C. elegans and the results may be compared to assess reproducibility and generalizability of the results. The present work and our prior screen (Chen et al., 2011) analyzed the effect of genetic mutations on PLM axon regeneration. In contrast, other studies have used RNAi or genetic mutants to analyze motor neuron regeneration (Nix et al., 2014). Differing results between the two screens (e.g. the opposite requirement for nex-1 in PLM and motor neurons) may reflect cell-type-specific roles of the regulators in axon regeneration. A recent genome-wide in vitro axon regeneration screen in mouse cortical neurons revealed significant overlap with orthologous genes identified from C. elegans screen despite differences in neuron types, species, and experimental methods (Chen et al., 2011; Nix et al., 2014; Sekine et al., 2018), suggesting significant conservation of regenerative mechanisms.
Our screen approach is based on candidates and not random mutagenesis, and thus classical estimates of genetic saturation do not apply. However, it is notable that the frequency of positive and negative hits in the current screen does not differ from our previous screen. Our prior screen included many previously well-studied axon guidance and outgrowth pathways and thus might have been enriched for functionally important factors, but the present analysis suggests many genes not previously associated with the nervous system (e.g. ptps-1, tep-1, brap-2) also have functionally important roles in regrowth. One trend is that fewer mutants with dramatically reduced regrowth (<30% of wild type, such as dlk-1, unc-75, sdn-1) were identified, and thus the number of genes essential for initiation of regrowth may be limited. On the other hand, the present screen identified new mutants with drastically enhanced regrowth (>140% of wild type, such as efa-6 and pxn-2 from previous screen and nmat-2 and drag-1 from this screen). Interestingly, a recent genome-wide screen for enhanced regrowth in mouse cortical neurons reported a positive hit rate of 3% (Sekine et al., 2018), whereas we find 3.9% of genes displayed significantly elevated axon regrowth. The frequency of axon regrowth phenotypes may therefore be consistent across screening platforms.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Bacterial strain | E. coli: OP50 | Caenorhabditis Genetics Center | RRID: WB-STRAIN:OP50 | |
| Genetic reagent (C. elegans) | Strain wild type N2 | Caenorhabditis Genetics Center | RRID:WB-STRAIN:N2_ (ancestral) | |
| Genetic reagent (C. elegans) | CZ10969: Pmec-7-GFP(muIs32) II | Considered as‘WT’ in many axotomy experiments | ||
| Genetic reagent (C. elegans) | CZ10175: Pmec-4-GFP(zdIs5) I | Considered as ‘WT’ in many axotomy experiments | ||
| Genetic reagent (C. elegans) | CZ25411: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ25415: nmat-2(ju1514) I/hT2 I, III; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ17633: nmat-2(tm2905) I/hT2 I, III; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ24324: Pmec-7-GFP(muIs32) II; qns-1(ju1563) IV/mIs11 sd IV | |||
| Genetic reagent (C. elegans) | CZ25642: Pmec-4-GFP(zdIs5) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C.elegans) | CZ25534: nmrk-1(ok2571) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ24241: Pmec-4-GFP(zdIs5) I; nprt-1(tm6342) IV | |||
| Genetic reagent (C. elegans) | CZ24242: Pmec-4-GFP(zdIs5) I; pnc-1(tm3502) IV | |||
| Genetic reagent (C. elegans) | CZ24802: Pmec-4-GFP(zdIs5) I; pnc-2(tm6438) IV | |||
| Genetic reagent (C. elegans) | CZ25466: Pmec-7-GFP(muIs32) II; juSi347[nmat-2 gDNA] IV | |||
| Genetic reagent (C. elegans) | CZ25469: nmat-2(tm2905) I;Pmec-7-GFP(muIs32) II; juSi347[nmat-2 gDNA] IV | |||
| Genetic reagent (C. elegans) | CZ26216: Pmec-7-GFP(muIs32) II; Ex[Pmec-4-nmat-2(juEx7834)] | |||
| Genetic reagent (C. elegans) | CZ26217: Pmec-7-GFP(muIs32) II; Ex[Pmec-4-nmat-2(juEx7835)] | |||
| Genetic reagent (C. elegans) | CZ26220: Pmec-7-GFP(muIs32) II; Ex[Pcol-12-nmat-2(juEx7838)] | |||
| Genetic reagent (C. elegans) | CZ26221: Pmec-7-GFP(muIs32) II; Ex[Pcol-12-nmat-2(juEx7839)] | |||
| Genetic reagent (C. elegans) | CZ26218: Pmec-7-GFP(muIs32) II; Ex[Pmtl-2-nmat-2(juEx7836)] | |||
| Genetic reagent (C. elegans) | CZ26219: Pmec-7-GFP(muIs32) II; Ex[Pmtl-2-nmat-2(juEx7837)] | |||
| Genetic reagent (C. elegans) | CZ26222: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmec-4-nmat-2(juEx7840)] | |||
| Genetic reagent (C. elegans) | CZ26223: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmec-4-nmat-2(juEx7841)] | |||
| Genetic reagent (C. elegans) | CZ26224: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pcol-12-nmat-2(juEx7842)] | |||
| Genetic reagent (C. elegans) | CZ26225: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pcol-12-nmat-2(juEx7843)] | |||
| Genetic reagent (C. elegans) | CZ26310: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmtl-2-nmat-2(juEx7836)] | |||
| Genetic reagent (C. elegans) | CZ26311: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmtl-2-nmat-2(juEx7837)] | |||
| Genetic reagent (C. elegans) | CZ26332: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmtl-2, col-12::nmat-2(juEx7853)] | |||
| Genetic reagent (C. elegans) | CZ26333: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmtl-2, col-12::nmat-2(juEx7854)] | |||
| Genetic reagent (C. elegans) | CZ26285: nmat-2(ju1512) I/hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmtl-2, col-12, mec-4::nmat-2(juEx7850)] | |||
| Genetic reagent (C. elegans) | CZ26286: nmat-2(ju1512) I / hT2 I, III; Pmec-7-GFP(muIs32) II; Ex[Pmtl-2, col-12, mec-4::nmat-2(juEx7851)] | |||
| Genetic reagent (C. elegans) | CZ26391: jph-1(ok2823) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ22032: Pmec-4-GFP(zdIs5) I; anoh-1(tm4762) III | |||
| Genetic reagent (C. elegans) | CZ22033: Pmec-4-GFP(zdIs5) I; anoh-2(tm4796) IV | |||
| Genetic reagent (C. elegans) | CZ26325: Pmec-4-GFP(zdIs5) I; anoh-1(tm4762) III; anoh-2(tm4796) IV | |||
| Genetic reagent (C. elegans) | CZ26069: Pmec-4-GFP(zdIs5) I; obr-1(xh16) III | |||
| Genetic reagent (C. elegans) | CZ24555: Pmec-4-GFP(zdIs5) I; obr-2 (xh17) V | |||
| Genetic reagent (C. elegans) | CZ24556: Pmec-4-GFP(zdIs5) I; obr-3(tm1087) X | |||
| Genetic reagent (C. elegans) | CZ24557: obr-4(tm1567) I; Pmec-4-GFP(zdIs5) I | |||
| Genetic reagent (C. elegans) | CZ25696: obr-4(tm1567) I; Pmec-4-GFP(zdIs5) I; obr-1(xh16) III; obr-2(xh17) V; obr-3(tm1087) X | |||
| Genetic reagent (C. elegans) | CZ26375: Pmec-4-GFP(zdIs5) I; esyt-2(ju1409) III | |||
| Genetic reagent (C. elegans) | CZ26570: juIs540 [Pmec-4-mKate2-ESYT-2]; juEx7807[Pmec-4-GFP-PISY-1] | |||
| Genetic reagent (C. elegans) | CZ24897: juEx7604 [Pmec-4-GFP-ESYT-2] | |||
| Genetic reagent (C. elegans) | CZ22087: Pmec-7-GFP(muIs32) II; atgl-1(tm3116) III / hT2 I, III | |||
| Genetic reagent (C. elegans) | CZ22536: Pmec-7-GFP(muIs32) II; hosl-1(gk278589) X | |||
| Genetic reagent (C. elegans) | CZ22006: Pmec-7-GFP(muIs32) II; abhd-5.1(ok3722) V | |||
| Genetic reagent (C. elegans) | CZ21968: Pmec-7-GFP(muIs32) II; abhd-5.2(ok3245) V | |||
| Genetic reagent (C. elegans) | CZ22007: lid-1(gk575511) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ22163: Pmec-7-GFP(muIs32) II; abhd-5.2(ok3245) V abhd-5.1(ju1282) V | |||
| Genetic reagent (C. elegans) | CZ22166: lid-1(gk575511) I; Pmec-7-GFP(muIs32) II; abhd-5.2(ok3245) abhd-5.1(ju1282) V | |||
| Genetic reagent (C. elegans) | CZ22686: Pmec-7-GFP(muIs32) II; lipl-1(tm1954) V | |||
| Genetic reagent (C. elegans) | CZ22688: Pmec-7-GFP(muIs32) II; lipl-3(tm4498) V | |||
| Genetic reagent (C. elegans) | CZ22535: Pmec-7-GFP(muIs32) II; lipl-4(tm4417) V | |||
| Genetic reagent (C. elegans) | CZ24364: Pmec-4-GFP(zdIs5) I; lgg-1(tm3489) II/ mIn1 II | |||
| Genetic reagent (C. elegans) | CZ23325: Pmec-7-GFP(muIs32) II; lgg-2(tm6474) IV | |||
| Genetic reagent (C. elegans) | CZ23322: Pmec-7-GFP(muIs32) II; hlh-30(tm1978) IV | |||
| Genetic reagent (C. elegans) | CZ14408: Pmec-7-GFP(muIs32) II; mxl-3(ok1947) X | |||
| Genetic reagent (C. elegans) | CZ22541: nhr-49(nr2041) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ22510: Pmec-7-GFP(muIs32) II; nhr-80(tm1011) III | |||
| Genetic reagent (C. elegans) | CZ25587: Pmec-4-GFP(zdIs5) I; cka-1(tm1241) IV | |||
| Genetic reagent (C. elegans) | CZ25549: Pmec-4-GFP(zdIs5) I; cka-2(tm841) X | |||
| Genetic reagent (C. elegans) | CZ25403: Pmec-4-GFP(zdIs5) I; cka-1(tm1241) IV; cka-2(tm841) X. | |||
| Genetic reagent (C. elegans) | CZ25370: Pmec-4-GFP(zdIs5) I; pcyt-1(et9) X | |||
| Genetic reagent (C. elegans) | CZ25790: Pmec-4-GFP(zdIs5) I; pcyt-2.1(gk440213) I | |||
| Genetic reagent (C. elegans) | CZ25368: Pmec-4-GFP(zdIs5) I; pcyt-2.2(ok2179) X | |||
| Genetic reagent (C. elegans) | CZ25992: Pmec-4-GFP(zdIs5) I; pcyt-2.1(gk440213) I; pcyt-2.2(ok2179) X | |||
| Genetic reagent (C. elegans) | CZ26521: Pmec-4-GFP(zdIs5) I; pcyt-2.1(gk440213) I; pcyt-1(et9) X | |||
| Genetic reagent (C. elegans) | CZ25369: Pmec-4-GFP(zdIs5) I; cept-1(et10) X | |||
| Genetic reagent (C. elegans) | CZ26423: Pmec-4-GFP(zdIs5) I; cept-2(ok3135) V | |||
| Genetic reagent (C. elegans) | CZ19835: Pmec-4-GFP(zdIs5) I; kel-8(tm5214) V | |||
| Genetic reagent (C. elegans) | CZ23911: kel-20(tm3676) I; mec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ18224: Pmec-4-GFP(zdIs5) I; ivns-1(ok3171) X | |||
| Genetic reagent (C. elegans) | CZ18225: Pmec-4-GFP(zdIs5) I; ivns-1(gk252) X | |||
| Genetic reagent (C. elegans) | CZ25508: Pmec-4-GFP(zdIs5) I; ivns-1(ok3171) X; Ex[ivns-1_gDNA(juEx7673)] | |||
| Genetic reagent (C. elegans) | CZ25509: Pmec-4-GFP(zdIs5) I; ivns-1(ok3171) X; Ex[ivns-1_gDNA(juEx7674)] | |||
| Genetic reagent (C. elegans) | CZ25510: Pmec-4-GFP(zdIs5) I; ivns-1(gk252) X; Ex[ivns-1_gDNA(juEx7673)] | |||
| Genetic reagent (C. elegans) | CZ25511: Pmec-4-GFP(zdIs5) I; ivns-1(gk252) X; Ex[ivns-1_gDNA(juEx7674)] | |||
| Genetic reagent (C. elegans) | CZ24755: juEx7584[Pesyt-2-GFP] | |||
| Genetic reagent (C. elegans) | CZ21465: Pmec-4-GFP(zdIs5) I; epi-1(gm121) IV | |||
| Genetic reagent (C. elegans) | CZ21463: Pmec-7-GFP(muIs32) II; emb-9(tk75) III | |||
| Genetic reagent (C. elegans) | CZ21198: Pmec-7-GFP(muIs32) II; adt-1(cn30) X | |||
| Genetic reagent (C. elegans) | CZ20937: Pmec-4-GFP(zdIs5) I; adt-2(wk156) X | |||
| Genetic reagent (C. elegans) | CZ21004: Pmec-4-GFP(zdIs5) I; adt-3 (T19D2.1) (ok923) X | |||
| Genetic reagent (C. elegans) | CZ26611: Pmec-7-GFP(muIs32) II; gon-1(e1254) IV / + | |||
| Genetic reagent (C. elegans) | CZ23908: rab-8(tm2526) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ23909: rab-11.2(tm2081) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ20682: Pmec-7-GFP(muIs32) II; nex-1(gk148) III | |||
| Genetic reagent (C. elegans) | CZ20683: Pmec-7-GFP(muIs32) II; nex-2(ok764) III | |||
| Genetic reagent (C. elegans) | CZ20684: Pmec-7-GFP(muIs32) II; nex-3(gk385) III | |||
| Genetic reagent (C. elegans) | CZ20685: Pmec-7-GFP(muIs32) II; nex-4(gk102) V | |||
| Genetic reagent (C. elegans) | CZ14006: Pmec-7-GFP(muIs32) II; mec-17(ok2109) IV | |||
| Genetic reagent (C. elegans) | CZ14008: Pmec-7-GFP(muIs32) II; atat-2(ok2415) X | |||
| Genetic reagent (C. elegans) | CZ14848: mec-17(ok2109) IV; atat-2(ok2415) X; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ17720: Pmec-7-GFP(muIs32) II; mec-12(tm5083) III | |||
| Genetic reagent (C. elegans) | CZ9247: tba-1(ok1135) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ26688: Pmec-4-GFP(zdIs5) I; tba-7(gk787939) III | |||
| Genetic reagent (C. elegans) | CZ26833: Pmec-4-GFP(zdIs5) I; tba-7(u1015) III | |||
| Genetic reagent (C. elegans) | CZ26635: Pmec-4-GFP(zdIs5) I; mec-7(ok2152) X | |||
| Genetic reagent (C. elegans) | CZ10615: Pmec-4-GFP(zdIs5) I; tbb-2(gk129) III | |||
| Genetic reagent (C. elegans) | CZ11083: Pmec-4-GFP(zdIs5) I; tbb-4(ok1461) X | |||
| Genetic reagent (C. elegans) | CZ10810: Pmec-4-GFP(zdIs5) I; tbb-6(tm2004) V | |||
| Genetic reagent (C. elegans) | CZ21461: Pmec-4-GFP(zdIs5) I; fli-1(ky535) III | |||
| Genetic reagent (C. elegans) | CZ21199: Pmec-7-GFP(muIs32) II; gsnl-1(ok2979) V | |||
| Genetic reagent (C. elegans) | CZ10888: viln-1(ok2413) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ13606: Pmec-4-GFP(zdIs5) I; twf-2(ok3564) X | |||
| Genetic reagent (C. elegans) | CZ20063: Pmec-4-GFP(zdIs5) I; mfsd-6(ju833) III | |||
| Genetic reagent (C. elegans) | CZ19827: Pmec-4-GFP(zdIs5) I; mfsd-6(tm3356) III | |||
| Genetic reagent (C. elegans) | CZ24417: Pmec-4-GFP(zdIs5) I; mfsd-6(tm3356) III; Prgef-1-mfsd-6(juEx6079) | |||
| Genetic reagent (C. elegans) | CZ21030: Pmec-4-GFP(zdIs5) I; C05D9.7(ok2931) X | |||
| Genetic reagent (C. elegans) | CZ25317: Pmec-4-GFP(zdIs5) I;dpy-10(e128) II | |||
| Genetic reagent (C. elegans) | CZ23667: Pmec-4-GFP(zdIs5) I;F35G2.1(ok1669) IV | |||
| Genetic reagent (C. elegans) | CZ23772: Pmec-4-GFP(zdIs5) I;gly-2(tm839) I | |||
| Genetic reagent (C. elegans) | CZ17890: Pmec-4-GFP(zdIs5) I;osm-11(rt142) X | |||
| Genetic reagent (C. elegans) | CZ17021: Pmec-4-GFP(zdIs5) I;zig-1(ok784) II | |||
| Genetic reagent (C. elegans) | CZ17023: Pmec-4-GFP(zdIs5) I;zig-3(gk33) X | |||
| Genetic reagent (C. elegans) | CZ17024: Pmec-4-GFP(zdIs5) I;zig-3(ok1476) X | |||
| Genetic reagent (C. elegans) | CZ22031: abts-1(ok1566) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ21461: Pmec-4-GFP(zdIs5) I; fli-1(ky535) III | |||
| Genetic reagent (C. elegans) | CZ17435: Pmec-7-GFP(muIs32) II;mec-12(e1605) III | |||
| Genetic reagent (C. elegans) | CZ17637: Pmec-4-GFP(zdIs5) I;tba-9(ok1858) X | |||
| Genetic reagent (C. elegans) | CZ20033: vab-10(e698) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ17099: Pmec-4-GFP(zdIs5) I; plk-1(or683ts) III | |||
| Genetic reagent (C. elegans) | CZ17285: Pmec-4-GFP(zdIs5) I; svh-2(tm737) X | |||
| Genetic reagent (C. elegans) | CZ19343: Pmec-4-GFP(zdIs5) I; cpr-1(ok1344) V | |||
| Genetic reagent (C. elegans) | CZ19200:dhhc-11(gk1105) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ22823: Pmec-4-GFP(zdIs5) I;eat-3(tm1107) II | |||
| Genetic reagent (C. elegans) | CZ16134: Pmec-4-GFP(zdIs5) I;npr-20(ok2575) II | |||
| Genetic reagent (C. elegans) | CZ23845: Pmec-4-GFP(zdIs5) I; ptps-1(tm1984) I | |||
| Genetic reagent (C. elegans) | CZ17995: supr-1(ju1118) I;Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ12031: Pmec-4-GFP(zdIs5) I;rsef-1(ok1356) X | |||
| Genetic reagent (C. elegans) | CZ17629: Pmec-4-GFP(zdIs5) I; osm-7(tm2256) III | |||
| Genetic reagent (C. elegans) | CZ17098: Pmec-4-GFP(zdIs5) I; par-2(or373ts) III | |||
| Genetic reagent (C. elegans) | CZ18676: Pmec-4-GFP(zdIs5) I; rgl-1(ok1921) X | |||
| Genetic reagent (C. elegans) | CZ20056: Pmec-7-GFP(muIs32) II; wdfy-3(ok912) IV | |||
| Genetic reagent (C. elegans) | CZ19721: Pmec-4-GFP(zdIs5) I wdr-23(tm1817) I | |||
| Genetic reagent (C. elegans) | CZ22063: Pmec-4-GFP(zdIs5) I; brap-2(tm5132) II | |||
| Genetic reagent (C. elegans) | CZ21217: Pmec-4-GFP(zdIs5) I; brap-2(ok1492) II | |||
| Genetic reagent (C. elegans) | CZ19337: Pmec-4-GFP(zdIs5) I; cdc-48.1(tm544) II | |||
| Genetic reagent (C. elegans) | CZ19725: Pmec-4-GFP(zdIs5) I; ced-9(n1950sd) III | |||
| Genetic reagent (C. elegans) | CZ21651: Pmec-4-GFP(zdIs5) I; dnj-23(tm7102) II | |||
| Genetic reagent (C. elegans) | CZ21356: Pmec-4-GFP(zdIs5) I; fbxc-50(tm5154) II | |||
| Genetic reagent (C. elegans) | CZ16950: Pmec-7-GFP(muIs32) II; math-33(ok2974) V | |||
| Genetic reagent (C. elegans) | CZ16951: Pmec-7-GFP(muIs32) II; skr-5(ok3068) V | |||
| Genetic reagent (C. elegans) | CZ21010: tep-1(tm3720) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ22796: mec-8(e398) I;Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ14510: Pmec-4-GFP(zdIs5) I;rict-1(mg360) II | |||
| Genetic reagent (C. elegans) | CZ22570: rtcb-1(gk451) I / [bli-4(e937) let-?(q782) qIs48](hT2) I, III; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ21655: Pmec-4-GFP(zdIs5) I; skn-1(ok2315) IV/nT1(qIs51) IV; V | |||
| Genetic reagent (C. elegans) | CZ23377: Pmec-4-GFP(zdIs5) I; smg-3(r930) IV | |||
| Genetic reagent (C. elegans) | CZ13997: Pmec-7-GFP(muIs32) II; syd-9(ju49) X | |||
| Genetic reagent (C. elegans) | CZ21723: Pmec-4-GFP(zdIs5) I; tdp-1(ok803) II | |||
| Genetic reagent (C. elegans) | CZ23133: Pmec-4-GFP(zdIs5) I; wdr-5.1(ok1417) III | |||
| Genetic reagent (C. elegans) | CZ21194: Pmec-7-GFP(muIs32) II; mig-17(k174) V | |||
| Genetic reagent (C. elegans) | CZ22792: Pmec-7-GFP(muIs32) II; ZC116.3(ok1618) V | |||
| Genetic reagent (C. elegans) | CZ19193: Pmec-7-GFP(muIs32) II;lgc-12(ok3546) III | |||
| Genetic reagent (C. elegans) | CZ18217: Pmec-4-GFP(zdIs5) I;tmc-1(ok1859) X | |||
| Genetic reagent (C. elegans) | CZ17639: Pmec-4-GFP(zdIs5) I;nud-1(ok552) III | |||
| Genetic reagent (C. elegans) | CZ17841: Pmec-4-GFP(zdIs5) I;mgl-1(tm1811) X | |||
| Genetic reagent (C. elegans) | CZ17843: Pmec-4-GFP(zdIs5) I;mgl-3(tm1766) IV | |||
| Genetic reagent (C. elegans) | CZ17848: Pmec-4-GFP(zdIs5) I;npr-25(ok2008) V | |||
| Genetic reagent (C. elegans) | CZ22890: Pmec-4-GFP(zdIs5) I;ucr-2.3(ok3073) III | |||
| Genetic reagent (C. elegans) | CZ15607: drag-1(tm3773) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ17393: Pmec-4-GFP(zdIs5) I; snb-6(tm5195) II | |||
| Genetic reagent (C. elegans) | CZ17018: Pmec-4-GFP(zdIs5) I; drag-1(tm3773) I | |||
| Genetic reagent (C. elegans) | CZ18617: Pmec-4-GFP(zdIs5) I; ect-2(ku427) II | |||
| Genetic reagent (C. elegans) | CZ18818: Pmec-4-GFP(zdIs5) I; lin-2(e1309) X | |||
| Genetic reagent (C. elegans) | CZ18817: Pmec-4-GFP(zdIs5) I; magi-1(zh66) IV | |||
| Genetic reagent (C. elegans) | CZ20673: Pmec-4-GFP(zdIs5) I; prmt-5(gk357) III | |||
| Genetic reagent (C. elegans) | CZ18816: Pmec-4-GFP(zdIs5) I; rap-1(tm861) IV | |||
| Genetic reagent (C. elegans) | CZ18460: Pmec-4-GFP(zdIs5) I; smz-1(ok3576) IV | |||
| Genetic reagent (C. elegans) | CZ22544: Pmec-4-GFP(zdIs5) I; trxr-1(sv47) IV | |||
| Genetic reagent (C. elegans) | CZ24963: Pmec-4-GFP(zdIs5) I; natb-1(ju1405) V/nT1 IV; V | |||
| Genetic reagent (C. elegans) | CZ16946: Pmec-7-GFP(muIs32) II; rnf-5(tm794) III | |||
| Genetic reagent (C. elegans) | CZ23068: ulp-5/tofu-3(tm3063) I; Pmec-7-GFP(muIs32) II | |||
| Genetic reagent (C. elegans) | CZ23091: Pmec-7-GFP(muIs32) II; csr-1(fj54) IV/nT1 IV; V | |||
| Genetic reagent (C. elegans) | CZ17638: Pmec-4-GFP(zdIs5) I; elpc-3(ok2452) V | |||
| Genetic reagent (C. elegans) | CZ12938: Pmec-4-GFP(zdIs5) I; hda-6(tm3436) IV | |||
| Recombinant DNA reagent | Plasmid: pCZGY3260:nmat-2 genomic DNA | This work | N/A | nmat-2 genomic DNA (~1500 bp upstream;~670 bp downstream); modified pCFJ201 plasmid for modified MosSCI on ChIV |
| Recombinant DNA reagent | Plasmid: pCZ993:Pmec-4-nmat-2 gDNA-let-858 3'UTR | This work | N/A | nmat-2 expression driven by mec-4 promoter in the mechanosensory neurons |
| Recombinant DNA reagent | Plasmid: pCZ994: Pmtl-2-nmat-2 gDNA-let-858 3'UTR | This work | N/A | nmat-2 expression driven by mtl-2 promoter in the intestine |
| Recombinant DNA reagent | Plasmid: pCZ995: Pcol-12-nmat-2 gDNA-let-858 3'UTR | This work | N/A | nmat-2 expression driven by col-12 promoter in the epiderdims |
| Recombinant DNA reagent | Plasmid: pCZGY3329: Pmec-4-GFP-ESYT-2 | This work | N/A | |
| Recombinant DNA reagent | Plasmid: pCZGY3344: Pmec-4-mKate2-ESYT-2 | This work | N/A | |
| Recombinant DNA reagent | Plasmid: pCZGY3342: Pmec-4-mKate2-PISY-1 | This work | N/A | |
| Recombinant DNA reagent | Plasmid: pCZGY3302: ivns-1 genomic DNA | This work | N/A | ivns-1 genomic DNA (2 kb upstream; 800 bp downstream) |
| Recombinant DNA reagent | Plasmid: pCZGY3347: Prgef-1-mfsd-6 | This work | N/A | |
| Recombinant DNA reagent | Plasmid: pCZGY3346:Pesyt-2-GFP | This work | N/A | GFP expression driven by esyt-2 promoter |
| Sequence- based reagent | crRNA: nmat-2:/AltR1/rCrGrArGrU rCrGrC rUrCrU rUrCrU rUrGrC rCrGrUrGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to makenmat-2(ju1512) and nmat-2(ju1514) |
| Sequence- based reagent | crRNA: nmat-2:/AltR1/rCrGrUrGrU rUrGrA rArCrU rArArC rUrCrC rArCrU rGrUrU rUrUrA rGrArG rCrUrArUrGrC rU/AltR2/ | IDT | N/A | crRNA to makenmat-2(ju1512) |
| Sequence- based reagent | crRNA: nmat-1:/AltR1/rArA rCrUrU rUrUrU rCrGrGrUrCrC rCrCrA rUrArG rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to mak e nmat-1(ju1565) |
| Sequence- based reagent | crRNA: nmat-1:/AltR1/rArU rGrUrA rCrUrU rGrArU rUrArC rGrGrA rArUrC rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to make nmat-1(ju1565) |
| Sequence- based reagent | crRNA: qns-1:/AltR1/rGrGrUrGrU rUrArU rUrCrA rCrGrU rGrUrU rArCrA rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to make qns-1(ju1563) |
| Sequence-based reagent | crRNA: qns-1:/AltR1/rGrA rUrArA rCrUrG rArArA rUrCrU rGrGrA rUrArG rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to make qns-1(ju1563) |
| Sequence-based reagent | crRNA: esyt-2:/AltR1/rGrG rUrUrU rCrArG rUrArA rUrUrGrUrGrG rGrCrU rGrUrUrUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to make esyt-2(ju1409) |
| Sequence-based reagent | crRNA: esyt-2:/AltR1/rGrU rGrCrA rCrUrU rArCrG rGrGrU rUrGrU rArGrGrGrUrU rUrUrA rGrArGrCrUrA rUrGrC rU/AltR2/ | IDT | N/A | crRNA to makeesyt-2(ju1409) |
| Peptide, recombinant protein | Protein: Cas9-NLS purified protein | QB3 MacroLab, UC Berkley | N/A | |
| Peptide, recombinant protein | Phusion High-Fidelity DNA polymerases | Thermo Scientific | Cat#F530L | |
| Peptide, recombinant protein | DreamTaq DNA polymerases | Thermo Scientific | Cat#EP0705 | |
| Commercial assay or kit | ||||
| Chemical compound, drug | 5-fluoro-2-deoxy uridine | Sigma-Aldrich | Cat#50-91-9 | |
| Software, algorithm | ImageJ | NIH image | RRID:SCR_003070 | |
| Software, algorithm | ZEN | Zeiss | https://www.zeiss.com/microscopy/us/downloads/zen.html | |
| Software, algorithm | Zeiss LSM Data Server | Zeiss | https://www.zeiss.com/microscopy/us/downloads/lsm-5-series.html | |
| Software, algorithm | GraphPad Prism 5 | GraphPad Software, Inc. | RRID:SCR_002798 |
Experimental model
Request a detailed protocolThe nematode Caenorhabditis elegans was used as the experimental model for this study. All experiments were performed with hermaphrodite animals; males were used only for crosses. Unless otherwise indicated, all experiments were carried out with L4 stage animals. Strains were maintained under standard conditions on Nematode Growth Media (NGM) plates seeded with E. coli OP50 bacteria unless mentioned. Wild type was the N2 Bristol strain (Brenner, 1974). New strains were constructed using standard procedures and all genotypes confirmed by PCR or sequencing. Extrachromosomal array transgenic lines were generated as described (Mello et al., 1991).
Laser microsurgery of axons (axotomy)
Request a detailed protocolWe cut PLM axons and quantified the length of regrown axons as previously described (Wu et al., 2007).
Axotomy imaging with MicroPoint laser
Request a detailed protocolL4 stage animals were immobilized using 2.5 mM levamisole in M9 buffer on 5% agar pads. Using a MicroPoint laser on an Andor spinning disk confocal unit (CSU-W1) with Leica DMi8 microscope, laser axotomy was performed on the PLM axon ~45 μm away from the cell body. Images were taken immediately before and immediately after axotomy (0.81 s) with iXon ultra 888 EMCCD camera.
Confocal imaging with Airyscan
Request a detailed protocolL4 stage animals were immobilized using 2.5 mM levamisole in M9 buffer on 5% agar pads. PLM mechanosensory neuron cell bodies were imaged using a Zeiss LSM800 equipped with Airyscan. Z-stack planes were taken at 0.2 μm intervals in both mKate2 and GFP channels using Airyscan.
CRISPR-Cas9 gene editing
Request a detailed protocolWe generated the nmat-2(ju1512), nmat-1(ju1564), qns-1(ju1563), and esyt-2(ju1409) deletion alleles using co-CRISPR (Arribere et al., 2014; Friedland et al., 2013). We generated the nmat-2(ju1514) point mutation allele using the homology-directed genome editing and single-strand oligodeoxynucleotide repair method (Paix et al., 2017).
FUdR treatment
Request a detailed protocolWe transferred worms onto plates containing 50 µg/ml 5-fluoro-2-deoxy uridine (FUdR) immediately after axotomy. No offspring were observed after 2 days, confirming FUDR-induced sterility.
Quantification and statistical analysis
Request a detailed protocolStatistical analysis was performed using GraphPad Prism 5. Significance was determined using unpaired t-tests for two samples, one-way ANOVA followed by multiple comparison tests for multiple samples. p<0.05 (*) was considered statistically significant. *p<0.05; **p<0.01; ***p<0.001. Data are shown as mean ± SEM. ‘n’ represents the number of animals and is shown in graphs.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responsesJournal of Neuroscience 29:11674–11685.https://doi.org/10.1523/JNEUROSCI.1542-09.2009
-
Extracellular matrix: functions in the nervous systemCold Spring Harbor Perspectives in Biology 3:a005108.https://doi.org/10.1101/cshperspect.a005108
-
Cytoskeleton dynamics in axon regenerationCurrent Opinion in Neurobiology 51:60–69.https://doi.org/10.1016/j.conb.2018.02.024
-
Annexins in plasma membrane repairBiological Chemistry 397:961–969.https://doi.org/10.1515/hsz-2016-0171
-
Assembly of a new growth cone after axotomy: the precursor to axon regenerationNature Reviews Neuroscience 13:183–193.https://doi.org/10.1038/nrn3176
-
The Neuronal and Peripheral Expressed Membrane-Bound UNC93A Respond to Nutrient Availability in MiceFrontiers in Molecular Neuroscience 10:351.https://doi.org/10.3389/fnmol.2017.00351
-
Peripheral regenerationAnnual Review of Neuroscience 30:209–233.https://doi.org/10.1146/annurev.neuro.30.051606.094337
-
Caenorhabditis elegans nicotinic acetylcholine receptors are required for nociceptionMolecular and Cellular Neuroscience 59:85–96.https://doi.org/10.1016/j.mcn.2014.02.001
-
Transcription, biochemistry and localization of nematode annexinsJournal of cell science 112:1901–1913.
-
Heritable genome editing in C. elegans via a CRISPR-Cas9 systemNature Methods 10:741–743.https://doi.org/10.1038/nmeth.2532
-
Rescue of peripheral and CNS axon defects in mice lacking NMNAT2Journal of Neuroscience 33:13410–13424.https://doi.org/10.1523/JNEUROSCI.1534-13.2013
-
Actin dynamics in growth cone motility and navigationJournal of Neurochemistry 129:221–234.https://doi.org/10.1111/jnc.12506
-
An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory responseCell Death & Differentiation 16:1323–1331.https://doi.org/10.1038/cdd.2009.55
-
Membrane turnover and receptor trafficking in regenerating axonsEuropean Journal of Neuroscience 43:309–317.https://doi.org/10.1111/ejn.13025
-
A deficiency of the small GTPase rab8 inhibits membrane traffic in developing neuronsMolecular and Cellular Biology 15:918–924.https://doi.org/10.1128/MCB.15.2.918
-
Growing the growth cone: remodeling the cytoskeleton to promote axon regenerationTrends in Neurosciences 35:164–174.https://doi.org/10.1016/j.tins.2011.11.002
-
Causes and Consequences of Microtubule AcetylationCurrent Biology 27:R1287–R1292.https://doi.org/10.1016/j.cub.2017.10.044
-
Caenorhabditis elegans gelsolin-like protein 1 is a novel actin filament-severing protein with four gelsolin-like repeatsJournal of Biological Chemistry 283:26071–26080.https://doi.org/10.1074/jbc.M803618200
-
The junctophilin family of proteins: from bench to bedsideTrends in Molecular Medicine 20:353–362.https://doi.org/10.1016/j.molmed.2014.02.004
-
Neural activity promotes long-distance, target-specific regeneration of adult retinal axonsNature Neuroscience 19:1073–1084.https://doi.org/10.1038/nn.4340
-
The tubulin repertoire of C. elegans sensory neurons and its context-dependent role in process outgrowthMolecular Biology of the Cell pp. 3717–3728.https://doi.org/10.1091/mbc.e16-06-0473
-
Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric geneNature Neuroscience 4:1199–1206.https://doi.org/10.1038/nn770
-
Enzymology of NAD+ synthesisAdvances in Enzymology and Related Areas of Molecular Biology 73:135–182.
-
Intrinsic mechanisms of neuronal axon regenerationNature Reviews Neuroscience 19:323–337.https://doi.org/10.1038/s41583-018-0001-8
-
Novel Mutations in Synaptic Transmission Genes Suppress Neuronal Hyperexcitation in Caenorhabditis elegansG3; Genes|Genomes|Genetics 7:2055–2063.https://doi.org/10.1534/g3.117.042598
-
Axon outgrowth enhanced by a previous nerve injuryArchives of Neurology 29:53–55.https://doi.org/10.1001/archneur.1973.00490250071008
-
Annexins family: insights into their functions and potential role in pathogenesis of sarcoidosisJournal of Translational Medicine 14:89.https://doi.org/10.1186/s12967-016-0843-7
-
Annexins as intracellular calcium sensorsCell Calcium 41:207–219.https://doi.org/10.1016/j.ceca.2006.06.008
-
Twinfilin is an actin-filament-severing protein and promotes rapid turnover of actin structures in vivoJournal of Cell Science 119:1547–1557.https://doi.org/10.1242/jcs.02860
-
Axon regeneration genes identified by RNAi screening in C. elegansJournal of Neuroscience 34:629–645.https://doi.org/10.1523/JNEUROSCI.3859-13.2014
-
MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availabilityNature Cell Biology 15:668–676.https://doi.org/10.1038/ncb2741
-
The Caenorhabditis elegans R13A5.9 gene plays a role in synaptic vesicle exocytosisBiochemical and Biophysical Research Communications 463:994–998.https://doi.org/10.1016/j.bbrc.2015.06.048
-
Twinfilin, a molecular mailman for actin monomersJournal of Cell Science 115:881–886.
-
The kelch protein NS1-BP interacts with alpha-enolase/MBP-1 and is involved in c-Myc gene transcriptional controlBiochimica et Biophysica Acta (BBA) - Molecular Cell Research 1773:1774–1785.https://doi.org/10.1016/j.bbamcr.2007.09.002
-
The novel membrane-bound proteins mfsd1 and mfsd3 are putative slc transporters affected by altered nutrient intakeJournal of Molecular Neuroscience 61:199–214.https://doi.org/10.1007/s12031-016-0867-8
-
Structure and function of ER membrane contact sites with other organellesNature Reviews Molecular Cell Biology 17:69–82.https://doi.org/10.1038/nrm.2015.8
-
Axon-soma communication in neuronal injuryNature Reviews Neuroscience 15:32–42.https://doi.org/10.1038/nrn3609
-
Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neuronsMolecular Biology of the Cell 13:1778–1791.https://doi.org/10.1091/mbc.01-10-0514
-
Control of plasma membrane lipid homeostasis by the extended synaptotagminsNature Cell Biology 18:504–515.https://doi.org/10.1038/ncb3339
-
Taxol facilitates axon regeneration in the mature CNSJournal of Neuroscience 31:2688–2699.https://doi.org/10.1523/JNEUROSCI.4885-10.2011
-
Regulation of body fat in Caenorhabditis elegansAnnual Review of Physiology 77:161–178.https://doi.org/10.1146/annurev-physiol-021014-071704
-
Rab GTPases as coordinators of vesicle trafficNature Reviews Molecular Cell Biology 10:513–525.https://doi.org/10.1038/nrm2728
-
ADAMTS: a novel family of extracellular matrix proteasesThe International Journal of Biochemistry & Cell Biology 33:33–44.https://doi.org/10.1016/S1357-2725(00)00061-3
-
Shaping neurodevelopment: distinct contributions of cytoskeletal proteinsCurrent Opinion in Neurobiology 51:111–118.https://doi.org/10.1016/j.conb.2018.02.022
-
Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regenerationCurrent Opinion in Neurobiology 42:118–127.https://doi.org/10.1016/j.conb.2016.12.005
-
Nmnat exerts neuroprotective effects in dendrites and axonsMolecular and Cellular Neuroscience 48:1–8.https://doi.org/10.1016/j.mcn.2011.05.002
-
The structure of the ventral nerve cord of Caenorhabditis elegansPhilosophical Transactions of the Royal Society B: Biological Sciences 275:327–348.https://doi.org/10.1098/rstb.1976.0086
-
Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channelsThe Journal of Neuroscience 21:1923–1930.https://doi.org/10.1523/JNEUROSCI.21-06-01923.2001
-
NS1-Binding protein (NS1-BP): a novel human protein that interacts with the influenza A virus nonstructural NS1 protein is relocalized in the nuclei of infected cellsJournal of Virology 72:7170–7180.
-
Structural biology of the major facilitator superfamily transportersAnnual Review of Biophysics 44:257–283.https://doi.org/10.1146/annurev-biophys-060414-033901
-
Important role of junctophilin in nematode motor functionBiochemical and Biophysical Research Communications 289:234–239.https://doi.org/10.1006/bbrc.2001.5951
-
Distinct effects of tubulin isotype mutations on neurite growth in Caenorhabditis elegansMolecular Biology of the Cell 28:2786–2801.https://doi.org/10.1091/mbc.e17-06-0424
Article and author information
Author details
Funding
American Heart Association (13POST14800057)
- Kyung Won Kim
Hallym University Research Fund (HRF-201809-014)
- Kyung Won Kim
Canadian Institutes of Health Research (MFE-146808)
- Matthew G Andrusiak
National Institutes of Health (NS057317)
- Yishi Jin
- Andrew D Chisholm
National Institutes of Health (NS093588)
- Yishi Jin
- Andrew D Chisholm
National Institute of Health (K99 NS097638)
- Salvatore J Cherra III
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank our members for valuable discussions and Laura Toy for assistance in strain construction. We thank Dr. S Mitani and National Bioresource Project of Japan, and the Caenorhabditis Genetics Center (funded by NIH Office of Research Infrastructure Programs P40 OD010440) for strains. KWK received an American Heart Association postdoctoral fellowship and Hallym University research funds (HRF-201809–014), MGA received a Canadian Institutes of Health Research Postdoctoral Fellowship (MFE-146808), and SJC was a receipient of NIH K99 (NS097638). This work was supported by NIH R01 grants to YJ, and ADC (NS057317 and NS093588).
Copyright
© 2018, Kim et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,640
- views
-
- 565
- downloads
-
- 43
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 43
- citations for umbrella DOI https://doi.org/10.7554/eLife.39756
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Expanded genetic screening in Caenorhabditis elegans identifies new regulators and an inhibitory role for NAD+ in axon regeneration
eLife 7:e39756.
https://doi.org/10.7554/eLife.39756




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}