Environmentally-induced epigenetic conversion of a piRNA cluster
- Sorbonne Université, CNRS, Institut de Biologie Paris-Seine, France
- Université Clermont Auvergne, CNRS, INSERM, BP 10448, France
Abstract
Transposable element (TE) activity is repressed in animal gonads by PIWI-interacting RNAs (piRNAs) produced by piRNA clusters. Current models in flies propose that germinal piRNA clusters are functionally defined by the maternal inheritance of piRNAs produced during the previous generation. Taking advantage of an inactive, but ready to go, cluster of P-element derived transgene insertions in Drosophila melanogaster, we show here that raising flies at high temperature (29°C) instead of 25°C triggers the stable conversion of this locus from inactive into actively producing functional piRNAs. The increase of antisense transcripts from the cluster at 29°C combined with the requirement of transcription of euchromatic homologous sequences, suggests a role of double stranded RNA in the production of de novo piRNAs. This report describes the first case of the establishment of an active piRNA cluster by environmental changes in the absence of maternal inheritance of homologous piRNAs.
Editorial note: This article has been through an editorial process in which the authors decide how to respond to the issues raised during peer review. The Reviewing Editor's assessment is that all the issues have been addressed (see decision letter).
https://doi.org/10.7554/eLife.39842.001Introduction
Transposable element (TE) activity needs to be repressed to avoid severe genome instability and gametogenesis defects. In humans, growing evidence has implicated TE in several disorders such as cancers defining a new field of diseases called transposopathies (Wylie et al., 2016a; Wylie et al., 2016b). In the animal germline, TE activity is controlled at both transcriptional and post-transcriptional levels by small RNAs called piRNAs associated with the PIWI clade of germline Argonaute proteins (Piwi, Aub and Ago3 in Drosophila) (Brennecke et al., 2007; Gunawardane et al., 2007; Le Thomas et al., 2013; Sienski et al., 2012). piRNAs are processed from transcripts produced from specific heterochromatic loci enriched in TE fragments, called piRNA clusters (Brennecke et al., 2007; Gunawardane et al., 2007). These loci undergo non-canonical transcription, ignoring splicing and transcription termination signals, licensed by specific protein complexes such as Rhino-Deadlock-Cutoff (Mohn et al., 2014; Zhang et al., 2014) and Moonshiner-TRF2 (Andersen et al., 2017). Thus, when a new TE inserts into a naive genome, it will freely transpose until one copy gets inserted into a piRNA cluster leading to the production of homologous new TE piRNAs that will then repress transposition (Brennecke et al., 2008). In support of this idea, exogenous sequences inserted into preexisting piRNA clusters lead to the production of matching piRNAs (de Vanssay et al., 2012; Hermant et al., 2015; Marie et al., 2017; Muerdter et al., 2012; Pöyhönen et al., 2012). The specificity of the efficient repression mediated by piRNAs appears to be determined solely by the piRNA cluster sequences. Thus, it raises the question of how piRNA cluster loci are themselves specified. Histone H3 lysine nine tri-methylation (H3K9me3) that is recognized by Rhino, a paralog of heterochromatin protein HP1 (Klattenhoff et al., 2009), is a shared feature of piRNA clusters. Enrichment of H3K9me3, however, is not specific to piRNA clusters and tethering Rhino onto a transgene leads to the production of piRNAs only when both sense and antisense transcripts are produced (Zhang et al., 2014). This suggests that neither H3K9me3 marks nor having Rhino-bound is sufficient to induce piRNA production. One current model proposes that piRNAs clusters are defined and activated at each generation by the deposition in the egg of their corresponding piRNAs from the mother (Huang et al., 2017). In support of this model, we previously described the first case of a stable transgenerational epigenetic conversion known as paramutation in animals (de Vanssay et al., 2012). This phenomenon was first described in plants and defined as "an epigenetic interaction between two alleles of a locus, through which one allele induces a heritable modification of the other allele without modifying the DNA sequence" (Brink, 1956; Chandler, 2007). In our previous study, we showed that an inactive non-producing piRNA cluster of P transgene insertions inherited from the father can be converted into a piRNA-producing cluster by piRNAs inherited from the mother (de Vanssay et al., 2012). This attractive model, however, does not answer the question of how the first piRNAs were produced.
To address this paradox, we used the same BX2 cluster of seven P(lacW) transgenes, which resulted from multiple and successive P(lacW) transposition events, thus resembling the structure of natural piRNA clusters (Dorer and Henikoff, 1994). The key advantage of the BX2 locus is that it can exist in two epigenetic states for the production of germline piRNAs: 1) the inactive state (BX2OFF) does not produce any piRNAs and thus is unable to repress the expression of homologous sequences, and 2) the active state (BX2ON) produces abundant piRNAs that functionally repress a homologous reporter transgene in the female germline (de Vanssay et al., 2012; Hermant et al., 2015). We therefore used BX2 in an inactive state to search for conditions that would convert it into an active piRNA-producing locus, without pre-existing maternal piRNAs. In this report, we describe how culturing flies at high temperature, 29°C instead of 25°C, induces the conversion of an inactive BX2 locus (BX2OFF) into a stable piRNA cluster exhibiting repression properties (BX2ON). It should be noted that flies in their natural habitat exhibit this range of temperature, especially in the context of global warming. These data provide the first report of a de novo piRNA cluster establishment independent of maternal inheritance of homologous piRNAs and highlight how environmental changes can stably induce transgenerational modification of the epigenome.
Results
Germline silencing induced at high temperature
Earlier studies of hybrid dysgenesis reported that high temperature enhances P-element repression (Ronsseray et al., 1984) and that thermic modification of P repression can persist over several generations (Ronsseray, 1986). Moreover, P-element repression in a strain carrying two P-elements inserted into a subtelomeric piRNA cluster can be stimulated by heat treatment (Ronsseray et al., 1991). Very recently, the tracking of natural invasion of P elements in Drosophila simulans confirmed the key role of high temperature in the establishment of repression through generations (Kofler et al., 2018). These results suggested that temperature may influence the activity of some piRNA clusters. To investigate whether high temperature (29°C) could affect the stability of BX2 epialleles (BX2OFF and BX2ON) across generations, we generated flies carrying, on the same chromosome, each of the BX2 epialleles and an euchromatic reporter transgene sharing P and lacZ sequences with BX2 (made of seven P(lacW), Figure 1—figure supplement 1A). This transgene promotes the expression of ß-Galactosidase both in the germline and in the somatic cells of the ovary and thus will hereinafter be referred to as 'P(TARGET)GS' (Figure 1—figure supplement 1B). As was previously described (de Vanssay et al., 2012), at 25°C BX2OFF does not synthesize functional piRNAs complementary to P(TARGET)GS resulting in ß-Galactosidase expression in whole ovaries of BX2OFF, P(TARGET)GS lines (Figure 1A). Whereas in BX2ON, P(TARGET)GS lines, functional lacZ piRNAs are synthesized in the germline where they specifically repress the P(TARGET)GS ß-Galactosidase expression (Figure 1B). Both BX2OFF, P(TARGET)GS and BX2ON, P(TARGET)GS lines incubated at 25°C for 23 generations maintained their epigenetic state, showing that both epialleles are stable (Figure 1D, Supplementary file 1). At 29°C, the repression capacity of BX2ON, P(TARGET)GS lines remained stable through 25 generations. Among the BX2OFF, P(TARGET)GS lines, 24.7% of females analyzed during 25 generations showed a complete and specific germline ß-Galactosidase repression (n = 3812, Figure 1E and Supplementary file 1), suggesting a conversion of the BX2OFF epiallele into BX2ON. Interestingly, the appearance of females showing ß-Galactosidase repression was gradual and stochastic, resulting in a global frequency that increased with the number of generations (Supplementary file 1). To test whether the temperature-induced conversion was stable, a set of five lines showing full repression capacity after 23 generations at 29°C, obtained from an independent experiment, were transferred to 25°C and tested for their silencing capacities for several generations. In all cases, the silencing capacities of the BX2ON epiallele induced at 29°C remained stable during 50 additional generations at 25°C (Supplementary file 2). These stable BX2ON lines converted by high temperature were named hereafter BX2Θ (Greek theta for temperature) to distinguish them from the BX2* lines converted by maternally inherited piRNAs (de Vanssay et al., 2012). Taken together, our data show that BX2OFF can be functionally converted by high temperature (29°C), strongly suggesting that de novo piRNA production can occur in the absence of maternal inheritance of homologous piRNAs.
Figure 1 with 3 supplements see all

Functional assay of the BX2 epigenetic state.
Females carrying either one of the BX2 epialleles and P(TARGET)GS were analyzed. (A) When BX2 is OFF for production of piRNAs (BX2OFF), no repression of P(TARGET)GS occurs, allowing expression of ß-Galactosidase in both germline and in somatic lineages in ovaries (ß-Galactosidase staining). BX2OFF is illustrated by a blue fly. (B) When BX2 is ON for production of piRNAs (BX2ON), repression of P(TARGET)GS occurs only in the germline lineage. BX2ON is illustrated by a light brown fly. (C) Drawing of an intermediate egg chamber showing germ cells (nurse cells and oocyte in blue) surrounded by somatic follicular cells (in pink), adapted from Figure 1A from Frydman and Spradling (2001). (D) At 25°C, BX2OFF and BX2ON are stable over generations. (E) At 29°C, BX2OFF can be converted into BX2ON, while BX2ON is stable over generations.
BX2 lines converted by high temperature or by maternal homologous piRNA inheritance present identical functional and molecular properties
We further characterized the functional and molecular properties of BX2Θ activated by temperature and compared them to BX2* activated by maternal inheritance of homologous piRNAs. Firstly, we compared the maternal and paternal BX2 locus inheritance effect of three BX2Θ lines and three BX2* at 25°C (Figure 1—figure supplement 2A). Maternal inheritance of either BX2Θ or BX2* loci leads to complete and stable repression of ß-Galactosidase expression (n flies = 152 and 159, respectively, Supplementary file 3), whereas paternal inheritance of either BX2Θ or BX2* loci, that is in absence of maternal piRNA deposition, results in ß-Galactosidase expression, and thus a definitive loss of BX2 silencing capacities (n flies = 156 and 155, respectively, Supplementary file 3). Secondly, we previously showed that progeny with a paternally inherited BX2OFF locus and maternally inherited BX2* piRNAs, but lacking the maternally BX2* genomic locus, have 100% conversion (de Vanssay et al., 2012). This process of recurrent conversions of an allele that is heritable without DNA modification is known as paramutation, thus BX2* females are paramutagenic, that is able to trigger paramutation. To test this property on BX2 lines converted by temperature, BX2Θ females were crossed with BX2OFF males. The progeny that inherited the paternal BX2OFF locus but not the maternal BX2Θ locus were selected and three independent lines were established (Figure 1—figure supplement 2B). Silencing measured over 20 generations revealed 100% of repression capacity showing that BX2Θ is also paramutagenic (n flies = 159, Supplementary file 4).
To determine whether the silencing capacities of BX2Θ involved piRNAs, small RNAs from BX2OFF, BX2Θ and BX2* ovaries were extracted and sequenced (Supplementary file 5). Unique reads matching the P(lacW) sequences were identified only in the BX2Θ and BX2* libraries (Figure 2A). Most of these small RNAs display all of the characteristics of bona fide germline piRNAs, that is a high proportion of 23–29 nt with a strong U bias on the first 5' nucleotide, an enrichment of a 10 nucleotide overlap between sense and antisense piRNAs, also known as the ping-pong signature, and a high proportion of reads with A at the tenth position among the 10 nt overlapped reads (Figure 2B–C) (Brennecke et al., 2007; Gunawardane et al., 2007). As a control, the 42AB piRNA cluster, a canonical germline dual-strand piRNA cluster, presented no significant difference between the three genotypes (Figure 2—figure supplement 1). Therefore, these results show that high temperature can initiate piRNA production from BX2 naive sequences (BX2OFF) and strongly suggest that once a piRNA cluster is activated for piRNA production, the 'ON' state is maintained at each generation by maternal inheritance of piRNAs.
Figure 2 with 3 supplements see all
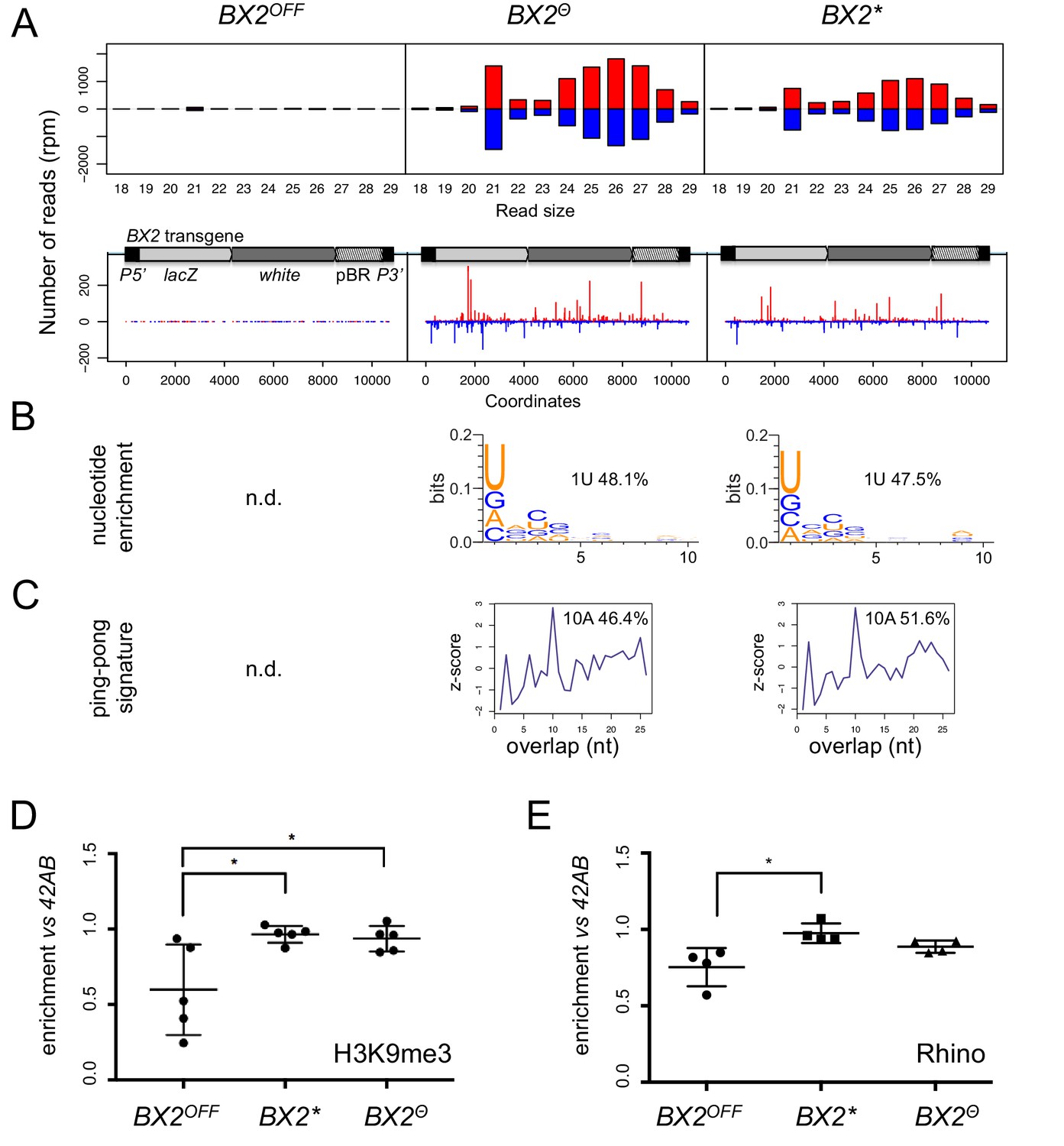
BX2Θ and BX2* produce piRNAs and are enriched in H3K9me3.
(A) Size distribution of ovarian small RNAs matching BX2 transgene sequences reveals that both BX2Θ and BX2* but not BX2OFF produce 21 nt siRNAs and 23–29-nt piRNAs (upper panels, pBR = backbone plasmid pBR322). Positive and negative values correspond to sense (red) and antisense (blue) reads, respectively. Unique 23–29 nt mappers are shown on the BX2 transgene sequences (lower panels). (B) Percentage of 23–29 nt small RNAs from BX2Θ and BX2* matching transgene sequence with a U at the first position are shown. n.d.: not determined due to low number of reads. (C) Relative frequency (z-score) of overlapping sense-antisense 23–29 nt RNA pairs reveals an enrichment of 10 nt overlapping corresponding to the ping-pong signature. (D) H3K9me3 and (E) Rhino binding on the BX2 transgene in ovaries of BX2OFF, BX2Θ and BX2* strains revealed by chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR) on specific white sequences. In both ‘ON’ strains, BX2Θ and BX2*, H3K9me3 and Rhino levels over the transgene are very similar and higher than in the BX2OFF strain (unpaired t-test was used to calculate significance of the differences (p<0.05, n = 5).
BX2 is inserted into the first intron of the AGO1 gene (de Vanssay et al., 2012) and we looked at the piRNA production from this region in the different BX2 epigenetic contexts. No significant amount of piRNAs coming from the AGO1 gene region can be detected whatever the BX2 state (Figure 2—figure supplement 2). These findings indicate that the AGO1 gene region is not a natural piRNA cluster. To test whether other non-piRNA producing genomic loci have started to produce piRNAs following high temperature treatment, we looked for specific piRNAs (23–29 nt) matching at unique positions on Drosophila chromosomes and compared them between BX2Θ and BX2OFF. The reads were then resampled per 50 kilobases windows. To eliminate background noise, only regions that produced more than five piRNAs per kilobase on average in both libraries were considered. Only exons of the white gene present in the P(lacW) transgenes of BX2 showed differential piRNA expression (log2 ratio >8.5, Figure 2—figure supplement 3). This analysis revealed that the activation of piRNA production after thermic treatment is restricted to the BX2 locus, suggesting that all other loci able to produce piRNAs are already active.
Previous studies had suggested that the chromatin state plays a role in the differential activity of BX2 (Le Thomas et al., 2014). We therefore profiled H3K9me3 marks and Rhino binding on the P(lacW) transgene in ovaries from BX2OFF, BX2Θ and BX2* strains by chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR). In both strains BX2Θ and BX2*, H3K9me3 and Rhino were similarly enriched over the P(lacW) transgene compared to the BX2OFF strain, significantly for H3K9me3 (Figure 2D–E). Taken together, these results show that de novo activation of BX2OFF by 29°C treatment (BX2Θ) or paramutation by maternal inheritance of homologous piRNAs (BX2*) lines leads to similar functional and molecular properties.
Epigenetic conversion at 29°C occurs at a low rate from the first generation
To explain the low occurrence and the generational delay of BX2 conversion at 29°C (Supplementary file 1), we propose that conversion is a complete but rare event occurring in a small number of egg chambers at each generation. Under this hypothesis, the sampling size of tested females should be crucial to observe such stochastic events. We therefore increased the number of analyzed females raised at 29°C during one generation. For this, eggs laid by females maintained at 25°C carrying the P(TARGET)GS reporter transgene and the BX2OFF locus were collected during three days. These eggs were then transferred at 29°C until adults emerged (Figure 3A). To follow their offspring, we individually crossed 181 G1 females with two siblings and let them lay eggs for three days at 25°C. These 181 G1 females were then stained for ß-Galactosidase expression. Strikingly, repression occurred only after one generation at 29°C in a few of the egg chambers of 130 G1 females (≈2.7% of the estimated total number of G1 egg chambers n≈21700, Figure 3B, right panel). These results support our hypothesis whereby epigenetic conversion of BX2OFF into BX2Θ is an instantaneous and complete event occurring at a low frequency per egg chamber and at each generation that is kept at 29°C.
Figure 3 with 3 supplements see all
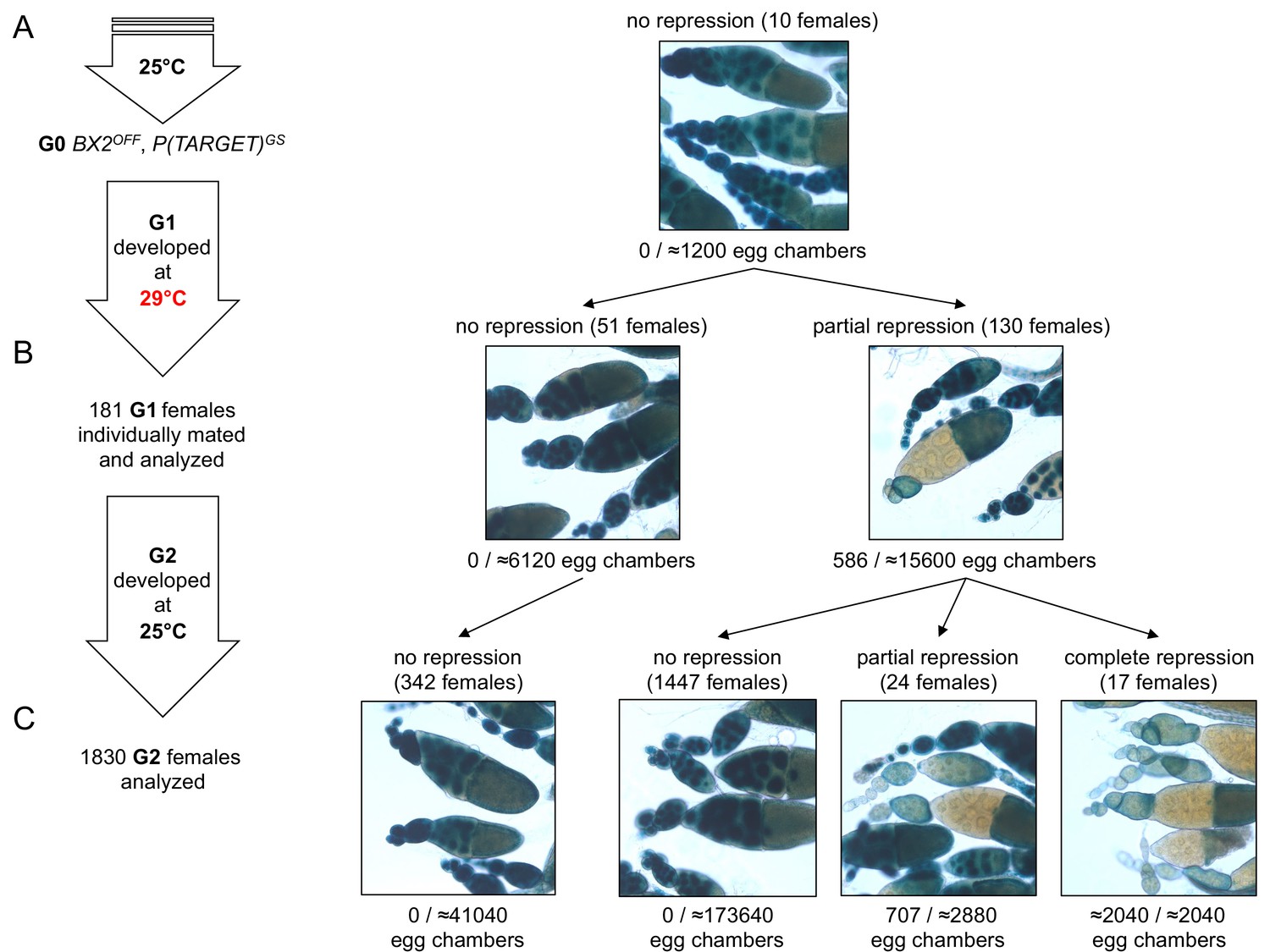
BX2 conversion at 29°C occurs in one generation at a low rate.
(A) G0 females carrying the P(TARGET)GS reporter and BX2OFF laid eggs at 25°C during three days. The BX2OFF state of these females was confirmed after the three days at 25°C by ß-Galactosidase staining (number of egg chambers ≥ 1200). (B) Their eggs were allowed to develop at 29°C until emergence of the next generation. G1 females (n = 181) were individually mated with two siblings and left to lay for three days at 25°C. G1 females were then individually stained for ß-Galactosidase expression. Strikingly, 130 females (71.8%) show ß-Galactosidase repression in some egg chambers (586 among ≈21700 - estimation of the total egg chamber number among 181 females). The BX2OFF into BX2ON conversion frequency is ≈2.7%. (C) Analysis of each G1 female progeny developed at 25°C by ß-Galactosidase staining. The progeny of the 51 G1 females that did not present repression maintained BX2OFF state (n flies = 342). Most of the progeny of the 130 G1 females presenting conversion show no repression (97.2%, n flies = 1488) while 41 females present partial (n = 24) or complete (n = 17) repression of the germline expression of ß-Galactosidase.
To test the stability of the epigenetic BX2Θ states observed in G1 females, offspring daughters (G2) were raised at 25°C and their ovaries examined for ß-Galactosidase expression. Among G2 females, partial (n = 24) or complete (n = 17) repression of ß-Galactosidase expression in the germline was observed only in the progeny of those 130 G1 females in which partial repression was previously detected (Figure 3C). The proportion of 2.2% of converted G2 females (41/1830) is reminiscent with the proportion of repressed egg chambers observed in G1. The progeny of the 51 G1 females that did not present repression (Figure 3B left panel) did not show spontaneous conversion (Figure 3C). Taken together, these observations strongly suggest that newly converted BX2ON egg chambers give rise to adult females with complete or partial silencing capacities. The low conversion rate observed in thousands of flies after one generation raised at high temperature and its stability through the next generation might explain the apparent delay of BX2ON conversion of dozens of flies continuously raised at 29°C observed in the first set of experiments (see Figure 1E and Supplementary file 1). We more finely analyzed the silencing capacities of eight independent BX2OFF lines throughout generations at 29°C (Supplementary file 1) by monitoring P(TARGET)GS repression in each egg chamber instead of whole ovaries (Supplementary file 6). BX2 conversion occurred in each tested line with various dynamics (Figure 1—figure supplement 3A), likely reflecting a genetic drift due to the low conversion occurrence coupled to an important sampling effect at each generation. Globally, the mean of repression frequencies seems to indicate a progressive increase of BX2 conversion over generations (Figure 1—figure supplement 3B). Altogether, our results illustrate how environmental modifications like high temperature experienced during one generation might stably modify the epigenome of the future ones. Such a newly acquired epigenetic state may spread in a given population within a few generations.
High temperature increases BX2 antisense RNA but not piRNAs
Previously, we showed that BX2OFF and BX2ON produce similar amounts of sense and antisense transcripts (de Vanssay et al., 2012). However, these transcripts do not lead to ß-Galactosidase expression in the germline nor piRNA production in the BX2OFF line. We wondered if high temperature might change the RNA steady-state level of BX2. To ensure that we were detecting RNA specifically from BX2, qRT-PCR experiments targeting the lacZ gene were carried out on ovarian RNA extracted from the BX2OFF line that did not contain P(TARGET)GS. We observed a significant increase in the steady-state BX2 RNA levels at 29°C compared to 25°C (Figure 4A). Remarkably, strand-specific qRT-PCR experiments revealed that only BX2 antisense transcripts, corresponding to antisense lacZ transcripts, increase at 29°C (Figure 4B). As BX2 is inserted into the AGO1 gene in a convergent transcription manner (de Vanssay et al., 2012), Figure 4C), we compared AGO1 steady-state RNA level at 29°C and 25°C. AGO1 transcript isoforms that are initiated upstream the BX2 insertion point are significantly increased at 29°C (Figure 4D and Figure 4—figure supplement 1A–B). Thus, it is possible that, at 29°C, an increase of transcription from the AGO1 promoters located upstream the BX2 insertion point could lead to an increase of BX2 antisense RNA transcription.
Figure 4 with 1 supplement see all
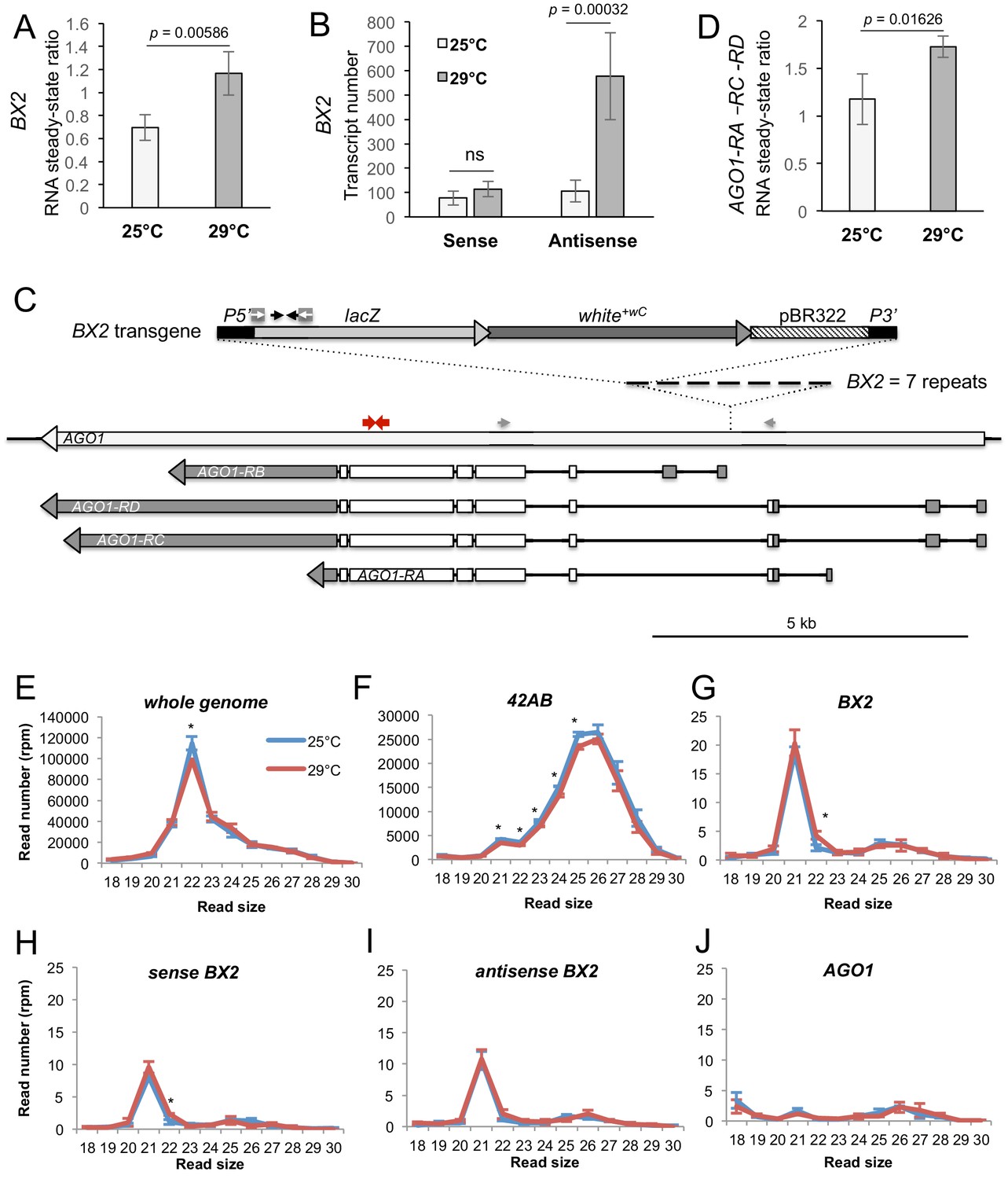
BX2OFF antisense RNA increase at 29°C.
(A) RT-qPCR experiments revealed that the steady-state level of ovarian lacZ RNAs from BX2 is more abundant at 29°C (n = 5) compared to 25°C (n = 6). (B) Sense-specific RT-qPCR experiments revealed that only antisense transcripts from BX2, corresponding to antisense lacZ transcripts, are increased (25°C n = 6, 29°C n = 4). Significant p-values are given (bilateral Student's t-test). ns: not significant. (C) Map of the BX2 locus containing seven P(lacW) transgenes inserted into the AGO1 gene. P(lacW) and AGO1 are drawn to scale. The lacZ gene contained in P(lacW) and AGO1 are transcriptionally in opposite directions. Black arrows show lacZ primers used for (A) and (B) experiments. White arrows show primers used for sense-specific reverse transcription in (B) experiment. Grey arrows show AGO1 primers used for (D) experiment: these primers are specific for AGO1 transcripts (RA, RC and RD) that originate from promoters located upstream the BX2 insertion point and, thus, are potentially convergent to BX2. Red arrows show primers used to measure steady-state of all AGO1 isoforms (see Figure 4—figure supplement 1D). (D) RT-qPCR experiments performed on flies devoid of P(lacW) transgenes (w1118 context) revealed that the steady-state level of ovarian AGO1-RA, -RC and -RD RNA isoforms is more abundant at 29°C (n = 5) compared to 25°C (n = 6). (E–I) To compare small RNAs at 25 versus 29°C, total RNAs were extracted from BX2OFF ovaries dissected from adults incubated at 25°C or 29°C. Three samples were tested for each temperature. Small RNAs from 18 to 30 nt were deep sequenced. For each library, normalization has been performed for 1 million reads matching the Drosophila genome (rpm, Supplementary file 7). Size distributions of unique reads that match reference sequences are given. (E) Small RNAs matching the Drosophila genome present similar profiles in both temperatures except for 22 nt RNAs that are more represented at 25°C. (F) The 21 to 25 nt reads matching the 42AB piRNA cluster that range from 21 to 25 nt are slightly more abundant at 25°C. (G) Strikingly, almost only 21 nt RNAs match BX2 sequence. They are equally distributed among sense (H) and antisense (I) sequences at both temperatures. (J) No small RNAs corresponding to the AGO1 gene can be detected whatever the temperature. *=p < 0.05, bilateral Student's t-test.
We then examined whether the increase of BX2 antisense RNAs leads to an increase of antisense small RNAs. Ovarian small RNAs (18 to 30 nucleotides) of BX2OFF flies (without P(TARGET)GS) raised at 25°C and at 29°C for one generation were sequenced and the read numbers normalized (Supplementary file 7). A slight, yet statistically significant, decrease is observed at 29°C for small RNAs matching the whole genome (Figure 4E) and for the 42AB piRNA cluster (Figure 4F). Strikingly, no piRNAs were produced from the BX2 locus at 25°C nor after one generation at 29°C. Thus, the increase of BX2 antisense transcripts observed at 29°C (Figure 4B) did not correlate with an increase of corresponding antisense piRNAs. At 25°C and 29°C, BX2OFF produced the same low amount of 21 nt small RNAs, equally distributed between sense and antisense (Figure 4G,H,I), suggesting that BX2 transcripts are processed into siRNAs. These results confirm that AGO1 is not a piRNA producing locus (as shown in Figure 2—figure supplement 2) and showed that, at 29°C, AGO1 is still not producing small RNAs (Figure 4J). These data indicate that 21 nt small RNA production was restricted to BX2 sequences.
Heat conversion requires a transcribed homologous sequence in trans
Quantitative RT-PCR experiments described above were carried out on flies bearing only the BX2 locus while all conversion experiments at 29°C were done with flies bearing the BX2 and the P(TARGET)GS locus. Interestingly, the amount of P(TARGET)GS transcripts is affected by temperature but in the opposite way to BX2, as less transcripts were measured at 29°C compared to 25°C (Figure 4—figure supplement 1C). We next asked whether the P(TARGET)GS transgene could participate in the conversion process of BX2. For this, 'heat-activated-conversion' experiments of BX2 were done in flies not carrying the P(TARGET)GS (Figure 3—figure supplement 1). To assess the BX2 epigenetic state of the G1 raised at 29°C, 157 G1 females were individually crossed at 25°C with males harboring the P(TARGET)GS transgene. Among the 1137 G2 females analyzed, only one female presented partial repression of the ß-Galactosidase expression and none presented complete repression (Figure 3—figure supplement 1). If we compare these results with those obtained with the BX2, P(TARGET)GS lines (Figure 3), the difference was highly significant (p=8.5×10−6, homogeneity χ2 = 23.35 with 2 degrees of freedom, Supplementary file 8). To further validate the requirement of the euchromatic homologous transgene P(TARGET)GS in establishing the temperature-dependent BX2 conversion, we generated eight independent lines in which BX2 was recombined into the same P(TARGET)GS genetic background but without the P(TARGET)GS transgene. After 30 generations at 29°C, no female showing ß-Galactosidase repression was observed (Supplementary file 9). A homogeneity χ2 test comparing, at G13, the repression occurrence in BX2, P(TARGET)GS lines (31/161, Supplementary file 1) and in recombined BX2 lines (0/975, Supplementary file 9) is highly significant (p=7.04×10−44, homogeneity χ2 = 192.9 with 1 degree of freedom), arguing against a background effect of the P(TARGET)GS line in the conversion phenomenon. Additionally, we reproduced the experiment with an euchromatic P(TARGET) expressed only in the germline and referred to hereinafter as 'P(TARGET)G' (Figure 1—figure supplement 1A–B). In the presence of P(TARGET)G, BX2 was converted at 29°C at a rate comparable with that observed with P(TARGET)GS (Supplementary file 10). These data show that BX2 conversion by temperature cannot be attributed to any specificity linked to the P(TARGET)GS insertion. To know if the transcription of the P(TARGET)GS (or of the P(TARGET)G) is required, we reproduced the same experiment with another euchromatic transgene that is not expressed in the germline but in the somatic cells surrounding the germ cells and therefore referred to hereinafter as 'P(TARGET)S' (Figure 1—figure supplement 1A–B). In the presence of P(TARGET)S, BX2 was not converted at 29°C (0/784, Figure 3—figure supplement 2). A homogeneity χ2 test comparing these results with those obtained with P(TARGET)GS considering only the complete BX2ON G2 females (17/1464 females, Figure 3) is significant (p=0.0055, homogeneity χ2 = 7.7 with 1 degree of freedom). We conclude that the transcription of a reporter transgene sharing homologous sequences (i.e. lacZ) with BX2 is required in the germline for BX2 conversion at 29°C.
To summarize, in the absence of P(TARGET) sequences, at 29°C BX2OFF produces an elevated number of antisense transcripts, no piRNAs and is unable to be converted to BX2ON. In contrast, when a P(TARGET) is present and transcribed in the germline, BX2 conversion and piRNA production are observed at 29°C. Although much of the mechanistic aspects of BX2 conversion remain unknown, these findings lead us to propose that, at 29°C, double strand RNA (dsRNA) made of the excess of BX2 antisense transcripts and the sense P(TARGET) transcripts could be a prerequisite for the production of de novo piRNAs and the conversion of BX2 into an active piRNA cluster (see recapitulative model Figure 5).
Figure 5
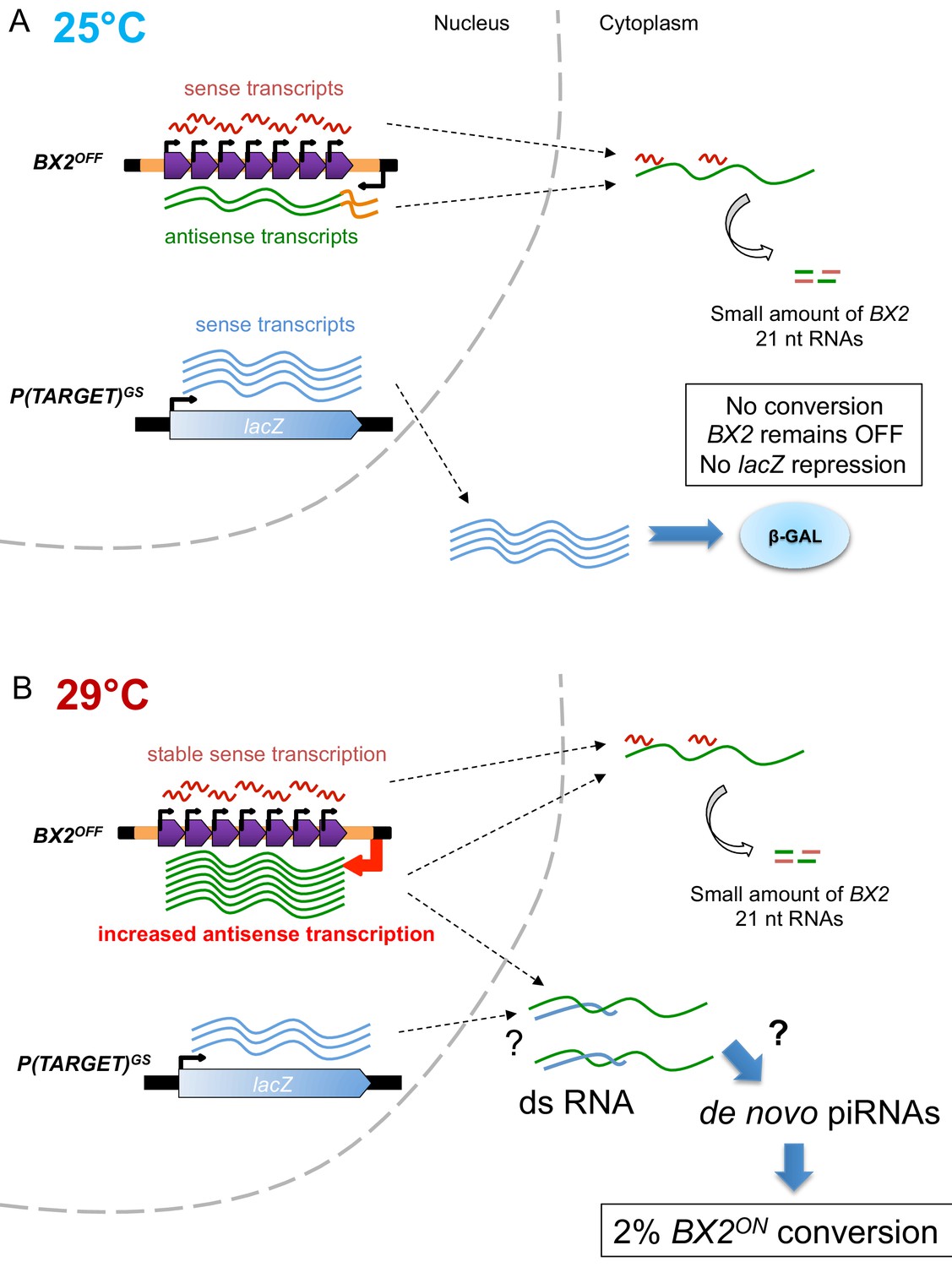
Model of BX2 activation at 29°C.
(A) At 25°C, a low bidirectional transcription of the BX2 cluster leads to a small production of 21 nt RNAs. BX2OFF is stable at 25°C, no conversion event is observed and the lacZ reporter gene from P(TARGET)GS remains active throughout generations. (B) At 29°C, a specific increase in antisense transcription occurs, presumably due to a higher activity of the promoter of the AGO1 gene (orange box). This excess of BX2 antisense RNA could interact with sense P(TARGET)GS transcripts to produce double stranded RNA. Through a yet unknown mechanism, such dsRNA could lead to the formation of de novo BX2 piRNAs. These piRNAs could in turn trigger the conversion of BX2OFF into an active piRNA cluster, a phenomenon observed based on the repression of the lacZ reporter gene of P(TARGET)GS. The BX2 conversion is a rare event (≈2% per generation) but once achieved, BX2ON remains active throughout generations due to the maternal inheritance of homologous piRNAs and the paramutation of the paternal BX2OFF allele.
Discussion
Here, we report on the heritable establishment of a new piRNA cluster associated with silencing properties induced by high temperature during development. The epigenetic response to heat exposure has been studied in several model species: in Arabidopsis for instance, increasing temperature induces transcriptional activation of repetitive elements (Ito et al., 2011; Pecinka et al., 2010; Tittel-Elmer et al., 2010). Whether these changes involve chromatin modifications is not clear but none of these modifications have been found to be heritable through generations except in mutants for siRNA biogenesis where high frequency of new TE insertions was observed in the progeny of stressed plants (Ito et al., 2011). In animals, response to heat can result in modification of DNA methylation at specific loci in reef building coral (Dimond and Roberts, 2016), chicken (Yossifoff et al., 2008) and wild guinea pigs (Weyrich et al., 2016). In the latter, modifications affecting ≈50 genes are inherited in G1 progeny (Weyrich et al., 2016). The mechanisms of this heritability, however, are not yet understood. In Drosophila, heat-shock treatment of 0–2 hr embryos for one hour at 37°C or subjecting flies to osmotic stress induce phosphorylation of dATF-2 and its release from heterochromatin (Seong et al., 2011). This defective chromatin state is maintained for several generations before returning to the original state. We have tested if such stresses were able to convert BX2OFF into BX2ON in one generation but neither heat-shock nor osmotic stress induces BX2 conversion (Supplementary file 11), suggesting that BX2 activation does not depend on dATF-2. In a more recent paper, Fast et al. (Fast et al., 2017) found less piRNAs at 29°C than at 18°C. However, RNAseq analyses of differentially expressed genes involved in the piRNA pathway were not conclusive, as some piRNA genes (ago3, aub, zuc, armi) were more expressed at 29°C while others (shu, hsp83, Yb) were less expressed as compared to the levels at 18°C (Fast et al., 2017). Overall, the enhancement of the piRNA ping-pong amplification loop observed at 29°C was attributed to RNA secondary structures, because of a lack of specificity for any particular class of TE (Fast et al., 2017). Furthermore, in contrast to our RT-qPCR results obtained at 25°C and 29°C (Figure 4D and Figure 4—figure supplement 1A–B), AGO1 was not differentially expressed between 18°C and 29°C (Fast et al., 2017). This difference can be explained, because, in our experiments, we checked for specific spliced transcripts of AGO1 (RA, RC and RD) that originate upstream the BX2 insertion point (Figure 4C). In agreement with Fast et al., no statistical difference in the global amount of AGO1 transcripts between 25°C and 29°C is observed using primers located downstream the BX2 insertion point (Figure 4—figure supplement 1D). This suggests that the AGO1 promoter located downstream the BX2 insertion point, and responsible for the production of the RB isoform (see Figure 4), is not sensitive to temperature. Taken together, these observations show that temperature modification might induce epigenetic changes in several species but the underlying mechanisms remain largely unsolved.
Whole genome comparison of small RNA sequencing between BX2OFF and newly BX2ON heat-converted flies (BX2Θ) did not reveal additional regions stably converted for piRNA production (Figure 2—figure supplement 3). This suggests that no other loci are metastable for piRNA production, that is all potential piRNA clusters are already active at 25°C. This observation raises the question of what makes BX2 locus competent for piRNA activation at high temperature. The BX2 locus is the result of successive induced transpositions of P(lacW) used to screen for white-variegating phenotype (Dorer and Henikoff, 1994). Thus, BX2 resembles natural TE clusters where TEs have the capacity to transpose into each other assembling structures named nested TE, as described in numerous genomes (Gao et al., 2012; Liu et al., 2007; Zanni et al., 2013). Some nested TE loci might have the capacity to be activated and respond to a new TE invasion. In Drosophila, BX2-like tandemly inserted transgenes were shown to be new sites of HP1 enrichment in larval salivary glands, emphasizing a heterochromatic structure at the BX2 locus in somatic cells (Fanti et al., 1998), and to cause pairing-dependent silencing (Dorer and Henikoff, 1997). When tested for repressing capacities, however, this strain is inactive for BX2 piRNA production (de Vanssay et al., 2012; Josse et al., 2008). We have shown that maternally inherited P(lacW) piRNAs are able to paramutate with complete and stable penetrance from an inactive BX2 locus into an active locus for piRNA production (de Vanssay et al., 2012). The paramutated BX2 locus appeared to be a genuine piRNA cluster since it is sensitive to a number of factors known to be involved in piRNA biogenesis such as aub, rhi, cuff, zuc (Hermant et al., 2015) and moonshiner (Figure 3—figure supplement 3). The number of transgene copies appears to be crucial in the process since smaller number of transgenes results in less somatic heterochromatinization (Dorer and Henikoff, 1994; Fanti et al., 1998), less pairing-dependent silencing (Dorer and Henikoff, 1997) and unstable paramutation (de Vanssay et al., 2012). Taken together, these data suggest that the heterochromatic structure of a cluster precedes piRNA production. This is supported by our ChIP experiments showing that H3K9me3 levels on BX2OFF are slightly below the H3K9me3 level of piRNA producing states (BX2ON and BX2Θ, Figure 2D). The same observation can be made for Rhino (Figure 2E), suggesting that Rhino may be already present on the BX2OFF locus but below the threshold required for piRNA production as suggested by Akulenko et al. (2018). Thus, a locus made of repeated sequences and being likely heterochromatic (H3K9me3, Rhino) is a necessary but not sufficient condition to specify an active piRNA cluster.
In the germline, piRNA clusters produce piRNAs from both strands and it was recently shown that, in most cases, transcription initiates within clusters on both strands through the interaction of Rhino and Moonshiner (Andersen et al., 2017). In few cases, however, piRNA cluster transcription may take advantage of the read-through from a flanking promoter (Andersen et al., 2017). Zhang et al. (Zhang et al., 2014) have shown that tethering Rhino onto a transgene leads to its repression but the production of piRNA depends on the presence of another transgene producing antisense RNA. Moreover, in the context of the Pld promoter deletion, a gene flanking the 42AB piRNA cluster, flies can produce Pld piRNAs only if a Pld cDNA is expressed in trans (Andersen et al., 2017). From all of these observations, a model emerges predicting that simultaneous production of sense and antisense RNA is a shared requirement for piRNA production. However, even if BX2OFF is transcribed on both strands, without additional signals, it still remains inactive for piRNA production.
In addition to having a number of heterochromatic repeats and a double stranded transcription, the production of de novo piRNAs from BX2 requires a triggering signal. From our experiments, BX2 conversion relies on the simultaneous increase of both sense and antisense RNAs. An active role of euchromatic copies in the establishment of new piRNA clusters by high temperature appears to be consistent with what would naturally happen during the invasion of a naive genome by new TEs or when chromosomal breakages occur leading to the loss of piRNA cluster loci (Asif-Laidin et al., 2017). At first, uncontrolled euchromatic TE transposition takes place before the establishment of repression. Such repression would occur after a copy integrates into a preexisting piRNA cluster or by the generation of a new cluster made by successive insertion of nested copies. Consequently, clusters of elements cannot exist without transcriptionally active euchromatic copies. The increase of germline antisense transcripts upon stress or environmental factors, depending on the neighboring genomic environment, and the concomitant presence of numerous sense transcripts from euchromatic active copies, appear to be the starting signals for new piRNA production. These piRNAs can then be inherited at the next generation where they will stably paramutate the corresponding DNA locus with repetitive nature. At that time, the triggering signal is no longer necessary since BX2 remains activated once flies get back at 25°C. Future generations thus remember what was once considered a threat only through the legacy of maternal piRNAs.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Drosophila melanogaster) | AGO1 | NA | FLYB: FBgn0262739 | |
| Gene (D. melanogaster) | RpL32 | NA | FLYB: FBgn0002626 | |
| Gene (D. melanogaster) | eEF5 | NA | FLYB: FBgn0285952 | |
| Gene (D. melanogaster) | Moonshiner | NA | FLYB: FBgn0030373 | |
| Strain, strain background (D. melanogaster) | w1118 | Laboratory Stock | FLYB: FBal0018186 | |
| Strain, strain background (D. melanogaster) | BX2 | Dorer and Henikoff, 1994 PMID:8020105 | FLYB: FBti0016766 | |
| Strain, strain background (D. melanogaster) | P(TARGET)GS | Bloomington Drosophila Stock Center | FLYB: FBst0011039 | also called P-1039 |
| Strain, strain background (D. melanogaster) | P(TARGET)G | Bloomington Drosophila Stock Center | FLYB: FBti0003435 | also called BQ16 |
| Strain, strain background (D. melanogaster) | P(TARGET)S | Bloomington Drosophila Stock Center | FLYB: FBti0003418 | also called BA37 |
| Strain, strain background (D. melanogaster) | nosGAL4 | Bloomington Drosophila Stock Center | FLYB: FBti0131635, RRID:BDSC_32180 | |
| Genetic reagent (D. melanogaster) | P(lacW) | PMID: 2558049 | FLYB: FBtp0000204 | |
| Genetic reagent (D. melanogaster) | P(PZ) | Mlodzik and Hiromi, 1992 doi: 10.1016/B978-0-12-185267-2.50030–1 | FLYB: FBtp0000210 | |
| Genetic reagent (D. melanogaster) | P(A92) | PMID: 2827169 | FLYB: FBtp0000154 | |
| Genetic reagent (D. melanogaster) | Moon shRNA PA61 | Andersen et al., 2017 doi:10.1038/nature23482 | Dr. Julius Brennecke (Institute of Molecular Biotechnology, Vienna) | |
| Genetic reagent (D. melanogaster) | Moon shRNA PA62 | Andersen et al., 2017 doi:10.1038/nature23482 | Dr. Julius Brennecke (Institute of Molecular Biotechnology, Vienna) | |
| Antibody | Mouse IgG polyclonal antibody | Merck (ex-Millipore) | Cat# 12-371B, RRID:AB_2617156 | |
| Antibody | Rabbit polyclonal antibody against H3K9me3 | Merck (ex-Millipore) | Cat# 07–442 | |
| Antibody | Rabbit polyclonal antibody against Rhino | PMID: 19732946 | Dr. William Theurkauf (University of Massachusetts Medical School, Worcester) | |
| Sequence-based reagent | RT-qPCR primers | Sigma-Aldrich | ||
| Sequence-based reagent | RT-qPCR primers | Eurogentech | ||
| Commercial assay or kit | RNeasy kit | Qiagen | Cat# 74104 | |
| Commercial assay or kit | Illumina TruSeq Small RNA library preparation kits | Fasteris | http://www.fasteris.com | |
| Commercial assay or kit | Revertaid RT | Thermo Scientific | EP0442 | |
| Commercial assay or kit | Random Hexamers | Invitrogen | N8080127 | |
| Commercial assay or kit | DNaseI (Rnase free) | New Englands Biolabs | M0303S | |
| Commercial assay or kit | dNTPs solution Mix | New Englands Biolabs | N0447S | |
| Commercial assay or kit | Ribolock RNA inhibitor | Thermo Scientific | EO0381 | |
| Commercial assay or kit | Ssofast Evagreen Supermix | Biorad | Cat# 172–5204 | |
| Commercial assay or kit | qPCR kit | Roche | Cat# 04887352001 | |
| Chemical compound, drug | TRIzol | Invitrogen | Cat# 15596026 | |
| Chemical compound, drug | Chloroform | Carlo Erba Reagents | Cat# 438601 | |
| Chemical compound, drug | Chloroform | Sigma-Aldrich | C2432 | |
| Chemical compound, drug | Ethanol (EtOH) | Merck millipore | Cat# 100983 | |
| Chemical compound, drug | Ethanol (EtOH) | Honeywell | Cat# 32221 | |
| Chemical compound, drug | Glycerol | VWR AnalaR NORMAPUR | Cat# 24388.295 | |
| Chemical compound, drug | Glutaraldehyde | Sigma Aldrich | G-5882 | |
| Chemical compound, drug | Potassium hexacyanoferrate(III) | Sigma Aldrich | P3667 | |
| Chemical compound, drug | Potassium hexacyanoferrate(II) trihydrate | Sigma Aldrich | P3289 | |
| Chemical compound, drug | X-Gal | Dutscher | Cat# 895014 | |
| Chemical compound, drug | NaCl | VWR AnalaR NORMAPUR | Cat# 27810.295 | |
| Chemical compound, drug | NaCl | Sigma-Aldrich | Cat# 31432 | |
| Chemical compound, drug | Formaldehyde | Sigma-Aldrich | Cat# 252549 | |
| Chemical compound, drug | Schneider Medium | Gibco | Cat# 21720–024 | |
| Chemical compound, drug | Insulin | Sigma-Aldrich | I4011 | |
| Chemical compound, drug | PBS | Ambion | AM9625 | |
| Chemical compound, drug | Triton | Sigma-Aldrich | T8787 | |
| Chemical compound, drug | KCl | Ambion | AM9640G | |
| Chemical compound, drug | HEPES | Fisher Scientific | BP299 | |
| Chemical compound, drug | IPEGAL | Sigma-Aldrich | Cat# 18896 | |
| Chemical compound, drug | DTT | Fisher Scientific | R0861 | |
| Chemical compound, drug | Na Butyrate | Sigma-Aldrich | Cat# 07596 | |
| Chemical compound, drug | EDTA free protease inhibitor | Roche | Cat# 04693159001 | |
| Chemical compound, drug | N lauryl sarkosyl | Sigma-Aldrich | L-5125 | |
| Chemical compound, drug | BSA | Fisher Scientific | BP9703 | |
| Chemical compound, drug | SDS 20% | Euromedex | EU0660-B | |
| Chemical compound, drug | Tris HCl | Invitrogen | Cat# 15504–020 | |
| Chemical compound, drug | Dynabeads A | Invitrogen | 10002D | |
| Chemical compound, drug | Glycine | Sigma-Aldrich | G8898 | |
| Chemical compound, drug | Isopropanol | VWR | Cat# 20842.298 | |
| Software, algorithm | Galaxy Server | ARTBIO | https://mississippi.snv.jussieu.fr/ | |
| Software, algorithm | Weblogo | Crooks et al., 2004 doi:10.1101/gr.849004 |
Transgenes and strains
Request a detailed protocolAll transgenes are in the w1118 background. The BX2 line carries seven P-lacZ-white transgenes, (P(lacW), FBtp0000204) inserted in tandem and in the same orientation at cytological site 50C on the second chromosome (Dorer and Henikoff, 1994). The transgene insertion site is located in an intron of the AGO1 gene (de Vanssay et al., 2012). Homozygous individuals are rare and sterile and the stock is maintained in heterozygous state with a Cy-marked balancer chromosome. ß-Galactosidase activity from these transgenes cannot be detected in the germline. P(TARGET)GS corresponds to P(PZ) (FBtp0000210), a P-lacZ-rosy enhancer-trap transgene inserted into the eEF5 gene at 60B7 and expressing ß-Galactosidase in the germline and somatic cells of the female gonads (Bloomington stock number 11039 (FBst0011039). Homozygous flies are not viable and the stock is maintained over a Cy-marked balancer chromosome. P(A92) (FBtp0000154) is another P-lacZ-rosy enhancer-trap transgene that has been used in this study: P(TARGET)G corresponds to BQ16 (FBti0003435) expressing lacZ only in the germline and P(TARGET)S corresponds to BA37 (FBti0003418) expressing lacZ only in the somatic follicle cells that surround the nurse cells. In both lines, homozygous flies are viable. The nosGAL4 transgene used is from the w[*]; PBac{w[+mW.hs]=GreenEye.nosGAL4}Dmel6 line (FBti0131635). Modified miRNA against moonshiner (lines PA61 and PA62) were a kind gift from Julius Brennecke (Andersen et al., 2017). Additional information about stocks are available at Flybase: ‘http://flybase.bio.indiana.edu/".
Thermic and osmotic treatments
Request a detailed protocolSince maintaining flies at high temperature (29°C) decreases viability, we used the following procedure at each generation: fertilized adult females (G0) were allowed to lay eggs for three days at 25°C on standard cornmeal medium. Adults were then discarded or tested for ovarian ß-Galactosidase expression. Vials containing progeny were transferred at 29°C for the rest of the development until complete emergence of G1 adults. Young adults were then transferred into a new vial where they were allowed to lay eggs for three days at 25°C. For heat-shock treatment, embryos (0–2 hr) were incubated at 37°C for 1 hr as described in Seong et al. (2011) and then transferred at 25°C until adult emergence. For osmotic treatments, culturing flies on 300 mM NaCl, as described in Seong et al. (2011), leads to either a large increased time of development or lethality and did not allow us to perform conversion measurements. Accordingly, flies were incubated on 150 mM NaCl for one generation before dissection and ß-Galactosidase staining.
ß-Galactosidase staining
Request a detailed protocolOvarian lacZ expression assays were carried out using X-gal (5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside) overnight staining at 37°C as previously described (Lemaitre et al., 1993), except that ovaries were fixed afterwards for 10 min. After mounting in glycerol/ethanol (50/50), the germline lacZ repression was then calculated by dividing the number of repressed egg chambers by the total number of egg chambers. Most of the time, the total number of egg chambers was estimated by multiplying the number of mounted ovaries by 60, corresponding to an average of three to four egg chambers per ovariole and 16 to 18 ovarioles per ovary. Images were acquired with an Axio-ApoTome (Zeiss) and ZEN2 software.
Fly dissection and RNA extraction
Request a detailed protocolFor each genotype tested, 20 pairs of ovaries were manually dissected in 1X PBS. For small RNA sequencing, total RNA was extracted using TRIzol (Life Technologies) as described in the reagent manual (http://tools.lifetechnologies.com/content/sfs/manuals/trizol_reagent.pdf). For the RNA precipitation step, 100% ethanol was used instead of isopropanol. For RT-qPCR experiments, total RNA was extracted using TRIzol for BX2 and w1118 females or RNeasy kit (Qiagen) for P(TARGET) females. Up to six biological replicates were used for each genotype.
Small RNA sequencing analyses
Request a detailed protocolA small RNA fraction of 18 nt to 30 nt in length was obtained following separation of total RNA extracted from dissected ovaries on a denaturing polyacrylamide gel. This fraction was used to generate multiplexed libraries with Illumina TruSeq Small RNA library preparation kits (RS-200–0012, RS200-0024, RS-200–036 or RS-200–048) at Fasteris (http://www.fasteris.com). A house protocol based on TruSeq, which reduces 2S RNA (30 nt) contamination in the final library, was performed. Libraries were sequenced using Illumina HiSeq 2000 and 2500. Sequence reads in fastq format were trimmed from the adapter sequence 5’-TGGAATTCTCGGGTGCCAAG-3’ and matched to the D. melanogaster genome release 5.49 using Bowtie (Langmead et al., 2009). Only 18–29 nt reads matching the reference sequences with 0 or one mismatch were retained for subsequent analyses. For global annotation of the libraries (Supplementary files 5 and 7), we used the release 5.49 of fasta reference files available in Flybase, including transposon sequences (dmel-all-transposon_r5.49.fasta) and the release 20 of miRNA sequences from miRBase (http://www.mirbase.org/).
Sequence length distributions, small RNA mapping and small RNA overlap signatures were generated from bowtie alignments using Python and R (http://www.r- project.org/) scripts, which were wrapped and run in Galaxy instance publicly from ARTbio platform available at http://mississippi.fr. Tools and workflows used in this study may be downloaded from this Galaxy instance. For library comparisons, read counts were normalized to one million miRNA (Supplementary files 5 and 7). A second normalization, performed using the total number of small RNAs matching the D. melanogaster genome (release 5.49), gave similar results (Supplementary files 5 and 7). For small RNA mapping (Figures 2 and 4, Figure 2—figure supplements 1 and 2), we took into account only 23–29 nt RNA reads that uniquely aligned to reference sequences. Logos were calculated using Weblogo (Crooks et al., 2004) from 3' trimmed reads (23 nt long) matching either P(lacW) (Figure 2B) or 42AB (Figure 2—figure supplement 1B). The percentage of reads containing a ‘U’ at the first position was calculated with all 23–29 nt RNA matching the reference sequence (BX2 transgene in Figure 2B and 42AB in Figure 2—figure supplement 1B). Distributions of piRNA overlaps (ping-pong signatures, Figure 2C and Figure 2—figure supplement 1C) were computed as first described in Klattenhoff et al. (2009) and detailed in Antoniewski (2014). Thus, for each sequencing dataset, we collected all of the 23–29 nt RNA reads matching P(lacW) or the 42AB locus whose 5’ ends overlapped with another 23–29 nt RNA read on the opposite strand. Then, for each possible overlap of 1 to 29 nt, the number of read pairs was counted. To plot the overlap signatures, a z-score was calculated by computing, for each overlap of 1 to i nucleotides, the number O(i) of read pairs and converting the value using the formula z(i) = (O(i)-mean(O))/standard deviation (O). The percentage of reads containing a ‘A’ at the tenth position was calculated within the paired 23–29 nt RNA matching the reference sequence as described in de Vanssay et al. (2012) (BX2 transgene in Figure 2C and 42AB in Figure 2—figure supplement 1B). GRH49 (BX2*) was previously analyzed in Hermant et al. (2015). Small RNA sequences and project have been deposited at the GEO under accession number GSE116122.
ChIP experiments
Request a detailed protocol100 ovaries were dissected in Schneider medium supplemented with insulin at room temperature. Cross-linking was performed for 10 min at room temperature in 1X PBS 1% formaldehyde (Sigma). The cross-linking reaction was stopped by adding glycine to a final concentration of 0.125 mM in PBS 0.1% Triton and incubating 5 min on ice. The cross-linked ovaries were washed with 1 ml of PBS 0.1% Triton and crushed in a dounce A potter 20 times. Then a centrifugation at 400 g for 1 min was performed. The pellet was suspended with 1 ml of cell lysis buffer (KCl 0.085 M, HEPES 5 mM, IGEPAL 0.5%, DTT 0.5 mM, Na butyrate 10 mM, 0.01 M EDTA free protease inhibitor cocktail Roche) and crushed in a dounce B potter 20 times, then 2 mL of cell lysis buffer were added. Centrifugation at 2000 g for 5 min was performed and the pellet was resuspended in 0.5 mL nucleus buffer (HEPES 0.05 M, EDTA 0.01 M, N lauryl Sarcosyl 0.5%, Na butyrate 0.01 M EDTA free protease inhibitor cocktail Roche) and incubated 15 min in a cold room on a rotator. Sonication was performed with Bioruptor (Diagenode) set to high power for 10 cycles (15 s on and 15 s off). A centrifugation was performed 15 min at 16000 g at 4°C. Five µg of chromatin was used for each immunoprecipitation. A preclear of 4 hr was performed with 25 µL of dynabead Protein A. The immunoprecipitation reaction was performed with 50 µL of dynabead Protein A coated with 5 µg of antibodies (H3K9me3 polyclonal antibody C1540030 diagenode or Normal Mouse IgG polyclonal antibody 12–371 Millipore), or 20 µL of serum for the Rhino antibody (kindly provided by Dr W. Theurkauf) over night in the cold room on a rotator. Three washes of 10 min in a high salt buffer (Tris HCl pH 7.5 0.05 M, NaCl 0.5 M, Triton 0.25%, IGEPAL 0.5%, BSA 0.5%, EDTA 5 mM) were performed and the elution of chromatin was performed 30 min with 500 µL of elution buffer (Tris pH 7.5 0.05 M, NaCl 0.05 M, EDTA 5 mM, SDS 1%); RNase treatment was omitted; H3K9me3 and Rhino ChIP were respectively done on 5 and 4 independent biological samples followed by qPCR (Roche light Cycler) on each sample. Values were normalized to respective inputs and to two genomic regions known to be enriched in H3K9me3 and Rhino (42AB): region 1 (chr2R: 6449409–6449518) and region 2 (chr2R: 6288809–6288940). An unpaired t-test was used to calculate significance of the differences (p<0.05). Error bars represent the standard deviation.
RT-qPCR experiments
Request a detailed protocolFor each sample, 10 µg of total RNA was treated with DNase (Fermentas). For classical RT-qPCR experiments, 1 µg of DNase-treated RNA was used for reverse transcription using random hexamer primers (Fermentas). Real-time qPCR was performed on triplicates of each sample. RpL32 was used as reference. The same series of dilution of a mix of different RT preparations was used to normalize the quantity of transcripts in all RT preparations leading to standard quantity (Sq) values. Variations between technical triplicates was very low when compared to variations between biological replicates. The mean of the three technical replicates was then systematically used (meanSq). For each biological sample, we calculated the ratio meanSq(gene)/meanSq(RpL32) to normalize the transcript quantity. Then, the mean of each sample ratio was used to compare the two conditions. For sense-specific RT-qPCR experiments, three reverse transcription were performed using 1 μg of DNase-treated RNA (Fermentas): first without primer (control RT), second with a lacZ sense primer (antisense transcript specific RT) and third with a lacZ antisense primer (sense transcript specific RT). qPCR was then performed on technical triplicates of each RT using a primer pair specific for lacZ sequence. A series of dilutions - ranging from 50 × 10−15 g.µl−1 to 0.08 × 10−15 g.µl−1 - of a plasmid containing the P(lacW) transgene was used as reference to normalize the quantity of lacZ transcripts (Sq values). The number of molecules was estimated by considering that P(lacW) is 11191 bp long and that the average weight of a base pair is 650 g/mol. Using Avogadro's number, the number of copies was estimated as equal to the dsDNA amount (in g) times 6.022 × 1023 divided by the dsDNA length times 650. For example, 50 × 10−15 g corresponds to approximately 4139 molecules. Variations between technical triplicates were very low when compared to variations between biological replicates. The mean of the three technical replicates was then systematically used (meanSq). The measure of the quantity of transcripts (sense or antisense) for a biological sample was then calculated as the (meanSq(sense or antisense specific) - meanSq(control)). This allowed us to eliminate background noise due to unspecific RT amplification for both sense or antisense without specific primer. The mean of each sample ratio was used to compare the two conditions.
Primer sequences
Request a detailed protocolFor classical RT-qPCR experiments, primers used were for w (ChIP experiment): 5'-GTCAATGTCCGCCTTCAGTT-3' and 5'-GGAGTTTTGGCACAGCACTT-3', these primers are specific of the P(lacW) transgene in a w1118 background; for 42AB regions, 5'-TGGAGTTTGGTGCAGAAGC-3' and 5'-AGCCGTGCTTTATGCTTTACT-3' (region 1) and 5'-AAGACCCAATTTTTGCGTCGC-3' and 5'-CAAGGATAGGGATTTGGTCC-3' (region 2); for RpL32: 5’-CCGCTTCAAGGGACAGTATCTG-3’ and 5’-ATCTCGCCGCAGTAAACGC-3’; for lacZ: 5’-GAGAATCCGACGGGTTGTTA-3’ and 5’-AAATTCAGACGGCAAACGAC-3’; for eEF5: 5’-TAACATGGATGTGCCCAATG-3’ and 5’-AACGCAATTGTTCACCCAAT-3’; for AGO1, primers have been chosen in order to detect spliced forms of transcripts coming upstream of the insertion point of BX2 and encoding AGO1-RA, -RD and -RC isoforms (Figure 4C): 5’-GGATCTCCAGATGACCTCCA-3’ and 5’-GGACACTTGTCCGGCTGTAT-3’. For detecting all AGO1 transcripts isoforms, including the AGO1-RB isoform that originates from a promoter located downstream the BX2 insertion point: 5'-ATGAGCCGGTCATCTTTTTG-3' and 5'-GGCAATCGATGGTTTCTTGT-3'. For sense-specific RT-qPCR experiments, we used specific primers during the reverse transcription step: 5’-AGTACGAAATGCGTCGTTTAGAGC-3’ for detection of antisense lacZ transcripts and 5’-AATGCGCTCAGGTCAAATTC-3’ for detection of sense lacZ transcripts.
Data availability
Small RNA sequences and project have been deposited at the GEO under accession number GSE116122.
-
NCBI Gene Expression OmnibusID GSE116122. Environmentally-induced epigenetic conversion of a piRNA cluster.
References
-
Computing siRNA and piRNA overlap signaturesMethods in Molecular Biology 1173:135–146.https://doi.org/10.1007/978-1-4939-0931-5_12
-
A genetic change associated with the R locus in maize which is directed and potentially reversibleGenetics 41:872–889.
-
Transgene repeat arrays interact with distant heterochromatin and cause silencing in cis and transGenetics 147:1181–1190.
-
Heterochromatin protein 1 binds transgene arraysChromosoma 107:286–292.https://doi.org/10.1007/s004120050310
-
The receptor-like tyrosine phosphatase lar is required for epithelial planar polarity and for axis determination within Drosophila ovarian folliclesDevelopment 128:3209–3220.
-
piRNA biogenesis in Drosophila melanogasterTrends in Genetics 33:882–894.https://doi.org/10.1016/j.tig.2017.09.002
-
Molecular dissection of a natural transposable element invasionGenome Research 28:824–835.https://doi.org/10.1101/gr.228627.117
-
Maternal repression of the P element promoter in the germline of Drosophila melanogaster: a model for the P cytotypeGenetics 135:149–160.
-
A GeneTrek analysis of the maize genomePNAS 104:11844–11849.https://doi.org/10.1073/pnas.0704258104
-
From embryo to adult: pirna-mediated silencing throughout germline development in DrosophilaG3: Genes|Genomes|Genetics 7:505–516.https://doi.org/10.1534/g3.116.037291
-
Enhancer trap method in Drosophila: its application to neurobiologyMethods in Neurosciences pp. 397–414.https://doi.org/10.1016/b978-0-12-185267-2.50030-1
-
Homology-dependent silencing by an exogenous sequence in the Drosophila germlineG3: Genes|Genomes|Genetics 2:331–338.https://doi.org/10.1534/g3.111.001925
-
Hybrid dysgenesis in Drosophila melanogaster: influence of temperature on cytotype determination in the P-M systemMGG Molecular & General Genetics 196:17–23.https://doi.org/10.1007/BF00334086
-
P-M system of hybrid dysgenesis inDrosophila melanogaster: thermic modifications of the cytotype can be detected for several generationsMGG Molecular & General Genetics 205:23–27.https://doi.org/10.1007/BF02428028
-
The maternally inherited regulation of P elements in Drosophila melanogaster can be elicited by two P copies at cytological site 1A on the X chromosomeGenetics 129:501–512.
-
p53 genes function to restrain mobile elementsGenes & Development 30:64–77.https://doi.org/10.1101/gad.266098.115
Article and author information
Author details
Funding
Fondation ARC pour la Recherche sur le Cancer (SFI20131200470)
- Stéphane Ronsseray
Fondation pour la Recherche Médicale (DEP20131128532)
- Stéphane Ronsseray
Agence Nationale de la Recherche (plastisipi)
- Stéphane Ronsseray
Pierre and Marie Curie University (EME1223)
- Laure Teysset
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Doug Dorer, Steve Henikoff, Julius Brennecke and the Bloomington Stock Center for providing stocks. We thank Bill Theurkauf for providing antibodies. We thank flybase.org for providing databases. We thank Ritha Zamy for technical assistance. We thank Clément Carré, Lori Pile, Ana Maria Vallès and Jean-René Huynh for critical reading of the manuscript. We thank Christophe Antoniewski for helpful advices and development of the ARTbio platform (http://artbio.fr/).
Copyright
© 2019, Casier et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,491
- views
-
- 329
- downloads
-
- 34
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 34
- citations for umbrella DOI https://doi.org/10.7554/eLife.39842
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Environmentally-induced epigenetic conversion of a piRNA cluster
eLife 8:e39842.
https://doi.org/10.7554/eLife.39842




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}