A calcium transport mechanism for atrial fibrillation in Tbx5-mutant mice
- University of Chicago, United States
- University of Wisconsin-Madison School of Medicine and Public Health, United States
Abstract
Risk for Atrial Fibrillation (AF), the most common human arrhythmia, has a major genetic component. The T-box transcription factor TBX5 influences human AF risk, and adult-specific Tbx5-mutant mice demonstrate spontaneous AF. We report that TBX5 is critical for cellular Ca2+ homeostasis, providing a molecular mechanism underlying the genetic implication of TBX5 in AF. We show that cardiomyocyte action potential (AP) abnormalities in Tbx5-deficient atrial cardiomyocytes are caused by a decreased sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA2)-mediated SR calcium uptake which was balanced by enhanced trans-sarcolemmal calcium fluxes (calcium current and sodium/calcium exchanger), providing mechanisms for triggered activity. The AP defects, cardiomyocyte ectopy, and AF caused by TBX5 deficiency were rescued by phospholamban removal, which normalized SERCA function. These results directly link transcriptional control of SERCA2 activity, depressed SR Ca2+ sequestration, enhanced trans-sarcolemmal calcium fluxes, and AF, establishing a mechanism underlying the genetic basis for a Ca2+-dependent pathway for AF risk.
https://doi.org/10.7554/eLife.41814.001eLife digest
The human heart contains four distinct chambers that work together to pump blood around the body. In individuals with a condition called atrial fibrillation, two of the chambers (known as the atria) beat irregularly and are unable to push all the blood they hold into the other two chambers of the heart. This can cause heart failure and increases the likelihood of blood clots, which may lead to stroke and heart attacks.
Small molecules called calcium ions play a crucial role in regulating how and when the atria contract by driving electrical activity in heart cells. To contract the atria, a storage compartment within heart cells known as the sarcoplasmic reticulum releases calcium ions into the main compartment of the cells. Calcium ions also enter the cell from the surrounding tissue. As the atria relax, calcium ions are pumped back into the sarcoplasmic reticulum or out of the cell by specific transport proteins.
Individuals with mutations in a gene called Tbx5 are more likely to develop atrial fibrillation than other people, but it was not clear how such gene mutations contribute to the disease. Here, Dai, Laforest et al. used mice with a mutation in the Tbx5 gene to study how defects in Tbx5 affect electrical activity in heart cells.
The experiments found that the Tbx5 gene was critical for calcium ions to drive normal electrical activity in mouse heart cells. Compared with heart cells from normal mice, the heart cells from the mutant mice had decreased flow of calcium ions into the sarcoplasmic reticulum and increased flow of calcium ions out of the cell.
These findings provide a direct link between atrial fibrillation and the flow of calcium ions in heart cells. Together with previous work, these findings indicate that multiple different mechanisms could lead to atrial fibrillation, but that many of these involve changes in the flow of calcium ions. Therefore, personalized medicine, where clinicians uncover the specific mechanisms responsible for atrial fibrillation in individual patients, may play an important role in treating this condition in the future.
https://doi.org/10.7554/eLife.41814.002Introduction
Atrial fibrillation (AF) is the most common arrhythmia in humans, characterized by irregularly irregular atrial electrical activity, resulting in asynchronous atrial contraction. AF is a global problem, affecting more than 33 million people and approximately 25% of Americans over the age of forty (Nishida and Nattel, 2014; Weng et al., 2018). AF is associated with significant morbidity and mortality due to thromboembolic events, heart failure, and sudden cardiac death. AF also significantly complicates overall health care management, with AF patients costing five times more to treat than patients without AF (Andrade et al., 2014). The total annual cost to treat AF patients in the US is on the order of 26 billion dollars (Nishida and Nattel, 2014). AF is a highly significant and growing public health concern.
A genetic basis for AF risk has been described in the last decade. Large community-based cohort studies indicate that heritability provides between 40% and 62% of AF risk (Nishida and Nattel, 2014; Christophersen et al., 2009). An emerging paradigm describes AF as a multifactorial disease with genetic predisposition that will determine the propensity of secondary clinical insults to cause AF. This model highlights the importance of understanding the molecular mechanisms underlying the genetic predisposition to AF. Genome-wide association studies (GWAS) studies have identified common risk variants and familial mutations at the T-box transcription factor 5 (TBX5) locus that result in increased risk for AF (McDermott et al., 2008; Sinner et al., 2014). Adult-specific Tbx5 knockout mice demonstrate primary spontaneous and sustained AF, providing evidence supporting the genetic implication at this locus. GWAS have also implicated multiple genes involved in cardiomyocyte calcium handling, including Atp2a2, encoding the sarcolemmal calcium ATPase SERCA2, and Sln and Pln, encoding direct binding SERCA2 inhibitors sarcolipin and phospholamban, respectively. We have previously demonstrated that these cardiomyocyte calcium control genes are direct TBX5 targets (Nadadur et al., 2016). These observations suggested that tight transcriptional control of SERCA2 activity may be central to atrial rhythm robustness and that variation in SERCA2 expression and activity may contribute to AF risk.
The cellular mechanisms causing the irregular electrical activity in AF are believed to include an abnormal myocardial substrate and formation of an ectopic trigger. Abnormal substrate refers to altered electrical conduction between cardiomyocytes. Ectopic trigger refers to cardiomyocyte ectopy, or initiation of electrical activity at regions outside of the sinoatrial node. Both of these cellular phenomena are observed in Tbx5 adult-specific mutant mice and have been associated with abnormal cellular calcium handling (Dobrev, 2010; Voigt et al., 2012; Voigt et al., 2014; Vest et al., 2005; Shanmugam et al., 2011; Neef et al., 2010; Macquaide et al., 2015; Liang et al., 2008; Lenaerts et al., 2009; Hove-Madsen et al., 2004; Greiser et al., 2011; El-Armouche et al., 2006; Brundel et al., 1999). We described the TBX5-dependent gene regulatory network essential for atrial rhythm control and identified downstream ion channels and transporters potentially important to rhythm control (Nadadur et al., 2016; Yang et al., 2017). Triggered activity in the form of early and delayed afterdepolarizations (EADs and DADs) observed in Tbx5-deficient atrial cardiomyocytes could be rescued by heavy buffering of cytoplasmic calcium (Nadadur et al., 2016). Tbx5-dependent calcium handling has thereby emerged as a potential mediator of the myocardial physiologic abnormalities resulting in AF.
We sought to define the Tbx5-dependent cellular mechanisms responsible for abnormal calcium-dependent electrical activity. We found that Tbx5-dependent AF is associated with abnormal sarcoplasmic reticulum (SR) calcium uptake due to depressed SERCA2 expression, depressed SERCA function, and increased phospholamban expression. Decreased SR calcium uptake is compensated by increased Ca2+ extrusion from cardiomyocytes via sodium-calcium exchanger (NCX) current (INCX), which provides a mechanism for TBX5-dependent action potential (AP) prolongation and the propensity for triggered cellular ectopy. In the setting of enhanced NCX mediated Ca2+ efflux and depressed SR uptake, compensatory increases in L-type calcium current (ICaL) balance calcium extrusion to maintain steady state calcium homeostasis. Together these calcium handling alterations contribute to AP prolongation and triggered activity.
We further demonstrated that calcium handling abnormalities, AP alterations, and triggered activity are all normalized by knockout of phospholamban, which prevents Tbx5-dependent AF. These results establish a direct link between depressed SR Ca2+ sequestration, enhanced NCX activity, and AF. This model suggests that targeting calcium handling pathways may be a treatment approach for a subpopulation of AF patients.
Results
Cytoplasmic calcium is responsible for AP prolongation in Tbx5-mutant atrial cardiomyocytes
We previously reported that Tbx5-deficient atrial cardiomyocytes demonstrated AP prolongation and myocardial ectopy. We hypothesized that these defects were caused by cellular calcium handling abnormalities. We therefore surveyed the expression of known calcium handling genes in the adult-specific Tbx5 knockout model. We assessed gene expression in Tbx5fl/fl;R26CreERT2 and control R26CreERT2 mice at 10 weeks of age following tamoxifen (TM) treatment at 8 weeks of age. Consistent with previous observations, the adult Tbx5fl/fl;R26CreERT2 but not control mice developed spontaneous AF, showing an irregularly irregular heartbeat, by telemetric electrocardiogram (ECG) recordings (Figure 1A,B). As previously shown, APs and [Ca]i transients were prolonged in Tbx5fl/fl;R26CreERT2 (Figure 1C) (Nadadur et al., 2016). We assessed expression of genes important to cellular calcium handling in the left atrium by quantitative PCR (Figure 1D). mRNA transcripts for RyR2 (Ryr2) and SERCA2 (Atp2a2), two of the main determinants of sarcoplasmic reticulum (SR) calcium flux, were decreased by 61% and 71% respectively in Tbx5fl/fl;R26CreERT2 mice compared to R26CreERT2 controls (p=0.026 and p=0.001 for Ryr2 and Atp2a2 respectively) consistent with previous studies (Nadadur et al., 2016). In addition, phospholamban (Pln) mRNA expression was increased by 69% in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 (p=0.023), which would be expected to further depress SERCA2 activity. There was no significant difference in mRNA expression of the alpha 1C subunit of the L-type calcium channel (Cacna1c), the cardiac sodium calcium exchanger (Ncx1), or any of the calmodulins 1–3 (Calm1, Calm2, Calm3) (Figure 1C). These data are consistent with the hypothesis that the myocardial electrophysiology deficits in the Tbx5-deficient AF model may be due to abnormal calcium handling.
Figure 1 with 1 supplement see all
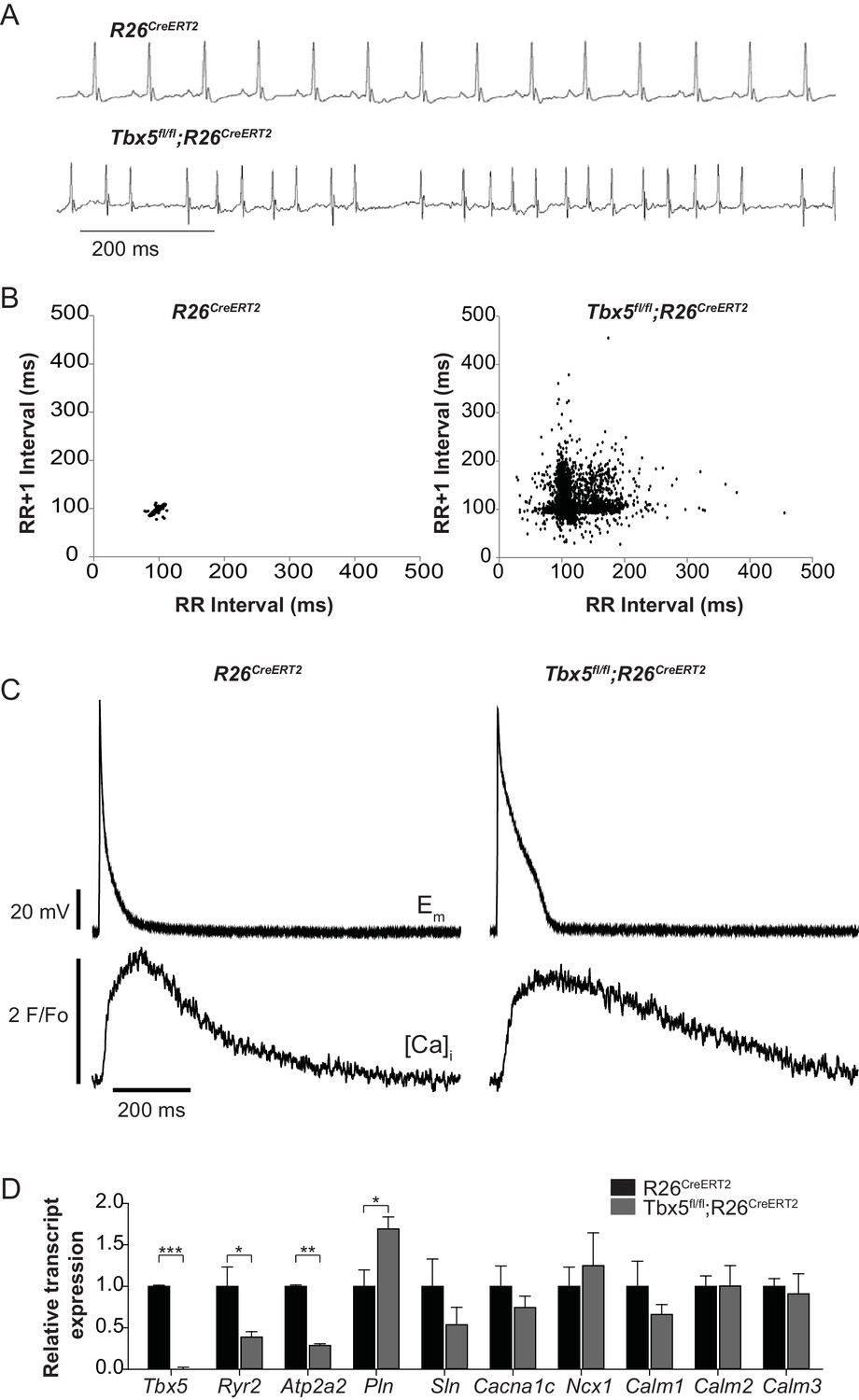
Atrial fibrillation in Tbx5fl/fl;R26CreERT2 mice is associated with altered expression of genes important to cellular calcium handling.
(A) Tbx5fl/fl;R26CreERT2 mice developed spontaneous AF as assessed by surface ECG compared to R26CreERT2. Traces are representative of 15 animals per genotype. (B) Poincaré plot shows irregularly irregular rhythm in Tbx5fl/fl;R26CreERT2, consistent with AF, compared to normal sinus rhythm in R26CreERT2 mice. Poincaré plots are each from one animal, andrepresentative of 15 animals per genotype. (C) Simultaneous AP and [Ca]i recordings show prolonged AP duration and slowed [Ca2+]i transient decay in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes compared to R26CreERT2. Recordings are representative of simultaneous [Ca]i and Em recordings (myocytes/mice; dual Em and [Ca]i from 5/5 R26CreERT2 and 17/5 Tbx5fl/fl;R26CreERT2, Em only from 23/9 R26CreERT2 and 20/9 Tbx5fl/fl;R26CreERT2, and [Ca]i only from 27/6 R26CreERT2 and 28/6 Tbx5fl/fl;R26CreERT2). (D) Quantitative PCR was performed on RNA isolated from left atrial tissue of 3–5 animals per genotype. mRNA expression of a panel of calcium handling genes potentially important for rhythm regulation was determined. Ryr2 and Atp2a2 expression were decreased and Pln expression was increased in Tbx5fl/fl;R26CreERT2 relative to R26CreERT2 atria. (***p<0.001, **, p<0.01, *, p<0.05).
-
Figure 1—source data 1
qPCR data for Figure 1D.
- https://doi.org/10.7554/eLife.41814.005
We examined the relationship between myocardial electrophysiology deficits and calcium flux in Tbx5-mutant atria. In steady state, with each cardiomyocyte contraction cycle, calcium entering the cardiomyocyte (L-type calcium channel, ICaL) is extruded from the cell (predominantly via inward INCX). Similarly, calcium leaving the SR via RyR2 release or SR leak pathways is taken back up into the SR via SERCA2. We examined the effect of altered TBX5-dependent gene expression on these aspects of cardiomyocyte calcium flux. Given the observed changes in Ryr2 and Atp2a2 mRNA abundance, we hypothesized that AP prolongation in Tbx5 deficient cardiomyocytes was due to calcium handling defects downstream of initial Ca2+ entry through ICaL. To test this, we recorded APs in the presence and absence of the L-type Ca2+ channel blocker nifedipine. This approach blocks Ca2+ entry into the cell and indirectly removes the effect of Ca2+ entry on downstream Ca2+ handling pathways, including SR Ca2+ release/reuptake as well as the electrogenic effect of calcium transport out of the cell via inward INCX. 30 μM nifedipine completely inhibited L-type calcium current, preventing Ca2+ entry or release of SR calcium in control R26CreERT2 and Tbx5fl/fl;R26CreERT2 (Figure 2—figure supplement 1). In control R26CreERT2 atrial cardiomyocytes, the effect of nifedipine on AP duration (APD) was small, with 19 ± 4% shortening of APD at 90% repolarization (APD90) (p=0.008) (Figure 2A). However, in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes, nifedipine had a profound effect: APD at 50% repolarization (APD50) was shortened by 16 ± 6% and APD90 by 61 ± 6% (p=0.02 and 0.007 respectively) (Figure 2B,C). Western blot with densitometry analysis for CaV1.2 showed no significant difference in protein expression (Figure 2D), in line with the qPCR data (Figure 1D), consistent with no TBX5-driven direct transcriptional regulation of L-type calcium channels. However, peak ICaL current was increased 92 ± 34% (p=0.027) in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes compared to control R26CreERT2 (Figure 2E and F). The inactivation kinetics at peak ICaL were accelerated Tbx5fl/fl;R26CreERT2 compared to control R26CreERT2 ( τ = 26.7 ± 3.4 ms vs. τ = 40.0 ± 3.0 ms; p=0.05). Steady-state ICaL inactivation was unchanged (Figure 2—figure supplement 2). These data suggest that increased ICaL may contribute to TBX5-loss associated AP prolongation and EADs. However, nifedipine also blocks SR Ca2+ release as well as downstream Ca2+ extrusion pathways, which also affect AP duration. Further, since late AP repolarization is dramatically prolonged (negative to −30 mV where ICaL is largely inactive) we hypothesized that Tbx5-deficiency disrupts Ca2+ handling pathways downstream of ICaL.
Figure 2 with 2 supplements see all
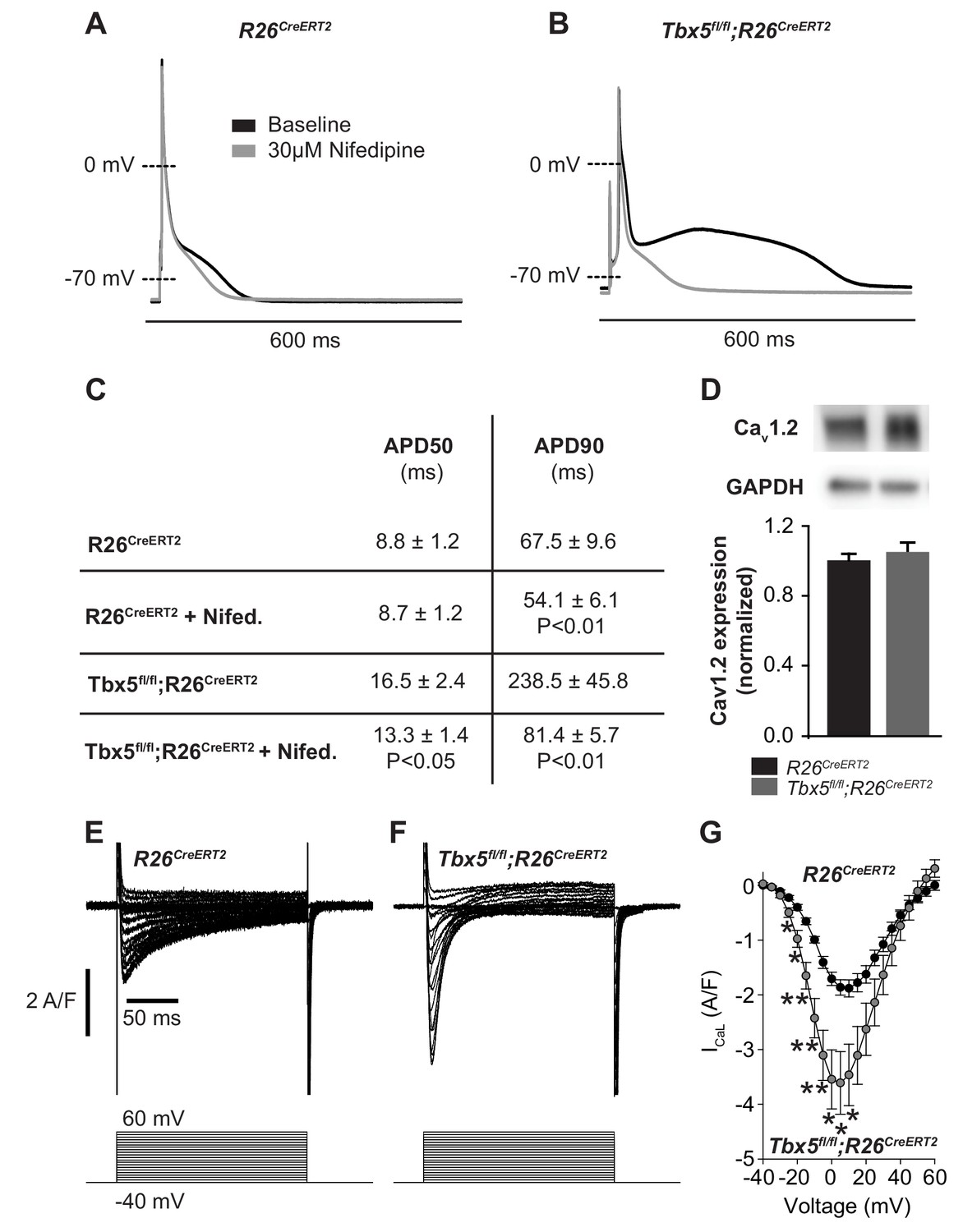
Calcium current blockade dramatically shortened the AP in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes, consistent with the [Ca]i dependence of AP prolongation following TBX5 loss.
(A) Representative recording of an AP from R26CreERT2 atrial cardiomyocytes before and after 30 µM nifedipine treatment. (B) Representative recording of a Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes before and after nifedipine treatment. (C) Paired APD properties before and after treatment with 30 µM nifedipine (myocytes/mice; n = 8/3Tbx5fl/fl;R26CreERT2 and n = 6/4 R26CreERT2). In R26CreERT2 cardiomyocytes, the effect of nifedipine on APD90 was small, but significant 19 ± 4%. A much larger nifedipine effect was observed in Tbx5fl/fl;R26CreERT2 cardiomyocytes. APD50 decreased by 16 ± 6% and APD90 decreased by 61 ± 6% in the presence of nifedipine. (D) Western blot of atrial tissue in five animals for each genotype showed protein expression for the alpha 1C subunit of the L-type calcium channel (Cav1.2) was unchanged. (normalized to GAPDH) (E,F) Representative ICaL recordings show Peak L-type calcium current was increased in Tbx5fl/fl;R26CreERT2 cardiomyocytes compared to R26CreERT2 (G) Average IV relationship of L-type calcium current (myocytes/mice; n = 22/7 R26CreERT2 and 20/5 Tbx5fl/fl;R26CreERT2). (***p<0.001, **, p<0.01, *, p≤0.05).
-
Figure 2—source data 1
AP and ICaL parameters for Figure 2C,G.
- https://doi.org/10.7554/eLife.41814.010
-
Figure 2—source data 2
Western blot for Figure 2D.
- https://doi.org/10.7554/eLife.41814.011
Removal of Tbx5 results in decreased Ca2+ sparks and RyR2 expression, but no overall reduction in RyR2 open probability
Because RyR2 is a critically important sarcolemmal calcium extrusion channel and Ryr2 mRNA was downregulated in Tbx5-mutant atria, we investigated the Tbx5 dependent regulation of RyR2 protein expression and function. RyR2 protein expression was significantly decreased in left atria of Tbx5fl/fl;R26CreERT2 mice compared to R26CreERT2 mice by western blot (Figure 3A), consistent with the observed downregulation of Ryr2 mRNA (Figure 1B). We hypothesized that decreased RyR2 contributed to abnormal Ca2+ release from the SR and tested this by measuring local spontaneous RyR2-mediated Ca2+ release events (Ca2+ sparks) using confocal linescans (Figure 3B). The frequency of Ca2+ sparks in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes was decreased in comparison with R26CreERT2 atrial cardiomyocytes at different pacing frequencies from 0 to 2 Hz (Figure 3C). A decrease in calcium sparks can be due to either decreased RyR2 open probability or a reduced SR calcium load. To differentiate these possibilities, we first examined RyR2 function in the setting of reduced RYR2 expression by performing a [3H]-ryanodine binding assay. [3H]-ryanodine binding to RyR2 correlates with RyR2 open probability (Dobrev, 2010). Despite reduced ryanodine receptor expression, overall ryanodine binding was unchanged over the majority of the physiological range of calcium values, with no shift in calcium sensitivity (Figure 3D). This observation suggests that the alterations in spark frequency were not due to changes in total RyR2 open probability. Instead, it may be caused by diminished SR Ca2+ uptake, a SERCA-dependent property.
Figure 3
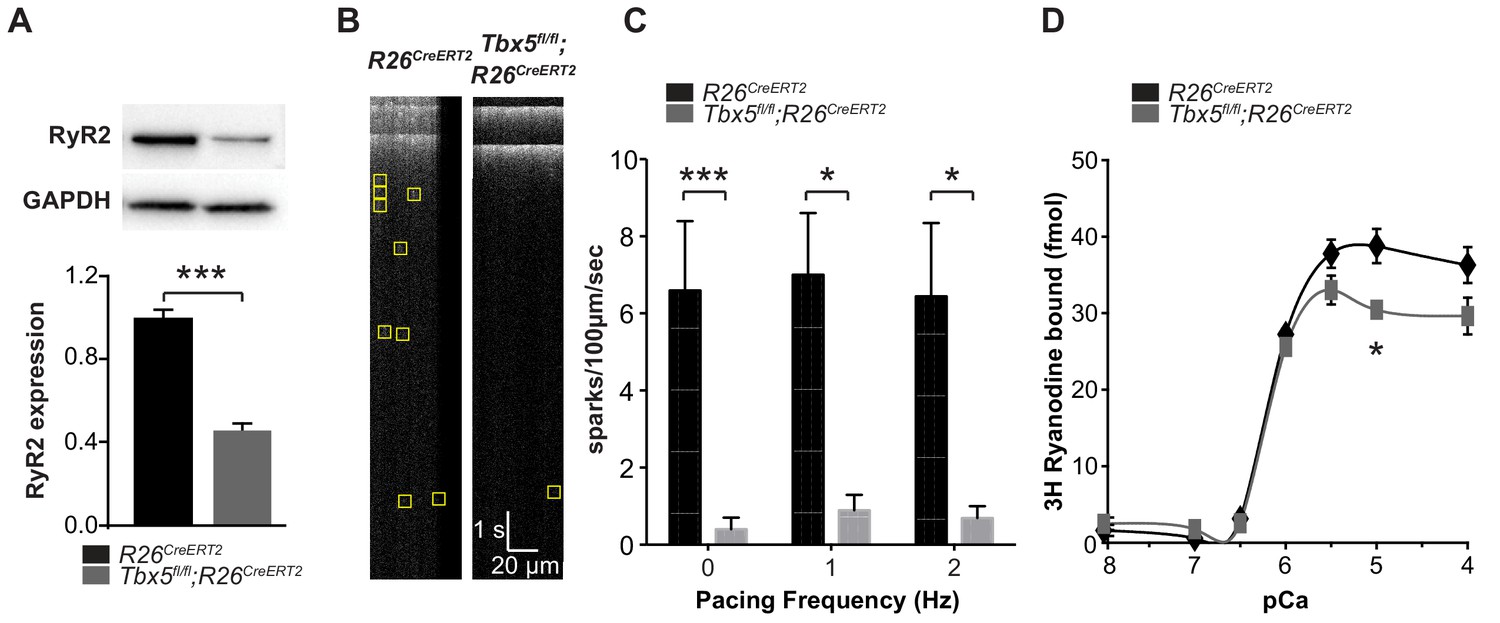
Spark frequency is reduced in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes.
(A) Western blot from atrial tissue from 10 animals per genotype was used to measure RyR2 expression. RyR2 was significantly decreased in Tbx5fl/fl;R26CreERT2 atria compared to R26CreERT2 atria (normalized to GAPDH). (B) Fluo-4 loaded cardiomyocytes demonstrated reduced spark frequency in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 atrial cardiomyocytes (representative recordings). (C) Spark frequency was reduced at rest and after steady state pacing at different frequencies (myocytes/mice; n = 12/4 Tbx5fl/fl;R26CreERT2 and n = 12/3 R26CreERT2). (D) Ryanodine binding assay (without normalization) demonstrated no significant difference over the physiologic range of [Ca]i in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 (Weng et al., 2018). Each measure corresponds to an assay performed on pooled atria from 8 to 10 mice with three independent measures per condition (*p<0.05, **p<0.01, ***p<0.001).
-
Figure 3—source data 1
Western blot for Figure 3A.
- https://doi.org/10.7554/eLife.41814.013
-
Figure 3—source data 2
Spark analysis for Figure 3C.
- https://doi.org/10.7554/eLife.41814.014
-
Figure 3—source data 3
Ryanodine binding assay for Figure 3D.
- https://doi.org/10.7554/eLife.41814.015
Adult-specific Tbx5 deficiency reduces SERCA activity and SR load while increasing sodium-calcium exchanger activity
We next focused on the balance of diastolic calcium efflux pathways as potential mediators of Ca2+ mishandling by measuring SR Ca2+ content and protein expression and function of SERCA2 and NCX1. We observed that SERCA2 protein expression was decreased while NCX1 protein expression was increased in Tbx5fl/fl;R26CreERT2 in comparison with R26CreERT2 atria (Figure 4A,B). To define steady state SR Ca2+ content, we loaded cardiomyocytes with Fluo-4 AM and paced with a train of field stimuli to achieve a steady state Ca2+ content, peak Ca2+ content, and rate of Ca2+ removal were measured (Figure 4C,D). The [Ca2+]i transient peaks were unchanged, but [Ca2+]i transient decay rates, corresponding to SR Ca2+ uptake and cellular Ca2+ extrusion, were slowed in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 atrial cardiomyocytes (Figure 4E,F) (Nadadur et al., 2016), consistent with defective Ca2+ removal from the cytosol. We also measured [Ca2+]i transients in voltage clamp mode using 40 ms square wave voltage clamp pulses from −80 to 0 mV (Figure 4—figure supplement 1). Similar to the field stimulation experiments, [Ca2+]i transient decay rates were slowed, but [Ca2+]i transient peaks were decreased by 23 ± 4% (p=0.02) in Tbx5fl/fl;R26CreERT2 cardiomyocytes compared to R26CreERT2, which suggests that AP prolongation is essential to maintaining peak twitch [Ca2+]i. The latter experiment is also consistent with depressed SR loads in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 atrial myocytes.
Figure 4 with 1 supplement see all
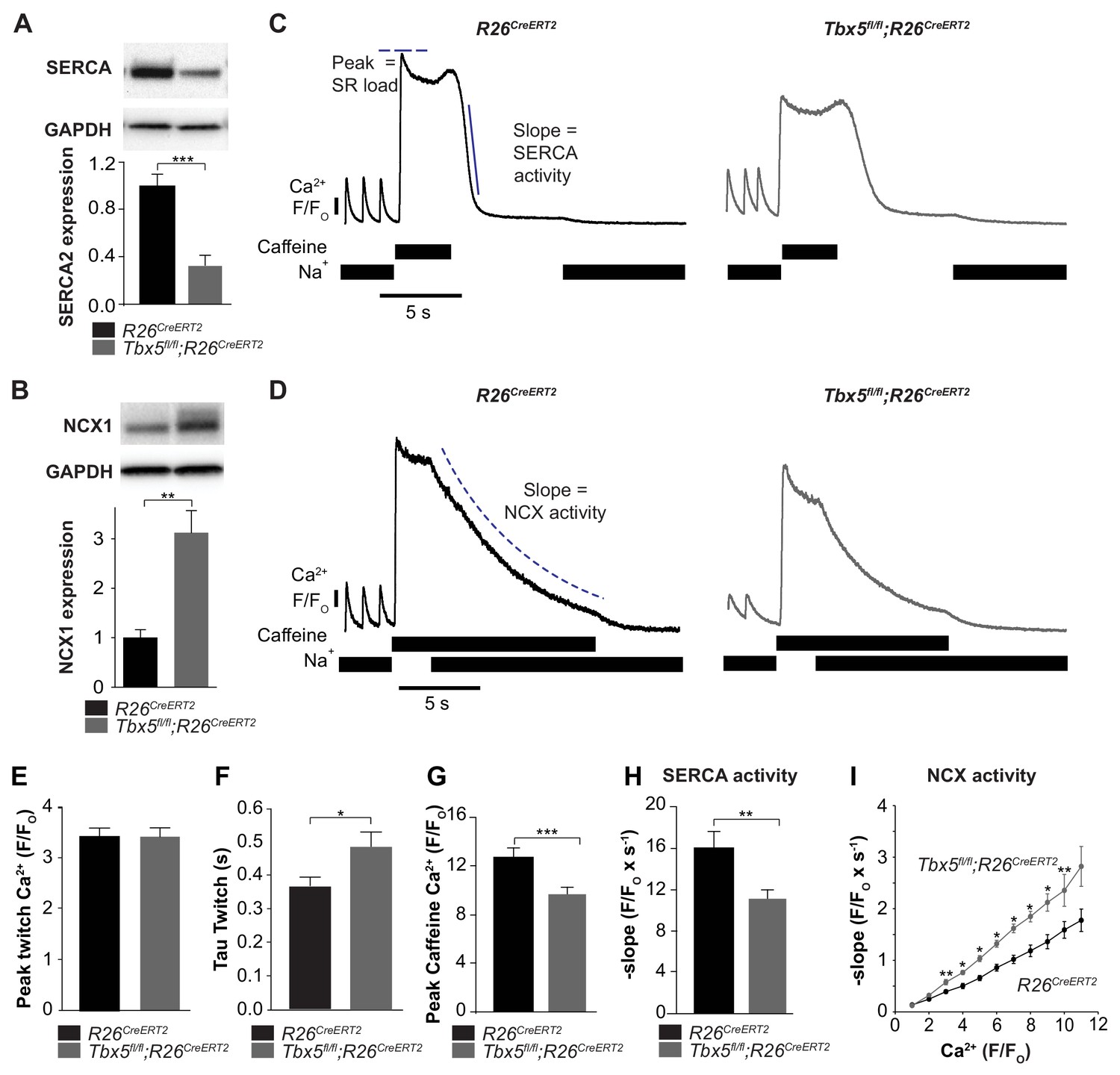
SERCA function is decreased while NCX function is increased in Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes.
(A) Expression of SERCA2 was significantly decreased (normalized to GAPDH) while (B) expression of NCX1 was significantly increased in Tbx5fl/fl;R26CreERT2 atria compared to R26CreERT2 atria as measured by western blot in 10 animals per genotype. (normalized to GAPDH) (C) Application of Na+ free caffeine solution after pacing to steady state at 1 Hz provided a measurement of SR load. In the absence of extracellular Na+, [Ca2+]i plateaued at high levels due to negligible role of non-NCX mediated extrusion in R26CreERT2 and Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes. Removal of caffeine in the absence of external Na+ provided a measure of SERCA mediated SR calcium uptake (representative traces). (D) Restoration of external Na+, in the presence of sustained extracellular caffeine provided a measure of NCX mediated calcium efflux (representative traces). (E) The peak of steady state twitch [Ca2+]i transients was similar but (F) tau of [Ca2+]i decay, determined from twitch [Ca2+]i transients, was increased in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 cardiomyocytes (myocytes/mice; n = 27/6 R26CreERT2, n = 28/6 Tbx5fl/fl;R26CreERT2). (G) SR load, determined from peak caffeine transients was decreased in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 cardiomyocytes (myocytes/mice; n = 34/6 R26CreERT2, n = 32/6 Tbx5fl/fl;R26CreERT2). (H) SERCA activity, determined from the maximal rate of calcium decay was diminished in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 cardiomyocytes (myocytes/mice; n = 29/3 R26CreERT2, n = 32/3 Tbx5fl/fl;R26CreERT2). (I) NCX activity (decay slope), was increased at all levels of calcium in Tbx5fl/fl;R26CreERT2 cardiomyocytes (myocytes/mice; n = 35/3 R26CreERT2, n = 21/3 Tbx5fl/fl;R26CreERT2). (*p<0.05, **p<0.01, ***p<0.001).
-
Figure 4—source data 1
Western blot for Figure 4A,B.
- https://doi.org/10.7554/eLife.41814.019
-
Figure 4—source data 2
Ca2+ handling parameters for Figure 4E–H.
- https://doi.org/10.7554/eLife.41814.020
-
Figure 4—source data 3
NCX activity for Figure 4I.
- https://doi.org/10.7554/eLife.41814.021
We hypothesized that decreased SERCA2 expression caused decreased SR load. We examined SERCA activity by synchronizing the opening of RyR2 channels while preventing Ca2+ extrusion through NCX using caffeine containing, sodium-free, Tyrode solution. This provides a measurement of the maximum release of Ca2+ into the cytosol from the SR, a measure of the SR Ca2+ load (Figure 4C,D). SR [Ca2+] was reduced by 24 ± 8% (p=0.0005) in Tbx5fl/fl;R26CreERT2 compared with R26CreERT2 atrial cardiomyocytes (Figure 4G). SERCA activity was assessed from [Ca2+]i decay rate after SR release in the absence of external sodium (NCX inactive). Peak SERCA activity was reduced by 31 ± 9% (p=0.006) in Tbx5fl/fl;R26CreERT2 compared with R26CreERT2 atrial cardiomyocytes (Figure 4C,H). NCX activity was assessed as the rate of change in [Ca2+]i decay in Na+ containing caffeine solution, preventing net SR uptake. Since NCX activity depends on [Ca]i, we plotted NCX as a function of the [Ca]i signal. NCX activity was ~60% higher in Tbx5fl/fl;R26CreERT2 in comparison with R26CreERT2 atrial cardiomyocytes (Figure 4D,I). Thus, removal of Tbx5 causes decreased SR Ca2+ load and decreased SERCA function, but increased NCX mediated Ca2+ extrusion. Increased inward NCX activity promotes cardiomyocyte depolarization, providing a mechanism for prolonged APs and increased ectopy in Tbx5-mutant atrial cardiomyocytes.
Genetic augmentation of SERCA activity and normalization of SR load eliminates susceptibility to AF
We hypothesized that Tbx5 deficiency reduces SERCA activity by decreasing SERCA2 protein expression (Figure 4A) and increasing expression of phospholamban (Pln), a negative regulator of SERCA2 (Figure 1D). If these were the primary causes of decreased SERCA function in Tbx5-mutant atria, reduced PLN or PLN phosphorylation (relieving inhibition of SERCA2) would be expected to normalize SERCA function. Western blot analysis showed that PLN expression was significantly increased in Tbx5fl/fl;R26CreERT2 compared with R26CreERT2 atria (Figure 5A). In addition, PLN phosphorylation was also increased at serine 16 in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2. These data suggest PLN phosphorylation may be a compensatory mechanism in response to decreased SERCA expression and activity, but is insufficient to normalize SERCA function (Figure 4). Thus, we hypothesized that reduction of Pln gene expression would be more effective in restoring SERCA function.
Figure 5
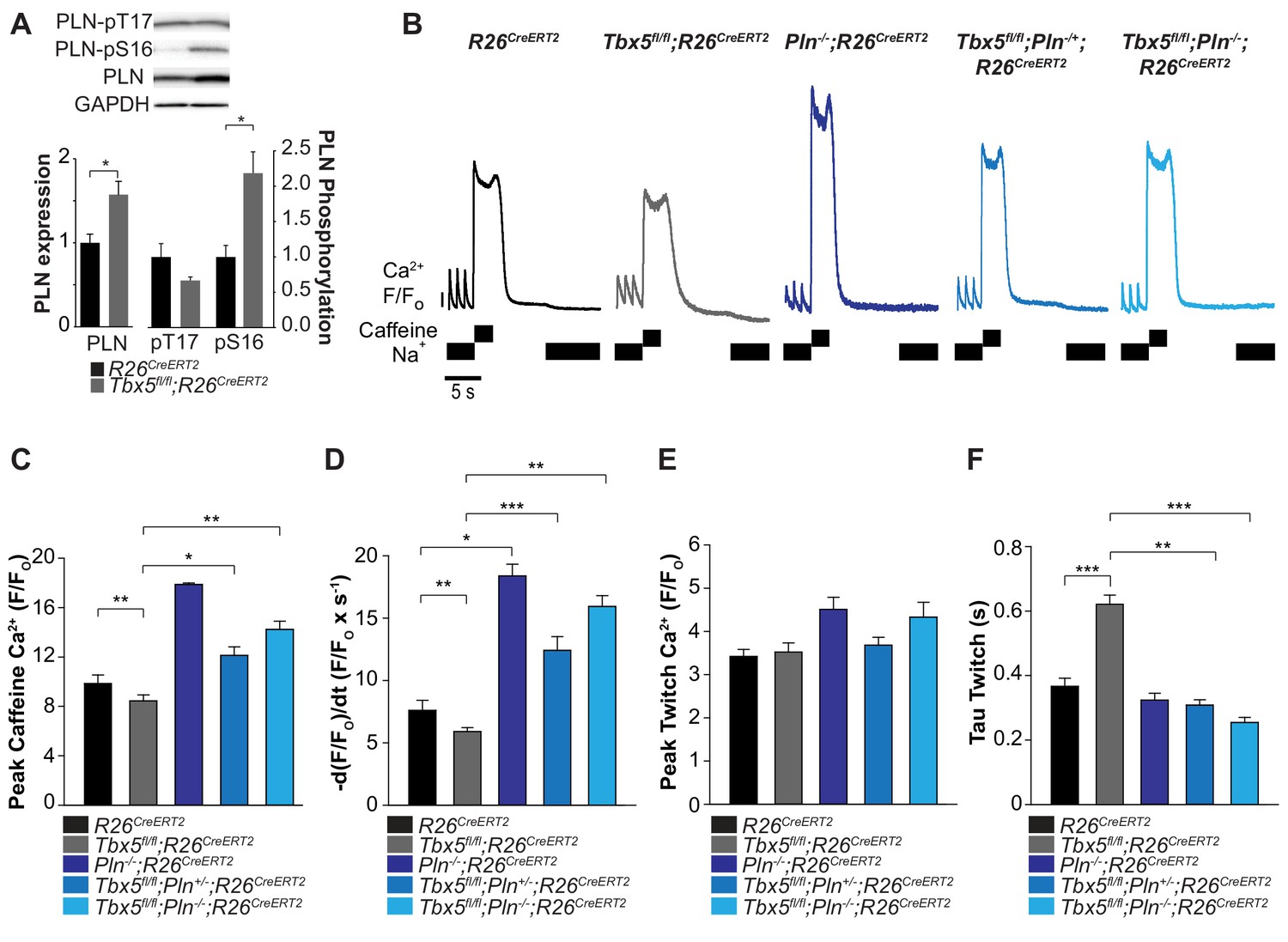
Phospholamban knockout normalized SERCA function in Tbx5fl/fl;R26CreERT2.
(A) PLN expression was increased in Tbx5fl/fl;R26CreERT2 compared to R26CreERT2 as measured by western blot with five animals per genotype. PLN expression was normalized to GAPDH. The proportion of PLN S16, but not T17 phosphorylation was also increased (normalized to PLN). (B) Representative SR load and SERCA measurements in R26CreERT2, Tbx5fl/fl;R26CreERT2, Pln-/-;R26CreERT2, Tbx5fl/fl;Pln-/+;R26CreERT2 and Tbx5fl/fl;Pln-/-;R26CreERT2 atrial cardiomyocytes were collected as described in Figure 4. (C, D) SR load and SERCA function were significantly higher in Tbx5fl/fl;Pln-/+;R26CreERT2 and Tbx5fl/fl;Pln-/-;R26CreERT2 compared to Tbx5fl/fl;R26CreERT2 cardiomyocytes and comparable to R26CreERT2 cardiomyocytes. (E) [Ca2+]i transient peaks were unchanged in Tbx5fl/fl;R26CreERT2, Tbx5fl/fl;Pln-/+;R26CreERT2 and Tbx5fl/fl;Pln-/-;R26CreERT2, but increased in Pln-/-;R26CreERT2 cardiomyocytes. (F) [Ca2+]i transient decay rate in Tbx5fl/fl;Pln-/+;R26CreERT2 and Tbx5fl/fl;Pln-/-;R26CreERT2 cardiomyocytes were normalized to that of R26CreERT2 cardiomyocytes (myocytes/mice; n = 34/3 R26CreERT, n = 36/3 Tbx5fl/fl;R26CreERT2, n = 30/3 Pln-/-;R26CreERT2, n = 21/3 Tbx5fl/fl;Pln-/+;R26CreERT2, n = 27/3 Tbx5fl/fl;Pln-/- atrial cardiomyocytes). (*p<0.05, **p<0.01, ***p<0.001).
-
Figure 5—source data 1
Western blot for Figure 5A.
- https://doi.org/10.7554/eLife.41814.023
-
Figure 5—source data 2
Ca2+ Handling parameters for Figure 5C–F.
- https://doi.org/10.7554/eLife.41814.024
We assessed if Pln deficiency can affect SERCA function in a dose dependent manner by crossing the Tbx5fl/fl;R26CreERT2 with germline Pln knockout mice (Pln-/-;R26CreERT2) (Luo et al., 1994). We compared SR load and SERCA function in adult-specific Tbx5; Pln double mutant mice versus Tbx5 mutant mice. We measured SR load and SERCA function using caffeine-induced SR release in atrial cardiomyocytes from R26CreERT2, Tbx5fl/fl;R26CreERT2, Pln-/-;R26CreERT2, Tbx5fl/fl;Pln-/+;R26CreERT2 mice, and Tbx5fl/fl;Pln-/-;R26CreERT2 mice. Control Pln deficient mice (Pln-/-;R26CreERT2) had increased steady state SR load and SERCA activity relative to R26CreERT2 (Figure 5C,D). The decreased SR load and SERCA function observed in Tbx5 mutant mice (Tbx5fl/fl;R26CreERT2) was converted to elevated SR load and SERCA function after the removal of Pln (Tbx5fl/fl;Pln-/-; R26CreERT2) (Figure 5C,D). Pln loss alone increased peak twitch calcium. However, in the setting of combined Tbx5;Pln deficiency, peak twitch calcium and tau twitch were normalized to R26CreERT2 values (Figure 5E,F).
We next tested the possibility that decreased SERCA function was the mechanism of TBX5-deficiency-driven AP prolongation and triggered activity and that decreased Pln may rescue these defects. As we previously showed, Tbx5fl/fl;R26CreERT2 atrial cardiomyocytes exhibited significantly prolonged APs and frequent EADs and DADs compared to R26CreERT2 atrial cardiomyocytes (Figure 6A,B) (Nadadur et al., 2016). APs of Pln-/-;R26CreERT2 atrial cardiomyocytes were similar to R26CreERT2 controls (Figure 6C). The prolonged AP duration observed Tbx5fl/fl;R26CreERT2 was rescued in both Tbx5fl/fl;Pln+/-;R26CreERT2 and Tbx5fl/fl;Pln-/-;R26CreERT2 atrial cardiomyocytes (43 ± 10% and 38 ± 5% shorter than Tbx5fl/fl;R26CreERT2 respectively; p=0.01, 0.0007) (Figure 6D,E). Along with normalization of AP duration, we observed significantly fewer EADs and DADs in Tbx5fl/fl;Pln-/-;R26CreERT2 cardiomyocytes (Figure 6F). The data demonstrate the importance of TBX5-driven SERCA activity on cellular electrophysiology and triggered activity in atrial cardiomyocytes and decreased Pln rescues both SERCA function and cardiomyocyte electrophysiological abnormalities in Tbx5-mutant mice.
Figure 6
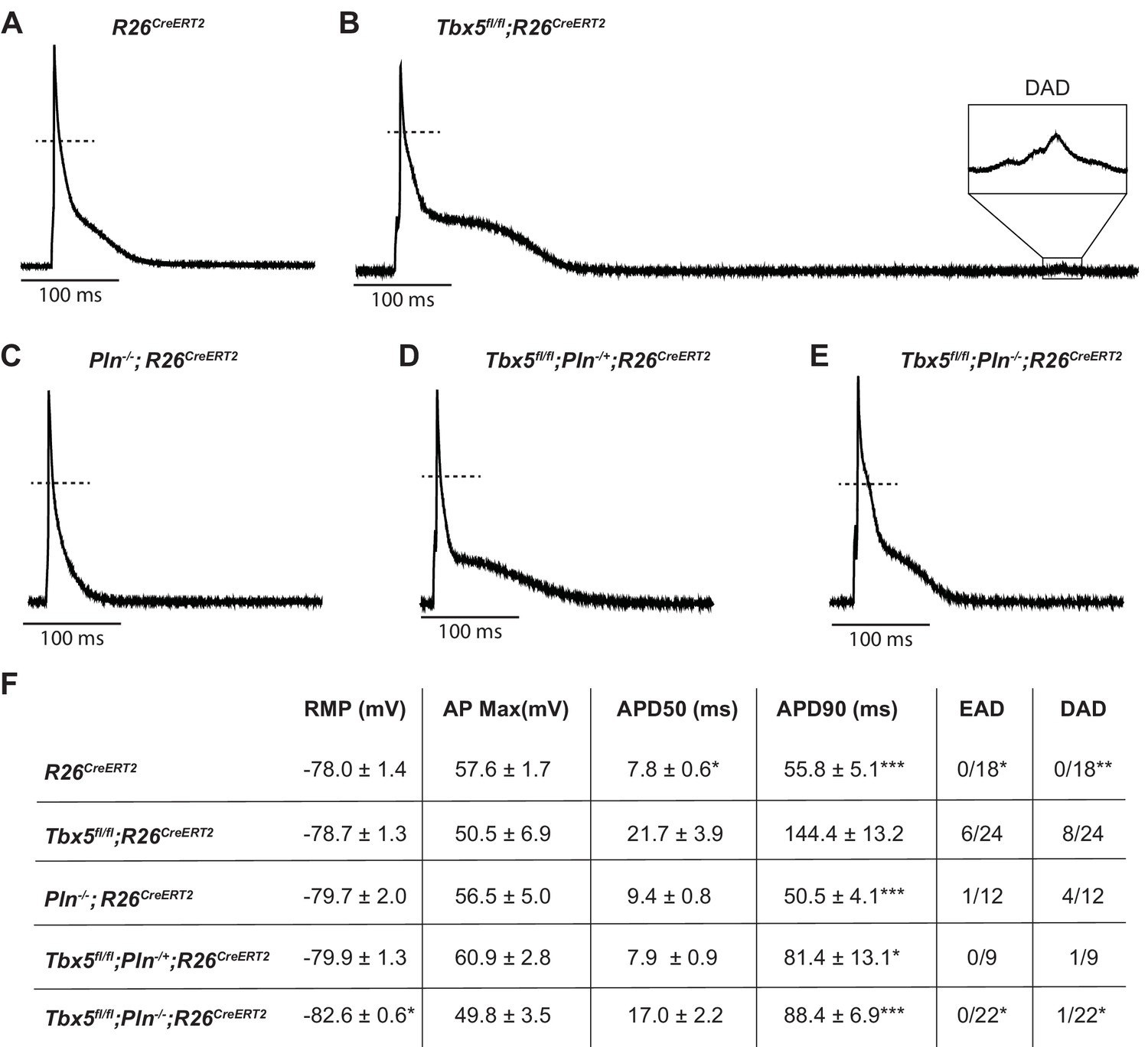
PLN knockout normalized AP duration and prevented triggered activity in Tbx5fl/fl;R26CreERT2.
Representative APs recorded from (A) R26CreERT2, (B)Tbx5fl/fl;R26CreERT2, (C)Pln-/-;R26CreERT2, (D)Tbx5fl/fl;Pln-/+;R26CreERT2, (E)Tbx5fl/fl;Pln-/-;R26CreERT2 atrial cardiomyocytes as described previously in Figure 2. (F) TBX5-loss dependent AP prolongation and frequency of triggered activity was normalized by phospholamban knockout (myocytes/mice: n = 18/7 R26CreERT2, n = 24/12 Tbx5fl/fl;R26CreERT2, n = 12/5 Pln-/-;R26CreERT2, n = 9/3 Tbx5fl/fl;Pln-/+;R26CreERT2, and n = 22/3 Tbx5fl/fl;Pln-/-;R26CreERT2). (*p<0.05, **p<0.01, ***p<0.001).
-
Figure 6—source data 1
AP Parameters for Figure 6F.
- https://doi.org/10.7554/eLife.41814.026
The data above show reducing Pln gene dosage rescues calcium handling defects, AP prolongation and triggered activity observed in Tbx5-mutant atrial cardiomyocytes. We hypothesized that normalizing these cardiomyocyte cellular defects would reduce AF susceptibility in Tbx5 knockout mice (Figure 7). We performed intracardiac burst pacing. All Tbx5fl/fl; R26CreERT2 mice (6/6) paced into AF, compared to none of the R26CreERT2 (0/5) or Pln-/-;R26CreERT2 littermate controls (0/7). Consistent with our hypothesis, AF susceptibility was significantly decreased in Tbx5; Pln compound knockouts: only 1/11 of Tbx5fl/fl;Pln-/-;R26CreERT2 paced into AF (Figure 7F). Thus, Tbx5-deficiency induced AF is due to calcium handling abnormalities, specifically decreased SR load and SERCA activity, and that modulation of the SERCA2 inhibitor, Pln, normalized SERCA activity and AF susceptibility caused by Tbx5 loss.
Figure 7
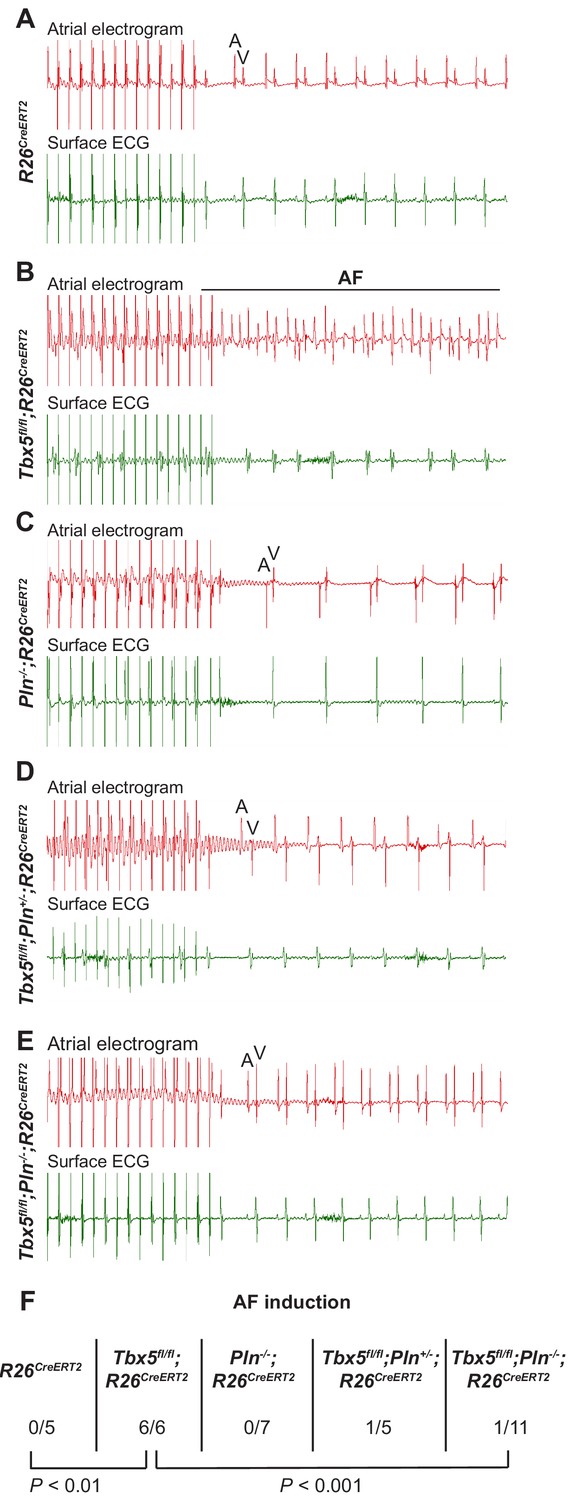
PLN deficiency protected against TBX5-loss associated AF Intra-atrial pacing was used to induce AF.
Representative intracardiac atrial electrogram recordings and corresponding surface ECG are shown from (A) R26CreERT2, (B)Tbx5fl/fl;R26CreERT2, (C)Pln-/-;R26CreERT2, (D)Tbx5fl/fl;Pln-/+;R26CreERT2, (E)Tbx5fl/fl;Pln-/-;R26CreERT2 atrial cardiomyocytes. A, atrial electrical signal; V, far field ventricular electrical signal. (F) AF was reproducibly demonstrated in 6/6 Tbx5 knockouts in contrast to 1/11 Pln/Tbx5 double knockouts, indicating rescue of atrial arrhythmogenesis. P values were determined by Fisher’s exact test (n = 5 R26CreERT2, n = 6 Tbx5fl/fl;R26CreERT2, n = 7 Pln-/-;R26CreERT2, and n = 5 Tbx5fl/fl;Pln-/+;R26CreERT2, n = 11 Tbx5fl/fl;Pln-/- mice).
Discussion
AF initiation has been linked to calcium handling abnormalities in computational and in vivo disease models (Grandi et al., 2011; Heijman et al., 2014). Ectopic or triggered activity in the form of EADs and DADs are often due to calcium handling abnormalities that increase NCX activity and promote AF initiation (Heijman et al., 2014). However, the mechanism underlying genetic predispositions for AF remain poorly understood. Genetic variants and mutations at the Tbx5 locus are associated with increased risk for human AF and Tbx5-mutant mice show both spontaneous and burst pacing-induced AF (Nadadur et al., 2016). We report a calcium transport mechanism for Tbx5-dependent ectopic activity. We show that TBX5 is a critical regulator of SERCA-mediated SR calcium handling and that Tbx5-deficient mice have increased NCX-mediated Ca2+ extrusion, balanced by increased ICaL mediated Ca2+ influx. These calcium handling abnormalities provide a mechanism explaining the frequent triggered activity observed after TBX5 knockout. We show that decreasing phospholamban dosage can normalize TBX5-loss associated cellular calcium handling abnormalities, shorten AP duration, prevent triggered activity, and diminish AF susceptibility.
Decreased SERCA activity and SR load in TBX5-loss associated atrial fibrillation
We and others have hypothesized that ectopic triggers of AF can be due to abnormal atrial calcium handling (Greiser et al., 2011; Bers and Grandi, 2011). Here we define this relationship in a model of spontaneous AF. We analyzed the major calcium transport pathways in atrial myocytes and demonstrated that the critical calcium handling deficit associated with Tbx5-loss is depressed SERCA-mediated SR calcium uptake. We report significant reduction of SERCA2 protein expression and function, consistent with human paroxysmal or chronic AF (Voigt et al., 2012; Voigt et al., 2014; El-Armouche et al., 2006; Brundel et al., 1999).
The mechanism causing cardiomyocyte depolarizations from depressed SERCA activity must be indirect, given that SERCA2 is localized to the intracellular SR membrane and therefore does not directly contribute to membrane potential itself. Instead, slowed SR calcium uptake from depressed SERCA activity provides higher cytosolic calcium driving force for calcium extrusion from the cell via electrogenic inward INCX.
Increased NCX activity in Tbx5- mutant atrial cardiomyocytes drives ectopic activity
We demonstrate increased NCX1 protein expression with Tbx5 knockout, a finding also observed in human and other animal models of AF (Neef et al., 2010; Lenaerts et al., 2009; Greiser et al., 2011; El-Armouche et al., 2006). Since protein expression, electrochemical driving force, and allosteric calcium regulation can all affect amplitude of inward INCX (Blaustein and Lederer, 1999), we measured NCX activity following loss of Tbx5. NCX activity was significantly increased at all levels of calcium (Figure 4I). Thus, increased NCX function coupled with prolonged [Ca2+]i transients, drives increased inward INCX, providing additional depolarizing current during the AP, contributing to its prolongation. While increased NCX function may partially compensate for the depressed SERCA function to bring down calcium levels, it may also promote calcium-induced DADs in the setting of inappropriately timed SR calcium release events. Previous modeling in ventricular cardiomyocytes predicted countervailing functions of SERCA and NCX (Li et al., 2011), which we observed in Tbx5 knockout mice. Our data further support that DADs, which are classically thought to relate to SR calcium overload can still occur with depressed SR loads in the appropriate context of depressed SERCA and elevated NCX function (Voigt et al., 2012).
Modeling suggests that compensatory increases in L-type calcium current in the setting of depressed SERCA function could be required to maintain systolic and diastolic calcium levels (Li et al., 2011). In line with the modeling, we observed enhanced peak ICaL with loss of Tbx5. This may account for early AP prolongation as well as EADs. It is interesting that peak [Ca]i is depressed using controlled square wave voltage clamp pulses (Figure 4—figure supplement 1), which suggests that 40 ms is insufficient to maintain calcium entry in Tbx5 knockout, even in the setting of enhanced ICaL. However, in the setting of AP prolongation (Figure 2E–G) peak twitch calcium levels are maintained. Additionally, the increase in calcium entering the cell through ICaL during the AP would be expected to balance a net increase in NCX mediated calcium extrusion (Figure 4I), a requirement for steady state [Ca2+]i homeostasis. However, our observations that L-type calcium channel expression is TBX5-independent (Figures 1D and 2D) and that genetically targeting only the Ca2+ efflux pathways in our model is sufficient to restore normal electrical activity (Figures 5–7) suggest that the ICaL change is not a primary TBX5-dependent effect. Furthermore, although ICaL is increased, it quickly inactivates in TBX5 knockout cardiomyocytes (Figure 2F). Together with our observation that nifedipine normalizes the APD, this supports that enhanced calcium entry impacts APD via secondary [Ca]i dependent mechanisms.
TBX5 loss results in reduced RyR2 expression
In addition to identifying the role of altered NCX and SERCA function, we assessed the importance of TBX5-driven RyR2 expression. RYR2 is a known susceptibility locus for AF and RYR2 mutations are correlated with AF (Fatkin et al., 2017; Di Pino et al., 2014). We observed that RyR2 protein expression was significantly depressed following Tbx5 loss . Defective RyR2 function has also been associated with AF (Vest et al., 2005; King et al., 2013). Despite TBX5-dependent RyR2 expression, the ryanodine binding assay (Xu et al., 1998) suggested that RyR2 function is generally preserved over the physiologic range of calcium in Tbx5-mutant atria (Figure 3D). This suggests a compensatory mechanism must occur allowing for preserved RyR2 open probability in the setting of depressed protein expression. For example, CaMKII is a potential regulator of RyR2 function which could increase the open probability and thereby increase steady leakage or favor spontaneous local Ca2+ release events from the SR (Vest et al., 2005; Neef et al., 2010; Fischer et al., 2015) in Tbx5-deficient mice. In line with such compensation, we could not detect any differences in the calcium rise kinetics during controlled square wave voltage clamp pulses (Figure 4—figure supplement 1). Nevertheless, RyR2 compensation in the setting of reduced expression could contribute to abnormal triggered activity in the setting of Tbx5 loss, and is an important topic for further investigation.
Abnormalities of the TBX5/SERCA2/PLN regulatory axis drive AF formation
We found that depressed SERCA function in TBX5 knockout was completely normalized with heterozygous or homozygous phospholamban knockout, which normalized AP duration, decreased frequency of afterdepolarizations, and reduced AF inducibility. This finding demonstrates the importance of SERCA2 to the pathophysiology of AF in the Tbx5-loss model. While phospholamban has been associated with AF by GWAS (Fatkin et al., 2017; Federico et al., 2017), its functional role is less clear. Although PLN is predominantly found in the ventricle (Bhupathy et al., 2007), we showed not only its expression is increased in the atria in the context of Tbx5 loss, but also PLN participates in rheostatic control of SERCA activity in the atria, which is sufficient to protect against AF inducibility (Figures 5–7). Our findings are further supported by patient studies. For example, in patients who experience post-operative AF, SERCA2 is significantly decreased in the atrial tissue (Zaman et al., 2016), but those with PLN mutations have decreased AF susceptibility in the context of arrhythmogenic right ventricular cardiomyopathy (Bourfiss et al., 2016). Thus, AF is a heterogeneous disease and there can be variability in how the calcium handling proteins are expressed (Dai et al., 2016) in different disease settings. The genetic background of an individual may be a critical determinant of how calcium handling moieties are disrupted to result in AF.
In summary, the most important features of the Tbx5-dependent SERCA2 and PLN regulatory axis are reduced SR uptake and load (Figure 8). In this setting, enhanced inward INCX and ICaL contributes to AP prolongation, and, more importantly, to cardiomyocyte ectopy. Finally, we demonstrate PLN as a potential means to augment SERCA function, restoring normal atrial myocyte electrical activity and normal sinus rhythm in Tbx5 knockout mice. Thus, the Tbx5 knockout model represents an excellent system to study pharmacologic rescue of SERCA activity, prevention of cardiomyocyte ectopy, and AF.
Figure 8
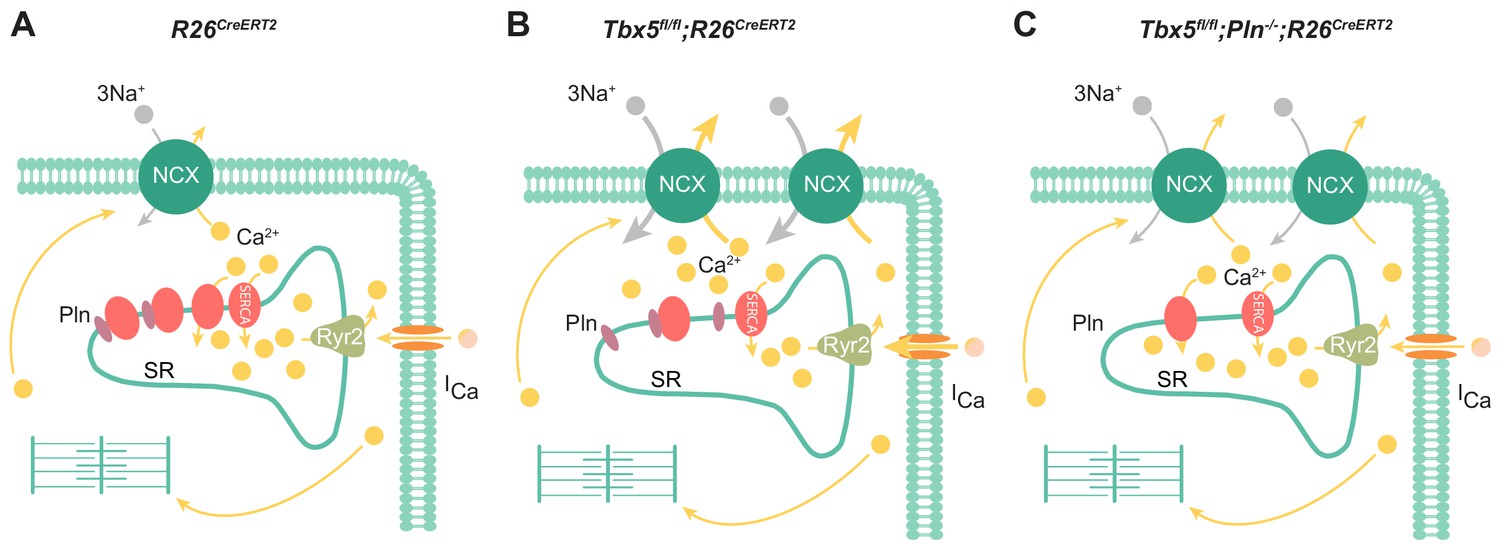
Model of TBX5-dependent calcium regulation in atrial cardiomyocytes.
(A) Excitation-contraction coupling of atrial cardiomyocytes is achieved through regulation of intracellular calcium handling. (B) Adult-specific Tbx5 knockout leads to decreased expression of SERCA2 and increased expression of PLN, leading to decreased SR Ca2+ load. In addition, removal of Tbx5 is associated with increased NCX1 expression and activity, thereby increasing Ca2+ extrusion, which is balanced by increased L-type calcium entry. (C) Combined Tbx5/Pln knockout relieves repression of SERCA2. This results in normalization of SERCA activity and rescue of cardiomyocyte ectopy, triggered activity, and AF observed with Tbx5 deficiency.
Broader implications for clinical treatment of AF
AF has become an increasingly common cause of morbidity and mortality, underlying over one-third of stroke cases and significantly increases the risk for heart failure (Nishida and Nattel, 2014). Consequently, AF poses a significant socioeconomic burden. AF does not always exist in isolation, but rather in conjunction with other predisposing factors such as obesity, thyroid hormone alterations, or heart failure. Interestingly, disruptions in calcium handling proteins such as the SERCA2-PLN regulatory axis are implicated as predisposing factors. In AF compounded by heart failure, decreased SERCA2 and phosphorylated PLN, and increased NCX1 expression were observed (Lugenbiel et al., 2015). Decrease in phosphorylated PLN coupled with an increase in total PLN has been found in animal models of obesity, potentially increasing risk of AF (Lima-Leopoldo et al., 2014; Lima-Leopoldo et al., 2008). These findings suggest a need to evaluate an individual’s genetic background as well as changes in calcium handling proteins when considering predisposing factors for AF.
Currently, there are few effective and targeted AF therapies, in part due to an incomplete understanding of the mechanisms underlying AF. Recent studies of specific genetic loci for AF susceptibility have opened new opportunities to identify specific mechanisms at play in subpopulations of AF patients. Understanding these specific mechanisms may facilitate more effective personalized therapies to target specific atrial Ca2+ handling abnormalities. Our data is consistent with the knowledge that pharmacologic regulators of NCX1 or SERCA2 may normalize defects in cellular calcium handling in the atrium (Dobrev, 2010; Jost et al., 2013; Nagy et al., 2014; Ferrandi et al., 2013; Parikh et al., 2012). For example, a selective NCX1 inhibitor, ORM-10103, was shown to prevent cellular Ca2+ handling abnormalities in ischemic ventricular cardiomyocytes, possibly by limiting calcium entry through outward INCX (Jost et al., 2013; Kormos et al., 2014). The benefit of NCX inhibition might also be considered in human cases of AF with increased NCX activity. Further, resveratrol, which increases SERCA2 activity, has been shown to decrease AF, suggesting that targeting SERCA2 activity may be a viable therapeutic approach (Bai et al., 2016; Chong et al., 2015). In addition to providing specific insight into treating TBX5-loss associated AF, our findings may be more broadly applied. These data suggest that pharmacological treatment of AF may be improved by assessing for a defect in the TBX5-SERCA2-PLN axis followed by specifically targeting the defect to restore normal cardiomyocyte electrical activity. We expect this work and continued efforts to uncover mechanisms responsible for AF in subpopulations of patients will play a key role in advancing personalized therapeutics for AF.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (M. musculus) | Tbx5fl/fl (Tbx5tm1Jse) | PMID: 11572777, 27582060 | MGI:2387850 | Dr. Jonathan G Seidman (Harvard) |
| Genetic reagent (M. musculus) | Pln-/- (Plntm1Egk) | PMID: 8062415 | MGI:2158357 | Dr. Evangelia Kranias (University of Cincinnati) |
| Genetic reagent (M. musculus) | Rosa26CreERT2(Gt(ROSA)26Sortm1(cre/ERT2)Tyj) | PMID: 17251932, 27582060 | MGI:3790674 | Dr. Tyler Jacks (Massachusetts Insititute of Technology) |
| Antibody | Mouse anti-RyR2 | ThermoFisher | Cat. #: MA3-925 | WB (1:2000) |
| Antibody | Mouse anti-SERCA2 | ThermoFisher | Cat. #: MA3-919 | WB (1:1000) |
| Antibody | Mouse anti-NCX | ThermoFisher | Cat. #: MA3-926 | WB (1:1000) |
| Antibody | Mouse anti-PLN | Badrilla | Cat. #: A010-14 | WB (1:5000) |
| Antibody | Rabbit anti-pT17-PLN | Badrilla | Cat. #: A010-13 | WB (1:5000) |
| Antibody | Rabbit anti-pS16-PLN | Badrilla | Cat. #: A010-12 | WB (1:5000) |
| Antibody | Rabbit anti-Cav1.2 | Alomone | Cat. #: ACC-003 | WB (1:200) |
| Antibody | Mouse anti-GAPDH | Millipore | Cat. #: MAB374 | WB (1:10000) |
| Antibody | Goat anti-mouse-HRP | Thermofisher | Cat. #: 31437 | WB (1:5000) |
| Antibody | Goat anti-rabbit-HRP | Thermofisher | Cat. #: 31463 | WB (1:5000) |
| Chemical compound, drug | Fluo-4 AM | Thermofisher | Cat. #: 14201 | 10 µM x 20 min |
| Chemical compound, drug | Nifedipine | Sigma | Cat. #: N7634 | 30 µM |
| Chemical compound, drug | Collagenase Type 2 | Worthington Biochemical | Cat. # LS004177 | 1 g/L |
| Chemical compound, drug | Tamoxifen | MP Biomedicals | Cat#: 156738 | 2 mg/injection x three doses |
| Chemical compound, drug | Laminin | Invitrogen | Cat. #: 2039175 | 0.5 mg/ml |
| Chemical compound, drug | [3H]ryanodine | PerkinElmer | Cat. #: NET950250UC | |
| Chemical compound, drug | TRIzol | Invitrogen | Cat. #: 15596026 | |
| Chemical compound, drug | Ryanodine | MP Biomedicals | SKU #:0215377001 | |
| Chemical compound, drug | Caffeine | Sigma | Cat. #: C0750 | 10 mM |
| Software, algorithm | Clampex/Clampfit Data acquisition and analysis | Molecular Devices | Version 10.3.2.1 | |
| Software, algorithm | LabChart for electrophysiology studies | ADInstruments | Version 5 and 8 | |
| Software, algorithm | Buffering analyses using MaxChelator | Stanford | WEBMAXCLITE v1.15 | Chris Patton, Stanford University |
| Software, algorithm | Western Blot quantification ImageJ | NIH | Version 1.48 | |
| Software, algorithm | Hierarchical Statistical technique using R | R Core Team | Script from PMID: 29016722 | Ken Macleod, Imperial College London |
| Commercial Assay or Kit | qScript cDNA synthesis kit | Quanta | ||
| Commercial Assay or Kit | Power SYBR Green PCR Master Mix | Applied Biosystems |
Generation of mice
Request a detailed protocolThe Tbx5fl/fl, Pln-/- and Rosa26CreERT2 lines have all been previously described and were kept in a mixed genetic background (Luo et al., 1994; Bruneau et al., 2001; Ventura et al., 2007). Double knockout mice were generated by crossing Tbx5fl/fl;R26CreERT2 mice with germline Pln-/- mice. After two generations, we obtained Tbx5fl/fl;R26CreERT2 mice with either loss of one (Pln+/-) or both (Pln-/-) copies of Pln. All experiments were done using age- and genetic strain-matched littermate controls. Tamoxifen was administered for three consecutive days at a dose of 0.167 mg/kg body weight by intraperitoneal injection at 6–10 weeks of age, as previously described (Nadadur et al., 2016). All experiments were performed in accordance to The University of Chicago Institutional Animal Care and Use Committee (IACUC) approved protocol.
ECG recordings
View detailed protocol8-to 10- week-old mice were anesthetized using isoflurane, and telemetry transmitters (ETA-F10, Data Science International) were implanted in the back with leads tunneled to the right upper and left lower thorax, as previously described (Wheeler MT et al., JCI 2004). Baseline recordings were obtained for 24 hr after a post-implant recovery period of one day. ECG data was analyzed using LabChart 8 (AD Instruments).
Intracardiac electrophysiology studies
Request a detailed protocolDetailed protocols for intracardiac electrograms have been previously described (Nadadur et al., 2016). Briefly, 8- to 10- week-old mice were anesthetized with isoflurane and a vertical skin cut-down at the right jugular vein was performed. A 1.1 F octapolar catheter (EPR-800, Millar Instruments) was advanced in the right jugular vein to perform electrical stimulation. The catheter was connected to ADI BioAmp and PowerLab apparatus and signals were recorded using LabChart Software (ADInstruments). Atrial induction pacing was performed using burst pacing and the presence of at least three cycles of atrial tachycardia or fibrillation at least twice was considered positive.
[Ca2+]i transient measurement
Request a detailed protocolLangendorff perfusion with 2 mg/mL of Collagenase Type 2 (Worthington Biochemical) at 5 ml/min was used to isolate atrial cardiomyocytes. Cardiomyocytes were then plated on laminin coated glass bottom dishes for 30 min prior to incubation with 10 µM Fluo-4/AM (Molecular Probes/Invitrogen) in normal Tyrode’s solution containing (in mM): 140 NaCl, 4 KCl, 10 glucose, 10 HEPES, and 1 MgCl2, 1 CaCl2 pH 7.4 using NaOH for 20 min at room temperature. Cells were perfused with prewarmed Tyrode for 10 min prior to imaging. Imaging was performed on an Olympus microscope with a 20x objective lens, a LAMBDA DG-4 power source with 488 nm excitation and 515 nm emission filters and a PMT (photomultiplier tube) to record whole cell signal. Electrical field stimulation (Grass stimulator; Astro-Med) was performed at 1 Hz. SERCA and NCX measurements were performed by flowing sodium free Tyrode with 10 mM caffeine followed by sodium free Tyrode alone or Tyrode with caffeine respectively. Cells were returned to normal Tyrode in both cases at the end of the recording. [Ca2+]i transients are presented as total fluorescence intensity normalized to resting fluorescence (F/F0) obtained from steady-state resting conditions before field stimulation. [Ca2+]i transients and sparks were acquired in line-scan mode (3 ms per scan; pixel size 0.12 µm) using a Zeiss confocal microscope.
Whole-cell electrophysiological recordings
Request a detailed protocolAPs and voltage clamp recordings were recorded using standard ruptured patch protocol (Nadadur et al., 2016). We used current clamp mode with 0.5 nA ×2 ms current clamp pulses to measure APs. Voltage clamp mode was used to measure capacitance transients and to study [Ca2+]i transients with fixed duration depolarizations. Cardiomyocytes are kept at 37°C and perfused with Tyrode solution (140 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 Glucose, and pH 7.4 with NaOH). Internal pipette solution composition was (in mM): 20 KCl, 100 K-glutamate, 10 HEPES, 5 MgCl2, 10 NaCl, 5 Mg-ATP, 0.3 Na-GTP. Patch pipettes (World Precision Instruments) were pulled to have a mean resistance of 3.5–5 MΩ. An Ag–AgCl pellet and 3M KCl agar bridge was used to ground the bath. Liquid junction potentials, were always corrected after cell rupture.
External solution for ICaL contained (in mM): 120 Tetraethylammonium-chloride, 10 CsCl, 10 Glucose, 10 HEPES, 1.5 MgCl2, 1 CaCl2, pH 7.4 with CsOH. Internal pipette solution contained (in mM): 100 Cs-methanesulfonate, 30 CsCl, 10 HEPES 5 EGTA, 2 MgCl2, 5 Mg-ATP, pH 7.2 with CsOH. ICaL was recorded during 200 ms voltage clamp pulses from a holding potential of −40 mV to test potentials ranging from −40 to 60 mV, with pulses applied every 2 s in 5 mV increments. Peak current amplitudes were normalized to the cell capacitance (Cm) and presented as current density (A/F). Steady-state inactivation of ICaL was investigated using two-pulse protocol. Holding potential was −80 mV. The first pulse depolarized membrane from −60 to 20 mV with 10 mV increments during 500 ms, the second pulse depolarized the membrane to 10 mV for 50 ms. The inactivation curves were fit to a Boltzmann distribution.
Acquisition was performed using an Axopatch-200B amplifier connected to a Digidata1550A acquisition system (Axon Instruments, Foster City, CA, USA). In recording filtering at 2 kHz was performed using the amplifier Bessel and sampled at 10 kHz. Analysis was performed using pCLAMP10 (Axon Instruments) and a home written analysis code.
Western blots
Request a detailed protocolAtrial tissue was collected and homogenized as described previously (Alvarado et al., 2017), in a buffer containing 0.9% NaCl, 10 mM Tris-HCl pH 6.8, 20 mM NaF and protease inhibitors. Equal amounts of protein, as determined by Bradford assay, were loaded. 50 µg of tissue homogenate, in Laemmli buffer, was separated by SDS-PAGE in 4–20% TGX or AnyKD precast gels (Bio-Rad). Proteins were transferred to PVDF membrane using the iblot2 transfer system (ThermoFisher) or wet transfer. Primary antibodies were as follows: anti-RyR2 (1:2000; MA3-925, ThermoFisher), SERCA2 (1:1000; MA3-919, ThermoFisher), NCX (1:1000; MA3-926, ThermoFisher), PLN (1:5000; A010-14, Badrilla), pT17-PLN (1:5000; A010-13, Badrilla), pS16-PLN (1:5000; A010-12, Badrilla), Cav1.2 (1:200; ACC-003, Alomone), GAPDH (1:10000; MAB374, Millipore). Secondary antibodies were: goat anti-mouse-HRP (1:5000; 31437, ThermoFisher) or goat anti-rabbit-HRP (1:5000; 31463, ThermoFisher). Secondary antibody concentrations were 5x higher when using the ibind Flex system. SuperSignal ECL reagent (ThermoFisher) was used to develop membranes followed by imaging with a ChemiDoc MP apparatus (Bio-Rad). Band intensities were quantified with the ImageLab software (Bio-Rad) or using ImageJ (NIH).
[3H]Ryanodine binding assay
Request a detailed protocolBinding assays were carried out following a protocol previously described (Federico et al., 2017). Binding mixtures contained 100 µg of protein from homogenates prepared from pooled atria (5–7 mice), 0.2 M KCl, 20 mM Hepes (pH 7.4), 6.5 nM [3H]ryanodine (PerkinElmer), 1 mM EGTA and enough CaCl2 to set free [Ca2+] between 10 nM (pCa2+ 8) and 100 µM (pCa2+ 4). The ratio between Ca2+ and EGTA was determined using MaxChelator (WEBMAXCLITE v1.15 http://maxchelator.stanford.edu/webmaxc/webmaxclite115.htm). Following a 2 hr incubation at 36°C, reactions were filtered through Whatman GF/B Filters using a Brandel M24-R Harvester. [3H]ryanodine binding was determined using a Beckman LS6500 scintillation counter and BioSafe II scintillation cocktail (RPI Corp). Non-specific binding was quantified in the presence of 2 µM unlabeled ryanodine (MP Biomedicals) and subtracted.
Quantitative real time PCR
Request a detailed protocolLeft atrial tissue of Tbx5fl/fl;R26CreERT2 and R26CreERT2 mice was removed two weeks after receiving tamoxifen and RNA was isolated using a Trizol (Invitrogen) based method. Reverse transcription reaction was carried out using the qScript cDNA synthesis kit (Quanta) according to the manufacturer’s protocol. Quantitative RT-PCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems) and run on an Applied Biosystems AB7500 machine. Relative fold changes were calculated using the comparative threshold cycle method (2-ΔΔCt), using glyceraldehyde-3-phosphate dehydrogenase (Gapdh) gene expression level as internal control.
PCR primers
Request a detailed protocol| Gene | F primer | R primer |
|---|---|---|
| Tbx5 | GGCATGGAAGGAATCAAGGT | CTAGGAAACATTCTCCTCCCTGC |
| Ryr2 | CAAATCCTTCTGCTGCCAAG | CGAGGATGAGATCCAGTTCC |
| Atp2a2 | CTGGTGATATAGTGGAAATTGCTG | GGTCAGGGACAGGGTCAGTA |
| Pln | TTATGCCAGGACGGCAAAAG | CACTGTGACGATCACCGAAG |
| Sln | CTGAGGTCCTTGGTAGCCTG | GGTGTGTCAGGCATTGTGAG |
| Cacna1c | CTACAGAAACCCATGTGAGCAT | CAGCCACGTTGTCAGTGTTG |
| Ncx1 | TTCTCATACTCCTCGTCATCG | TTGAGGACACCTGTGGAGTG |
| Calm1 | TGGGAATGGTTACATCAGTGC | CGCCATCAATATCTGCTTCTCT |
| Calm2 | ACGGGGATGGGACAATAACAA | TGCTGCACTAATATAGCCATTGC |
| Calm3 | GATGGCACCATTACCACCAAG | CGCTGTCTGTATCCTTCATCTTT |
Statistical analysis
Request a detailed protocolValues are represented as mean ±standard error of the mean (±SEM). Statistical significance for quantitative metrics of APs, SERCA, NCX, SR load, ICaL, spark frequency, and [Ca2+]i transients were determined using hierarchical statistical methods (Sikkel et al., 2017). Statistical significance for mRNA, and protein expression studies was determined using Student’s t-test. Statistical significance of the nifedipine effect on AP duration was determined using two-tailed paired t-test. A two-tailed Fisher’s exact test was used for statistical significance of count-based analysis of AF inducibility and EAD and DAD count. Statistical significance is designated as *p<0.05, **p<0.01, and ***p<0.001.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Ablation of the cardiac ryanodine receptor phospho-site Ser2808 does not alter the adrenergic response or the progression to heart failure in mice. Elimination of the genetic background as critical variableJournal of Molecular and Cellular Cardiology 103:40–47.https://doi.org/10.1016/j.yjmcc.2017.01.001
-
Resveratrol protects against lipopolysaccharide-induced cardiac dysfunction by enhancing SERCA2a activity through promoting the phospholamban oligomerizationAmerican Journal of Physiology-Heart and Circulatory Physiology 311:H1051–H1062.https://doi.org/10.1152/ajpheart.00296.2016
-
Human atrial fibrillation: insights from computational electrophysiological modelsTrends in Cardiovascular Medicine 21:145–150.https://doi.org/10.1016/j.tcm.2012.04.004
-
Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPaseJournal of Molecular and Cellular Cardiology 42:903–911.https://doi.org/10.1016/j.yjmcc.2007.03.738
-
Sodium/calcium exchange: its physiological implicationsPhysiological Reviews 79:763–854.https://doi.org/10.1152/physrev.1999.79.3.763
-
Influence of Genotype on Structural Atrial Abnormalities and Atrial Fibrillation or Flutter in Arrhythmogenic Right Ventricular Dysplasia/CardiomyopathyJournal of Cardiovascular Electrophysiology 27:1420–1428.https://doi.org/10.1111/jce.13094
-
Familial aggregation of atrial fibrillation: a study in Danish twinsCirculation. Arrhythmia and electrophysiology 2:378–383.https://doi.org/10.1161/CIRCEP.108.786665
-
Characterization of Ca2+ handling proteins and contractile proteins in patients with lone atrial fibrillationInternational Journal of Cardiology 202:749–751.https://doi.org/10.1016/j.ijcard.2015.10.010
-
Atrial Ca2+ signaling in atrial fibrillation as an antiarrhythmic drug targetNaunyn-Schmiedeberg's Archives of Pharmacology 381:195–206.https://doi.org/10.1007/s00210-009-0457-1
-
Genetics of Atrial Fibrillation: State of the Art in 2017Heart, Lung and Circulation 26:894–901.https://doi.org/10.1016/j.hlc.2017.04.008
-
Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibitionBritish Journal of Pharmacology 169:1849–1861.https://doi.org/10.1111/bph.12278
-
Late INa increases diastolic SR Ca2+-leak in atrial myocardium by activating PKA and CaMKIICardiovascular Research 107:184–196.https://doi.org/10.1093/cvr/cvv153
-
Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillationCirculation research 109:1055–1066.https://doi.org/10.1161/CIRCRESAHA.111.253955
-
Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillationCardiovascular Research 89:722–733.https://doi.org/10.1093/cvr/cvq389
-
ORM-10103, a novel specific inhibitor of the Na+/Ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heartBritish Journal of Pharmacology 170:768–778.https://doi.org/10.1111/bph.12228
-
Efficacy of selective NCX inhibition by ORM-10103 during simulated ischemia/reperfusionEuropean Journal of Pharmacology 740:539–551.https://doi.org/10.1016/j.ejphar.2014.06.033
-
Obesity induces upregulation of genes involved in myocardial Ca2+ handlingBrazilian Journal of Medical and Biological Research 41:615–620.https://doi.org/10.1590/S0100-879X2008000700011
-
Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythmScience Translational Medicine 8:ra115.https://doi.org/10.1126/scitranslmed.aaf4891
-
Selective Na+/Ca2+ exchanger inhibition prevents Ca2+ overload-induced triggered arrhythmiasBritish journal of pharmacology 171:5665–5681.https://doi.org/10.1111/bph.12867
-
Decreased sarcolipin protein expression and enhanced sarco(endo)plasmic reticulum Ca2+ uptake in human atrial fibrillationBiochemical and Biophysical Research Communications 410:97–101.https://doi.org/10.1016/j.bbrc.2011.05.113
-
Post-operative atrial fibrillation is associated with a pre-existing structural and electrical substrate in human right atrial myocardiumInternational Journal of Cardiology 220:580–588.https://doi.org/10.1016/j.ijcard.2016.06.249
Article and author information
Author details
Francisco J Alvarado

Christopher R Weber
Funding
National Institutes of Health (HL126509)
- Ivan P Moskowitz
American Heart Association (Collaborative Sciences Award)
- Ivan P Moskowitz
National Institutes of Health (T32HL007381)
- Wenli Dai
National Institutes of Health (HL114010)
- Ivan P Moskowitz
National Institutes of Health (R33 HL123857)
- Ivan P Moskowitz
National Institutes of Health (T32GM007281)
- Wenli Dai
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Evangalia Kranias for providing the phospholamban knockout mice (NIH HL26057). The project described was supported by R01 HL126509 and HL114010, AHA Collaborative Sciences Award, R33 HL123857, and T32HL007381.
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (#71737) of the University of Chicago. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Chicago. All surgery was performed under isoflurane anesthesia, and every effort was made to minimize suffering.
Copyright
© 2019, Dai et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,738
- views
-
- 312
- downloads
-
- 38
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 38
- citations for umbrella DOI https://doi.org/10.7554/eLife.41814
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A calcium transport mechanism for atrial fibrillation in Tbx5-mutant mice
eLife 8:e41814.
https://doi.org/10.7554/eLife.41814




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}