Multiple pairs of allelic MLA immune receptor-powdery mildew AVRA effectors argue for a direct recognition mechanism
- Max Planck Institute for Plant Breeding Research, Germany
- RWTH Aachen University, Germany
- University of Zurich, Switzerland
- Cluster of Excellence on Plant Sciences, Germany
Abstract
Nucleotide-binding domain and leucine-rich repeat (NLR)-containing proteins in plants and animals mediate intracellular pathogen sensing. Plant NLRs typically detect strain-specific pathogen effectors and trigger immune responses often linked to localized host cell death. The barley Mla disease resistance locus has undergone extensive functional diversification in the host population and encodes numerous allelic NLRs each detecting a matching isolate-specific avirulence effector (AVRA) of the fungal pathogen Blumeria graminis f. sp. hordei (Bgh). We report here the isolation of Bgh AVRa7, AVRa9, AVRa10, and AVRa22, which encode small secreted proteins recognized by allelic MLA7, MLA9, MLA10, and MLA22 receptors, respectively. These effectors are sequence-unrelated, except for allelic AVRa10 and AVRa22 that are co-maintained in pathogen populations in the form of a balanced polymorphism. Contrary to numerous examples of indirect recognition of bacterial effectors by plant NLRs, co-expression experiments with matching Mla-AVRa pairs indicate direct detection of the sequence-unrelated fungal effectors by MLA receptors.
https://doi.org/10.7554/eLife.44471.001eLife digest
Powdery mildews are fungal diseases that affect many plants, including important crops such as barley. The fungi behind these diseases deliver molecules known as effectors inside plant cells, which manipulate the plants' biology and help the fungus to invade the plants' tissues. In response, some plants have evolved immune receptors encoded by so-called R genes (short for resistance genes) that detect the effectors inside the plant cell and trigger an immune response. The response often kills the plant cell and those nearby to limit the spread of the fungus. Effectors that are recognized by host immune receptors are termed avirulence effectors (or AVRs for short).
Scientists tend to assume that most effectors do not bind directly to their immune receptors. Instead, it is thought that the immune receptors are more likely to be detecting a change in some other plant protein that is caused by the effectors' activities.
In barley populations, one R gene that protects against powdery mildew encodes an immune receptor known as MLA. Different plants can carry subtly different versions of this R gene meaning that they make similar but different variants of the same receptor. Each MLA variant confers immunity only to strains of powdery mildew that carry the matching AVR effector. A few AVR effectors from powdery mildews have been identified, but most AVR effectors from powdery mildews remain unknown.
Saur et al. looked for new AVR effectors from powdery mildew fungi collected in the field, and found four that were recognized by barley plants carrying MLA variants. Two of these new effectors were fairly similar to each other, but they were all unlike those that had been identified previously.
When Saur et al. engineered barley cells to make these new AVRs alongside their matching MLA receptors, the cells died – which is consistent with the expected immune response. Similar experiments with distantly related tobacco plants agave the same results. This suggested that the immune receptors did not need any other barley proteins to recognize the effectors, indicating that the interaction between the two may be direct. Indeed, two other techniques that test for direct protein-protein interactions, – one that involved extracts from tobacco leaves, and another that involved yeast, – gave results consistent with a direct interaction between the MLA receptor variants and the fungal effectors.
Plant disease is still a major cause of loss of yield in crops. Transferring an R gene from one plant species to another is a potentially powerful approach to help crops resist disease. The discovery that multiple variants of the same resistance gene can bind to dissimilar effectors from a disease-causing fungus in distantly related plant species underlines the potential of this approach.
https://doi.org/10.7554/eLife.44471.002Introduction
The NLR family of immune receptors is structurally conserved between animals and plants, perceives non-self and modified-self molecules inside host cells, and mounts potent innate immune responses to terminate microbial pathogenesis (Maekawa et al., 2011a). Animal NLRs are normally activated by conserved microbe- or damage-associated molecular patterns (MAMPs/DAMPs), whereas plant NLRs typically detect strain-specific pathogen effectors, designated avirulence effector proteins (AVRs) (Maekawa et al., 2011a; Jones et al., 2016). Plant NLRs recognize either the effector structure or sense effector-mediated modifications of host proteins (Jones et al., 2016). Single plant NLRs or NLR pairs can also detect the manipulation of effector target mimics, called decoys, and a variation of the latter mechanism involves the direct integration of decoy domains mimicking host targets within NLRs, termed integrated decoy domains (Jones et al., 2016; Cesari et al., 2014; Kroj et al., 2016). The selective forces shaping the evolution of the comparatively small complement of NLRs in vertebrates are incompletely understood (~20 family members, (Lange et al., 2011; Meunier and Broz, 2017)), but in plant species co-evolution with host-adapted pathogens has strongly influenced the expansion and diversification of NLR repertoires (Jacob et al., 2013; Meyers et al., 2005).
Plant disease resistance (R) genes to host-adapted pathogens often encode NLRs, are frequently members of large gene families organized in complex clusters of paralogous genes, and can rapidly evolve through a range of natural gene diversification mechanisms (Jacob et al., 2013; Meyers et al., 2005). There are several examples of allelic series of NLR-type R genes known in plants (Ellis et al., 1999; Allen et al., 2004; Srichumpa et al., 2005; Seeholzer et al., 2010; Kanzaki et al., 2012). In these cases, multiple distinct recognition specificities evolved in the host population at a single R gene with each allele detecting a corresponding strain-specific AVR in the pathogen population. Such multi-allelic NLR-type R genes are particularly instructive for studying the underlying co-evolutionary process between host and pathogen.
Ascomycete powdery mildews are widespread pathogens of thousands of angiosperm plant species in temperate climates, including economically relevant crops (Glawe, 2008). They are obligate biotrophic pathogens, meaning that their growth and reproduction is entirely dependent on living host cells. The haploid barley powdery mildew pathogen Blumeria graminis forma specialis hordei (Bgh) multiplies mainly clonally and is a member of the species Blumeria graminis that is specialized for its host plant barley (Hordeum vulgare). There are various specialized forms (formae speciales or fs. spp.) of B. graminis, each of which is capable of infecting the respective host plant species belonging to the grass (Poaceae) family, including cereals such as barley and wheat (Wyand and Brown, 2003). Within each forma specialis, numerous isolates (strains) can be distinguished in the pathogen population, based on their respective virulence or avirulence infection phenotypes vis-à-vis particular genotypes of the host population (Lu et al., 2016). The genomes of powdery mildews are characterized by the loss of several, otherwise widely conserved Ascomycete genes with functions related to carbohydrate degradation and primary and secondary metabolism (Spanu et al., 2010; Wicker et al., 2013), and this is believed to explain their strict dependence on living plant cells. Similar to other filamentous phytopathogens, grass-infecting powdery mildew genomes harbor hundreds of candidate secreted effector protein (CSEP)-coding genes, which are assumed to contribute to fungal pathogenesis (Wicker et al., 2013; Pedersen et al., 2012; Guttman et al., 2014). Pathogen effectors often work by subverting innate immune responses, thereby facilitating host colonization and disease (Rovenich et al., 2014).
Domesticated barley and wheat contain numerous powdery mildew R gene loci that were often introgressed from their corresponding wild relatives (Jørgensen and Wolfe, 1994; Lutz et al., 1995; Maekawa et al., 2019). In both barley and its close relative wheat, one of these powdery mildew R loci, designated mildew locus a (Mla) and powdery mildew 3 (Pm3), respectively, has been subject to exceptional functional diversification, resulting in large numbers of Mla or Pm3 recognition specificities (Seeholzer et al., 2010; Lutz et al., 1995; Bhullar et al., 2010). Although wheat Pm3 and barley Mla loci each span a cluster of NLR genes, known Pm3 and Mla recognition specificities to the B. graminis pathogen appear to have arisen from allelic diversification of a single NLR gene in the corresponding NLR clusters (Seeholzer et al., 2010; Maekawa et al., 2019; Shen, 2003). Isolate-specific disease resistance to Bgh mediated by MLA receptors is invariably linked to the activation of localized host cell death, and this immune response likely terminates growth of the biotrophic pathogen by shutting off its nutrient supply (Boyd et al., 1995). Of note, the Mla orthologs Sr33 in wheat (Periyannan et al., 2013) and Sr50 in rye (Mago et al., 2015) confer disease resistance to the stem rust pathogen Puccinia graminis f. sp. tritici (Pgt) isolate Ug99, a major threat to global wheat production. Pgt and Bgh belong to the Basidiomycota and Ascomycota phyla, respectively, indicating that MLA receptors can detect the presence of independently evolved avirulence effectors. Barley Mla and wheat Pm3 both encode intracellular NLRs with an N-terminal coiled-coil (CC) domain but lack significant sequence relatedness (Zhou et al., 2001), whereas 23 allelic barley MLA resistance proteins exhibit >91% amino acid (aa) sequence identity (Seeholzer et al., 2010), and 17 deduced allelic wheat Pm3 receptors share >97% aa sequence identity (Bhullar et al., 2010). Diversifying selection among resistance alleles of Mla and Pm3 is largely confined to regions encoding the C-terminal leucine-rich repeats (LRR) of the receptors (Seeholzer et al., 2010; Maekawa et al., 2019; Bhullar et al., 2010). The polymorphic MLA LRR is critical for effector detection as shown by a series of reciprocal domain swaps between MLA1 and MLA6 (Shen, 2003). Barley Mla1 confers race-specific disease resistance to a Bgh isolate carrying the cognate avirulence gene AVRa1 in transgenic Arabidopsis thaliana, suggesting ∼ 150 million years of evolutionary conservation of the underlying immune mechanism and potentially pointing to a direct recognition mechanism of AVRA effectors that is highly conserved between barley and A. thaliana (Maekawa et al., 2012).
Recently, the Bgt avirulence gene AvrPm3a2/f2, recognized by the wheat Pm3a and Pm3f alleles, was identified by a map-based cloning approach and found to encode a typical CSEP that belongs to an effector family of 24 members (Bourras et al., 2015). The clonal nature of the haploid Bgh pathogen facilitated the identification of isolate-specific sequence variation in the transcriptomes of a global collection of 17 Bgh strains. These were associated with Bgh pathotypes on Mla1- and Mla13-containing near-isogenic barley lines to identify sequence-unrelated AVRa1- and AVRa13-encoding CSEPs (Lu et al., 2016). However, it remains unclear whether these powdery mildew avirulence effectors bind directly to the corresponding NLR receptors or whether the receptors sense the presence of the pathogen indirectly through effector-mediated modifications of host proteins.
To reveal the molecular mechanism underlying the co-evolutionary diversification of barley MLA NLRs with host-adapted Bgh, we first described the natural AVRa gene diversity in a local Bgh population. This allowed us to isolate four additional AVRA effectors by applying a transcriptome-wide association study (TWAS) with high-confidence single nucleotide polymorphisms (SNPs) underlying disease resistance/susceptibility phenotypes in barley. We then employed co-transfection experiments to show that only matching Mla-AVRa pairs activate cell death in barley leaf protoplasts. Agrobacterium tumefaciens-mediated co-delivery of Mla-AVRa pairs in Nicotiana benthamiana leaves demonstrated that their co-expression is sufficient to trigger cell death in this heterologous plant species. The AVRa avirulence effectors encode CSEPs that are sequence-unrelated except for allelic AVRA10 and AVRA22. These findings are inconsistent with a co-evolutionary arms race model describing iterative cycles of MLA receptor and AVRA effector adaptations. Moreover, we failed to detect cell death activity for the previously reported EKA_AVRa10, a member of the EKA gene family that is derived from part of a class-1 LINE retrotransposon (Ridout, 2006; Amselem et al., 2015). Using co-expression experiments with matching Mla-AVRa pairs, including previously reported AVRa13, we present evidence for direct receptor-avirulence effector interactions. Our findings imply that MLA receptors have an exceptional propensity to directly detect sequence-unrelated and likely structurally diverse pathogen effectors and that this feature might have facilitated the functional diversification of the receptor in the host population.
Results
Natural variation of AVRa genes within a local Bgh population
In 2016 we collected 13 Bgh field isolates from leaves of barley plants within the same locality near Cologne, Germany (GPS 5˚57′N, 6˚51′E 5; isolates designated K2, K3, K4, S11, S15, S16, S19, S20, S21, S22, S23, S25 and S26). We purified the isolates by serial propagation of conidiospores collected from single powdery mildew colonies (Materials and methods). Then, we determined their pathotypes (virulence/avirulence profiles) together with a global collection of 17 previously described Bgh isolates (Lu et al., 2016) on a panel of near-isogenic barley lines (NILs) in the cultivar (cv.) Pallas genetic background (Kolster et al., 1986) carrying Mla1 (P01), Mla3 (P02), Mla6 (P03), Mla7 (P04B), Mla9 (P08B), Mla10 (P09), Mla12 (P10), Mla22 (P12), or Mla23 (P13) recognition specificities (Supplementary file 1). To assess infection types (ITs) independently, we additionally employed a panel of NILs in the genetic background of cv. Manchuria (Moseman, 1972) carrying introgressed Mla1 (CI16137), Mla6 (CI16151), Mla7 (CI16147 and CI16153), Mla10 (CI16149), or Mla13 (CI16155) (Supplementary file 1). Similar to previous studies (Seeholzer et al., 2010; Lu et al., 2016), we scored five macroscopically distinguishable ITs at 9 d after conidiospore inoculation on leaves, ranging from 1 to 3 (=avirulent) or 4 to 5 (=virulent). Unambiguous assignment of virulent and avirulent interactions was possible for all isolates on Mla1, Mla6, Mla9, Mla12, Mla13, and Mla22 NILs that displayed only IT 1, 2 or 5. A subset of the Bgh strains displayed IT 3 (avirulent) or 4 (virulent) on Mla3, Mla7, Mla10, and Mla23 NILs, which were in some cases difficult to distinguish from each other and thus potentially complicating the discrimination of avirulent and virulent interactions with our experimental setup (Supplementary file 1). For two of the tested Bgh isolates different avirulent/virulent interactions were recorded between Pallas and Manchuria NILs carrying Mla7 (isolates K4 and S22) and, similarly, two other isolates exhibited different avirulent/virulent interactions on Pallas and Manchuria NILs carrying Mla10 (isolates CC88 and S20; Supplementary file 1). These few discrepancies of avirulent/virulent interactions might be explained by differences in the size of introgressed chromosomal segments from the respective donor lines in Pallas and Manchuria NILs that might contain other R genes besides Mla. Therefore, we omitted the IT data of isolates K4 and S22 on Mla7 NILs and of CC88 and S20 isolates on Mla10 NILs in the TWAS (see below) to identify AVRa7 and AVRa10 candidates, respectively. Among the 13 newly described Bgh isolates collected from the same geographic site, isolates S19 and S21, as well as S23, S25, and S26 exhibited identical avirulence/virulence patterns on the NIL test panel (Supplementary file 1). Thus, 10 out of 13 isolates showed unique avirulece/virulence patterns and each isolate carries at least five different AVRa genes (Supplementary file 1). These findings point to a high level of genetic variation among AVRa genes within the local Bgh population.
We obtained deep fungal transcriptomes (RNA-Seq) for the ten Cologne-derived field isolates with unique avirulent/virulent patterns during pathogenesis on susceptible barley leaves at 16 and 48 hr post conidiospore inoculation, resulting in 0.8 to 6 million sequenced and mapped Bgh read pairs (fragments) per sample (Materials and methods). RNA-seq reads of these isolates, combined with RNA-seq reads of the previously characterized global collection of Bgh isolates (Lu et al., 2016) and of reference isolate DH14, were aligned against the recently assembled near chromosome-level DH14 genome supplemented with updated gene models (Frantzeskakis et al., 2018). Subsequently, a collective set of sequence polymorphisms for all isolates was identified from the combined alignment data. To assess the potential influence of population structure among the isolates prior to TWAS, we performed a population structure analysis, for which we extracted a set of high-quality diallelic and synonymous single-nucleotide polymorphisms (SNPs) from the complete set of polymorphisms (Methods). A principal component analysis (PCA) plot and a neighbour-joining tree including all isolates confirmed the Japanese isolate RACE1 to be exceptionally divergent and assigned the three Australian isolates to a distinct clade, whereas the ten new field isolates clustered mostly together with other European, Japanese, and USA isolates (Figure 1A, Figure 1—figure supplement 1). Although all newly described isolates were collected from a single geographic location, three of these (K2, K3, and S16) appear to form a subgroup that is distinct from the other European isolates (Figure 1A, Figure 1—figure supplement 1). Interestingly, the avirulence profiles of isolates K3 and S16 on the 11 tested Mla barley recognition specificities belong to distinct pathotype clusters (Figure 1B), despite an exceptionally high similarity of K3 and S16 at the transcriptome level (Figure 1A).
Figure 1 with 1 supplement see all
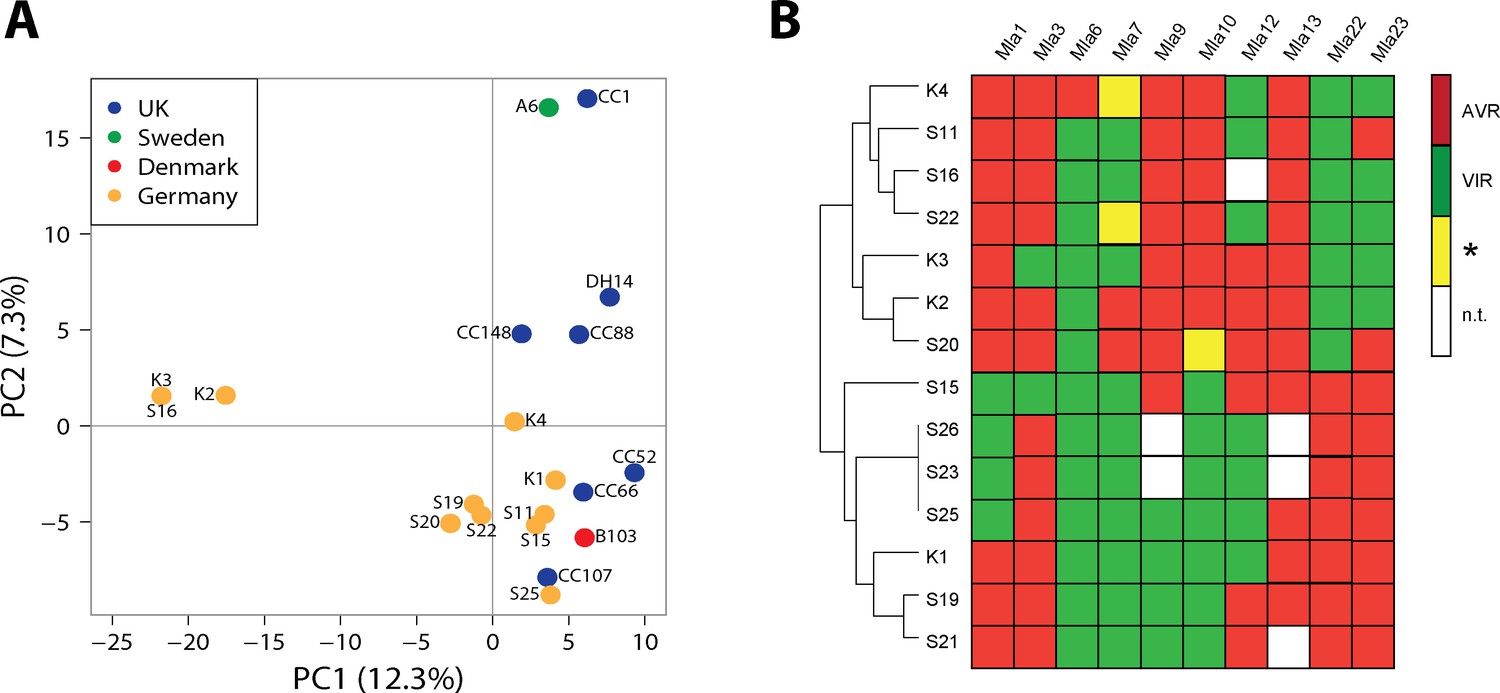
Population structure and avirulence profiles of Blumeria graminis f. sp. hordei (Bgh) isolates.
(A) Principal Component Analysis (PCA) of the indicated European Bgh isolates, including ten newly collected strains from a local pathogen population in Germany, based on a set of 5170 diallelic high-quality synonymous SNPs. (B) Hierarchical clustering (R package 'pheatmap') of avirulence profiles from 14 Bgh isolates collected within the same area near Cologne, Germany (GPS 5˚57’N, 6˚51’E 5). Numbers correspond to infection types (ITs) 1, 2 and 3 = avirulent, red; infection types 4 and 5 = virulent, green. *denotes differences of ITs between cultivars Pallas and Manchuria, yellow. n.t.: not tested, white.
Combined TWAS and Bgh genome analysis identified candidates for AVRa7, AVRa9, AVRa10, and AVRa22
Our TWAS analysis (Supplementary file 2 - 4) allowed us to identify several AVRa candidates. Manual re-inspection of these candidates (Lu et al., 2016), revealed that only transcript polymorphisms of the top candidates AVRa7, AVRa9, AVRa10, and AVRa22 exhibit tight linkage to the avirulence phenotypes of our Bgh strain panel on Mla7, Mla9, Mla10, and Mla22 NILs, respectively. Two genes, BLGH_06689 and BLGH_06672, encoding two identical copies of CSEP0059, were identified as top-ranking candidates for AVRa7 (p=1.78E-05, Supplementary file 4). This is consistent with previous reports in which two AVRa7 loci were inferred based on segregation analysis among the progeny of crosses between Bgh strains (Skamnioti et al., 2008; Pedersen et al., 2002). In the near chromosome-level genome assembly of isolate DH14 (Frantzeskakis et al., 2018) the two CSEP0059 copies are physically separated by 141 kb on scaffold 44 and, together with four other CSEPs, form a CSEP cluster in this region (Figure 2). We designated the two identical CSEP0059 copies in DH14 as candidate gene AVRa7-1 (Figure 2A). One of the DH14 CSEPs in the cluster is a CSEP0059 paralog, CSEP060 (BLGH_06671), which at the protein level shares 77% aa identity with CSEP0059 (Figure 2B and C). All virulent strains on Mla7 NILs carry a SNP in CSEP0059, which results in a L51P substitution in the deduced candidate effector protein, and the corresponding variant was thus named AVRA7-V1 (Figure 2A and B). Interestingly, only about half of the transcripts from the virulent isolate B103 harbor a SNP leading to the L51P substitution in the deduced protein. The other half of the B103 transcripts harbor a SNP resulting in a L28F substitution in the deduced protein (designated AVRA7-V2), suggesting that in this strain the two CSEP0059 copies differ. Similarly, approximately half of the transcript reads of all three avirulent Australian isolates (Art, Aby, and Will) harbor a SNP leading to an I65T substitution in the deduced protein (designated AVRA7-AUS; Figure 2A and B), suggesting that these isolates also contain an even number of non-identical CSEP0059 copies. Finally, the most divergent Bgh isolate RACE1 is clearly avirulent on Mla7 NILs (Supplementary file 1), and in its transcriptome we found two non-synonymous SNPs in all CSEP0059 transcripts corresponding to S47R and A96V aa substitutions in the deduced protein (designated AVRA7-2; Figure 2A and B). Inspection of the near chromosome-level RACE1 genome (Frantzeskakis et al., 2018) revealed two identical CSEP0059 gene copies (BGHR1_17217 and BGHR1_17236) separated by 151 kb on a single contig within a CSEP cluster that is largely syntenic with the corresponding DH14 CSEP cluster (Figure 2C; Frantzeskakis et al., 2018). RACE1 CSEP0060 (BGHR1_16533) is located on the neighbouring contig tig00005324 and is possibly syntenic to the CSEP0060 location in DH14. Additionally, RACE1 harbors a third CSEP0059 copy (BGHR1_17237) with S5N, M9T, T11A, L12W, L17F, S47R, L51P, N62S, A96V, and R105Q substitutions in the deduced effector polypeptide (designated AVRA7-3) (Figure 2A, B and C). Taken together, TWAS and the comparative genome analysis revealed the potential existence of multiple AVRA7 variants, partly encoded by paralogous genes, and two virulent AVRA7 variants.
Figure 2
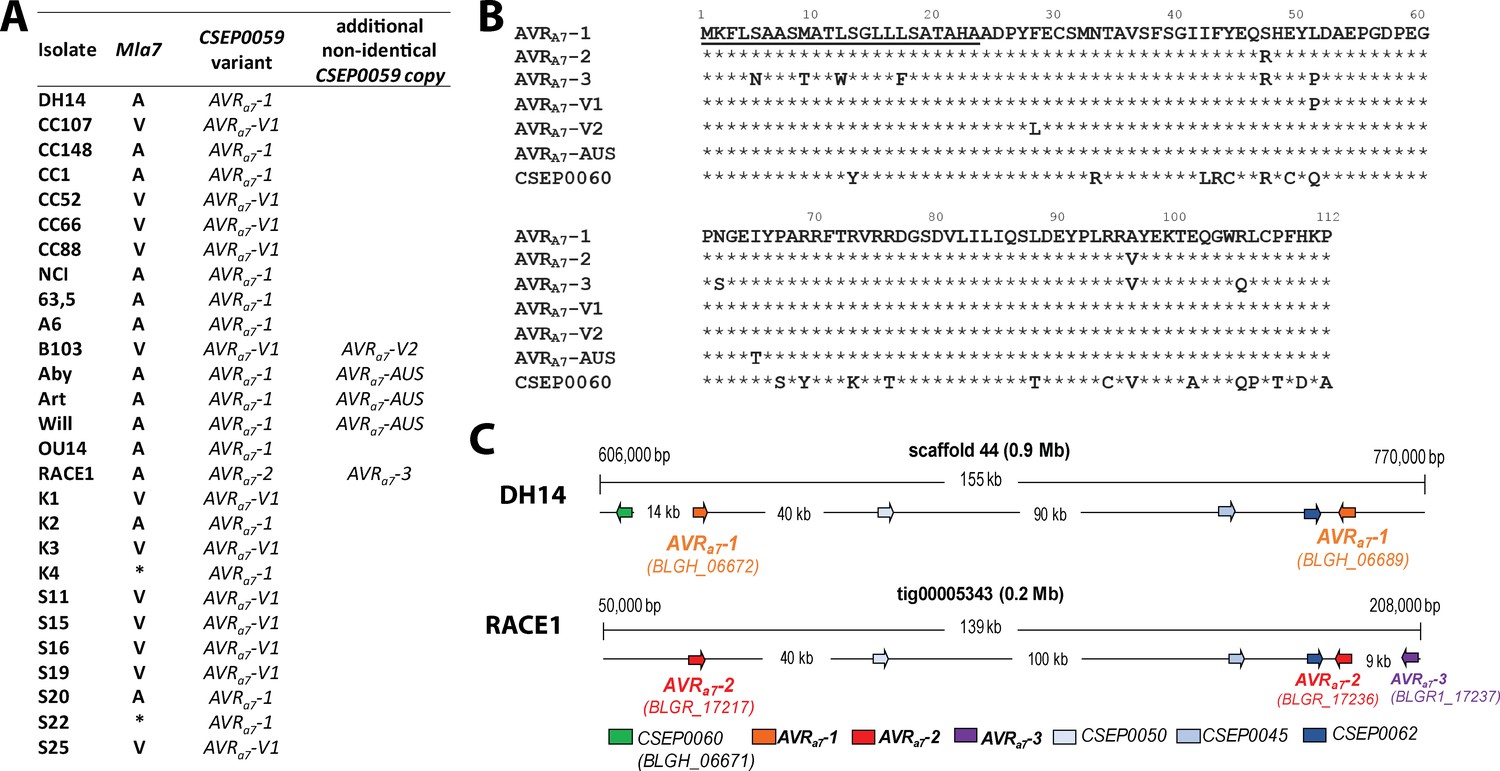
Identification of CSEP0059 as an AVRa7 candidate by association of avirulence profiles with transcript polymorphisms and integration in the physical Bgh map.
(A) AVRa7 transcript variants encoded by the indicated Bgh isolates with corresponding avirulence profiles. (B) Alignment of deduced AVRA7 amino acid sequences with all variants highlighted. (C) Visualization of the chromosomal regions harboring CSEP0059/AVRa7 candidate variants with corresponding gene IDs in the genomes of Bgh isolates DH14 and RACE1. All CSEPs are depicted by arrows. * denotes different infection types between cultivars Pallas and Manchuria.
TWAS identified CSEP0174 (BLGH_04994 in DH14 and BGHR1_10042 in RACE1) as the top-ranking AVRa9 candidate (p=1.84E-05; Supplementary file 4, Figure 3A). Analysis of the transcriptome reads showed that isolates virulent on Mla9 carry AVRa9 variants with either H28D (AVRA9-V1) or G22E (AVRA9-V2) substitutions (Figure 3A and B). Furthermore, we identified CSEP0141 as the top-ranking candidate for both AVRa10 (BGHR1_10013, p=4.50E-07) and AVRa22 (p=1.22E-05) (Supplementary file 4, Figure 3C). All Bgh isolates avirulent on Mla10 plants encode a CSEP0141 variant with an I77F substitution (AVRA10) compared to the deduced effector protein in reference isolate DH14 (BLGH_05021; Figure 3C and D). By contrast, all isolates avirulent on Mla22 plants carry a CSEP0141 variant with 13 deduced aa substitutions (V11A, F13L, D45G, D53E, Q55H, D58N, G59D, Q61P, H64Y, I77Y, V93L, W96L, and I111N; AVRA22 in Figure 3C and D). DH14 is the only tested isolate that is virulent on both Mla10- and Mla22-expressing plants, and we therefore named the CSEP0141 variant encoded by DH14 AVRa10-V/AVRa22-V. CSEP0141 belongs to the CSEP family 64 with CSEP0266 being the only other family member that shares significant similarity with candidates AVRA10 and AVRA22 (59% and 63% identical aa sequences, respectively). Collectively, these data raised the possibility that MLA10 and MLA22 each recognize one of the two major natural variants of CSEP0141 in the Bgh population.
Figure 3 with 1 supplement see all
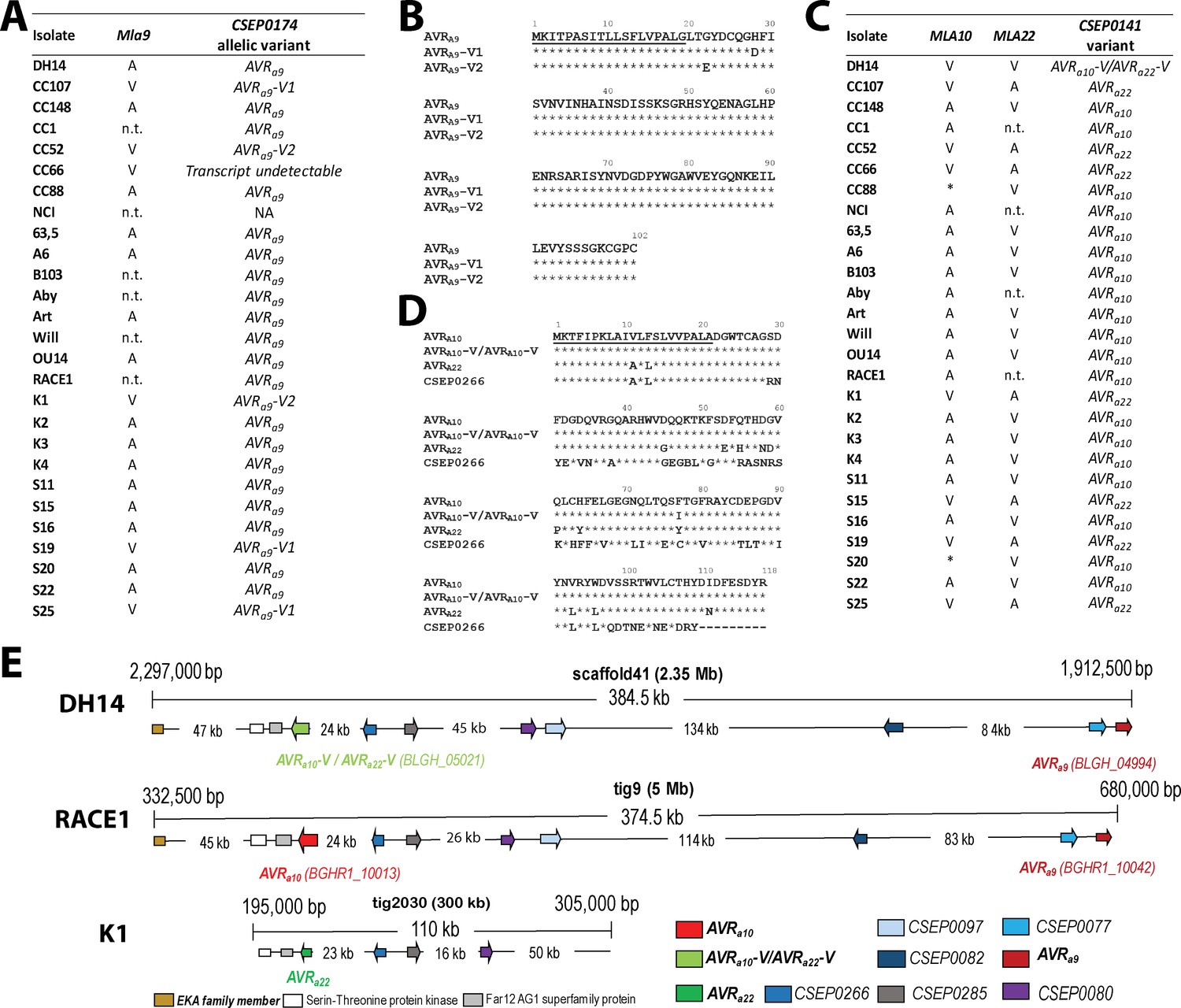
Identification of CSEP0174 as an AVRa9 candidate and CSEP0141 as an AVRa10 and AVRa22 candidate by association of avirulence profiles with transcript polymorphisms as well as integration in the physical Bgh map.
(A) AVRa9 variants encoded by each Bgh isolate with corresponding avirulence profiles. (B) Alignment of deduced AVRA9 amino acid sequences with variants highlighted. (C) AVRa10/AVRa22 variants encoded by each Bgh isolate with corresponding avirulence profiles. (D) Alignment of AVRA10, AVRA22, and AVRA10-V/AVRA22-V amino acid sequences. (E) Visualization of the chromosomal regions harboring CSEP0174/AVRa9 and CSEP0141/AVRa10/AVRa22 candidates with corresponding gene IDs as well as a copy of the EKA family class-1 retrotransposon and other CSEPs in the genomes of Bgh isolates DH14, RACE1, and K1. *denotes differences of infection types between cultivars Pallas and Manchuria. n.t.: not tested.
AVRa9 and AVRa10/AVRa22 candidates are each single-copy genes in both DH14 and RACE1 Bgh genomes (Frantzeskakis et al., 2018). Notably, candidates AVRa10-V/AVRa22-V (BLGH_05021) in DH14 and AVRa10 (BGHR1_10013) in RACE1 physically locate approximately 300 kb away from the 3’ end of AVRa9 (BLGH_04994 and BGHR1_10042) on the respective contiguous chromosomal DNA stretches (Figure 3E). The physical linkage of these candidate avirulence genes is consistent with a tight genetic coupling of the AVRa10 and AVRa22loci and their linkage to the AVRa9 locus (Skamnioti et al., 2008). In addition, we found the transcript of candidate AVRa22 to map to a physical contig in the Bgh K1 genome assembly (tig19704; Hacquard et al., 2013) that is syntenic with the genomic region encompassing candidate AVRa10-V/AVRa22-V in DH14 and candidate AVRa10 in RACE1 (Figure 3E). The findings lend further support to the notion that AVRa10 and AVRa22 represent two alleles of a single Bgh effector gene (Figure 3—figure supplement 1). Candidate AVRa10, which encodes a CSEP0141 variant, maps only 24 kb away from the 3’ end of the only other family member CSEP0226 (BLGH_05020 and BGHR1_10014 in DH14 and RACE1 genomes, respectively) and approximately 60 kb away from the 5’ end of one of the EKA class-1 LINE retrotransposon family members (Figure 3E, Amselem et al., 2015). Reminiscent of the AVRa7-containing cluster of sequence-unrelated effector genes in the Bgh genome (Figure 2c), the genomic region encompassing CSEP0174 (AVRa9) and CSEP0141 (AVRa10/AVRa22) contains sequence-unrelated CSEP0077, CSEP0080, CSEP0082, CSEP0085, CSEP0097, and CSEP0266, and therefore represents another cluster of candidate secreted effector genes in the fungal genome.
Functional analysis of AVRa candidates in barley leaf protoplasts
We examined the capability of the identified AVRA candidates to trigger MLA-dependent cell death upon transient co-expression with matching MLA receptors in barley leaf protoplasts. All gene constructs were expressed from the Zea mays ubiquitin promotor together with the firefly luciferase (LUC) reporter gene (Lu et al., 2016), and relative LUC activity was quantified as a proxy for protoplast viability. As epitope tag sequences can interfere with signal-noise ratios of LUC activity in this assay (Lu et al., 2016), we refrained from fusion of constructs with epitope sequences.
When compared to the empty vector (EV) control, co-expression of previously reported AVRa1(Lu et al., 2016) with Mla1 reduced LUC activity in a Mla1-dependent manner by 71% (Figure 4A) and co-expression of AVRa7-1 with Mla7 significantly reduced the reporter activity by 37% but not when co-expressed with Mla1 (Figure 4A). Interestingly, co-expression of Mla7 with either of the two AVRa7 variants present in RACE1, AVRa7-2 and AVRa7-3, resulted in a 91% and 79% reduction of LUC activity, respectively, whereas their co-expression with Mla1 did not significantly alter reporter activity (Figure 4A, Figure 4—figure supplement 1A). Taken together these data indicate that naturally occurring AVRa7variants in the Bgh population differ in their propensity to activate MLA7-mediated cell death. Neither expression of AVRa7-V1 (CSEP0059) nor CSEP0060, the paralog located adjacent to AVRa7-1 in the DH14 genome (Figure 3C), resulted in a significant Mla7-dependent reduction in reporter activity (Figure 4A, Figure 4—figure supplement 1B). We detected a 30% reduction in LUC activity when AVRa7-AUS was co-expressed with Mla7 but not when co-expressed with Mla1. Co-expression of AVRa7-V2 with Mla7 did not result in significantly reduced LUC activity when compared to co-expression with Mla1 (Figure 4—figure supplement 1A). Barley accessions expressing the MLA7 polypeptide sequences CI1647 and CI1653 from Manchuria MLA7 NILs were found to differ slightly in two independent earlier studies (MLA7_AAQ55540, Halterman and Wise, 2004; MLA7 Seeholzer et al., 2010; Figure 4—figure supplement 1B). Therefore, we also tested the capability of MLA7_AAQ55540 to recognize AVRa7 and found that co-expression of MLA7_AAQ55540 with AVRa7-2 reduced LUC activity by only 68% (as compared to 91% when co-expressed with Mla7). Luciferase activity in protoplasts co-expressing MLA7_AAQ55540 and AVRa7-1 (30% LUC reduction compared to EV) did not differ significantly from protoplasts co-expressing MLA7_AAQ55540 and AVRa7-V1 (15% LUC reduction compared to EV; Figure 4—figure supplement 1C).
Figure 4 with 2 supplements see all
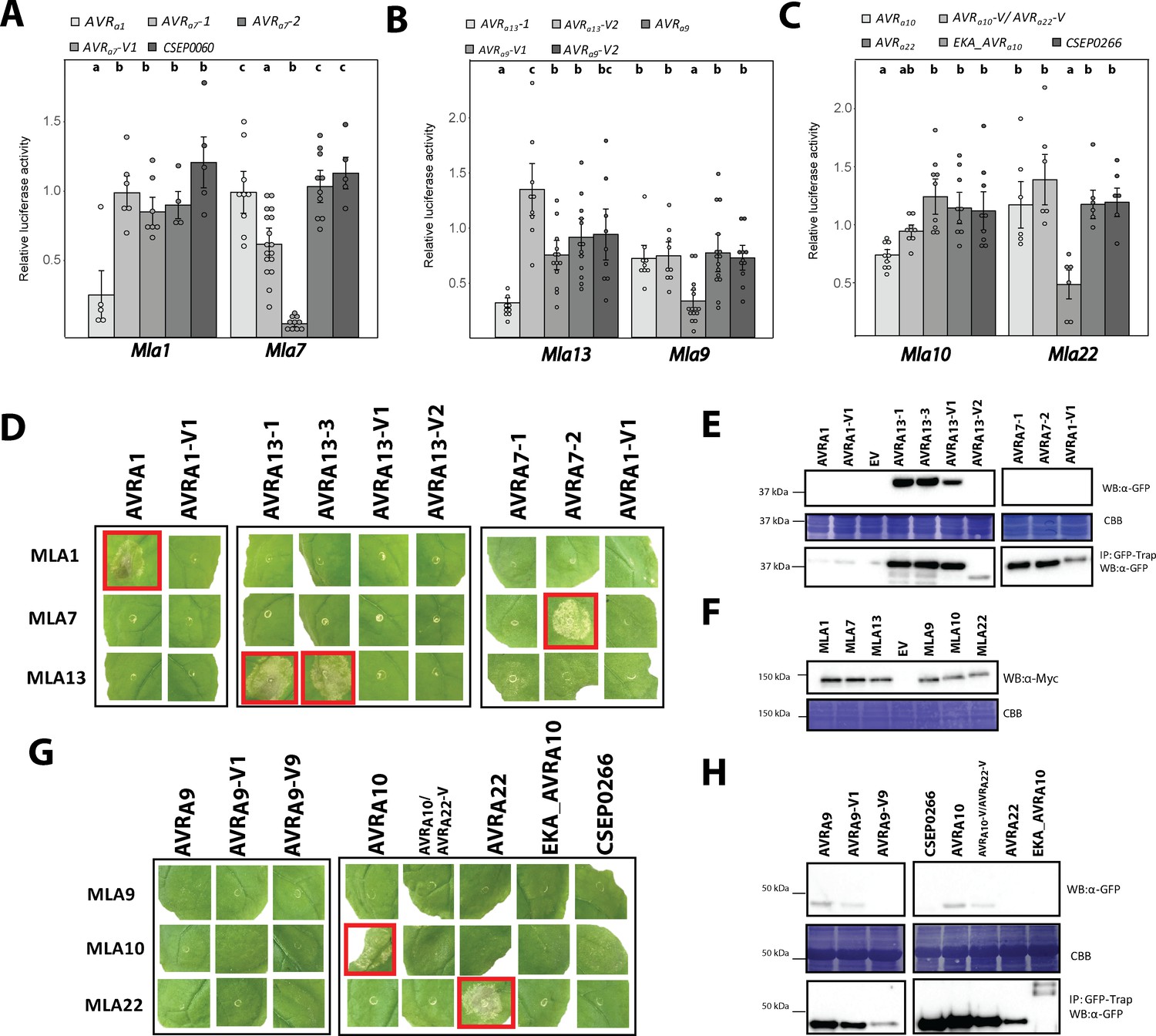
AVRA candidates induce MLA-specified cell death in transient gene expression assays.
(A-C) Barley protoplasts were transfected with pUBQ:luciferase and either an piPKb002 empty vector control (EV) or piPKb002 containing cDNAs of (A) AVRa1, AVRa7 variants and CSEP0060, all lacking their respective signal peptides (SPs) together with either Mla1 or Mla7; (B) AVRa9 variants and AVRa13 variants, all lacking their respective SPs together with either Mla9 or Mla13; (C) AVRa10, AVRa10-V/AVRa22-V, AVRa22, and CSEP0266 without SPs and EKA_AVRa10, together with either Mla10 or Mla22. Luciferase activity was determined ~16 hr post transfection as a proxy for cell death and normalized for each Mla construct by setting the detected luminescence for the corresponding EV transfection to 1. All values obtained in at least four independent experiments are indicated by dots, error bars = standard deviation. Differences between samples were assessed by analysis of variance (ANOVA) and subsequent Tukey post hoc tests for each Mla construct. Calculated P values were as follows: Mla7: p=1.2e-13 and Mla1: p=1.9e-03 (A); Mla13: p=3.2e-06 and Mla9: p=1.3e-04 (B); Mla10: p=4.2e-04 and Mla22: p=5.5e-04 (C). Samples marked by identical letters in the plots do not differ significantly (p<0.05) in the Tukey test for the corresponding transfected Mla. (D–H) Nicotiana benthamiana plants were transformed transiently as indicated. cDNAs lacking stop codons were fused in between the 35S promotor sequence and 4xMyc (Mla variants) or mYFP (CSEPs and AVRa variants lacking SPs and EKA_AVRa10) epitope sequences. Cell death (D, G) was determined three days post transformation and figures shown are representatives of at least three independently performed experiments with at least three transformations per experiment. Protein levels (E, F, H) of MLA-4xMyc, CSEP-mYFP, AVRA-mYFP and EKA_AVRA-mYFP in Nicotiana benthamiana corresponding to constructs of D and G. Leaf tissue was harvested two days post infiltration. Total protein was extracted and recovered by GFP-Trap pull-down as indicated. Extracts and immunoprecipitates were separated by gel electrophoresis and probed by anti-Myc or anti-GFP western blotting (WB) as indicated. IP: Immunoprecipitated fraction. CBB: Coomassie brilliant blue.
-
Figure 4—source data 1
Data points indicating relative luciferase activity of Figure 4A.
- https://doi.org/10.7554/eLife.44471.011
-
Figure 4—source data 2
Data points indicating relative luciferase activity of Figure 4B.
- https://doi.org/10.7554/eLife.44471.012
-
Figure 4—source data 3
Data points indicating relative luciferase activity of Figure 4C.
- https://doi.org/10.7554/eLife.44471.013
Co-expression of Mla9 with the AVRa9 candidate, but not its variants AVRa9-V1 and AVRa9-V2 or AVRa13-1 and AVRa13-V2 (Lu et al., 2016), resulted in a significant 67% relative reduction of LUC activity. This cell death activity was specific to Mla9 as co-expression of AVRa9 with Mla13 did not significantly reduce LUC activity (Figure 4B). Co-expression of the previously reported AVRa13 (Lu et al., 2016) with Mla13 reduced LUC activity in a Mla13-dependent manner by 69% (Figure 4B).
Similar to the moderate reduction of LUC activity when AVRa7-1 was co-expressed with Mla7 (Figure 4A), co-expression of the AVRa10 candidate with Mla10 reduced the reporter activity by only 25% (Figure 4C). LUC reduction was not observed when AVRa10-V/AVRa22-V, AVRa22, CSEP0266, or EKA_AVRa10 were co-expressed with Mla10. Co-expression of EKA_AVRa10, AVRa10, and Mla10 had no impact on LUC reduction when compared to co-expression of AVRa10 and Mla10 alone (Figure 4—figure supplement 2D). Co-expression of Mla22 with AVRa22, but not AVRa10, AVRa10-V/AVRa22-V, CSEP0266, or EKA_AVRa10, reduced relative LUC activity by 53% when compared to co-expression of Mla22 with an EV control (Figure 4C). These findings provide functional evidence that CSEP0141 alleles define AVRa10 and AVRa22 avirulence effectors and are specifically recognized by allelic MLA10 and MLA22 receptors, respectively.
Co-expression of matching Mla and AVRa pairs is necessary and sufficient to trigger cell death in N. benthamiana
Next we tested whether Agrobacterium tumefaciens-mediated delivery and co-expression of matching Mla and AVRa pairs can trigger cell death in heterologous N. benthamiana leaves. In addition to newly isolated AVRA7, AVRA9 and AVRA10/AVRA22, we included the previously reported AVRA1 and AVRA13 variants as additional specificity controls in these experiments (Figure 4D–4H) (Lu et al., 2016). For protein expression and stability analysis in this heterologous system, the constructs were designed to express C-terminally 4xMyc-tagged MLA receptors and C-terminally mYFP-tagged AVRA variants without signal peptide sequences.
Delivery of the AVRa1-mYFP construct but not the construct for its virulent variant, AVRa1-V1-mYFP, conferred cell death in N. benthamiana when co-expressed with Mla1-4xMyc, but not when co-expressed with Mla7-4xMyc or Mla13-4xMyc (Figure 4D, Figure 4—figure supplement 2). We also observed statistically significant cell death intensity when AVRa13-1-mYFP and AVRa13-3-mYFP, but not the virulent variants AVRa13-V1-mYFP or AVRa13-V2-mYFP when co-expressed with Mla13-4xMyc (Figure 4D; Figure 4—figure supplement 2). Cell death was not seen when the same AVRa effector constructs were co-expressed with Mla1-4xMyc or Mla7-4xMyc (Figure 4D; Figure 4—figure supplement 2), indicating retained recognition specificity of MLA1 and MLA13 receptors in this heterologous plant species, respectively.
AVRa7-1 mediates moderately reduced LUC reporter activity in barley protoplasts when co-expressed with Mla7, whereas AVRa7-2 expression leads to a strong reduction of reporter activity in the same experiment (Figure 4A). Correspondingly, in N. benthamiana we observed MLA7-dependent cell death only when expressing AVRa7-2-mYFP together with Mla7-4xMyc, but not when Mla7-4xMyc was co-expressed with AVRa7-1-mYFP or AVRa7-V1-mYFP variants (Figure 4D, Figure 4—figure supplement 2). The lack of AVRA7-1 - MLA7 cell death in N. benthamiana is not due to AVRA7-1-mYFP protein stability (Figure 4E) but may be due to other unknown aspects of the heterologous system.
Major differences in protein steady-state levels were found between individual AVRA effectors, whereas protein levels of all MLA receptors were comparable in α-Myc western blots (Figure 4E and F). mYFP-tagged AVRA1 variants were barely detectable even after enrichment by GFP-Trap (Figure 4E), whilst AVRA13-1, AVRA13-3, and AVRA13-V2 were detectable in N. benthamiana extracts without GFP-Trap pull-down (Figure 4E). AVRA13-V1-mYFP protein was barely detectable even after GFP-Trap enrichment (Figure 4E) suggesting that loss of MLA13-mediated cell death activity for AVRA13-V1 may be due to protein instability. Notably, we found a Western blot signal corresponding to the expected size of ~40 kDa for the AVRA13-V2-mYFP fusion protein (Figure 4E). This differs from the expression of FLAG-AVRa13-V2 in barley protoplasts, where most of the FLAG-AVRA13-V2 fusion protein was visible as a cleaved protein product (Lu et al., 2016). mYFP-tagged AVRA7 variants were only detectable in N. benthamiana leaf extracts after GFP-Trap pull-downs (Figure 4E).
AVRa9 elicited a 67% reduction in LUC activity when co-expressed with Mla9 in barley protoplasts (Figure 4B). Surprisingly however, in N. benthamiana, neither AVRA9-mYFP nor its virulent variants AVRA9-V1-mYFP or AVRA9-V2-mYFP triggered MLA9-dependent cell death (Figure 4G; Figure 4—figure supplement 2) although mYFP-tagged AVRA9 was detectable in N. benthamiana protein extracts (Figure 4H). We postulate either that a third barley protein other than MLA9 and AVRA9 is needed for MLA9-mediated cell death activation in N. benthamiana or that the chosen combination of epitope tags prevents effective AVRA9 recognition in this plant species.
Although AVRa10 reduced LUC reporter activity in barley protoplasts only moderately when co-expressed with Mla10 (Figure 4C), we detected a clearly visible cell death response when AVRa10-mYFP was co-expressed with Mla10-4xMyc in N. benthamiana leaves in multiple independent experiments (Figure 4G; Figure 4—figure supplement 2). The MLA10-triggered cell death specifically required the presence of AVRA10-mYFP because leaf cells remained alive upon co-expression with AVRA10-V/AVRA22-V-mYFP or AVRA22-mYFP (Figure 4G; Figure 4—figure supplement 2). Consistent with data obtained with barley leaf protoplasts, cell death in heterologous N. benthamiana was induced upon co-expression of Mla22-4xMyc with AVRa22-mYFP, but not AVRa10-mYFP or AVRa10/-V/AVRa22-V-mYFP (Figure 4G; Figure 4—figure supplement 2). Furthermore, transient expression of CSEP0266-mYFP or EKA_AVRa10-mYFP together with either Mla10-4xMyc or Mla22-4xMyc failed to trigger cell death in N. benthamiana (Figure 4G; Figure 4—figure supplement 2), corroborating our conclusion that allelic AVRA10 and AVRA22 avirulence effectors are specifically recognized by allelic MLA10 and MLA22 receptors, respectively. Only AVRA10-mYFP and AVRA10-V/AVRA22-V-mYFP were detectable in N. benthamiana protein extracts and these two fusion proteins as well as CSEP0266-mYFP and AVRA22-mYFP ran at the expected size of ~40 kDa after enrichment by GFP-Trap pull-downs (Figure 4H). In these experiments the AVRA22-mYFP protein appears less stable than AVRA10-mYFP (Figure 4H). EKA_AVRA10-mYFP is expected to migrate at ~70 kDa. We detected a western blot signal at this expected size and one faster-migrating variant (Figure 4H). In conclusion, we failed to detect cell death activity in response to the previously reported recognition of EKA_AVRa10 by MLA10 (Ridout, 2006). This contrasts with the functional validation of the AVRa10 candidate identified here by TWAS.
Candidate AVRA proteins interact with matching MLA receptors in plant extracts and in yeast
AVRA1 is recognised by MLA1 in barley, A. thaliana and, as shown here, also in N. benthamiana (Figure 4D; Lu et al., 2016). The retention of MLA1-dependent recognition of AVRA1 in three divergent plant families (Triticeae, Brassicaceae, Solanaceae) suggests direct interactions of matching MLA and AVRA pairs or an indirect recognition mechanism involving highly evolutionarily conserved AVRA host target(s). The wheat Mla ortholog Sr50 interacts with its cognate effector AvrSr50 of Puccinia graminis f. sp. tritici in yeast (Chen et al., 2017). Despite the lack of sequence conservation between most of the identified AVRA effectors and AvrSr50, we tested whether barley MLA directly interacts with cognate Bgh effectors. So far it has been impossible to purify large quantities of recombinant full-length MLA receptors for in vitro AVRA-MLA association studies, possibly because of MLA-triggered cell death and receptor oligomerisation (Maekawa et al., 2011b). We thus focused on quantitatively measuring putative AVRA-MLA associations in plant extracts using the highly sensitive split-luciferase (split-LUC) complementation assay (Paulmurugan et al., 2002; Luker et al., 2004). Whereas barley protoplasts can undergo cell death upon expression of matching Mla and AVRa pairs at ~16 hr post transfection (Figure 4A–4C), expression in N. benthamiana leaves permitted MLA and AVRA interaction analysis at two days post A. tumefaciens leaf infiltration and prior to the appearance of macroscopically visible MLA-mediated cell death. To examine MLA and AVRA associations by luciferase activity of protein extracts from A. tumefaciens-infiltrated leaf area, we generated gene constructs in which MLA and AVRA were fused at the C-terminus to cLUC and nLUC, respectively (Mla-cLUC and AVRa-nLUC), and used these for A. tumefaciens-mediated transient gene expression experiments in N. benthamiana leaves (Materials and methods). We focused on AVRa13, AVRa7, and AVRa10/AVRa22 variants and their cognate Mla receptors for split-LUC assays (Figure 5A–5C), because AVRA1 protein levels were barely detectable (Figure 4E), and co-expression of Mla9 and AVRa9 failed to trigger a cell death response in N. benthamiana leaves (Figure 4G).
Figure 5
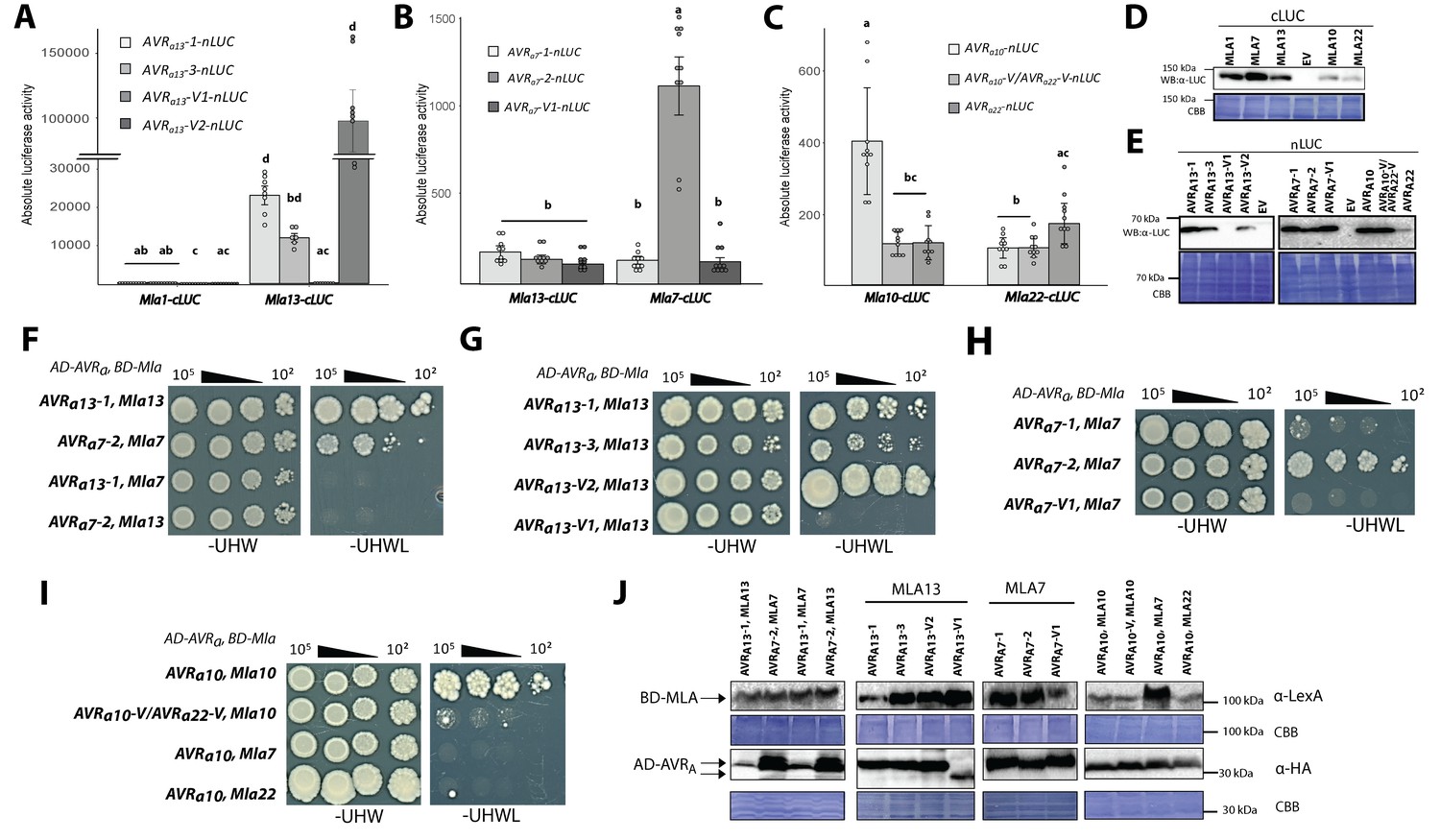
Association of candidate AVRA proteins with MLA in plant extracts (A–E) and in yeast (F–J).
(A-C) Nicotiana benthamiana plants were transformed transiently with constructs encoding (A) Mla1-cLUC or Mla13-cLUC together with cDNAs of AVRa13-1, AVRa13-3, AVRa13-V1, AVRa13-V2 lacking signal peptides (SPs) and fused C-terminally in frame with nLUC, (B) Mla1-cLUC, or Mla7-cLUC together with cDNAs of AVRa7-1, AVRa7-2, and AVRa7-V1 lacking SPs and fused C-terminally in frame with nLUC, (C) Mla10-cLUC or Mla22-cLUC together with cDNAs of AVRa10, AVRa10-V/AVRa22-V, and AVRa22 without SPs fused C-terminally in frame with nLUC, all under the control of the 35S promotor. Luciferase activity was determined two days post transfection. All values obtained in at least six experiments are indicated by dots, error bars = standard deviation. For each graph, differences between samples were assessed using non-paramatric analysis of variance (Kruskal-Wallis) and subsequent Dunn’s post hoc tests. Calculated P values were as follows: (A) p=6.8e-10, (B) p=1.2e-04, (C) p=8.0e-07. Samples marked by identical letters in the plot did not differ significantly (p<0.05) in Dunn’s test. (D–E) Protein levels of MLA-cLUC (D) and AVRA-nLUC (E) variants in Nicotiana benthamiana corresponding to constructs of 5A to 5C. Leaf tissue was harvested two days post infiltration. Total protein was extracted, separated by gel electrophoresis and probed by anti-LUC western blotting (WB). (E–I) Yeast cells were co-transformed with Mla alleles fused N-terminally to the LexA binding domain (BD) and AVRa constructs lacking SPs fused N-terminally to the B42 activation domain (AD) and 1xHA tag as indicated. Growth of transformants was determined on selective growth media containing raffinose and galactose as carbon sources but lacking uracil, histidine and tryptophan (-UHW), and interaction of proteins was determined by leucine reporter activity reflected by growth of yeast on selective media containing raffinose and galactose as carbon sources but lacking uracil, histidine, tryptophan and leucine (-UHWL). Figures shown are representatives of at least three independent experiments with yeast clones obtained from three independent yeast transformation experiments and pictures were taken 12 days after drop out. (J) Protein levels of BD-MLA and AD-AVRA variants corresponding to yeast of Figure 5D–5G. Yeast transformants were grown in glucose containing selective media lacking uracil, tryptophan, and histidine to OD600 = 1. Cells were harvested, total protein extracted, separated by gel electrophoresis, and western blots (WB) were probed with anti-LexA or anti-HA antibodies as indicated.
-
Figure 5—source data 1
Data points indicating absolute luciferase activity of Figure 5A.
- https://doi.org/10.7554/eLife.44471.020
-
Figure 5—source data 2
Data points indicating absolute luciferase activity of Figure 5B.
- https://doi.org/10.7554/eLife.44471.021
-
Figure 5—source data 3
Data points indicating absolute luciferase activity of Figure 5C.
- https://doi.org/10.7554/eLife.44471.022
We detected high LUC activities (>10,000 units) in extracts of leaves expressing Mla13-cLUC together with AVRa13-1-nLUC and AVRa13-3-nLUC but not when Mla13-cLUC was exchanged to Mla1-cLUC (Figure 5A), although the two MLA-cLUC proteins were similarly stable (Figure 5D). This was not the case for samples expressing AVRa13-V1-nLUC, possibly due to lack of detectable AVRa13-V1-nLUC protein (Figure 5E). Unexpectedly, the highest LUC activity was seen when Mla13-cLUC was expressed together with AVRa13-V2-nLUC (>100,000 units; Figure 5A). The high LUC activities were dependent on the MLA13 receptor because only low LUC activities (<300 units) were observed when the same AVRa13 variants were co-expressed with Mla1-cLUC (Figure 5A).
We detected LUC activity when co-expressing Mla7-cLUC with AVRa7-2-nLUC, which was at least 7-fold higher when the latter construct was replaced by AVRa7-1-nLUC or AVRa7-V1-nLUC (1000 units; Figure 5B), despite comparable protein levels of all AVRA7-nLUC variants (Figure 5E). The LUC activities were much lower compared to the LUC complementation of Mla13-cLUC and AVRa13-1-nLUC (Figure 5A). The higher LUC reporter activity was not detected when co-expressing Mla13-cLUC with AVRa7-2-nLUC. This proxy for in planta receptor-avirulence effector interaction is in agreement with our observation that AVRA7-2-mYFP but not AVRA7-1-mYFP or AVRA7-V1-mYFP is capable of inducing MLA7-dependent cell death in N. benthamiana (Figure 4D).
Co-expression of AVRa10-nLUC with Mla10-cLUC resulted in the detection of a 3.5-fold higher LUC activity when compared to expression of its virulent variants AVRa10-V/AVRa22-V-nLUC and AVRa22-nLUC (Figure 5C). LUC activity upon expression of AVRa22-nLUC together with Mla22-cLUC was only slightly higher (1.6-fold) when compared to its virulent variants AVRa10-nLUC and AVRa10-V/AVRa22-V-nLUC in the same experiment (Figure 5C). This marginal increase in LUC activity may partly reflect differences in protein stability, as the AVRA22-nLUC protein is barely detectable when compared to AVRA10-nLUC (Figure 5E) and because both, MLA10-cLUC and MLA22-cLUC are seemingly less stable when compared to MLA7-cLUC and MLA13-cLUC.
Although the LUC complementation assay is suggestive of direct receptor – avirulence effector associations, we cannot fully exclude the involvement of other plant proteins in this association. We thus tested MLA and AVRA interactions in a yeast two-hybrid assay using leucine reporter gene activity (Figure 5F–5I). Similar to results obtained with the split-LUC assay, yeast growth on leucine-deficient medium was observed when we co-expressed AD (B42 activation domain)-AVRa13-1 with BD (LexA binding domain)-Mla13 and AD-AVRa7-2 with BD-Mla7, but not when BD-Mla13 and BD-Mla7 were swapped in these two interaction experiments, indicating the interactions are specific for matching MLA receptor and avirulence effector pairs (Figure 5F). Interactions in yeast were detectable when BD-Mla13 was co-expressed with either AVRa13-1 or AVRa13-3, but undetectable upon co-expression with AVRa13-V1, suggesting specific MLA13 interactions with the avirulent AVRA13 variants (Figure 5G). However, clear interactions in yeast were also detectable upon co-expression of BD-Mla13 with AD-AVRa13-V2, even at a cell plating density of 10² (Figure 5E), which is reminiscent of the split-LUC result with the corresponding construct pair (Figure 5A). In contrast to AVRA13-V1-mYFP (Figure 4E) and AVRA13-V1-nLUC (Figure 5E) protein in N. benthamiana leaves, AD-AVRA13-V1 protein level in yeast was comparable to other AD-AVRA13 variants (Figure 5J). Robust interaction in yeast was found upon co-expression of BD-Mla7 with AD-AVRa7-2, but not when the former was co-expressed with AD-AVRa7-V1 (Figure 5H). Only sporadic yeast colony growth was found when BD-Mla7 was co-expressed with AD-AVRa7-1 (Figure 5H). This again mirrors the findings with the corresponding gene pairs in the LUC complementation assay (Figure 5B). We conclude that specific interactions can be detected for the matching MLA7 and AVRA7-2 pair in yeast and specific associations for the corresponding protein pair in plant extracts. In sum, yeast two-hybrid and split-LUC experiments suggest direct detection of the sequence-unrelated avirulence effectors AVRA7 and AVRA13 by matching MLA7 and MLA13 receptors, but the strong association of the virulent effector AVRA13-V2 with MLA13 represents one case in which receptor-effector association is uncoupled from receptor activation, that is from cell death induction (see Discussion).
Consistent with a direct interaction of matching MLA and AVRA pairs, co-expression of BD-Mla10 with AD-AVRa10 in yeast resulted in leucine reporter gene activation and this was undetectable when Mla10 was replaced by either Mla7 or Mla22, and minor when AD-AVRa10 was replaced by its virulent variant AD-AVRa10-V/AVRa22-V (Figure 5I). The latter virulent effector differs only by a single amino acid from the avirulence effector AD-AVRA10 (Figure 3D).
Evolutionary history of Bgh AVRa genes and population-level AVRa10/AVRaa22 sequence variation in B. graminis formae speciales
We used a high-quality genome assembly of the wheat powdery mildew Bgt reference isolate 96224 to investigate the evolutionary history of Bgh AVRa genes and to potentially identify distinctive selection pressures on these effector genes exerted by wheat and barley hosts. AVRa7 (sequence identical BLGH_06672 and BLGH_06689; Figure 2) belongs to a larger CSEP gene family in the Bgh DH14 genome and, together with three closely related members, defines one sublineage (Figure 6—figure supplement 1A). Although we identified four genes sharing between 40% and 47% aa sequence similarity with AVRa7 (Figure 6—figure supplement 1A), the flanking genes of AVRa7 differ from those adjacent to the four Bgt homologs and therefore do not permit conclusions on potential orthologous relationships after the split of the two formae speciales.
We found three Bgt genes sharing between 52% and 57% identical polypeptide sequences with Bgh AVRa9 (BLGH_04994) (Figure 6—figure supplement 1C and D). These genes are closely located to each other in a region of the wheat powdery mildew genome that is largely collinear between Bgt and Bgh genomes (Figure 6—figure supplement 1E and F). We conclude that either local gene duplications of an ancestral ortholog of BLGH_04994 gave rise to the extant Bgt gene organization, or that these duplications were already present in the last common ancestor of Bgt and Bgh and that paralogs were lost in the Bgh genome.
Applying a phylogenetic approach to the wheat powdery mildew genome, we identified BgtE-5921 as the ortholog of Bgh CSEP0141 (with haplotypes AVRa10 and AVRa22), which shares 68% and 65% identical deduced polypeptide sequences with AVRA10 and AVRA22, respectively (Figure 6—figure supplement 1G; Table 1; Figure 6A). In addition, we found a single CSEP0141 candidate ortholog in each of the genomes of a world-wide collection of other B. graminis formae speciales (Table 1, Supplementary file 5), for which short-read genome sequences are available (Spanu et al., 2010; Wicker et al., 2013; Hacquard et al., 2013; Menardo et al., 2016; Praz et al., 2017; Müller et al., 2019). With these short-read genome sequences from multiple isolates of each of these B. graminis f. sp. (Supplementary file 5) we were able to assess genome-wide nucleotide diversity (π) and population-level sequence diversification of the CSEP0141 orthologs and compared them with the diversification pattern of CSEP0141 in the Bgh population. Based on 1141 neutral markers, we calculated a genome-wide per-site nucleotide diversity (π) of 0.022 for the f. sp. hordei, 0.013 for the f. sp. secalis, 0.050 for the f. sp. tritici2, 0.062 for the f. sp. tritici, and 0.040 for the f. sp. triticale isolates (Supplementary file 5). Unlike the two major CSEP0141 haplotypes in Bgh (AVRa10 and AVRa22), a single dominant BgtE-5921 haplotype was found in the Bgt population (29 out of 40 isolates; Table 1, Figure 6A). The remaining 11 Bgt isolates represent six further BgtE-5921 haplotypes encoding effector variants with at most four aa polymorphisms in the deduced proteins compared to the dominant BgtE-5921 variant (Table 1). Three of the latter haplotypes are exclusively present in Bgt isolates that were collected from tetraploid wheat and represent a distinctive Bgt sublineage, designated B. graminis f. sp. tritici2 (Menardo et al., 2016). A single haplotype of the CSEP0141 ortholog was detected among 22 B. graminis f. sp. triticale isolates and a single haplotype was also found in five B. graminis f. sp. secalis isolates (Table 1), indicating either limited or no natural variation of this effector in populations of wheat, triticale, and rye powdery mildews (up to four aa substitutions in wheat powdery mildews). This underlines the exceptional level of polymorphism between the two major CSEP0141 haplotypes, AVRa10 and AVRa22, in the Bgh population with 13 deduced aa changes. Bgh CSEP0141 defines one of 190 core effector genes that are conserved across B. graminis ff. spp. (Frantzeskakis et al., 2018) and exhibits the highest frequency of non-synonymous SNPs in the Bgh population (4.2 non-synonymous SNPs/100 bp coding sequence; Figure 6C). Virulence functions of core effectors are likely important for B. graminis pathogen fitness. Thus, we hypothesize that in the Bgh population the two dominant CSEP0141 haplotypes, AVRa10 and AVRa22, have emerged from the action of two opposing selective pressures: sequence conservation that maintains pathogen fitness through retention of effector virulence activity and sequence diversification to escape recognition by MLA10 and MLA22 receptors, respectively.
Figure 6 with 1 supplement see all
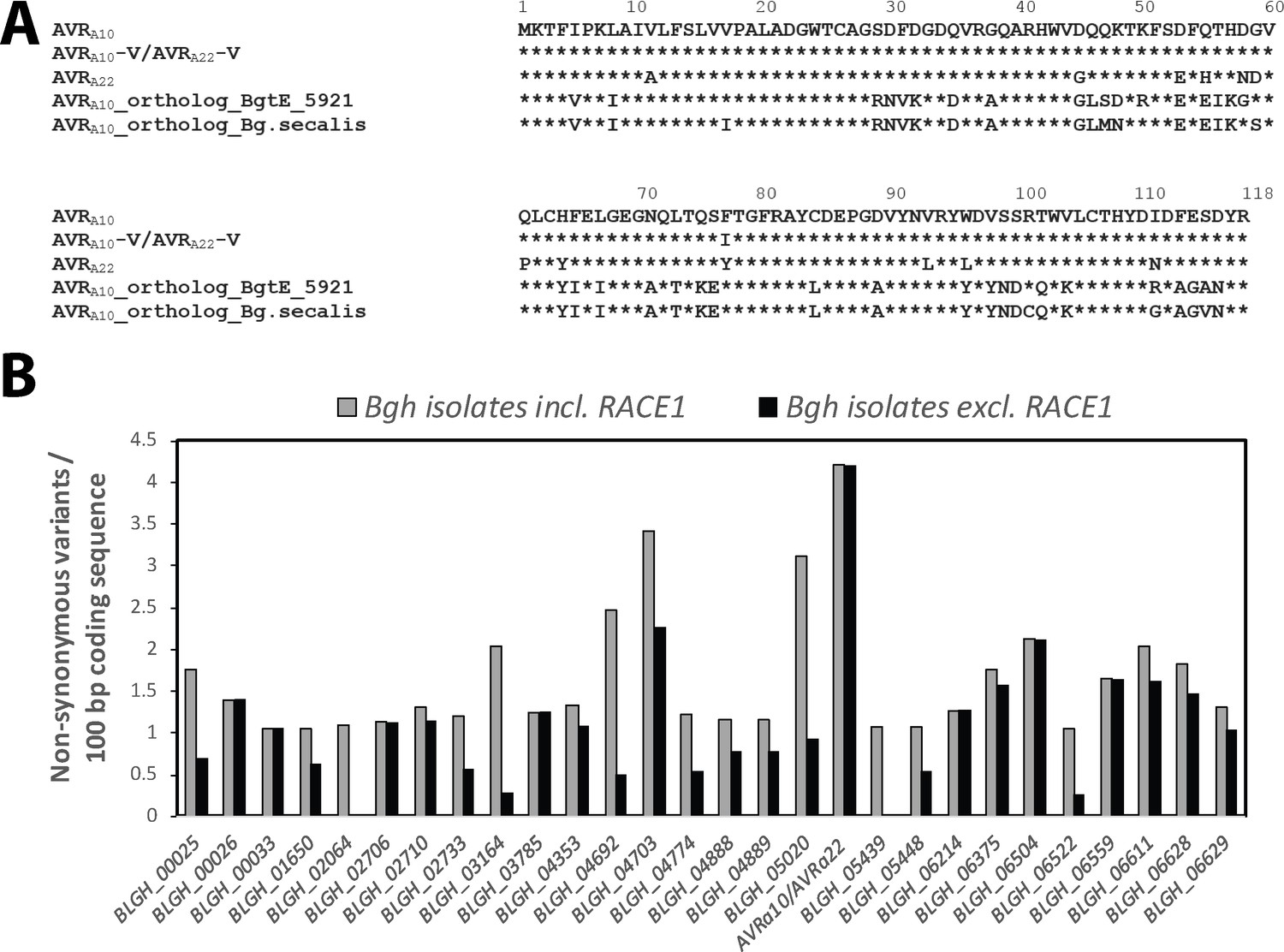
Conservation of AVRa10/AVRa22 orthologs between Blumeria graminis formae speciales.
(A) Alignment of protein sequences (AVRA10/AVRA22) encoded by Bgh CSEP0141 and orthologs detected in B. graminis f. sp. tritici and B. graminis f. sp. secalis. (B) Number of non-synonymous sequence variants was determined for 190 core effectors (Frantzeskakis et al., 2018) among all Bgh isolates described in this study and is displayed for all core effectors with ≥1 non-synonymous variants/100 bp coding sequence. Grey bars, including all Bgh isolates; black bars, all Bgh isolates excluding RACE1.
Table 1
AVRa10/AVRa22 ortholog BgtE-5921 variants in different Blumeria graminis formae speciales.
https://doi.org/10.7554/eLife.44471.025| Number of variants in Blumeria graminis f. sp. | |||||
|---|---|---|---|---|---|
| AVRa10/AVRa22 ortholog BgtE-5921 | tritici | dicocci/tritici2 | triticale | secalis | Total |
| Hap_96224 | 29 | 22 | 51 | ||
| Hap_96224 R111G | 2 | 2 | |||
| Hap_96224 V5I, R111G | 2 | 2 | |||
| Hap_96224 R111G, A115G | 1 | 1 | |||
| Hap_96224 F77L, R111G | 4 | 4 | |||
| Hap_96224 F77L, R111G, Y117H | 1 | 1 | |||
| Hap_96224 V5I, I8L, F77L, R111G | 1 | 1 | |||
| Hap_96224 V17I, S47M, D48N, R50K, G58D, G59S, R101C, R111G, A115V | 5 | 5 | |||
| total number of isolates | 34 | 6 | 22 | 5 | 67 |
Discussion
Previous pathotyping studies of Bgh field isolates with barley varieties carrying different powdery mildew R genes suggested that the Central European pathogen population can be considered as a single epidemiological unit (Limpert, 1987). We have shown here that among 13 Bgh isolates from a local population in Germany ten virulence combinations can be distinguished in interactions with a panel of Mla NILs, suggesting potential adaptation of the pathogen population to multiple Mla resistance specificities. Our findings are congruent with a recent study describing a high complexity of Central European Bgh virulence pathotypes on a panel of 50 differential barley varieties carrying Mla or other powdery mildew R genes (Dreiseitl, 2014). The same study also provided evidence for a complete separation of the Central European and Australian Bgh populations with non-overlapping pathotypes and estimated an almost three-fold higher virulence complexity for the European population. This is believed to be due to the cultivation of barley varieties carrying distinct powdery mildew R gene combinations on the two continents, which leads to differential intensities of directed selection on Bgh populations. Analysis of our isolate collection comprising, among others, 13 newly characterized Bgh strains from a local population in Germany and three representative Australian isolates, is supportive of a significantly greater virulence complexity of the Central European over the Australian isolates even when considering virulence patterns only on Mla NILs (Supplementary file 1). The complexity of avirulence and virulence alleles in the Central European population suggests that the fungus adapts by balancing selection of AVRa genes among strains rather than selective sweeps to maintain pathogenicity.
Specific activation of cell death upon transient gene expression of matching AVRa and Mla gene pairs in barley protoplasts and heterologous N. benthamiana provided evidence for the function of candidate AVRa7, AVRa9, AVRa10, and AVRa22 as avirulence effectors, all of which encode Bgh CSEPs with a predicted signal peptide. This is different from the previously reported EKA_AVRA10, which lacks a canonical signal peptide, belongs to the large EKA gene family that is derived from part of a class-1 LINE retrotransposon (Ridout, 2006; Amselem et al., 2015), and was used as an elicitor of induced MLA10 nuclear interaction with a WRKY transcription factor (Shen et al., 2007). However, we were unable to detect MLA10-mediated cell death activity for EKA_AVRA10. Our findings demonstrate that virulent Bgh isolates escape recognition of corresponding MLA receptors predominantly by non-synonymous SNPs but also loss of expression of the corresponding genes (Figure 3A; Lu et al., 2016). Upon in silico removal of the signal peptide, phylogenetic analysis for the 805 predicted secreted proteins of Bgh (Frantzeskakis et al., 2018) and comparative sequence analysis of AVRa7, AVRa9, AVRa10, AVRa22, and previously reported AVRa1 and AVRa13, also representing CSEPs, failed to detect evolutionary conservation (Figure 3—figure supplement 1) or significant sequence similarity (at most 8% sequence identity) between any pair of these polypeptides except for allelic AVRa10 and AVRa22 (Figure 3C and E). By contrast, MLA7, MLA9, MLA10 and MLA13 receptors are >96% identical in aa sequence to each other, whereas MLA1 and MLA22 are more diverged and share 91% identical polypeptide sequences with this receptor group (Seeholzer et al., 2010). This raises questions regarding the evolutionary history of MLA10- AVRA10 and MLA22-AVRA22 receptor-effector pairs. AVRa10 and AVRa22 are notable for several reasons: (i) these allelic effectors define two dominant haplotypes of CSEP0141 in Bgh, (ii) CSEP0141 belongs to a core of 190 effectors that are conserved among different B. graminis f. sp. and, therefore, likely contributes to pathogen fitness, (iii) CSEP0141 represents the core effector with the highest frequency of non-synonymous SNPs among the tested global collection of Bgh isolates, and (iv) a single dominant haplotype of its Bgt ortholog, designated BgtE-5921, was found in the wheat powdery mildew population. Collectively, this suggests a model in which the two dominant CSEP0141 haplotypes, AVRa10 and AVRa22, evolved in Bgh by the action of two opposing selective forces: functional conservation that maintains pathogen fitness through retention of effector virulence activity and sequence diversification to escape recognition by MLA10 and MLA22 receptors, respectively. AVRa10 and AVRa22 likely represent a balanced polymorphism in the extant pathogen population because Bgh isolates containing one or the other haplotype do not form discrete subgroups (Figure 1—figure supplement 1A). However, the likely source of many, if not all, Mla recognition specificities in domesticated barley is wild barley, H. spontaneum (Jørgensen and Wolfe, 1994). Thus, it is possible that Mla10, Mla22 and their matching avirulence effector genes have not diversified during a co-evolutionary arms race (Ravensdale et al., 2011) but have rather evolved independently in separate host and pathogen populations. In such a scenario, the balanced AVRA10 and AVRA22 polymorphism in the extant pathogen population is the consequence of concurrent cultivation of domesticated barley varieties with these Mla resistance specificities. Besides an apparently balanced AVRa10 and AVRa22 polymorphism at CSEP0141 in the Bgh field population one would expect sporadic strains that are virulent on both Mla10- and Mla22-harboring host varieties, which is the case for Bgh isolate DH14 carrying a SNP that introduces a single aa substitution in AVRa10 (Figure 3). In such strains, a fitness penalty for the pathogen might be mitigated by residual virulence activity of the CSEP0141 variant.
Our split-LUC and yeast two-hybrid experiments provided evidence for direct and specific interactions between multiple matching MLA/AVRA pairs (Figure 5). More than 50 years after the original discovery of multi-allelic race-specific disease resistance at Mla (Moseman and Schaller, 1960), these findings now imply that the co-evolutionary functional diversification of these immune receptors is at least in part mechanistically underpinned, and was perhaps driven by direct interactions with sequence-unrelated Bgh avirulence effectors. Direct receptor-avirulence effector interactions have been described for flax L and rice Pik multi-allelic disease resistance genes, which encode NLR-type receptors, (Kanzaki et al., 2012; Dodds et al., 2006). In flax, a subset of allelic L receptors (L5, L6, and L7) interact with a specific subset of highly sequence-related flax rust AvrL567 proteins, and in rice, allelic Pik receptors interact with specific variants of highly sequence-related AVR-Pik proteins. Rice Pik immune receptors contain an integrated heavy metal-associated RATX1/HMA domain, which binds directly to AVR-Pik and Pik functional diversification is driven by polymorphisms in this integrated domain (Kanzaki et al., 2012; Maqbool et al., 2015; De la Concepcion et al., 2018). Barley MLA and flax L proteins lack detectable integrated domains, and diversifying selection in allelic MLA receptors is largely confined to predicted solvent-exposed residues of leucine-rich repeats 7 to 15 (Seeholzer et al., 2010). Thus, to the best of our knowledge, a co-evolutionary functional diversification of multi-allelic NLR-type receptors in plants with directly recognized sequence-unrelated avirulence effectors, as described here for matching MLA, is without precedence. Effectors AVR1-CO39 and AVR-Pia of the ascomycete Magnaporthe oryzae are sequence-unrelated but have very similar 6 β-sandwich structures that are stabilized in both cases by a disulfide bridge and are both recognized by the rice NLR pair RGA4-RGA5 through the integrated RATX1/HMA domain located at the C-terminus of RGA5 (de Guillen et al., 2015; Cesari et al., 2013). Structural similarity searches then showed that AVR1-CO39 and AVR-Pia are founders of a family of sequence-unrelated but structurally conserved fungal effectors in a broad range of ascomycete phytopathogens (de Guillen et al., 2015). Consistent with structural modelling (IntFOLD Version 3.0 (McGuffin et al., 2015)), the recently resolved NMR-based and crystal structure of Bgh CSEP0064 revealed a ribonuclease-like fold (Pennington et al., 2019) with the absence of canonical catalytic residues in the substrate-binding pocket, and the gene products of ~120 additional Bgh CSEPs very likely adopt a similar structure (Pennington et al., 2019). Structural similarity searches (IntFOLD Version 3.0) also suggested a ribonuclease-like fold for AVRA7 and AVRA13 (high and certain confidence at p=3.739E-3, and 6.174E-4, respectively), whereas no significant structural similarities were detected between CSEP0064 and AVRA1, AVRA9, AVRA10, or AVRA22 (low and medium confidence at p>0.01). Instead, we find that AVRA9 may adopt a structural fold that is similar to an antimicrobial peptide called microplusin (p=6.014E-3). In addition, no obvious structural similarities were predicted between AVRA1, AVRA9, and AVRA10/AVRA22, suggesting that allelic MLA receptors are capable of detecting the presence of structurally unrelated Bgh effectors. This assumption is consistent with the recent finding that the wheat Mla ortholog Sr50 directly binds to the Basidiomycete stem rust (Pgt) avirulence effector AvrSr50 (Chen et al., 2017). This Basidiomycete effector most likely evolved independently from the Ascomycete Bgh effectors and lacks significant sequence and predicted structural similarity with the known AVRa effectors. We speculate that MLA receptors might have an exceptional propensity to directly detect unrelated pathogen effectors and that this feature might have facilitated the functional diversification of the receptor in the host population.
In a whole leaf context, race-specific disease resistance specified by MLA receptors to Bgh is invariably linked to the activation of localized host cell death (Boyd et al., 1995). NLR-mediated cell death likely contributes to the termination of biotrophic fungal pathogenesis, including that of powdery mildews, because this class of pathogens feeds on living plant cells. A striking feature of the functional diversification at Mla is the enormous variation in microscopic and macroscopic resistance-associated Bgh infection phenotypes as shown with barley NILs carrying different Mla resistance specificities (Boyd et al., 1995). For instance, the onset of detectable host cell death can vary dramatically and can be both rapid and limited to the first attacked leaf epidermal cell, terminating early fungal pathogenesis, or can occur at later stages of fungal pathogenesis and involve numerous leaf mesophyll cells that subtend Bgh-infected epidermal cells (Boyd et al., 1995). Here we have employed co-transfection experiments of barley leaf protoplasts with Mla-AVRa pairs and protoplast cell death as a proxy for receptor activation, excluding the possibility that additional Bgh-derived molecules associated with Pattern-triggered immunity influence the timing of immune receptor-mediated cell death in this system. Although based on overexpression data, the significant variation in cell death phenotypes reported here could partly reflect variable Bgh infection phenotypes on different MLA NILs (Supplementary file 1, Boyd et al., 1995). In turn, these differences of infection phenotypes are possibly due to variations in the steady-state levels of the MLA receptors during Bgh infection, timing of Bgh-mediated AVRA secretion and/or AVRA steady-state levels in planta, or MLA-AVRA pair-dependent receptor binding affinities. Whilst etablishing the relevance of the the latter requires future biochemical characterization of MLA-AVRA complexes, our work revealed a very strong binding of AVRA13-V2 to MLA13 both in the split-LUC and yeast two-hybrid experiments (Figure 5), thereby uncoupling AVRA binding to the receptor from receptor activation, that is immune receptor-mediated cell death activation. Future biochemical and genetic experiments will clarify whether the naturally occurring AVRA13-V2 effector variant acts as a dominant negative ligand when co-expressed with the AVRA13 avirulence effector.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain (Blumeria graminis f. sp. hordei) | CC107, CC148, CC1, CC52, CC66, CC88, NCI, 63.5, A6, B103, Aby, Art, Will, OU14, RACE1, K1 | Lu et al. (2016) doi:10.1073/pnas.1612947113. | GEO:GSE83237 | |
| Strain (Blumeria graminis f. sp. hordei) | DH14 | Frantzeskakis et al. (2018) doi:10.1186/s12864-018-4750-6. | GEO:GSE106282 | |
| Strain (Blumeria graminis f. sp. hordei) | K2, K3, K4, S11, S15, S16, S19, S20, S21, S22, S23. S25, S26 | this paper | GEO:GSE110266 | collected in 2017 on cv. Meridian and Keeper barley at the Max Planck Institute for Plant Breeding Research, Cologne, Germany (GPS 5˚57′N, 6˚51′E 5) |
| Recombinant DNA reagent | pIPKb002 | Himmelbach et al. (2007) doi:10.1104/pp.107.111575. | NCBI:EU161568.1 | pZmUBQ:GW, SpcR |
| Recombinant DNA reagent | pGWB517 | Nakagawa et al. (2007) doi:10.1263/jbb.104.34. | NCBI:AB294484.1 | p35S:GW-4Myc, SpcR |
| Recombinant DNA reagent | pXCSG-GW-mYFP | García et al. (2010) doi:10.1371/journal.ppat.1000970. | NA | p35S:GW-mYFP, CarbR |
| Recombinant DNA reagent | pB42AD-GW | Shen et al. (2007) doi:10.1126/science.1136372. | NA | pGal1:B42-AD-−1xHA-GW, TRP |
| Recombinant DNA reagent | pLexA-GW | Shen et al. (2007) doi:10.1126/science.1136372. | NA | pADH1:LexA-BD-GW, HIS3 |
| Recombinant DNA reagent | pDest-GW-nLUC | Gehl et al. (2011) doi:10.1111/j.1365-313X.2011.04607.x. | NA | p35S:GW-NterminusLuciferase, KanR |
| Recombinant DNA reagent | pDest-GW-cLUC | Gehl et al. (2011) doi:10.1111/j.1365-313X.2011.04607.x. | NA | p35S:GW-CterminusLuciferase, KanR |
| Gene (Blumeria graminis f. sp. hordei) | AVRa7 variants | Frantzeskakis et al. (2018) doi:10.1186/s12864-018-4750-6. | csep0059; BLGH_06689; BLGH_06672; BGHR1_17217; BGHR1_17236; BGHR1_17237 | |
| Gene (Blumeria graminis f. sp. hordei) | AVRa9 variants | Frantzeskakis et al. (2018) doi:10.1186/s12864-018-4750-6. | csep0174; BLGH_04994; BGHR1_10042 | |
| Gene (Blumeria graminis f. sp. hordei) | AVRa10/AVRaa22 variants | Frantzeskakis et al. (2018) doi:10.1186/s12864-018-4750-6 | csep0141; BLGH_05021; BGHR1_10013 | |
| Gene (Blumeria graminis f. sp. hordei) | AVRa1 variants | Lu et al. (2016) doi:10.1073/pnas.1612947113; Frantzeskakis et al. (2018) doi:10.1186/s12864-018-4750-6 | csep0008; BLGH_03023; BLGH_03022; BGHR1_11142 | |
| Gene (Blumeria graminis f. sp. hordei) | AVRa13 variants | Lu et al. (2016) doi:10.1073/pnas.1612947113; Frantzeskakis et al. (2018) doi:10.1186/s12864-018-4750-6 | csep0372; BLGH_02099; BGHR1_12484 | |
| Gene (Hordeum vulgare) | Mla9 | Seeholzer et al. (2010) doi.org/10.1094/MPMI-23-4-0497 | NCBI: GU245941.1 | |
| Gene (Hordeum vulgare) | Mla22 | Seeholzer et al. (2010) doi.org/10.1094/MPMI-23-4-0497 | NCBI:GU245946 | |
| Gene (Hordeum vulgare) | Mla10 | Seeholzer et al. (2010) doi.org/10.1094/MPMI-23-4-0497 | Mla10 | Different from NCBI:AY266445.1 |
| Gene (Hordeum vulgare) | Mla7 | Seeholzer et al. (2010) doi.org/10.1094/MPMI-23-4-0497; Lu et al. (2016) doi:10.1186/s12864-018-4750-6 | Mla7 | Different from NCBI:AY266444.1 |
| Gene (Hordeum vulgare) | Mla7 (AAQ55540_Halterman et al., 2004) | Halterman and Wise (2004) doi:10.1111/j.1365-313X.2004.02032.x | NCBI:AY266444.1 | |
| Gene (Hordeum vulgare) | Mla1 | Seeholzer et al. (2010) doi.org/10.1094/MPMI-23-4-0497; Lu et al. (2016) | NCBI:GU245961 | |
| Gene (Hordeum vulgare) | Mla13 | Seeholzer et al. (2010), Lu et al. (2016) doi:10.1073/pnas.1612947113. | AF523678.1 | |
| Antibody | monoclonal rat anti-HA | Merck | 3F10, RRID:AB_390914 | 1:2000 |
| Antibody | monoclonal mouse anti-LexA | Santa Cruz Biotechnology | sc7544, RRID:AB_627883 | 1:1000 |
| Antibody | polyclonal rabbit anti-c-myc | Abcam | ab9106, RRID:AB_307014 | 1:5000 |
| Antibody | polyclonal rabbit anti-GFP | Abcam | ab6556, RRID:AB_305564 | 1:5000 |
| Antibody | polyclonal rabbit anti-LUC | Sigma | L0159, RRID:AB_260379 | 1:2000 |
| Antibody | polyclonal goat anti-rat IgG-HRP | Santa Cruz Biotechnology | sc2065, RRID:AB_631756 | 1:100 000 |
| Antibody | polyclonal goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc2005, RRID:AB_631736 | 1:100 000 |
| Antibody | polyclonal donkey anti-rabbit IgG-HRP | Santa Cruz Biotechnology | sc-2313, RRID:AB_641181 | 1:100 000 |
| Antibody | monoclonal rabbit anti-GFP | Santa Cruz Biotechnology | sc-8334, RRID:AB_641123 | 1:5000 |
Plant materials and growth conditions
Request a detailed protocolThe barley cultivars (cv.) Golden Promise, Manchuria, and Pallas and their near isogenic lines (Kolster et al., 1986; Moseman, 1972), were grown at 19°C, 70% relative humidity, and under a 16 h photoperiod. Nicotiana benthamina plants were grown in standard greenhouse conditions under a 16 h photoperiod.
Fungal isolates
Request a detailed protocolBarley leaves suspected of being infected by Bgh were collected from the cv. Meridian and Keeper barley fields at the Max Planck Institute for Plant Breeding Research, Cologne, Germany (GPS 5˚57′N, 6˚51′E 5). Spores of different infected field leaves were transferred onto one-week-old barley leaves of the cv. Manchuria (lacking any Mla resistance specificity). Inoculated Manchuria leaves were incubated on 1% Bacto Agar plates supplemented with 1 mM benzimidazole at 20°C, 70% humidity, and long-day conditions for one week until Bgh conidiospore growth was visible. Subsequent single spore propagation was applied three (S11, S15, S19, S20, S21, S23, S25, S26) or six (K2, K3, K4, S16 and S22) times for isolation of single Bgh isolate genotypes. In total, we collected 13 Bgh isolates, which were tested at least three times on a panel of cv. Pallas and cv. Manchuria Mla near-isogenic lines. Maintenance of fungal isolates and other Bgh isolates in this study was carried out as described previously (Lu et al., 2016).
RNA sequencing
Request a detailed protocolThe new RNA-seq data generated for this study are deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (accession no. GSE110266). The previously generated RNA-seq data for DH14 and all other isolates can also be found at GEO (accession nos. GSE106282 and GSE83237, respectively).
RNA-seq read alignment and variant calling
Request a detailed protocolThe RNA-seq reads for all datasets were mapped to the new Bgh DH14 reference genome assembly (version 4.0) taking into account exon-intron structures using the splice-aware aligner Tophat2 (Kim et al., 2013), which considers known splice sites based on the new DH14 gene models (Frantzeskakis et al., 2018). Read length was 100 bp for previously sequenced isolates (GSE83237), 150 bp for DH14 (GSE106282), S20 and S25, and 250 bp for all other isolates (GSE110266). To allow for adequate alignment efficiency also for those isolates with higher sequence divergence from the reference genome, we adjusted the alignment settings as follows: --read-mismatches 10 --read-gap-length 10 --read-edit-dist 20 --read-realign-edit-dist 0 --mate-inner-dist 260 --mate-std-dev 260 --min-anchor 5 --splice-mismatches 2 --min-intron-length 30 --max-intron-length 10000 --max-insertion-length 20 --max-deletion-length 20 --num-threads 10 --max-multihits 10 --coverage-search --library-type fr-firststrand --segment-mismatches 3 --min-segment-intron 30 --max-segment-intron 10000 --min-coverage-intron 30 --max-coverage-intron 10000 --b2-very-sensitive. Using the SAMtools suite (Version 0.1.18) (Li et al., 2009), the generated alignment files were subsequently filtered to retain only properly paired reads with mapping quality >0 for the downstream analyses.
To assess the expression levels of individual genes, we obtained the fragment counts per gene for each isolate and time point from the mapped RNA-seq reads after filtering using the Subread function ‘featureCounts’ (version 1.5.0) (Liao et al., 2014) with options -t CDS -s 2 M –p. Subsequently, the raw counts were summarized over both time-points for each isolate and normalized to FPKM (fragments per kilobase of transcript per million mapped reads) values for better comparability of expression levels.
In parallel, sequence variants were identified from the mapped RNA-seq reads using two different tools. In both cases, the variant calling was performed on a combined alignment dataset that was obtained by merging the mapped RNA-seq reads from all isolates using the merge function of the SAMtools suite (Li et al., 2009). In one approach, single nucleotide polymorphisms (SNPs) were identified using the mpileup function in the SAMtools suite (Li et al., 2009) with options -A, -u, -D, -d 30000 –L 7000. The resulting mpileup variants were filtered using SnpSift (Version 3.4) (Cingolani et al., 2012a) with filter settings “(AF1 >= 0.01852) and ((DP >= 30) | ((DP >= 10) and (GEN[*].DP>=5))) and (QUAL >= 50) and (GEN[*].GQ>=10) and ((na PV4) | ((PV4[0]>1e-10) and (PV4[3]>1e-5)))” to extract high-quality variants with sufficiently high allele frequency (≥0.01852; that is alternate allele present in at least ~50% of the reads of one isolate), sufficient read coverage (≥30 reads in total, or ≥10 reads in total and at least one isolate with ≥5 reads), a SNP calling quality score ≥50, at least one isolate with a genotype quality score ≥10, and absence of extreme placement bias. In the other approach, variants were called using freebayes (version 9.9.2) (Garrison and Marth, 2012) with options --ploidy 1 --use-duplicate-reads --min-mapping-quality 0 --min-base-quality 20 --min-coverage 30 --genotype-qualities. To allow correct variant calling from our RNA-seq data with freebayes, the mapped reads in this case were preprocessed using the function SplitNCigarReads in the GenomeAnalysis Toolkit (version 3.4.0) (McKenna et al., 2010) with options -U ALLOW_N_CIGAR_READS -fixNDN -maxOverhang 10 to split any reads with splice junctions. An additional, independent freebayes variant calling was also performed with --ploidy 2, to allow processing of cases where an isolate contains additional gene copies that differ from each other at some residue(s). The resulting freebayes variant sets were filtered using SnpSift (Cingolani et al., 2012a) with filter settings “(AF[*]>=0.01852) and ((exists GEN[*].GQ) and (GEN[*].GQ[*]>=10)) and ((exists GEN[*].AO) and (GEN[*].AO[*]>=3)) and ((DP >= 30) | ((DP >= 10) and (exists GEN[*].DP) and (GEN[*].DP>=5))) and (SAR[*]>0) and (SAF[*]>0) and (RPR[*]>1) and (RPL[*]>1)’ to extract high-quality variants fulfilling the same criteria as described above for mpileup. The subsequent variant annotation and effect prediction for all datasets was performed using snpEff (Version 3.4; default settings) (Cingolani et al., 2012b) based on the new DH14 genome and gene models.
Population structure and genetic association analysis of Bgh isolates
Request a detailed protocolTo obtain a suitable set of single nucleotide polymorphisms (SNPs) for population structure analysis, we further filtered the set of high-quality variants obtained with mpileup from the combined alignment data as described above. We extracted only silent (synonymous) SNPs in coding regions for which exactly two different alleles were found (diallelic SNPs), and complete genotype information was available for all isolates (i.e., no missing data). The resulting set of 6286 high-quality diallelic synonymous SNPs was used to examine the genotype data for the presence of any obvious population structure using the R packages adegenet (Version 2.0.1) (Jombart and Ahmed, 2011) and ape (Version 3.4) (Paradis et al., 2004). To this end, we created a PCA plot from the genotype data using the function glPca (R package adegenet) and additionally computed a neighbour-joining tree based on the pairwise Euclidean distances between the isolate genotypes using the function nj (R package ape). Additionally, another PCA plot was generated for the European isolates only, based on a set of 5170 high-quality diallelic synonymous SNPs found in the European isolates.
For the association analysis we focused on the high-quality variants obtained with freebayes in the haploid SNP calling, which we filtered further to extract only non-synonymous coding variants predicted to change the protein sequence and generated a simplified genotyping table listing all of these variants. Additionally, we also screened the results from the diploid freebayes variant calling for ‘heterozygous’ positions with a minor allele frequency of at least 1/3, as in this case the different ‘alleles’ are likely derived from differing paralog copies, and added these positions to the genotyping table. This procedure was implemented in R (Supplementary file 2) and resulted in a set of 22,838 high-confidence variants with predicted effect on the protein sequence, which we tested for their association with the observed avirulence phenotypes using Fisher’s exact test.
Loss of avirulence might be caused by different variants in different isolates. Therefore, we integrated all high-confidence non-synonymous variants over each gene to obtain gene-wise genotypes. Moreover, to also include presence/absence polymorphisms we considered the complete absence of a transcript as a ‘missing’ genotype. Finally, these gene-wise genotypes were tested for association with the observed avirulence phenotypes using Fisher’s exact test. This gene-wise integration of variants and the subsequent association test were implemented in R. As further technical validation, we additionally performed the same association test also on gene-wise genotypes obtained in the same way from the high-quality mpileup SNPs (Supplementary file 3). All identified AVRa candidates were picked up using both tools; the p values mentioned are based on the freebayes variants (Supplementary file 4).
Genome sequencing, assembly and annotation
Request a detailed protocolFor the improvement of the Bgh isolate K1 assembly, genomic DNA was extracted as described (Feehan et al., 2017) and sequenced using the Oxford Nanopore MinION platform according to the manufacturer’s instructions for library generation and flow-cell handling. Base-Calling of the resulting long-reads was performed with the Albacore Sequencing Pipeline Software (version 2.0.2, Oxford Nanopore) yielding 3,09 GB of data in 781831 reads, and subsequently assembled with Canu (v1.4, Koren et al., 2017). The 2238 assembled contigs were corrected using Illumina short reads (SRR650349 and SRR654727; Hacquard et al., 2013) with Pilon (v1.18, Walker et al., 2014) in four iterations. Re-annotation of the new genome assembly was performed as described in Frantzeskakis et. al., and CSEPs were then manually curated using WebApollo (v2.0.6, Lee et al., 2013). Data are deposited under the accession number PRJEB30373 at EBI-ENA.
Genome-wide nucleotide diversity of B.g. ff. spp. isolates
Request a detailed protocolIllumina short-read sequences of different B.g. ff. ssp. (SRA accession number: SRP062198, Supplementary file 5) were mapped to the reference genome of isolate 96224 (t ENA accession number: PRJEB28180) as described (Müller et al., 2019). For extraction of neutral markers, SNP calling was done with freebayes with default parameters (Garrison and Marth, 2012) and SNPs located within genes were excluded (Müller et al., 2019). Vcftools (Danecek et al., 2011) was used to filter SNPs with the following options: vcf --remove-indels --max-alleles 2 --min-alleles 2 --minDP 8 --maxDP 100 --max-missing 1 --recode --maf 0.01 --minGQ 20. Per-site nucleotide diversity was calculated with vcftools –sites-pi command.
Maximum likelihood phylogeny for predicted secreted proteins
Request a detailed protocolIn order to generate a maximum-likelihood phylogenetic tree based on the mature peptide sequences of the Bgh DH14 SPs, the 805 predicted secreted protein sequences were aligned using MAFFT v7.310 (Katoh and Standley, 2013) with the settings --maxiterate 1000 --localpair. The alignment was then passed to IQTree v1.6.beta4 (Nguyen et al., 2015) with the settings -nt 10 -mem 12G -bb 1000. The resulting tree was then visualized using iTOL (Letunic and Bork, 2016).
Generation of expression constructs
Request a detailed protocolAll genes with or without stop codons, were amplified from the cDNA of Bgh isolates (Lu et al., 2016) or plasmid templates (Seeholzer et al., 2010) using Phusion Hot Start II high-fidelity DNA polymerase (Thermo Scientific) and subsequently cloned into pENTR/D-TOPO (KmR) (Thermo Scientific) or synthesized as pDONR221 (KmR) entry clones from GeneArt (Thermo Scientific). The sequence integrity of all clones was confirmed by Sanger sequencing (Eurofins). Primers for AVRa amplification were designed to replace the signal peptide with the ATG start codon (Supplementary file 6).
For transient gene expression assays in planta and for yeast 2-hybrid interaction studies, respective genes were transferred from entry or donor vectors into the expression vectors pIPKb002 (SpcR) (Himmelbach et al., 2007), pGWB517 (SpcR) (Nakagawa et al., 2007), pXCSG-GW-mYFP (CarbR) (García et al., 2010), pLexA-GW (CarbR), or pB42AD-GW (CarbR) (Shen et al., 2007) as indicated using LR Clonase II (Thermo Scientific).
For the split-LUC assay, genes of interest were transferred from expression vectors into pDONR207 (GmR) using BP clonase II (Thermo Scientific) and subsequently cloned into pDEST-GW-nLUC (KmR) or pDEST-GW-cLUC (KmR) (Gehl et al., 2011) using LR Clonase II. Alternatively, pENTR/D-TOPO or pDONR221 entry clones were double-digested using PvuI and NruI (NEB) to remove KmR and linearized constructs were transferred directly into pDEST-GW-nLUC (KmR) or pDEST-GW-cLUC (KmR) using LR clonase II; the integrity of the resulting expression constructs was examined by Sanger sequencing (Eurofins).
Transient gene expression and cell death assay in barley protoplasts
Request a detailed protocolAssessment of protoplast cell death using a luciferase activity as a proxy for cell viability was adapted from (Lu et al., 2016) with the following modifications: cDNAs of the AVRa candidate genes lacking their respective signal peptides (SPs) were co-expressed together with cDNAs of the corresponding MLA receptors from the same strong ubiquitin promotor in the same barley genetic background to directly compare cell death activities mediated by different MLA and AVRA pairs. For this, the epidermis of the second leaves from seven to eight-day-old plants of the cultivar Golden Promise was removed before leaves were immersed in the enzyme solution. A total volume of 35 µl water containing 5 µg of the luciferase reporter plasmid, 12 µg of the respective Mla construct, and 6 µg of the respective effector construct or an EV was transfected into 300 µL barley protoplasts at a concentration of 3 × 105 protoplasts/ml solution. For co-transfection of AVRa10 and EKA_AVRa10, 5 µg of the luciferase reporter plasmid, 10 µg of the Mla10 construct, 4 µg of AVRa10, and either 4 µg of EKA AVRa10 or 4 µg of EV were transfected. At 16 hr after transfection, protoplasts were collected by centrifugation at 1000 × g, the supernatant was discarded, and 200 µl 2x cell culture lysis buffer were added (Promega, E1531). Luciferase activity was determined by mixing 50 µl of protoplast lysate with 50 µl luciferase substrate (Promega, E1501) in a white 96-well plate and light emission was measured 1 s/well using a microplate luminometer (Centro, LB960). Relative luciferase reads (Figure 4—source datas 1–3, Figure 4—figure supplement 1—source datas 1–3), were calculated by setting the control empty vector sample read of each individual experiment to 1.
Transient gene expression by Agrobacterium-mediated transformation of Nicotiana benthamiana leaves
Request a detailed protocolAgrobacterium tumefaciens GV3101::pMP90 and A. tumefaciens GV3101::pMP90K were freshly transformed with respective constructs of interest and grown from single colonies in liquid Luria broth medium containing appropriate antibiotics for ~24 hr at 28°C to an OD600 not higher than 1.5. Bacterial cells were harvested by centrifugation at 2500 × g for 15 min followed by resuspension in infiltration medium (10 mM MES, pH 5.6, 10 mM MgCl2, and 200 µM acetosyringone) to a final OD600 = 1.2. Cultures were incubated for 2 to 4 hr at 28°C with 180 rpm shaking before infiltration into leaves from three to five-week-old N. benthamiana plants. Bacteria carrying AVRa constructs or EV plasmid were mixed equally with Mla plasmid-carrying bacteria. Tissue for immunodetection analysis was harvested two days post infiltration and cell death scores (Figure 4—figure supplement 2—source datas 4,5) were assessed three days post infiltration throughout.
Plant protein extraction and pull-down for fusion protein detection by immunoblotting
Request a detailed protocolFrozen leaf material was ground to a fine powder using pre-cooled adapters in a bead beater (Retsch) and thawed in cold plant protein extraction buffer (150 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM EDTA, 10% (v/v) glycerol, 5 mM DTT, 2% (v/v) plant protease inhibitor cocktail (Sigma), 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and 0.5% (v/v) IGEPAL) at a ratio of 150 mg fresh tissue/1 ml of extraction buffer. Extracts were centrifuged twice at 15,000 × g for 15 min at 4°C. For SDS-PAGE, extracts were diluted 4:1 with 4x SDS loading buffer and heated to 95°C for 5 min.
For pull-down of mYFP-tagged proteins, GFP-Trap-MA (Chromotek) beads were incubated in equilibration buffer (Saur et al., 2015) for 1 hr at 4°C and subsequently mixed with protein extracts for 2 to 3 hr at 4°C with slow but constant rotation. Then, conjugated GFP-Trap beads were washed five times in 1 ml of cold wash buffer (Saur et al., 2015) at 4°C before interacting proteins were stripped from the beads by boiling in 25 μl of 4x SDS loading buffer for 5 min.
Samples were separated on 10% SDS-PAGE gels, blotted onto PVDF membrane, and probed with anti-GFP (Santa Cruz Biotechnology sc-8334, RRID:AB_641123; or abcam ab6556, RRID:AB_305564), anti-LUC (Sigma L0159) or anti-c-Myc (abcam ab9106, RRID:AB_307014), followed by anti-rabbit IgG-HRP (Santa Cruz Biotechnology sc-2313, RRID:AB_641181) secondary antibodies. Proteins were detected by the HRP activity on SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher 34095) using a Gel Doc XR +Gel Documentation System (Bio-Rad).
Split-luciferase complementation assay
Request a detailed protocolTo obtain protein extracts for luciferase measurements, one leaf disk with a diameter of 0.38 cm was harvested from three different leaves at two days post transformation, resulting in three leaf disks/sample. Samples were frozen in liquid nitrogen and ground to a fine powder using a pre-cooled adapter in a Retsch bead beater. Each sample was thawed in 100 µl 2x cell culture lysis buffer (Promega, E1531) supplemented with Tris-HCl, pH 7.5 to a final concentration of 150 mM. Luciferase activity was determined by mixing 50 µl of leaf extract with 50 µl luciferase substrate (Promega, E1501) in a white 96-well plate and light emission (Figure 5—source datas 1–3) was measured 1 s/well in a microplate luminometer (Centro, LB960). Complementation of a functional LUC protein by genetic fusion of bait/prey with the nucleotides encoding the N-terminal 416 aa of LUC (nLUC) and C-terminal 152 aa of LUC (cLUC) allows the detection of a real‐time and reversible signal for direct interaction (Bosmans et al., 2016; Chen et al., 2008).
Yeast 2-hybrid assay and yeast protein extraction
Request a detailed protocolMla variants were cloned into the pLexA-GW vector (Shen et al., 2007) for expression with an N-terminal LexA activation domain under the control of a constitutive ADH1 promoter (BD-MLA). The AVRa variants were cloned into pB42AD-GW (Shen et al., 2007) for expression with an N-terminal B42 activation domain followed by the HA-tag under the control of an inducible GAL1 promoter (AD-AVRA). Using the lithium acetate method (Gietz and Woods, 2002), Mla bait constructs and AVRa13 prey constructs were co-transformed into the yeast strain EGY4.8 p8op-lacZ and successful transformants were selected by colony growth on SD-UHW/Glu (4% (w/v) Glucose, 0.139% (w/v) yeast synthetic drop-out medium pH 5.8 without uracil, histidine, tryptophan, 0.67% (w/v) BD Difco yeast nitrogen base, 2% (w/v) Bacto Agar). Yeast transformants were grown to OD600 = 1 in liquid SD-UHW/Glu before harvesting cells for drop out of the dilution series on SD-UHW/Gal/Raf media (SD-UHW without glucose but with 2% (w/v) Galactose 1% (w/v) Raffinose, with (-UHW) or without Leucine (-UHWL)) and incubated for six days at 30°C followed by room temperature incubation for another six days.
For protein detection, yeast strains were grown to OD600 = 1 in SD-UHW/Gal/Raf liquid medium at 30°C and 200 rpm shaking, and proteins were extracted using 200 mM NaOH (NaOH method; Zhang et al., 2011). Total protein samples were separated on 9% or 12% SDS-PAGE gels, blotted onto PVDF membrane, and probed with anti-HA (Merck, clone 3F10, RRID:AB_390914) or anti-LexA (Santa Cruz Biotechnology, sc7544, RRID:AB_627883) primary antibodies followed by anti-rat (Santa Cruz Biotechnology, sc2065, RRID:AB_631756) or anti-mouse IgG-HRP (Santa Cruz Biotechnology, sc2005, RRID:AB_631736) secondary antibodies as appropriate. HA and LexA fusion proteins were detected by HRP activity on SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher 34095) using a Gel Doc XR +Gel Documentation System (Bio-Rad).
Data availability
RNA sequencing data have been deposited in GEO under accession code GSE110266 and improved Blumeria graminis f.sp. hordei isolate K1 assembly is deposited under the accession number PRJEB30373 at EBI-ENA. All data generated or analysed during this study are included in the manuscript and supporting files.
-
European Nucleotide ArchiveID PRJEB30373. Improved Blumeria graminis f.sp. hordei isolate K1 assembly.
-
NCBI Gene Expression OmnibusID GSE110266. Identification of the AVRa7,AVRa9, AVRa10 and AVRa22 effector genes from barley powdery mildew fungus (Bgh) association analysis between transcript polymorphisms and AVRa phenotypes from 27 Bgh isolates.
-
NCBI Gene Expression OmnibusID GSE83237. Identification of avirulence genes in the barley powdery mildew fungus (Bgh) by RNA-sequencing and transcriptome-wide association analysis on a set of Bgh isolates.
References
-
Supramolecular control over Split-Luciferase complementationAngewandte Chemie 55:02807.https://doi.org/10.1002/anie.201602807
-
The variant call format and VCFtoolsBioinformatics 27:2156–2158.https://doi.org/10.1093/bioinformatics/btr330
-
Pathogenic divergence of central european and australian populations of blumeria graminis f. sp. hordeiThe Annals of Applied Biology 165:.https://doi.org/10.1111/aab.12141
-
Purification of high molecular weight genomic DNA from powdery mildew for Long-Read sequencingJournal of Visualized Experiments 121:.https://doi.org/10.3791/55463
-
The powdery mildews: a review of the world's most familiar (yet poorly known) plant pathogensAnnual Review of Phytopathology 46:27–51.https://doi.org/10.1146/annurev.phyto.46.081407.104740
-
Microbial genome-enabled insights into plant-microorganism interactionsNature Reviews Genetics 15:797–813.https://doi.org/10.1038/nrg3748
-
A set of modular binary vectors for transformation of cerealsPlant Physiology 145:1192–1200.https://doi.org/10.1104/pp.107.111575
-
Evolution and Conservation of Plant NLR FunctionsFrontiers in Immunology 4:297.https://doi.org/10.3389/fimmu.2013.00297
-
Genetics of powdery mildew resistance in barleyCritical Reviews in Plant Sciences 13:97–119.https://doi.org/10.1080/07352689409701910
-
MAFFT multiple sequence alignment software version 7: improvements in performance and usabilityMolecular Biology and Evolution 30:772–780.https://doi.org/10.1093/molbev/mst010
-
Defining the origins of the NOD-like receptor system at the base of animal evolutionMolecular Biology and Evolution 28:1687–1702.https://doi.org/10.1093/molbev/msq349
-
The sequence alignment/Map format and SAMtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
Frequencies of virulence and fungicide resistance in the european barley mildew population in 1985Journal of Phytopathology 119:298–311.https://doi.org/10.1111/j.1439-0434.1987.tb04401.x
-
NLR functions in plant and animal immune systems: so far and yet so closeNature Immunology 12:817–826.https://doi.org/10.1038/ni.2083
-
Subfamily-Specific specialization of RGH1/MLA immune receptors in wild barleyMolecular Plant-Microbe Interactions 32:107–119.https://doi.org/10.1094/MPMI-07-18-0186-FI
-
IntFOLD: an integrated server for modelling protein structures and functions from amino acid sequencesNucleic Acids Research 43:W169–W173.https://doi.org/10.1093/nar/gkv236
-
Evolutionary convergence and divergence in NLR function and structureTrends in Immunology 38:744–757.https://doi.org/10.1016/j.it.2017.04.005
-
Evolving disease resistance genesCurrent Opinion in Plant Biology 8:129–134.https://doi.org/10.1016/j.pbi.2005.01.002
-
Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformationJournal of Bioscience and Bioengineering 104:34–41.https://doi.org/10.1263/jbb.104.34
-
IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogeniesMolecular Biology and Evolution 32:268–274.https://doi.org/10.1093/molbev/msu300
-
Filamentous pathogen effector functions: of pathogens, hosts and microbiomesCurrent Opinion in Plant Biology 20:96–103.https://doi.org/10.1016/j.pbi.2014.05.001
-
The N-terminal domain of the tomato immune protein prf contains multiple homotypic and pto kinase interaction sitesJournal of Biological Chemistry 290:11258–11267.https://doi.org/10.1074/jbc.M114.616532
-
Diversity at the mla powdery mildew resistance locus from cultivated barley reveals sites of positive selectionMolecular Plant-Microbe Interactions 4:.https://doi.org/10.1094/MPMI-23-4-0497
Article and author information
Author details
Paul Schulze-Lefert

Funding
Deutsche Forschungsgemeinschaft (SFB670)
- Barbara Kracher
- Takaki Maekawa
- Paul Schulze-Lefert
Max-Planck-Gesellschaft (Open-access funding)
- Saskia Bauer
- Paul Schulze-Lefert
European Molecular Biology Organization (ALTF 368-2016)
- Isabel ML Saur
Cluster of Excellence in Plant Sciences (CEPLAS 1028)
- Paul Schulze-Lefert
Deutsche Forschungsgemeinschaft (SPP1819)
- Lamprinos Franzeskakis
- Ralph Panstruga
Daimler und Benz Stiftung
- Isabel ML Saur
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Sabine Haigis for maintaining the Bgh isolates; Petra Köchner for technical assistance; and the Max Planck Genome Centre Cologne for RNA-seq. This work was supported by the Max-Planck Society (IMLS, SB, BK, XL and PS-L); German Research Foundation in the Collaborative Research Centre Grant SFB670 (to BK, TM, and PS-L); Cluster of Excellence on Plant Sciences (CEPLAS 1028 to PS-L), European Molecular Biology Organization (ALTF 368–2016 to IMLS); Daimler and Benz Foundation (to IMLS) and German Research Foundation-funded Priority Programme SPP1819 (PA 861/14–1 to RP).
Copyright
© 2019, Saur et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 6,392
- views
-
- 1,082
- downloads
-
- 131
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 131
- citations for umbrella DOI https://doi.org/10.7554/eLife.44471
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Multiple pairs of allelic MLA immune receptor-powdery mildew AVRA effectors argue for a direct recognition mechanism
eLife 8:e44471.
https://doi.org/10.7554/eLife.44471




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}