Multi-enhancer transcriptional hubs confer phenotypic robustness
- European Molecular Biology Laboratory, Germany
- Collaboration for joint PhD degree between EMBL and Heidelberg University, Faculty of Biosciences, Germany
Abstract
We previously showed in Drosophila melanogaster embryos that low-affinity Ultrabithorax (Ubx)-responsive shavenbaby (svb) enhancers drive expression using localized transcriptional environments and that active svb enhancers on different chromosomes tended to colocalize (Tsai et al., 2017). Here, we test the hypothesis that these multi-enhancer ‘hubs’ improve phenotypic resilience to stress by buffering against decreases in transcription factor concentrations and transcriptional output. Deleting a redundant enhancer from the svb locus led to reduced trichome numbers in embryos raised at elevated temperatures. Using high-resolution fluorescence microscopy, we observed lower Ubx concentration and transcriptional output in this deletion allele. Transcription sites of the full svb cis-regulatory region inserted into a different chromosome colocalized with the svb locus, increasing Ubx concentration, the transcriptional output of svb, and partially rescuing the phenotype. Thus, multiple enhancers could reinforce a local transcriptional hub to buffer against environmental stresses and genetic perturbations, providing a mechanism for phenotypical robustness.
https://doi.org/10.7554/eLife.45325.001Introduction
During embryogenesis, transcriptional regulation controls precise patterns of gene-expression, leading to cell-fate specification (Long et al., 2016; Mallo and Alonso, 2013; Reiter et al., 2017; Spitz and Furlong, 2012). This involves coordinating a complex series of interactions between transcription factors and their target binding sites on DNA, leading to the recruitment or exclusion of active RNA polymerases, which determines the transcriptional state of the gene. Live imaging experiments have shown that transcription factor binding in eukaryotic cell lines and embryos is dynamic but transient, occurring frequently but with each event lasting for at most a few seconds (Chen et al., 2014; Izeddin et al., 2014; Liu et al., 2014; Normanno et al., 2015). Additionally, recent studies have shown that many developmental enhancers harbor functionally important low-affinity binding sites (Antosova et al., 2016; Crocker et al., 2010; Crocker et al., 2015; Crocker et al., 2016; Farley et al., 2015; Farley et al., 2016; Gaudet and Mango, 2002; Lebrecht et al., 2005; Lorberbaum et al., 2016; Rister et al., 2015; Rowan et al., 2010; Tanay, 2006). One example is the Homeobox (Hox) family that is responsible for body segment identity along the anterior-posterior axis in animals. Because Hox transcription factors descended from a common ancestor, their preferences for binding sequences are very similar (Berger et al., 2008; McGinnis and Krumlauf, 1992; Noyes et al., 2008). To select for specific Hox factors, several enhancers in the shavenbaby (svb) locus make use of low-affinity binding sequences for Ultrabithorax (Ubx) (Crocker et al., 2015). Svb is a transcription factor that drives the formation of trichomes, epidermal projections on the surface of the segmented fly embryo (Chanut-Delalande et al., 2006; Delon et al., 2003; Payre et al., 1999). Thus, a key question was how low affinity binding sequences are able to drive strong transcriptional activation in developing embryos.
We have previously shown in living Drosophila melanogaster embryos that Ubx transiently but repeatedly explores the same physical locations in a nucleus, which are likely clusters of binding sites (Tsai et al., 2017). We have additionally shown that transcriptional microenvironments of high local Ubx and cofactor concentrations surround active transcription sites driven by low-affinity svb enhancers. As the distributions of many transcription factors in the nucleus are highly heterogeneous, the transcriptional activity of low-affinity enhancers would depend on the local microenvironments. Interestingly, we observed that transcriptionally active, minimalized versions of two of the three ventral svb enhancers (E3 and 7) are Ubx-responsive and preferentially appear near or overlap spatially with transcription sites of the endogenous svb gene, despite being on different chromosomes (Crocker et al., 2015; Tsai et al., 2017). This colocalization suggests microenvironments could be shared between related enhancers to increase transcriptional output, where enhancers would synergistically form a larger local trap for transcription factors than each could alone. Retaining multiple enhancers within a microenvironment could also provide redundancy in case individual enhancers are compromised and buffer negative impacts when the system is subjected to stress. This idea is consistent with the observed phenotypic robustness of svb enhancers in maintaining sufficient trichome numbers even under temperature stress (Crocker et al., 2015; Frankel et al., 2010). These results are consistent with multi-component transcriptional ‘hubs’ that are local areas enriched for components of the transcriptional machinery and transcription factors through multiple attractive and cooperative interactions (Boija et al., 2018; Cisse et al., 2013; Furlong and Levine, 2018; Ghavi-Helm et al., 2014; Lim et al., 2018; Mir et al., 2017; Mir et al., 2018). One potential building block for these ‘hubs’ are multiple, long-range, enhancer-to-enhancer interactions. However, it is not yet understood how such multivalent interactions function mechanistically, and how they contribute to phenotypic robustness.
To understand the mechanistic implications of having multiple enhancers in a shared microenvironment, here we examined the ability of the svb locus to maintain transcriptional output and produce the correct phenotype under temperature-induced stress in flies harboring a deletion of a partially redundant enhancer—the DG3 ventral enhancer. When embryos were raised at high temperatures, we observed phenotypical defects in ventral trichome formation for the DG3-deletion svb allele but not for the wild-type. At the molecular level, Ubx concentrations around transcription sites of the DG3-deletion allele decreased. The transcriptional output of svb without DG3 also decreased. To test the hypothesis that shared microenvironments modulate transcriptional output and provide buffering under stress, we sought to rescue the DG3-deletion allele through inserting the complete svb cis-regulatory region on a BAC (svbBAC) on a different chromosome. We observed that Ubx concentration around active transcription sites of the DG3-deletion allele and their transcriptional output increased when the svbBAC is physically nearby. Moreover, we found that trichome formation was partially rescued at high temperature. As a result, our findings support the hypothesis that shared microenvironments provide a mechanism for phenotypic robustness.
Results
The DG3 enhancer responds specifically to Ubx in the A1 segment
The ventral svb enhancers DG3, E3 and 7 (Figure 1A) contain low-affinity Ubx binding sites and have been shown to be transcribed in microenvironments of high Ubx concentrations in the first abdominal (A1) segment on the ventral surface of the embryo (Tsai et al., 2017). Each of these enhancers produces ventral stripes of expression along segments A1-A7 in the embryo, resembling the endogenous expression pattern of svb (Figure 1B). Each enhancer contributes to different but partially overlapping portions of the total expression pattern. Furthermore, they have different Ubx ChIP enrichment profiles (Figure 1—figure supplement 1). Whereas the interaction of E3 and 7 with Ubx had been previously explored in detail (Crocker et al., 2015), DG3 remained unexplored. Therefore, we tested the response of the DG3 enhancer to Ubx by altering Ubx levels and measuring the transcriptional output with a reporter gene (lacZ). In wild-type embryos, the DG3 reporter gene was expressed ventrally in stripes along segments A1-A7, in addition to narrow thoracic stripes in T1-T3 (Figure 1C). Expression from DG3 on the ventral surface in the T2 and T3 segments was weak with wild-type Ubx expression and was primarily seen on the sides. In the absence of Ubx, DG3 reporter expression was almost completely lost on the ventral side of A1 and reduced between A2-A7 (Figure 1D), consistent with the responses of E3, 7 and the full svb locus (Crocker et al., 2015). The incomplete loss of expression in A2-A7 suggests that additional factors influence the expression of ventral trichomes in those segments. Ubiquitous expression of Ubx increased the expression levels in A1-A7, in addition to generating ectopic expressions on the ventral side of the thoracic segments T2-T3 and A8 (Figure 1E). In summary, we showed that Ubx is necessary for DG3 expression in the ventral region of the abdominal segments (completely in A1 and partially in A2-7) and can induce ectopic expressions when overexpressed. These data are consistent with our previous observation of the localization of DG3-driven transcription sites within Ubx microenvironments (Tsai et al., 2017).
Figure 1 with 1 supplement see all
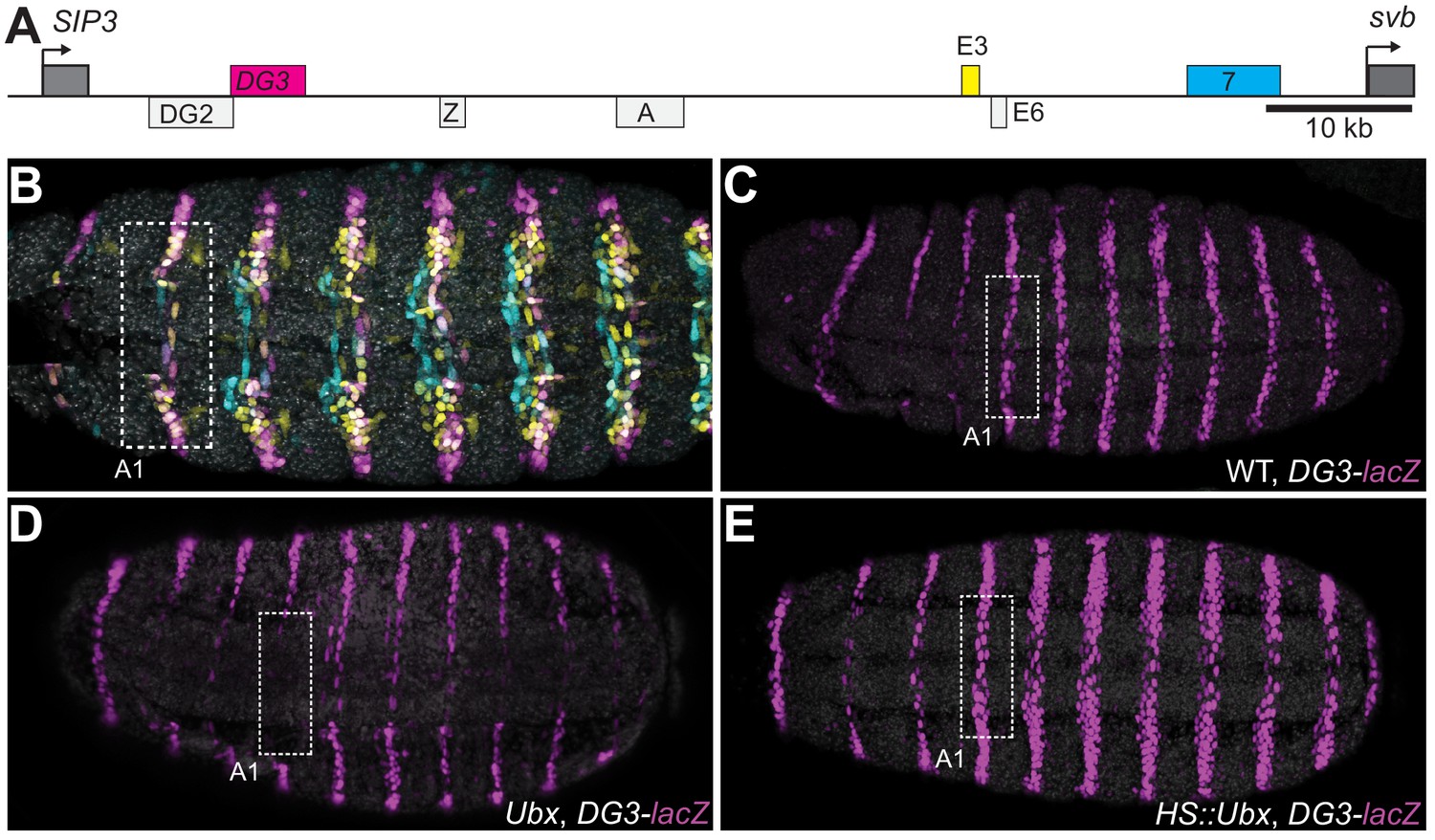
Ubx drives the expression of the DG3 shavenbaby enhancer along the ventral abdominal segments.
(A) The cis-regulatory region of the shavenbaby (svb) gene contains three enhancers expressing stripes on the ventral side of the abdominal segments: DG3, E3 and 7. (B) The expression patterns of the three enhancers are partially overlapping. The color scheme corresponds to (A), where DG3 is magenta, E3 is yellow and 7 is cyan. (C) Expression pattern of a reporter construct with the DG3 enhancer driving LacZ expression in an embryo with wild-type Ubx expression, as visualized using immunofluorescence staining. (D) DG3 reporter in Ubx null mutant shows no expression in A1 and significantly weakened expression in the other abdominal segments. (E) Overexpression of Ubx driven through a heat shock promoter induces overexpression of DG3 reporter in all abdominal segments and ectopic expression on the ventral surface of the thoracic segments T2 and T3.
Deletion of a region including DG3 enhancer causes defects in ventral trichome formation specifically at elevated temperatures
Given the clear ventral stripes that DG3 generated in the abdominal segments, we next explored the phenotypic impact of its activity in driving trichome formation. It had been previously shown that deleting a region in the svb locus containing DG3 (Df(X)svb108) led to reduced phenotypic robustness of svb under non-optimal temperatures, with reduced numbers of trichomes produced (Frankel et al., 2010). This svb DG3-deletion allele encompasses the enhancers DG2, DG3 and Z (Figure 2A)—of which only DG3 is a ventral enhancer.
Figure 2 with 1 supplement see all
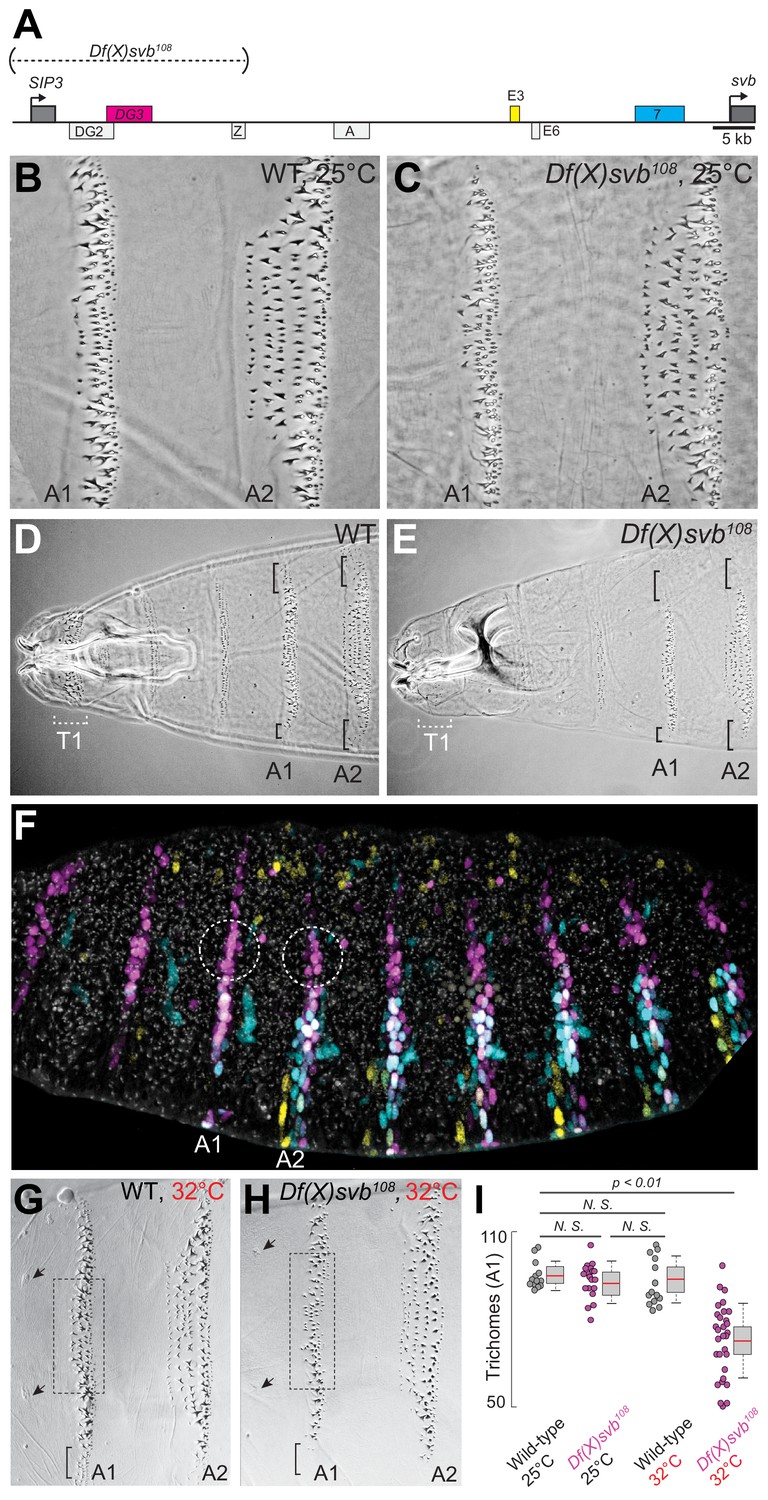
Deletion of a region from the svb locus containing DG3 reduces ventral trichome numbers under heat-induced stress.
(A) The Df(X)svb108 allele contains a deletion in the cis-regulatory of svb spanning three enhancers: DG2, DG3 and Z. Of those, only DG3 expresses on the ventral side. (B) Wild-type phenotype of trichomes along the A1 and A2 segments at 25°C. (C) At 25°C, the Df(X)svb108 deletion allele did not show a mutant phenotype along the A1 and A2 segments. (D) Zoomed out shot of the anterior region of a cuticle preparation of a wild-type (w1118) larva. (E) Zoomed out shot of the anterior region of a cuticle preparation of a larva carrying Df(X)svb108. The lack of trichomes along the T1 segment is a recessive marker used in subsequent experiments to select for embryos/larvae carrying this deletion allele. Even at 25°C, where the overall trichome numbers in A1 and A2 for the Df(X)svb108 deletion mutant is indistinguishable from wild-type svb, the trichomes at the side of the ventral stripe for A1 and A2 were lost, as marked by the black brackets. (F) Within the overall expression pattern of svb enhancers, DG3 provides exclusive coverage in the circled regions in segments A1 and A2. These regions correspond to the black brackets in panels D and E. (G) Wild-type phenotype of trichomes along the A1 and A2 segments at 32°C. (H) At 32°C, the Df(X)svb108 deletion allele showed a mutant phenotype. (I) A1 trichomes in the dashed boxes bounded by the two sensory cells, as indicated by the arrows, as shown in panels G and H, were counted. Deficiencies of the deletion allele only become clear when the animal is subjected to elevated temperature at 32°C, showing reduced trichome numbers in the A1 segment. The number of larvae counted was: 13 for wild-type at 25°C, 19 for Df(X)svb108 at 25°C, 14 for wild-type at 32°C and 30 for Df(X)svb108 at 32°C. Two-tailed t-test was applied for each individual comparison. In box plots, center line is the mean, upper and lower limits are standard deviation and whiskers show 95% confidence intervals.
In the A1 and A2 segments at 25°C, deletion of the DG3 enhancer did not result in a clear change in ventral trichome formation in the abdominal segments compared to the wild-type (Figure 2B and C), perhaps due to the redundancy provided by overlapping expression patterns from other svb enhancers. However, the T1 trichomes were missing in larvae homozygous for the deletion (Df(X)svb108) allele (Figure 2D and E), which we subsequently used as a homozygous marker to select for larvae homozygous for the deletion allele when crossing Df(X)svb108 flies to other lines (See ‘Cuticle preparations and trichome counting’ in Materials and Methods). Also, we observed defects in trichome formation in the dorsal edges of the stripe pattern, which are exclusively covered by DG3 (Figure 2D and E, the black brackets at A1 and A2). This is consistent with a lack of redundancy in enhancer usage in these areas (Figure 2F, white dotted circles). The trichome number in regions covered by the overlapping expression of the E3, 7 and DG3 enhancers in the A1 segment did not significantly reduce at 25°C upon the deletion of DG3 (Figure 2B,C and I). However, larvae homozygous for the Df(X)svb108 allele developed at 32°C produced fewer trichomes compared to wild-type larvae (Figure 2G,H and I). We also observed similar effects in the A2 segment (Figure 2—figure supplement 1). These results are similar to those shown with quartenary A5 trichomes (Frankel et al., 2010). However, the mechanisms behind this loss of phenotypic robustness under heat-induced stress are yet to be understood in detail.
Transcription sites from the DG3-deletion allele have weaker Ubx microenvironment and lower transcriptional output
To address molecular sources that may lead to the reduced number of ventral trichomes we observed for the Df(X)svb108 deletion allele, we imaged Ubx distributions and the transcriptional output of the svb gene in fixed Drosophila melanogaster embryos using high-resolution confocal microscopy. We reasoned that the defect could be due to changes in the transcription factor concentration around the enhancers (input) and/or the transcriptional output of the gene (output). As Ubx is specifically needed to drive DG3 and svb expression on the ventral surface of the A1 segment, we used it as a metric for the transcription factor distributions around svb transcription sites. The samples were stained with immunofluorescence (IF) for Ubx and RNA fluorescence in situ hybridization (FISH) for svb transcription sites as previously described (Tsai et al., 2017). We imaged both embryos containing the wild-type svb allele or the Df(X)svb108 allele, raised at either 25°C or 32°C. For all imaging experiments involving the svb allele with the deletion, we selected only homozygous embryos for imaging (See ‘Imaging fixed embryos’ in Materials and Methods). Because DG3 expression in the A1 segment showed a clear link to changes in Ubx level, we focused most of our subsequent imaging quantifications in this segment.
To gauge the Ubx concentration around a transcription site, we counted the averaged intensity in the Ubx IF channel within a circle four pixels in diameter (170 nm, roughly the resolution limit of AiryScan) centered on the transcription site (Figure 3A and B, see ‘Analysis of microenvironment and svb transcription intensity’ in materials and methods). In nuclei from the A1 segment, Ubx intensities around svb transcription sites with the wild-type allele did not significantly change between 25°C and 32°C (Figure 3C, bottom right panel). Measuring Ubx intensity from random locations within nuclei of wild-type embryos expressing svb at 25°C using the same method showed that Ubx concentrations around svb transcription sites were in general higher than the nuclear average (Figure 3—figure supplement 1), consistent with our previous findings (Tsai et al., 2017). Transcription sites in embryos with the DG3-deletion (Df(X)svb108) allele had a local Ubx concentration that is slightly lower than wild-type at 25°C (Figure 3C, bottom right panel). However, there was a clear decrease in Ubx intensity compared to the wild-type when we subjected the DG3-deletion embryos to heat-stress (Figure 3B, right panel, and 3C, bottom right panel). To measure the transcriptional output of svb, we adopted the same approach, but quantified the intensity in the svb RNA FISH channel (Figure 3C, upper left panel). Interestingly, we detected clear decreases in transcriptional output when the embryos are heat-stressed at 32°C, even with the wild-type allele. The Df(X)svb108 allele at 25°C showed reduced levels of transcriptional output slightly lower than the wild-type under heat-shock. At 32°C, the transcriptional output further decreased in the mutant. When the various conditions were plotted by their svb transcriptional output and Ubx intensity (Figure 3C, center panel), they displayed a weak positive correlation with Ubx concentration (R-square = 0.4331). In sum, stress conditions reduced the transcriptional output of enhancers and the correlation to Ubx concentrations was positive but weak.
Figure 3 with 1 supplement see all
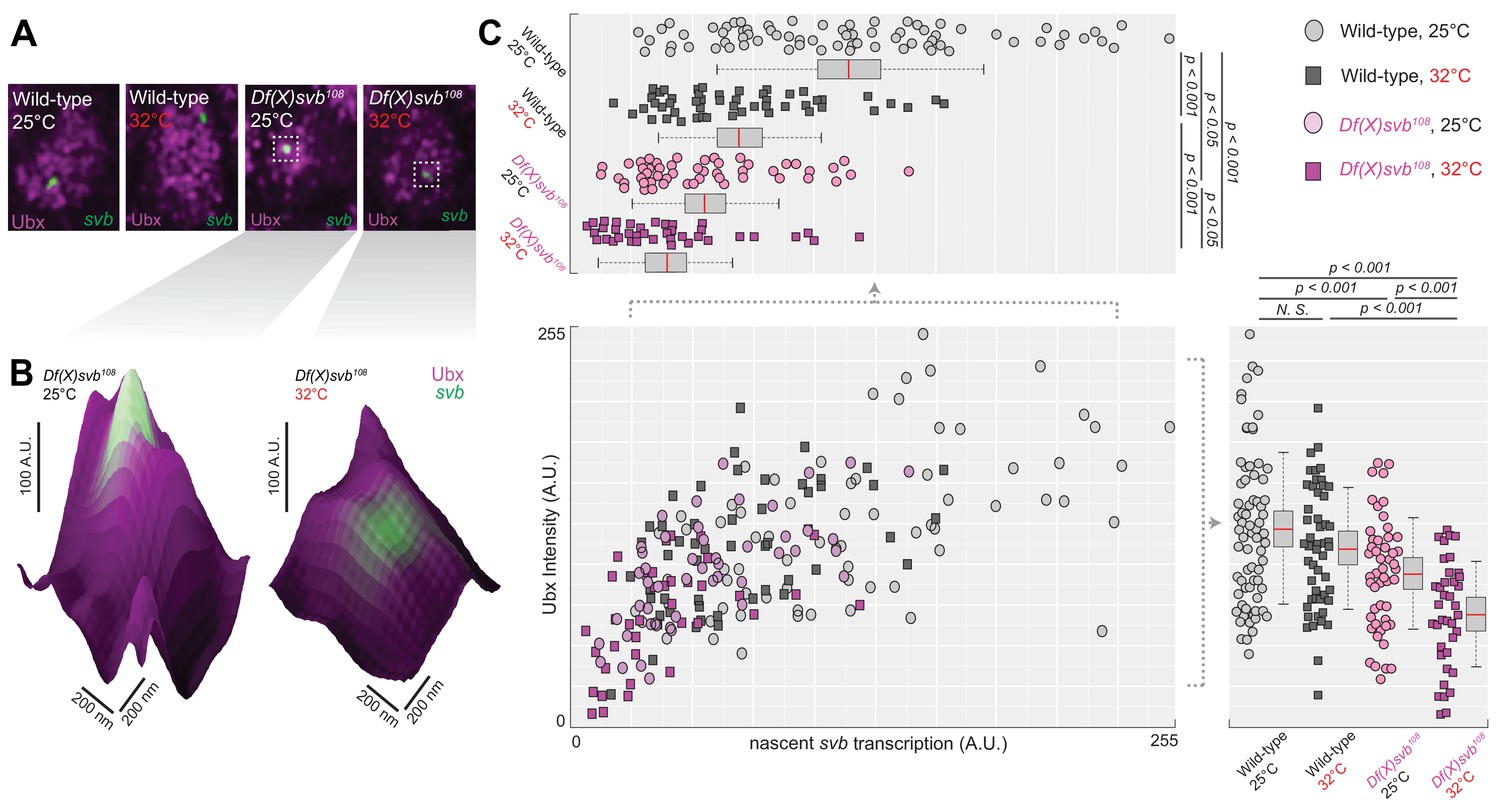
Deletion of the cis-region of svb containing DG3 led to defects in the Ubx microenvironment and svb transcriptional output.
(A) Panels showing a nucleus from embryos with either the wild-type (w1118) or Df(X)svb108 deletion svb allele at either at normal (25°C) or elevated temperature (32°C), imaged using confocal fluorescence microscopy. Ubx (shown in magenta) is stained using immunofluorescence (IF) and the svb transcription sites (shown in green) are stained using fluorescence in situ hybridization (FISH). (B) Zoomed-in panes centered on svb transcription sites, with the height of the surface plots representing the Ubx intensity. (C) Correlation between Ubx and svb transcription intensities at different conditions. The number of transcription sites quantified was: 71 for wild-type at 25°C, 51 for wild-type at 32°C, 50 for Df(X)svb108 at 25°C and 38 for Df(X)svb108 at 32°C. Bottom right panel: Integrating the Ubx intensity surrounding transcription sites shows a small defect in the Ubx concentration around the deletion allele. The drop in Ubx increased at elevated temperature. Upper left panel: The integrated intensity of svb transcriptional output shows that there is a drop in transcriptional output for the deletion allele compared to the wild-type at both 25°C and 32°C. Even the wild-type showed reduced transcriptional output at elevated temperature (32°C). We analyzed four embryos for each genotype/temperature combination. Two-tailed t-test was applied for each individual comparison. In box plots, center line is mean, upper and lower limits are standard deviation and whiskers show 95% confidence intervals.
Df(X)svb108 deficiencies are rescued upon insertion of the full svb cis-regulatory region in a different chromosome
Having observed in the past that transcriptional microenvironments can be shared between related svb enhancers on different chromosomes (Tsai et al., 2017), we wondered whether this phenomenon could enhance transcriptional output and thus buffer against adverse environmental conditions. Therefore, we tested the capacity of a DNA sequence containing the full svb cis-regulatory region to rescue the described molecular and developmental defects of the Df(X)svb108 allele (Figure 4A). For this purpose, we used a transgenic fly line, where a bacterial artificial chromosome (BAC) carrying the complete cis-regulatory region of svb (Preger-Ben Noon et al., 2018) was integrated into chromosome 2. To exclude svb mRNA from effecting the rescue, this svbBAC construct drives a dsRed reporter gene instead of another copy of svb. We confirmed that DsRed protein expression driven by this regulatory sequence recapitulated the svb expression patterns (Figure 4B) in D. melanogaster embryos and was responsive to Ubx—the lack of Ubx led to a decrease of expression in the A1 segment (Figure 4C).
Figure 4 with 1 supplement see all
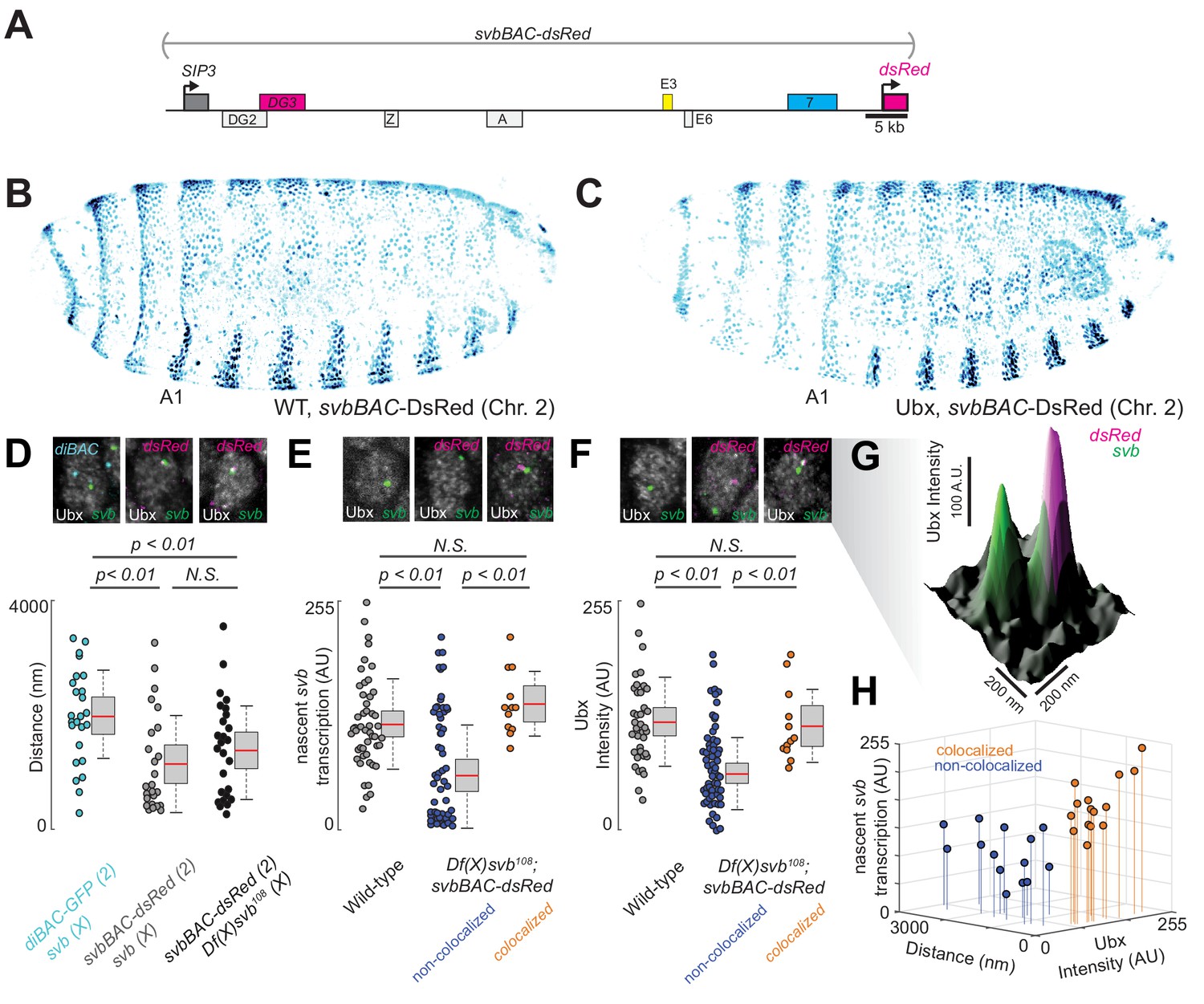
Introduction of the cis-regulatory region of svb on another chromosome rescues the microenvironment deficiencies of the Df(X)svb108 deletion mutant.
(A) The svbBAC construct encompasses the entire cis-regulatory region of svb, driving the expression of dsRed. (B and C) The svbBAC driving the expression of DsRed inserted into the second chromosome drives a similar expression pattern as the wild-type svb locus and responds similarly to Ubx. (D) In nuclei having both svb (for both the wild-type and the Df(X)svb108 allele) and svbBAC-dsRed transcription sites at 32°C, the distances between them areon average closer than between that of svb and a reporter construct of an unrelated gene, diachete (diBAC-gfp, at 25°C), inserted into the same location as svbBAC on the second chromosome. The pairs of distance quantified were: 25 between diBAC-gfp and wild-type svb, 25 between svbBAC-dsRed and wild-type svb and 26 between svbBAC-dsRed and Df(X)svb108. (E) The svb FISH intensity (representing transcriptional output) in Df(X)svb108 x svbBAC-dsRed embryos at 32°C recovered to wild-type levels when the svb transcription site is close to a dsRed transcription site (colocalized). FISH intensity of svb in the same embryos in nuclei without a dsRed transcription site or where svb and dsRed transcription sites were not near each other (non-colocalized) did not recover. The center image panel was stained for dsRed but the particular nucleus does not show dsRed signal. The number of transcription sites quantified was: 49 for wild-type, 53 for Df(X)svb108 not near a dsRed transcription site (including cells where there were no dsRed transcription sites) and 12 for Df(X)svb108 near a dsRed transcription site. (F) At 32°C, Ubx concentration around svb transcription sites recovered to wild-type levels in nuclei containing colocalized svb and dsRed transcription sites (colocalized) in Df(X)svb108 x svbBAC-dsRed embryos. Ubx levels around transcription sites of svb in the same embryos in nuclei without a dsRed transcription site or where svb and dsRed transcription sites were not near each other (non-colocalized) did not recover. The number of transcription sites quantified was: 38 for wild-type, 60 for Df(X)svb108 not near a dsRed transcription site (including cells where there were no dsRed transcription sites) and 12 for Df(X)svb108 near a dsRed transcription site. Two-tailed t-test was applied for each individual comparison. In box plots, center line is mean, upper and lower limits are standard deviation and whiskers show 95% confidence intervals. (G) A surface plot (the height representing Ubx intensity) showing two svb transcription sites in a nucleus, with the one on the right overlapping with a svbBAC-dsRed transcription site and showing higher Ubx concentration. (H) At 32°C, in nuclei having both svb (from the Df(X)svb108 allele) and svbBAC-dsRed transcription sites, the distances between them are plotted against svb FISH intensity and Ubx intensity. There are two clusters separated by a threshold of 360 nm in distance. Co-localized pairs below this distance threshold present higher intensities for both Ubx intensity and nascent svb transcription. The pairs quantified were 15 for colocalized and 14 for non-colocalized between svbBAC-dsRed and Df(X)svb108. We analyzed four embryos for diBAC-gfp x w1118, four embryos for svbBAC-dsRed x Df(X)svb108 and for five embryos for svbBAC-dsRed x w1118.
To test the rescue, embryos or larvae with a svbBAC-dsRed crossed into them were incubated at 32°C. We observed that many active dsRed transcription sites were close to svb transcription sites in nuclei expressing both svb and dsRed in embryos from crosses between svbBAC-dsRed and wild-type (w1118) flies (Figure 4D). Similar observations were previously seen for the endogenous svb locus with itself and with the other two ventral enhancers (E3 and 7) (Tsai et al., 2017). This observation was also true for embryos from crosses between svbBAC-dsRed and Df(X)svb108 flies, suggesting that the co-localization of related regulatory regions could occur under stressed conditions. This effect was not observed for the unrelated regulatory region of diachete driving the expression of gfp, which was inserted on a BAC in the same chromosomal location as svbBAC.
We observed that the introduction of the svb regulatory region was able to rescue both molecular and functional defects observed from the loss of the region containing DG3. Both svb transcriptional output (Figure 4E) and local Ubx concentration around svb transcription sites (Figure 4F and G) were restored to wild-type levels, but only when they co-localized with an active svbBAC-dsRed transcription site in the same nucleus (within 360 nm from each other, see ‘Analysis of distances between transcription spots’ in materials and methods for the definition of colocalization). The pairs of transcription sites under and above this threshold clustered in two groups distinguishable also by Ubx and svb transcription levels (Figure 4H). Wild-type (w1118) x svbBAC-dsRed embryos and larvae were similar to wild-type in both trichome number and Ubx levels around svb transcription sites (Figure 4—figure supplement 1).
The phenotype, ventral trichome formation on the A1 segment (Figure 5A–C), which is reduced with the DG3-deletion allele, was partially rescued by the introduction of svbBAC (Figure 5D). The loss of the outer edge trichomes in A1 (in the black brackets in Figure 5A–C, where only DG3 provides coverage Figure 2F) with the DG3-deletion allele was not rescued with svbBAC. Additionally, introducing only the DG3 enhancer as opposed to svbBAC did not rescue trichome formation under heat-stress (Figure 5D).
Figure 5

The complete cis-regulatory region of svb rescues trichome number.
(A–C) Cuticle preparations from larvae developed at 32°C with wild-type svb, Df(X)svb108 and Df(X)svb108 x svbBAC-dsRed. The bracket at the edge of the A1 stripe marks a region where trichome growth is exclusively covered by DG3, which disappeared with the deletion of DG3 and did not recover with the introduction of svbBAC-dsRed. (D) The trichome number in larvae developed at 32°C with Df(X)svb108 partially recovered to wild-type levels with the introduction of svbBAC-dsRed. The number of larvae counted was: 12 for wild-type svb, 28 for Df(X)svb108, 14 for Df(X)svb108 x svbBAC-dsRed and 13 for Df(X)svb108 x DG3-lacZ. Two-tailed t-test was applied for each individual comparison. In box plots, center line is mean, upper and lower limits are standard deviation and whiskers show 95% confidence intervals.
Discussion
Transcriptional regulation is a complex and dynamic process which requires coordinated interactions between transcription factors and chromatin. Given the transient nature of these interactions, using multiple binding sites to ensure efficient and consistent transcriptional regulation under different environmental conditions appears to be a preferred strategy among many developmental enhancers (Frankel, 2012; Perry et al., 2010). Genes such as shavenbaby add another layer of redundancy on top of this through long cis-regulatory regions containing multiple enhancers whose expression patterns overlap. Previous works have shown that this redundancy ensures proper phenotype development when systems are subjected to stress (Crocker et al., 2015; Frankel et al., 2010; Osterwalder et al., 2018). However, the mechanism underlying this phenotypic robustness was not clear.
In this work, we took advantage of the high-resolution imaging and analysis techniques we had developed to observe transcriptional microenvironments around transcription sites (Tsai et al., 2017) and investigated how the DG3 enhancer contributes to the phenotypic robustness of the svb locus at the molecular level. Deletion of the DG3 enhancer from svb did not lead to clear defects in ventral trichome formation unless the embryos were subjected to heat-induced stress, as shown here and previously with the deletion of ‘shadow enhancers’ (Hong et al., 2008) for lateral svb expression (Frankel et al., 2010). Nevertheless, we observed that the DG3-deletion allele showed reduced transcriptional output even at normal temperature. Wild-type embryos did not show phenotypic defects under either normal or stressed conditions, but heat-induced stress led to slightly lower transcriptional output from the wild-type svb allele (Figure 6A and B). However, the mutant svb locus, starting with lower transcriptional output even under ideal conditions (Figure 6C), drops below a threshold and the system fails (Figure 6D). We observed that Ubx concentration and svb transcriptional output in the A1 segment initially have a weak positive correlation that quickly dissipated at higher Ubx concentrations and svb outputs. While this data must be interpreted with some caution, as multiple stages of the transcription cycle are included (initiation, elongation or termination), Ubx concentration increases after a certain point were not clearly coupled to increases in svb transcriptional output. Additional regulatory mechanisms could be at play beyond transcription factor retention that determines the final transcriptional output of the locus. Determining the complete response function would be complicated, due to reasons such as the enhancers each having clusters of factor binding sites and the overlap of expression patterns from related enhancers. As svb also accepts inputs from many additional transcription factors (Stern and Orgogozo, 2008), especially in the other body segments, the total response of the system would also depend on many factors not observed in this study.
Figure 6
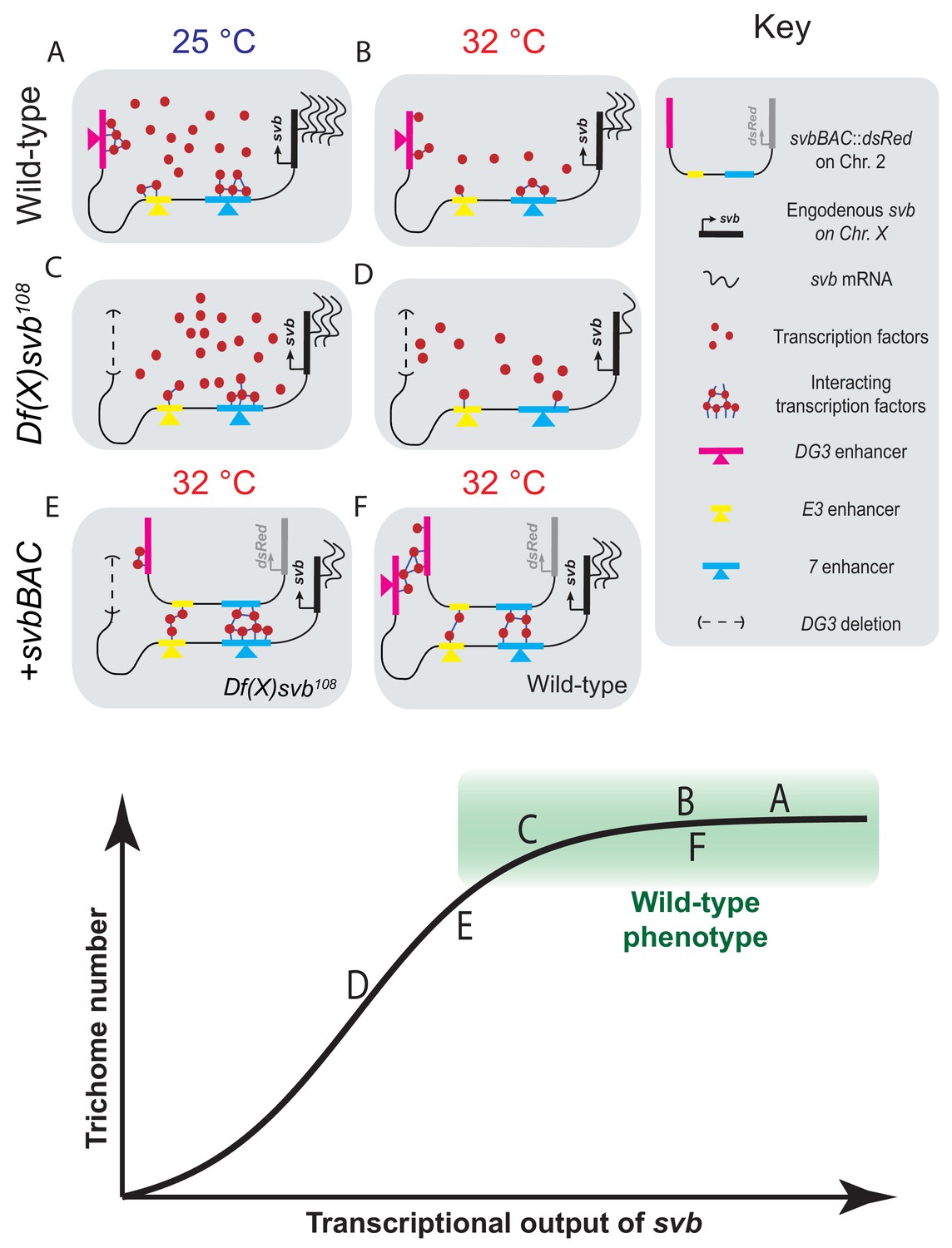
Summary of model including a sigmoidal relationship between svb transcriptional output and the number of cells fated to become trichomes.
(A–F) Schematic representations of the various genotypes, temperatures and rescue conditions as tested in this work. Bottom panel: Schematic of the proposed sigmoidal relationship between svb transcriptional output and the number of cells fated to become trichomes. Note that A and C are at 25°C and the rest are at 32°C. The different conditions we have studied are represented by letters A-F along the sigmoidal curve. The part of the curve corresponding to wild-type phenotype is shaded in green.
We previously observed that transcription sites of reporter genes driven by minimal svb enhancers tended to colocalize with the endogenous svb locus when it is transcriptionally active (Tsai et al., 2017). This is true also for entire cis-regulatory regions, as we observed that the svb locus did the same with svbBAC, which implies that they potentially share a common microenvironment. Homologous regions were shown to pair over long distances, between homologous chromosomal arms (Lim et al., 2018), translocated domains and even different chromosomes (Gemkow et al., 1998; Johnston and Desplan, 2014; Peifer and Bender, 1986). Our observations could be related to them and share similar mechanisms. However, our constructs may not contain sites for structural elements such as insulators, which have been described as important supporters of trans-interactions (Postika et al., 2018). On the other hand, our observations are in line with transcription-dependent associations of interchromosomal interactions (Branco and Pombo, 2006; Joyce et al., 2016; Lomvardas et al., 2006; Maass et al., 2018; Monahan et al., 2019). It is possible that such long range interactions are driven, or reinforced, through shared microenvironments.
We were able to partially rescue the DG3-deletion svb allele with svbBAC, which contains the cis-regulatory region of svb but not the svb gene itself. High-resolution imaging showed that colocalizing with a svbBAC increases the local Ubx concentration and transcriptional output of the DG3-deletion allele (Figure 6E). This supports a mechanism where transcriptional microenvironments sequestered around large and related cis-regulatory regions in physical proximity can work in trans to increase transcriptional output of other genes, even on different chromosomes. The introduction of a shorter reporter construct containing the DG3 enhancer alone did not rescue trichome expression perhaps because it is not able to effectively pair with the svb locus. It is possible that structural elements, such as insulator proteins (Lim et al., 2018) or other topologically associated elements (Furlong and Levine, 2018) could overcome this by increasing pairing efficiency. This is consistent with recent findings for long-range interactions that are dependent on specific topologically associating domains (TADs), where pairing is necessary but not sufficient for transvection (Viets et al., 2018). Interestingly, embryos with the DG3 deletion allele and svbBAC could not produce trichomes at the dorsal edges of the ventral trichome patches, where DG3 provides exclusive coverage. As the rescue only occurred on regions where other ventral svb enhancers provided overlapping coverage, it likely is the result of compensation from the additional ventral enhancers (E3 and 7) at the svb locus instead of the DG3 enhancer on the svbBAC driving svb expression in trans. Interactions in trans may serve an auxiliary role through influencing the properties of the local transcriptional environments.
Summarizing our observations, we hypothesize that the relationship between svb transcriptional output and the number of cells fated to become trichomes is sigmoidal (Figure 6, bottom panel). Below a certain threshold, the number of trichomes would drop with decreasing svb mRNA; however, any additional transcriptional output above this threshold (the green box in the figure) would not lead to significant changes in trichome production and would appear to be wild-type in phenotype. The cis-regulatory region of wild-type svb under ideal conditions likely already saturates the system, as evidenced by the lack of change in Ubx concentration and trichome numbers even with the addition of svbBAC (Figure 6F). Although operating under saturation renders this system relatively insensitive to changes in svb transcription, the risk of developing defective phenotypes when conditions are no longer ideal likely selected for this strategy to buffer against stresses. Overall, the system remains phenotypically robust and develops the same number of trichomes despite fluctuations in transcriptional outputs. In the future, it would be important to understand mechanistically how the phenotype can tolerate significant drops in transcriptional output before defects appear. It requires the direct observation of intermediate steps between svb transcription and phenotype production to understand how this buffering is achieved. Furthermore, our current technique only allows us to probe environments around active transcription sites without knowing which phase of transcription (initiation, elongation or termination) it is in and how the gene loci positioned themselves into these locations. Future works to visualize and track genes, regardless of their transcriptional state, in fixed and living embryos would answer key questions on how they find, form and interact with transcriptional microenvironments.
We previously proposed that transcriptional microenvironments form across multiple enhancers through scaffolding interactions to ensure efficient transcription from developmental enhancers. By investigating the mechanisms of how correct transcriptional regulation is maintained under stress using a DG3-deletion allele of svb, we have shown that transcriptional microenvironments could span multiple enhancers that share similar transcription factor binding sites. These microenvironments of transcription factors could form the protein core of transcription factor ‘hubs’ that have been proposed to form through phase-separation mediated through protein-protein interactions between disordered domains (Cisse et al., 2013; Furlong and Levine, 2018; Ghavi-Helm et al., 2014; Mir et al., 2017). Thus, they add another layer of redundancy on top of using multiple enhancers with overlapping expression patterns in a cis-regulatory region to ensure a sufficient margin to buffer against the negative effects of environmental stresses (Frankel et al., 2010; Perry et al., 2010). This extra margin of safety preserves phenotypical development even when environmental conditions are not ideal. Integrating multiple noisy and low-affinity elements into a coherent and synergistic network would also reduce the variance stemming from the transient and stochastic transcription factor binding dynamics observed in eukaryotic cells (Cisse et al., 2013; Ghavi-Helm et al., 2014; Mir et al., 2017; Tsai et al., 2017). In sum, specialized transcriptional microenvironments could be a critical element to ensure that gene expression occurs specifically and consistently in every embryo. Given that shadow enhancers are widespread features of gene regulatory networks (Cannavò et al., 2016; Osterwalder et al., 2018), it is likely that high local concentrations of transcription factors are a widespread feature that provides an effective regulatory buffer to prevent deleterious phenotypic consequences to genetic and environmental perturbations.
Materials and methods
Fly strains
Request a detailed protocolAll fly strains used have been previously described: DG3-lacZ (Tsai et al., 2017); ubx1 (Crocker et al., 2015); HS::ubx-1: (Crocker et al., 2015); Df(X)svb108 (Frankel et al., 2010); svbBAC-dsRed (Preger-Ben Noon et al., 2018); diBAC-gfp is CH322-35A16 EGFP tagged in VK37, covering D (Venken et al., 2009). Unless otherwise noted, they are generated from w1118 stock, which is referred to as wild-type.
Preparing Drosophila embryos for staining and cuticle preps
Request a detailed protocolD. melanogaster strains were maintained under standard laboratory conditions, reared at 25 °C, unless otherwise specified. For heat-shock experiments, these conditions were followed: for staining with fluorescent antibodies, flies were allowed to lay eggs on apple-juice agar plates for 5 hr at 25 °C and then kept in an incubator at 32 °C for 7 hr before fixation; for cuticle preps, dechorionated embryos were kept at 32 °C until they emerged as larvae. Df(X)svb108 embryos/larvae with svbBAC-dsRed are readily discernable by the loss of svb and trichomes in the T1 segment (see Figure 2D & E).
Cuticle preparation and trichome counting
Request a detailed protocolLarvae collected for cuticle preparations were mounted according to a published protocol (Stern and Sucena, 2011). A phase-contrast microscope was used to image the slides. In the case of crosses involving the Df(X)svb108 allele, only larvae lacking trichomes in the T1 segment were imaged (Figure 2E). This is a homozygous marker for the deletion locus as larvae carrying any wild-type svb allele will produce trichomes in T1 (Figure 2D). Ventral trichomes in larval A1 or A2 segments were counted in Fiji/ImageJ by find using the find maximum function (Schindelin et al., 2012; Schneider et al., 2012).
Immuno-fluorescence staining of transcription factors and in situ hybridization to mRNA
Request a detailed protocolStandard protocols were used for embryo fixation and staining (Crocker et al., 2015; Tsai et al., 2017). Secondary antibodies labeled with Alexa Fluor dyes (1:500, Invitrogen) were used to detect primary antibodies. In situ hybridizations were performed using DIG, FITC or biotin labeled, antisense RNA-probes against a reporter construct RNA (lacZ, dsRed, gfp) or the first intron and second exon (16 kb) of svb. See Supplementary file 1 for primer sequences. DIG-labeled RNA products were detected with a DIG antibody: Thermofisher, 700772 (1:100 dilution), biotin-labeled RNA products with a biotin antibody: Thermofisher, PA1-26792 (1:100) and FITC-labeled RNA products with a FITC antibody: Thermofisher, A889 (1:100). Ubx protein was detected using Developmental Studies Hybridoma Bank, FP3.38-C antibody at 1:20 dilution, DsRed protein using MBL anti-RFP PM005 antibody at 1:100, LacZ protein using Promega anti-ß-Gal antibody at 1:250 and GFP protein using Aves Labs chicken anti-GFP at 1:300.
Imaging fixed embryos
Request a detailed protocolMounting of fixed Drosophila embryos was done in ProLong Gold + DAPI mounting media (Molecular Probes, Eugene, OR). Fixed embryos were imaged on a Zeiss LSM 880 confocal microscope with FastAiryscan (Carl Zeiss Microscopy, Jena, Germany). Excitation lasers with wavelengths of 405, 488, 561 and 633 nm were used as appropriate for the specific fluorescent dyes. For imaging in embryos carrying Df(X)svb108, only embryos without svb mRNA expression in the T1 segment were imaged, following the same reason described in the section on ‘Cuticle preparations and trichome counting’. Unless otherwise stated, all images were processed with Fiji/ImageJ (Schindelin et al., 2012; Schneider et al., 2012) and Matlab (MathWorks, Natick, MA, USA).
Analysis of microenvironment and svb transcription intensity
Request a detailed protocolInside nuclei with svb transcription sites, the center of the transcription site was identified using the find maximum function of Fiji/ImageJ. A circle with a diameter of 4 pixels (170 nm, roughly the lateral resolution limit of AiryScan in 3D mode) region of interest (ROI) centered on the transcription site is then created. The integrated fluorescent intensity inside the ROI from the Ubx IF channel and the RNA FISH channel are then reported as the local Ubx concentration and the transcriptional output, respectively. The intensity presented in the figures is the per-pixel average intensity with the maximum readout of the sensor normalized to 255.
Analysis of distances between transcription spots
Request a detailed protocolInside nuclei with svb and dsRed/gfp transcription sites, the centers of the transcription site were identified using the find maximum function of Fiji/ImageJ. The distance between the transcription sites were then computed using the coordinates of the transcription sites. Two sites are considered colocalized when they are within 360 nm of each other.
Ubx ChIP profile
Request a detailed protocolThe ChIP profile for Ubx around the svb cis-regulatory region is from Choo et al. (2011), using whole Drosophila melanogaster embryos between stages 10 and 12.
Data availability
The original images (cuticle preparations and embryo images, organized into zip files) are available for download and are indexed at: https://www.embl.de/download/crocker/svb_enhancer_colocalization/index.html. Please note that the raw AiryScan images must be processed though the Zen software from Zeiss before they can be opened/analyzed using standard image processing softwares. These files are large, totaling up to approximately 180 GB in size. We can also send these files directly if a means of transfer (hard drives, etc.) is provided.
References
-
The soft touch: low-affinity transcription factor binding sites in development and evolutionCurrent Topics in Developmental Biology 117:455–469.https://doi.org/10.1016/bs.ctdb.2015.11.018
-
Multiple layers of complexity in cis-regulatory regions of developmental genesDevelopmental Dynamics : An Official Publication of the American Association of Anatomists 241:1857–1866.https://doi.org/10.1002/dvdy.23871
-
Homologous association of the Bithorax-Complex during embryogenesis: consequences for transvection in Drosophila MelanogasterDevelopment 125:4541–4552.
-
Pairing and anti-pairing: a balancing act in the diploid genomeCurrent Opinion in Genetics & Development 37:119–128.https://doi.org/10.1016/j.gde.2016.03.002
-
The regulation of hox gene expression during animal developmentDevelopment 140:3951–3963.https://doi.org/10.1242/dev.068346
-
Dense bicoid hubs accentuate binding along the morphogen gradientGenes & Development 31:1784–1794.https://doi.org/10.1101/gad.305078.117
-
Shadow enhancers foster robustness of Drosophila gastrulationCurrent Biology 20:1562–1567.https://doi.org/10.1016/j.cub.2010.07.043
-
Combinatorial function of transcription factors and cofactorsCurrent Opinion in Genetics & Development 43:73–81.https://doi.org/10.1016/j.gde.2016.12.007
-
Fiji: an open-source platform for biological-image analysisNature Methods 9:676–682.https://doi.org/10.1038/nmeth.2019
-
NIH image to ImageJ: 25 years of image analysisNature Methods 9:671–675.https://doi.org/10.1038/nmeth.2089
-
Transcription factors: from enhancer binding to developmental controlNature Reviews Genetics 13:613–626.https://doi.org/10.1038/nrg3207
-
Preparation of cuticles from unhatched first-instar Drosophila larvaeCold Spring Harbor Protocols 2011:pdb.prot065532.https://doi.org/10.1101/pdb.prot065532
Article and author information
Author details
Funding
Damon Runyon Cancer Research Foundation (DRG 2220-15)
- Albert Tsai
European Molecular Biology Organization
- Albert Tsai
- Mariana R P Alves
- Justin Crocker
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The fly line containing diBAC-gfp (CH322-35A16 EGFP) was a gift from Schulze, Karen Lynn and Bellen, Hugo J. The Df(X)svb108 flies were a gift from Stern DL and the svbBAC flies were a gift from Preger Ben-Noon E and Frankel N (Preger-Ben Noon et al., 2018). We thank Rafael Galupa, Nicolas Frankel and Ella Preger Ben-Noon for suggestions and discussions. We thank the entire Crocker lab for discussion and feedback. We would also like to thank the reviewers for their constructive input. Albert Tsai is a Damon Runyon Fellow of the Damon Runyon Cancer Research Foundation (DRG 2220–15). Albert Tsai, Mariana R P Alves and Justin Crocker are supported by the European Molecular Biological Laboratory (EMBL).
Copyright
© 2019, Tsai et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,706
- views
-
- 736
- downloads
-
- 93
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 93
- citations for umbrella DOI https://doi.org/10.7554/eLife.45325
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Multi-enhancer transcriptional hubs confer phenotypic robustness
eLife 8:e45325.
https://doi.org/10.7554/eLife.45325




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}