Genome plasticity in Candida albicans is driven by long repeat sequences
- Creighton University Medical School, United States
- Bowdoin College, United States
Figures
Figure 1 with 2 supplements
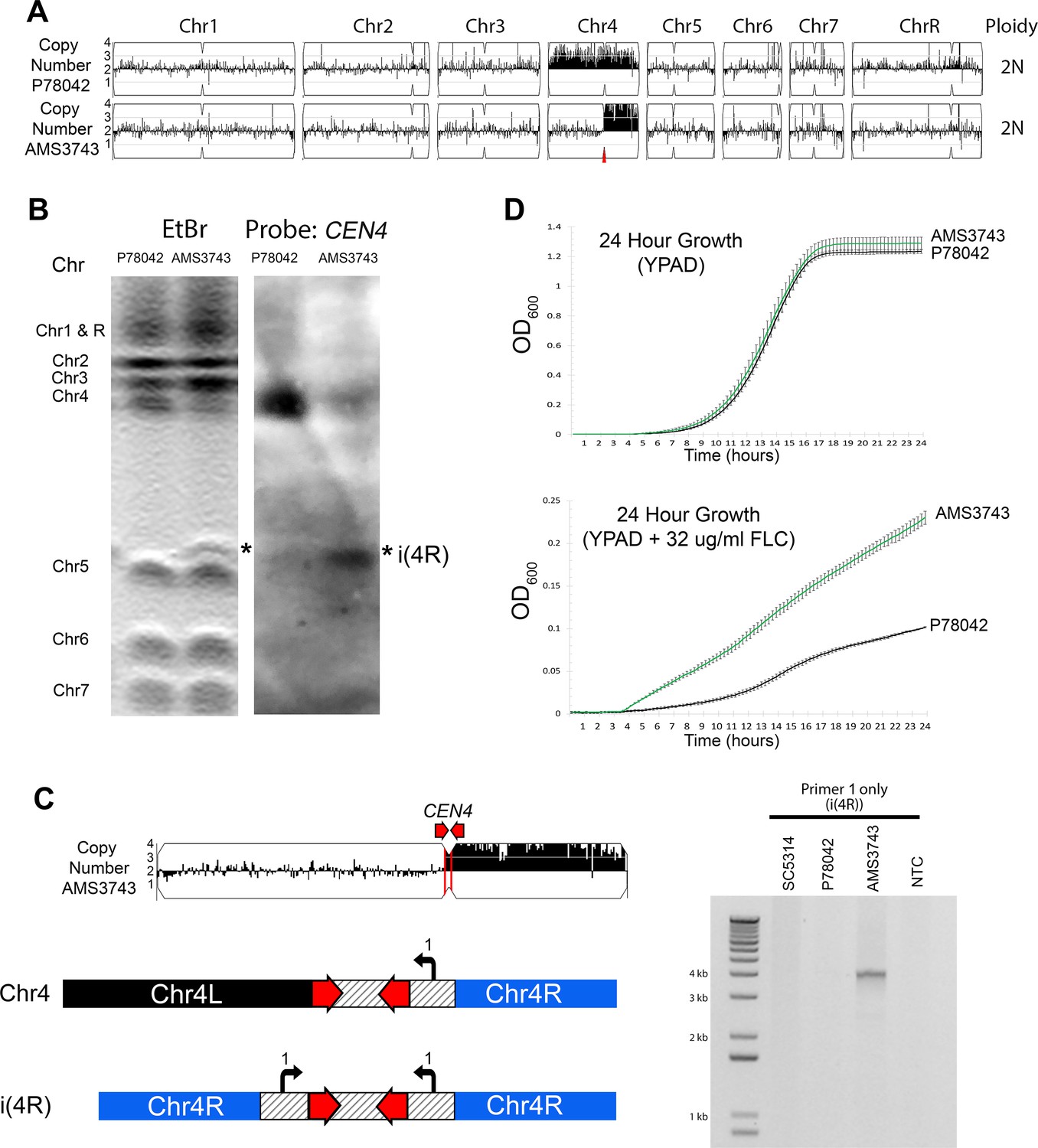
Inverted repeat at CEN4 causes a novel isochromosome leading to increased fluconazole resistance.
(A) Whole genome sequence data plotted as a log2 ratio and converted to chromosome copy number (Y-axis) and chromosome location (X-axis) using YMAP, for the progenitor clinical isolate (P78042) and an isolate obtained after 100 generations in FLC (AMS3743). The copy number breakpoint in AMS3743 occurs at CEN4 (red arrow). (B) CHEF karyotype gel stained with ethidium bromide (left panel) identifies a novel band (asterisk) above Chr5. Southern blot analysis (right panel) of the same gel using a DIG-labeled CEN4 probe identifies the full-length Chr4 homolog in P78042 and AMS3743, and the novel band in AMS3743 that is twice the size of the right arm of Chr4 in an isochromosome structure (asterisk, i(4R)). (C) PCR validation of i(4R). Schematic representation of the Chr4 homologue (top) and i(4R), where the location of a single primer sequence (Primer 1, Supplementary file 7) that flanks the CEN4 inverted repeat is indicated. PCR with Primer 1 amplified the expected product of i(4R) in AMS3743. (D) 24 hr growth curves in YPAD (top panel) and YPAD +32 µg/ml FLC (bottom panel) for P78042 (black line) and AMS3743 (green line). Average slope and standard error of the mean for three biological replicates is indicated. The average maximum slope (n = 3) of P78042 and AMS3743 in YPAD was not significantly different (0.046 and 0.046, respectively, p>0.75, t-test). The average maximum slope (n = 3) of P78042 and AMS3743 was significantly different in FLC (0.002 and 0.003, respectively, p<0.0006, t-test). OD, optical density (Figure 1—source data 1).
-
Figure 1—source data 1
Growth curve analysis.
- https://doi.org/10.7554/eLife.45954.005
Figure 1—figure supplement 1
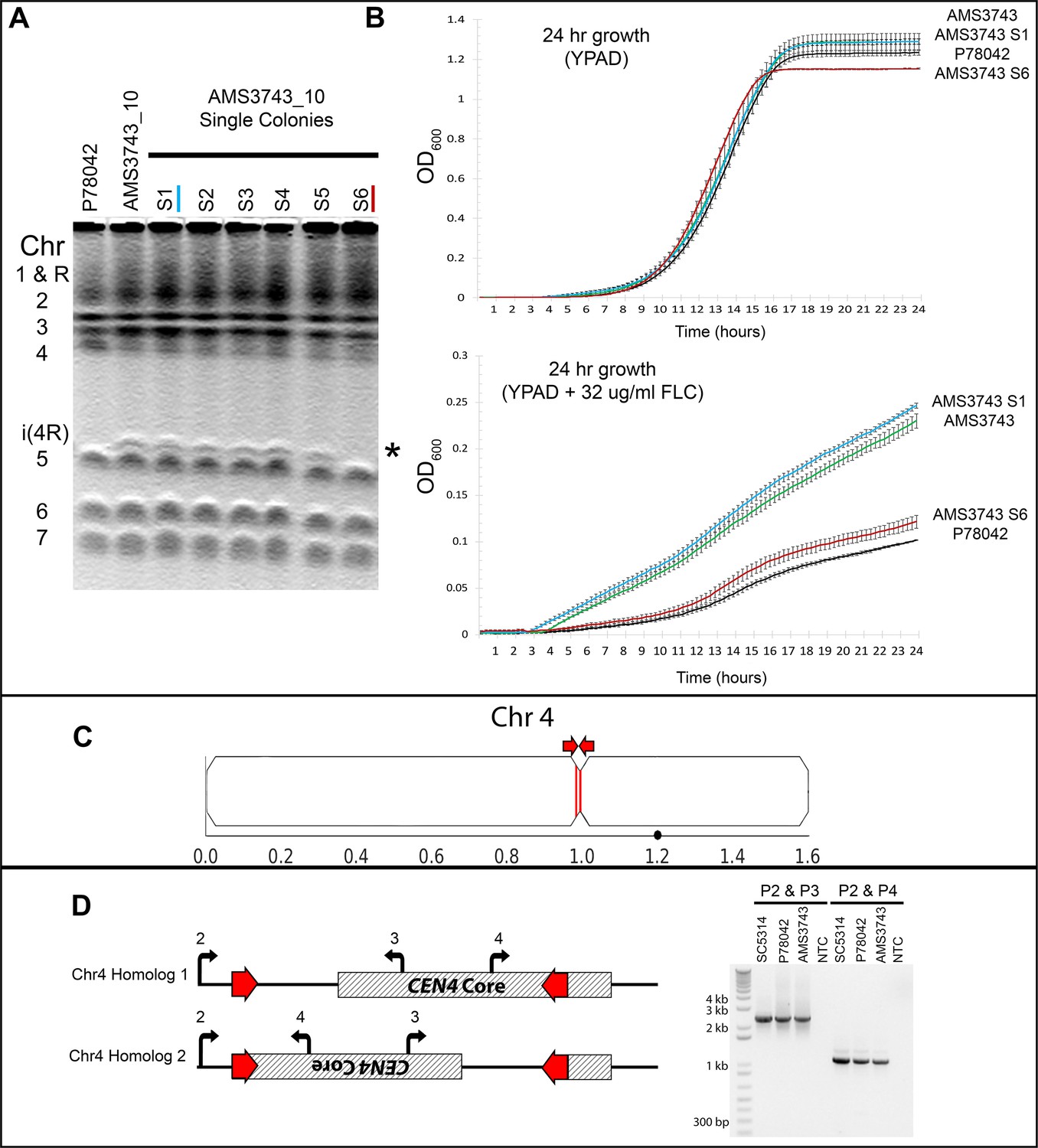
Long inverted repeats on Chr4 are associated with a centromere inversion and an isochromosome that confers increased fitness in FLC.
(A) CHEF karyotype gel stained with ethidium bromide. Passage of AMS3743_10 for 30 days in YPAD alone followed by single colony selection identified one single colony that had lost the i(4R) band (AMS3743_S6). (B) 24 hr growth curves in YPAD (top panel) and YPAD +32 µg/ml FLC (bottom panel) of P78042 (black line), AMS3743 with i(4R) (green line), AMS3734_S1 with i(4R) (blue line), and AMS3743_S6 which lost the i(4R) (red line). There was no significant difference in average max slope between P78042, AMS3743, AMS3743_S1, and AMS3746_S6 in YPAD (p>0.96, one-way ANOVA with Tukey’s multiple comparison). The average maximum slope in FLC was significantly higher in isolates containing i(4R) (0.003 for both AMS3743 and AMS3743_S1) than in the isolates not containing i(4R) (0.002 for both P78042 and AMS3742_S6) (p>0.05, one-way ANOVA with Tukey’s multiple comparison). OD, optical density (Figure 1—source data 1). (C) Location of the CEN4 inverted repeat (red arrows and lines). Location of the Major Repeat Sequence on Chr4 (black circle). (D) CEN4 is comprised of a ~ 3.6 kb CENP-A-binding core sequence (hatched box) asymmetrically flanked by a 524 bp inverted repeat sequence (red) separated by ~3.1 kb. PCR analysis with primers anchored outside or inside the inverted repeat (Primers 2, 3, and 4, see Supplementary file 7), identified two different orientations of CEN4 (denoted Chr4A and Chr4B) that arose due to an inversion between the repeat sequences on one homologue, in the reference isolate SC5314 all isolates analyzed.
Figure 1—figure supplement 2

Sanger sequencing of CEN4 in SC5314.
Unique PCR fragments flanking the left side of the CEN4 inverted repeat were obtained for the reference isolate SC5314. PCR products were amplified for both the reference and inverted orientations of CEN4. Primers are indicated as in Figure 1—figure supplement 1D and Supplementary file 7. Sanger sequencing was performed with both forward and reverse primers.
Figure 2 with 3 supplements
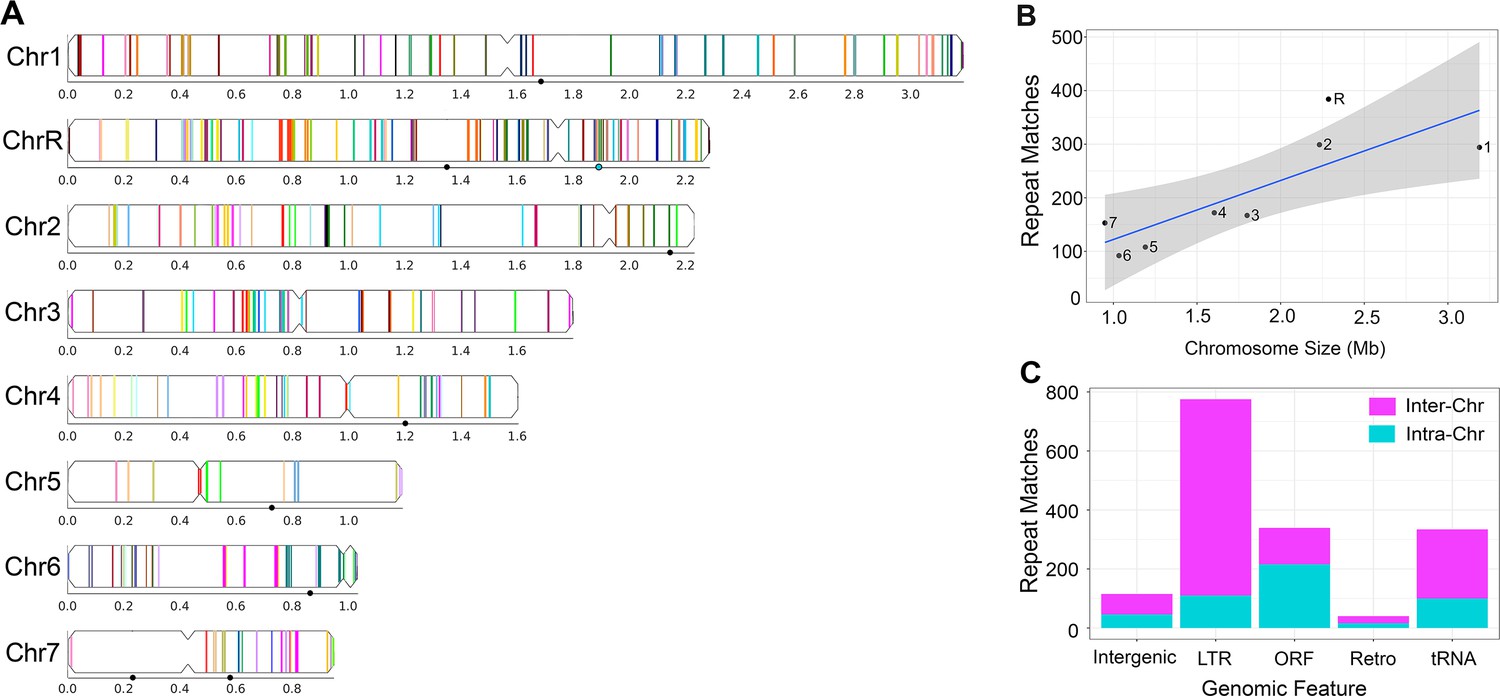
Long repeat sequences are found across the C. albicans genome.
Detailed results for all long intra- and inter-chromosomal repeat positions, orientations, and gene features are found in Supplementary file 2. Repeats associated with the rDNA, major repeat sequences (MRS), and sub-telomeric repeats were removed prior to the analysis. (A) Representative image of the long intra-chromosomal repeat positions (colored lines – not to scale). Each repeat family is assigned a unique color within its respective chromosome. Numbers and symbols below each chromosome indicate chromosomal position (Mb), MRS position (black circles), and rDNA locus (blue circle, ChrR). (B) Number of all repeat matches (excluding the complex tandem repeat genes) on each chromosome, ordered by chromosome size (R2 = 0.65, p<0.016, gray indicates 95% confidence interval, Figure 2—source data 1). (C) The number of intra-chromosomal (Intra-Chr) and inter-chromosomal (Inter-Chr) repeat matches assigned to each genomic feature: Intergenic, LTR, ORF (excluding the complex tandem repeat genes), retrotransposon (Retro), and tRNA (Figure 2—source data 1).
-
Figure 2—source data 1
Distribution, features, and coverage of long repeat sequences in C. albicans.
- https://doi.org/10.7554/eLife.45954.010
-
Figure 2—source data 2
Analysis of long repeat spacer length in C. albicans.
- https://doi.org/10.7554/eLife.45954.011
-
Figure 2—source data 3
Analysis of key features of long repeat sequences in C. albicans.
- https://doi.org/10.7554/eLife.45954.012
Figure 2—figure supplement 1
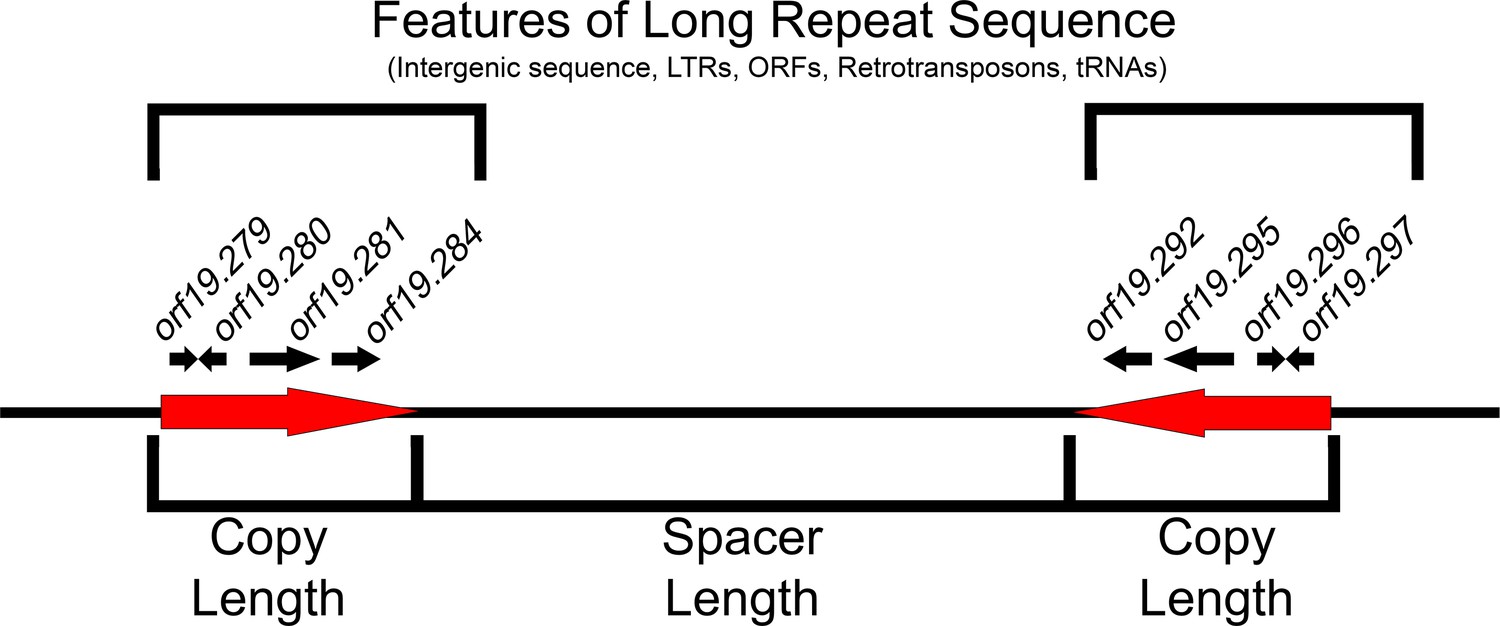
Features of long repeat sequences.
Schematic of a previously uncharacterized long repeat sequence (repeat family 124). The repeat sequence (red arrows) is described in terms of copy length (bp) and shared sequence identity (% of exact nucleotide matches) between the two matched sequences. The distance between intra-chromosomal repeat matches is the spacer length, and their orientation can be inverted (reverse complement located on the opposite DNA strand), mirrored (reverse complement located on the same DNA strand), or tandem. Long repeat sequences are further characterized by the genomic features contained within the repeat. Long repeats that contain ORFs include partial ORF sequences, single complete ORF sequences (paralogs) or multiple ORFs and intergenic sequences. Repeat family 124 contains four complete ORFs (black arrows) and flanking intergenic sequences in each copy of the long repeat sequence. Other repeat sequences contain lone LTRs, retrotransposons, tRNAs, and intergenic sequences. Details of all repeat sequence matches are found in Supplementary file 2.
Figure 2—figure supplement 2
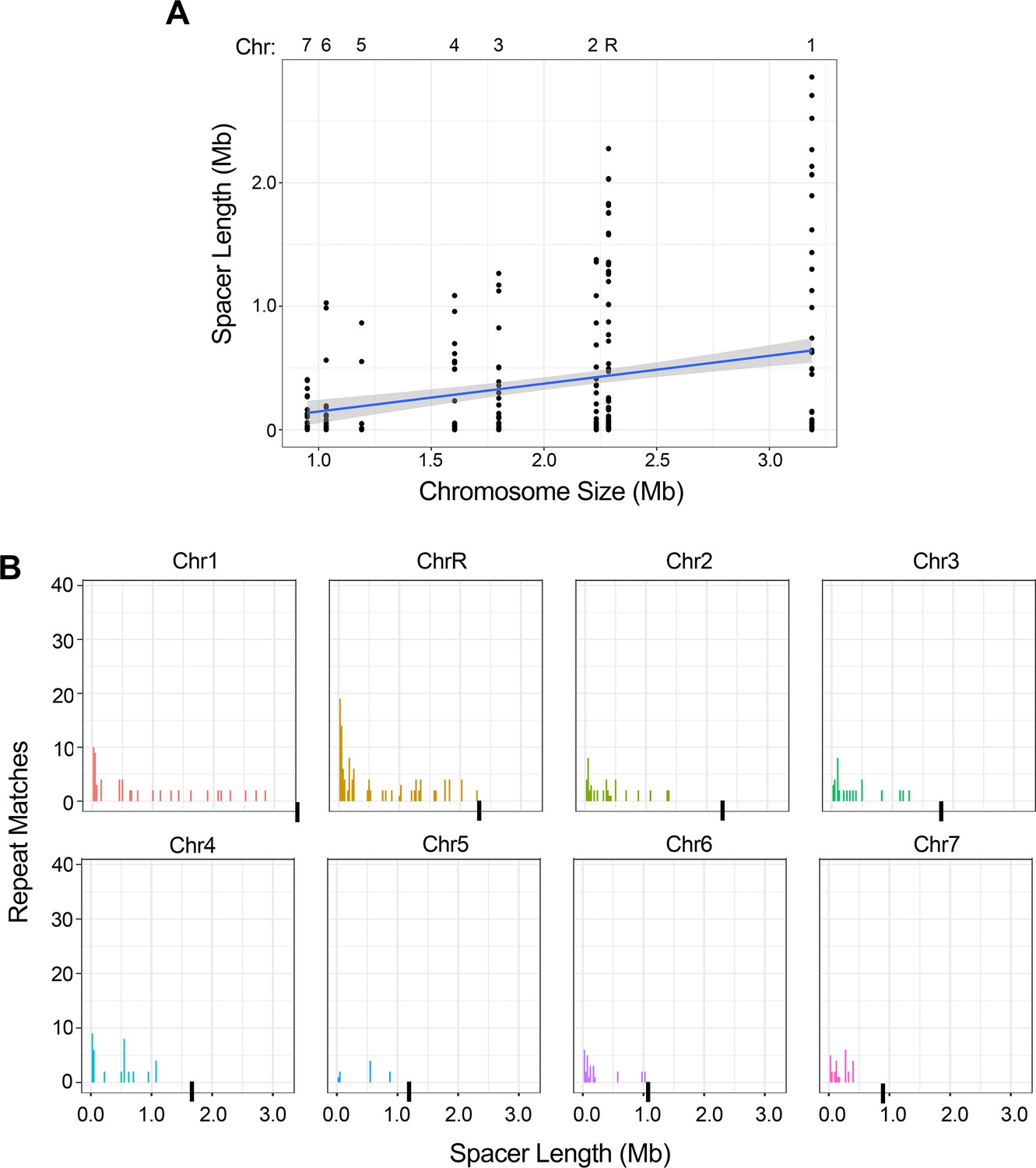
The intra-chromosomal repeats with the longest spacer length are found on the longer chromosomes.
(A) The spacer length for all intra-chromosomal repeat matches (excluding the complex tandem repeat genes) for each chromosome, ordered by chromosome size in bp (R2 = 0.06, p<0.0001, Figure 2—source data 2). (B) Distribution of intra-chromosomal spacer length for each of the eight C. albicans chromosomes (chromosome ends indicated with a black bar). There is a significant difference in the distributions of repeat spacer lengths between chromosomes (p<0.035, Kruskal-Wallis test with Dunn’s multiple comparison), with the longest chromosomes having more repeat matches that are separated by greater spacer length than the smallest chromosomes (Figure 2—source data 2).
Figure 2—figure supplement 3
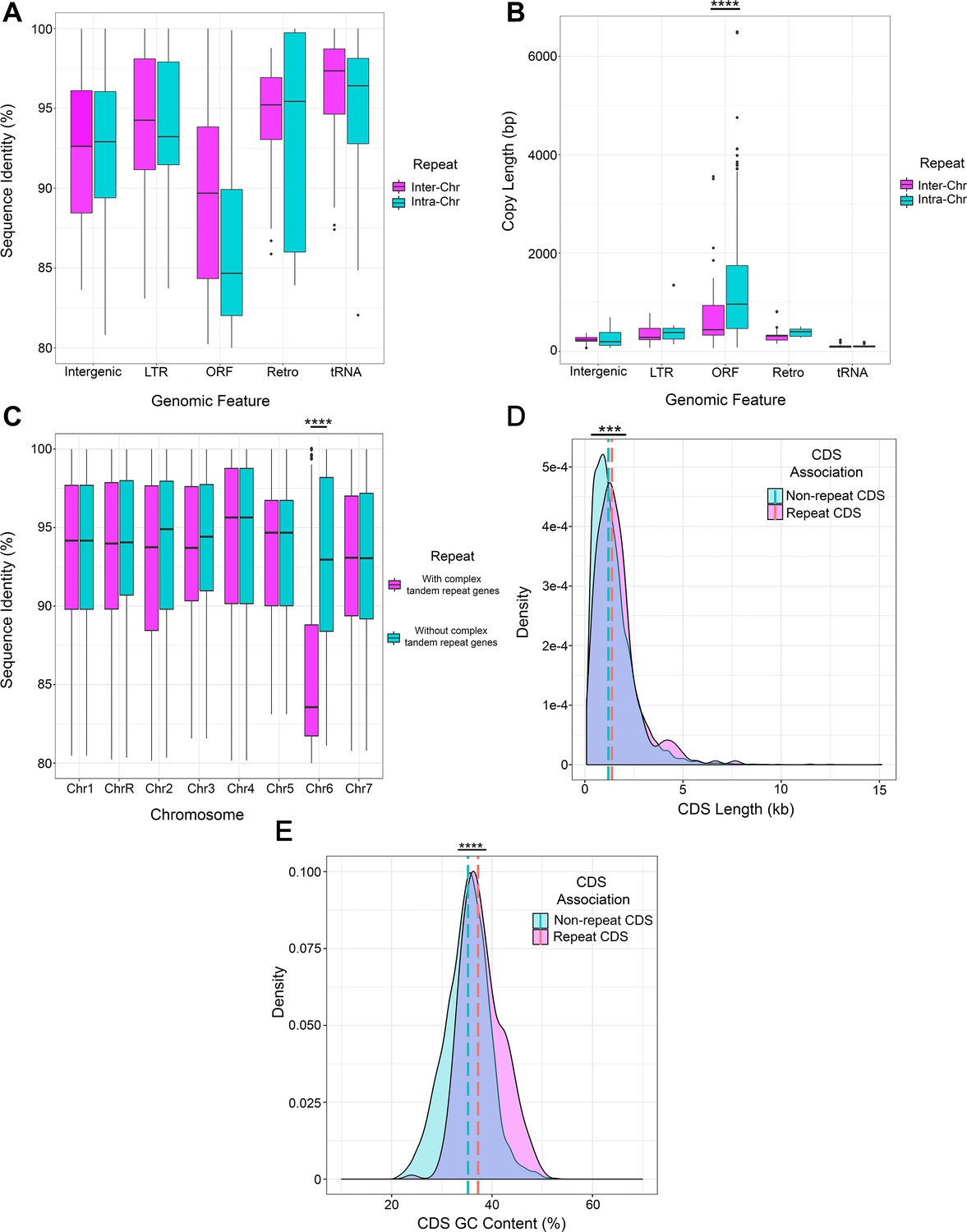
Key features of long repeat sequences in C. albicans.
The percent shared identity (A) and repeat copy length (B) of intra-chromosomal (Intra-Chr) or inter-chromosomal (Inter-Chr) repeat matches containing each genomic feature: Intergenic, LTR, ORF (excluding the complex tandem repeat genes), Retrotransposon (Retro), and tRNA (Supplementary file 2). Copy length is significantly different between repeats containing ORFs compared to repeats containing other features (p<0.0001, Kruskal-Wallis with Dunn’s multiple comparisons). (C) Percent sequence identity of repeat matches for each chromosome both before (pink) and after (blue) removal of the complex tandem repeat genes. The median sequence identity of repeats on Chr6 is significantly increased after removal of the complex tandem repeat genes (p<0.0001, Kruskal-Wallis with Dunn’s multiple comparisons). The length (D) and percent GC content (E) of the full-length ORF coding sequences (CDS) within long repeat sequences (pink) and all other full-length CDSs not contained in long repeat sequences (blue). Dashed lines represent median values. The full-length CDSs contained in long repeats are significantly longer (p<0.0008, Kolmogorov-Smirnov test) and have a significantly higher percent GC content (p<0.0001, Kolmogorov-Smirnov test) than all full-length CDSs not contained in long repeat sequences. ***p<0.001, ****p<0.0001 (see Materials and methods, Figure 2—source data 3).
Figure 3 with 1 supplement

All copy number breakpoints resulting in segmental aneuploidy occur at repeat sequences.
(A) Whole genome sequence data plotted as a log2 ratio and converted to chromosome copy number (Y-axis) and chromosome location (X-axis) using YMAP. The source of each isolate is indicated in color: in vivo evolution experiments in a murine model of oropharyngeal candidiasis (OPC) (green), in vitro evolution experiments in the presence of azole antifungal drugs (red), and clinical isolates (blue). Ploidy, determined by flow cytometry, is indicated on the far right. Every copy number breakpoint occurred at a repeat sequence (red arrow), additional details are in Supplementary file 3. Location of the Major Repeat Sequences (black circle) and rDNA array (blue circle) are shown below. Example copy number breakpoints for two isolates (B–C). (B) Isolate AMS3053 underwent a complex rearrangement on Chr3L at a long inverted repeat (Repeat 124, red lines). Read depth (top panel) and allele frequency (IGV panel) data indicate the copy number breakpoint coincided with LOH (blue region) telomere proximal to the breakpoint. The inverted repeat copies (~3.2 kb, 99.5% sequence identity, separated by ~11.3 kb) each contain four complete ORFs and intergenic sequences. (C) Read depth (top panel) and allele frequency (IGV panel) data for isolate CEC2871 shows an internal chromosome deletion on ChrR with copy number breakpoints (red lines) and LOH (blue) that occur between a long tandem repeat (Repeat family 201, red arrows). The tandem repeat copies (~1.4 kb, 93.8% sequence identity, separated by ~55 kb) each contain one ORF.
Figure 3—figure supplement 1
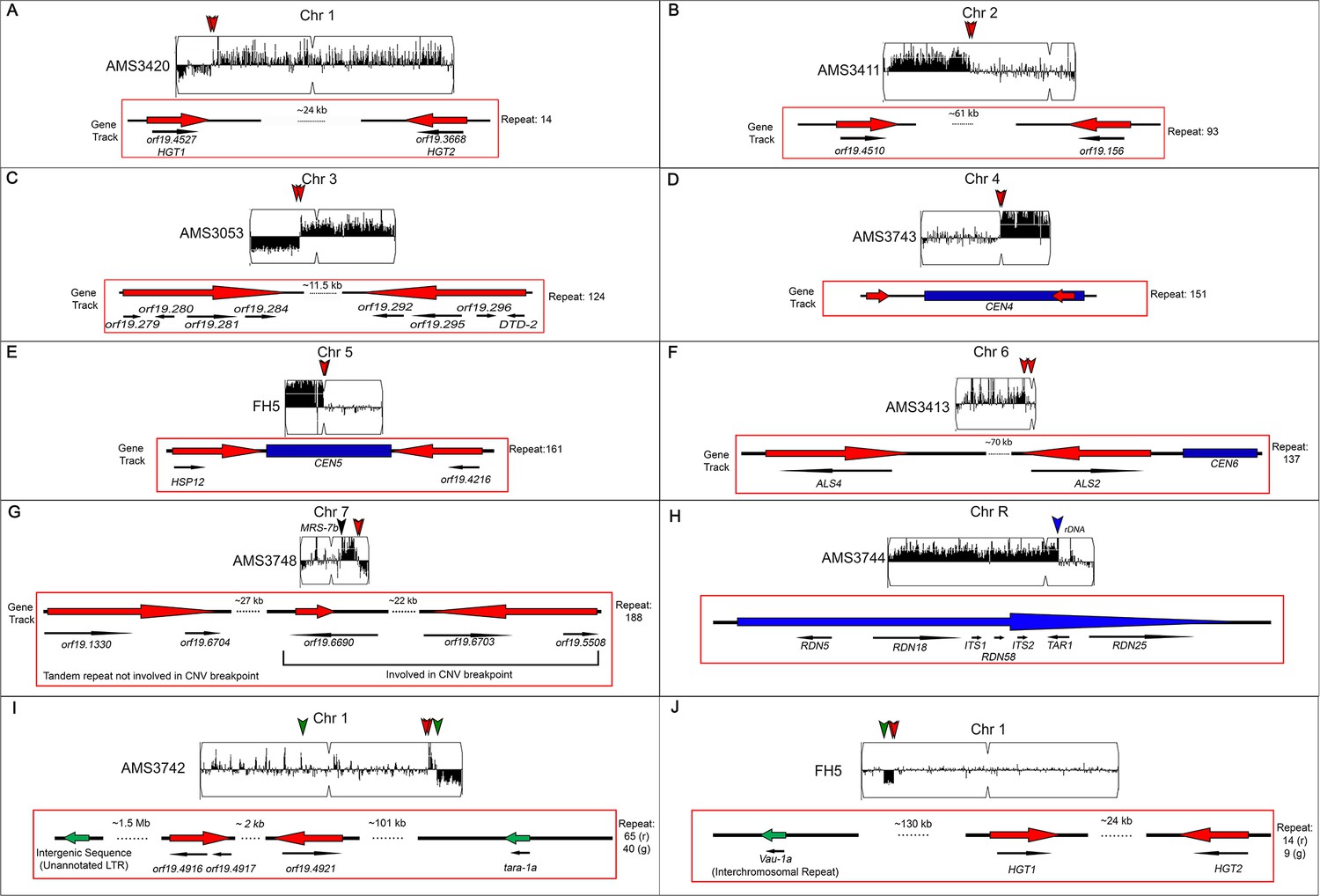
Segmental aneuploidies occur at previously characterized and uncharacterized long repeat sequences.
Representative segmental aneuploidy breakpoints from Figure 3. Whole genome sequence data plotted as a log2 ratio and converted to chromosome copy number (Y-axis) and chromosome location (X-axis) using YMAP. Copy number variation breakpoints (red and green arrowheads) are indicated. Each breakpoint is associated with a long repeat sequence (red or green arrow) shown in the gene track, and annotated genomic features are indicated with black arrows, below the gene track (A-J, Supplementary file 3). Segmental chromosome aneuploidies from the indicated isolates occur within (A) Chr1 repeat family 14, containing ORFs HGT1 and HGT2; (B) Chr2 repeat family 93, containing two uncharacterized ORFs; (C) Chr3 repeat family 124 containing eight ORFs and associated intergenic sequences; (D) CEN4 repeat family 151; (E) CEN5 repeat family 161, containing two ORFs; (F) Chr6 repeat family 137, containing the ALS gene family; (G) a complex repeat region on Chr7 with both inverted and tandem repeat sequences containing five uncharacterized ORFs; and (H) ChrR repeat region containing the rDNA array. Two examples of complex segmental aneuploidies involving more than one repeat family (I and J). (I) Chr1 repeat family 65 is associated with a centromere proximal amplification, while repeat family 40 is associated with a chromosome truncation event. (J) Example of a segmental aneuploidy flanked by two different repeat families. An internal deletion is flanked by repeat family 14 and family 9 in clinical isolate FH5. Family 9 is the only inter-chromosomal repeat associated with any observed copy number breakpoint.
Figure 4 with 1 supplement
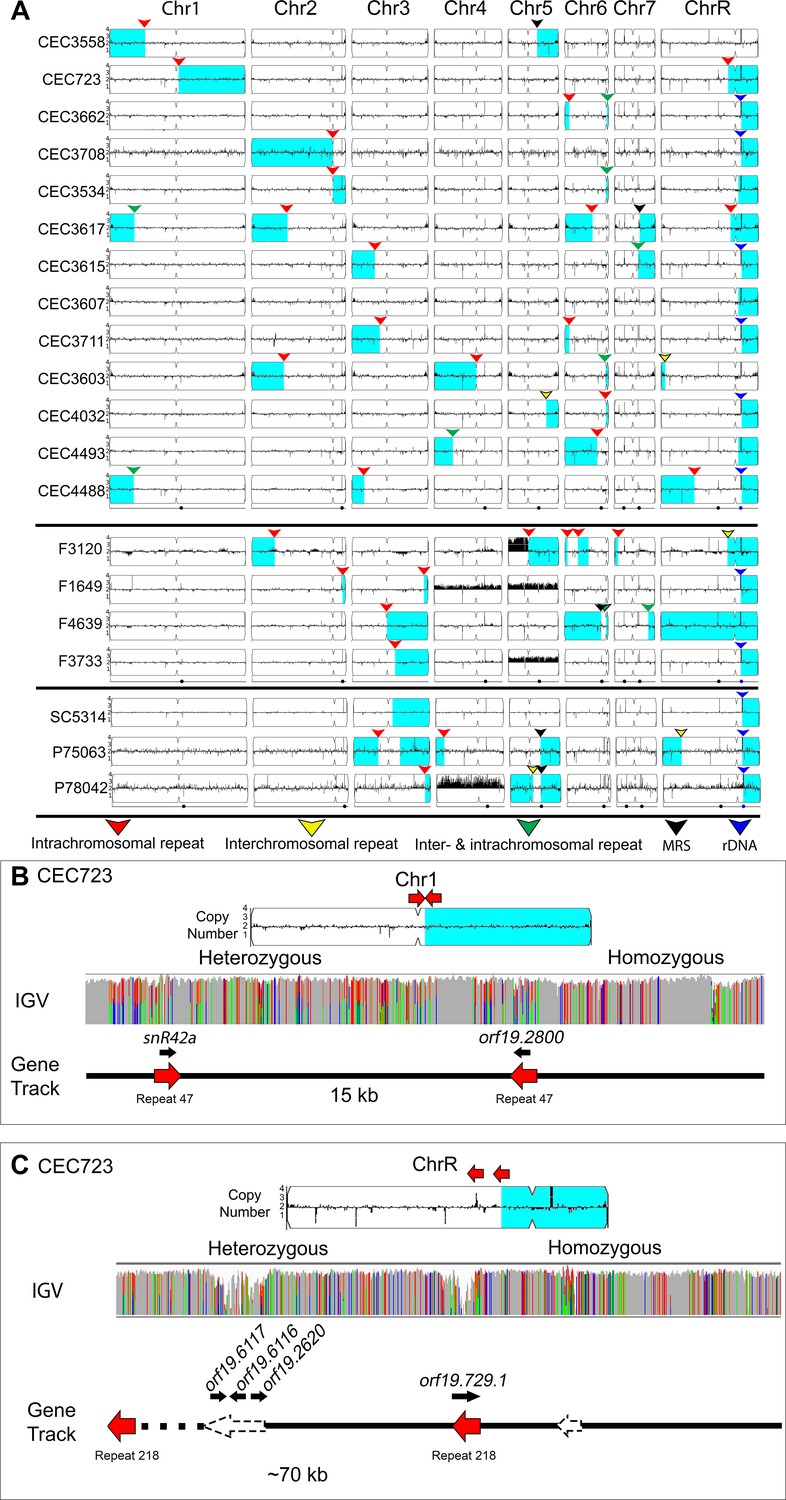
Many LOH breakpoints occur at long intra- and inter-chromosomal repeat sequences.
Whole genome sequence data plotted as a log2 ratio and converted to chromosome copy number (Y-axis) and chromosome location (X-axis) using YMAP. (A) All long-range homozygous regions (light blue) that are associated with long repeat sequences (colored arrows) are indicated for 20 diverse C. albicans isolates. LOH breakpoints and isolate information are detailed in Supplementary files 1 and 4. The type of long repeat is indicated with colored arrows: intra-chromosomal (red), inter-chromosomal (yellow), both intra- and inter-chromosomal (green), rDNA repeat (blue), and MRS (black). (B–C) Two example LOH breakpoints in isolate CEC723 that occur at long repeats (red arrows) on (B) Chr1 (repeat copy length ~1.1 kb), and (C) ChrR (repeat copy length ~3.3 kb) and continue to the right telomere of the respective chromosomes. Heterozygous and homozygous allele ratios are indicated in the IGV track. The position, orientation, and spacer length of the long repeat sequence is indicated in the gene track. ORFs (black arrows) contained within the long repeat sequences are indicated above the gene track. The LOH breakpoint on ChrR is within a repeat-dense region; additional long repeats in the region are indicated (dashed arrows).
Figure 4—figure supplement 1
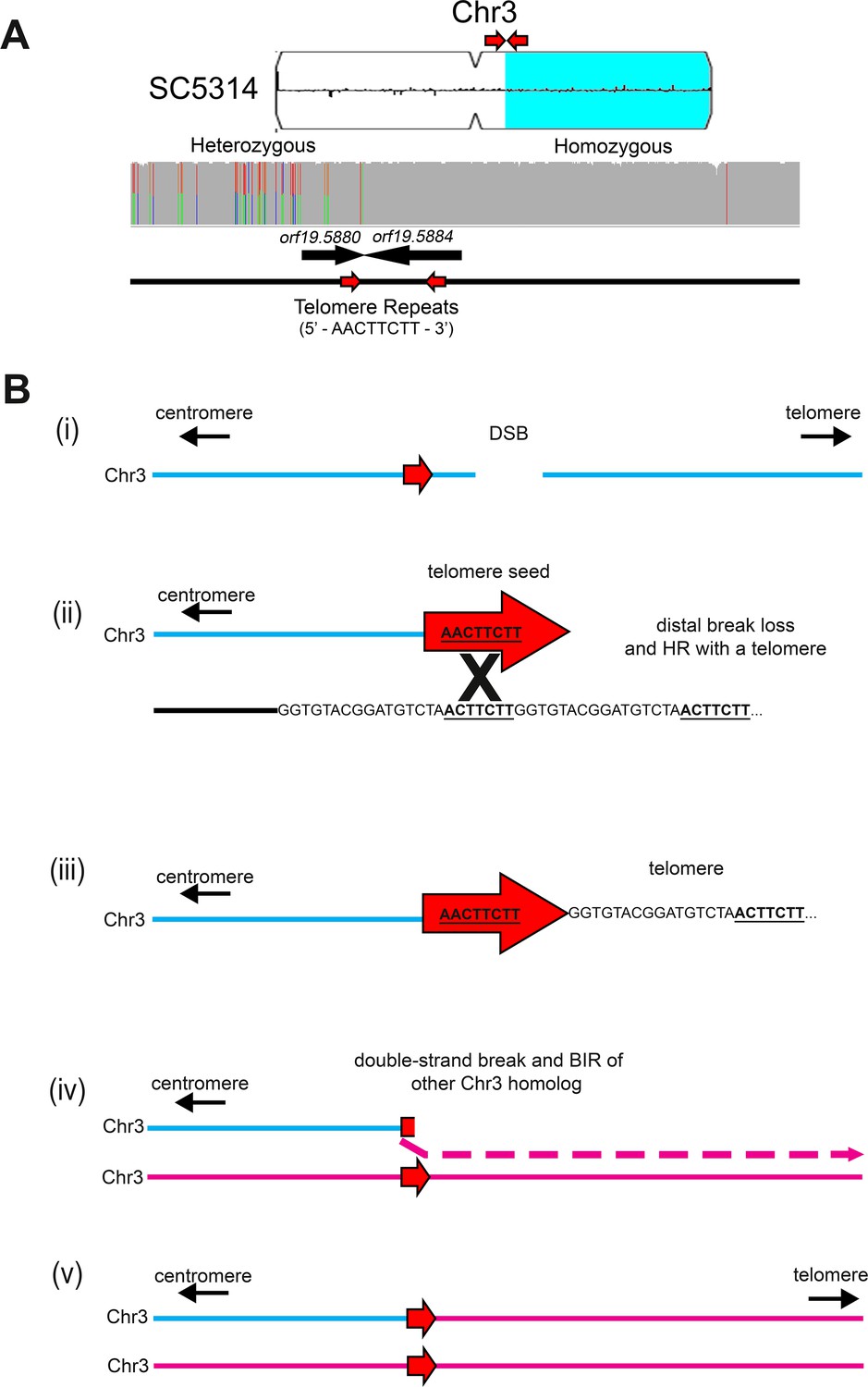
Long-track homozygosis occurs on Chr3L at telomere seed sequences.
(A) Homozygosis of the right arm of Chr3 in SC5314 occurred near a telomere repeat sequence. Chromosome plot indicating the location of homozygosis (light blue) on Chr3R in SC5314. An 8 bp unit of the C. albicans telomere repeat sequence (5’ – AACTTCTT – 3’) indicated by the two red arrows. Read depth and allele ratios above the gene track indicates that homozygosis occurs near the 8 bp telomere seed sequence in the 3’ end of orf19.5880 and continues to the Chr3R telomere. (B) Proposed model of telomere addition and subsequent homozygosis of the right arm of Chr3 in SC5314. (i) A double-strand DNA break occurs on one homolog of Chr3 (blue) near the 8 bp telomere seed sequence (red arrow). (ii) Recombination between the 8 bp telomere seed sequence on Chr3 and a telomere on another chromosome end (iii) leads to the formation of a truncated Chr3 capped with a new telomere. (iv) A secondary break within the newly added telomere sequence and BIR of the opposite Chr3 homolog results in (v) formation of a full-length disomic Chr3 that is homozygous for the right arm.
Figure 5

Long repeat sequences are associated with chromosomal inversions.
(A) Whole genome sequence read depth plotted as a log2 ratio and converted to chromosome copy number (Y-axis) and chromosome location (X-axis) using YMAP. Long-range homozygous regions (blue) on Chr4 are indicated for the isolate P75063. IGV allele ratio track indicates multiple homozygous to heterozygous transitions between a long inverted repeat (red arrows, repeat 144, copy length ~1.7 kb). Primers (5, 6, and 7, Supplementary file 7) were designed to unique sequences flanking repeat 144. (B) PCR amplification between Primers 6 and 7 identifies a ~32 kb chromosomal inversion in both the reference isolate SC5314 and P75063; the reference orientation did not amplify (Primers 5 and 6).
Figure 6 with 1 supplement
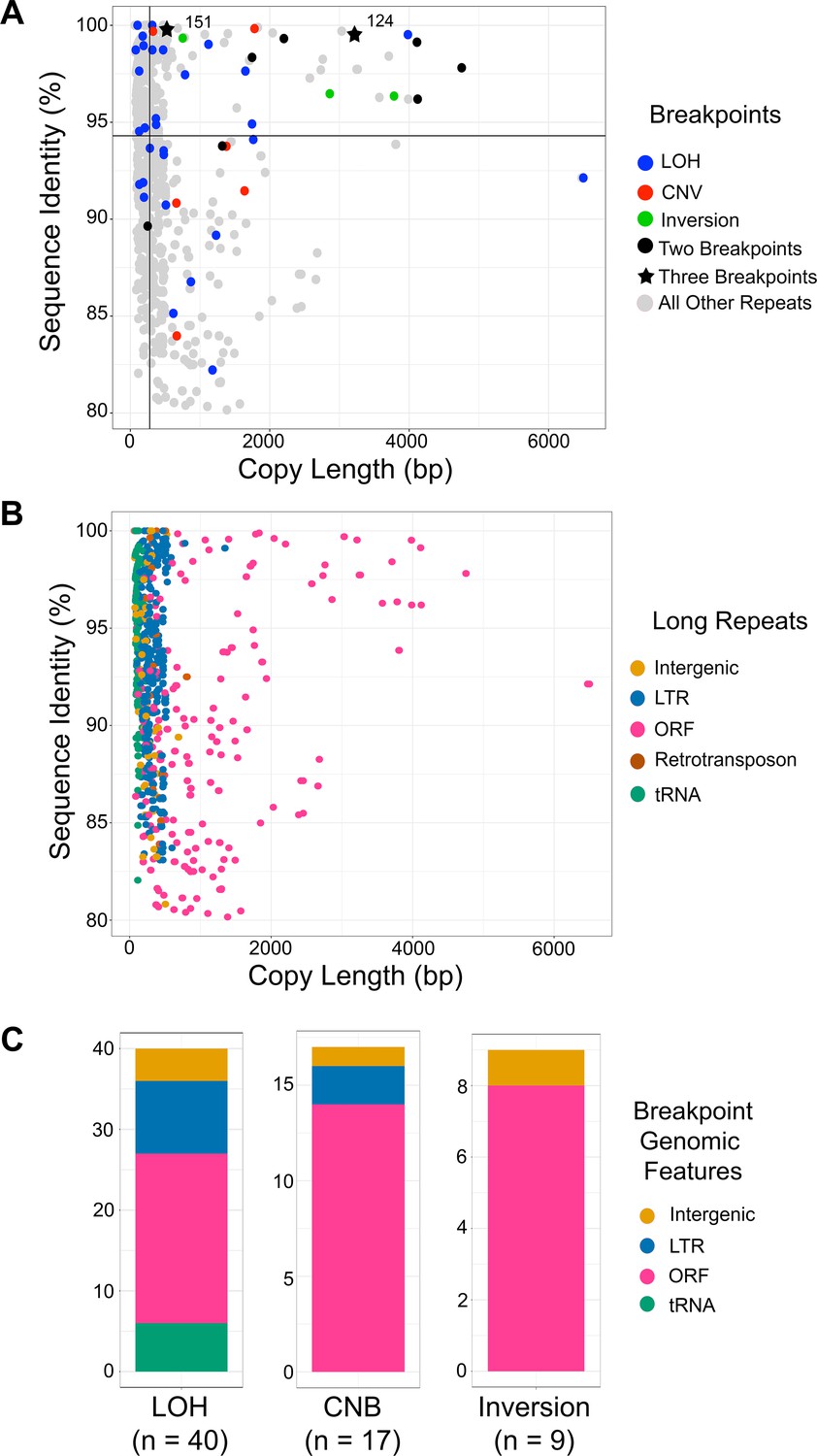
Breakpoints associated with CNV, LOH, and inversion predominantly occur at long repeats that contain ORFs.
(A) Scatterplot of percent sequence identity and copy length of all long repeat matches in Supplementary file 2, excluding the complex tandem repeat genes. All long repeats are indicated in gray, and repeats associated with the observed breakpoints are indicated as follows: LOH (blue), CNV (red), and inversion (green). Six repeats (black circle) were associated with more than one type of breakpoint, and two repeats (black star) were associated with all three types of breakpoints. Solid black lines indicate the median repeat copy length (278 bp, vertical black line) and median percent sequence identity (94.3%, horizontal black line). Repeats associated with LOH, CNV, and inversion breakpoints have a significantly higher median copy length (p<0.0001, Kolmogorov-Smirnov test) and median sequence identity (p<0.036, Kolmogorov-Smirnov test) than all other long repeat sequences (excluding the complex tandem repeat genes, Figure 6—source data 1). (B) Scatterplot as in Figure 6A, where genomic features contained within long repeats are indicated: intergenic sequence (light brown), lone LTR (blue), ORF (pink), retrotransposon (dark brown), and tRNA (green). (C) The distribution of genomic features contained within long repeats at LOH, CNV, and inversion breakpoints. Colors indicated as in Figure 6B.
-
Figure 6—source data 1
Analysis of long repeat sequences associated with CNV, LOH, and sequence inversion.
- https://doi.org/10.7554/eLife.45954.020
Figure 6—figure supplement 1
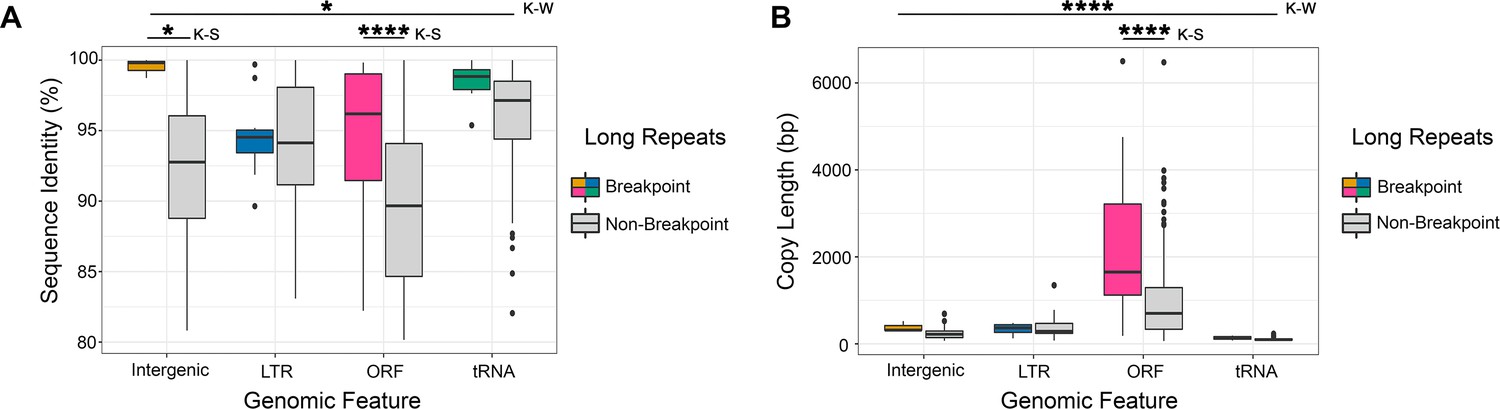
Breakpoint-associated repeats containing ORFs have both high sequence identity and long copy length.
(A) Percent sequence identity of long repeat matches (excluding the complex tandem repeat genes) associated with an observed breakpoint, or not associated with an observed breakpoint (gray) for each genomic feature contained within the long repeat (intergenic sequence [light brown], lone LTR [blue], ORF [pink], and tRNA [green]). Breakpoint-associated repeats containing intergenic sequences (n = 3) have significantly higher identity than all other breakpoint-associated repeats combined (p<0.036, Kruskal-Wallis [K–W] test). The sequence identity of breakpoints containing ORFs and intergenic sequence are significantly higher than non-breakpoint associated repeats containing the same genomic features (intergenic sequence p<0.023, ORF p<0.0001, Kolmogorov-Smirnov [K–S] test). (B) The copy length of repeats associated with an observed breakpoint (color as in A) or not associated with an observed breakpoint (gray) for each genomic feature contained within the long repeat. Breakpoint-associated repeats containing ORFs are significantly longer than all other repeats (p<0.0001, Kruskal-Wallis [K-W] test, Figure 6—source data 1). Breakpoint-associated repeats containing ORFs are significantly longer than non-breakpoint associated repeats containing ORFs (p<0.0001, Kolmogorov-Smirnov [K-S] test). Importantly, breakpoint-associated repeats containing ORFs were the only repeats with both significantly higher identity and significantly longer copy length than non-breakpoint associated repeats (Figure 6—figure supplement 1).
Figure 7
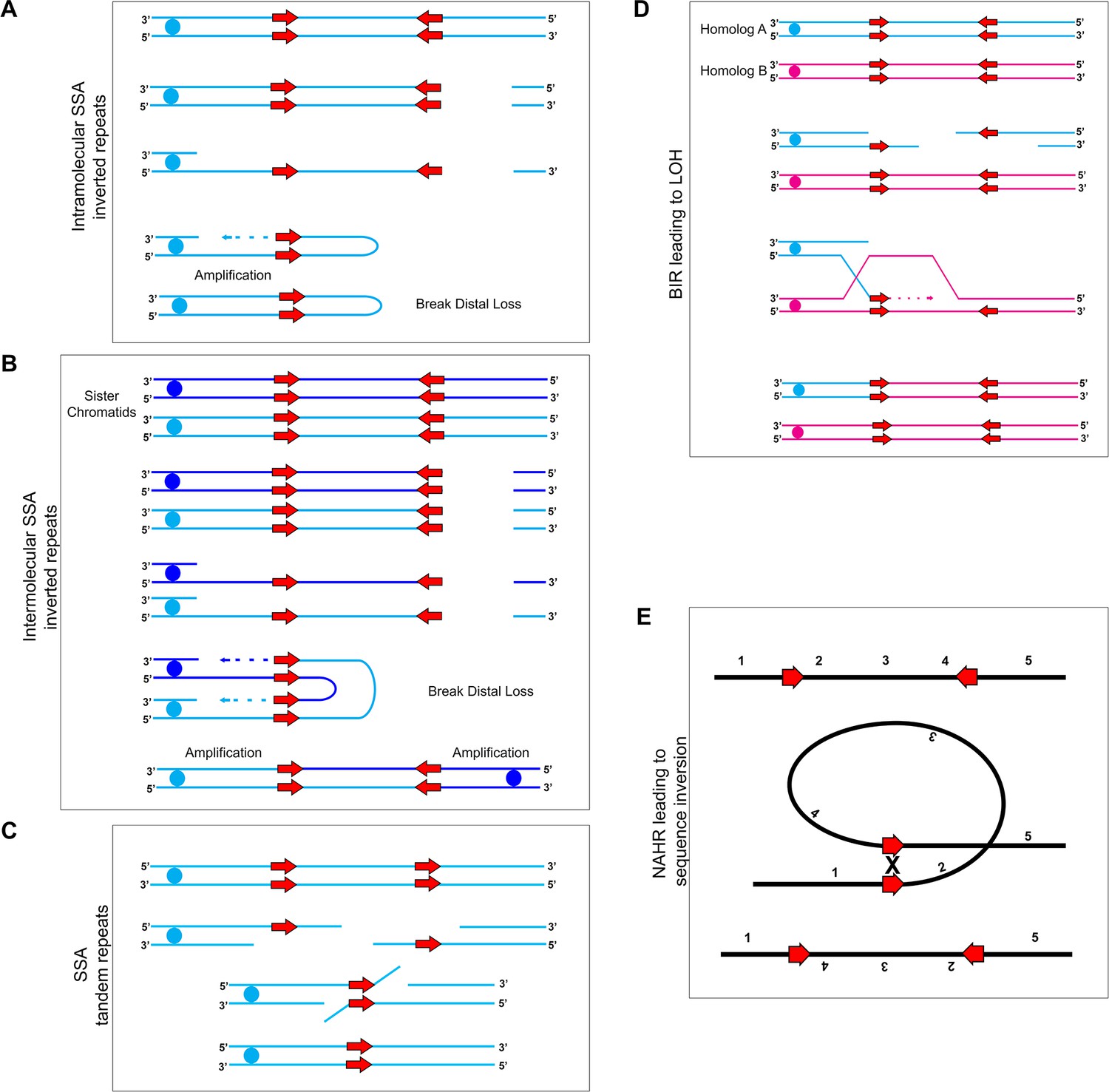
Mechanisms for recombination between long repeats that result in segmental amplification, deletion, LOH, and/or inversion.
(A) Intra-molecular single-strand annealing occurs after a double strand break (DSB) on a single DNA molecule undergoes 5’−3’ resection exposing two copies of an inverted repeat on the single-stranded 3’ overhang. Annealing of the two inverted repeat copies occurs followed by DNA synthesis resulting in a fold-back structure and partial chromosome truncation. (B) Inter-molecular single-strand annealing occurs when a DSB occurs on two separate DNA molecules. After 5’−3’ resection, annealing between the single-stranded inverted repeat copies of the two different DNA molecules results in the formation of a dicentric chromosome and partial chromosome truncation. (C) A single DNA molecule (blue) containing two tandem repeats (red arrows) undergoes a DSB leading to 5’−3’ resection that exposes the tandem repeats. The homologous sequences anneal and non-homologous 3’ tails are removed. The remaining gap is filled producing an intact chromosome that has undergone an internal deletion. (D) Break-Induced Replication (BIR) induces LOH between repeat sequences found on opposite homologs: Two homologs, homolog A (blue) and homolog B (magenta), contain inverted repeat sequences (red arrows). A DSB occurring on homolog A leads to strand invasion and DNA synthesis. Upon termination of synthesis of both the leading and lagging strands, all sequences to the right of the DSB are homozygous. (E) Inversion events occur due to intra-molecular recombination between inverted repeats (red arrows) flanking a unique sequence. The orientation of the reference sequence is indicated above chromosome (1-2-3-4-5). Non-Allelic Homologous Recombination (NAHR) between the inverted repeats leads to an inversion of the sequence between the repeats (1-4-3-2-5).
Author response image 1
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Candida albicans) | SC5314 | Hirakawa et al., 2015 (doi:10.1101/gr.174623.114) | RRID:SCR_013437 | |
| Strain, strain background (C. albicans) | P78042 | Hirakawa et al., 2015 (doi:10.1101/gr.174623.114) | ||
| Strain, strain background (C. albicans) | AMS3743 | This Study | In vitro evolution of P78042 in 128 ug/ml FLC for 100 generations | |
| Strain, strain background (C. albicans) | AMS3743_10 | This Study | In vitro evolution of AMS3743 in rich medium for 300 generations | |
| Strain, strain background (C. albicans) | AMS3743_10_S6 | This Study | Single colony from AMS3743_10 | |
| Antibody | Anti-Digoxigenin-AP Fab Fragments | Roche | 11093274910 RRID:AB_2734716 | (1:5000) |
| Sequenced-based reagent | PCR Primers | This Study | Supplementary file 7 | |
| Commercial assay or kit | Illumina Nextera XT Library Prep Kit | Illumina | 105032350 | |
| Commercial assay or kit | Illumina Nextera XT Index Kit | Illumina | 105055294 | |
| Commercial assay or kit | Illumina MiSeq v2 Reagent Kit | Illumina | 15033625 | 2 × 250 cycles |
| Commercial assay or kit | Blue Pippin 1.5% agarose gel dye-free cassette | Sage Science | 250 bp - 1.5 kb DNA size range collections, Marker R2 | Target of 900 bp |
| Commercial assay or kit | Qubit dsDNA HS kit | Life Technologies | Q32854 | |
| Commercial assay or kit | PCR DIG Probe Synthesis Kit | Roche | 11636090910 | |
| Commercial assay or kit | Agilent 2100 Bioanalyzer High Sensitivity DNA Reagents | Agilent Technologies | 5067–4626 | |
| Chemical compound, drug | Fluconazole (FLC) | Alfa Aesar | J62015 | |
| Software, algorithm | MUMmer Sutie | Kurtz et al., 2004 (doi:10.1186/gb-2004-5-2-r12) | v3.0 RRID:SCR_001200 | |
| Software, algorithm | Trimmomatic | Bolger et al., 2014 (doi:10.1093/bioinformatics/btu170) | v0.33 RRID:SCR_011848 | |
| Software, algorithm | BWA | Li, 2013 (doi:10.1093/bioinformatics/btp324) | v0.7.12 RRID:SCR_010910 | |
| Software, algorithm | Samtools | Li et al., 2009 (doi:10.1093/bioinformatics/btp352) | v0.1.19 RRID:SCR_002105 | |
| Software, algorithm | Genome Analysis Toolkit | McKenna et al., 2010 (doi:10.1101/gr.107524.110) | v3.4–46 RRID:SCR_001876 | |
| Software, algorithm | REPuter | Kurtz et al., 2001 (doi:10.1093/nar/29.22.4633) | V1.0 https://bibiserv.cebitec.uni-bielefeld.de/reputer | |
| Software, algorithm | Yeast Analysis Mapping Pipeline | Abbey et al., 2014 (doi:10.1186/s13073-014-0100-8) | v1.0 | |
| Software, algorithm | Graphpad Prism | https://www.graphpad.com | v6.0 RRID:SCR_002798 | |
| Software, algorithm | ImageJ | https://imagej.nih.gov/ij/? | v2.0.0-rc-30/1.49 s RRID:SCR_003070 | |
| Software, algorithm | Integrative Genomics Viewer | Thorvaldsdóttir et al., 2013 (doi:10.1093/bib/bbs017) | v2.3.92 RRID:SCR_011793 | |
| Software, algorithm | R | https://www.r-project.org | v3.5.2 RRID:SCR_001905 | |
| Software, algorithm | Candida Genome Database | http://Candidagenome.org | RRID:SCR_002036 | |
| Other | Propidium Iodide | Invitrogen | P3566 | 25 ug/ml final concentration |
| Other | Ribonuclease A | MP Biomedicals | 101076 | 0.5 mg/ml final concentration |
Additional files
-
Supplementary file 1
Strains used in this study.
- https://doi.org/10.7554/eLife.45954.022
-
Supplementary file 2
Long repeat sequences in the Candida albicans genome.
- https://doi.org/10.7554/eLife.45954.023
-
Supplementary file 3
Copy number variation breakpoints in diverse C. albicans isolates.
- https://doi.org/10.7554/eLife.45954.024
-
Supplementary file 4
Loss of heterozygosity breakpoints in diverse C. albicans isolates.
- https://doi.org/10.7554/eLife.45954.025
-
Supplementary file 5
Location of telomere-seed sequences throughout the C. albicans genome.
- https://doi.org/10.7554/eLife.45954.026
-
Supplementary file 6
Predicted inversion breakpoints in diverse C. albicans isolates.
- https://doi.org/10.7554/eLife.45954.027
-
Supplementary file 7
Primers used in this study.
- https://doi.org/10.7554/eLife.45954.028
-
Transparent reporting form
- https://doi.org/10.7554/eLife.45954.029
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Genome plasticity in Candida albicans is driven by long repeat sequences
eLife 8:e45954.
https://doi.org/10.7554/eLife.45954




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}