TMEM16B regulates anxiety-related behavior and GABAergic neuronal signaling in the central lateral amygdala
- University of California, San Francisco, United States
- Howard Hughes Medical Institute, University of California, San Francisco, United States
- Gladstone Institute of Neurological Disease, United States
Abstract
TMEM16B (ANO2) is the Ca2+-activated chloride channel expressed in multiple brain regions, including the amygdala. Here we report that Ano2 knockout mice exhibit impaired anxiety-related behaviors and context-independent fear memory, thus implicating TMEM16B in anxiety modulation. We found that TMEM16B is expressed in somatostatin-positive (SOM+) GABAergic neurons of the central lateral amygdala (CeL), and its activity modulates action potential duration and inhibitory postsynaptic current (IPSC). We further provide evidence for TMEM16B actions not only in the soma but also in the presynaptic nerve terminals of GABAergic neurons. Our study reveals an intriguing role for TMEM16B in context-independent but not context-dependent fear memory, and supports the notion that dysfunction of the amygdala contributes to anxiety-related behaviors.
https://doi.org/10.7554/eLife.47106.001Introduction
Anxiety disorders are marked by excessive fear. Hence, it is important to study the basic fear circuitry in animal models. In rodent animal models, amygdala, hippocampus, ventromedial hypothalamus, nucleus accumbens, periaqueductal gray and prefrontal cortex have been identified as key brain regions in the neurocircuitry associated with fear responses (Davis, 2006; Maren, 2008; Quirk and Mueller, 2008). Among these brain regions, amygdala play a central role in fear acquisition and expression of fear responses. Investigations of the amygdala microcircuits via optogenetics reveal the importance of inhibitory inputs from central lateral amygdala (CeL) to central medial amygdala (CeM), as well as disinhibition of various output neurons of the central nucleus of the amygdala (CeA), in mediating conditioned fear or anxiolytic processes to reduce anxiety (Ciocchi et al., 2010; Haubensak et al., 2010; Janak and Tye, 2015); acute anxiolytic responses may result from excitatory inputs from basolateral amygdala (BLA) to the CeA, leading to activation of CeL neurons and the ensuing feed-forward inhibition (Janak and Tye, 2015; Paré et al., 2004; Tye et al., 2011). These studies underscore the importance of characterizing neuronal signaling in the amygdala to better understand the physiological basis of fear and anxiety disorders.
Ano2 encodes an evolutionarily conserved Ca2+-activated Cl- channel (CaCC). As members of the enigmatic TMEM16 (ANO2) family of transmembrane proteins with various physiological functions (Yang and Jan, 2016), TMEM16B (ANO2) and TMEM16A (ANO1) are pore forming subunits of the Ca2+-activated Cl- channels (Dang et al., 2017; Paulino et al., 2017; Peters et al., 2015). Ano2 mRNA expression is enriched in brain regions including the amygdala, lateral septum, and hippocampus according to the Allen Brain Atlas, raising the intriguing question regarding the role of the TMEM16B Ca2+-activated Cl- channel in anxiety-related behaivor. To test whether alteration of TMEM16B activity may contribute to anxiety-related behaviors, we carried out behavioral studies of the knockout mice we generated (Zhang et al., 2017). Our finding that Ano2 knockout mice exhibit impaired anxiety-related behaviors and fear memory indicates that the physiological function of TMEM16B is important for the processing and expression of anxiety.
TMEM16B is known to serve important functions in various brain regions including the hippocampus (Ha et al., 2016; Huang et al., 2012; Neureither et al., 2017; Wang et al., 2019; Zhang et al., 2015; Zhang et al., 2017), however, whether it also modulates neuronal signaling in the amygdala remains an open question. We found that TMEM16B-CaCC is highly enriched in central lateral amygdala (CeL). Loss of TMEM16B function results in action potential broadening in SOM+ CeL neurons that express somatostatin, increase of the amplitude and frequency of GABAergic spontaneous inhibitory postsynaptic current (sIPSC), and reduction of the frequency of miniature IPSC (mIPSC) that depends on presynaptic voltage-gated Ca2+ channel activity. Our results provide evidence for a critical role of TMEM16B in neuronal signaling in the amygdala as well as fear and anxiety-like behavior.
Results
Ano2 KO mice display reduced anxiety-related behaviors
To look into the physiological relevance of TMEM16B in anxiety, we performed a battery of anxiety-related behavior tests of 3–4 months old Ano2 knockout (KO) mice that have sequences for farnesylated mCherry inserted into the coding sequence for Ano2, and used their wildtype littermates as controls. First, we subjected each animal to the open field test (OFT) where we could simultaneously record their anxiety-related behaviors and locomotion. As shown in Figure 1A, the control (Con) mouse but not the KO mouse tended to avoid the center of the open field. There was a significant increase in the center exploration behavior in Ano2 KO mice as compared to their control littermates (Figure 1B) (unpaired t test in (B), p<0.001, n = 26 for KO and n = 23 for control). This abnormal behavior was not accompanied by any detectable locomotive defects in Ano2 KO mice, as evident from measurements of the total distance traveled (Figure 1C,D).
Figure 1
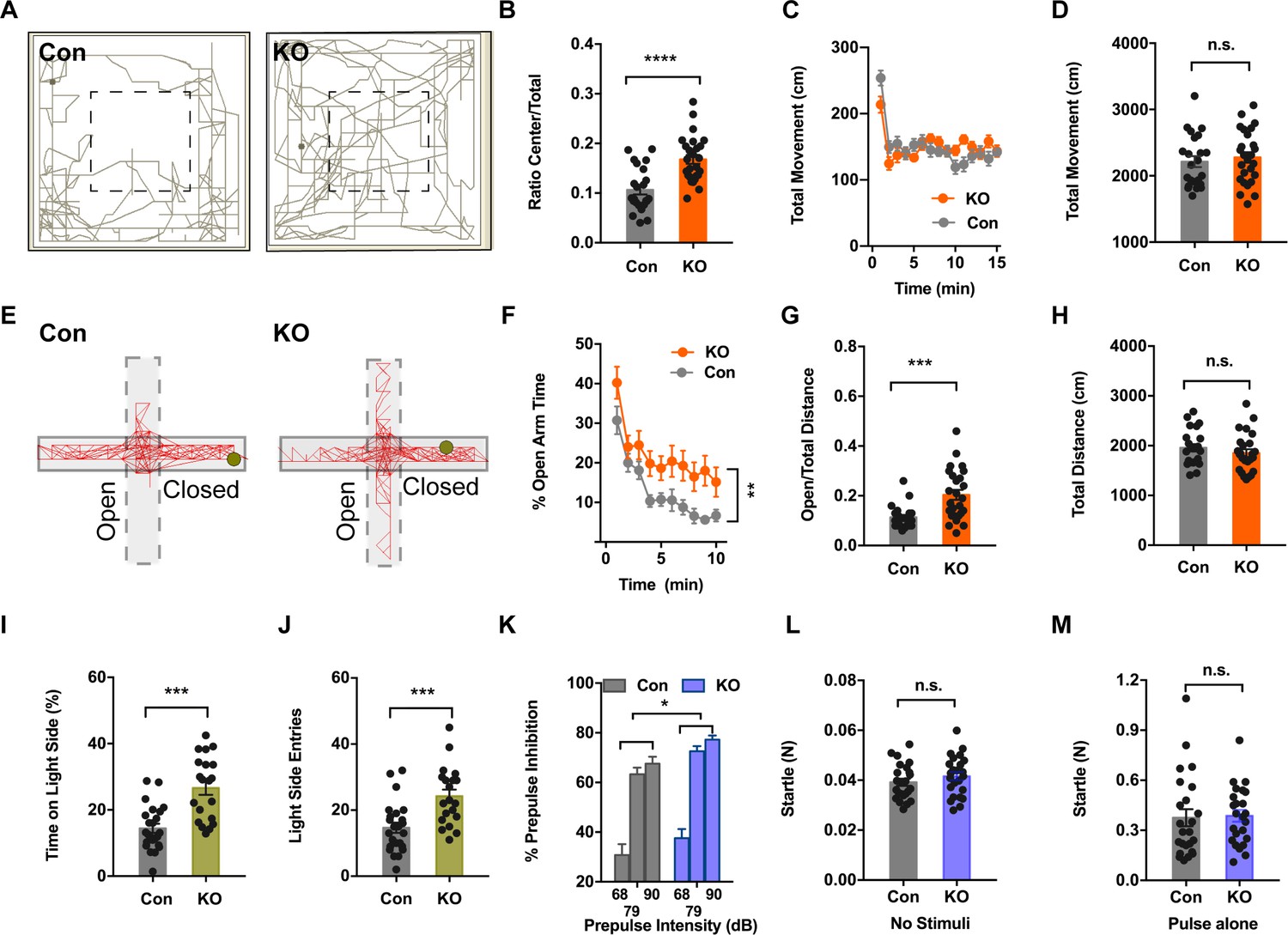
Ano2 Mutant Mice Display Reduced Anxiety-related Behavior and Impaired Pre-pulse Inhibition.
(A) Representative traces of Ano2 KO mice and control (Con) littermates in open field. (B–D) Ano2 mutant mice display reduced anxiety-related behavior in the open field test, shown as time spent at the center normalized by total time (B). Overall exploratory behavior in the open-field test, shown as distance traveled per minute as function of time (C) and total distance traveled (D), is comparable between mutant and control mice. Two-tail unpaired t test in (B), t = 4.744, df = 46.7, p<0.0001. Two-way ANOVA in (C), F (1,47)=0.04778, p=0.8279. Two-tail unpaired t test in (D), t = 0.5721, df = 47, p=0.5700, n = 26 for KO and n = 23 for control (number of mice tested) for (B–D). (E) Representative elevated plus maze (EPM) tracks of control and Ano2 KO mice. (F–H) Ano2 mutant mice display reduced anxiety-related behavior in the EPM test, plotted as time spent in the open arms as function of time (F) and distance traveled in open arms normalized by the total distance traveled (G), with the total distance traveled in the elevated plus maze (H) as control. Two-way ANOVA in (F), F (1, 47)=9.067, p=0.0042. Mann Whitney test in (G), U = 120, p=0.0002. Two-tail unpaired t test in (H), t = 0.9944 df=46, p=0.3252, n = 26 for KO and n = 23 for control (number of mice tested) for F–H. (I and J) Ano2 mutant mice show reduced anxiety-related behavior in the light-dark box test, shown as the percentage of total time spent in the bright compartment (I) and number of entries to the bright compartments (J) over a 10 min period. Two-tail unpaired t test in (I), t = 4.804, df = 32.94, p<0.0001. Two-tail unpaired t test in (J), t = 3.79, df = 37.77, p=0.0005, n = 20 for KO and n = 24 for control for I and J. (K–M) Ano2 mutant mice show reduced pre-pulse inhibition (PPI) of startle responses induced by pre-pulses at 68 dB, 79 dB and 90 dB(K). Startle responses to no stimuli (L) and startle response to 120 dB stimuli without pre-pulse (M) are comparable between mutant and control mice. Two-way ANOVA in (K). F (1, 46)=4.612, p=0.0370. Two-tail unpaired t test in (L), t = 1.119, df = 46, p=0.2688. Two-tail unpaired t test in (M), t = 0.1889, df = 46, p=0.8510, n = 24 for KO and n = 24 for control (number of mice tested) for K–M. Data are presented as mean ± SEM. *p<0.05, **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference.
To further examine anxiety-related behaviors of Ano2 KO mice, we tested for their performance in the elevated plus maze (EPM). Each individual mouse was given the choice of spending time in the open and unprotected arms or the enclosed and protected arms of the maze. Figure 1E shows representative EPM tracks of control and KO mice. Consistent with the OFT results, mice lacking Ano2 showed significantly greater open-arm exploration (Two-way ANOVA in (F), p=0.0042; Mann Whitney test in (G), p=0.0002 n=26 for KO and n = 23 for control), reflecting a reduction in anxiety-related behaviors as compared to control littermates (Figure 1F,G). As in the case of OFT tests, the total distance traveled by Ano2 KO mice and their control littermates were comparable (Figure 1H).
Next, we exposed these mice to a novel environment with protected (dark compartment) and unprotected (light compartment) areas. We found that mice with loss of TMEM16B function entered the light side more frequently and spent more time in this unprotected area as compared to control mice (Figure 1I,J) (unpaired t test in (I,J), p<0.001(I), p=0.005 (J), n = 20 for KO and n = 24 for control). Collectively, all these three different assays indicate that Ano2 KO mice are deficient in anxiety-related behaviors.
Pre-pulse inhibition (PPI) provides a measure of sensorimotor gating that is reduced in several neuropsychiatric disorders. Panic disorder patients have difficulties in progressively dampening their response to sensory and cognitive stimuli, suggesting that they have reduced PPI (Ludewig et al., 2002). In our assays, Ano2 KO mice showed significantly greater pre-pulse inhibition of startle responses as compared with control mice (Figure 1K) (Two-way ANOVA in (K), p=0.0370, n = 20 for KO and n = 24 for control). The amplitudes of startle responses induced by stimuli without pre-pulse were comparable between KO and control, as were the control startle response in the absence of any stimuli (Figure 1L,M). Thus, Ano2 KO mice display enhanced PPI and reduced anxiety-related behavior.
Ano2 knockout mice exhibit altered fear expression
Disturbances in fear learning may contribute to disorders of fear and anxiety, such as panic disorder and specific phobias (Rosen et al., 1998). Pavlovian fear conditioning and fear expression has become an important model for investigating the learning and memory processes related to fear in rodents (Maren, 2001). We tested for both context-dependent and context-independent fear learning.
Exposing mice to aversive foot shock (Unconditioned Stimuli, US) in an area with contextual stimuli (Conditioned Stimuli, CS) allowed for the testing of context-dependent fear learning and memory. Ano2 KO and control mice displayed comparable freezing levels during context fear acquisition, which increased as pairings of CS and US were repeatedly presented (Figure 2A,C) (Two-way ANOVA, p=0.3928. n = 24 for KO and n = 24 for control). When the same context was presented 24 hr after fear conditioning, Ano2 KO and control mice exhibited no significant difference in context-dependent fear expression (Figure 2B,C) (Two-way ANOVA, p = 0.1472. n = 24 for KO and n = 24 for control), indicating that Ano2 is not required for context-dependent fear acquisition and expression.
Figure 2
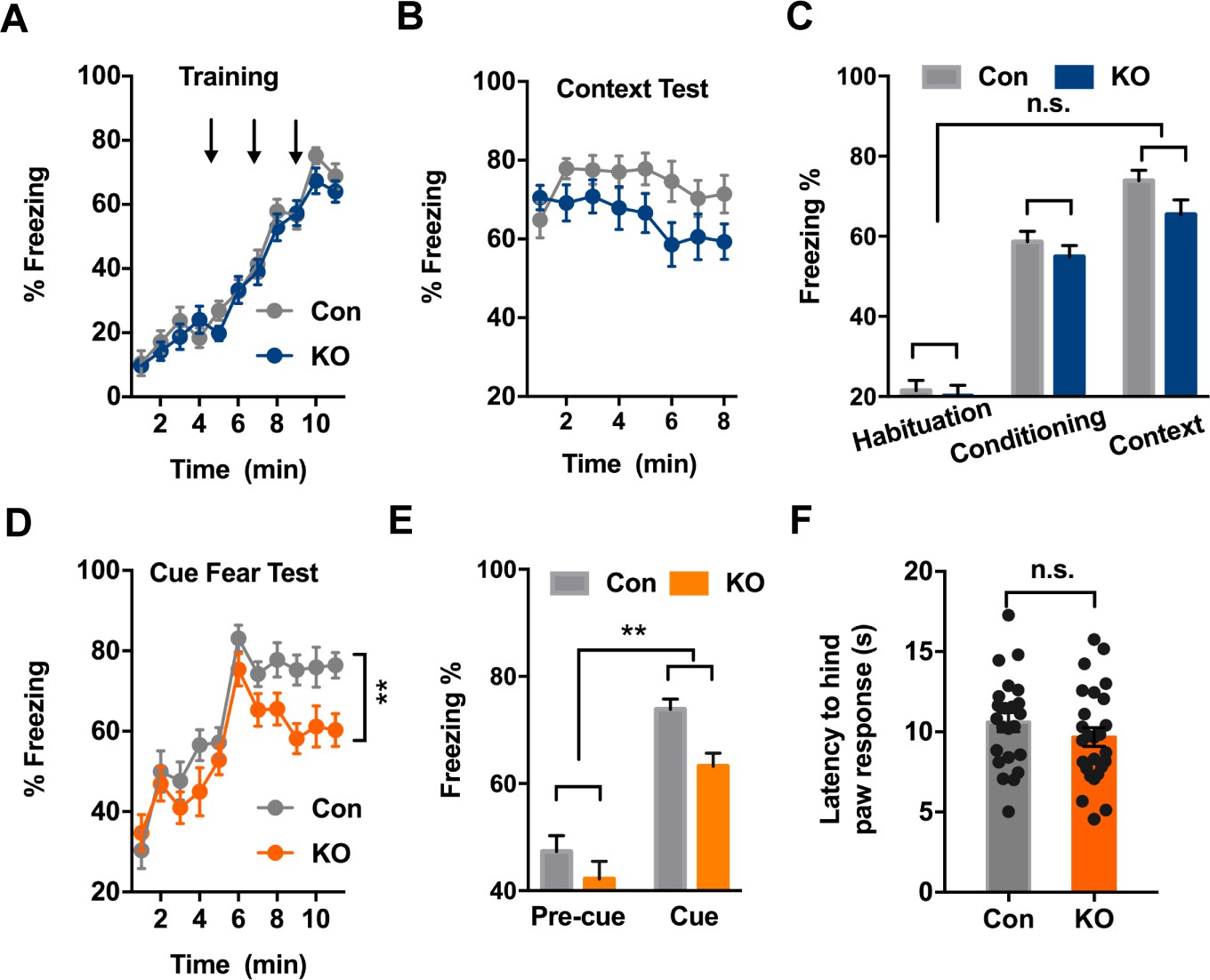
Ano2 Knockout Mice Exhibit Reduced Context-independent Cued Fear Expression.
(A) Ano2 KO and control mice exhibited comparable freezing levels during fear acquisition. After habituation for 5 min, freezing levels during fear training with three 30 s 80 dB tones that each was terminated at the same time as a 2 s, 0.45 mA foot shock, separated by 120 s interval for monitoring. Two-way ANOVA in (A), F (1, 46)=0.7443, p=0.3928. (B) Context-dependent fear recall tested 24 hr after fear conditioning. Mice were placed in the fear conditioning apparatus for 8 min. No shock was presented. Two-way ANOVA in (B), F (1, 46)=3.601, p=0.0640. (C) Ano2 KO and control mice exhibited comparable context-dependent fear expression, shown as averaged freezing levels during habituation, conditioning and context test. Two-way ANOVA in (C), F (1, 46)=2.173, p=0.1472. (D and E) The context-independent cue fear test took place 5–24 hr after the context-dependent test in (B). After a 5 min baseline (pre-cue), three 30 s 80 dB tone cues not accompanied with foot shock were presented with 120 s interval for monitoring (cue), in the absence of context cues. Two-way ANOVA in (D), F (1, 46)=9.151, p=0.004. Two-way ANOVA in (E), F (1, 46)=9.007, p=0.0043. TEMEM16 KO mice displayed lower levels of freezing than control mice during cued fear expression. (F) Pain threshold was comparable between control and Ano2 KO mice.Two tail unpaired t test in (F), t = 1.114, df = 46, p=0.2711, n = 24 for KO and n = 24 for control (number of mice tested for A–F). Data are presented as mean ± SEM. *p<0.05, **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference.
Exposing mice to the aversive foot shock (US) paired with a tone allowed for the testing of context-independent auditory fear learning and memory. Ano2 KO mice displayed lower levels of freezing than control mice during cued fear expression (Figure 2D,E) (Two-way ANOVA, p=0.004, n = 24 for KO and n = 24 for control) while the mutant and control mice displayed similar pain threshold (Figure 2F) and similar freezing levels during the acquisition of cued fear learning (Figure 2A), highlighting the involvement of TMEM16B for controlling context-independent cued fear expression.
Ano2 expression in somatostatin-positive GABAergic neurons in the central lateral amygdala
To examine the cellular expression pattern of Ano2, we used RNA scope for its superior sensitivity to detect low abundance transcripts. We found enrichment of Ano2 mRNA expression in brain regions including the central lateral amygdala, lateral septum and hippocampus, consistent with the expression pattern deposited at the Allen Brain Atlas (Figure 3A; Figure 3—figure supplement 1). Given that anxiety and fear conditioning involves multiple brain regions including amygdala and hippocampus (Chen et al., 2016; Hall et al., 2001; Kochli et al., 2015; Vazdarjanova and McGaugh, 1999; Weeden et al., 2015; Xu et al., 2016; Yin et al., 2002), it is important to examine Ano2 expression and physiological function in amygdala to assess the potential relevance to anxiety. Ano2 is mainly expressed in central lateral amygdala (CeL), but not other sub nuclei of amygdala such as basal lateral amygdala (BLA) or the medial part of central amygdala (CeM) (Figure 3B,C). CeL, which is composed of several classes of GABAergic inhibitory neurons (Cassell and Gray, 1989; Ehrlich et al., 2009; Haubensak et al., 2010), modulates conditioned fear response and anxiety-related behavior via inhibition of neurons in the central lateral and medial subdivisions of CeA, which provide the major output of amygdala (Ciocchi et al., 2010; Janak and Tye, 2015; LeDoux et al., 1988; Tye et al., 2011). GABAergic inhibitory neurons in CeL can be classified by expressions of distinct neurochemical markers (Ehrlich et al., 2009; Haubensak et al., 2010). Among these subtypes, somatostatin-positive (SOM+/Sst) neurons constitute a major population, and are intermingled with protein kinase C-δ expressing (PKC-δ+/Prkcd) neurons and neurons containing corticotropin-releasing hormone (Crh), neurotensin (Nts) and tachykinin 2 (Tac2); neurons expressing various combinations of these markers account for the majority (96%) of the GABAergic neurons in CeL (Kim et al., 2017). Of these markers, Prkcd and Sst almost have no overlap with each other (2%–13%) (Kim et al., 2017; Li et al., 2013), while Sst exhibits high levels of overlap (>50%) with Crh+/Nts+/Tac2+. Our RNA scope study revealed that the majority (92.6%) of SOM+ (Sst) neurons expressed Ano2, and the majority (71.4%) of Ano2 positive cells displayed Sst immunoreactivity (Figure 3E). We further confirmed that SOM+ (Sst) neurons expressed glutamate decarboxylase 2 (Gad2), a marker of inhibitory neurons (Figure 3D).
Figure 3 with 1 supplement see all
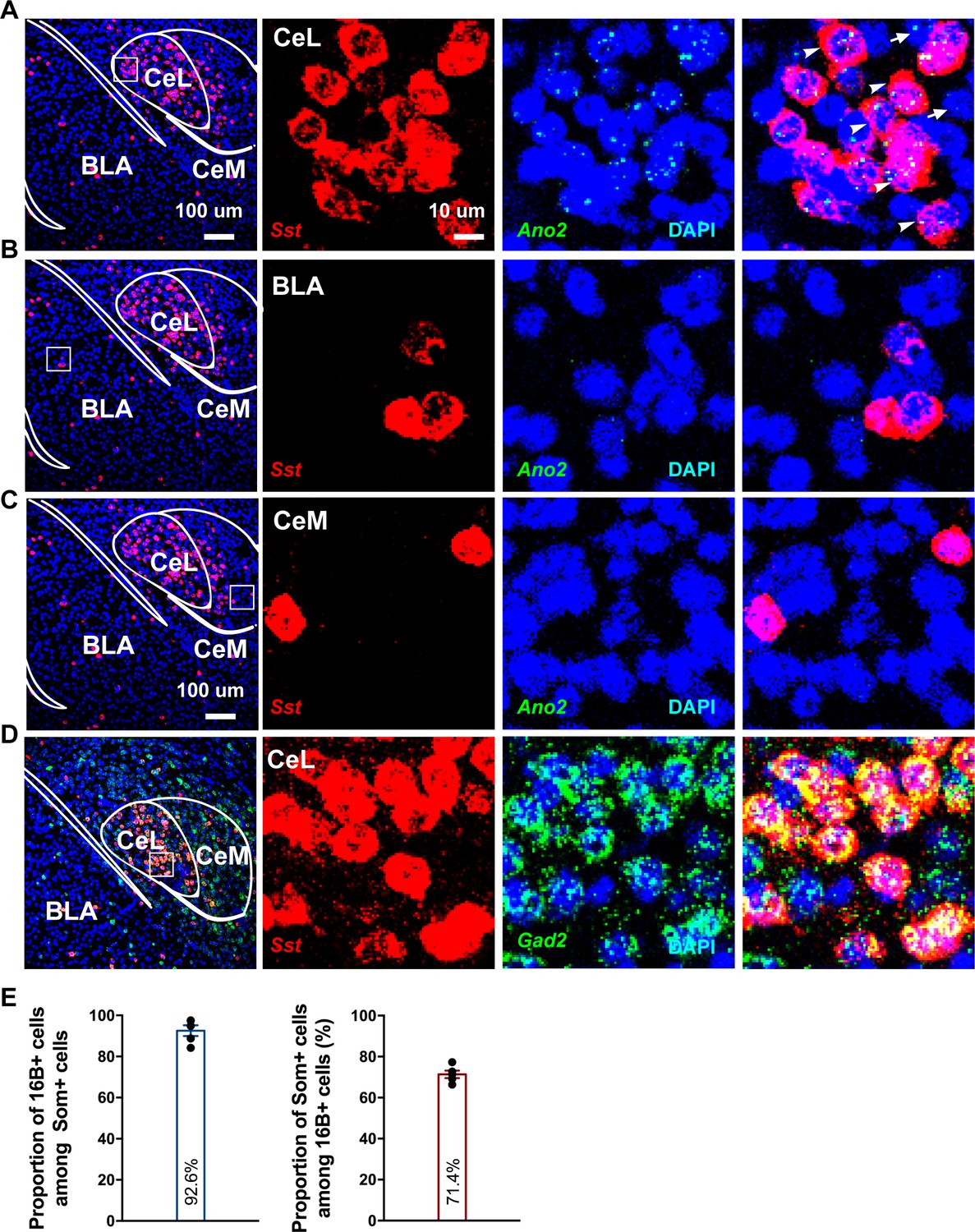
Ano2 mRNA Expression in Somatostatin-positive GABAergic Neurons in the Central Lateral Amygdala.
(A–C) RNA scope studies reveal that Ano2 is expressed in central lateral amygdala (CeL), but not in basal lateral amygdala (BLA) or central medial amygdala (CeM). Representative images of immunofluorescence staining for Sst (red) and RNA scope labeling of Ano2 mRNA (green). Cell nuclei are stained with DAPI (blue). (D) Representative images of double immunofluorescence staining for Gad2 (green) and Sst (red). Cell nuclei are stained with DAPI (blue). (E) Left, percentage of Sst-positive neurons that express Ano2 mRNA. Right, percentage of Ano2 mRNA-positive neurons that express Sst. n = 653 neurons from five mice.
TMEM16B-mediated Ca2+-activated Cl– current in somatostatin-positive neurons of central lateral amygdala
For studies of TMEM16B-mediated Ca2+-activated Cl- current (CaCC) in SOM+ cells, we used a Sst-IRES-Cre knock-in mouse line, with Cre driven by the endogenous promoter of the Somatostatin (Sst) gene (Taniguchi et al., 2011). SOM+ neurons in Sst-IRES-Cre;Ai14 mice can be readily identified by the red fluorescence of the tdTomato reporter of Ai14 (Figure 4A).
Figure 4 with 1 supplement see all
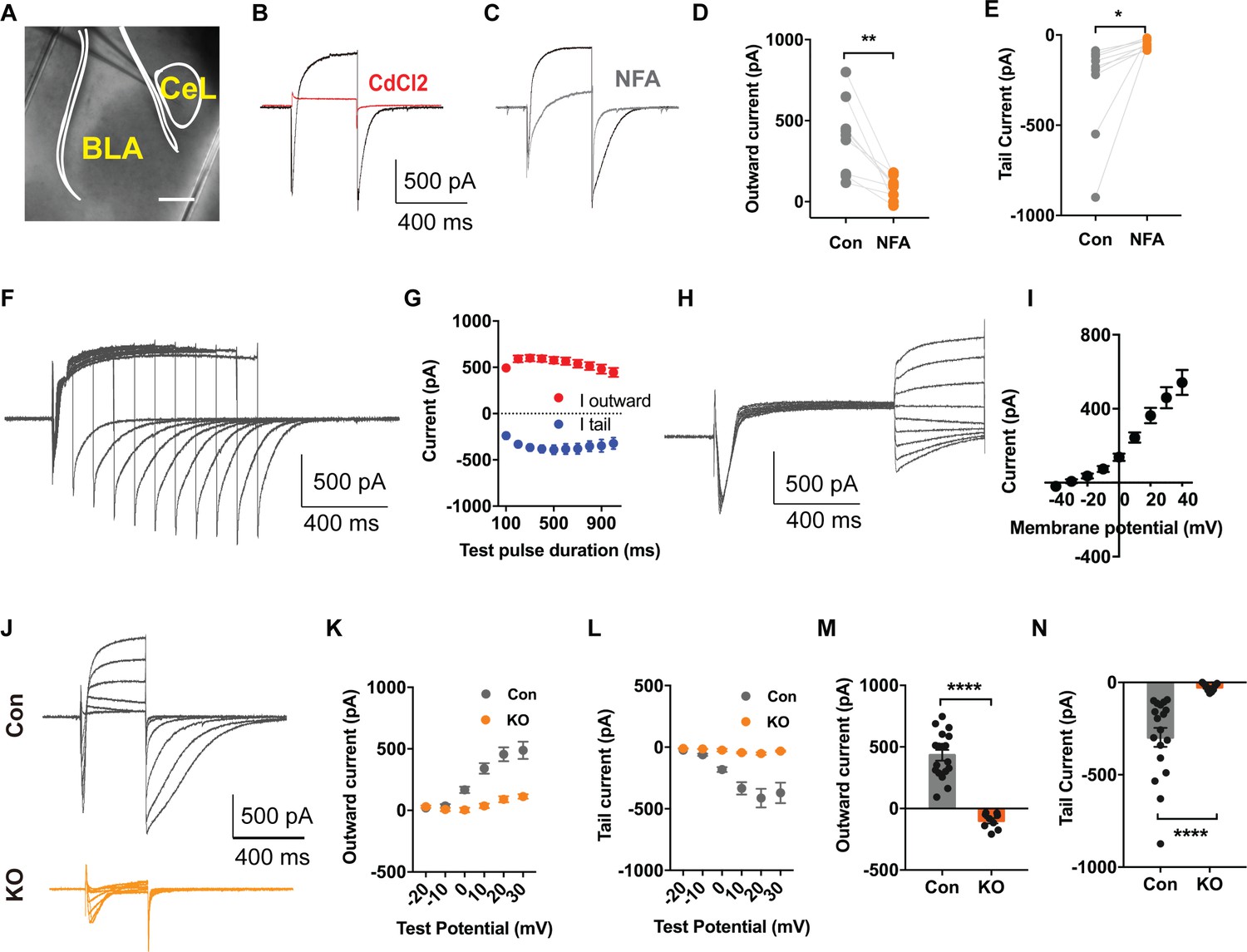
TMEM16B Mediated Ca2+-activated Cl- Currents in Somatostatin-positive Neurons of the Central Lateral Amygdala.
(A) Brain slice of Sst-IRES-Cre and Ai14 reporter mice with fluorescently marked somatostatin-positive (SOM+) neurons in the amygdala. Scale bar, 200 μm. (B and C) Depolarization of SOM+ neurons to +10 mV elicited a Ca2+ current superimposed with a large outward current that developed over time, followed with a tail current upon hyperpolarization to −80 mV. The outward current and tail current can be inhibited by 100 μM niflumic acid (NFA) (C), and it is completely abolished by 200 μM CdCl2, a blocker of voltage-gated Ca2+ channels, indicating that it is induced by Ca2+ influx (B). (D and E) NFA reduced the outward current and tail current. Paired t test in (D), t = 4.392, df = 8, p=0.0023. Paired t test in (E), t = 2.55, df = 8, p=0.0342, n = 9 cells for each group from three mice per group for D and E. (F and G) Summary plot of the peak amplitude of the outward current and tail current as a function of the duration of the test pulse to +10 mV. n = 7 cells from three mice. (H and I) Measurements of reversal potential indicating that the current induced by Ca2+ influx in SOM+ neurons arises from Ca2+-activated Cl- channels (CaCCs). Membrane potential was depolarized to +10 mV to activate Ca2+ channels and then held from −40 mV to +40 mV in 10 mV increments for reversal potential measurement, followed with repolarization to −80 mV. n = 15 cells from four mice. (J–L) Genetic ablation of Ano2 completely eliminated the outward current and tail current in SOM+ neurons, indicating that TMEM16B CaCC mediates the Ca2+-sensitive outward current and tail current in SOM+ neurons. Summary plot of the outward current (K) and the tail current (L) at various membrane potentials. Membrane potential was depolarized from −20 mV to +30 mV in 10 mV increments to activate Ca2+ channels, and then repolarized to −80 mV. (M and N) Comparison of the amplitudes of the outward current and tail current of SOM+ neurons from Ano2 KO mice and control mice. Mann Whitney test in (M), U = 0, p<0.0001. Mann Whitney test in (N), U = 0, p<0.0001. Cells were held at −80 mV. n = 18 cells from 5 KO and n = 9 cells from four control for K, L, M and N. Data are presented as mean ± SEM. *p<0.05, **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference.
Given that TMEM16B-CaCC is expressed in the hippocampus, lateral septum, and Inferior olive neurons, where it is activated by Ca2+ influx through voltage-gated Ca2+ channels (Huang et al., 2012; Wang et al., 2019; Zhang et al., 2017), we tested whether TMEM16B can also be activated by Ca2+ influx through voltage-gated Ca2+ channels in neurons of central lateral amygdala. First, we confirmed that Ca2+ currents were elicited in SOM+ neurons with depolarizations lasting 60 ms (Figure 4—figure supplement 1A). Next, we carried out whole cell recording in the presence of 5 mM QX314, 110 mM Cs+, and 20 mM tetraethylammonium (TEA), to block voltage-gated Na+ channels and the majority of K+ channels including Ca2+-activated K+ channels. We found that depolarization to +10 mV for 500 ms elicited a transient inward current as a result of the superimposition of an inward Ca2+ current and an outward CaCC that developed over time leading to the appearance of a net outward current following the transient inward current, followed by a slowly deactivating tail CaCC current upon repolarization to −80 mV (Figure 4B). The feature of CaCC we recorded is similar to that reported previously (Zhang et al., 2017). Treatment of brain slices with CdCl2, a Ca2+ channel blocker, abolished both the inward Ca2+ current and the CaCC (Figure 4B), indicating that CaCC activation is triggered by Ca2+ influx through voltage-gated Ca2+ channels. To verify that the outward current superimposed on the inward Ca2+ current during depolarization and the tail current upon repolarization are Cl- currents, we first showed that NFA, a Cl- channel blocker, reduced both the outward current and the tail current (Figure 4C,D,E) (Paired t test in (D), p=0.0023; Paired t test in (E), p=0.0342, n = 9 cells for each group from three mice per group). For further validation, we varied the duration of the Ca2+ channel activation and hence the extent of Ca2+ entry. The peak amplitudes of the outward current and the tail current increased and reached a plateau with prolonged Ca2+ channel activation (Figure 4F,G). We further measured the reversal potential of the tail current and found that it corresponds to the Cl- equilibrium potential (Figure 4H,I). Moreover, no obvious CaCC current is elicited in non-SOM+ neurons (Figure 4—figure supplement 1B,C,D,E). These findings support the notion that the Ca2+-sensitive outward current in SOM+ neurons is mediated by Ca2+-activated Cl- channels (CaCCs).
Finally, we recorded from brain slices of Sst-IRES-Cre;Ai14 mice lacking TMEM16B to determine whether the outward current is eliminated in Ano2 KO mice. We held the SOM+ neurons at different membrane potentials and measured the outward current (Figure 4J,K) and tail current (Figure 4J,L). In contrast to control brain slices, depolarization of SOM+ neurons lacking TMEM16B elicited a fast inward calcium current without the outward current and tail current (Figure 4M,N) (Mann Whitney test in (M, N), p<0.0001(M,N), n = 18 cells from 5 KO and n = 9 cells from four control for K, L, M and N), indicating that TMEM16B is solely responsible for CaCC in those GABAergic neurons.
TMEM16B-CaCC regulates action potential waveform of somatostatin-positive neurons in central lateral amygdala
To investigate whether TMEM16B-mediated CaCC activity regulates the physiological property of SOM+ cells, we first performed whole-cell current-clamp recordings with acute CeL slices from control Sst-IRES-Cre;Ai14 mice and Sst-IRES-Cre;Ano2-/-;Ai14 mutants. We assessed neuronal excitability by applying a series of current injections each lasting 500 ms. Whereas Ano2 KO and control littermates had comparable numbers of action potentials generated, firing frequency adaptation, threshold, spike amplitude, after hyperpolarization (AHP), resting membrane potential (RMP), and passive membrane properties (Figure 5A,B,C,G; Figure 5—figure supplement 1), there was a significant increase in the half width, half width adaptation, and the width of the action potential at 90% from the peak (APD90%) in Ano2 KO mice as compared with control littermates (Figure 5A,D,E,F) (Mann Whitney u test in (D, E, F), p=0.0145 (D), p=0.0091 (E), p=0.0003 (F), n = 13 cells for KO and 14 cells for control, from three mice each).
Figure 5 with 1 supplement see all
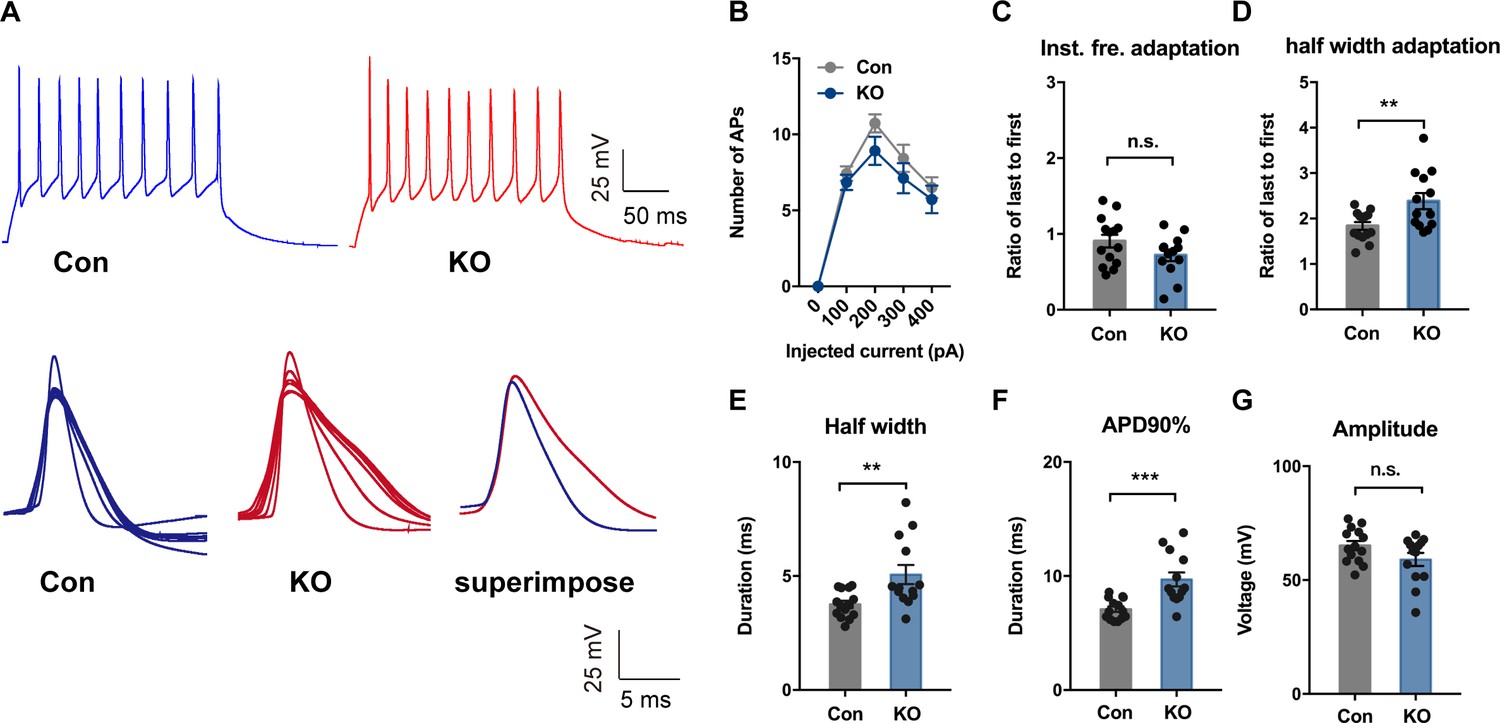
TMEM16B-CaCC Regulates Action Potential Waveform of Somatostatin-positive Neurons in the Central Lateral Amygdala.
(A) upper panel, representative traces of action potentials in SOM+ neurons from control and Ano2 KO mice in response to 100 pA current pulse lasting for 500 ms. Lower panel, overlay of action potentials induced by the current pulse of neurons with (left) or without (middle) Ano2, with superimposition of the averaged spikes from neurons with or without Ano2 shown on the right. (B) Number of spikes induced by step current injection in SOM+ neurons from control and Ano2 KO mice. Two-way ANOVA in (B), F (1, 49)=1.804, p=0.1855. n = 25 cells for KO and 26 cells for control, from seven mice each. (C) Ratio of the last to the first instantaneous firing frequency, from control and Ano2 KO brain slices. Two tail Unpaired t test, t = 1.635, df = 25, p=0.1146. n = 13 cells for KO and 14 cells for control, from three mice each. (D) Ratio of the last to the first half width, from control and Ano2 KO brain slices. Mann Whitney u test in (D), U = 41, p=0.0145. n = 13 cells for KO and 14 cells for control, from three mice each. (E) Half width of action potentials in SOM+ neurons with or without Ano2. Mann Whitney test, U = 38, p=0.0091. n = 13 cells for KO and 14 cells for control, from three mice each. (F) The averaged action potential duration at 90% repolarization (APD90%) in SOM+ neurons from control and Ano2 KO mice. Mann Whitney u test in (F), U = 21, p=0.0003. n = 13 cells for KO and 14 cells for control, from three mice each. (G) Average amplitude of action potential in SOM+ neurons with or without Ano2. Two tail Unpaired t test, t = 1.771, df = 25, p=0.0888. n = 13 cells for KO and 14 cells for control, from three mice each. Data are presented as mean ± SEM. *p<0.05, **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference.
Altered inhibitory neurotransmission in central lateral amygdala of Ano2 mutant mice
Abnormalities in GABAergic neurotransmission are associated with the neurobiology of anxiety (Makkar et al., 2010; Sanders and Shekhar, 1995). Central to the circuits of fear and anxiety, local inhibition in central lateral amygdala (CeL) plays a crucial role in the processing of emotions (Ciocchi et al., 2010; Tye et al., 2011). To date, however, little is known about the molecular mechanisms of inhibitory synapses within CeL that may contribute to the pathophysiology of anxiety disorders. As TMEM16B-CaCC is largely present in SOM+ GABAergic inhibitory neurons in CeL, we wondered whether TMEM16B-CaCC may affect GABAergic neurotransmission in CeL, which is involved in fear and anxiety-related behaviors (Janak and Tye, 2015).
Previous studies employing optogenetics have shown that CeL SOM+ and SOM- neurons mutually inhibit one another (Li et al., 2013). Likewise, CeL PKC-δ+ and PKC-δ- neurons innervate one another (Haubensak et al., 2010). In addition, CeL neurons are highly interconnected, and the majority of common local synaptic connections are between SOM+ neurons, as revealed with paired recording (Hunt et al., 2017). Whereas CeL PKC-δ+ interneurons project to CeM (Haubensak et al., 2010), CeL SOM+ neurons provide potent inhibition within CeL rather than direct inhibition of CeM neurons (Li et al., 2013). To determine whether inhibitory transmission is altered in Ano2 KO mice, we measured the spontaneously occurring action potential dependent inhibitory postsynaptic currents (sIPSCs) and action potential independent miniature inhibitory postsynaptic currents (mIPSCs) of SOM+ cells in CeL slices from Ano2 KO mice and control littermates. Given that the waveform of action potential controls synaptic efficacy in pyramidal neurons (Kole et al., 2007), it is possible that the action potential broadening in Ano2 KO mice could lead to an increase in transmitter release. Indeed, loss of TMEM16B function led to an increase of sIPSC amplitude and frequency (Figure 6) (Mann Whitney test in (B), p=0.0004 for amplitude; p=0.0021 for frequency. n = 31 cells for KO and n = 29). As both the amplitude and frequency of sIPSC were increased in KO neurons, we considered two possible underlying mechanisms. First, the effect may reflect increased presynaptic transmitter release. Second, It may include both increased presynaptic transmitter release and alteration of the postsynaptic response. To distinguish between these two possibilities, we evaluated the GABA receptor function in control and Ano2 KO SOM+ neurons, by puffing 100 μm GABA (50 ms, 4–6 psi) to generate evoked inhibitory postsynaptic currents (eIPSC). This eIPSC can be eliminated by applying PTX in the bath solution along with GABA puffing (Figure 6—figure supplement 1A). The amplitude of eIPSC was comparable between control and TMEM16B knockout SOM+ neurons (Figure 6—figure supplement 1B) (Unpaired t test, p=0.2628. n = 9 cells for each genotype from three mice each), indicating that TMEM16B activity does not alter GABA receptor function. Our findings thus support the notion that the potentiation of sIPSC could have arisen from the broadening of action potentials in presynaptic neurons lacking TMEM16B. These studies reveal that the inhibitory neurotransmission is abnormal in the CeL of Ano2 KO mice, as evident from the potentiation of the sIPSC due to spontaneous network activities of the CeL.
Figure 6 with 1 supplement see all
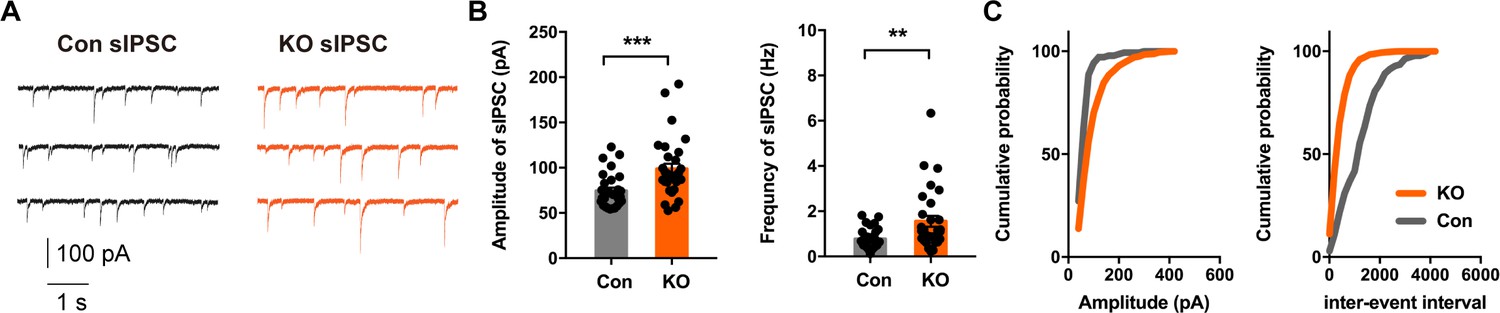
Altered Inhibitory Neurotransmission in the Central Lateral Amygdala of Ano2 KO mice.
(A) Representative traces of sIPSCs from SOM+ neurons of Ano2 KO and control mice. (B) The SOM+ neurons of Ano2 KO mice showed increased sIPSC frequency and sIPSC amplitude. Mann Whitney test in (B), U = 215, p=0.0004 for amplitude of sIPSC. U = 245, p=0.0021 for frequency of sIPSC. n = 31 cells for KO and n = 29 cells for control, from 10 mice each. (C) Representative cumulative probability plot of sIPSC frequency and amplitude of SOM+ neurons in CeL from Ano2 KO and control mice. Data are presented as mean ± SEM. *p<0.05, **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference.
Voltage-gated Ca2+ channels are involved in the presynaptic regulation of spontaneous transmitter release by TMEM16B-CaCC
Not only do voltage-gated Ca2+ channels mediate Ca2+ influx to trigger transmitter release that is evoked by an action potential, Ca2+ channels have also been found to be physiological triggers for spontaneous transmitter release at mouse inhibitory synapses (Williams et al., 2012). By blocking action potential generation with the Na+ channel blocker TTX, we examined spontaneous release of GABA as revealed by mIPSC recordings. Whereas the amplitude of mIPSC appeared normal in CeL slices of Ano2 KO mice, the frequency of mIPSC was significantly reduced (Figure 7A,B) (Mann Whitney test in (B, left panel), p = 0.0016; unpaired t test in (B, right panel), p = 0.2283, n = 16 cells for KO and n = 15 cells for control, from 5 mice each). As revealed by the plot of the cumulative probability of inter-event interval and mIPSC amplitude in Figure 7C, the frequency of spontaneous transmitter release was reduced in the Ano2 KO mice. Consistent with a previous study (Williams et al., 2012), application of Cd2+ (200 μM), a Ca2+ channel blocker, reduced the mIPSC frequency by 54.74% ± 4.032% without affecting the mIPSC amplitude (Figure 7D-H), thus implicating Ca2+ channels in spontaneous GABA release in central lateral amygdala. On average, about 46% of the mIPSCs were Cd2+ resistant, suggesting that other mechanisms also contribute to the regulation of spontaneous GABA release. CdCl2 had a much smaller effect on the mIPSC frequency in KO mice (22.52% ± 6.326%, Figure 7F, H), while the frequency of CdCl2 insensitive mIPSC was comparable between control and KO (Figure 7F). These findings provide evidence for TMEM16B function in the nerve terminal, including regulation of spontaneous GABA release that depends on Ca2+ channel activity.
Figure 7
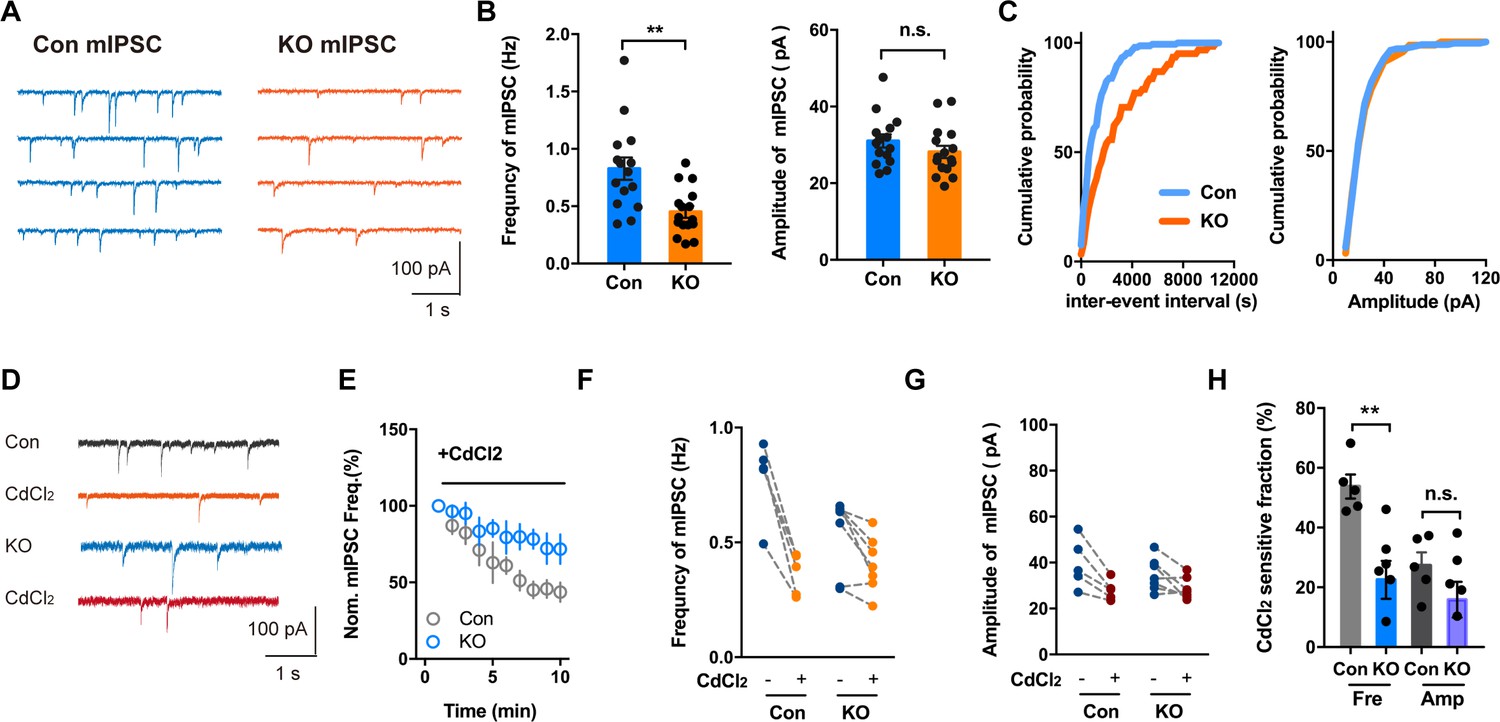
Voltage-activated Ca2+ Channel Involvement in mIPSC in Brain Slices With or Without Ano2.
(A) Representative traces of mIPSCs from SOM+ neurons of Ano2 KO and control mice. (B) The SOM+ neurons of Ano2 KO mice showed a decrease of mIPSC frequency, and normal mIPSC amplitude. Mann Whitney test in (B, left panel), U = 42.5, p=0.0016 for frequency of mIPSC. Two tail unpaired t test in (B, right panel), t = 1.231, df = 29, p=0.2283 for amplitude of mIPSC. n = 16 cells for KO and n = 15 cells for control, from five mice each. (C) Representative cumulative probability plot of mIPSC frequency and amplitude of SOM+ neurons in CeL from Ano2 KO and control mice. (D) Representative traces of mIPSCs from Ano2 KO and control brain slices in basal solution with or without Cd2+. (E) Cd2+ treatment reduced the mIPSC frequency of SOM+ neurons in the central lateral amygdala from KO mice and control littermates, as shown in the plot of normalized mIPSC frequency as function of time. Data are normalized to the baseline of mIPSC recorded in the first 3 min before Cd2+ application. (F) The effects of Cd2+ treatment on the mIPSC frequency in SOM+ neurons from control and KO mice. (G) The effects of Cd2+ treatment on the mIPSC amplitude in SOM+ neurons from control and KO mice. (H) The Cd2+-sensitive fraction of mIPSCs in SOM+ neurons from central lateral amygdala of KO mice and control littermates. Two tail Unpaired t test, t = 3.765, df = 10, p=0.0037 for Cd2+ sensitive mIPSC frequency. t = 1.424, df = 10, p=0.1848 for Cd2+ sensitive mIPSC amplitude. n = 7 cells from KO and n = 5 cells from control, three mice each. Data are presented as mean ± SEM. *p<0.05, **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference.
Discussion
In this study, we report that loss of TMEM16B (Ano2) function leads to behavioral abnormalities, including impaired anxiety-related behaviors and context-independent auditory fear expression. At the cellular level, TMEM16B constitutes a previously unknown Ca2+-activated Cl- channel in CeL GABAergic neurons with somatostatin expression. In addition, we found that SOM+ neurons in central lateral amygdala of Ano2 mutant mice have broadened action potential. The lengthening of action potential duration is associated with potentiation of the action potential evoked inhibitory postsynaptic current (sIPSC). Our finding that ablation of TMEM16B also causes a decrease of the mIPSC frequency provides further evidence for the functional significance of TMEM16B in the presynaptic nerve terminal. Intriguingly, we found that TMEM16B regulates miniature inhibitory postsynaptic current (mIPSC) by modulating the frequency of spontaneous GABA release that depends on voltage-gated Ca2+ channel activity. In summary, our results provide evidence for a critical role of TMEM16B in the regulation of SOM+ GABAergic neuronal signaling and inhibitory transmission in central lateral amygdala, as well as the involvement of TMEM16B in fear and anxiety-like behaviors.
Otowa and colleagues conducted GWAS studies of panic disorder (PD) in the Japanese population and found seven SNPs that are significantly associated with PD, which are located in or adjacent to genes such as PKP1, PLEKHG1, TMEM16B, CALCOCO1, SDK2 and CLU (Otowa et al., 2009). However, two follow up studies from the same group failed to show any significant association between SNPs in TMEM16B and PD (Otowa et al., 2012; Otowa et al., 2010). Our finding that loss of TMEM16B function results in a reduction of context-independent fear memory and anxiety-related behaviors in mice allows us to look into the physiological functions of TMEM16B of potential relevance to anxiety and panic disorders.
TMEM16B expression is mainly confined to the nervous system, including the olfactory neurons (Billig et al., 2011; Pietra et al., 2016), retina (Mercer et al., 2011; Stöhr et al., 2009), hippocampus (Huang et al., 2012), lateral septum (Wang et al., 2019), and inferior olive neurons (Zhang et al., 2017). In the hippocampus, lateral septum and inferior olive, TMEM16B modulates action potentials and neuronal excitability. In the photoreceptor terminals of mouse retina, a region where TMEM16B forms a complex with the anchor protein PSD95, TMEM16B may regulate glutamate release. Our study focused on the physiological functions of TMEM16B in the amygdala, the site for the formation and storage of fear memories crucial for anxiety (Davis, 1992; LeDoux et al., 1988). Alterations in the structure (Hayano et al., 2009) and activity (Pfleiderer et al., 2007; Shin and Liberzon, 2010) of the amygdala have been reported in patients with anxiety and panic disorder (PD), indicating that the amygdala has a pivotal role in the pathogenesis of these diseases. Our finding that Ano2 KO mice displayed impaired auditory fear expression, raises the intriguing possibility that TMEM16B expressing neurons in the amygdala contribute to the regulation of fear and anxiety-related behaviors. It remains possible that TMEM16B-CaCC in other brain areas may also contribute to fear and anxiety-like behaviors. In fact, ventral hippocampus is involved in defensive fear related behavior which is revealed by elevated plus maze (Kjelstrup et al., 2002). It would be of interest to conduct a more targeted knockout approach and test how removal Ano2 from SOM+ neurons of CeL affects anxiety-related behaviors. Ablation and pharmacological experiments implicate both hippocampus and amygdala in context-dependent and context-independent fear conditioning (Chen et al., 2016; Hall et al., 2001; Kochli et al., 2015; Vazdarjanova and McGaugh, 1999; Weeden et al., 2015; Xu et al., 2016; Yin et al., 2002). Intriguingly, loss of TMEM16B function affected context-independent but not context-dependent fear memory, indicating that different forms of fear memory may involve circuitry with different molecular features of neuronal regulation.
GABA agonists and antagonists can modulate fear (Makkar et al., 2010) and anxiety-like behaviors (Barbalho et al., 2009; Sanders and Shekhar, 1995). Central lateral amygdala has emerged as a primary site for the diazepam anxiolytic action (Griessner et al., 2018). The diazepam-binding GABAA-receptor subunits (Olsen and Sieghart, 2009) are expressed at higher levels in SOM+ neurons than in SOM- neurons. In addition, application of diazepam increases both the frequency and the size of spontaneous inhibitory postsynaptic currents (sIPSCs) of SOM+/PKCδ− neurons but not SOM−/PKCδ+ neurons (Griessner et al., 2018). Moreover, we found that elimination of TMEM16B-CaCC resulted in prolonged action potentials and increased inhibitory postsynaptic current in SOM+ neurons of the central lateral amygdala, reminiscent of the effect of diazepam on SOM+ neurons. This alteration in neuronal signaling may alter the inhibitory tone in the central amygdala (CeA), thereby affecting disinhibition of CeA output neurons that are involved in anxiety and conditioned fear memory in Ano2 KO mice. For example, increased synaptic inhibition of SOM+ CeL neurons could dampen the excitatory input from BLA to SOM+ neurons that drives fear expression (Figure 8) (Janak and Tye, 2015).
Figure 8
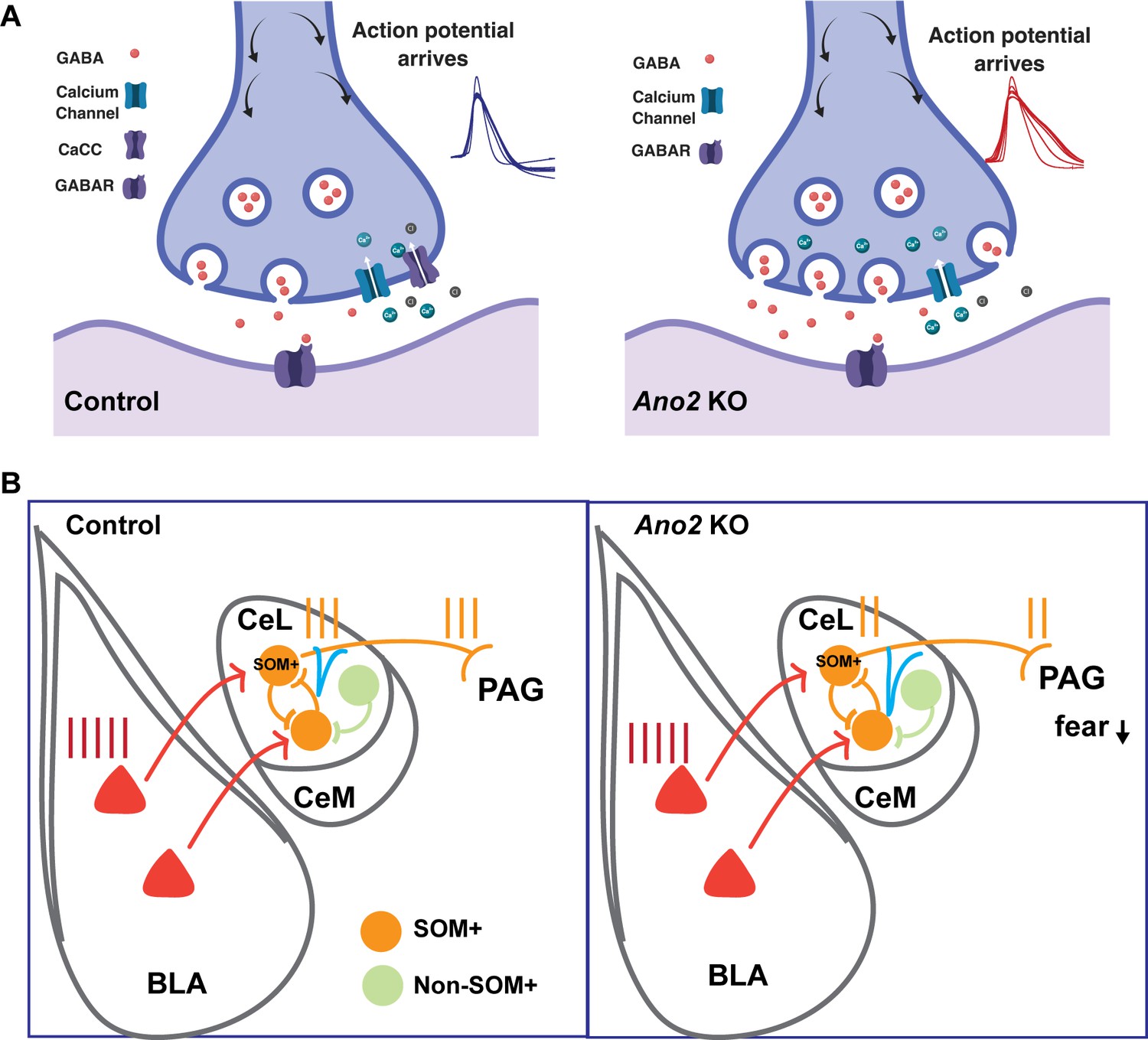
Mechanistic model for how presynaptic loss of function of TMEM16B, a Ca2+-activated Cl- channel, in central lateral amygdala (CeL) SOM+ GABAergic neurons mediates the electrophysiological and behavioral phenotypes.
(A) Loss of TMEM16B mediated CaCC in SOM+ CeL neurons results in broadening of action potential, thereby increasing presynaptic calcium channel activity and GABA release evoked by spike firing. (B) Increased inhibition tone onto SOM+ CeL neurons could contribute to the reduction of fear and anxiety-related behaviors in Ano2 KO mice, by reducing the extent that SOM+ CeL neurons are excited by neurons in the basolateral complex of the amygdala (BLA) as well as reducing the extent that SOM+ CeL neurons exert inhibition of the periaqueductal gray (PAG).
SOM+ CeL neurons receive excitatory inputs from BLA neurons that display learning-related plasticity during fear conditioning (Li et al., 2013). These SOM+ neurons project to the periaqueductal gray (PAG) and the paraventricular nucleus of the thalamus (PVT) and form part of the circuit for fear (Janak and Tye, 2015; Penzo et al., 2014). TMEM16B-CaCC modulates inhibition of SOM+ CeL neurons, as evident from the increased sIPSCs (Figure 6) associated with action potential broadening that is caused by loss of TMEM16B function (Figure 8A). Increased inhibition of SOM+ CeL neurons in Ano2 KO mice could reduce the extent of their excitation by BLA neurons as well as their ability to inhibit downstream neurons in PAG, thus resulting in a reduction of fear and anxiety-related behaviors (Figure 8B). This scenario is in accordance with the reduced auditory fear expression and anxiety-like behaviors of Ano2 KO mice. The correlation between alteration in the SOM+ neuronal activity and GABAergic transmission and the impaired fear and anxiety-like behaviors in Ano2 KO mice is consistent with the notion that inhibitory transmission in CeL plays a key role in the modulation of fear expression and anxiety-like behaviors.
CaCC can be activated by high threshold Cav channel in inferior olive neurons (Zhang et al., 2017). In our case, SOM+ neurons exhibited CaCC when the membrane potential was depolarized beyond −10 mV (Figure 4J,K,L), indicating that TMEM16B-CaCC activation is triggered by Ca2+ influx through dendritic high-threshold CaV channels. The R type Ca2+ channel is a major source of dendritic Ca2+ influx in response to action potentials (Magee and Johnston, 1995; Sabatini and Svoboda, 2000). Interestingly, the R type Ca2+ channel is highly enriched in central amygdala neurons (Zhang et al., 1993), and mice without R type Ca2+ channels have deficits in fear response (Lee et al., 2002).
In summary, we found that Ano2, is expressed in somatostatin-positive GABAergic neurons of central lateral amygdala. TMEM16B-CaCC regulates action potential waveform and GABAergic neurotransmission in these neurons as well as fear and anxiety-like behaviors; the physiological function of TMEM16B is important for expression of anxiety and context-independent fear memory. Better understanding of TMEM16B-CaCC regulation of neuronal signaling could lead to novel strategies for treating anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD).
Materials and methods
Animals
Adult mice aged 12–16 weeks were used for behavioral experiments. Ano2 knockout mice and control littermates were bred in the on a C57BL6 background as described previously (Zhang et al., 2017). To assist electrophysiological recording of somatostatin interneurons, we used the Sst-IRES-Cre; Ai14 mice expressing Tdtomato in Somatostatin interneurons to fluorescently mark somatostatin expressing cells. Ai14 mice were crossed with Ano2+/- mice to generate Ano2+/-; Ai14 mice, and further crosses with Sst-IRES-Cre; Ano2+/-; Ai14 mice resulted in fluorescently marking SOM+ cells in Ano2 KO mice and control mice for slice recording. Mice aged 16–21 days were used for brain slice recording. All mice were socially housed four per cage under a 12 hr light/dark cycle with food and water ad libitum. The use and care of the mice complied with the guidelines of the Institutional Animal Care and Use Committee of UCSF (IACUC protocol AN181236), in accordance with the US National Institute of Health guidelines. Two cohorts were used in the behavioral assays. The first cohort was tested with open field, elevated plus maze, fear conditioning and hot plate. The second cohort was tested with light/dark box and pre-pulse inhibition. There is at least one week between consecutive assays, starting from assays with weak stimulation and followed with tests with progressively stronger stimulation.
Behavioral assays
Open Field Test
Request a detailed protocolMice are placed in the center area of open field chamber (16 in x 16 in) with dim light in the dark phase for 10 min. Mice trajectory was recorded with photobeam assays (San Diego Instrument, San Diego, CA). Center area is a 8 in x eight in square in the center part of the chamber. Anxiety-related behavior was defined by ratio of traveled in the center area to total area.
Elevated Plus Maze
Request a detailed protocolThe elevated plus maze consists of two open and two enclosed arms elevated 63 cm above the ground (Hamilton-Kinder, Poway, CA). Mice are placed at the intersection between the open and closed arms and allowed to explore for 10 min. Anxiety-related behavior was measured by the time and distance traveled in the open arm.
Light/Dark Box
Request a detailed protocolA dark box with a small opening in the middle is inserted into the open field such that it divides the chamber into two equal portions, a light side and a dark side. Mice are placed inside the dark box in plastic chambers and allowed to explore for 10–30 min (Cryan and Holmes, 2005).
Fear conditioning
Request a detailed protocolTraining
Request a detailed protocolMice are placed in the fear conditioning apparatus with light set at 3, fan on, trays sprayed with Windex, and even-barred metal floor grid exposed. Test starts with a 5 min baseline period to access baseline freezing level. Then a combination of a 30 s 80 dB tone that co-terminates with a 2 s, 0.45 mA foot shock is presented three times, separated by 120 s intervals during which freezing is monitored.
Context Test
Request a detailed protocolThe Context Test takes place 24 hr after Training. Setup is the exactly the same as for training. Mice are placed in the fear conditioning apparatus for 8 min. No shock is presented. Freezing is monitored.
Cued Fear Test
Request a detailed protocolThe Cued Fear Test takes place 5–24 hr after the Context Test. The context is altered by changing floor grid, turning the fan off and spraying with Simple Green. Mice are placed in the fear conditioning apparatus for 12 min. After a 5 min baseline period four tones are delivered as described in Training, separated by 120 s intervals during which freezing is monitored. No shock is presented (Kopec et al., 2007).
Pre-pulse inhibition
Habituation
Request a detailed protocolMice are given 5 min to acclimate to the restraining chamber and the 64 dB background noise. After 5 min, mice are introduced to a series of acoustic pulses lasting for 20 min in which weaker acoustic signal (pre-pulse) is ahead of some pulses at random intervals. Trials with no auditory stimulus are also included to provide a measure of baseline activity. The test is comprised of a total of 80 trials (twenty-four 40 ms/120 dB trials, fourteen each of the 4dbpp/40 ms/120 dB, 15dbpp/40 ms/120 dB, 26dbpp/40 ms/120 dB, and no stim trials). The inter-stimulus interval (ISI) is variable with a mean of 15 s, and a range of 8–22 s.
Data extraction
Request a detailed protocolWe calculated the mean startle amplitude for each trial type: 40 ms/120 dB, 4dbpp/40 ms/120 dB, 15dbpp/40 ms/120 dB, 26dbpp/40 ms/120 dB, and no stimulus across the entire session. To calculate the % pre-pulse inhibition, we used this formula: 100 * ((mean of 40 ms/120 dB trials-mean of pre-pulse trial type)/(mean of 40 ms/120 dB)) (Vitucci et al., 2016).
In situ hybridization
Request a detailed protocolMice were euthanized with isoflurane and mice brains were removed without perfusion. The specimen was frozen on dry ice or in liquid Nitrogen within 5 min of tissue harvest. Frozen Brain was embedded in cryo-embedding medium (OCT), and then sectioned with a Cryostat. Sections were fixed with chilled 4% PFA for 15 min, and then washed with 0.1 M PBS twice. Sections were then dehydrated with 50% ethanol, 70% ethanol, and 100% ethanol sequentially. Slides were treated with protease and then incubated with customized probe for 2 hr. Signal was amplified by using the ACD RNAscope Fluorescent Multiplex Kit. RNAscope Probe-Mm-Ano2 (Cat No. 441951), Probe-Mm-Gad2-C3 (Cat No. 439371-C3), Probe-Mm-Sst (Cat No. 404631-C2). Image is analyzed by Fuji.
Slice preparation
Request a detailed protocolSlices (300 µm) were prepared as previously described (Li et al., 2012). In brief, brain slices from P16-P20 were sliced with a Vibroslice (Leica VT 1000S) in ice-cold artificial cerebrospinal fluid (ACSF), which consisted of (in mM) 125 NaCl, 3 KCl, 1.25 NaH2PO4, 1.3 MgCl2, 2.6 CaCl2, 25 NaHCO3 and 10 glucose. Slices were recovered for ~30 min at 33°C, incubated in ACSF for ~60 min at 22°C, then transferred to the recording chamber and perfused (3 ml min−1) with ACSF at 22°C. All external solutions were saturated with 95% O2, 5% CO2.
Electrophysiology
Request a detailed protocolNeurons were visualized with a 60x water-immersion lens (Zeiss) and recorded using whole-cell techniques (MultiClamp 700B Amplifier, Digidata 1322A analog-to-digital converter) and pClamp 9.2 software (Axon Instruments/Molecular Devices). The solution for action potential recording containing (in mM): 125 potassium gluconate, 15 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 disodium phosphocreatine and 0.2 EGTA, pH 7.2 with KOH, 288 mOsm. Voltage threshold was defined as dV/dt = 10 mV ms−1. Spike amplitude was measured as the voltage difference between the peak amplitude and the threshold of the action potential. After afterhyperpolarization potential (AHP) was measured as the difference between the spike threshold and the minimum voltage after the action potential peak. Spike half width was measured at half the spike amplitude. APD90% was defined as width of the action potential repolarization to 90%. All analysis was performed using pClamp/Clampfit.
To measure the calcium activated chloride currents in voltage clamp, the patch pipette (3–4 MΩ) was filled with a solution (Zhang et al., 2017) containing (in mM): 110 Cs-methanesulphonate, 20 tetraethylammonium chloride (TEACl), 8 KCl, 10 HEPES, 4 MgATP, 0.3 NaGTP, 0.2 EGTA, 5 QX-314-Br, pH 7.25 with CsOH, 290 mOsm. For pharmacological experiments, 200 μM Cd2+ or 100 μM niflumic acid (NFA) was applied to ACSF. The holding potential for CaCC recordings was −80 mV.
To record evoked postsynaptic currents, the patch pipette (3–4 MΩ) was filled with a solution same as the one for CaCC recording. The puff micropipette (4–5 MΩ) was placed 20–40 μm away from the neuron recorded. A low gas pressure (4–6 psi) was applied to the puff micropipette containing GABA by Picrospritzer. 100 μM GABA is dissolved in ACSF freshly. The holding potential for eIPSC recording is 0 mV.
To record GABAergic postsynaptic currents in voltage clamp, a high chloride internal solution (in mM) was used: 110 CsCl, 30 Potassium gluconate, 10 HEPES, 2 MgCl2, 4 Mg-ATP, 0.3 Na-GTP 10 disodium phosphocreatine and 0.2 EGTA, pH 7.2 with KOH, 288 mOsm. IPSCs were recorded with 50 µM DL-AP5 and 20 µM DNQX in ACSF. mIPSCs were recorded in the presence of 50 µM DL-AP5, 20 µM DNQX and 1 µM TTX. The holding potential for both action potential and IPSCs recordings was −70 mV. The threshold amplitude for mIPSCs is 15 pA. As most mIPSCs are less than 40 pA in amplitude, we analyzed all the events in sIPSC recordings that are greater than 40 pA, hence unlikely to arise from spontaneous GABA release. During recording, access resistance was compensated by up to 80%. Neurons with series resistance above 20 MΩ and >20% of changes throughout the recording were discarded, otherwise were used for analysis. Bridge balance in current clamp and whole-cell compensation in voltage clamp were adjusted throughout the recording. Recordings were sampled at 100 kHz for action potentials, sampled at 10 kHz for IPSCs and CaCC recordings and filtered at 3 kHz. IPSCs were analyzed using Mini Analysis Program (Synaptosoft).
Quantification and statistical analysis
Request a detailed protocolAll data are represented as mean ± SEM. All statistical analyses were performed with Prism (GraphPad) with appropriate method which indicated in the figure legend. Paired or unpaired t tests and Mann Whitney tests were used for two group comparisons. Two-way repeated-measures ANOVA was used to perform group comparisons. Data are considered to be statistically significant if p<0.05. Model illustration is created by https://biorender.com/.
Reagents
Pharmacological drugs used included 100 μM Picrotoxin (Tocris, Cat. No. 1128/1G), 50 µM DL-AP5 (Tocris, Cat. No. 0105/10), 20 µM DNQX (Tocris, Cat. No. 2312/10), Tetrodotoxin (abcam, ab120054), QX-314 (Tocris, Cat. No. 1014/100), Cadmium chloride (Sigma Aldrich, 10108-64-2), Niflumic acid (Sigma Aldrich, 4394-00-7) and 100 μM GABA (Sigma Aldrich, A2129).
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Similar anxiolytic-like effects following intra-amygdala infusions of benzodiazepine receptor agonist and antagonist: evidence for the release of an endogenous benzodiazepine inverse agonist in mice exposed to elevated plus-maze testBrain Research 1267:65–76.https://doi.org/10.1016/j.brainres.2009.02.042
-
Ca2+-activated cl− currents are dispensable for olfactionNature Neuroscience 14:763–769.https://doi.org/10.1038/nn.2821
-
Morphology of peptide-immunoreactive neurons in the rat central nucleus of the amygdalaThe Journal of Comparative Neurology 281:320–333.https://doi.org/10.1002/cne.902810212
-
The ascent of mouse: advances in modelling human depression and anxietyNature Reviews Drug Discovery 4:775–790.https://doi.org/10.1038/nrd1825
-
The role of the amygdala in fear and anxietyAnnual Review of Neuroscience 15:353–375.https://doi.org/10.1146/annurev.ne.15.030192.002033
-
Neural systems involved in fear and anxiety measured with fear-potentiated startleAmerican Psychologist 61:741–756.https://doi.org/10.1037/0003-066X.61.8.741
-
Cellular imaging of zif268 expression in the Hippocampus and amygdala during contextual and cued fear memory retrieval: selective activation of hippocampal CA1 neurons during the recall of contextual memoriesThe Journal of Neuroscience 21:2186–2193.https://doi.org/10.1523/JNEUROSCI.21-06-02186.2001
-
Smaller amygdala is associated with anxiety in patients with panic disorderPsychiatry and Clinical Neurosciences 63:266–276.https://doi.org/10.1111/j.1440-1819.2009.01960.x
-
Intrinsic circuits in the lateral central amygdalaEneuro 4:ENEURO.0367-16.2017.https://doi.org/10.1523/ENEURO.0367-16.2017
-
Experience-dependent modification of a central amygdala fear circuitNature Neuroscience 16:332–339.https://doi.org/10.1038/nn.3322
-
Prepulse inhibition deficits in patients with panic disorderDepression and Anxiety 15:55–60.https://doi.org/10.1002/da.10026
-
Neurobiology of pavlovian fear conditioningAnnual Review of Neuroscience 24:897–931.https://doi.org/10.1146/annurev.neuro.24.1.897
-
Pavlovian fear conditioning as a behavioral assay for Hippocampus and amygdala function: cautions and caveatsEuropean Journal of Neuroscience 28:1661–1666.https://doi.org/10.1111/j.1460-9568.2008.06485.x
-
Lateral mobility of presynaptic L-type calcium channels at photoreceptor ribbon synapsesJournal of Neuroscience 31:4397–4406.https://doi.org/10.1523/JNEUROSCI.5921-10.2011
-
Genome-wide association study of panic disorder in the japanese populationJournal of Human Genetics 54:122–126.https://doi.org/10.1038/jhg.2008.17
-
Replication of a genome-wide association study of panic disorder in a japanese populationJournal of Human Genetics 55:91–96.https://doi.org/10.1038/jhg.2009.127
-
New vistas on amygdala networks in conditioned fearJournal of Neurophysiology 92:1–9.https://doi.org/10.1152/jn.00153.2004
-
fMRI amygdala activation during a spontaneous panic attack in a patient with panic disorderThe World Journal of Biological Psychiatry 8:269–272.https://doi.org/10.1080/15622970701216673
-
The Ca2+-activated cl- channel TMEM16B regulates action potential firing and axonal targeting in olfactory sensory neuronsThe Journal of General Physiology 148:293–311.https://doi.org/10.1085/jgp.201611622
-
Neural mechanisms of extinction learning and retrievalNeuropsychopharmacology 33:56–72.https://doi.org/10.1038/sj.npp.1301555
-
Regulation of anxiety by GABAA receptors in the rat amygdalaPharmacology Biochemistry and Behavior 52:701–706.https://doi.org/10.1016/0091-3057(95)00153-N
-
The neurocircuitry of fear, stress, and anxiety disordersNeuropsychopharmacology 35:169–191.https://doi.org/10.1038/npp.2009.83
-
Basolateral amygdala is involved in modulating consolidation of memory for classical fear conditioningThe Journal of Neuroscience 19:6615–6622.https://doi.org/10.1523/JNEUROSCI.19-15-06615.1999
-
Rasd2 modulates Prefronto-Striatal phenotypes in humans and 'Schizophrenia-Like Behaviors' in MiceNeuropsychopharmacology 41:916–927.https://doi.org/10.1038/npp.2015.228
-
The role of the ventral dentate gyrus in anxiety-based behaviorsNeurobiology of Learning and Memory 118:143–149.https://doi.org/10.1016/j.nlm.2014.12.002
-
Coactivation of multiple tightly coupled calcium channels triggers spontaneous release of GABANature Neuroscience 15:1195–1197.https://doi.org/10.1038/nn.3162
-
Ion Channels in Health and Disease165–197, TMEM16 Membrane Proteins in Health and Disease, Ion Channels in Health and Disease, Elsevier.
-
Kainic acid lesions disrupt fear-mediated memory processingNeurobiology of Learning and Memory 77:389–401.https://doi.org/10.1006/nlme.2001.4037
Article and author information
Author details
Funding
National Institute for Health Research (RO1 NS069229)
- Lily Yeh Jan
Eunice Kennedy Shriver National Institute of Child Health and Human Development (F32HD089639)
- Mu He
Howard Hughes Medical Institute
- Yuh Nung Jan
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank for Dr. Andrea Hasenstaub for providing Som-ires-Cre and Ai14 mice, Dr. Chin Fen Teo, Dr. Yanmeng Guo, Dr. Tun Li for stimulating discussions and critical comments. This study is supported by the NIH RO1 grant NS069229 to LYJ and the F32HD089639 to MH. YNJ and LYJ are Howard Hughes Medical Institute investigators.
Ethics
Animal experimentation: The use and care of the mice complied with the guidelines of the Institutional Animal Care and Use Committee of UCSF (IACUC protocol AN181236), in accordance with the US National Institute of Health guidelines.
Copyright
© 2019, Li et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,594
- views
-
- 313
- downloads
-
- 24
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 24
- citations for umbrella DOI https://doi.org/10.7554/eLife.47106
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
TMEM16B regulates anxiety-related behavior and GABAergic neuronal signaling in the central lateral amygdala
eLife 8:e47106.
https://doi.org/10.7554/eLife.47106




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}